

Abstract
Most of the genetic information has been lost or transferred to the nucleus during the evolution of mitochondria. Nevertheless, mitochondria have retained their own genome that is essential for oxidative phosphorylation (OXPHOS). In mammals, a gene‐dense circular mitochondrial DNA (mtDNA) of about 16.5 kb encodes 13 proteins, which constitute only 1% of the mitochondrial proteome. Mammalian mtDNA is present in thousands of copies per cell and mutations often affect only a fraction of them. Most pathogenic human mtDNA mutations are recessive and only cause OXPHOS defects if present above a certain critical threshold. However, emerging evidence strongly suggests that the proportion of mutated mtDNA copies is not the only determinant of disease but that also the absolute copy number matters. In this review, we critically discuss current knowledge of the role of mtDNA copy number regulation in various types of human diseases, including mitochondrial disorders, neurodegenerative disorders and cancer, and during ageing. We also provide an overview of new exciting therapeutic strategies to directly manipulate mtDNA to restore OXPHOS in mitochondrial diseases.
Keywords: ageing, Alzheimer’s disease, cancer, mitochondria, mitochondrial diseases, mtDNA, mtDNA copy number, neurodegenerative disorders, Parkinson’s disease, TFAM

Abbreviations
AD, Alzheimer’s disease
AICAR, 5‐aminoimidazole‐4‐carboxamide ribonucleotide
AMPK, AMP‐dependent Kinase
ANT1, ADP/ATP translocase 1
CSF, cerebrospinal fluid
DA, dopaminergic
ddPCR, droplet digital PCR
DGUOK, deoxyguanosine kinase
DSB, double‐strand break
FBXL4, F‐box leucine‐rich repeat protein 4
LHON, Leber's hereditary optic neuropathy
MDS, mitochondrial DNA depletion syndrome
MELAS, mitochondrial encephalopathy, lactic acidosis and stroke‐like episodes
MERRF, myoclonic epilepsy with ragged‐red fibres
MFN2, mitofusin 2
mtDNA, mitochondrial DNA
mTORC1, mammalian target of rapamycin complex 1
mtSSB, mitochondrial single‐stranded DNA‐binding protein
NAD, nicotinamide adenine dinucleotide
NGS, next‐generation sequencing
NR, nicotinamide riboside
OH , heavy‐strand origin of replication
OL , light‐strand origin of replication
OXPHOS, oxidative phosphorylation
PD, Parkinson’s disease
PGC1α, peroxisome proliferator‐activated receptor gamma coactivator 1 α
POLGA, DNA polymerase gamma, subunit A
POLGB, DNA polymerase gamma, subunit B
POLRMT, mitochondrial DNA‐directed RNA polymerase
PPAR, peroxisome proliferator‐activated receptor
qPCR, quantitative real‐time PCR
REs, restriction endonucleases
SB, southern blot
SIRT1, sirtuin 1
SN, substantia nigra
SUCLA2, succinate‐CoA ligase [ADP‐forming] subunit beta
SUCLG1, succinate‐CoA ligase [ADP/GDP‐forming] subunit alpha
TALEN, transcription activator‐like effector nuclease
TFAM, mitochondrial transcription factor A
TK2, thymidine kinase 2
TWNK, TWINKLE helicase
TYMP, thymidine phosphorylase
WES, whole‐exome sequencing
WGS, whole‐genome sequencing
ZFN, zinc finger nuclease
Mitochondrial DNA: genetics, packaging and maintenance
Mitochondria are double‐membrane organelles present in almost all eukaryotic cells and regulate a wide variety of cellular processes, including ATP production by oxidative phosphorylation (OXPHOS), apoptosis, β‐oxidation of fatty acids and biogenesis of iron–sulfur clusters. Mitochondria house their own genome, the mitochondrial DNA (mtDNA) and the molecular machinery responsible for its maintenance and expression, that is several hundred factors controlling replication, transcription, transcript maturation and mitochondrial translation [1]. The mammalian mtDNA is a circular double‐stranded DNA (dsDNA) molecule present in multiple copies per cell. In the mitochondrial matrix, mtDNA molecules are not naked but packaged into slightly elongated DNA–protein structures known as mitochondrial nucleoids (Fig. 1A) [2]. Superresolution microscopy techniques have revealed that mitochondrial nucleoids have a diameter of ~ 100 nm and mainly contain one single copy of mtDNA [3, 4, 5]. The most abundant structural component of nucleoids is the mitochondrial transcription factor A (TFAM) (Box 1) [6, 7]. When bound to mtDNA, the high‐mobility group (HMG) box domains of TFAM facilitate mtDNA compaction [8], thereby regulating the accessibility of the genome to the replication and transcription machineries [9].
Fig. 1.
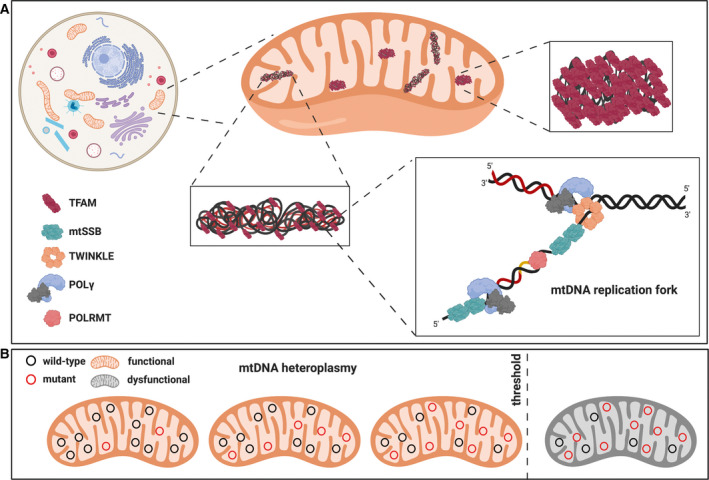
Schematic representation of mtDNA maintenance, packaging and genetics. (A) mtDNA nucleoids and the replisome. mtDNA is present in multiple copies within the cell. It is compacted by TFAM into structures known as nucleoids. Mitochondrial nucleoids can be found in a compacted or relaxed state depending on the local TFAM concentration. In the relaxed state, mtDNA is accessible for replication by the mitochondrial replisome which is formed by the mitochondrial RNA polymerase POLRMT, the hexameric DNA helicase TWINKLE, the tetrameric mtSSB and the mtDNA polymerase gamma POLγ. POLγ is a heterotrimer formed by a catalytic subunit with DNA polymerase and 3′‐5′ exonuclease activities (encoded by POLGA), and by two accessory subunits (encoded by POLGB) required for the tight DNA‐binding and processive DNA synthesis. During replication, TWINKLE unwinds and proceeds on the DNA in a 5’ to 3’ direction, and the single‐stranded DNA generated by TWINKLE activity is protected by the mtSSB binding. The RNA primers for replication initiation are generated by POLRMT. (B) The concept of heteroplasmy. Mutations affecting the mtDNA can coexist with the wild‐type molecules, a condition known as heteroplasmy. mtDNA mutations are tolerated until they exceed a certain level (threshold). Therefore, defects in the OXPHOS system will manifest only when the proportion of mutated mtDNA molecules exceeds the biochemical threshold, which is known to be tissue‐ and mutation‐specific.
Box 1. Many faces of TFAM: one protein with multiple functions.
TFAM is a highly abundant mitochondrial protein, present in about 1000 copies per mtDNA molecule or 1 TFAM molecule per 15–20 bp of mtDNA [5], and it is essential for mtDNA transcription initiation, nucleoid formation and mtDNA maintenance.
TFAM is a member of the HMG‐box protein superfamily, contains two HMG‐box domains, HMGA and HMGB, separated by a linker, and a charged C‐terminal tail [10]. The crystal structures of TFAM have shown that each of the two HMG boxes bend the DNA nearly 90° inducing U turn in the DNA minor groove and bending it towards the major groove [8, 11].
TFAM modulates the interactions with mtDNA thanks to a fine‐tuning of its DNA‐binding function. TFAM can bind, unwind and bend DNA without sequence specificity. It coats and packages individual mtDNA molecules to form mitochondrial nucleoids. In addition, TFAM can also bind DNA in a sequence‐specific manner to facilitate unwinding of the promoter regions during transcription initiation [12]. Notably, the C‐terminal tail of TFAM has been shown to prime the interaction with the transcription machinery [13], whereas the HMGB domain is responsible for the interaction with POLRMT to anchor it to the promoter [14].
TFAM is also essential for mtDNA maintenance and homozygous disruption of Tfam is embryonically lethal in the mouse [15]. Several genetic models have demonstrated a direct link between TFAM protein levels and mtDNA copy number in vitro and in vivo. Heterozygous ablation of Tfam causes ~ 50% decrease in mtDNA levels [15, 16], whereas the overexpression of Tfam results in ~ 50% increase of mtDNA copy number [16, 17, 18]. TFAM is therefore a multifunctional protein, which is necessary for mtDNA genome maintenance and expression. To date, our knowledge related to the regulation of TFAM activity in different processes remains very limited.
In mammals, mtDNA is an intronless molecule of ~ 16.5 kb containing 37 genes encoding 13 protein subunits of the OXPHOS system, two rRNAs (12S and 16S) and a complete set of 22 tRNAs for mitochondrial translation [19, 20]. Unlike the nuclear chromosomes, mtDNA replication is independent of the cell cycle and also occurs in postmitotic cells, a process often referred to as relaxed replication [21]. A minimal mtDNA replisome has been defined and consists of the hexameric helicase TWINKLE, the heterotrimeric DNA polymerase gamma (POLγ) and the tetrameric mitochondrial single‐stranded DNA‐binding protein (mtSSB), in addition to the replication primer forming mitochondrial RNA polymerase (POLRMT) [22] (Fig. 1A). During replication of the leading strand of mtDNA, TWINKLE moves in a 5′ to 3′ direction while unwinding the dsDNA [23]. The single‐stranded DNA (ssDNA) is protected through mtSSB binding, which also further stimulates the dsDNA unwinding by TWINKLE and the DNA synthesis by POLγ [23, 24]. The RNA primers required for the initiation of the synthesis of both DNA strands are produced by POLRMT [25]. It has been recognised for more than 45 years that replication of mammalian mtDNA occurs asymmetrically [26, 27, 28]. The replication process is understood in great detail, although not completely, and depends on dedicated origins on each DNA strand, the leading (heavy‐) and the lagging (light‐) strand origin of replication (OH and OL), respectively [1, 29]. According to this strand‐displacement mode, the replication of the leading strand initiates at OH and proceeds unidirectionally until about two‐thirds of the genome has been replicated. At this point, the replication of the leading strand proceeds beyond OL, which is activated to form a stem–loop structure [24, 30] that recruits POLRMT for RNA primer synthesis to initiate the replication of the lagging strand [31]. The synthesis of the two strands then proceeds continuously until two full‐length, dsDNA molecules are formed. To complete mtDNA replication, the mitochondrial genome maintenance exonuclease 1 and other nucleases help forming ligatable DNA ends and the concatenated newly replicated mtDNA molecules are released from each other by the action of topoisomerase 3α [32].
Given that mtDNA expression is a prerequisite for biogenesis of the OXPHOS system [33], mtDNA content in cells and tissues must be adjusted according to metabolic needs [34]. Therefore, mtDNA levels are tissue‐ and developmental‐stage‐specific and finely regulated by a balance between replication and turnover. Studies on human samples have revealed that mtDNA copy number per cell can vary by several orders of magnitude, ranging from ~ 1 × 105 mtDNA copies in oocytes [35], ~ 4–6 × 103 in heart to 0.5–2 × 103 in lungs, liver and kidney [36]. Consistent with its key role in packaging mtDNA into nucleoids, TFAM also serves as a key regulator of mtDNA copy number (Box 1). Heterozygous disruption of Tfam causes a decrease in mtDNA levels of ~ 50% [15, 16], whereas a moderate TFAM overexpression increases the amount of mtDNA by ~ 50–100% [16, 17, 18].
Our knowledge of mtDNA replication and maintenance has dramatically increased in recent years, but how the cellular mtDNA copy number is adjusted to and maintained at a certain level is currently not completely understood. Several models, based on ATP requirements, nucleotide availability and replication origin regulation, have been proposed to control mtDNA content [37]. In addition, genetic screens in Saccharomyces cerevisiae have been performed to identify novel regulators of mtDNA levels [38, 39]. Unfortunately, these studies have provided little new mechanistic insights as loss of mtDNA is a common secondary effect of mitochondrial dysfunction in budding yeast. Genetic studies in humans have identified a region on chromosome 10, which contains TFAM and other genes, to have a notable impact on mtDNA levels [40].
Causes and consequences of mtDNA mutations
Mutations of mtDNA can in principle be generated either by spontaneous errors during DNA replication or by incorrect repair of damaged DNA bases. In spite of being very popular, the free radical theory of ageing, which proposes that oxidative stress is the main cause for mtDNA mutations [41, 42], is much questioned. In fact, sequence analyses of mutations in a large number of mtDNA genomes and experimental studies of mice argue that the vast majority of mtDNA mutations are the result of spontaneous replication errors introduced by POLγ during mtDNA synthesis [43, 44, 45, 46, 47, 48]. The explanation for the paucity of damage‐induced mtDNA mutations may reside in the compacted nature of the mitochondrial nucleoid, which provides a protected environment that makes the mtDNA less accessible to chemical damage, including oxidation.
Pathogenic mtDNA mutations have been identified in mitochondrial tRNA, rRNA and protein‐coding genes, and they invariably compromise mitochondrial gene expression causing various degrees of OXPHOS deficiency. Due to the multicopy nature of the mitochondrial genome, mtDNA mutations may be present either in all (homoplasmy) or only in a subset of all (heteroplasmy) copies of mtDNA. Many severe mtDNA mutations cannot be tolerated in the homoplasmic state and are therefore only found in the heteroplasmic state in patients. Heteroplasmic pathogenic mtDNA mutations are typically functionally recessive [49], meaning that they cause respiratory‐chain deficiency only when present above a certain threshold level [50] (Fig. 1B). This implies that only cells containing levels of mutated mtDNA that exceeds a critical threshold will develop respiratory‐chain dysfunction, whereas adjacent cells with a lower mutation load can sustain normal respiratory‐chain function. The threshold level is highly dependent on the affected tissue and the type of mtDNA mutation present. For single large deletions of mtDNA, the threshold is ~ 50–60% [50], whereas some point mutations in tRNA genes have thresholds exceeding 90% mutated mtDNA [51, 52, 53]. During the life of an individual, the level of heteroplasmy can shift either up or down due to mitotic segregation, which is a consequence of relaxed replication and random partitioning of mitochondria between daughter cells. Furthermore, deleterious mtDNA alleles may be actively eliminated in proliferating tissues by purifying selection, which has been seen in blood for some types of mtDNA mutations [54, 55, 56].
