

Abstract
Small noncoding RNAs deep sequencing (sncRNA-Seq) has become a routine for sncRNA detection and quantification. However, the software packages currently available for sncRNA annotation can neither recognize sncRNA variants in the sequencing reads, nor annotate all known sncRNA simultaneously. Here, we report a novel anchor alignment-based small RNA annotation (AASRA) software package (https://github.com/biogramming/AASRA). AASRA represents an all-in-one sncRNA annotation pipeline, which allows for high-speed, simultaneous annotation of all known sncRNA species with the capability to distinguish mature from precursor microRNAs, and to identify novel sncRNA variants in the sncRNA-Seq sequencing reads.
Keywords: small RNA annotation, sequence alignment, bioinformatics, RNA-Seq, precursor microRNA
Introduction
Given their critical regulatory roles, small noncoding RNAs (sncRNAs) have become a major focus in biomedical research [1, 2]. The next-generation sequencing technologies have allowed for the identification of hundreds thousands of sncRNAs, which have been categorized into many unique sncRNA species, e.g., microRNAs (miRNAs) [3–5], endogenous small interference RNAs (endo-siRNAs) [6, 7], PIWI-interacting RNAs (piRNAs) [8–11], small nucleolar RNAs (snoRNAs) [12], tRNA-derived small RNAs (tsRNAs) [13, 14], mitochondrial genome encoded small RNAs (mitosRNAs) [15], etc. Among these sncRNAs, miRNAs and piRNAs have been studied extensively for the past decade largely because they were discovered first [8–11, 16]. To help investigators identify known and to predict novel miRNAs or piRNAs based on small noncoding RNAs deep sequencing (sncRNA-Seq) data, several software packages have been developed, e.g., ShortStack [17], miRanalyzer [18], miRDeep [19], PIANO [20], etc. Using these pipelines, researchers have not only validated previously reported sncRNAs, but also predicted sncRNAs based on their unique structural (e.g., length, stem-loop structure, etc.) and genomic features (e.g., repetitive sequences). Currently, there are many sncRNA databases, e.g., miRBase [21], piRNABank [22], piRNA Cluster Database [23], Rfam [24–26], snoRNA-LBME-db [27], etc., where known and predicted sncRNAs (for some of the databases) are collected. These databases serve as important resources because investigators can download these sncRNAs and use them as reference sequences to annotate their own sncRNA-Seq data for sncRNA identification and quantitation. The most popular sequence alignment software packages, e.g., Bowtie [28], SOAP [29], or BWA [30], are designed for mapping large RNA sequencing reads directly to the genome. However, these methods are not ideal for small RNA alignment analyses for two reasons. First, the library construction methods for large and small RNAs are fundamentally different (Figure 1A). The Illumina sequencers perform the so-called short-read sequencing, which requires shorter DNA fragments (~200–800 bp). Therefore, large RNAs have to be fragmented either physically (via heating or shearing) or enzymatically, followed by adaptor ligation (Figure 1A). After sequencing, the shorter reads (~50–150 nt) need to be aligned to the genome using Bowtie2-based TopHat followed by assembly using Cufflinks [31]. Fragmentation can generate numerous homologous fragments, which differ from each other by only a few nucleotides at either or both ends. Since they are all derived from the same transcripts, the downstream annotation will categorize these homologous fragments as single transcripts. In contrast, adaptors are ligated directly to small RNAs without fragmentation during sncRNA library preparation (Figure 1A) and thus, homologous fragments represent unique sncRNAs and should, therefore, be counted as individual sncRNAs. Second, mathematically, the possibility for shorter reads (~20–40 nt) to have multiple alignment in the genome is much greater, compared to that of longer reads (50–150 nt); multiple mapping leads to repetitive counting during alignment, causing quantification bias (Figure 1B). A straightforward solution would be to align the sequencing reads to the corresponding sncRNA reference sequences instead of the genome. However, this direct, RNA-to-RNA mapping strategy leads to multiple alignment due to the existence of homologous sncRNAs in both the reference databases and the sequencing reads. For example, the sequencing reads of a mature miRNA would align to both the mature and its homologous precursor miRNAs in the reference dataset, leading to double counting (Figure 1C). Many sncRNAs, e.g., MIWI2-associated piRNAs (i.e., pre-pachytene piRNAs), endo-siRNAs, and mitosRNAs, contain a large number of homologs with only a few nucleotide differences in either or both ends (Figure 1C). Thus, one such sncRNA would align to its multiple homologs, causing repetitive counting and quantification bias (Figure 1C). Moreover, the existing alignment programs would only select the perfectly matched reads and eliminate those with minor mismatches, although those may represent the sncRNAs synthesized by the cells. To overcome these problems, we developed a universal sncRNA annotation software package, called anchor alignment-based small RNA annotation (AASRA) based on our unique C/G repeat anchor alignment algorithm (Figure 1D). AASRA can annotate sncRNAs of all known species collected in various sncRNA databases with a much higher mapping rate and accuracy, as well as speed, compared to all existing software packages currently available for sncRNA annotation.
Figure 1 .
