

Abstract
Purpose:
Natural killer (NK) cell recognition and function against NK-resistant cancers remains substantial barriers to the broad application of NK cell immunotherapy. Potential solutions include bispecific engagers that target NK cell activity via an NK activating receptor when simultaneously targeting a tumor-specific antigen, as well as enhancing functionality using IL-12/15/18 cytokine pre-activation.
Experimental Design:
We assessed single-cell NK cell responses stimulated by the tetravalent bispecific antibody AFM13 that binds CD30 on leukemia/lymphoma targets and CD16A on various types of NK cells using mass cytometry and cytotoxicity assays. The combination of AFM13 and IL-12/15/18 pre-activation of blood and cord-blood-derived NK cells was investigated in vitro and in vivo.
Results:
We found heterogeneity within AFM13-directed conventional blood NK cell (cNK) responses, as well as consistent AFM13-directed polyfunctional activation of mature NK cells across donors. NK cell source also impacted the AFM13 response, with cNK cells from healthy donors exhibiting superior responses to those from Hodgkin lymphoma patients. IL-12/15/18-induced memory-like NK cells from peripheral blood exhibited enhanced killing of CD30+ lymphoma targets directed by AFM13, compared to cNK cells. Cord-blood NK cells pre-activated with IL-12/15/18 and ex vivo expanded with K562-based feeders also exhibited enhanced killing with AFM13 stimulation via upregulation of signaling pathways related to NK cell effector function. AFM13-NK complex cells exhibited enhanced responses to CD30+ lymphomas in vitro and in vivo.
Conclusions:
We identify AFM13 as a promising combination with cytokine-activated adult blood or cord blood NK cells to treat CD30+ hematologic malignancies, warranting clinical trials with these novel combinations.
Introduction
Cancer progression is associated with an ineffective antitumor response, mediated by multiple mechanisms, including downregulation of HLA antigens (1), upregulation of checkpoints, and secretion of suppressive cytokines (2–4). Therefore, promising immunotherapy strategies have been developed to enhance the activity of T and NK cells against cancer targets, including the use of cytokines, checkpoint inhibitors, genetic engineering of cells to express a chimeric antigen receptor, or the use of bispecific antibodies (5–12).
Natural killer (NK) cells are cytotoxic innate lymphoid cells that play an important role in immunosurveillance and display potent effector responses against infected, stressed and malignant cells (13). Unlike T cells, NK cells do not express antigen-specific receptors to recognize their target; instead, their activation and effector functions are mediated either by interactions between germline encoded activating or inhibitory receptors and their respective ligands on target cells (14,15), including CD16A (FcγRIIIa)-mediated antibody-dependent cellular cytotoxicity (ADCC) and cytokine production (16,17). NK cell functionality and activation thresholds are also modulated via cytokine receptors (18).
Deficiency in NK number and function is associated with cancer development (19). NK cells from patients with cancer have impaired effector function as shown in multiple disease settings (2,10,20). Mechanisms include downregulation of activating receptors, loss of CD16 expression, upregulation of inhibitory receptors, and induction of exhaustion by the tumor microenvironment (21–23). For these reasons, many investigators have explored the use of allogeneic NK cells for immunotherapy and have shown these cells to be safe and effective at killing cancers, especially myeloid malignancies (6,24,25).
Allogeneic NK cells can be derived from multiple sources, each with their unique biology (5,26). Some of the most commonly used sources of allogeneic NK cells currently being investigated in the clinic include peripheral blood (PB) from healthy donors and cord blood (CB) (26,27). PB-NK cells may be used with minimal cytokine activation (24), following expansion ex vivo in the presence of cytokines with or without feeder cells (28,29), or can be briefly activated with IL-12, IL-15, and IL-18 to induce differentiation into memory-like NK cells (30), with enhanced responses when stimulated through cytokine or activating receptors, including CD16 (31,32). This is a promising strategy that was shown to be safe and induced remissions in patients with high-risk AML (6,12). CB-derived NK cells are usually expanded ex vivo to generate sufficient number of cells for infusion (27–29). A novel approach with CAR19/IL15-engineered CB-NK cells was recently reported as safe and able to induce complete remission in patients with relapsed or refractory B cell malignancies (33).
Immune cell engagers have been developed to overcome barriers related to deficient cancer recognition and NK cell activation. AFM13 is a tetravalent bispecific antibody construct with two binding sites for each CD30 and CD16A, designed for the treatment of CD30+ malignancies. It binds CD16A on the surface of NK cells, thus activating and recruiting them to CD30 expressing tumor cells to mediate tumor cell killing (34,35). Here, we investigate the use of AFM13 to endow NK cells with anti-CD30 specificity coupled with a high affinity CD16A trigger, analyzing single NK cell responses via mass cytometry. Furthermore, we evaluate combining AFM13 with cytokine-induced memory-like differentiation with both adult PB-NK cells and with expanded CB-NK cells, to identify promising strategies to translate into the clinic for CD30+ leukemias and lymphomas.
Materials and Methods
NK cells and cell lines
Human PBMC from anonymous healthy donors were obtained from leukoreduction filters after platelet apheresis and isolated by Ficoll density gradient centrifugation. Cord blood (CB) units for research were obtained from the MD Anderson Cancer Center Cord Blood Bank. PBMC from patients with Hodgkin lymphoma were collected on an IRB-approved clinical protocol (Lab00–099). CB mononuclear cells were isolated from fresh CB units by Ficoll density gradient centrifugation. The protocol for the ex vivo expansion of CB NK cells using K562 feeder cells was previously published (7,33,36). Cytokine induced memory-like (ML) NK cells were generated from conventional peripheral blood NK cells (cNK) utilizing activation (16 hours) with rhIL-12, rhIL-15, and rhIL-18 as previously described (6). After the activation, NK cells were washed, and cultured for 6 days to allow memory-like differentiation. Pre-activated expanded (P+E) CB-NK cells were generated following the same pre-activation protocol with IL-12, IL-15 and IL-18 prior to expansion with uAPC and IL-2 as above.
The tumor cell lines Karpas 299 and HuT-78 cells were chosen for this study based on their high expression of CD30. Karpas 299 was purchased from Sigma-Aldrich (St. Louis, MO). Daudi, K562 and Raji cells were obtained from American Type Culture Collection (ATCC) (Manassas, VA). The HuT-78 cell line was provided by John DiPersio (Washington University, St Louis). The cell lines were cultured in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin and 1% L-glutamine. Karpas 299 and Daudi cells were used as targets for the cytotoxicity assays. All cells lines were maintained according to ATCC instructions and were Mycoplasma negative.
NK cell functional assays
Details of the flow cytometry to measure cytokine production and degranulation, 51Chromium (51Cr)-release assay and Incucyte real-time assay are described in details in the Supplementary Methods.
Detection of AFM13
For immunofluorescence detection of AFM13 on the surface of NK cells, CB-NK cells were loaded with AFM13 for 1h, washed, and cultured for another 24 hours to 72 hours at 37°C in the presence of IL-2, harvested, and stained with rat anti-AFM13 (clone 7, Affimed), followed by staining with goat anti-rat IgG (Alexa Fluor 647, Jackson ImmunoReseaech). Images were obtained using imaging flow cytometry (Amnis Image Stream Mark II) and analyzed by the Ideas software (Luminex). The retention of AFM13 on the surface of NK cells was determined by the co-localization of CD16 and AFM13 signals. Imaging flow cytometry (Amnis Image Stream Mark II) was also used to assess immunological synapse formation between AFM13-loaded CB-NK cells and Karpas 299 by staining the cells with a biotinyalated anti-pericentrin primary antibody (Novusbio) followed by streptavidin-conjugated APC-Cy7 (Biolegend) and AF 488-conjugated phalloidin (Thermofisher) to measure F-actin. Images were analyzed with the IDEAS software (Miltenyi Biotech).
RNA sequencing
Total RNA was collected from 5 million expanded CB-NK cells (with or without pre-activation) and extracted and purified using RNeasy Plus Mini Kit (Qiagen). SmartSeq2 RNA-seq libraries were prepared and data analyzed as described in the Supplementary Methods.
Mass Cytometry
Supplementary Tables S1 and S2 list antibodies used for the characterization of PB-NK cells and CB-NK cells respectively, and custom conjugations were described (6,37). Mass cytometry data were collected on a CyTOF2 mass cytometer (Fluidigm) and analyzed using Cytobank as previously described (6,38–40). Detailed information regarding staining, acquisition, and analysis is provided in Supplementary Methods.