Besides being the direct cause of primary mitochondrial diseases, typically characterised by severe mitochondrial impairment in multiple tissues [57], mtDNA mutations have also been implicated in the pathophysiology of common age‐associated human diseases [57, 58, 59, 60, 61] and in the naturally occurring ageing process [62]. It is well established that point mutations in tRNA genes or single large deletions of mtDNA lead to functional impairment or lack of one or several tRNAs. However, these types of pathogenic mtDNA mutations have no dominant effects and high levels of mutated mtDNA can therefore be tolerated. The recessive nature of most human pathogenic mtDNA mutations leads to a deficiency of wild‐type gene products, which, in turn, impairs OXPHOS and causes the disease phenotypes.
Almost 25 years ago, Attardi and colleagues hypothesised that both the fraction of wild‐type mtDNA and the mtDNA copy number play a role in the phenotypic manifestations of mtDNA mutations. The authors demonstrated a correlation between mtDNA copy number and oxygen consumption rate and also experimentally proved that low levels of wild‐type mtDNA can protect cybrid cell lines from the deleterious effects of a pathogenic mtDNA mutation in the tRNALeu gene [63]. In recent years, increasing evidence based on human correlative data and experimental mouse genetics has confirmed that high absolute levels of wild‐type mtDNA genomes indeed may counteract the consequences of pathogenic mtDNA mutations [64, 65, 66, 67].
In this review, we provide a comprehensive overview of the current knowledge on the contribution of mtDNA copy number regulation to clinical manifestations of mitochondrial disorders, neurodegenerative diseases, cancer and ageing. We critically assess available data on mtDNA copy number regulation and experimental challenges. Finally, we discuss the rationale and the applications of current therapeutic strategies to improve mitochondrial function.
mtDNA copy number in human disease
Quantification of mtDNA copy number
To understand the implication of mtDNA variations in human diseases, mtDNA copy number must be accurately assessed to avoid biases that confound interpretation of results.
Measurements of mtDNA levels can be performed with a variety of techniques. For a long time, southern blot (SB) hybridisation was used as the gold standard method to assess mtDNA levels and integrity in patient samples [68, 69, 70] and in model organisms [71, 72, 73, 74]. Despite being highly reliable in assessing mtDNA content, SB techniques are time‐consuming, only semi‐quantitative and require a relatively large amount of DNA, which combined represent a major disadvantage when studying human tissues.
Fluorescent in situ hybridisation approaches have also been employed to spatially visualise mtDNA content with single‐cell resolution but have proven to be only partially informative [75, 76, 77]. In fact, this multistep protocol can be quite laborious and it provides only a rough estimate of changes in mtDNA amount.
The most popular current standard techniques to measure mtDNA copies are PCR‐based methods. Due to its simplicity, quantitative real‐time PCR (qPCR) is the most widely used approach to assess relative or absolute mtDNA levels. However, the large majority of protocols assess the relative mtDNA copy number as a ratio between mtDNA levels and levels of a selected nuclear gene, which makes comparison between studies very difficult. It is also possible to engage qPCR‐based assays to determine the absolute mtDNA copy number by using either a standard curve based on the mtDNA region of interest inserted into a reference plasmid [78, 79] or droplet digital PCR (ddPCR). Although the limited effective range of the ddPCR method might affect precision when measuring high copy number, this technology can provide a rigorous quantification of mtDNA copies without the use of external standards [80, 81, 82].
Recent studies have shown that the quantification of mtDNA copy number can be inferred from next‐generation sequencing (NGS) data, including whole‐exome sequencing (WES) and whole‐genome sequencing (WGS) [83, 84, 85, 86]. The output from NGS datasets relies on the ratio of sequencing reads from nuclear DNA and mtDNA and allows analysis of hundreds of thousands of available data sets shared by research consortia. Although a series of normalisations are needed to correct for counts, ploidy, purity and batch biases [36, 87, 88], the NGS technology enables high‐throughput, high‐sensitivity and accurate assessment of mtDNA levels. Thus, WES/WGS approaches will likely be increasingly employed as gold standard tools to assess mtDNA content and heteroplasmy levels in the future.
mtDNA copy number in mitochondrial diseases
Mitochondrial diseases are a group of heterogenous hereditary disorders characterised by OXPHOS deficiency. These disorders can affect different cell types and organs, and can therefore cause a wide range of symptoms. Mitochondrial diseases have been classified according to their clinical manifestations and can be caused either by mutations in nuclear genes, leading to reduced mtDNA expression, or by primary mtDNA mutations directly impairing the function or abundance of mtDNA‐encoded gene products [59, 60].
mtDNA depletion syndromes (MDSs) [89] are autosomal recessive disorders characterised by a strong tissue‐specific reduction in mtDNA levels due to mutations in genes involved in various mtDNA maintenance processes, ranging from mitochondrial nucleotide metabolism, mtDNA replication, mitochondrial dynamics and quality control (Table 1). Thus, MDSs establish a causal and direct link between low mtDNA content and pathological conditions. However, the importance of mtDNA copy number in the pathophysiology of other mitochondrial diseases often remains enigmatic.
Table 1.
mtDNA copy number variation among mitochondrial diseases. Top panel: mitochondrial diseases caused by mutations in nuclear genes. n.a. not available in the original work. Bottom panel: diseases caused by primary mtDNA mutations.
| Disease | Gene (nuclear) | Sample type | mtDNA levels | Quantification method | Reference |
|---|---|---|---|---|---|
| MDS | ANT1 | Skeletal muscle | Down | qPCR | [232] |
| TK2 | Skeletal muscle | Down | SB; qPCR | [91, 233] | |
| DGUOK | Liver | Down | SB; qPCR | [91, 234] | |
| TYMP | Gastrointestinal tract | Down | qPCR | [235] | |
| MPV17 | Liver | Down | SB; qPCR | [91, 236] | |
| SUCLA2 | Skeletal muscle | Down | qPCR | [91, 237] | |
| SUCLG1 | Liver/Skeletal muscle | Down | qPCR | [91, 238] | |
| MFN2 | Skeletal muscle | Unchanged/down | n.a | [239, 240] | |
| FBXL4 | Skeletal muscle | Down | qPCR | [241, 242] | |
| POLGA | Liver/blood | Unchanged/ down | qPCR | [91, 243] | |
| POLGB | Liver/skeletal muscle/blood | Down | qPCR | [244] | |
| TWNK | Liver | Down | qPCR | [245, 246] | |
| mtSSB | Muscle/blood/kidney | Down | qPCR | [247] | |
| Disease | mtDNA mutation | Sample type | mtDNA levels | Quantification method | Reference |
| Pearson’s syndrome | Deletion | Blood | Up | qPCR | [95] |
| KSS | Deletion | Blood/muscle | Up | qPCR | [95] |
| MELAS | m.3243A>G | Leucocytes | Up/unchanged/down a | qPCR | [96] |
| MERRF | m.8344A>G | Leucocytes | Up/unchanged/down a | qPCR | [96] |
| LHON | m.11778G>A | Peripheral blood cells | Up | qPCR | [65] |
| LHON | m.11778G>A | Blood | Up/unchanged | qPCR | [97, 98] |
| LHON | m.3460G>A | Blood | Up/unchanged | qPCR | [97, 98] |
Copy number found to be increased in younger patients but unchanged or even decreased in older patients.
The POLGA gene, encoding the catalytic subunit of mitochondrial polymerase POLγ, is one of the several nuclear genes associated with MDS. However, the over 300 mutations identified so far in the POLGA gene can cause a variety of mtDNA defects leading to disparate symptoms, disease severity and age of onset of disease (https://tools.niehs.nih.gov/polg/) [90]. Patients affected by POLGA‐related disorders can manifest either severe reduction in mtDNA copy number or accumulation of mtDNA mutations and deletions with no effect on mtDNA levels [91]. Even though the crystal structure of POLγ has shed some light on how different mutations can impact enzyme activity and replication fidelity [92], the relation between genotype and phenotype is not always straightforward. The strong phenotypic variability in POLGA patients can even occur between subjects of the same family [93]. Interestingly, a mutation markedly impairing the proofreading activity of DNA polymerase gamma, subunit A (POLGA) in mice resulted in numerous mtDNA mutations, but had no impact on copy number [94].
Homoplasmic and heteroplasmic pathogenic mtDNA mutations are responsible for diverse mitochondrial syndromes, including mitochondrial encephalopathy, lactic acidosis and stroke‐like episodes (MELAS), Leber's hereditary optic neuropathy (LHON), myoclonus epilepsy with ragged‐red fibres (MERRF), Pearson's syndrome or Kearns–Sayre syndrome (KSS). mtDNA copy number has been assessed only in small cohorts of patients affected by either Pearson’s or KSS, caused by single large heteroplasmic mtDNA deletions. The results of these investigations showed that most patients presented an increased mtDNA copy number compared with the control individuals, although mtDNA levels did not correlate with the size or position of the deletion [95].
The copy number variations in patients with mtDNA point mutations have been more extensively investigated. A recent study of a large group of MELAS patients carrying the heteroplasmic m.3243A>G mutation showed that high mtDNA copy number and low heteroplasmy levels correlated with less severe disease [66]. Other studies with smaller patient cohorts reported that MELAS and MERRF patients with moderately high mtDNA copy number showed milder phenotypes [96]. Also, in LHON patients, that mostly carry homoplasmic mutations, mtDNA copy number was reported to play a role in the penetrance of the disease, as the asymptomatic carriers of the m.11778G>A or m.3460G>A mutations had higher mtDNA copy number than visually impaired patients [65, 97, 98].
The data discussed above and additional data summarised in Table 1 strongly suggest a direct relation between the absolute mtDNA copy number and mitochondrial diseases onset and progression. Particularly intriguing is the idea that an increase in the absolute mtDNA copy number could be a compensatory mechanism aimed at sustaining OXHPOS activity. This endogenous upregulation of mtDNA content might be able to efficiently overcome the bioenergetic defects caused by some mtDNA mutations. In a well‐documented mouse model harbouring a pathogenic heteroplasmic tRNA gene mutation in mtDNA [99], affected animals spontaneously upregulate mtDNA copy number in several tissues, which may delay onset of disease manifestations [64].
mtDNA levels in ageing and age‐related neurodegenerative disorders
Ageing is a biological process characterised by the slow and progressive decline of the physiological functions of an organism, which ultimately determines multimorbidity and lethality. This process includes a decay of mitochondrial function, concomitant with alterations in mitochondrial morphology [100, 101], mitochondrial content (number and protein levels) [102] and OXPHOS capacity [103]. As previously mentioned, pathogenic mtDNA mutations, both large deletions and point mutations, have been identified in postmitotic and proliferating tissues of aged individuals [62, 104, 105, 106]. There is experimental evidence suggesting that the somatic mtDNA mutations seen in ageing humans and other mammals have the capacity to cause at least some ageing phenotypes. In mice, proofreading‐deficient POLγ caused a progressive accumulation of mtDNA mutations and a premature ageing syndrome with reduced lifespan, decreased fertility, anaemia, hair greying, hair loss, hearing impairment and stem cell dysfunction [94, 107]. Interestingly, increasing evidence suggests that somatic mtDNA mutations may contribute to age‐related neurodegenerative disorders, such as Parkinson’s disease (PD) and Alzheimer’s disease (AD). The accumulation of mtDNA mutations was observed in the Substantia nigra (SN) of PD patients [108] and in frontal cortex and hippocampus of AD patients [109]. Furthermore, in these brain regions the presence of high levels of mtDNA point mutations or deletions correlated with severe mitochondrial biochemical defects, for example in complex I [110] or complex IV [111].
Correlative studies in humans with age‐associated degenerative disease and ageing have thus strongly implicated mtDNA mutations and OXPHOS dysfunction in these conditions, but a possible role for mtDNA copy number is less clear. Multiple studies have assessed the mtDNA content in tissues of individuals at different ages (Table 2). The majority of these investigations showed a reduction in mtDNA copy number in aged individuals. For instance, mtDNA levels measured in lymphocytes from over 2000 Sardinians were found modestly, but significantly, decreased with age [112]. A more pronounced reduction in mtDNA copy number was found in blood samples and was first observed in individuals in their 50s. The decline in mtDNA levels was even more dramatic in older subjects [113, 114], and a loss of a few per cent of all copies per decade has been estimated [115]. Remarkably, in the old group (> 58 years of age), low mtDNA copy number in peripheral blood was associated with high mortality and poor health, including a decline in cognitive and physical performance [114]. Studies performed in long‐lived families comprising nonagenarian and centenarian individuals have often shown puzzling and contradictory outcomes. mtDNA copy number in blood samples was reported to be clearly reduced in individuals between 50 and 70 years of age, whereas mtDNA levels were either lower [116] or higher [117] in nonagenarians and centenarians in comparison with middle‐aged controls. Therefore, these results do not clarify whether low mtDNA copy number correlates with a negative or positive impact on longevity.
Table 2.
mtDNA copy number variation in ageing and neurodegenerative disorders.
| Disease | Sample type | mtDNA levels | Quantification method | References |
|---|---|---|---|---|
| Ageing | Lymphocytes | Down | WGS | [112] |
| Blood | Down | qPCR; qPCR; WGS; qPCR; qPCR | [113, 114, 115, 116, 117] | |
| Heart | Unchanged | qPCR; SB | [118, 119] | |
| Skeletal muscle | Unchanged | qPCR; SB | [118, 119] | |
| Skeletal muscle | Down | NGS and ddPCR | [120] | |
| Liver | Up | NGS and ddPCR | [120] | |
| Caudate nucleus | Unchanged | SB | [118] | |
| Frontal lobe cortex | Unchanged | SB | [118] | |
| Cerebellar cortex | Unchanged | SB | [118] | |
| SN a | Up | qPCR | [121] | |
| PD | SN a | Down | qPCR; qPCR | [121, 122] |
| Cerebellum | Unchanged | WES | [124] | |
| Cerebellar cortex | Unchanged | WES | [124] | |
| Frontal cortex | Unchanged | qPCR | [123] | |
| SN | Down | qPCR | [123] | |
| Blood | Down | qPCR; qPCR | [123, 127] | |
| CSF | Down | qPCR | [130] | |
| AD | Frontal cortex | Down | qPCR | [109] |
| Cerebellum | Unchanged | qPCR | [125] | |
| Cerebellum | Down | WES | [124] | |
| Hippocampus | Unchanged | qPCR | [125] | |
| Hippocampus a | Down | qPCR | [126] | |
| Cerebellar cortex | Down | WES | [124] | |
| Blood | Unchanged | qPCR | [125, 128] | |
| CSF | Down | qPCR | [129] |
Analyses were performed on microdissected neurons from these specific brain regions.
A series of experiments performed on samples other than blood and lymphocytes also yielded mixed results. For instance, age‐related reduction in mtDNA levels were not observed in skeletal muscle and heart [118, 119], whereas a more recent study reported decreased mtDNA copy number in skeletal muscle and increased copy number in liver of aged subjects [120].