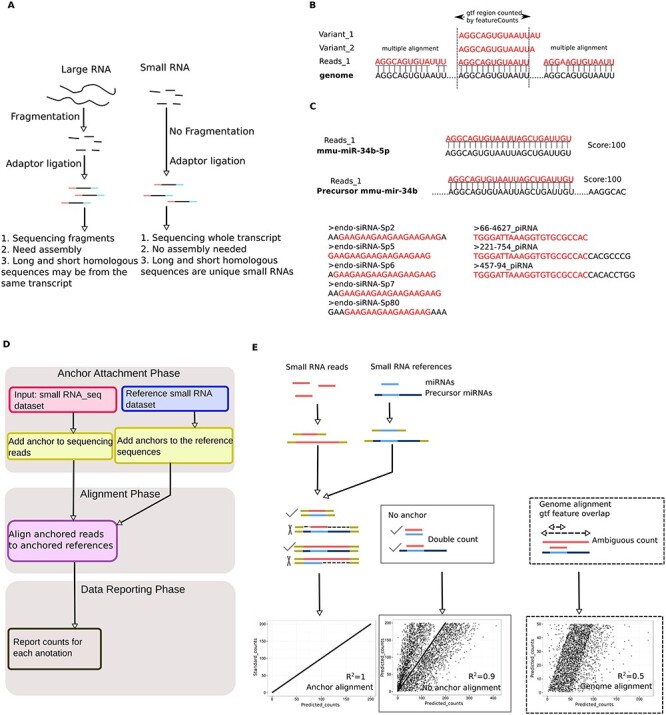
Development of the anchor alignment algorithm for sncRNA annotation. (A) Schematic illustration of the differences in large and small RNA library construction methods. Note that adaptors are directly added to the small RNAs for sncRNA-Seq, whereas fragmentation is needed before adaptor ligation for large RNA sequencing. (B) Issues associated with direct sncRNA alignment to the genome: multiple alignment of sncRNAs to the genome due to their small sizes (20–40 nt), and inability to recognize sncRNA variants (e.g., homologous piRNAs, endo-siRNAs, mitosRNAs, etc.). (C) Issues associated with the direct sncRNA–sncRNA alignment algorithm: repetitive counting of mature miRNA reads (because they can be mapped to both mature and precursor miRNA references), and certain sncRNA reads (e.g., endo-siRNAs and piRNAs, due to the presence of multiple, staggered sncRNA homologs in the reference databases, which differ by only several nucleotides). (D) Workflow of the AASRA pipeline. (E) Schematic illustration of anchor alignment algorithm. Anchors are added to both ends of the sequencing reads and the reference sncRNAs. Gap opening penalty can prevent mature miRNA sequence reads from mapping to the precursor miRNA reference sequences. Perfect alignment and correct annotation of both mature and precursor miRNAs were achieved for the simulation data using the anchor alignment algorithm (R2 = 1), whereas direct alignment of the simulation data to either the sncRNA references (R2 = 0.9), or the genome (R2 = 0.5) led to partial alignment.
Methods
SncRNA reference data source
The reference sncRNA datasets consist of mature and precursor miRNAs in the miRBase (release 21) [21], tRNAs in the Genomic tRNA Database [32], piRNAs in the piRNABank [22], and piRNA Cluster Database [23], rRNAs, snoRNAs, snRNAs, and mitochondrial RNAs in ENSEMBL (release 76) [33–35], and endo-siRNAs in DeepBase [36].
Simulation data
Simulation sequences were based upon sncRNA sequences from the known sncRNA databases. Small noncoding RNA variant sequences, including 1–2 nt overhangs, internal insertions, deletions, and mutations, were generated by randomly adding or changing 1–2 nts at either end or internally using R script of the Biostrings package and EMBOSS-msbar. To generate the simulation Fasta file, individual sncRNAs were randomly duplicated such that the counts for each ranged from 1 to 50. For the realistic simulation of miRNA sequencing reads, mouse miRNA reference from miRBase (release 21) [21] was used. From 1 to 100 counts were randomly generated for each miRNA. The length distribution of precursor miRNAs was randomly trimmed to the range of 40–80 nt and fitted to the normal distribution with mean close to 60 nt and standard deviation close to 3 nt. A total of 0.08% of two single nucleotide mismatches (SNMs), 0.001% of 1–4 nt insertion or deletion and 0.4% of 1–4 nt overhang were incorporated into the simulation reads by EMBOSS-msbar and custom scripts. Each read is tagged with true miRNA ID. The true count for each miRNA (standard count) was generated during the simulation.
Anchor alignment
Anchor sequences (1–10 bp) were added to both ends of the reference sncRNAs and the sequencing reads, as well as simulation sequences using the Python script. Bowtie2-build was employed to index all the anchored reference sncRNAs. The anchored sequencing reads/simulation sequences were then aligned to the indexed anchored reference sncRNAs using Bowtie2 [37] with the –norc -N 1 -L 16 -I S,0,0.2 as optimal settings for sncRNA alignment. The featureCounts [38] was used to summarize the counts in the alignment file. The same procedure was used to align the non-anchored sequencing reads or simulation sequences to the indexed, non-anchored reference sncRNA sequences. The 5′ C/3′ G repeat sequence anchor of 2 nt/3 nt length to the reads and 10 nt length to the reference was chosen as the optimal AASRA alignment anchor sequence length. All Bowtie2/Bowtie1 based alignment parameters are summarized in Supplementary Table S1.
Genome alignment
Bowtie2-build was used to index the mouse genome (NCBI_Assembly: GCA_000001635.2). The sequencing data were aligned to the indexed genome using Bowtie2. The Feature Count was used to summarize the reads in the alignment file based on mmu.gff3 (miRbase V21) [21].