Xenograft models
NSG mice (10–12 weeks old; Jackson Laboratories, ME) were irradiated with 300 cGy and inoculated intravenously (i.v.) with FFLuc-labeled Karpas cells (1×105) on day 0. Treatment groups received 10×106 CB-NK cells or CB-NK cells loaded with AFM13 or 100 μg/ml of AFM13 alone through tail vein injection on day 0. Mice received recombinant human IL-2 injection intraperitoneally (i.p.) three times per week (10,000 units/mouse). Mice were subjected to weekly bioluminescence imaging (BLI; Xenogen-IVIS 200 Imaging system; Caliper, Waltham, MA).
Statistical analysis
The Student’s t-test was used to compare quantitative variables. Two-way ANOVA was used for comparison between samples and longitudinal timepoints followed by Bonferroni’s or Sidak’s multiple comparisons. Probabilities of survival were calculated using the Kaplan Meier method. Statistical significance was assessed with the Prism 8.0 software (GraphPad Software, Inc.). p values <0.05 were considered significant.
Results
Mass cytometry reveals AFM13-induced polyfunctional conventional NK cell responses against CD30+ malignancies
The bispecific nature of AFM13 supports activation of NK cells through its CD16 binding domain along with an enhanced tumor recognition mediated by CD30 binding on the tumor cells. However, the quality of single NK cell responses within a heterogeneous NK cell population to respond to CD16 engagement with AFM13 remains unclear. To confirm the effect of AFM13 in enhancing tumor recognition and NK cell functionality (34,35,41), conventional NK cells (cNK) from healthy donor PBMC were incubated with HuT-78 (CD30+) with or without AFM13 labeling. Flow cytometry confirmed a specific and robust increase in IFN-γ, TNF, and surface CD107+ positive cells NK cells with the HuT-78 plus AFM13 condition (Supplementary Fig. S1A–D). Control stimulation with Raji cells (CD30 negative) or Raji plus AFM13 did not induce significant responses by cNK cells (Supplementary Fig. S1A–D). We hypothesized that the underlying NK cell repertoire and subsets affect AFM13-triggered responses. To define the multidimensional profiles of cNK cells responding to AFM13, mass cytometry was employed to assess the expression of phenotypic and function-relevant NK cell markers modulated upon AFM13 triggering (Supplementary Table S1). AFM12 (binding to CD16A and CD19) was used as negative control, as HuT-78 cells do not express CD19 (Fig. 1A). Density viSNE maps (Fig. 1B) demonstrate that HuT-78+AFM13 stimulation induced a unique multidimensional profile, which was markedly altered compared to control conditions (unstimulated, HuT-78, or HuT-78+AFM12). The composite viSNE plot highlights these differences, where HuT-78+AFM13 stimulated cells are predominantly located in a distinct island (Fig. 1B) and serves as a reference for the investigation of function (Fig. 1C). Functional mass cytometry analysis showed that HuT-78+AFM13 significantly induced IFN-γ, TNF, MIP1α and surface CD107a (as a surrogate of degranulation) in cNK cells (Fig. 1C–D). As expected, AFM12 did not upregulate cytokine secretion or degranulation (Fig. 1D), indicating that enhanced NK cell functionality upon AFM13 triggering is related to simultaneous binding of both CD16A on NK cells and CD30 on target cells, rather than CD16A binding alone on the NK cells. The short-term functional changes induced by AFM13-triggering correlated with activation-related decreases of CD16A and CD62L and upregulation of CD25 and more modestly, NKp44 (Fig. 1E). We also investigated the impact of AFM13 on the quality of the NK cell response measured via polyfunction (combinations of IFN-α, TNF, MIP1α and CD107a). HuT-78+AFM13 was found to upregulate polyfunctional responses in cNK cells while HuT-78+AFM12 activated NK cells with single functional features (Fig. 1F). Thus, mass cytometry functional assays identify highly activated NK cell islands upon AFM13 triggering, and an increase in polyfunctional NK cells, compared to control conditions.
Fig. 1.
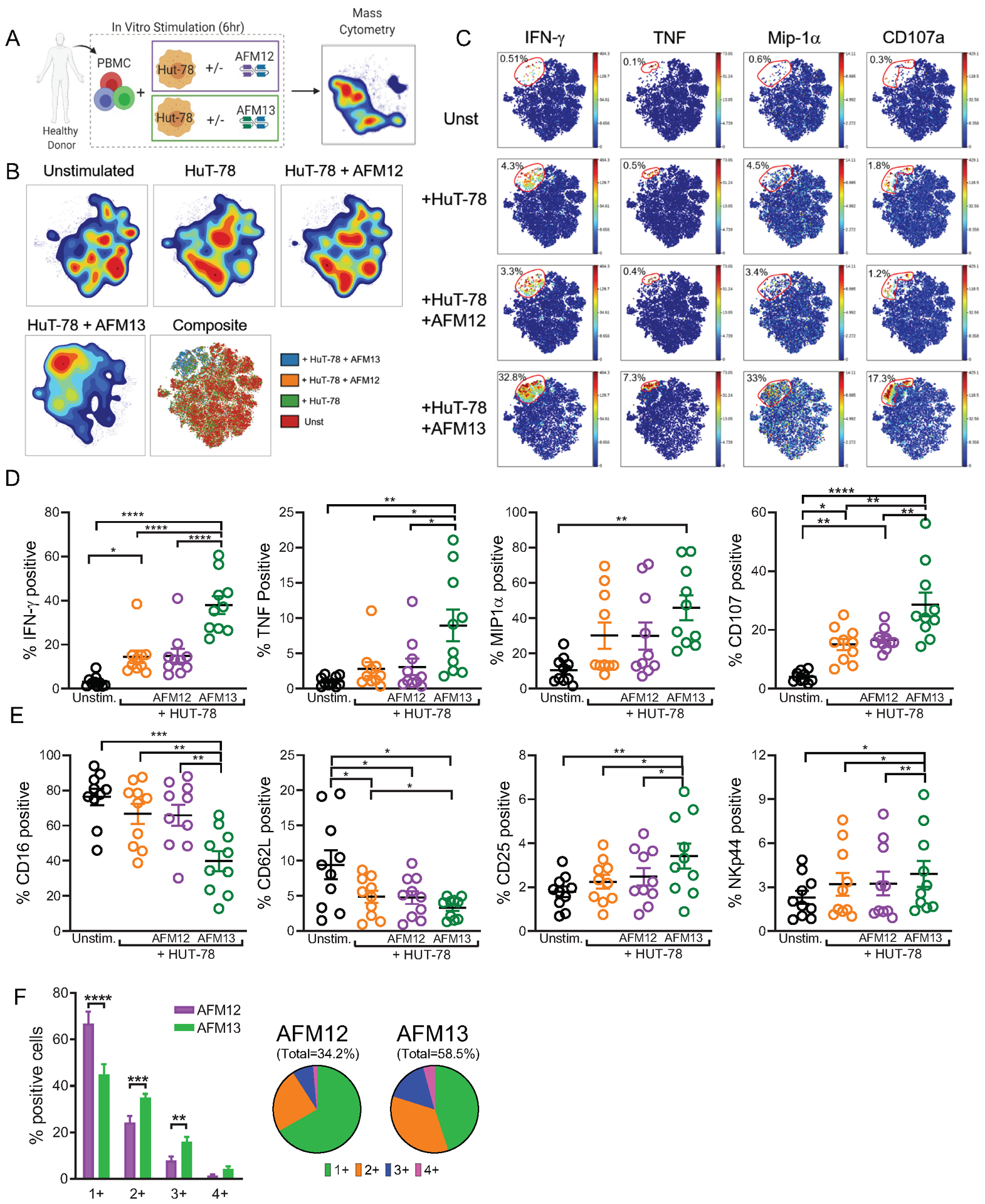
AFM13 triggering induces cell activation and polyfunctional responses of human cNK cells, mostly with a mature phenotype. (A) Schema of cell stimulation used to evaluate NK cells upon AFM13 triggering by CyTOF. (B) Representative density viSNE maps show NK cells unstimulated and stimulated with Hut-78 cells, Hut-78 cells + AFM12 and Hut-78 cells + AFM13. Composite map shows a differential clustering of HuT-78 cells + AFM13. (C) ViSNE maps of a representative donor and summary data (D) show IFN-γ, TNF, MIP1α, CD107 and CD16 expression on NK cells unstimulated or stimulated with HuT-78, HuT-78 + AFM12 and HuT-78 + AFM13. (E) Percent of CD16, CD62L, CD25 and NKp44 positive NK cells unstimulated and stimulated with HuT-78 cells, HuT-78 + AFM12 or HuT-78 + AFM13. (F) Percent of NK cells single or multiple producers of IFN-γ, TNF, MIP1α or CD107 after stimulation with HuT-78 + AFM12 and HuT-78 + AFM13. Numbers under each pie represent the frequency of positive cells for 1, 2, 3 or all the 4 molecules. Bars represent mean + SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. One-way ANOVA with Tukey pots-hoc test. n=10.