Significant differences in mtDNA copy number have been reported across different brain regions, and these variations were more pronounced in patients affected by neurodegenerative disorders. For instance, no alterations in mtDNA levels were identified in samples from caudate nucleus, frontal lobe and cerebellar cortex of aged individuals [118, 121]. Interestingly, microdissected dopaminergic (DA) neurons from SN of healthy aged subjects contained increased levels of mtDNA deletions and increased total mtDNA levels [121]. In contrast, DA neurons of PD patients had no increase in total mtDNA copy number despite having increased levels of deleted mtDNA [121]. It is possible that this selective depletion of functional wild‐type mtDNA molecules caused a bioenergetic deficiency in DA neurons of PD patients [121]. Other studies that analysed both total SN homogenates and microdissected DA neurons found a general reduction in mtDNA content in PD patients [122, 123]. Notably, mtDNA levels were unaltered in brain regions that were only mildly affected or not compromised in PD patients [123, 124].
Measurements of mtDNA levels were also performed in brains of subjects affected by AD. These investigations revealed that mtDNA levels were decreased by 30–50% in the frontal cortex of AD patients when compared to controls [109, 125]. This mtDNA depletion was also present in microdissected pyramidal neurons from the hippocampus and was concomitant with a disruption in mitochondrial biogenesis signalling pathways [126]. The correlation between reduction in mtDNA levels and neurodegeneration in AD has been further supported by a comprehensive in‐depth analysis of mtDNA sequence variation and abundance in over 1000 human brains [124].
The need for novel diagnostic and prognostic biomarkers for PD and AD has promoted significant research efforts to assess mtDNA copy number in peripheral blood and cerebrospinal fluid (CSF). In blood samples from PD patients, mtDNA levels were decreased when compared to healthy controls [123, 127]. Lower mtDNA copy number was more frequently observed in elderly PD subjects, suggesting that mtDNA content might even have a prognostic relevance in PD progression [127]. Different results were found in AD subjects, where mtDNA levels were unchanged [125, 128] in spite of altered mitochondrial gene expression [128]. Analyses performed in CSF, which often mirrors the pathological changes in brain metabolism, revealed a reduction in cell‐free mtDNA in both AD and PD samples, suggesting that the levels of cell‐free mtDNA may be used as biomarker for the early detection of both of these neurodegenerative diseases [129, 130].
The majority of the data discussed here suggest that the decline in mitochondrial function observed in age‐related disorders and ageing correlates with a progressive reduction in mtDNA copy number. Even if it is completely plausible that a gradual loss of functional mtDNA may have harmful effects on the brain, the results of these investigations might be deeply biased by the quality and the composition of the specimens (see further discussion below). Therefore, the possible existence of a causal relationship between mtDNA levels and age‐related disorders needs further experimental validation.
mtDNA variation in human cancer
Historically, the existence of a relationship between mitochondrial metabolism and cancer was first proposed by Otto Warburg in the early 1920s based on his observation that cancer cells can fulfil their energetic needs almost exclusively through aerobic glycolysis [131]. Warburg postulated that an impairment in mitochondrial respiration could drive tumorigenesis. This view became a milestone in cancer research but gradually faded away when the focus was shifted to oncogenes and the regulation of the cell cycle. However, during the last decades new evidence suggests important roles for mitochondria in cancer initiation and progression [132]. A recent in vivo characterisation of metabolic needs in tumours showed for instance that the conversion of pyruvate to lactate (aerobic glycolysis) was not essential for melanoma tumour growth [133]. Similarly, lung adenocarcinomas displayed high dependency on substrates for glucose oxidation and the tricarboxylic acid cycle to harvest energy [134] and were also reported to retain a high mitochondrial membrane potential [135]. In contrast, in the intestine, the ablation of the mitochondrial pyruvate carrier was sufficient to initiate tumour formation [136]. Therefore, the extent of the contribution of mitochondrial metabolism to tumour onset and progression is heterogeneous and it is likely to depend on both the cancer type and the tumour stage.
Mutations in the mtDNA can be used as a measure of mitochondrial fitness, and the accumulation of mtDNA mutations is well known to occur in multiple cancers [137, 138]. Several groups have characterised the mutational pattern of tumours by exploiting the recent high availability of NGS and WGS data [139, 140, 141, 142]. Although mutations in mtDNA seem to accumulate in virtually all cancer types tested [139, 142], the pathophysiological relevance, that is whether mtDNA mutations are causative or simply by‐products generated by rapid mtDNA replication in fast‐dividing cancer cells, is typically unclear. Mathematical models suggest that mtDNA mutations may accumulate due to the expansion of pre‐existing heteroplasmic mutations or polymorphisms and that these undergo passive clonal expansion during the multiple cell divisions in tumours [143]. There are also other arguments against a positive role for mtDNA mutations in tumour development because mtDNA mutations that truncate proteins are counterselected in the majority of tumours [142, 144, 145], with the exception of kidney, and colorectal and thyroid cancers [142]. Interestingly, in these tumours the levels of mtDNA were found upregulated, suggesting a compensatory mechanism to sustain mitochondrial function [142]. The study also assessed the mutational landscape of over 2000 cancer patients and identified a strong strand bias for mtDNA mutations, which strongly argues for a replication‐related mechanism in the generation of mutations [142]. A likely scenario for mtDNA mutations accumulation in tumours is that pre‐existing mutations or mutations created by POLγ replication errors can clonally expand to high levels in fast‐dividing cancer cells and become fixed in tumour subpopulations. Oncocytomas may represent an interesting confirmation of this hypothesis, as they are characterised by high levels of mtDNA mutations that result in a strong OXPHOS dysfunction and a compensatory increase in mitochondrial mass. Although pre‐existing mtDNA mutations may drive oncocytoma formation, these tumours are typically benign lesions, characterised by low‐invasiveness and a non‐aggressive phenotype [146, 147, 148, 149]. Mitochondrial dysfunction might represent a major boundary to cancer progression, but it is also possible that the accumulation of mtDNA mutations in some types of cancer provides a selective advantage to transformed cells and that mtDNA mutations thereby directly contribute to cancer development. Along these lines, a recent report has shown that metabolic alterations induced by mtDNA mutations may facilitate tumour formation in colon [150].
Besides the mutation load, mtDNA abundance is an important indicator of the reliance of a cell on OXPHOS. However, not many groups have looked at these two parameters at the same time and most studies only explore one of the two aspects. The levels of mtDNA have been associated with cancer, for either diagnostic or prognostic purposes. For diagnostic purposes, many attempts were directed towards finding a correlation between mtDNA copy number and disease onset, pursuing the idea that mtDNA levels in blood could be a marker for cancer risk. High mtDNA levels in blood were reported to correlate with an increased risk of developing lymphomas [151], breast [152, 153], skin [154], lung [155] and pancreatic tumours [156], whereas other studies reported a protective effect of high mtDNA copy number against bone, kidney and other cancer types [157]. The risk of developing colorectal carcinoma was associated with both higher [158] or lower [159] amount of mtDNA in peripheral blood.
The levels of mtDNA have also been assessed as a prognostic factor by comparing matched normal and tumour tissue samples in several cancer types. The data from such studies, summarised in Table 3, are highly heterogeneous, and the reports suggest that both increased and decreased mtDNA levels correlate with disease severity. Colorectal carcinoma progression was associated with both an increase [160, 161] and a decrease [162, 163, 164] in mtDNA copy number. Similar findings were reported for lung, and gastric and renal cell carcinomas, where some research groups found a correlation between high mtDNA levels and disease severity [142, 165, 166], whereas some earlier studies found the inverse relationship [163]. Data addressing multiple cancer types are not really clarifying the situation, as some tumours seem to upregulate mtDNA levels, including lymphomas, pancreatic, thyroid [142] and prostate cancers [141, 142] and renal oncocytomas [147], whereas tumours affecting the breast [166, 167, 168, 169], the brain [170], bones [171], the oral tract [166] and the liver [142, 166, 172] have been associated with a reduction in mtDNA levels.
Table 3.
mtDNA copy number variation in cancer.
| Tissue affected | Sample type | MtDNA levels | Cancer risk | Quantification method | References |
|---|---|---|---|---|---|
| Lymphocytes | Peripheral blood lymphocytes | Up | Increased risk | Meta‐analysis of literature search (qPCR) | [151] |
| Bone | Peripheral blood lymphocytes | Up | Decreased risk | Meta‐analysis of literature search (qPCR) | [151] |
| Brain (glioma) | Blood | Up | Increased risk | qPCR | [248] |
| Breast | Blood | Up | Increased risk | qPCR; qPCR | [152, 153] |
| Colon/rectum | Peripheral blood lymphocytes | Down | Increased risk | qPCR | [159] |
| Peripheral blood lymphocytes | Up; down | Increased risk | qPCR | [158] | |
| Kidney | Peripheral blood lymphocytes | Down | Increased risk | qPCR | [157] |
| Lung | Blood | Up | Increased risk | qPCR | [155] |
| Pancreas | Blood | Up | Increased risk | qPCR | [156] |
| Skin | Blood | Up | Increased risk | qPCR | [154] |
| Tissue affected | Sample type (matched) | mtDNA levels | Disease severity | Quantification method | References |
|---|---|---|---|---|---|
| Bladder | Bladder | Down | Increased | WGS and WES | [166] |
| Bone | Ewing sarcoma | Down | Increased | qPCR | [171] |
| Brain | Glioma | Down | Increased | qPCR | [170] |
| Breast | Primary breast tumours | Down | Increased | WGS; Other a qPCR; WGS and WES | [166, 167, 168, 169] |
| Colon/rectum | Colorectal carcinoma | Up | Increased | qPCR; qPCR; qPCR | [160, 161, 162] |
| Colorectal adenoma | Up | Increased | qPCR | [164, 249] | |
| Advanced colorectal carcinoma | Down | Increased | qPCR; qPCR | [163, 164] | |
| Head and neck | Squamous cells carcinoma | Down | Increased | WGS and WES | [166] |
| Kidney | Chromophobe renal cell carcinoma | Up | Increased | WGS and WES | [165] |
| Renal carcinoma (TCGA) | Down | Decreased | WGS; WGS and WES | [142, 166] | |
| Renal oncocytomas | Up | Decreased c | WES; qPCR | [147] | |
| Liver | Hepatocellular carcinoma | Down | Increased | qPCR; WGS and WES; WGS | [142, 166, 172] |
| Lung | Non‐small cell lung cancer | Down | Increased | Other b ; qPCR | [249, 250] |
| Adenocarcinoma, small cells lung cancer | Up | Increased | qPCR; WGS and WES; WGS | [142, 166, 251] | |
| Lymphocytes | Lymphocytic leukaemia | Up | Increased | WGS | [142] |
| Oral / digestive tract | Oesophagus | Down | Increased | WGS and WES | [166] |
| Stomach | Down | Increased | Other b | [252] | |
| Up | Increased | qPCR | [253] | ||
| Pancreas | Pancreas tumour (endocrine) | Up | Increased | WGS | [142] |
| Prostate | Primary prostate cancer | Up | Increased | qPCR; WGS | [141, 142] |
| Thyroid | Adenocarcinoma | Up | Increased | WGS | [142] |
Microarray data.
Competitive PCR method.
mtDNA presents high levels of mutations.
Generally, it is important to be careful when comparing data from different sources as it is possible that at least part of the above discrepancies could be explained by methodological and study design biases (see section below). Moreover, the composition of the samples from different tumour types and stages could well explain the differences reported by various research groups. In this respect, recent analyses of thousands of available NGS/WGS data from cancer patients with specific focus on mtDNA may unravel more robust correlations between mtDNA and onset and progression of different tumour types. For future studies along these lines, it is of crucial importance to simultaneously assess both mtDNA mutations and mtDNA copy number and to associate these parameters with assessment of mtDNA expression and OXPHOS capacity.
The challenges of the precise quantification of mtDNA in humans
An extensive review of the literature finds many attempts aimed at understanding the role of mtDNA copy number in human disease. These studies are correlative and descriptive in their nature and often limited by methodology, sample selection and study design. Herein, we discuss some potential issues that must be considered when interpreting the available literature and when designing novel studies.
Firstly, methodological bias can contribute to the variability emerging among studies. The accuracy of the measurements can be much influenced by DNA extraction methods. Traditional approaches based either on organic solvent extraction (phenol–phenol–chloroform–isoamyl alcohol) or on silica‐based solid‐phase columns (DNA extraction kits) can influence the mtDNA:nuclear DNA ratio and can affect the experimental outcomes [84, 173]. Likewise, different quantification methods, as discussed above in the section quantification of mtDNA copy number, may well account for differences between studies, laboratories or even among batches in the same study. The NGS technologies allow sensitive and high‐throughput assessment of mtDNA levels in big data sets; thus, they will likely be more and more employed in the near future to overcome some of the methodological limitations.
Secondly, specimen heterogeneity represents a major constraint to reproducibility, as it is well known that mtDNA copy number often varies significantly between different cell types, and therefore, the cellular composition of the investigated tissue should be a main concern. For instance, the use of blood samples has obvious advantages, including the minimally invasive sampling procedure and the low cost. However, mtDNA levels in blood do not necessarily reflect levels in other tissues and the measurements can be strongly biased by differences in blood cell composition. Platelets are known to have high mtDNA content compared with white blood cells, and they also lack the nuclear genome. As consequence, mtDNA content can be overestimated in blood samples where platelets are more abundant, and mtDNA levels should be hence normalised to the platelets/leucocyte ratio [174, 175].
Thirdly, the cell‐type composition of a given tissue can change due to pathology or ageing. For example, in age‐associated forms of neurodegeneration, the loss of certain neuronal populations is accompanied by reactive gliosis [176]. Thus, neurons will likely be underrepresented in tissue homogenates from patients with neurodegeneration. The naturally occurring ageing process can also severely influence tissue composition due to fibrotic remodelling of the heart or nerve cell loss in the brain. Similar problems are evident in cancer, because tumour tissues typically contain multiple cell types besides the cancer cells, for example fibroblasts and endothelial cells. To circumnavigate the heterogeneity bias, significant attempts to isolate specific cell types have been made with a certain degree of success by using laser‐capture microdissection in defined organs. However, the availability and time engagement of such technology remain serious limiting factors for its application to a large number of samples.
Finally, it is important to point out that the study design has a strong impact on reliability. Genetic and environmental factors, such as age, gender and lifestyle, must be considered when selecting the study cohort. For instance, gender differences are important and it has been reported that females have higher mtDNA content than males [112, 116]. Although most studies aim to use matched control groups, this is not always trivial, as in the case of studies addressing ageing, where controls are by definition nonaged individuals. Similar caution should be used for individuals undergoing long‐term drug treatments, for example chemotherapy, which may well influence mtDNA levels. Likewise, the lifestyles should be taken into account, as regular endurance training is known to induce an adaptive metabolic response in skeletal muscle that leads to a general increase in mitochondrial mass and mtDNA [177, 178]. Finally, another important limitation of many clinical studies is that they are by necessity mostly retrospective and do not include a longitudinal follow‐up. Hence, their outcomes are correlative and must be validated by experimental investigations in animal models in order to establish cause‐and‐effect relationships.