The use of miRDeep/miRDeep2 for miRNA annotation
The GRCm38 mouse genome was built according to the user manual of miRDeep [19, 39]. Both miRNA sequencing reads/simulation dataset and GRCm38 pre-built genome were loaded for alignment analyses using the default setting of miRDeep/miRDeep2. Scatter plots were generated to correlate the predicted counts (by miRDeep/miRDeep2) with the standard counts (simulation counts).
The use of ShortStack for miRNA annotation
The indexed mouse genome reference (NCBI_Assembly: GCA_000001635.2) was generated by Bowtie2-build. The simulation data were then aligned to the indexed genome using Bowtie2 (ShortStack –readfile –outdir –genomefile), and the hits were –locifile –outdir –genomefile). Scatter plots were generated to correlate the predicted counts (by ShortStack) with the standard counts (simulation counts).
The use of miRanalyzer/sRNAbench for miRNA annotation
The stand-alone version of miRanalyzer and sRNAbench were downloaded and installed according to the user manual [18, 40]. The pre-built, Bowtie2-indexed genome sequences (UCSC mm9) were used as the reference mouse genome in miRanalyzer. The mature and precursor miRNA sequences were used as the sncRNA reference dataset for miRanalyzer and sRNAbench. MicroRNA simulation data with or without overhangs and the realistic miRNA dataset were analyzed using the default parameters. Scatter plots were generated to correlate the predicted counts (by miRanalyzer/sRNAbench) with the standard counts (simulation counts).
Alignment accuracy assessment
The alignment results were summarized by featureCounts to generate predicted counts. Pearson correlation coefficient was calculated to measure the linear correlation between standard counts and predicted counts. The realistic simulated miRNA reads were aligned to the miRbase miRNA references. Single read alignment results were extracted from SAM file or CORE file annotated by featureCounts. The read tag indicating true miRNA of each read was compared with the assigned miRNA read ID. The reads were then classified as “correctly mapped” if true miRNA ID equals to the assigned miRNA ID; “unmapped” if the read was not mapped to any miRNA reference or miRNA genome locus; “incorrectly mapped” if the read was assigned to a miRNA ID, which is not equal to the true miRNA ID. Precision is defined as (correctly mapped reads/(correctly mapped reads + incorrectly mapped reads)). Recall is defined as (correctly mapped reads/(correctly mapped reads + unmapped reads)). The F1 score is a summary statistic of precision and recall (β = 1) that weights precision and recall equally.
Mouse sperm sncRNA-Seq
Mouse epididymal sperms were collected in the HEPES-HTF medium, and a “swim-up” procedure was performed so that only motile sperm were selected for sncRNA-Seq [41]. Total RNA was isolated using the mirVana miRNA Isolation Kit (Life Technologies) following the manufacturer’s instructions. Small noncoding RNA libraries were prepared using the Ion Total RNA-Seq Kit v2 (Life Technologies), followed by sequencing using the Ion P1 chips on an Ion Proton Sequencer (Life Technologies) [41].
Data management and graphics
All the data were processed using the R script and graphs were plotted using the R script of the ggplot2 package.
The software environment
Operating system: Ubuntu 12.04.5 LTS, 64-bit; Memory 11.5GiB; Processor Intel® Xeon(R) CPU E3–1225 v3 @ 3.20GHz ×4 Graphics ATI Radeon™ HD 5450.
Results
The anchor alignment algorithm
AASRA first processes both the sequencing reads and the reference sequences by adding C/G repeat anchors to both ends. Then, the anchored sequencing reads are aligned to the anchored sncRNA references using Bowtie2. Finally, FeatureCounts (Subread) is used to summarize the unique read counts (Figure 1D). The anchor alignment algorithm can avoid multiple and ambiguous alignments, which are common in the straight matching algorithms (i.e., direct alignment to reference sncRNAs or to the genome by Bowtie2, or miRanalyzer, miRDeep, etc.). For example, the anchored mature miRNA reads can only align to the anchored mature miRNA references. When the mature miRNA reads are aligned to the anchored reference precursor miRNAs, the gap-opening penalty would prevent double matching (Figure 1E). In this way, the Bowtie2 penalty score is tweaked by the artificial anchors such that the anchored mature miRNA reads get a lower penalty score when aligned to the corresponding anchored mature miRNA references than that when aligned to precursor miRNAs. Therefore, mature miRNAs can be readily distinguished from their corresponding precursor miRNAs during the alignment. As a proof of concept, we used AASRA to align the simulation dataset containing both mature and precursor miRNA sequences to the reference miRNA dataset downloaded from the miRBase. The anchor alignment algorithm resulted in a perfect mapping (R2 = 1), whereas the direct alignment to the reference miRNAs or to the genome led to partial alignments with R2 values of 0.9 and 0.5, respectively. Together, the anchor alignment algorithm can avoid erroneous counting and can also distinguish mature miRNA reads from precursor miRNA reads accurately.