NK subset and NK receptor repertoire impacts AFM13 triggered NK cell responses
To examine the phenotype of NK cells responding to AFM13, SPADE analysis was employed with clustering of 17 markers associated with NK cell maturation and differentiation (Fig. 2A and Table S1). Visualization trees from SPADE analysis were used to identify nodes with higher and lower IFN-γ median expression in response to AFM13 within individual NK cell donors (Fig. 2B–D). Within the IFN-γhigh nodes, we identified mature (KIR+CD57+) and immature NK cells (NKG2A/CD94+) responding to AFM13 triggering as demonstrated in three representative donors. In one donor, expression of KIR3DL1 marked NK cells with the highest production of IFN-γ (node 1, Fig. 2B), but minimal KIR2DL2/3. Since HuT-78 lack the HLA-Bw4 ligand for KIR3DL1, this result suggests that licensed KIR3DL1+ NK cells that lack their inhibitory ligand (HuT-78 lack HLA-Bw4 motifs) exhibited the strongest response when triggered with AFM13 in this individual. In addition, node 8 in this donor and nodes 4 and 10 in a second donor (Fig. 2C) also exhibited a robust IFN-γ expression and expressed markers of terminally matured NK cells (CD57+KIR2DL2/3+NKG2A−). In a different individual (Fig. 2D), nodes with the greatest IFN-γ production (2 and 3), expressed CD57, CD94, NKG2A, and variable KIR3DL2/3. This demonstrates that both immature and mature NK cells responded robustly to AFM13-triggering. CD16 expression was found to be similar in both CD56dim mature (NKG2A−CD57+) and CD56dim immature (NKG2A+CD94+KIR−CD57−) NK cells and both subsets were equally triggered by AFM13 to produce similar levels of IFN-γ (Supplementary Fig. S2). When analyzing cNK cells from all individuals tested in mass cytometry functional assays, IFN-γhigh nodes had additional functional markers (e.g., MIP1α, CD107a degranulation) (Fig. 2E), and were enriched for mature (CD57+KIR+) NK cells, compared to IFN-γlow NK cells (Fig. 2F). Thus, blood cNK cells exhibited heterogeneous responses within and across multiple individuals; advanced NK cell maturation state and potentially loss of iKIR ligands were associated with AFM13-triggered responses. In addition, the correlative evidence of KIR and HLA-Bw4 interaction supports a role for specific inhibitory receptor and ligand interactions impacting AFM13-triggered responses in vitro. This will be further evaluated in future studies that manipulate inhibitory receptors or their ligands within functional assays.
Fig. 2. AFM13-induced functional response is impacted by underlying NK cell subsets and NK repertoire.
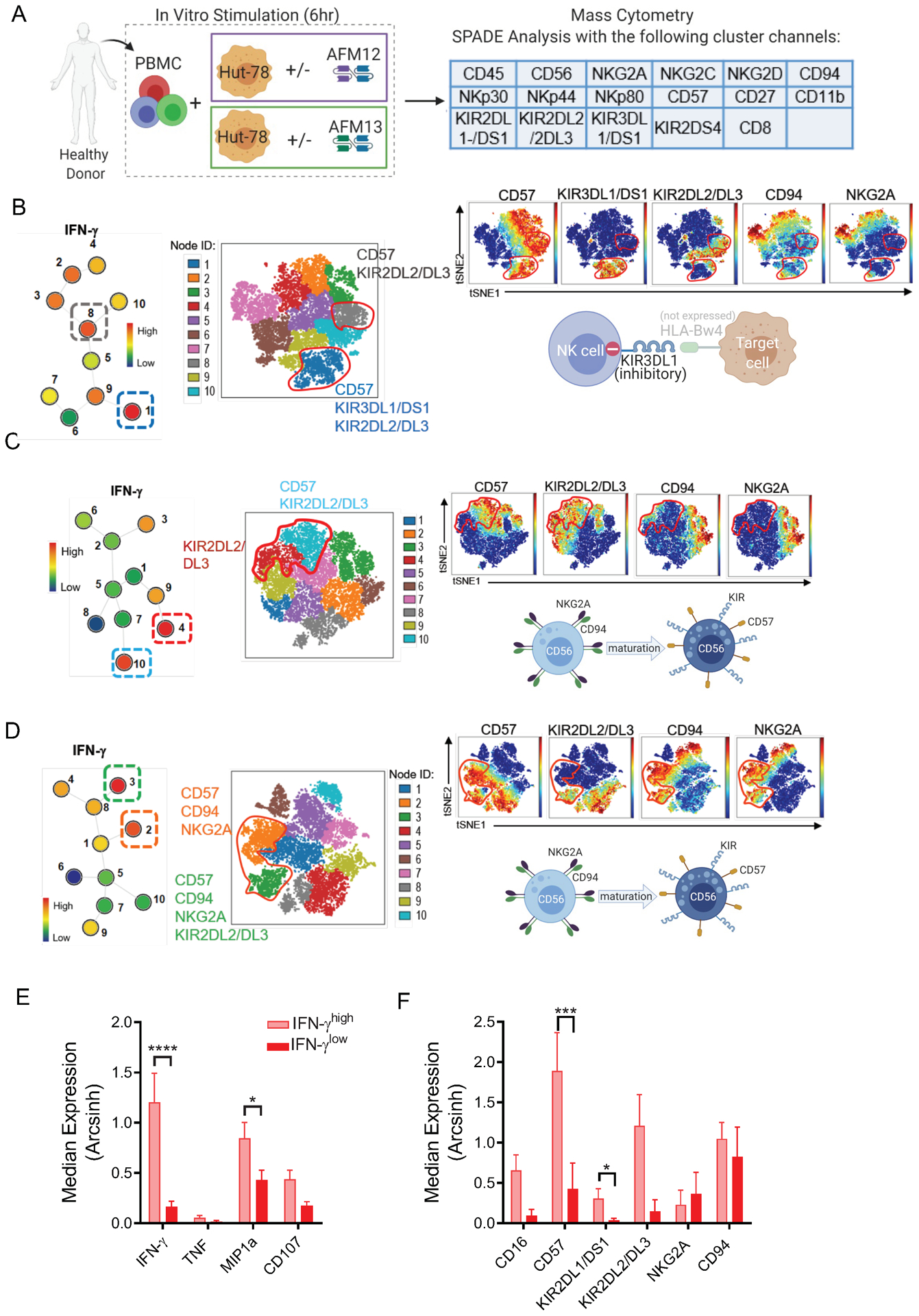
(A) Schema of cell stimulation to evaluate functionality and phenotype of cNK cells responding to AFM13 triggering. Clustering channels included for SPADE analysis are shown. B-D) Representative examples of SPADE analysis from 3 different donors showing the expression of KIRs, CD57 and CDK2A/CD94 in the top 2 nodes with highest IFN-γ expression in response to AFM13. Numbers next to each node represent the node ID and color indicates IFN-g median expression. For each donor, selected nodes were backgated to their own viSNE plots to identify their phenotype. (B) IFN-γ production (nodes 1, 8) in this donor was restricted to mature CD57+KIR2DL2/DL3+ NK cells. In this donor, lack of HLA-Bw4 expression in HuT-78 cells correlates with a robust response of KIR3DL1/DS1+ NK cells. (C) The highest IFN-γ production was restricted to terminally matured CD57+KIR2DL2/3+NKG2A- NK cells in this donor. (D) IFN-γ (nodes 2, 3) was produced by both mature and immature NK cells (CD57+NKG2A+CD94+KIR+/−) NK cells in this donor. (E) Median expression of IFN-γ, TNF, MIP1α, CD107a and (F) NK cell markers associated with cell maturation or differentiation in nodes with the highest and lowest IFN-γ median expression per donor. In F, all donors were positive for the depicted KIR. Consolidated data of 10 donors. Bars represent mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. One-way ANOVA with Tukey pots-hoc test. n=10.