Therapeutic approaches exploiting the modulation of mitochondrial fitness
As discussed above, there is good evidence that the absolute mtDNA levels can be a critical factor in human pathology and ageing. This has led to the development of strategies that directly or indirectly manipulate mtDNA levels to ameliorate or prevent disease progression. In essence, two strategies have been engaged to increase OXPHOS capacity and restore mitochondrial function, which employed either the manipulation of total mitochondrial mass or the selective manipulation of mtDNA to influence mtDNA copy number and/or heteroplasmy levels (Fig. 2). Although these therapeutic approaches have been mostly focused on treating primary mitochondrial disorders, if human trials prove successful, they might also be extended to treat various types of common age‐associated human diseases. Here, we briefly describe the rationale for each strategy and discuss applications, benefits and limitations.
Fig. 2.
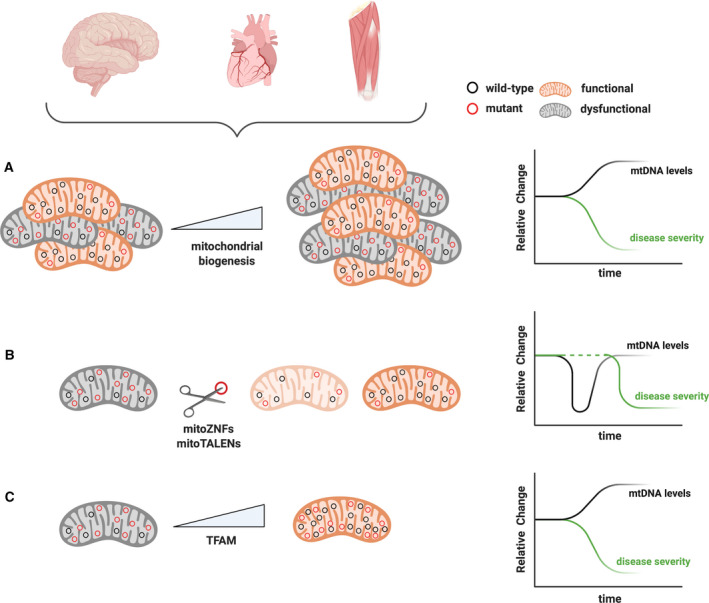
Therapeutic strategies to manipulate mtDNA and disease severity. (A) Boosting mitochondrial biogenesis represents an unspecific approach to decrease disease severity. With this approach, a general increase in all mitochondrial components, as well as mtDNA levels, is accomplished in order to rescue biochemical OXPHOS defects. (B) Mitochondrially directed ZNFs and TALENs selectively cut mtDNA in a sequence‐specific way. Mutant mtDNA is cleaved and the resulting linear molecules are quickly degraded generating a transient mtDNA reduction in the cell, which is restored by replication of the residual (wild‐type) mtDNA. (C) A moderate and selective increase in mtDNA copy number can be achieved by modulating TFAM levels. By doing so, mitochondrial function can be partially restored due to absolute increase of the functional mtDNA copies. Schematic representations of mtDNA levels (black lines) and their correlation with disease severity (green lines) are represented for each approach on the right‐hand side panels.
Manipulation of mitochondrial mass: autophagy
The selective degradation of impaired mitochondria certainly represents an appealing approach that could have the power of shifting the balance in favour of the functional mitochondria. Therefore, attempts to promote the mitochondrial turnover have been carried out by stimulation of autophagy, the cellular process whereby cells can self‐degrade and recycle different cellular components, including mitochondria [179]. Stimulation of bulk autophagy has been pursued by the inhibition of the mammalian target of rapamycin complex 1 (mTORC1), a master regulator of nutrient sensing and metabolism [180]. Inhibition of mTORC1 by using rapamycin improved mitochondrial function in some mouse models, but had no effect in others [181, 182, 183]. This may be explained by the complexity of mTORC1 pathway, which engages many targets and influences several cellular processes. In one study, it was reported that the beneficial effect of rapamycin treatment in a mouse model of the Leigh syndrome was related to the redirection of cellular metabolism towards amino acid catabolism, rather than through the activation of autophagy [183]. Thus, the therapeutic application of rapamycin and mTORC1 inhibitors might be strongly limited by possible off‐target effects. More specific autophagy stimulators, such as urolithin A, extended life span in Caenorhabditis elegans and increased muscle function of aged mice [184]. However, the mechanisms controlling bulk autophagy and the mitochondrial quality control are highly intricate and far from being understood, hence, their dysregulation might lead to an excessive mitochondrial clearance and potential long‐term harmful consequences. Along this line, it has been recently reported that mutations in FBXL4 in humans cause excessive mitochondrial turnover, which, at least partly, explains the severity of the disease in affected patients [185]. Therefore, the downstream effects of treatment strategies aiming to increase mitochondrial degradation must be carefully evaluated in preclinical studies before being safely translated into therapies for human diseases.
Manipulation of mitochondrial mass: boosting mitochondrial biogenesis
Increasing the overall amount of mitochondria by boosting mitochondrial biogenesis represents a valid rationale to counteract the bioenergetic defects caused by mutations in either mtDNA or the nuclear genome [186]. This hypothesis is supported by experimental evidence showing that an increase in mitochondrial mass was sufficient to restore the overall ATP production in skeletal muscle of mice with mitochondrial myopathy, although the individual mitochondria have a severely impaired function [187]. Mitochondrial biogenesis is a highly regulated physiological process controlled by both internal and external stimuli, such as exercise, calory intake, temperature, cell division and differentiation. The stimulation of mitochondrial biogenesis requires a synchronised expression of both nuclear and mtDNA‐encoded gene products. The peroxisome proliferator‐activated receptor gamma coactivator 1α (PGC1α) [188] is involved in making more mitochondria on demand and it interacts with several nuclear transcription factors, such as peroxisome proliferator‐activated receptors (PPARs), to induce the expression of nucleus‐encoded genes, including OXPHOS subunits [189, 190], Tfam and other genes involved in mtDNA gene expression [191]. PGC1α is post‐translationally activated via phosphorylation by the AMP‐dependent kinase (AMPK) [192] and through deacetylation by the nuclear deacetylase Sirtuin 1 (Sirt1) [193].
Both genetic and pharmacological interventions have been used to target PGC1α [194]. Overexpression of PGC‐1α ameliorated the myopathy phenotype in complex IV assembly‐deficient mice [195] and partially rescued the premature ageing phenotypes in the mtDNA mutator mouse [196], consistent with the hypothesis that increased mitochondrial biogenesis can alleviate mitochondrial diseases severity. In support of this view, studies in families carrying homoplasmic LHON‐causing mtDNA mutations have shown that asymptomatic carriers tend to have increased mitochondrial biogenesis and higher mtDNA copy number when compared with visually impaired maternal relatives [65].
PGC1α can also be activated by the pan‐PPAR agonist bezafibrate, by the AMPK agonist 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) and by Sirt1 activators and nicotinamide adenine dinucleotide (NAD)+ precursors, that is nicotinamide riboside (NR) and niacin. In spite of a few promising findings [197, 198], mouse studies have shown that bezafibrate treatment had no effect or even a deleterious impact on mitochondrial dysfunction [195, 199]. In contrast, the administration of AICAR or NR successfully rescued the pathological manifestations in different mouse models of mitochondrial diseases [195, 200]. Mitochondrial biogenesis boosters, such as bezafibrate, NR and niacin, have also been tested in human trials. A clinical trial with bezafibrate was not very encouraging and suggested that the prolonged use of this drug may worsen the metabolic signature of mitochondrial dysfunction in patients [201]. A recently published study with niacin supplementation was more promising and revealed increased mitochondrial biogenesis and improved muscle strength in patients with mitochondrial myopathy and NAD+ deficiency [202]. It should be pointed out that none of these clinical trials have yet been performed in the more rigorous double‐blind format.
Boosting mitochondrial biogenesis may provide a therapeutic strategy to treat mitochondrial diseases. Unfortunately, pharmacological interventions have produced mixed results and raised questions about the consequences of long‐term treatments [201]. PGC‐1α seems to be involved in the regulation a plethora of cellular processes by influencing the expression of a large number genes involved in metabolic pathways, such as gluconeogenesis, fatty acid synthesis and oxidation and glycolysis [203], and its activation must be finely tuned to avoid deleterious side effects. Indeed, detrimental impacts on cell physiology due to the over‐activation of PGC‐1α have been reported to occur in different organs of the mouse, for example heart [204], skeletal muscle [205] and DA neurons [206]. In addition, several studies in mouse models have explored whether the overexpression of PGC‐1α is a potential therapeutic strategy in PD, but the results of these investigations provided inconsistent and conflicting findings [207].
Selective manipulation of mtDNA: mitoTALENs and mitoZNFs
Currently, the most efficient strategy to shift the levels of heteroplasmy in favour of wild‐type mtDNA is the expression of mitochondrially targeted endonucleases [208, 209]. Unfortunately, the powerful CRISPR‐Cas9 system, which has revolutionised nuclear genome editing, will likely not work in mammalian mitochondria. Although Cas9 can be easily imported after addition of a mitochondrial targeting sequence (MTS), the import of guide RNA into mitochondria seems improbable. RNA import occurs in some metazoans, but several lines of evidence suggest that mammalian mitochondria lack RNA import system. Importantly, mammalian mtDNA encodes the full set of tRNAs required for mitochondrial translation overcoming the need for tRNA import from the cytosol. Furthermore, mammalian mitochondrial RNaseP is composed of three protein subunits and lacks a catalytic RNA component [210, 211]. Finally, the central protuberance of the large subunit of the mitochondrial ribosome contains a mtDNA‐encoded tRNA, and there is therefore no need to import 5S rRNA from the cytosol for mitoribosomal biogenesis [212, 213]. An additional hurdle, besides the lack of RNA import, preventing the use of the CRISPR‐Cas9 system in mammalian mitochondria, is the lack of a machinery for double‐strand break (DSB) repair and homologous recombination, whose activities are required to introduce a deletion or any other type of mutation after Cas9‐mediated DNA cleavage.
The import into mitochondria of the transcription activator‐like effector nucleases (mitoTALENs) and zinc finger nucleases (mitoZFNs) is straightforward, as well‐characterised machineries for protein import are engaged. Manipulation of mtDNA with mitoTALENs or mitoZFNs exploits distinctive features of mitochondrial genetics: (a) heteroplasmic pathogenic mtDNA mutations are typically functionally recessive [214], (b) mitochondria lack a DSB repair machinery [215], but have an efficient mechanism for degradation of linear mtDNA molecules [216], and (c) the mtDNA copy number is tightly regulated and after digestion of mutant molecules, the cells quickly recover from depletion by stimulating the replication of the residual mtDNA copies [37]. The earliest attempts to degrade mtDNA in cells were based on the use of bacterial restriction endonucleases targeted to mitochondria (mitoREs) with the addition of an amino‐terminal MTS. Once imported, the mitoREs shifted the levels of heteroplasmy both in cells [217, 218] and in mice [219, 220] by selectively binding and cleaving one of the mtDNA variants. However, the use of these enzymes for a wider range of mutations was hampered by their need of a unique restriction site in the mtDNA sequence, which only rarely is generated by pathogenic variants, and by the fact that restriction endonucleases (REs) cannot be engineered to recognise chosen DNA sites. These limitations were overcome by the use of the programmable mitoTALENs and mitoZFNs. Unlike REs, these endonucleases rely on sequence‐specific DNA‐binding domains coupled to a FokI endonuclease domain, which is active only as a dimer and creates a DSB when both monomers are bound to the target allele and interact. MitoTALENs and mitoZNFs have proved efficient in targeting numerous disease‐causing mtDNA variants in cells, where they cleave mutant genomes and thereby rescue the biochemical defect [221, 222, 223]. The in vivo feasibility of these technologies was recently demonstrated in a heteroplasmic mouse model carrying a pathogenic point mutation in the mitochondrial tRNAAla gene [99]. In these mice, either mitoTALENs or mitoZFNs, delivered by injection of adeno‐associated virus (AAV) vectors, shifted the heteroplasmy levels below the threshold leading to amelioration of the molecular phenotypes in heart and skeletal muscle [224, 225]. These data represent important proof of concept that mitoZFNs and mtTALENs have the potential to serve as therapeutic tools for heteroplasmic mitochondrial diseases. It is important to mention that the possible success of this strategy depends on vectors that efficiently can target cells in brain, skeletal muscle, heart and other organs. Some relevant AAV vectors already exist, and this field is under rapid development giving hopes for more efficient targeting of the brain and other tissues and for potential translation into the clinic. Other issues to be addressed are common limitations for any gene therapy approach, that is selectivity, efficiency, cost and potential off‐target effects. Lastly, it should be pointed out that the use of mitoTALENs and mitoZNFs is limited to heterosplamic mtDNA mutations, for example point mutations and large deletions, and cannot be used to treat some of the common homoplasmic pathogenic mutations, for example those causing LHON.
Selective manipulation of mtDNA: modulation of the absolute mtDNA levels
An increase in the absolute mtDNA copy number represents another possibility of treatment for human disease caused by heteroplasmic mtDNA mutations. In these pathological conditions, affected individuals always carry perfectly functional wild‐type mtDNA molecules, in spite of high levels of mutant mtDNA. It is widely recognised that the mutation load is an important determinant for disease severity, whereas the role of the absolute levels of mtDNA often has often been neglected. Over the last 15 years, only few studies have directly measured total mtDNA copy number in patients with pathogenic mtDNA mutations.
Although the molecular mechanisms underlying this regulation remain unclear, it is well established that the amount of mtDNA is directly proportional to TFAM protein levels (Box 1). As consequence, manipulation of mtDNA copy number can be genetically achieved through modulation of TFAM expression [17]. Notably, a moderate overexpression of human TFAM in wild‐type mice caused nearly 50% increase in mtDNA levels, without interfering with mtDNA transcription, mitochondrial respiratory‐chain function or mitochondrial mass [17]. Thus, the upregulation in mtDNA copy number can be dissociated from the mitochondrial biogenesis process, that is it is possible to selectively increase mtDNA copy number without increasing the amount of mitochondria.
Experimental studies have shown that an increase in mtDNA copy number, through TFAM overexpression, ameliorated phenotypes in mouse models with mitochondrial diseases caused by heteroplasmic mtDNA deletions [226] or a pathogenic point mutation in the tRNAALA gene [64]. These results were further supported by the observations that increasing total mtDNA levels partially rescued the male infertility in the mtDNA mutator mice, which carry a large number of different mtDNA point mutations and a linear deletion [67]. Remarkably, the results of these investigations also demonstrated that the increased mtDNA levels did not affect the proportion of pathogenic mtDNA mutations, that is the mutational load was unchanged. However, in absolute terms, the levels of wild‐type mtDNA segments were higher, which partly restored mitochondrial function despite the mutant mtDNA still being the predominant species [64, 67, 226].