Anchor optimization
To include sncRNA variants that bear small overhangs or internal insertions/deletions/mutations in the sncRNA-Seq reads, we tested a number of anchor sequences to see which ones gave the best alignment results. We first tested 5′/3′ 5 nt anchors by aligning the anchored simulation reads dataset against the reference sncRNA datasets downloaded from various sncRNA databases (Figure 2A). The simulation dataset containing all the known sncRNAs aligned perfectly to the sncRNA reference datasets (R2 = 1). However, when the simulation datasets containing 1–2 nt overhangs at either end were used, only partial alignment (R2 = 0.87) was achieved due to the gap-opening penalty caused by those miRNA variants (Figure 2A, Supplementary Figure S1). Since these miRNA variants are likely synthesized by the cell and the 1–2 nt mutations are probably due to sequencing errors, they should not be excluded from annotation. To accommodate theses sncRNA variants, we designed C/G repeat anchors of different lengths (5 nt for the reads and 10 nt for the references) based on the fact that C and G are the least common nucleotides at the ends of miRNAs and thus, can have higher specificity (Supplementary Figure S2). Using C/G repeat anchors for alignment, a 1–2 nt overhang in the read sequences would lead to a mismatch instead of a gap-opening penalty, which allows for inclusion of these sncRNA variants into the counts, leading to an increased alignment rate (R2 from 0.87 to 0.92) (Figure 2A, Supplementary Figure S1). We also examined the AG anchors as well as other possible single nucleotide anchors and found that anchors with the C/G combination consistently yielded the highest alignment rates (Supplementary Figure S2). By fine-tuning the parameters in AASRA, the optimal Bowtie2 setting was determined such that the sncRNA variants with 1–2 nt overhangs, internal insertions/deletions/mutations, could be included into the final counts (Supplementary Figure S3). For annotating sncRNA sequencing reads containing small internal insertions/deletions/mutations, AASRA (with the use of the CG anchors) consistently outperformed the Bowtie2-based direct sncRNA–sncRNA mapping method (Supplementary Figure S4). In addition, we evaluated AASRA alignment accuracy under different degrees of noise using simulation reads (Figure 3). The noise was generated by EMBOSS-msbar [42], with a gradient of 1–6 point mutations per read (Figure 3A), or a block mutation of 2–7 bp in length per read (Figure 3B). AASRA alignment shows drastic decrease in accuracy when the point mutation counts were greater than 3, and the size of the block mutations was greater than 5. The reduced accuracy was primarily due to unaligned mature miRNAs under the default setting of mismatch and gap opening penalty in Bowtie2. Moreover, the precursor miRNA alignment algorithm was not sensitive to the increased degree of variation. Compared to mature miRNA reads, precursor miRNA reads are relatively longer and consequently receive a lower minimum penalty score in Bowtie2 end-to-end alignment; this allows the precursor miRNA variant reads to be aligned to the corresponding references instead of being discarded. The penalty score parameters can be adjusted to increase or decrease the sensitivity of AASRA. Overall, these data indicate that the CG anchor-based alignment algorithm of AASRA allows for efficient mapping of not only perfect-matching sequencing reads, but also reads with small (1–2 nt) overhangs and internal insertions, deletions or mutations.
Figure 2 .
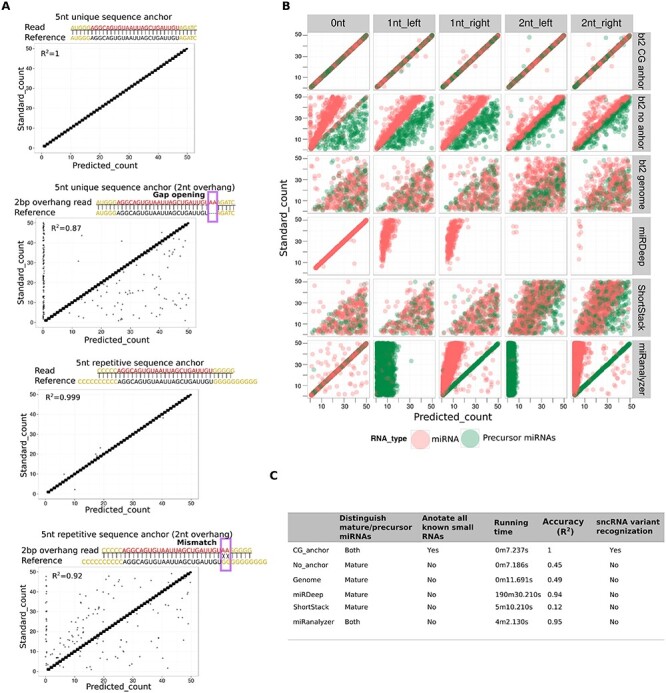
Anchor optimization and performance comparison between AASRA and three existing sncRNA annotation pipelines. (A) CG anchors outperformed other anchors because the CG anchors could turn the gap-opening penalty (causing exclusion) into mismatch penalty (leading to inclusion). The use of a non-CG anchors could align the simulation data without overhangs perfectly (R2 = 1), but simulation sequences with 2 nt overhangs were only aligned partially (R2 = 0.87) due to gap-opening penalty that excluded many miRNA variants. In contrast, the use of CG anchors aligned simulation datasets with or without 2 nt overhangs almost perfectly (R2 = 0.999 and 0.92, respectively) because those 2 nt overhangs were treated as mismatches rather than gaps and thus, those variants were counted and annotated. (B) Performance comparison between AASRA and existing sncRNA annotation software packages. Simulation datasets containing both mature (red dots) and precursor (green dots) miRNA sequences with 0–2 nt overhangs were aligned to the reference sncRNA dataset using AASRA alignment (CG_anchor), Bowtie2 no sample anchoring and 10 nt N repeats padding reference (No_anchor), direct alignment to the genome (genome), miRDeep, ShortStack, and miRanalyzer. (C) Summary of the performance of AASRA and other five sncRNA annotation pipelines tested.
Figure 3 .