NK cell source and IL-12/15/18 pre-activation influences AFM13-directed NK cell killing and cytokine production
As blood cNK cells exhibited varied AFM13-triggered responses within different NK cell maturation states and iKIR-defined subsets, several types of NK cells were evaluated for response to AFM13. These included healthy donor (HD) and Hodgkin lymphoma (HL) patient peripheral blood (PB) cNK cells, as well as cord-blood expanded (CB-NK) (27) and PB cytokine-induced memory-like (ML) (30) NK cells, which are currently being tested in cellular therapy clinical trials for hematologic malignancies. AFM13 significantly augmented PB cNK cytotoxicity against Karpas 299 lymphoma in HD and to a lesser extent PB cNK from relapsed/refractory HL patients who received multiple treatments including brentuximab. In contrast, AFM13-triggered CB-NK cells exhibited only a modest trend for increased cytotoxicity (Fig. 3A). Brief activation of PB NK cells with IL-12, IL-15, and IL-18 results in ML differentiation and improves cytokine production and killing of hematologic cancer targets (6,30). We hypothesized that IL-12/15/18 activation and consequent memory-like differentiation (Supplementary Fig. S3A–B) may positively impact AFM13-triggered responses. In a second set of experiments, PB cNK cells were compared to PB ML NK cells, with and without AFM13, for killing of CD30+ HuT-78 lymphoma targets. While cNK cells again demonstrated improved killing with AFM13 triggering, ML NK cells exhibited significantly enhanced killing compared to cNK cells (Fig. 3B and Supplementary Fig. S4A), consistent with previous studies showing their enhanced ADCC responses (31). We next evaluated the impact of IL-12/15/18 activation followed by expansion of CB-NK cells on AFM13-triggered Karpas 299 cell killing (Fig. 3C). At all concentrations of AFM13 tested, IL-12/15/18 pre-activated and expanded (P+E) CB-NK cells exhibited enhanced killing over control expanded CB-NK cells. Addition of AFM13 did not increase the cytotoxicity of PB cNK cells or P+E CB-NK cells against CD30− targets (Supplementary Fig. S4B–C). These data indicate that activation with IL-12/15/18 followed by memory-like differentiation of PB NK cells or expansion of CB NK cells enhanced AFM13 triggered killing. In addition, PB ML NK cells (Fig. 3D) and IL-12/15/18 P+E CB-NK cells (Fig. 3E) also produced IFN-γ, TNF, and degranulated (surface CD107a) effectively to CD30+ targets.
Fig. 3. AFM13 binding triggers enhanced cytotoxicity and cytokine secretion of cytokine induced ML NK cells and pre-activated-expanded CB NK cells.
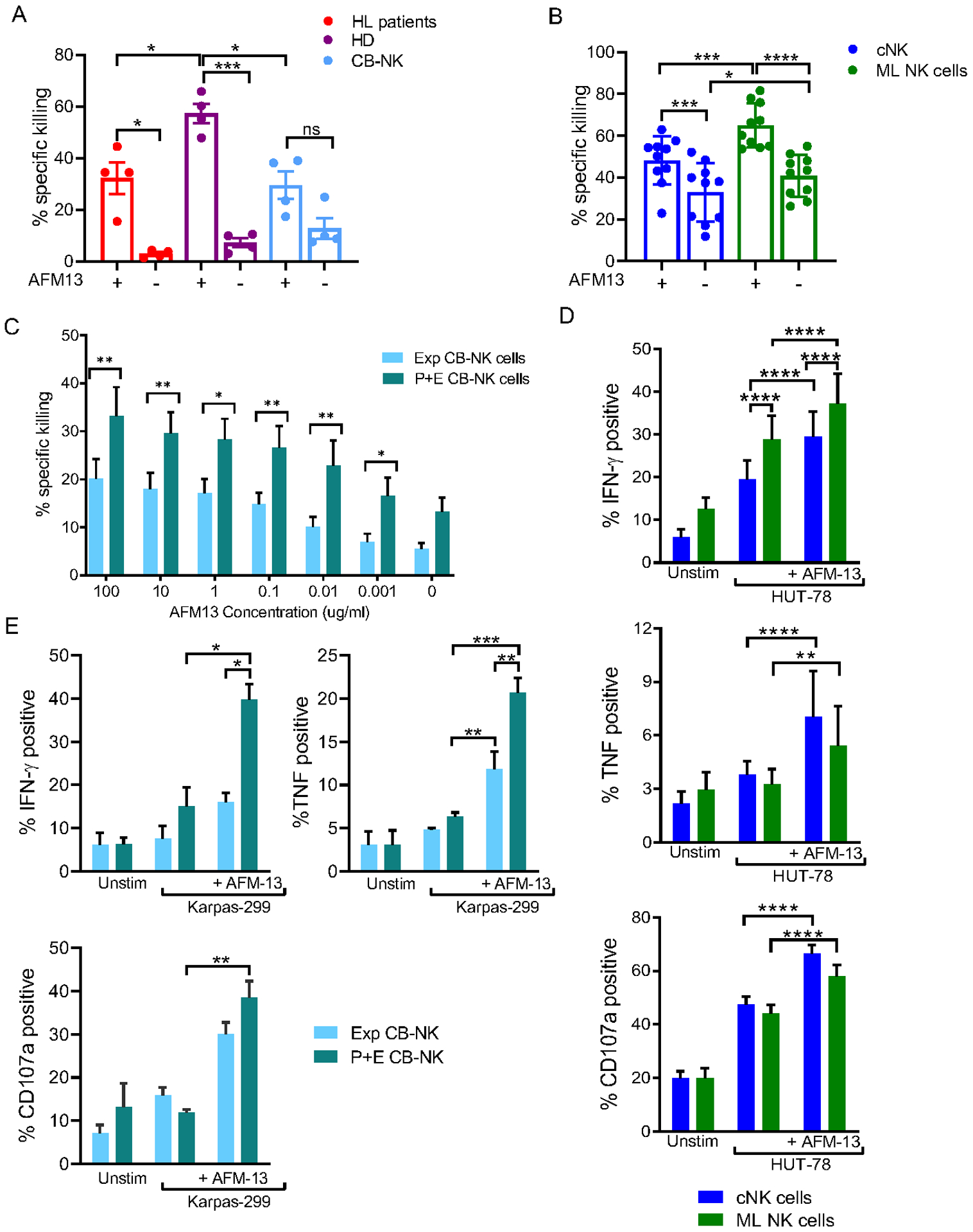
(A) Specific killing of NK cells from relapsed Hodgkin Lymphoma (HL), healthy donors (HD) or expanded cord blood (CB) against Karpas 299 targets in presence or absence of AFM13 (100 μg/ml). E:T ratio of 10:1. (B) Peripheral blood (PB) cNK but mainly ML NK cells from HD exhibit enhanced cytotoxicity against CD30+ HuT-78 lymphoma targets when triggered with AFM13. (C) Preactivated expanded (P+E) CB-NK cells shows enhanced cytotoxicity against Karpas 299 targets compared to expanded (Exp) CB-NK in presence of different concentrations of AFM13. E:T ratio 5:1. (D) PB cNK and ML NK cells were stimulated HuT-78 cells (5:1 E:T ratio) and the production of IFN-γ, TNF, and CD107a degranulation was evaluated by Flow Cytometry. PB ML NK cells exhibit enhanced cytokine production and degranulation in response HuT-78 cells compared to cNK. In both cNK and ML NK cells AFM13 triggering significantly upregulated the production of IFN-γ, TNF, and CD107 degranulation. (E) cytokine production (IFN-γ and TNF) and CD107a degranulation by AFM loaded (100 μg/ml) P+E CB-NK cells vs. AFM13 loaded (100 μg/ml) expanded (Exp) NK cells co-cultured with Karpas 299 cells for 6 hours. E:T ratio of 1:1 ratio. Bars represent mean ± SEM. Two-way ANOVA with (A, B, D, E) Tukey or (C) Sidak post-hoc test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. n=6–10.
IL-12/15/18 pre-activation of CB-NK cells enhances their effector function through upregulation of multiple inflammatory pathways
Since IL-12/15/18 pre-activation has not previously been reported with CB NK cell expansion, we next evaluated potential mechanisms responsible for their enhanced functionality. Flow cytometry analysis of P+E CB-NK cells compared to control expanded CB-NK cells showed similar CD16 expression (Supplementary Fig. S5), indicating that CD16 expression changes were not the underlying mechanism for the enhanced AFM13 response. RNA sequencing of purified CB-NK cells was performed comparing the transcriptome profiles of P+E CB-NK vs expanded CB-NK cells after 14 days of culture (Fig. 4A). P+E CB-NK cells had higher expression of select chemokines and cytokine genes, such as CCL3, CCL4, CCR5, and TNF (Fig. 4B–C). Gene set enrichment analysis (GSEA) confirmed enrichment of genes involved in the IFN-γ response, TNF signaling, IL-2/STAT5 signaling, IL-6/JAK/STAT3 signaling and mTOR pathway as well as genes related to inflammatory immune responses (Fig. 4D–E). Taken together, these data suggest that pre-activation of CB-NK cells with IL-12, IL-15 and IL-18 prior to expansion results in increased signaling pathways related to NK cell effector function, indicating a potential mechanism of enhanced AFM13 response.