Other studies have reported beneficial effects of an increase in mtDNA copy number in various types of pathology, primarily not of mitochondrial origin, for example in a mouse model of myocardial infarction [227]. TFAM overexpression has also been reported to improve cognitive function in an AD mouse model [228] and to reduce the memory impairment seen in aged mice [229]. The modulation of mtDNA levels seems to be a more selective way to improve OXPHOS compared with boosting the whole process of mitochondrial biogenesis. However, the degree of TFAM expression must be carefully considered. We and others have shown that moderate TFAM overexpression had beneficial effects on cellular functions in vivo [64, 67, 226]. In contrast, a marked TFAM overexpression can have detrimental consequences by impairing mtDNA transcription and causing a progressive OXPHOS dysfunction [230]. Similar negative outcomes have been reported in S. cerevisiae where high levels of ABF2, the yeast ortholog of TFAM, led to a dramatic loss of mtDNA, possibly due to hypercompaction of the genome preventing proper replication and distribution of genomes [231]. Modulation of mtDNA copy number through manipulation of TFAM levels may provide a future avenue to treat not only primary mitochondrial diseases but also to intervene in other human diseases characterised by mitochondrial impairment.
Concluding Remarks
Mitochondrial dysfunction is heavily implicated in variety of human pathological conditions and ageing, and it is therefore not surprising that a large number of studies have aimed to correlate the presence of mutations and the levels of mtDNA with decline of organ function. Although a key role for mtDNA mutations in inherited mitochondrial diseases is firmly established, the evidence for the involvement of mtDNA levels in common diseases and ageing remains merely correlative. In mitochondrial diseases, high mtDNA copy number mostly correlates with decreased disease severity, or even with incomplete disease penetrance [65, 66]. In contrast, the interpretation of the existing data related to mtDNA variations in neurodegenerative disorders, cancer and ageing is not equally straightforward, partly due to method‐, specimen‐ and study design‐related issues. Recent advances in NGS enable assessment of mtDNA mutation load and copy number in large‐scale data sets, which will much expand our knowledge about the impact of these factors on disease burden. Approaches exploiting the isolation and analysis of large numbers of selected cell types will also be required, for instance in studies on neurogenerative disorders, where dysfunctional cells driving the disease only represent a minority of the affected brain region.
In humans with mitochondrial diseases, the upregulation of mtDNA copy number, mostly concomitant with an overall increase in mitochondrial biogenesis, is frequently occurring and typically regarded as a compensatory mechanism to sustain cellular bioenergetics. Studies of animal models have provided an experimental proof of concept for selective increase in total mtDNA copy number induced by genetic manipulation of TFAM expression ameliorating pathogenic effects of heteroplasmic mtDNA mutations [64, 67] and the mitochondrial impairment associated with other diseases or with the physiological age‐related decline [227, 228, 229]. Unfortunately, the same rationale might not be applicable to cancer, or at least not for all forms of cancer. The available studies have indeed highlighted that the scenario in cancer is even more complex and varied, as mtDNA copy number can correlate with both increased and decreased disease burden. Future studies will be necessary to more exactly define the impact of mtDNA copy number on human health. Importantly, future development of therapies aimed at moderately increasing mtDNA copy number may provide a strategy to rescue the mitochondrial dysfunction not only in primary mitochondrial diseases but also in age‐associated degenerative diseases and ageing.
Acknowledgements
This review was supported by grants to NGL from the Swedish Research Council (2015.00418), the Knut and Alice Wallenberg Foundation (KAW 2016.0050), the European Research Council (Advanced Grant 2016‐741366), the Swedish Cancer Society (2018.602), Novo Nordisk Foundation and grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF agreement (SLL2018.0471). Figures have been created using BioRender.com.
Edited by Agnieszka Chacinska
References
- 1. Gustafsson CM, Falkenberg M and Larsson NG (2016) Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 85, 133–160. [DOI] [PubMed] [Google Scholar]
- 2. Bonekamp NA and Larsson NG (2018) SnapShot: mitochondrial nucleoid. Cell 172, 388.e1. [DOI] [PubMed] [Google Scholar]
- 3. Brown TA, Tkachuk AN, Shtengel G, Kopek BG, Bogenhagen DF, Hess HF and Clayton DA (2011) Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol Cell Biol 31, 4994–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kukat C, Davies KM, Wurm CA, Spåhr H, Bonekamp NA, Kühl I, Joos F, Polosa PL, Park CB, Posse V et al. (2015) Cross‐strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc Natl Acad Sci USA 112, 11288–11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson N‐G and Jakobs S (2011) Super‐resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci USA 108, 13534–13539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garrido N, Griparic L, Jokitalo E, Wartiovaara J, van der Bliek AM and Spelbrink JN (2003) Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell 14, 1583–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Legros F, Malka F, Frachon P, Lombès A and Rojo M (2004) Organization and dynamics of human mitochondrial DNA. J Cell Sci 117, 2653–2662. [DOI] [PubMed] [Google Scholar]
- 8. Rubio‐Cosials A, Sidow JF, Jiménez‐Menéndez N, Fernández‐Millán P, Montoya J, Jacobs HT, Coll M, Bernadó P and Solà M (2011) Human mitochondrial transcription factor A induces a U‐turn structure in the light strand promoter. Nat Struct Mol Biol 18, 1281–1289. [DOI] [PubMed] [Google Scholar]
- 9. Farge G, Mehmedovic M, Baclayon M, van den Wildenberg SMJL, Roos WH, Gustafsson CM, Wuite GJL and Falkenberg M (2014) In vitro‐reconstituted nucleoids can block mitochondrial DNA replication and transcription. Cell Rep 8, 66–74. [DOI] [PubMed] [Google Scholar]
- 10. Parisi M and Clayton D (1991) Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science 252, 965–969. [DOI] [PubMed] [Google Scholar]
- 11. Ngo HB, Kaiser JT and Chan DC (2011) The mitochondrial transcription and packaging factor Tfam imposes a U‐turn on mitochondrial DNA. Nat Struct Mol Biol 18, 1290–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ngo HB, Lovely GA, Phillips R and Chan DC (2014) Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat Commun 5, 3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dairaghi DJ, Shadel GS and Clayton DA (1995) Addition of a 29 residue carboxyl‐terminal tail converts a simple HMG Box‐containing protein into a transcriptional activator. J Mol Biol 249, 11–28. [DOI] [PubMed] [Google Scholar]
- 14. Hillen HS, Morozov YI, Sarfallah A, Temiakov D and Cramer P (2017) Structural basis of mitochondrial transcription initiation. Cell 171, 1072–1081.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Larsson N‐G, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS and Clayton DA (1998) Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 18, 231–236. [DOI] [PubMed] [Google Scholar]
- 16. Matsushima Y, Matsumura K, Ishii S, Inagaki H, Suzuki T, Matsuda Y, Beck K and Kitagawa Y (2003) Functional domains of chicken mitochondrial transcription factor A for the maintenance of mitochondrial DNA copy number in lymphoma cell line DT40. J Biol Chem 278, 31149–31158. [DOI] [PubMed] [Google Scholar]
- 17. Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM and Larsson N‐G (2004) Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 13, 935–944. [DOI] [PubMed] [Google Scholar]
- 18. Kanki T, Ohgaki K, Gaspari M, Gustafsson CM, Fukuoh A, Sasaki N, Hamasaki N and Kang D (2004) Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol Cell Biol 24, 9823–9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson S, Bankier AT, Barrell BG, De Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F et al. (1981) Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. [DOI] [PubMed] [Google Scholar]
- 20. Bibb MJ, Van Etten RA, Wright CT, Walberg MW and Clayton DA (1981) Sequence and gene organization of mouse mitochondrial DNA. Cell 26, 167–180. [DOI] [PubMed] [Google Scholar]
- 21. Bogenhagen D and Clayton DA (1977) Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell 11, 719–727. [DOI] [PubMed] [Google Scholar]
- 22. Wanrooij S and Falkenberg M (2010) The human mitochondrial replication fork in health and disease. Biochim Biophys Acta Bioenerg 1797, 1378–1388. [DOI] [PubMed] [Google Scholar]
- 23. Korhonen JA, Gaspari M and Falkenberg M (2003) TWINKLE Has 5’ ‐> 3’ DNA helicase activity and is specifically stimulated by mitochondrial single‐stranded DNA‐binding protein. J Biol Chem 278, 48627–48632. [DOI] [PubMed] [Google Scholar]
- 24. Miralles Fusté J, Shi Y, Wanrooij S, Zhu X, Jemt E, Persson Ö, Sabouri N, Gustafsson CM and Falkenberg M (2014) In vivo occupancy of mitochondrial single‐stranded DNA binding protein supports the strand displacement mode of DNA replication. PLoS Genet 10, e1004832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wanrooij S, Fuste JM, Farge G, Shi Y, Gustafsson CM and Falkenberg M (2008) Human mitochondrial RNA polymerase primes lagging‐strand DNA synthesis in vitro . Proc Natl Acad Sci USA 105, 11122–11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clayton DA (1991) Replication and transcription of vertebrate mitochondrial DNA. Annu Rev Cell Biol 7, 453–478. [DOI] [PubMed] [Google Scholar]
- 27. Kasamatsu H and Vinograd J (1973) Unidirectionality of replication in mouse mitochondrial DNA. Nat New Biol 241, 103–105. [DOI] [PubMed] [Google Scholar]
- 28. Robberson DL, Kasamatsu H and Vinograd J (1972) Replication of mitochondrial DNA. Circular replicative intermediates in mouse L cells. Proc Natl Acad Sci USA 69, 737–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Falkenberg M (2018) Mitochondrial DNA replication in mammalian cells: overview of the pathway. Essays Biochem 62, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martens PA and Clayton DA (1979) Mechanism of mitochondrial DNA replication in mouse L‐cells: localization and sequence of the light‐strand origin of replication. J Mol Biol 135, 327–351. [DOI] [PubMed] [Google Scholar]
- 31. Miralles Fusté J, Wanrooij S, Jemt E, Granycome CE, Cluett TJ, Shi Y, Atanassova N, Holt IJ, Gustafsson CM and Falkenberg M (2010) Mitochondrial RNA polymerase is needed for activation of the origin of light‐strand DNA replication. Mol Cell 37, 67–78. [DOI] [PubMed] [Google Scholar]
- 32. Nicholls TJ, Nadalutti CA, Motori E, Sommerville EW, Gorman GS, Basu S, Hoberg E, Turnbull DM, Chinnery PF, Larsson NG et al. (2018) Topoisomerase 3α is required for decatenation and segregation of human mtDNA. Mol Cell 69, 9–23.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kühl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A, Filipovska A and Larsson NG (2017) Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 6, e30952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dickinson A, Yeung KY, Donoghue J, Baker MJ, Kelly RD, McKenzie M, Johns TG and St John JC (2013) The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ 20, 1644–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen X, Prosser R, Simonetti S, Sadlock J, Jagiello G and Schon EA (1995) Rearranged mitochondrial genomes are present in human oocytes. Am J Hum Genet 57, 239–247. [PMC free article] [PubMed] [Google Scholar]
- 36. D’Erchia AM, Atlante A, Gadaleta G, Pavesi G, Chiara M, De Virgilio C, Manzari C, Mastropasqua F, Prazzoli GM, Picardi E et al. (2015) Tissue‐specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 20, 13–21. [DOI] [PubMed] [Google Scholar]
- 37. Moraes C (2001) What regulates mitochondrial DNA copy number in animal cells? Trends Genet 17, 199–205. [DOI] [PubMed] [Google Scholar]
- 38. Fukuoh A, Cannino G, Gerards M, Buckley S, Kazancioglu S, Scialo F, Lihavainen E, Ribeiro A, Dufour E and Jacobs HT (2014) Screen for mitochondrial DNA copy number maintenance genes reveals essential role for ATP synthase. Mol Syst Biol 10, 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang H and Singh KK (2014) Global genetic determinants of mitochondrial DNA copy number. PLoS One 9, e105242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Curran JE, Johnson MP, Dyer TD, Göring HHH, Kent JW, Charlesworth JC, Borg AJ, Jowett JBM, Cole SA, MacCluer JW et al. (2007) Genetic determinants of mitochondrial content. Hum Mol Genet 16, 1504–1514. [DOI] [PubMed] [Google Scholar]
- 41. Alexeyev MF (2009) Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J 276, 5768–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harman D (1972) The biologic clock: the mitochondria? J Am Geriatr Soc 20, 145–147. [DOI] [PubMed] [Google Scholar]
- 43. Kauppila JHK, Bonekamp NA, Mourier A, Isokallio MA, Just A, Kauppila TES, Stewart JB and Larsson NG (2018) Base‐excision repair deficiency alone or combined with increased oxidative stress does not increase mtDNA point mutations in mice. Nucleic Acids Res 46, 6642–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kennedy SR, Salk JJ, Schmitt MW and Loeb LA (2013) Ultra‐sensitive sequencing reveals an age‐related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet 9, e1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khrapko K, Coller HA, André PC, Li XC, Hanekamp JS and Thilly WG (1997) Mitochondrial mutational spectra in human cells and tissues. Proc Natl Acad Sci USA 94, 13798–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A and Larsson N‐G (2008) Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol 6, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Valente WJ, Ericson NG, Long AS, White PA, Marchetti F and Bielas JH (2016) Mitochondrial DNA exhibits resistance to induced point and deletion mutations. Nucleic Acids Res 44, 8513–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zheng W, Khrapko K, Coller HA, Thilly WG and Copeland WC (2006) Origins of human mitochondrial point mutations as DNA polymerase γ‐mediated errors. Mutat Res Fundam Mol Mech Mutagen 599, 11–20. [DOI] [PubMed] [Google Scholar]
- 49. Taylor RW and Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nat Rev Genet 6, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sciacco M, Bonilla E, Schon EA, DiMauro S and Moraes CT (1994) Distribution of wild‐type and common deletion forms of mtDNA in normal and respiration‐deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet 3, 13–19. [DOI] [PubMed] [Google Scholar]
- 51. Larsson NG, Tulinius MH, Holme E, Oldfors A, Andersen O, Wahlstrom J and Aasly J (1992) Segregation and manifestations of the mtDNA tRNA(Lys) A→G((8344)) mutation of myoclonus epilepsy and ragged‐red fibers (MERRF) syndrome. Am J Hum Genet 51, 1201–1212. [PMC free article] [PubMed] [Google Scholar]
- 52. Santorelli FM, Shanske S, Macaya A, DeVivo DC and DiMauro S (1993) The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh’s syndrome. Ann Neurol 34, 827–834. [DOI] [PubMed] [Google Scholar]
- 53. Yoneda M, Miyatake T and Attardi G (1995) Heteroplasmic mitochondrial tRNALys mutation and its complementation in MERRF patient‐derived mitochondrial transformants. Muscle Nerve 18, S95–S101. [DOI] [PubMed] [Google Scholar]
- 54. Larsson NG, Holme E, Kristiansson B, Oldfors A and Tulinius M (1990) Progressive increase of the mutated mitochondrial DNA fraction in kearns‐sayre syndrome. Pediatr Res 28, 131–136. [DOI] [PubMed] [Google Scholar]
- 55. Rahman S, Poulton J, Marchington D and Suomalainen A (2001) Decrease of 3243 A ‐‐> G mtDNA mutation from blood in MELAS syndrome: a longitudinal study. Am J Hum Genet 68, 238–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Walker MA, Lareau CA, Ludwig LS, Karaa A, Sankaran VG, Regev A and Mootha VK (2020) Purifying selection against pathogenic mitochondrial DNA in human T cells. N Engl J Med 383, 1556–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tuppen HAL, Blakely EL, Turnbull DM and Taylor RW (2010) Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 1797, 113–128. [DOI] [PubMed] [Google Scholar]
- 58. Greaves LC, Reeve AK, Taylor RW and Turnbull DM (2012) Mitochondrial DNA and disease. J Pathol 226, 274–286. [DOI] [PubMed] [Google Scholar]
- 59. La Morgia C, Maresca A, Caporali L, Valentino ML and Carelli V (2020) Mitochondrial diseases in adults. J Intern Med 287, 592–608. [DOI] [PubMed] [Google Scholar]
- 60. Rahman S (2020) Mitochondrial disease in children. J. Intern. Med. 287, 609–633. [DOI] [PubMed] [Google Scholar]
- 61. Reeve AK, Krishnan KJ and Turnbull D (2008) Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann N Y Acad Sci 1147, 21–29. [DOI] [PubMed] [Google Scholar]
- 62. Larsson N‐G (2010) Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 79, 683–706. [DOI] [PubMed] [Google Scholar]
- 63. Bentlage HACM and Attardi G (1996) Relationship of genotype to phenotype in fibroblast‐derived transmitochondrial cell lines carrying the 3243 mutation associated with the MELAS encephalomyopathy: shift towards mutant genotype and role of mtDNA copy number. Hum Mol Genet 5, 197–205. [DOI] [PubMed] [Google Scholar]