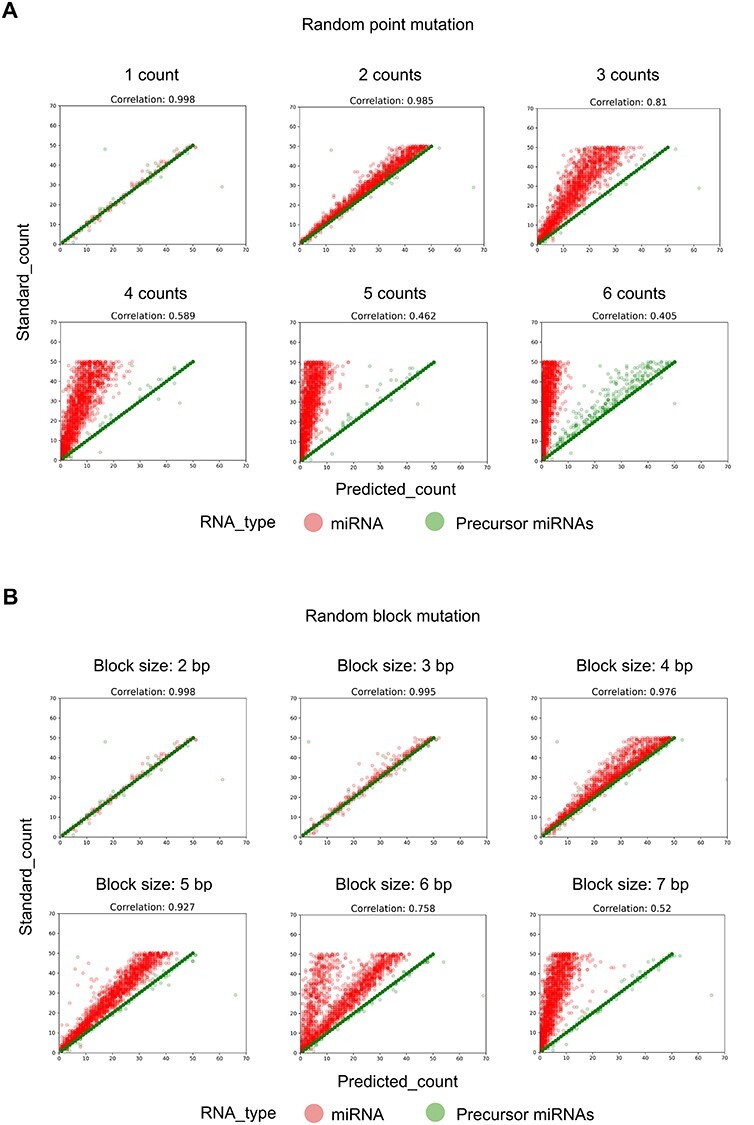
Evaluation of AASRA alignment accuracy in annotating reads with point or block mutations. (A) Random point mutations refer to point mutations randomly assigned to each simulation dataset generated by EMBOSS-msbar. Point mutations include insertion, deletion, mismatch, duplication, and move. Move means the block sequence copied from one region to another (without deletion of the original). Counts represent the number of point mutations in each read. (B) Various types of block mutations randomly assigned to each simulation dataset generated by EMBOSS-msbar. The types of block mutations include insertion, deletion, mismatch, duplication, and move. Block size refers to the length of block mutations in each read. Pearson product–moment correlation coefficients between the standard counts and the annotation software predicted counts are indicated.
Performance comparison between AASRA and existing sncRNA annotation software packages
To demonstrate the superior performance of AASRA, we generated simulation datasets containing mature and precursor miRNAs with 0, 1–2 nt overhangs at either end, and annotated the simulation sequence reads against the reference miRNA datasets downloaded from the miRBase using AASRA and the popular software packages for miRNA annotation, including ShortStack [17], miRDeep [19], miRDeep2 [43], Bowtie [28], and miRanalyzer [18] (Figure 2B, Supplementary Figure S5). The simulation sequences were aligned almost perfectly to the reference datasets using AASRA for both mature and precursor miRNAs with or without overhangs (R2 ≈ 1) (Figure 2B and C). Bowtie-based AASRA demonstrated very similar performance (Supplementary Figure S5). In contrast, default Bowtie2 mapping of the simulation miRNA and precursor miRNA sequences with or without overhangs to the mouse genome resulted in poor alignment rates (R2 = 0.45–0.49). The no sample anchoring Bowtie or Bowtie2 mapping without reference padding performed even worse with overhang datasets (R2 ≈ 0) (Supplementary Figure S5). The no sample anchoring Bowtie2 mapping to the reference miRNA datasets with 10 nt N repeats padding recovered a portion of the unaligned mature miRNAs when overhang was present (R2 = 0.8–0.92), but it could not rescue erroneously aligned mature miRNAs without overhang (R2 = 0.43) (Figure 2B, Supplementary Figure S5). The padding reference allowed overhang mature miRNA reads to receive a lower penalty score when aligned to mature than when they were aligned to precursor miRNAs. The non-overhang mature miRNA reads obtained the same alignment score when aligned to both mature and precursor miRNAs, even when the padding sequences were present in the reference. Although miRDeep could map sequences perfectly matching the known mature miRNAs efficiently (R2 = 0.94), it failed to align either precursor miRNA sequences or mature miRNA sequences with overhangs (Figure 2B and C), largely due to its strict length control criteria [19]. Thus, miRDeep cannot annotate precursor miRNAs, mature miRNAs with mismatches, or other sncRNAs with staggered sequence patterns (e.g., piRNAs, mitosRNAs, tsRNAs, etc.). miRDeep2 demonstrated improved mapping to known mature miRNAs while still showed the lack of accuracy at 1–2 nt overhangs (R2 = 0.662–0.966) (Supplementary Figure S5). ShortStack, similar to the direct genome alignment method, could only annotate a small fraction of the simulation sequences, largely due to repetitive and ambiguous counting. miRanalyzer utilizes a three-phase alignment procedure (i.e., mature miRNA alignment → pre-miRNA alignment → genome alignments) in conjunction with length control. miRanalyzer annotated the simulation data without overhangs as efficiently as AASRA (R2 = 0.95), but failed to annotate simulation data containing overhangs because it does not tolerate mismatches. In summary, AASRA appeared to be ideal for annotating known sncRNA species simultaneously with the capability of distinguishing mature and precursor miRNAs, and recognizing sncRNA variants with small overhangs and/or internal insertions/deletions, with a speed faster than any of the five pipelines tested (Figure 2C).