Fig. 4. Gene expression signature of IL-12/15/18 pre-activated and expanded CB-NK cells.

(A) Experimental schema describing the strategy to analyze gene expression of P+E CB NK cells. (B) Volcano plot showing significantly differentiated genes (red dots represent genes with higher expression; blue dots represent genes with lower expression) in P+E CB-NK cells compared to expanded CB-NK cells. (C) Heatmap of differentially expressed genes in P+E CB-NK cells comparing to Exp- CB-NK cells (Differences were identified at adjusted p-value <0.05 and absolute fold-change >1.5, -log10 adjusted p value in annotation padj). (D) Gene set enrichment analysis (GSEA) of differentially expressed genes between P+E CB-NK cells vs expanded CB-NK cells presented as a heatmap. Color scale indicates signal intensity, ranging from lower (blue) to higher (red) expression (for Hallmark). (E) GSEA plots showing enrichment of genes related to IFN-g response, TNF signaling, IL-2/STAT5 signaling, IL-6/JAK/STAT3 signaling and PI3K/AKT/MTOR signaling in P+E CB-NK cells compared to expanded NK cells. n=3
AFM13 activates IL-12/15/18 pre-activated and expanded CB-NK NK cells
Mass cytometry was used to investigate if AFM13 can induce changes in the phenotype of P+E CB NK cells using a second panel (Supplementary Table S2). P+E CB NK cells were co-cultured with AFM13 and tumor targets for 4 hours and then analyzed using CyTOF (Supplementary Fig. S6). viSNE based analysis revealed segregation in the cell clusters of P+E CB-NK cells treated with AFM13, with or without tumor targets (Supplementary Fig. S6A–B). Cells loaded with AFM13 had evidence of activation (increased expression of TRAIL, NKp44 and CD69) (Supplementary Fig. S6C). These data are consistent with the notion that through CD16 engagement, AFM13 can activate NK cells as it was previously shown in PB NK cells (41). No significant differences in NK cell associated markers other than the ones described above were observed in the studied conditions (Supplementary Table S2).
AFM13-loaded CB-NK cells retain their cytotoxicity against Karpas 299 lymphoma cells after washing
When combining AFM13 with donor NK cell therapy, one approach to maximize initial NK cell triggering is pre-loading effector cells with AFM13 prior to infusion. To address this, we investigated the feasibility and activity of pre-complexed AFM13-NK cells by testing the stability of AFM13 binding on P+E CB-NK cells over time. To test this, NK cells were incubated with different concentrations of AFM13 (1–100 μg/ml) for 1 hour and then washed twice with PBS prior to performing functional studies. Regardless of the AFM13 concentration used, AFM13-loaded and washed NK cells (washed) were as efficient as loaded and unwashed NK cells at killing Karpas 299 targets (Fig. 5A). Further, IncuCyte monitoring of NK cell cytotoxicity over 24 hours revealed similar killing kinetics of AFM13-loaded and washed and AFM13-loaded and unwashed NK cells against Karpas 299 targets (Fig. 5B). Thus, AFM13 binding to the surface of P+E CB NK cells is stable and retains activity to enhance tumor recognition and killing.
Fig. 5. Retention of AFM13 on NK cell surface enhances cytotoxicity against CD30 positive Karpas 299.
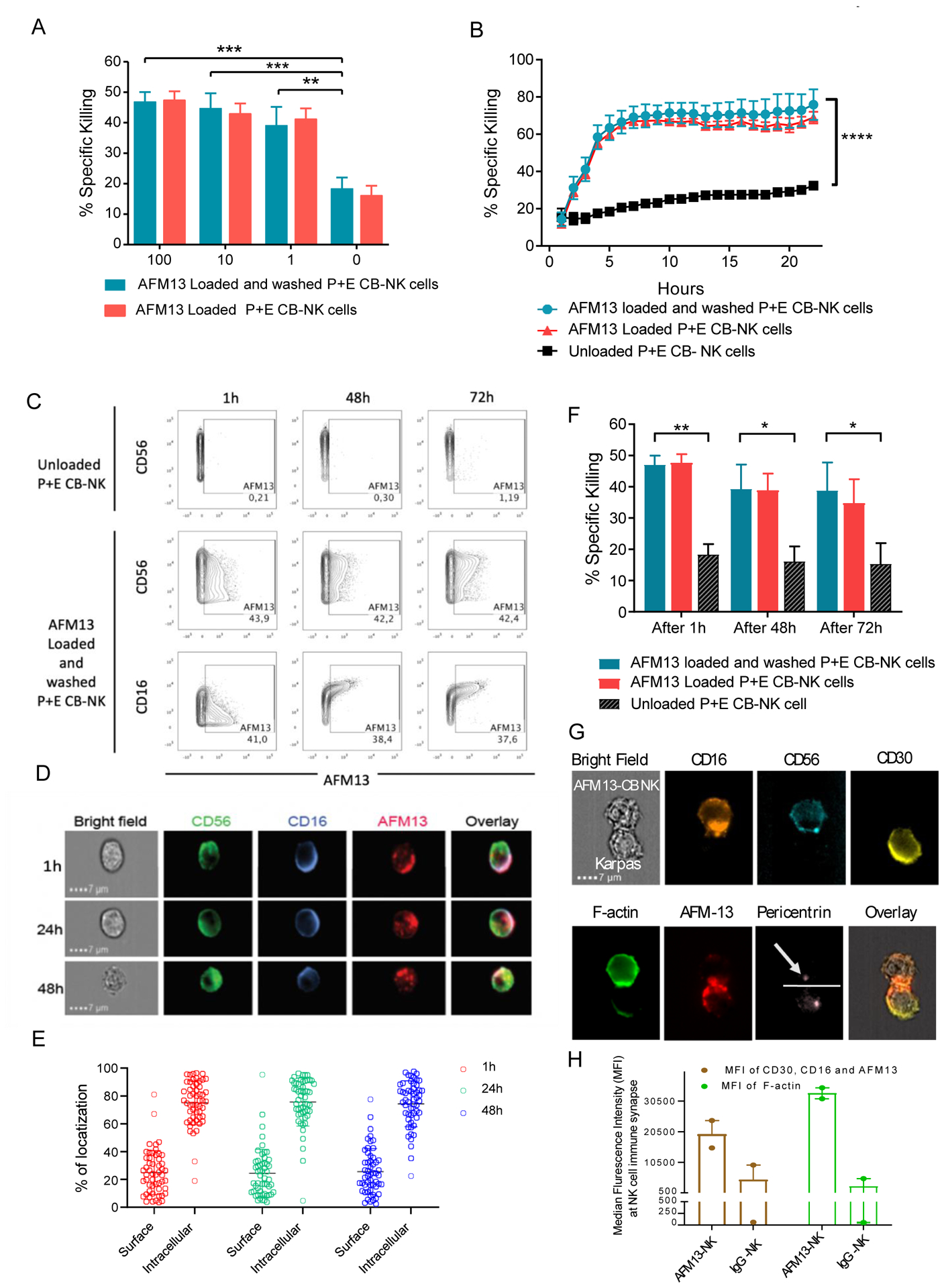
(A) Cytotoxicity (4 hour 51Cr-release assay) of CB-NK cells loaded with AFM13 (0–100 μg/ml) that were either washed twice with PBS (blue bars) or left unwashed (red bars) against Karpas 299 cells at E:T ratio 10:1. (B) Karpas 299 killing over a 24 hour period by AFM13 (100 μg/ml) loaded and washed (blue lines), or unwashed (red lines), along with unloaded (black lines) CB-NK cells as measured by incuCyte. (C) For cell surface retention assays, 1×106 human P+E CB-NK cells were labeled with 100 μg/ml of AFM13 at 37°C for one hour, cells were washed. At the indicated timepoints, the presence of AFM13 was measured using anti-AFM13 antibody followed by an APC-conjugated goat anti-rat IgG. The retention of AFM13 was evaluated by flow cytometry. (D) The retention of AFM13 on unwashed NK cell surface was observed by Image Stream at different timepoints. A representative example of NK cell from each of the time points stained for CD56 (green), CD16 (blue), and AFM13 (red) along with an overlay of all fluorescent channels and bright field is shown. Scale bar=7μm. (E) Percent distribution of total AFM13 fluorescent signal is summarized from the images of 50 cells, which were randomly sampled from 1 hour (red), 24 hours (green), and 48 hours (blue) time points. Each dot represents a cell, horizontal line the mean and error bars indicate +/− SD. (F) P+E CB-NK cells were cultured without or with AFM13 (100 ug/ml) for 1 hour at 37°C, followed by either a wash step or not. The cells were then cultured in media plus 100 IU/mL of rhIL-2 for 1 hour, 48 hours, or 72 hours and their cytotoxicity tested against Karpas 299 target cells at an E:T ratio of 5:1. (G) Bright field image showing AFM13 labelled NK cells were conjugated with Karpas 299 cells. Staining of the conjugates for CD16 (orange), CD56 (teal), CD30 (yellow), F-actin (green), AFM-13 (red), pericentrin (light pink) and an overlay of the BF images with AFM13, CD30 and CD16 are shown. The dotted line defines the synapse and the arrowhead is pointing to the pericentrin in NK cells. Conjugates were evaluated under a 60x objective in ImageStream; scale bar = 7mm. (H) Synaptic localization of CD30, CD16 and AFM13 (Brown), along with polymerization of F-actin (Green) at the immune synapse between NK cells and Karpas299 cells in the presence of AFM13 or IgG control antibody is shown. Horizontal bars represent the average and error bars indicate the range. Bars represent mean ± SEM. Two-way ANOVA with Sidak post-hoc test ** p ≤ 0.01, ***p ≤ 0.001, **** p ≤ 0.0001. n=3–4.