- 64. Filograna R, Koolmeister C, Upadhyay M, Pajak A, Clemente P, Wibom R, Simard ML, Wredenberg A, Freyer C, Stewart JB et al. (2019) Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse. Sci Adv 5, eaav9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, Valentino ML, Caporali L, Liguori R, Deceglie S, Roberti M et al. (2014) Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain 137, 335–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grady JP, Pickett SJ, Ng YS, Alston CL, Blakely EL, Hardy SA, Feeney CL, Bright AA, Schaefer AM, Gorman GS et al. (2018) mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol Med 10, e8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jiang M, Kauppila TES, Motori E, Li X, Atanassov I, Folz‐Donahue K, Bonekamp NA, Albarran‐Gutierrez S, Stewart JB and Larsson N‐G (2017) Increased total mtDNA copy number cures male infertility despite unaltered mtDNA mutation load. Cell Metab 26, 429–436.e4. [DOI] [PubMed] [Google Scholar]
- 68. Kornblum C, Nicholls TJ, Haack TB, Schöler S, Peeva V, Danhauser K, Hallmann K, Zsurka G, Rorbach J, Iuso A et al. (2013) Loss‐of‐function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat Genet 45, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Moraes CT, Shanske S, Tritschler HJ, Aprille JR, Andreetta F, Bonilla E, Schon EA and DiMauro S (1991) mtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet 48, 492–501. [PMC free article] [PubMed] [Google Scholar]
- 70. Ronchi D, Di Fonzo A, Lin W, Bordoni A, Liu C, Fassone E, Pagliarani S, Rizzuti M, Zheng L, Filosto M et al. (2013) Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am J Hum Genet 92, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Griffiths LM, Doudican NA, Shadel GS and Doetsch PW (2009) Mitochondrial DNA oxidative damage and mutagenesis in Saccharomyces cerevisiae . Methods Mol Biol 554, 267–286. [DOI] [PubMed] [Google Scholar]
- 72. Hance N, Ekstrand MI and Trifunovic A (2005) Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum Mol Genet 14, 1775–1783. [DOI] [PubMed] [Google Scholar]
- 73. Milenkovic D, Matic S, Kuhl I, Ruzzenente B, Freyer C, Jemt E, Park CB, Falkenberg M and Larsson N‐G (2013) TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D‐loop strands and complete mtDNA replication. Hum Mol Genet 22, 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tyynismaa H, Sembongi H, Bokori‐Brown M, Granycome C, Ashley N, Poulton J, Jalanko A, Spelbrink JN, Holt IJ and Suomalainen A (2004) Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum Mol Genet 13, 3219–3227. [DOI] [PubMed] [Google Scholar]
- 75. Alán L, Zelenka J, Ježek J, Dlasková A and Ježek P (2010) Fluorescent in situ hybridization of mitochondrial DNA and RNA. Acta Biochim Pol 57, 403–408. [PubMed] [Google Scholar]
- 76. Janes MS, Hanson BJ, Hill DM, Buller GM, Agnew JY, Sherwood SW, Cox WG, Yamagata K and Capaldi RA (2004) Rapid analysis of mitochondrial DNA depletion by fluorescence in situ hybridization and immunocytochemistry: potential strategies for HIV therapeutic monitoring. J Histochem Cytochem 52, 1011–1018. [DOI] [PubMed] [Google Scholar]
- 77. Lee CM, Lopez ME, Weindruch R and Aiken JM (1998) Association of age‐related mitochondrial abnormalities with skeletal muscle fiber atrophy. Free Radic Biol Med 25, 964–972. [DOI] [PubMed] [Google Scholar]
- 78. Bratic I, Hench J, Henriksson J, Antebi A, Bürglin TR and Trifunovic A (2009) Mitochondrial DNA level, but not active replicase, is essential for Caenorhabditis elegans development. Nucleic Acids Res 37, 1817–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Leung MCK, Rooney JP, Ryde IT, Bernal AJ, Bess AS, Crocker TL, Ji AQ and Meyer JN (2013) Effects of early life exposure to ultraviolet C radiation on mitochondrial DNA content, transcription, ATP production, and oxygen consumption in developing Caenorhabditis elegans . BMC Pharmacol. Toxicol. 14, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC et al. (2011) High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 83, 8604–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Memon AA, Zöller B, Hedelius A, Wang X, Stenman E, Sundquist J and Sundquist K (2017) Quantification of mitochondrial DNA copy number in suspected cancer patients by a well optimized ddPCR method. Biomol Detect Quantif 13, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pinheiro LB, Coleman VA, Hindson CM, Herrmann J, Hindson BJ, Bhat S and Emslie KR (2012) Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem 84, 1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chu H‐T, Hsiao WWL, Tsao TTH, Chang C‐M, Liu Y‐W, Fan C‐C, Lin H, Chang H‐H, Yeh T‐J, Chen J‐C et al. (2012) Quantitative assessment of mitochondrial DNA copies from whole genome sequencing. BMC Genom 13 (Suppl 7), S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Longchamps RJ, Castellani CA, Yang SY, Newcomb CE, Sumpter JA, Lane J, Grove ML, Guallar E, Pankratz N, Taylor KD et al. (2020) Evaluation of mitochondrial DNA copy number estimation techniques. PLoS One 15, e0228166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Qian Y, Butler TJ, Opsahl‐Ong K, Giroux NS, Sidore C, Nagaraja R, Cucca F, Ferrucci L, Abecasis GR, Schlessinger D et al. (2017) fastMitoCalc: an ultra‐fast program to estimate mitochondrial DNA copy number from whole‐genome sequences. Bioinformatics 33, 1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Theunissen TEJ, Nguyen M, Kamps R, Hendrickx AT, Sallevelt SCEH, Gottschalk RWH, Calis CM, Stassen APM, de Koning B, Mulder‐Den Hartog ENM et al. (2018) Whole exome sequencing is the preferred strategy to identify the genetic defect in patients with a probable or possible mitochondrial cause. Front Genet. 9, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Guo Y, Li J, Li C‐I, Shyr Y and Samuels DC (2013) MitoSeek: extracting mitochondria information and performing high‐throughput mitochondria sequencing analysis. Bioinformatics 29, 1210–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Samuels DC, Li C, Li B, Song Z, Torstenson E, Boyd Clay H, Rokas A, Thornton‐Wells TA, Moore JH, Hughes TM et al. (2013) Recurrent tissue‐specific mtDNA mutations are common in humans. PLoS Genet 9, e1003929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Viscomi C and Zeviani M (2017) MtDNA‐maintenance defects: syndromes and genes. J Inherit Metab Dis 40, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rahman S and Copeland WC (2019) POLG‐related disorders and their neurological manifestations. Nat Rev Neurol 15, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dimmock D, Tang L‐Y, Schmitt ES and Wong L‐JC (2010) Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin Chem 56, 1119–1127. [DOI] [PubMed] [Google Scholar]
- 92. Lee Y‐S, Kennedy WD and Yin YW (2009) Structural insight into processive human mitochondrial DNA synthesis and disease‐related polymerase mutations. Cell 139, 312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tzoulis C, Engelsen BA, Telstad W, Aasly J, Zeviani M, Winterthun S, Ferrari G, Aarseth JH and Bindoff LA (2006) The spectrum of clinical disease caused by the A467T and W748S POLG mutations: a study of 26 cases. Brain 129, 1685–1692. [DOI] [PubMed] [Google Scholar]
- 94. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly‐Y M, Gidlöf S, Oldfors A, Wibom R et al. (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. [DOI] [PubMed] [Google Scholar]
- 95. Bai R and Wong L (2005) Simultaneous detection and quantification of mitochondrial DNA deletion(s), depletion, and over‐replication in patients with mitochondrial disease. J Mol Diagn 7, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liu C‐S, Cheng W‐L, Lee C‐F, Ma Y‐S, Lin C‐Y, Huang C‐C and Wei Y‐H (2006) Alteration in the copy number of mitochondrial DNA in leukocytes of patients with mitochondrial encephalomyopathies. Acta Neurol Scand 113, 334–341. [DOI] [PubMed] [Google Scholar]
- 97. Bianco A, Bisceglia L, Russo L, Palese LL, D’Agruma L, Emperador S, Montoya J, Guerriero S and Petruzzella V (2017) High mitochondrial DNA copy number is a protective factor from vision loss in Heteroplasmic Leber’s Hereditary Optic Neuropathy (LHON). Invest Ophthalmol Vis Sci 58, 2193–2197. [DOI] [PubMed] [Google Scholar]
- 98. Bianco A, Valletti A, Longo G, Bisceglia L, Montoya J, Emperador S, Guerriero S and Petruzzella V (2018) Mitochondrial DNA copy number in affected and unaffected LHON mutation carriers. BMC Res Notes 11, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kauppila JHK, Baines HL, Bratic A, Simard M‐L, Freyer C, Mourier A, Stamp C, Filograna R, Larsson N‐G, Greaves LC et al. (2016) A phenotype‐driven approach to generate mouse models with pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep 16, 2980–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Beregi E and Regius O (1987) Comparative morphological study of age related mitochondrial changes of the lymphocytes and skeletal muscle cells. Acta Morphol Hung 35, 219–224. [PubMed] [Google Scholar]
- 101. Shigenaga MK, Hagen TM and Ames BN (1994) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA 91, 10771–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tauchi H and Sato T (1968) Age changes in size and number of mitochondria of human hepatic cells. J Gerontol 23, 454–461. [DOI] [PubMed] [Google Scholar]
- 103. Short KR, Bigelow ML, Kahl J, Singh R, Coenen‐Schimke J, Raghavakaimal S and Nair KS (2005) Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci USA 102, 5618–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Corral‐Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF and Wallace DC (1992) Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet 2, 324–329. [DOI] [PubMed] [Google Scholar]
- 105. Fayet G, Jansson M, Sternberg D, Moslemi AR, Blondy P, Lombès A, Fardeau M and Oldfors A (2002) Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord 12, 484–493. [DOI] [PubMed] [Google Scholar]
- 106. Yen TC, Su JH, King KL and Wei YH (1991) Ageing‐associated 5 kb deletion in human liver mitochondrial DNA. Biochem Biophys Res Commun 178, 124–131. [DOI] [PubMed] [Google Scholar]
- 107. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA et al. (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484. [DOI] [PubMed] [Google Scholar]
- 108. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T et al. (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38, 515–517. [DOI] [PubMed] [Google Scholar]
- 109. Coskun PE, Beal MF and Wallace DC (2004) Alzheimer’s brains harbor somatic mtDNA control‐region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci USA 101, 10726–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P and Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54, 823–827. [DOI] [PubMed] [Google Scholar]
- 111. Krishnan KJ, Ratnaike TE, De Gruyter HLM, Jaros E and Turnbull DM (2012) Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol Aging 33, 2210–2214. [DOI] [PubMed] [Google Scholar]
- 112. Ding J, Sidore C, Butler TJ, Wing MK, Qian Y, Meirelles O, Busonero F, Tsoi LC, Maschio A, Angius A et al. (2015) Assessing mitochondrial DNA variation and copy number in lymphocytes of ~2,000 sardinians using tailored sequencing analysis tools. PLoS Genet 11, e1005306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Knez J, Winckelmans E, Plusquin M, Thijs L, Cauwenberghs N, Gu Y, Staessen JA, Nawrot TS and Kuznetsova T (2016) Correlates of peripheral blood mitochondrial DNA content in a general population. Am J Epidemiol 183, 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Mengel‐From J, Thinggaard M, Dalgård C, Kyvik KO, Christensen K and Christiansen L (2014) Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum Genet 133, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhang R, Wang Y, Ye K, Picard M and Gu Z (2017) Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genom 18, 890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. van Leeuwen N, Beekman M, Deelen J, van den Akker EB, de Craen AJM, Slagboom PE and ’t Hart LM (2014) Low mitochondrial DNA content associates with familial longevity: the Leiden Longevity Study. Age 36, 9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. He Y‐H, Lu X, Wu H, Cai W‐W, Yang L‐Q, Xu L‐Y, Sun H‐P and Kong Q‐P (2014) Mitochondrial DNA content contributes to healthy aging in Chinese: a study from nonagenarians and centenarians. Neurobiol Aging 35, 1779.e1–1779.e4. [DOI] [PubMed] [Google Scholar]
- 118. Frahm T, Mohamed SA, Bruse P, Gemünd C, Oehmichen M and Meissner C (2005) Lack of age‐related increase of mitochondrial DNA amount in brain, skeletal muscle and human heart. Mech Ageing Dev 126, 1192–1200. [DOI] [PubMed] [Google Scholar]
- 119. Miller FJ, Rosenfeldt FL, Zhang C, Linnane AW and Nagley P (2003) Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR‐based assay: lack of change of copy number with age. Nucleic Acids Res 31, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wachsmuth M, Hübner A, Li M, Madea B and Stoneking M (2016) Age‐related and heteroplasmy‐related variation in human mtDNA copy number. PLoS Genet 12, e1005939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Dölle C, Flønes I, Nido GS, Miletic H, Osuagwu N, Kristoffersen S, Lilleng PK, Larsen JP, Tysnes O‐B, Haugarvoll K et al. (2016) Defective mitochondrial DNA homeostasis in the substantia Nigra in Parkinson disease. Nat Commun 7, 13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Grünewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M and Turnbull DM (2016) Mitochondrial DNA depletion in respiratory chain‐deficient parkinson disease neurons. Ann Neurol 79, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Pyle A, Anugrha H, Kurzawa‐Akanbi M, Yarnall A, Burn D and Hudson G (2016) Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol Aging 38, 216.e7–216.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wei W, Keogh MJ, Wilson I, Coxhead J, Ryan S, Rollinson S, Griffin H, Kurzawa‐Akanbi M, Santibanez‐Koref M, Talbot K et al. (2017) Mitochondrial DNA point mutations and relative copy number in 1363 disease and control human brains. Acta Neuropathol Commun 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Rodríguez‐Santiago B, Casademont J and Nunes V (2001) Is mitochondrial DNA depletion involved in Alzheimer’s disease? Eur J Hum Genet 9, 279–285. [DOI] [PubMed] [Google Scholar]
- 126. Rice AC, Keeney PM, Algarzae NK, Ladd AC, Thomas RR and Bennett JP Jr (2014) Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer’s Disease Hippocampi. J Alzheimer’s Dis 40, 319–330. [DOI] [PubMed] [Google Scholar]
- 127. Gui Y‐X, Xu Z‐P, Lv W, Zhao J‐J and Hu X‐Y (2015) Evidence for polymerase gamma, POLG1 variation in reduced mitochondrial DNA copy number in Parkinson’s disease. Parkinsonism Relat Disord 21, 282–286. [DOI] [PubMed] [Google Scholar]
- 128. Lunnon K, Keohane A, Pidsley R, Newhouse S, Riddoch‐Contreras J, Thubron EB, Devall M, Soininen H, Kłoszewska I, Mecocci P et al. (2017) Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol Aging 53, 36–47. [DOI] [PubMed] [Google Scholar]
- 129. Podlesniy P, Figueiro‐Silva J, Llado A, Antonell A, Sanchez‐Valle R, Alcolea D, Lleo A, Molinuevo JL, Serra N and Trullas R (2013) Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann Neurol 74, 655–668. [DOI] [PubMed] [Google Scholar]
- 130. Pyle A, Brennan R, Kurzawa‐Akanbi M, Yarnall A, Thouin A, Mollenhauer B, Burn D, Chinnery PF and Hudson G (2015) Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early‐stage Parkinson’s disease. Ann Neurol 78, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Warburg O (1956) On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- 132. Vasan K, Werner M and Chandel NS (2020) Mitochondrial metabolism as a target for cancer therapy. Cell Metab 32, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ždralević M, Brand A, Di Ianni L, Dettmer K, Reinders J, Singer K, Peter K, Schnell A, Bruss C, Decking S‐M et al. (2018) Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J Biol Chem 293, 15947–15961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hensley CT, Faubert B, Yuan Q, Kernstine K, Lenkinski RE and Deberardinis Correspondence RJ (2016) Metabolic heterogeneity in human lung tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Momcilovic M, Jones A, Bailey ST, Waldmann CM, Li R, Lee JT, Abdelhady G, Gomez A, Holloway T, Schmid E et al. (2019) In vivo imaging of mitochondrial membrane potential in non‐small‐cell lung cancer. Nature 575, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bensard CL, Wisidagama DR, Olson KA, Berg JA, Krah NM, Schell JC, Nowinski SM, Fogarty S, Bott AJ, Wei P et al. (2020) Regulation of tumor initiation by the mitochondrial pyruvate carrier. Cell Metab 31, 284–300.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Larman TC, DePalma SR and Hadjipanayis AG, Cancer Genome Atlas Research Network TCGAR , Protopopov A, Zhang J, Gabriel SB, Chin L, Seidman CE, Kucherlapati R et al. (2012) Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci USA 109, 14087–14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yadav N and Chandra D (2013) Mitochondrial DNA mutations and breast tumorigenesis. Biochim Biophys Acta Rev Cancer 1836, 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Gorelick AN, Kim M, Chatila WK, La K, Hakimi AA and Barry S (2020) Respiratory complex and tissue lineage drive mutational patterns in the tumor mitochondrial genome.