Mapping accuracy assessment using realistic simulated miRNA reads
To benchmark the performance of alignment methods with datasets that incorporate realistic miRNA read length and variable levels of noise, we generated simulation reads of mature miRNAs and precursor miRNAs. The length of precursor miRNA reads ranged from 50–150 nt (Supplementary Figure S6A). We trimmed the precursor miRNA references to simulate the reported length distribution of precursor miRNA reads observed in the small RNA next-generation sequencing datasets (Figure 4A) [44, 45]. Since overhangs are more common than SNMs and insertion/deletion (INS/DEL), we introduced realistic variation to miRNA reads based on the miRNA variation profile (Figure 4B) [45].
Figure 4 .
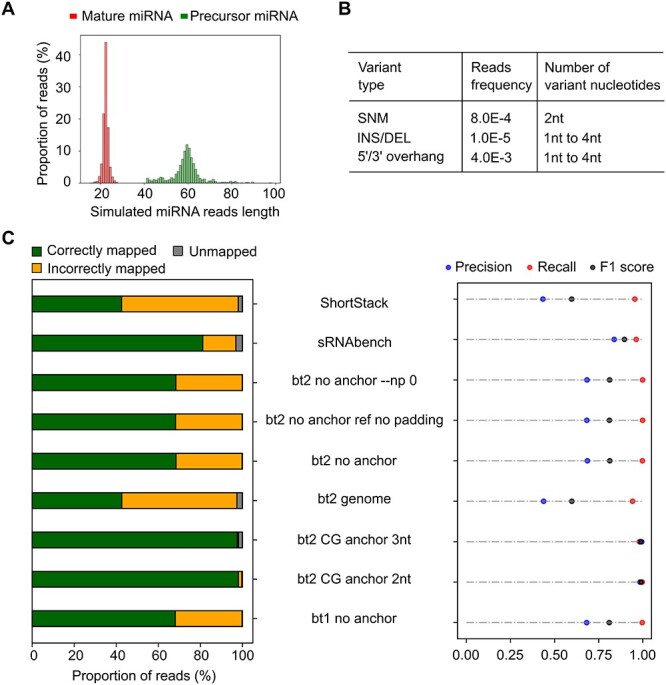
Performance comparison of miRNA alignment methods with realistic simulated miRNA reads. (A) Length distribution of the reads set, including mature miRNA (red) and precursor miRNA (green). (B) Reads variation profile of the read sets containing SNM, INS/DEL, and 5′/3′ overhang. (C) Assessment of alignment methods by percentage of correctly mapped reads (green), incorrectly mapped reads (yellow), and unmapped reads (grey) (left panel) and precision (correctly mapped/(correctly mapped + incorrectly mapped)), recall (correctly mapped/(correctly mapped + unmapped)), and F1 score.
We first assessed the mapping accuracy of Bowtie1/Bowtie2 CG anchor with different lengths of anchoring sequences (Supplementary Figures S6B and S7). Bowtie2 with 3 nt and 2 nt CG anchor achieved the best F1 scores of 0.99 and Pearson correlation coefficient of > 0.96. Both Bowtie2 and Bowtie1 showed increased unmapped rate with the increased length of anchoring sequences from 2 nt, due to the increased mismatch penalty introduced from the anchor sequence when anchored precursor miRNA fragment sequences aligned to the references (Supplementary Figure S6B). Reducing anchor sequence length to 1 nt showed close to 0 unmapped rate but increased incorrectly mapped reads compared with 2 nt anchor sequences. The scatter plot (Supplementary Figure S7) showed increased proportions of incorrect mapping came from the mature miRNAs falsely aligned to the precursor miRNAs due to the insufficient anchor length. Therefore, both 2 nt and 3 nt Bowtie2 CG anchor alignment strategies are ideal, specifically the 2 nt alignment gave slightly lower unmapped rate (0.3%) and 3 nt anchor yielded a relative lower incorrectly mapping rate (0.6%). The users could choose 3 nt CG anchor if precision is preferred over sensitivity, or choose the 2 nt for more sensitivity. Next, we compared the 2 nt/3 nt Bowtie2 CG anchor with other miRNA alignment methods (Figure 4 and Supplementary Figure S8). By aligning the reads to the genome, ShortStack and Bowtie2 received the lowest F1 score due to incorrect mapping and unmapped precursor miRNAs, as indicated in the scatter plots (Supplementary Figure S8). Bowtie2 and Bowtie1, with default setting to align the simulation reads to the 10 nt N repeats anchored miRNA references, showed mature miRNA reads mistakenly assigned to the precursor loci as the primary source of incorrect mapping, leading to the 32% incorrect mapping rate. Adjusting Bowtie2 alignment by adding –np 0 or removing the 10 nt N repeats padding sequence on the references led to a similar correlation score ~ 0.49 and F1 score of 0.81. sRNAbench is the updated version of miRanalyzer, which uses the same bowtie1-based alignment strategy as miRanalyzer, but added sequence variation detection [46]. sRNAbench showed improved mature miRNA alignment compared to miRanalyzer (Supplementary Figure S8A and Figure 2). However, both the F1 score and Pearson correlation score are slightly lower than those of the Bowtie2 2 nt/3 nt CG anchor method, which is likely due to the lack of precision and sensitivity in the presence of INS/DEL and 5′/3′ overhang over 2 nt (Figure 4, Supplementary Figure S8B and C). miRDeep/miRDeep2 are not included in the testing because it lacks the ability to align and annotate precursor miRNA reads (Figure 2B and Supplementary Figure S5). These results demonstrate that AASRA-based alignment with 2 nt/3 nt anchor setting is superior in accurately mapping real miRNA sequencing reads to miRNA references.