Retention of AFM13 on the surface of NK cells over time was evaluated using a monoclonal antibody directed against AFM13. Multiparameter flow cytometry showed that NK cells loaded with AFM13 (100 μg/ml) for one hour and then washed retained AFM13 on their surface for least 72 hours in culture (Fig. 5C). As expected, AFM13 was detected mainly on CD16+ NK cells (Fig. 5C). These results were confirmed using an imaging flow cytometer where we found that AFM13 was co-localized with CD16 on the surface of NK cells (Fig. 5D). Interestingly, AFM13 was partially internalized from the cell membrane into the cytoplasm (Fig. 5D–E). However, the retention of AFM13 on the surface of the NK cell remained the same overtime, depicting thereby that 1 hour incubation of AFM13 with NK cells (Fig. 5A) could have the same effect as incubating AFM13 with NK cells either for 24 hours or 48 hours. Nonetheless, to test whether AFM13 internalization affects the ability of NK cells to kill tumor targets, NK cells were loaded with AFM13, washed and incubated for 1 hour, 48 hours or 72 hours in media plus rhIL-2, with unwashed cells as control. All AFM13-loaded groups (washed and unwashed) exhibited significantly higher cytotoxicity against Karpas 299 lymphoma targets at all timepoints compared to unloaded P+E CB NK cell (Fig. 5F), suggesting that partial internalization of AFM13 does not affect the enhanced cytotoxic activity of NK cells. To provide additional mechanistic data to support the importance of combined CD16/AFM13 engagement in mediating the antitumor response, we used imaging flow cytometry to confirm significantly higher co-localization of CD16 at the immunological synapse between CB-NK cells and Karpas cells in the presence of AFM13 compared to a control IgG antibody (Fig. 5G–H). In addition, we confirmed that in the absence of NK cells, the addition of the CD30 bispecific engager failed to induce apoptosis of the Karpas tumor cells as demonstrated in Supplementary Fig. S7.
In vivo antitumor activity of the AFM13-NK cell complex in a mouse model of CD30+ lymphoma
To initially assess the safety of P+E CB-NK pre-loaded with AFM13 in vivo, immunodeficient NSG mice were injected i.v. with 10×106 AFM13-loaded (and washed as in Fig. 6) P+E CB-NK cells (AFM13-NK) and monitored for toxicity, including daily weight measurements for more than 10 months. We did not find any evidence of toxicity during the follow up or on histologic examination at necropsy (Supplementary Fig. S8A–B). Next, the antitumor activity of 10×106 AFM13-complexed NK cells in lymphoma bearing mice was tested (Fig. 6A). Mice received 1) Karpas 299 cells alone, 2) Karpas 299 cells plus unloaded P+E CB-NK cells (10×106), 3) Karpas 299 cells plus AFM13-complexed NK cells, or 4) Karpas 299 cells + 100 μg/ml of AFM13 (via i.v. without NK cells). All mice received human IL-2 (10,000 units/mouse) i.p on the day of NK cell injection and every 2–3 days to support human NK cell survival in NSG mice. Compared to control mice, infusion of the AFM13-NK complex improved tumor control measured via bioluminescent imaging (BLI, Fig. 6B) and survival (Fig. 6C). No differences in mouse weight was observed between the different mouse conditions, suggesting minimal cytokine release and no graft versus host disease (Fig. 6D). Collectively, these data support that the AFM13-complexed NK cells are capable of safely controlling tumor cells in vivo.
Fig. 6. In vivo antitumor activity of AFM13 loaded P+E CB-NK cells.
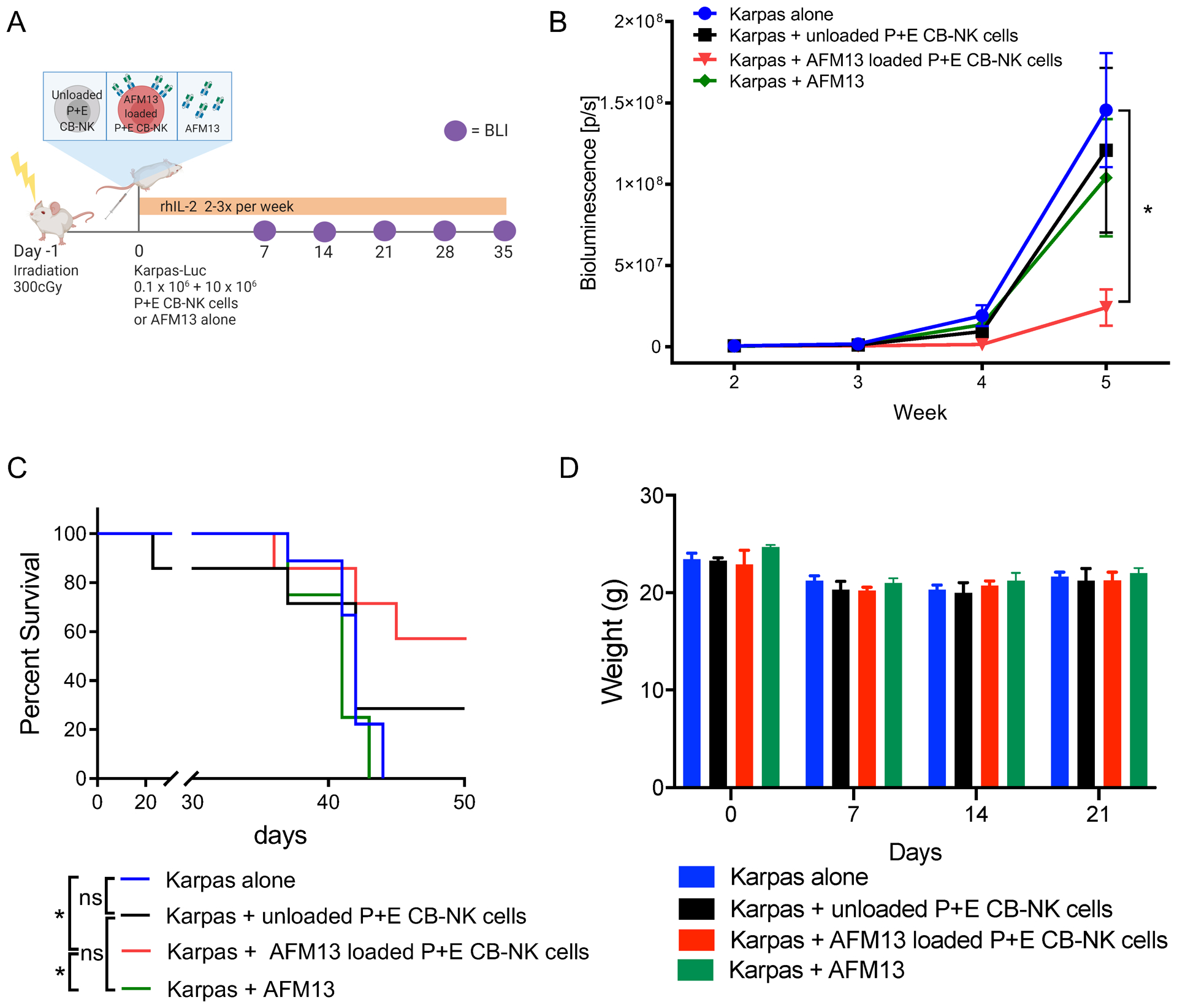
(A) Experimental schema applied to evaluate in vivo activity of AFM13. (B) Bioluminescence imagining (BLI) was used to monitor the growth of FFluc-labeled Karpas 299 tumor cells in NSG mice. The plot summarizes the bioluminescence data from four groups of mice treated with Karpas 299 alone, Karpas 299 plus one dose of unloaded NK cells (10 × 106), Karpas 299 plus one dose of AFM13 loaded (100 μg/ml) and washed NK cells (AFM13-NK cell complex; 10 × 106), or Karpas 299 and an injection of AFM13 (100 μg/ml). (C) Kaplan–Meier plots showing the survival of mice described in panel A. Mice treated with a single dose of 10 × 106 AFM13-NK cell complex (blue line) had better survival than control groups. (D) Bar plots summarize the weight of the animals over time as a measure of toxicity. Bars represent mean ± SEM. Two-way ANOVA with Tukey post-hoc test, *p ≤ 0.05; ns, not significant. n=4–9 mice per group, 2 independent experiments.