- 140. Grandhi S, Bosworth C, Maddox W, Sensiba C, Akhavanfard S, Ni Y and LaFramboise T (2017) Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue‐specific signals of relaxed and positive selection. Hum Mol Genet 26, 2912–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Schöpf B, Weissensteiner H, Schäfer G, Fazzini F, Charoentong P, Naschberger A, Rupp B, Fendt L, Bukur V, Giese I et al. (2020) OXPHOS remodeling in high‐grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat Commun 11, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Yuan Y, Ju YS, Kim Y, Li J, Wang Y, Yoon CJ, Yang Y, Martincorena I, Creighton CJ, Weinstein JN et al. (2020) Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet 52, 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Coller HA, Khrapko K, Bodyak ND, Nekhaeva E, Herrero‐Jimenez P and Thilly WG (2001) High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat Genet 28, 147–150. [DOI] [PubMed] [Google Scholar]
- 144. Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik‐Zainal S, Ramakrishna M, Davies HR, Papaemmanuil E, Gundem G, Shlien A et al. (2014) Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 3, e02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Stewart JB, Alaei‐Mahabadi B, Sabarinathan R, Samuelsson T, Gorodkin J, Gustafsson CM and Larsson E (2015) Simultaneous DNA and RNA mapping of somatic mitochondrial mutations across diverse human cancers. PLoS Genet 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Gasparre G, Romeo G, Rugolo M and Porcelli AM (2011) Learning from oncocytic tumors: why choose inefficient mitochondria? Biochim Biophys Acta Bioenerg 1807, 633–642. [DOI] [PubMed] [Google Scholar]
- 147. Gopal RK, Calvo SE, Shih AR, Chaves FL, McGuone D, Mick E, Pierce KA, Li Y, Garofalo A, Van Allen EM et al. (2018) Early loss of mitochondrial complex I and rewiring of glutathione metabolism in renal oncocytoma. Proc Natl Acad Sci USA 115, E6283–E6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Joshi S, Tolkunov D, Aviv H, Hakimi AA, Yao M, Hsieh JJ, Ganesan S, Chan CS and White E (2015) The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep 13, 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kürschner G, Zhang Q, Clima R, Xiao Y, Busch JF, Kilic E, Jung K, Berndt N, Bulik S, Holzhütter H‐G et al. (2017) Renal oncocytoma characterized by the defective complex I of the respiratory chain boosts the synthesis of the ROS scavenger glutathione. Oncotarget 8, 105882–105904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Smith ALM, Whitehall JC, Bradshaw C, Gay D, Robertson F, Blain AP, Hudson G, Pyle A, Houghton D, Hunt M et al. (2020) Age‐associated mitochondrial DNA mutations cause metabolic remodeling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 1, 976–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Mi J, Tian G, Liu S, Li X, Ni T, Zhang L and Wang B (2015) The relationship between altered mitochondrial DNA copy number and cancer risk: a meta‐analysis. Sci Rep 5, 10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lemnrau A, Brook MN, Fletcher O, Coulson P, Tomczyk K, Jones M, Ashworth A, Swerdlow A, Orr N and Garcia‐Closas M (2015) Mitochondrial DNA copy number in peripheral blood cells and risk of developing breast cancer. Cancer Res 75, 2844–2850. [DOI] [PubMed] [Google Scholar]
- 153. Shen J, Wan J, Song R and Zhao H (2015) Peripheral blood mitochondrial DNA copy number, length heteroplasmy and breast cancer risk: a replication study. Carcinogenesis 36, 1307–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Shen J, Gopalakrishnan V, Lee JE, Fang S and Zhao H (2015) Mitochondrial DNA copy number in peripheral blood and melanoma risk. PLoS One 10, e0131649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hosgood HD, Liu C‐S, Rothman N, Weinstein SJ, Bonner MR, Shen M, Lim U, Virtamo J, Cheng W, Albanes D et al. (2010) Mitochondrial DNA copy number and lung cancer risk in a prospective cohort study. Carcinogenesis 31, 847–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Lynch SM, Weinstein SJ, Virtamo J, Lan Q, Liu C‐S, Cheng W‐L, Rothman N, Albanes D and Stolzenberg‐Solomon RZ (2011) Mitochondrial DNA copy number and pancreatic cancer in the alpha‐tocopherol beta‐carotene cancer prevention study. Cancer Prev Res 4, 1912–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Xing J, Chen M, Wood CG, Lin J, Spitz MR, Ma J, Amos CI, Shields PG, Benowitz NL, Gu J et al. (2008) Mitochondrial DNA content: its genetic heritability and association with renal cell carcinoma. J Natl Cancer Inst 100, 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Thyagarajan B, Wang R, Barcelo H, Koh W‐P and Yuan J‐M (2012) Mitochondrial copy number is associated with colorectal cancer risk. Cancer Epidemiol Biomarkers Prev 21, 1574–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Huang B, Gao Y‐T, Shu X‐O, Wen W, Yang G, Li G, Courtney R, Ji B‐T, Li H‐L, Purdue MP et al. (2014) Association of leukocyte mitochondrial DNA copy number with colorectal cancer risk: results from the Shanghai Women’s Health Study. Cancer Epidemiol Biomarkers Prev 23, 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Feng S, Xiong L, Ji Z, Cheng W and Yang H (2011) Correlation between increased copy number of mitochondrial DNA and clinicopathological stage in colorectal cancer. Oncol Lett 2, 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang Y, He S, Zhu X, Qiao W and Zhang J (2016) High copy number of mitochondrial DNA predicts poor prognosis in patients with advanced stage colon cancer. Int J Biol Markers 31, 382–388. [DOI] [PubMed] [Google Scholar]
- 162. Cui H, Huang P, Wang Z, Zhang Y, Zhang Z, Xu W, Wang X, Han Y and Guo X (2013) Association of decreased mitochondrial DNA content with the progression of colorectal cancer. BMC Cancer 13, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Lin P‐C, Lin J‐K, Yang S‐H, Wang H‐S, Li AF‐Y and Chang S‐C (2008) Expression of β‐F1‐ATPase and mitochondrial transcription factor A and the change in mitochondrial DNA content in colorectal cancer: clinical data analysis and evidence from an in vitro study. Int J Colorectal Dis 23, 1223–1232. [DOI] [PubMed] [Google Scholar]
- 164. van Osch FHM, Voets AM, Schouten LJ, Gottschalk RWH, Simons CCJM, van Engeland M, Lentjes MHFM, van den Brandt PA, Smeets HJM and Weijenberg MP (2015) Mitochondrial DNA copy number in colorectal cancer: between tissue comparisons, clinicopathological characteristics and survival. Carcinogenesis 36, bgv151. [DOI] [PubMed] [Google Scholar]
- 165. Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC et al. (2014) The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 26, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Reznik E, Miller ML, Şenbabaoğlu Y, Riaz N, Sarungbam J, Tickoo SK, Al‐Ahmadie HA, Lee W, Seshan VE, Hakimi AA et al. (2016) Mitochondrial DNA copy number variation across human cancers. Elife 5, e10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. McMahon S and LaFramboise T (2014) Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 35, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Weerts MJA, Sieuwerts AM, Smid M, Look MP, Foekens JA, Sleijfer S and Martens JWM (2016) Mitochondrial DNA content in breast cancer: impact on in vitro and in vivo phenotype and patient prognosis. Oncotarget 7, 29166–29176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Yu M, Zhou Y, Shi Y, Ning L, Yang Y, Wei X, Zhang N, Hao X and Niu R (2007) Research communication reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in chinese breast cancer patients. IUBMB Life 59, 450–457. [DOI] [PubMed] [Google Scholar]
- 170. Zhang Y, Qu Y, Gao K, Yang Q, Shi B, Hou P and Ji M (2015) High copy number of mitochondrial DNA (mtDNA) predicts good prognosis in glioma patients. Am J Cancer Res 5, 1207–1216. [PMC free article] [PubMed] [Google Scholar]
- 171. Yu M, Wan Y and Zou Q (2010) Decreased copy number of mitochondrial DNA in Ewing’s sarcoma. Clin Chim Acta 411, 679–683. [DOI] [PubMed] [Google Scholar]
- 172. Yamada S, Nomoto S, Fujii T, Kaneko T, Takeda S, Inoue S, Kanazumi N and Nakao A (2006) Correlation between copy number of mitochondrial DNA and clinico‐pathologic parameters of hepatocellular carcinoma. Eur J Surg Oncol 32, 303–307. [DOI] [PubMed] [Google Scholar]
- 173. Guo W, Jiang L, Bhasin S, Khan SM and Swerdlow RH (2009) DNA extraction procedures meaningfully influence qPCR‐based mtDNA copy number determination. Mitochondrion 9, 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Hurtado‐Roca Y, Ledesma M, Gonzalez‐Lazaro M, Moreno‐Loshuertos R, Fernandez‐Silva P, Enriquez JA and Laclaustra M (2016) Adjusting MtDNA quantification in whole blood for peripheral blood platelet and leukocyte counts. PLoS One 11, e0163770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Urata M, Koga‐Wada Y, Kayamori Y and Kang D (2008) Platelet contamination causes large variation as well as overestimation of mitochondrial DNA content of peripheral blood mononuclear cells. Ann Clin Biochem 45, 513–514. [DOI] [PubMed] [Google Scholar]
- 176. Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T and Götz M (2008) Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci USA 105, 3581–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Dudley GA, Abraham WM and Terjung RL (1982) Influence of exercise intensity and duration on biochemical adaptations in skeletal muscle. J Appl Physiol Respir Environ Exerc Physiol 53, 844–850. [DOI] [PubMed] [Google Scholar]
- 178. Fink WJ, Costill DL and Pollock ML (1977) Submaximal and Maximal working capacity of elite distance runners. Part II. Muscle fiber composition and enzyme activities. Ann N Y Acad Sci 301, 323–327. [DOI] [PubMed] [Google Scholar]
- 179. Dikic I and Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19, 349–364. [DOI] [PubMed] [Google Scholar]
- 180. Rabanal‐Ruiz Y, Otten EG and Korolchuk VI (2017) mTORC1 as the main gateway to autophagy. Essays Biochem 61, 565–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Barriocanal‐Casado E, Hidalgo‐Gutiérrez A, Raimundo N, González‐García P, Acuña‐Castroviejo D, Escames G and López LC (2019) Rapamycin administration is not a valid therapeutic strategy for every case of mitochondrial disease. EBioMedicine 42, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Civiletto G, Dogan SA, Cerutti R, Fagiolari G, Moggio M, Lamperti C, Benincá C, Viscomi C and Zeviani M (2018) Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol Med 10, e8799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Johnson SC, Yanos ME, Kayser E‐B, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A et al. (2013) mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet‐dit‐Félix AA, Williams EG, Jha P, Lo Sasso G, Huzard D et al. (2016) Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med 22, 879–888. [DOI] [PubMed] [Google Scholar]
- 185. Alsina D, Lytovchenko O, Schab A, Atanassov I, Schober FA, Jiang M, Koolmeister C, Wedell A, Taylor RW, Wredenberg A et al. (2020) FBXL 4 deficiency increases mitochondrial removal by autophagy. EMBO Mol Med 12, e11659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Viscomi C (2016) Toward a therapy for mitochondrial disease. Biochem Soc Trans 44, 1483–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H and Larsson NG (2002) Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci USA 99, 15066–15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC et al. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC‐1. Cell 98, 115–124. [DOI] [PubMed] [Google Scholar]
- 189. Evans MJ and Scarpulla RC (1989) Interaction of nuclear factors with multiple sites in the somatic cytochrome c promoter. Characterization of upstream NRF‐1, ATF, and intron Sp1 recognition sequences. J Biol Chem 264, 14361–14368. [PubMed] [Google Scholar]
- 190. Evans MJ and Scarpulla RC (1990) NRF‐1: a trans‐activator of nuclear‐encoded respiratory genes in animal cells. Genes Dev 4, 1023–1034. [DOI] [PubMed] [Google Scholar]
- 191. Virbasius JV and Scarpulla RC (1994) Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci USA 91, 1309–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Jäer S, Handschin C, St‐Pierre J and Spiegelman BM (2007) AMP‐activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC‐1α. Proc Natl Acad Sci USA 104, 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM and Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC‐1α and SIRT1. Nature 434, 113–118. [DOI] [PubMed] [Google Scholar]
- 194. Viscomi C and Zeviani M (2020) Strategies for fighting mitochondrial diseases. J Intern Med 287, 665–684. [DOI] [PubMed] [Google Scholar]
- 195. Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C and Zeviani M (2011) In vivo correction of COX deficiency by activation of the AMPK/PGC‐1α axis. Cell Metab 14, 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Dillon LM, Williams SL, Hida A, Peacock JD, Prolla TA, Lincoln J and Moraes CT (2012) Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Hum Mol Genet 21, 2288–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Dillon LM, Hida A, Garcia S, Prolla TA and Moraes CT (2012) Long‐term bezafibrate treatment improves skin and spleen phenotypes of the mtDNA mutator mouse. PLoS One 7, e44335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Hofer A, Noe N, Tischner C, Kladt N, Lellek V, Schauß A and Wenz T (2013) Defining the action spectrum of potential PGC‐1α activators on a mitochondrial and cellular level in vivo . Hum Mol Genet 23, 2400–2415. [DOI] [PubMed] [Google Scholar]