AASRA-based annotation of sperm sncRNAs
Two advantages of AASRA over the existing sncRNA annotation software packages include the following: 1) it can identify novel sncRNA variants with small overhangs or internal insertions, deletions or mutations. 2) It can annotate not only miRNAs (both mature miRNAs and pre-miRNAs), but also all known sncRNA species collected in various databases. A key question remains: do those sncRNA variants exist in the sncRNA-Seq reads by a substantial proportion? If so, these sncRNA variants should not be overlooked in quantitative analyses. To answer this question, we annotated the sperm sncRNA-Seq data generated by both the Ion Proton and the Illumina sequencers using both AASRA and miRDeep. AASRA simultaneously annotated nine known species of sncRNAs from mouse sperm sncRNA-Seq reads (Figure 5A). By comparing the unique mature miRNA counts determined by miRDeep and AASRA, we found that AASRA identified 37% more unique mature miRNA counts than miRDeep (Figure 5B). Although miRDeep could not annotate precursor miRNAs, AASRA identified both mature and precursor miRNAs (Figure 5C). Interestingly, murine sperm appeared to contain numerous precursor miRNAs, which would not have been identified using miRDeep or other sncRNA annotation software packages (Figure 5C). Further examination of the alignment results for the four miRNAs (mir-376a, mir-361, mir-93, and mir-4660) revealed that AASRA not only identified more mature miRNAs than miRDeep, but also detected various miRNA variants, including those containing small (1–2 nt) overhangs, internal insertions, deletions or mutations, whereas these sncRNA variants were not detected by miRDeep (Figure 5D). For example, ~ 80% of the sequencing reads aligned to miR-93 all contained overhangs, which could be either biological variants of miR-93 or sequencing errors. Regardless, such a large number of miR-93 variants would have been totally ignored if other existing software packages were used (Figure 5D). If one wants to exclude these sncRNA variants, a more stringent alignment can be performed through adjusting the parameters, including anchor sequence and mismatch penalty. For example, four levels of specificity settings (high_specificity1, 2, 3, and ultra) (Supplementary Figure S9A) were tested for sequence alignment stringency. At the ultra-high specificity setting, AASRA could eliminate all the sequences with 1–2 nt overhangs in the simulation data (Supplementary Figure S9B). Under the same setting, perfectly matched miRNAs could be readily identified from a mixture of miRNA sequences with 1–2 nt overhangs (Supplementary Figure S9C). The ultra-high specificity setting made AASRA function similarly as miRDeep, whereas a less stringent setting allowed for the identification of miRNA variants (Supplementary Figure S9D). It will be up to the investigators to decide whether those sncRNA variants should be included or excluded in the final counts during sncRNA annotation depending on the nature of specific experiments conducted.
Figure 5 .
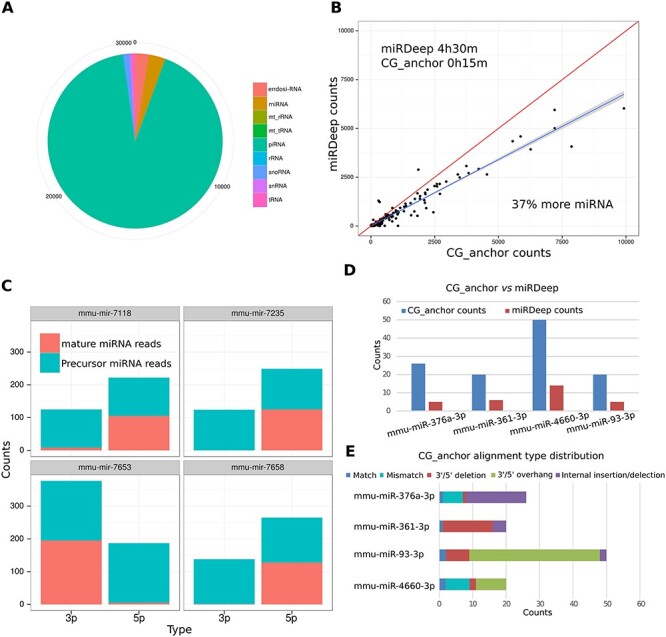
Annotation of sperm sncRNA-Seq data using AASRA. (A) Pie chart showing the count distribution of nine sncRNA species in murine sperm annotated using AASRA. (B) Scatter plot showing that AASRA could identify 37% more miRNAs than miRDeep in 1/12 of the time needed by miRDeep. (C) Counts of four miRNAs and their precursors in the sperm sncRNA-Seq data, as determined by AASRA. (D) Counts of four mature miRNAs in murine sperm sncRNA data, as determined by AASAR and miRDeep. (E) The contents of the AASRA counts of the four mature miRNAs shown in panel D. Note that mismatches, deletions, insertions, and overhangs appear to be common in the sncRNA sequencing reads.