Discussion
In this study, we have shown that AFM13, a tetravalent bispecific antibody directed against CD16A and CD30, potentiate NK cell cytotoxicity and cytokine production of IL-12/15/18 induced memory-like mature blood NK cells and preactivated and expanded CB-NK cells against CD30 expressing lymphoma targets both in vitro and in vivo. Multidimensional analysis of PB cNK cells triggered by AFM13 plus CD30+ lymphoma targets revealed responses that were polyfunctional, and exhibited a heterogeneity of response at the single cell and individual donor levels. However, a consistent pattern of response spanning analysis of all donors revealed the greatest responses occurring in mature PB cNK cells.
Comparisons of distinct types of NK cells revealed a source-dependent response profile to AFM13-based stimulation. While cNK blood NK cells exhibited enhanced cytotoxicity in response to AFM13 alone (41) or AFM13-dependent target recognition, this appeared decreased in cNK cells from Hodgkin lymphoma patients in vitro. Further, the established IL-12, IL-15, and IL-18-induced ML NK cell differentiation of PB NK cells resulted in significantly enhanced AFM13-dependent killing. This finding was also evident upon IL-12/15/18 activation followed by expansion of CB-NK cells, with enhancement of AFM13-triggered cytotoxicity. Thus, this is the first study that identifies immunotherapeutic strategies that combine activation via the IL-12, IL-15 and IL-18 receptors of two NK cell types with AFM13 triggering, resulting in augmented NK cell responses to CD30+ cancers pre-clinically.
Prior work has defined the induction of NK cells with memory-like properties following combined cytokine activation. Cytokine-induced memory-like responses were identified using a murine adoptive transfer system, where IL-12, IL-15, and IL-18 activated splenic NK cells differentiated in syngeneic mice, and produced increased IFN-γ in response to cytokine restimulation (42). Human PB NK cells briefly activated with IL-12, IL-15, and IL-18 and allowed to differentiate in vitro exhibited enhanced function when re-stimulated with cytokines, with activating receptors or with tumor targets (30), and may also be engineered with a CAR (11). Multiple pre-clinical studies have shown that human and murine NK cells exhibit enhanced anti-cancer properties of memory-like NK cells (6,30,31,43–45). PB ML NK cellular therapy was shown to safely induce complete remissions in 47% of evaluable relapsed/refractory AML patients with expansion and persistence of donor memory-like NK cells in the blood and bone marrow of patients for at least 21 days after infusion (6,12). Our finding that CB-NK cells pre-activated with IL-12, IL-15, and IL-18 prior to expansion had enhanced AFM13-mediated killing could be explained by the lack of complete maturation of expanded CB-NK cells (46). Further, while IL-2 can drive the proliferation of CB-NK cells, IL-15 was necessary for the cells to acquire full effector function (37). Here, RNA sequencing data demonstrated that activation with IL-12/15/18 prior to expansion of CB-NK cells induces a phenotype distinct from expanded CB-NK cells, and resemble aspects of cytokine-induced memory-like NK cells (6). RNA changes included upregulation of IFN-γ and TNF signaling as well as the STAT5 and mTOR pathways. Future studies will focus on unraveling molecular mechanisms that result in memory-like NK cell programs by various NK cell types.
Multiple studies have shown that NK cells from patients with cancer, including patients with HL, have impaired cytotoxic function (35,47,48). Indeed, we confirmed upregulation of multiple inhibitory receptors, including PD-1, TIM 3, TIGIT and Pan-KIR, and lower expression of Eomes on HL NK cells compared to PB-NK cells. In contrast, CD16 levels were equivalent in NK cells from patients with HL and HD (Supplementary Fig. S9), suggesting that the functional deficit observed in patients with HL is predominantly related to expression of multiple checkpoints, rather than to lower AFM13-binding to NK cells secondary to CD16 downregulation. This may in part explain the results of a phase 1 dose-escalation study of AFM13, where only 3 of 26 evaluable patients with heavily pretreated HL achieved partial remission (11.5%) (49). These data support the notion that AFM13 will be more effective when combined with allogeneic NK cells from healthy donors. One approach supported by our study is to pre-complex the NK cells with AFM13 ex vivo and to infuse the complexed product to the patient rather than to infuse the AFM13 and NK cells as two separate products. This strategy may prevent the in vivo dilution and uptake of AFM13 by the recipient endogenous NK cells, which are likely to have low cytotoxic activity. We have pre-activated, expanded and pre-complexed CB-NK cells with AFM13 and confirmed their cytotoxicity against CD30+ tumors both in vitro and in vivo. AFM13-complexed NK cells retain the engager on their surface for at least 3 days, which may provide sufficient time for the AFM13-complexed NK cells to engage and kill targets, as observed in our pre-clinical mouse model. CB-derived and PB memory-like NK cells are a ready source of cells to combine with AFM13, that have the potential for broad availability. Furthermore, we have shown that CB derived NK cells can be safely used in clinics even in the absence of HLA matching (33,50).
Our approach of loading NK cells with an anti-CD16 bispecific antibody can be extended to other targets, transforming the pre-complexed NK cells into de facto CAR-NK cells, thus providing a rapid approach to translate NK cells with CAR-like characteristics to the clinic. While antigen escape, including antigen loss or downregulation, may also occur after AFM13-complexed NK cell immunotherapy, the capacity of NK cells to recognize and target tumor cells through their endogenous receptors may make disease escape less likely.
In conclusion, we have developed a novel approach to immunotherapy using NK cells complexed with AFM13. A clinical trial with off-the-shelf AFM13-complexed pre-activated and expanded CB-NK cells in patients with relapsed/refractory CD30+ malignancies is underway and will elucidate if the promising in vitro and in vivo observations are reproducible in patients (Clinicaltrial.gov NCT04074746).
Supplementary Material
Translational Relevance Statement.
Limited persistence and modest anti-tumor properties of conventional NK cells are two barriers in the NK cell therapy field that limit their broad application as cancer immunotherapy. Here we demonstrated that AFM13, a CD16:CD30 immune cell engager, improves tumor recognition and targets the polyfunctional responses of blood NK cells to CD30+ targets. AFM13 directed the potent anti-tumor properties of blood and cord-blood-derived NK cells pre-activated with IL-12/15/18, resulting in enhanced responses against CD30+ tumors. AFM13 complexed with IL-12/15/18 pre-activated cord-blood NK cells formed a “CAR-like” stable complex with enhanced cytotoxic function and anti-CD30+ tumor activity in vitro and in vivo. These findings support the translation of combining AFM13-targeting with IL-12/15/18-activated blood or expanded cord-blood NK cells to target CD30+ malignancies, and warrants clinical trials with these novel combinations.
Acknowledgments
We thank the RNA sequencing core facility, which is supported by the Cancer Center Support (CORE), and the Research Animal Support Facility at MD Anderson Cancer Center. Figures created with biorender.com.