- 199. Yatsuga S and Suomalainen A (2011) Effect of bezafibrate treatment on late‐onset mitochondrial myopathy in mice. Hum Mol Genet 21, 526–535. [DOI] [PubMed] [Google Scholar]
- 200. Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J et al. (2014) NAD(+)‐dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab 19, 1042–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Steele H, Gomez‐Duran A, Pyle A, Hopton S, Newman J, Stefanetti RJ, Charman SJ, Parikh JD, He L, Viscomi C et al. (2020) Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol Med 12, e11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Pirinen E, Auranen M, Khan NA, Brilhante V, Urho N, Pessia A, Hakkarainen A, Kuula J, Heinonen U, Schmidt MS et al. (2020) Niacin cures systemic NAD+ deficiency and improves muscle performance in adult‐onset mitochondrial myopathy. Cell Metab 31, 1078–1090.e5. [DOI] [PubMed] [Google Scholar]
- 203. Fernandez‐Marcos PJ and Auwerx J (2011) Regulation of PGC‐1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93, 884S–890S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA and Kelly DP (2004) Cardiac‐specific induction of the transcriptional coactivator peroxisome proliferator‐activated receptor γ coactivator‐1α promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage‐dependent manner. Circ Res 94, 525–533. [DOI] [PubMed] [Google Scholar]
- 205. Miura S, Tomitsuka E, Kamei Y, Yamazaki T, Kai Y, Tamura M, Kita K, Nishino I and Ezaki O (2006) Overexpression of peroxisome proliferator‐activated receptor gamma co‐activator‐1alpha leads to muscle atrophy with depletion of ATP. Am J Pathol 169, 1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Ciron C, Lengacher S, Dusonchet J, Aebischer P and Schneider BL (2012) Sustained expression of PGC‐1α in the rat nigrostriatal system selectively impairs dopaminergic function. Hum Mol Genet 21, 1861–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Lindholm D, Eriksson O, Mäkelä J, Belluardo N and Korhonen L (2012) PGC‐1α: a master gene that is hard to master. Cell Mol Life Sci 69, 2465–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Jackson CB, Turnbull DM, Minczuk M and Gammage PA (2020) Therapeutic manipulation of mtDNA heteroplasmy: a shifting perspective. Trends Mol Med 26, 698–709. [DOI] [PubMed] [Google Scholar]
- 209. Nissanka N and Moraes CT (2020) Mitochondrial DNA heteroplasmy in disease and targeted nuclease‐based therapeutic approaches. EMBO Rep 21, e49612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C and Rossmanith W (2008) RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135, 462–474. [DOI] [PubMed] [Google Scholar]
- 211. Rackham O, Busch JD, Matic S, Siira SJ, Kuznetsova I, Atanassov I, Ermer JA, Shearwood AMJ, Richman TR, Stewart JB et al. (2016) Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep 16, 1874–1890. [DOI] [PubMed] [Google Scholar]
- 212. Amunts A, Brown A, Toots J, Scheres SHW and Ramakrishnan V (2015) The structure of the human mitochondrial ribosome. Science 348, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, Aebersold R and Ban N (2014) The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 515, 283–286. [DOI] [PubMed] [Google Scholar]
- 214. Sacconi S, Salviati L, Nishigaki Y, Walker WF, Hernandez‐Rosa E, Trevisson E, Delplace S, Desnuelle C, Shanske S, Hirano M et al. (2008) A functionally dominant mitochondrial DNA mutation. Hum Mol Genet 17, 1814–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Bayona‐Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA and Moraes CT (2005) Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc Natl Acad Sci USA 102, 14392–14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Peeva V, Blei D, Trombly G, Corsi S, Szukszto MJ, Rebelo‐Guiomar P, Gammage PA, Kudin AP, Becker C, Altmüller J et al. (2018) Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat Commun 9, 1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Alexeyev MF, Venediktova N, Pastukh V, Shokolenko I, Bonilla G and Wilson GL (2008) Selective elimination of mutant mitochondrial genomes as therapeutic strategy for the treatment of NARP and MILS syndromes. Gene Ther 15, 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Tanaka M, Borgeld H‐J, Zhang J, Muramatsu S, Gong J‐S, Yoneda M, Maruyama W, Naoi M, Ibi T, Sahashi K et al. (2002) Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J Biomed Sci 9, 534–541. [DOI] [PubMed] [Google Scholar]
- 219. Bacman SR, Williams SL, Duan D and Moraes CT (2012) Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9‐mediated delivery of a mitochondria‐targeted restriction endonuclease. Gene Ther 19, 1101–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Bacman SR, Williams SL, Garcia S and Moraes CT (2010) Organ‐specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria‐targeted restriction endonuclease. Gene Ther 17, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Bacman SR, Williams SL, Pinto M, Peralta S and Moraes CT (2013) Specific elimination of mutant mitochondrial genomes in patient‐derived cells by mitoTALENs. Nat Med 19, 1111–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Gammage PA, Rorbach J, Vincent AI, Rebar EJ and Minczuk M (2014) Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large‐scale deletions or point mutations. EMBO Mol Med 6, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Hashimoto M, Bacman SR, Peralta S, Falk MJ, Chomyn A, Chan DC, Williams SL and Moraes CT (2015) MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol Ther 23, 1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Bacman SR, Kauppila JHK, Pereira CV, Nissanka N, Miranda M, Pinto M, Williams SL, Larsson N‐G, Stewart JB and Moraes CT (2018) MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat Med 24, 1696–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Gammage PA, Viscomi C, Simard M‐L, Costa ASH, Gaude E, Powell CA, Van Haute L, McCann BJ, Rebelo‐Guiomar P, Cerutti R et al. (2018) Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo . Nat Med 24, 1691–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Nishiyama S, Shitara H, Nakada K, Ono T, Sato A, Suzuki H, Ogawa T, Masaki H, Hayashi J‐I and Yonekawa H (2010) Over‐expression of Tfam improves the mitochondrial disease phenotypes in a mouse model system. Biochem Biophys Res Commun 401, 26–31. [DOI] [PubMed] [Google Scholar]
- 227. Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K et al. (2005) Overexpression of mitochondrial transcription factor A ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation 112, 683–690. [DOI] [PubMed] [Google Scholar]
- 228. Oka S, Leon J, Sakumi K, Ide T, Kang D, LaFerla FM and Nakabeppu Y (2016) Human mitochondrial transcriptional factor A breaks the mitochondria‐mediated vicious cycle in Alzheimer’s disease. Sci Rep 6, 37889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Hayashi Y, Yoshida M, Yamato M, Ide T, Wu Z, Ochi‐Shindou M, Kanki T, Kang D, Sunagawa K, Tsutsui H et al. (2008) Reverse of age‐dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J Neurosci 28, 8624–8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Ylikallio E, Tyynismaa H, Tsutsui H, Ide T and Suomalainen A (2010) High mitochondrial DNA copy number has detrimental effects in mice. Hum Mol Genet 19, 2695–2705. [DOI] [PubMed] [Google Scholar]
- 231. Zelenaya‐Troitskaya O, Newman SM, Okamoto K, Perlman PS and Butow RA (1998) Functions of the high mobility group protein, Abf2p, in mitochondrial DNA segregation, recombination and copy number in Saccharomyces cerevisiae . Genetics 148, 1763–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Thompson K, Majd H, Dallabona C, Reinson K, King MS, Alston CL, He L, Lodi T, Jones SA, Fattal‐Valevski A et al. (2016) Recurrent de novo dominant mutations in SLC25A4 cause severe early‐onset mitochondrial disease and loss of mitochondrial DNA copy number. Am J Hum Genet 99, 860–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S and Elpeleg O (2001) Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet 29, 342–344. [DOI] [PubMed] [Google Scholar]
- 234. Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M et al. (2001) The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet 29, 337–341. [DOI] [PubMed] [Google Scholar]
- 235. Giordano C, Sebastiani M, De Giorgio R, Travaglini C, Tancredi A, Valentino ML, Bellan M, Cossarizza A, Hirano M, D’Amati G et al. (2008) Gastrointestinal dysmotility in mitochondrial neurogastrointestinal encephalomyopathy is caused by mitochondrial DNA depletion. Am J Pathol 173, 1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Spinazzola A, Viscomi C, Fernandez‐Vizarra E, Carrara F, D’Adamo P, Calvo S, Marsano RM, Donnini C, Weiher H, Strisciuglio P et al. (2006) MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet 38, 570–575. [DOI] [PubMed] [Google Scholar]
- 237. Elpeleg O, Miller C, Hershkovitz E, Bitner‐Glindzicz M, Bondi‐Rubinstein G, Rahman S, Pagnamenta A, Eshhar S and Saada A (2005) Deficiency of the ADP‐Forming Succinyl‐CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet 76, 1081–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Van Hove JLK, Saenz MS, Thomas JA, Gallagher RC, Lovell MA, Fenton LZ, Shanske S, Myers SM, Wanders RJA, Ruiter J et al. (2010) Succinyl‐CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy. Pediatr Res 68, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Renaldo F, Amati‐Bonneau P, Slama A, Romana C, Forin V, Doummar D, Barnerias C, Bursztyn J, Mayer M, Khouri N et al. (2012) MFN2, a new gene responsible for mitochondrial DNA depletion. Brain 135, e223. [DOI] [PubMed] [Google Scholar]
- 240. Rouzier C, Bannwarth S, Chaussenot A, Chevrollier A, Verschueren A, Bonello‐Palot N, Fragaki K, Cano A, Pouget J, Pellissier J et al. (2012) The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain 135, 23–24. [DOI] [PubMed] [Google Scholar]
- 241. Bonnen PEM, Yarham JW, Besse A, Wu P, Faqeih EA, Al‐Asmari AM, Saleh MAM, Eyaid W, Hadeel A, He L et al. (2013) Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet 93, 471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Gai X, Ghezzi D, Johnson MA, Biagosch CA, Shamseldin HE, Haack TB, Reyes A, Tsukikawa M, Sheldon CA, Srinivasan S et al. (2013) Mutations in FBXL4, encoding a mitochondrial protein, cause early‐onset mitochondrial encephalomyopathy. Am J Hum Genet 93, 482–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Davidzon G, Mancuso M, Ferraris S, Quinzii C, Hirano M, Peters HL, Kirby D, Thorburn DR and DiMauro S (2005) POLG mutations and Alpers syndrome. Ann Neurol 57, 921–923. [DOI] [PubMed] [Google Scholar]
- 244. Varma H, Faust PL, Iglesias AD, Lagana SM, Wou K, Hirano M, DiMauro S, Mansukani MM, Hoff KE, Nagy PL et al. (2016) Whole exome sequencing identifies a homozygous POLG2 missense variant in an infant with fulminant hepatic failure and mitochondrial DNA depletion. Eur J Med Genet 59, 540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A and Lonnqvist T (2007) Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain 130, 3032–3040. [DOI] [PubMed] [Google Scholar]
- 246. Sarzi E, Goffart S, Serre V, Chrétien D, Slama A, Munnich A, Spelbrink JN and Rötig A (2007) Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann Neurol 62, 579–587. [DOI] [PubMed] [Google Scholar]
- 247. Del Dotto V, Ullah F, Di Meo I, Magini P, Gusic M, Maresca A, Caporali L, Palombo F, Tagliavini F, Baugh EH et al. (2019) SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder. J Clin Invest 130, 108–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Shen J, Song R, Lu Z and Zhao H (2016) Mitochondrial DNA copy number in whole blood and glioma risk: a case control study. Mol Carcinog 55, 2089–2094. [DOI] [PubMed] [Google Scholar]
- 249. Lin C‐S, Wang L‐S, Tsai C‐M and Wei Y‐H (2008) Low copy number and low oxidative damage of mitochondrial DNA are associated with tumor progression in lung cancer tissues after neoadjuvant chemotherapy. Interact Cardiovasc Thorac Surg 7, 954–958. [DOI] [PubMed] [Google Scholar]
- 250. Lee H, Yin P, Lin J, Chen C, Chi C, Tam T and Wei Y (2005) Mitochondrial genome instability and mtDNA depletion in human cancers. Ann NY Acad Sci 1042, 109–122. [DOI] [PubMed] [Google Scholar]
- 251. Dasgupta S, Soudry E, Mukhopadhyay N, Shao C, Yee J, Lam S, Lam W, Zhang W, Gazdar AF, Fisher PB et al. (2012) Mitochondrial DNA mutations in respiratory complex‐I in never‐smoker lung cancer patients contribute to lung cancer progression and associated with EGFR gene mutation. J Cell Physiol 227, 2451–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Wu C‐W, Yin P‐H, Hung W‐Y, Li AF‐Y, Li S‐H, Chi C‐W, Wei Y‐H and Lee H‐C (2005) Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosom Cancer 44, 19–28. [DOI] [PubMed] [Google Scholar]
- 253. Datta S, Chattopadhyay E, Ray JG, Majumder M, Das RP and Roy B (2015) D‐loop somatic mutations and ∼5 kb “common” deletion in mitochondrial DNA: important molecular markers to distinguish oral precancer and cancer. Tumor Biol 36, 3025–3033. [DOI] [PubMed] [Google Scholar]