This work is supported by from the National Institutes of Health (1 R01 CA211044-01, 5 P01CA148600-03, and P50CA100632-16). The MD Anderson Cancer Center Flow Cytometry and Cellular Imaging Facility and the RNA sequencing core facility is supported in part by a grant from the National Institutes of Health, National Cancer Institute (CA016672). The Siteman Flow Cytometry Core and the Immune Monitoring Laboratory of the Siteman Cancer Center in Washington University, School of Medicine, St Louis, MO are supported by the NCI CCC support (P30CA091842). Additional support for the study includes National Institutes of Health (NIH): K12CA167540 (MMBE), SPORE in Leukemia P50CA171963 (TAF, MMBE), R01CA205239 (TAF). This study was partially supported by Affimed.
Footnotes
Disclosure of Potential Conflict of Interest:
TAF and MMBE are consultants and have equity interest in Wugen, and may receive royalty income based on a technology developed by TAF and MMBE and licensed by Washington University to Wugen. JK and WF are employees of Affimed. MT is an employee of Arjuna Therapeutics. TAF has received research support from ImmunityBio, Compass Therapeutics, and HCW Biologics, and advises Kiadis, Nkarta, Indapta, and Orca Biosystems. LNK, RB, EL, SOA, EJS, KR and The University of Texas MD Anderson Cancer Center has an institutional financial conflict of interest with Affimed GmbH. This institutional financial conflict of interest is related to the research reported in this publication. Because MD Anderson is committed to the protection of human subjects and the effective management of its financial conflicts of interest in relation to its research activities, MD Anderson is implementing an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MD Anderson’s conduct of any other ongoing or future research related to this relationship. LNK, PPB, RB, MD, EL, SOA, REC, EJS, KR and The University of Texas MD Anderson Cancer Center have institutional financial conflict of interest with Takeda Pharmaceutical for the licensing of the technology related to CAR-NK cell research. MD Anderson has implemented an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MDACC’s conduct of any other ongoing or future research related to this relationship. KR participates on Scientific Advisory Board for GemoAb, AvengeBio, Virogin, GSK and Bayer. The remaining authors declare no competing financial interests.
Data and material availability
Data may be shared after request to the corresponding authors by e-mail: Katayoun Rezvani: krezvani@mdanderson.org, Todd A. Fehniger: tfehnige@wustl.edu
References
- 1.Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rouce RH, Shaim H, Sekine T, Weber G, Ballard B, Ku S, et al. The TGF-β/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia. 2016;30:800–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018. page 1069–86. [DOI] [PubMed] [Google Scholar]
- 4.Garrido F, Cabrera T, Concha A, Glew S, Ruiz-Cabello F, Stern PL. Natural history of HLA expression during tumour development. Immunol. Today 1993. [Accessed 05/05/2020]. page 491–9. [DOI] [PubMed] [Google Scholar]
- 5.Berrien-Elliott MM, Romee R, Fehniger TA. Improving natural killer cell cancer immunotherapy. Curr Opin Organ Transplant. 2015;20:671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8:357ra123–357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr. Opin. Immunol 2018. [Accessed 05/05/2020]. page 146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol Biol. 2016. page 333–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morvan MG, Lanier LL. NK cells and cancer: You can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19. [DOI] [PubMed] [Google Scholar]
- 11.Gang M, Marin-Agudelo NM, Wong P, Neal CC, Marsala L, Foster M, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;doi: 10.1182/blood.2020006619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berrien-Elliott MM, Cashen AF, Cubitt CC, Neal CC, Wong P, Wagner JA, et al. Multidimensional Analyses of Donor Memory-Like NK Cells Reveal New Associations with Response after Adoptive Immunotherapy for Leukemia. Cancer Discov. 2020;10:1854–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10. [DOI] [PubMed] [Google Scholar]
- 14.Rezvani K, Rouce R, Liu E, Shpall E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther 2017. page 1769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–93. [DOI] [PubMed] [Google Scholar]
- 16.Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. [DOI] [PubMed] [Google Scholar]
- 18.Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica (Cairo). 2014; 10.1155/2014/205796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai K, Matsuyama S, Miyake S, Suga K, Yu H, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. 2000;356:1795–9. [DOI] [PubMed] [Google Scholar]
- 20.Bi J, Tian Z. NK cell dysfunction and checkpoint immunotherapy. Front. Immunol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. 2011;121:3609–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011;71:5412–22. [DOI] [PubMed] [Google Scholar]
- 23.Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–7. [DOI] [PubMed] [Google Scholar]
- 25.Bjorklund AT, Carlsten M, Sohlberg E, Liu LL, Clancy T, Bj AT, et al. Complete Remission with Reduction of High-Risk Clones following Haploidentical NK-Cell Therapy against MDS and AML. Clin Cancer Res. 2018;24:1834–45. [DOI] [PubMed] [Google Scholar]
- 26.Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The rise of allogeneic Natural killer cells as a platform for cancer immunotherapy: Recent innovations and future developments. Front. Immunol 2017. page 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehta RS, Shpall EJ, Rezvani K. Cord blood as a source of natural killer cells. Front. Med 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJN, et al. Antigen Presenting Cell-Mediated Expansion of Human Umbilical Cord Blood Yields Log-Scale Expansion of Natural Killer Cells with Anti-Myeloma Activity. PLoS One. 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ojo EO, Sharma AA, Liu R, Moreton S, Checkley-Luttge MA, Gupta K, et al. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci Rep. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine activation induces human memory-like NK cells. Blood. 2012;120:4751–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner JA, Berrien-Elliott MM, Rosario M, Leong JW, Jewell BA, Schappe T, et al. Cytokine-Induced Memory-Like Differentiation Enhances Unlicensed NK Cell Anti-Leukemia and FcγRIIIa-Triggered Responses. Biol Blood Marrow Transplant. 2016;23:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fehniger TA, Cooper MA. Harnessing NK Cell Memory for Cancer Immunotherapy. Trends Immunol. 2016;Epub Oct 2: 10.1016/j.it.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reusch U, Burkhardt C, Fucek I, Le Gall F, Le Gall M, Hoffmann K, et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs. 2014;6:728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reiners KS, Kessler J, Sauer M, Rothe A, Hansen HP, Reusch U, et al. Rescue of impaired NK cell activity in hodgkin lymphoma with bispecific antibodies in vitro and in patients. Mol Ther. 2013;21:895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu E, Ang SOT, Kerbauy L, Basar R, Kaur I, Kaplan M, et al. GMP-Compliant Universal Antigen Presenting Cells (uAPC) Promote the Metabolic Fitness and Antitumor Activity of Armored Cord Blood CAR-NK Cells. Front Immunol. 2021;12:626098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Chen H, Marin D, Xi Y, Miao Q, Lv J, et al. A novel immature natural killer cell subpopulation predicts relapse after cord blood transplantation. Blood Adv. 2019;3:4117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diggins KE, Ferrell PB, Irish JM. Methods for discovery and characterization of cell subsets in high dimensional mass cytometry data. Methods. 2015;82:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, et al. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom Part A. 2015;87:636–45. [DOI] [PubMed] [Google Scholar]
- 40.Kordasti S, Costantini B, Seidl T, Abellan PP, Llordella MM, McLornan D, et al. Deep phenotyping of tregs identifies an immune signature for idiopathic aplastic anemia and predicts response to treatment. Blood. 2016;128:1193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pahl JHW, Koch J, Gotz JJ, Arnold A, Reusch U, Gantke T, et al. Cd16a activation of nk cells promotes nk cell proliferation and memory-like cytotoxicity against cancer cells. Cancer Immunol Res. 2018;6:517–27. [DOI] [PubMed] [Google Scholar]
- 42.Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A. 2009;106:1915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med. 2012;209:2351–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leong JW, Chase JM, Romee R, Schneider SE, Sullivan RP, Cooper MA, et al. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transpl. 2014;20:463–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ni J, Hölsken O, Miller M, Hammer Q, Luetke-Eversloh M, Romagnani C, et al. Adoptively transferred Natural Killer cells maintain long-term anti-tumor activity by epigenetic imprinting and CD4 T cell help. Oncoimmunology. 2016;5:e1219009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verneris MR, Miller JS. The phenotypic and functional characteristics of umbilical cord blood and peripheral blood natural killer cells. Br. J. Haematol 2009. page 185–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frydecka I Natural killer cell activity during the course of disease in patients with Hodgkin’s disease. Cancer. 1985;56:2799–803. [DOI] [PubMed] [Google Scholar]
- 48.Chiu J, Ernst DM, Keating A. Acquired natural killer cell dysfunction in the tumor microenvironment of classic Hodgkin lymphoma. Front. Immunol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase I study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015;125:4024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah N, Li L, McCarty J, Kaur I, Yvon E, Shaim H, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2017;177:457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.