Discussion
The rapid advance of next-generation sequencing technologies has led to the discovery of hundreds thousands of sncRNAs [2]. Increasing lines of evidence suggest that these sncRNAs play regulatory roles critical to development and physiology [2]. Despite the rapid pace of sncRNA discovery, the bioinformatic tools for sncRNA annotation are very limited. None of the currently available sncRNA annotation pipelines can annotate simultaneously all known sncRNA species, nor can they tolerate sequences with mismatches, although these sncRNA variants are likely due to sequencing errors, but biologically relevant. AASRA utilizes a unique, anchor alignment-based algorithm, and is capable of annotating all known sncRNAs simultaneously. The specificity setting of AASR is adjustable such that small mismatches due to overhangs, insertions, deletions, or mutation, can be either included or excluded. AASRA can identify a much greater number of sncRNA counts (e.g., ~ 37% more identified from the murine sperm sncRNA-Seq data) compared to any of the existing pipelines because of the use of the anchor alignment algorithm. This feature offers the possibility of minimizing quantification bias caused by 1) over-counting (due to double and ambiguous alignments) and/or 2) exclusion of variant sequences in the sncRNA-Seq data (although these variants should be counted because they are produced by the cells, but simply slightly different from the main sncRNA sequences most likely due to sequencing errors). The fact that these variant sequences account for a large proportion of the total counts (e.g., up to 80% for mmu-miR-93), elimination of these variants would greatly skew the real expression profile, leading to inaccurate interpretation and conclusions. Since all existing sncRNA annotation software packages do not have these functions, AASRA will be very useful for investigators to revisit their sncRNA data to see how many variants were inadvertently excluded, and whether such exclusion had caused quantitation bias that would compromise their conclusions. Depending on the needs of the investigators, those variants can also be excluded by applying more strict alignment parameters.
The capability to annotate the precursor miRNAs is another useful feature of AASRA. Interestingly, a large number of precursor miRNAs appear to be present in sperm, which would not have been discovered if other existing programs were used. Although miRanalyzer and sRNAbench can annotate precursor miRNAs, they can only annotate those with perfect or near perfect matches although a significant proportion of reads do have overhangs of 2 nt or longer or mismatches in the sequencing reads. Mature miRNAs have been found in sperm of multiple species, including mouse [41, 47], rat [32, 48], cow [49], horse [50], monkey [41, 51], and human [51, 52]. However, sperm-borne precursor miRNAs have not been reported. Given that these precursor miRNAs can be potentially delivered into the eggs during fertilization, their potential regulatory roles would be an intriguing topic for future investigation.
AASRA represents the first universal sncRNA annotation software package, which allows for simultaneous annotation of all known sncRNAs with high speed and accuracy. AASRA can annotate not only known sncRNA species, but also sncRNA variants containing small overhangs, or internal deletions/insertions/mutations. AASRA provides another useful bioinformatic tool for studying sncRNA biology.
Supplementary Material
Acknowledgments
The authors would like to thank the rest of the Yan lab for helpful discussion during the development of AASRA.
Contributor Information
Chong Tang, BGI Genomics, BGI-Shenzhen, Shenzhen, China; Department of Physiology and Cell Biology, University of Nevada School of Medicine, Reno, NV, USA.
Yeming Xie, BGI Genomics, BGI-Shenzhen, Shenzhen, China; Department of Physiology and Cell Biology, University of Nevada School of Medicine, Reno, NV, USA.
Mei Guo, BGI Genomics, BGI-Shenzhen, Shenzhen, China.
Wei Yan, Department of Physiology and Cell Biology, University of Nevada School of Medicine, Reno, NV, USA; The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center, Torrance, CA, USA.
Authors’ contributions
CT and WY conceived and designed the study, CT wrote the software with the assistance from YX, and MG, CT, and WY wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declared no competing interest.
Data availability
The sncRNA-Seq datasets have been deposited into the NCBI GEO database with the accession number of GSE81216. Information of AASRA is as follows:
Project name: AASRA.
Source code: https://github.com/biogramming/AASRA.
Scripts used in the manuscript: https://github.com/biogramming/AASRA.
Operating system(s): Linux, Mac.
Programming language: Python.
License: UNR.
Any restrictions to use by non-academics: None.
References
- 1. Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet 2009; 10:94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barquist L, Vogel J. Accelerating discovery and functional analysis of small RNAs with new technologies. Annu Rev Genet 2015; 49:367–394. [DOI] [PubMed] [Google Scholar]
- 3. Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001; 294:862–864. [DOI] [PubMed] [Google Scholar]
- 4. Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001; 294:858–862. [DOI] [PubMed] [Google Scholar]
- 5. Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science 2001; 294:853–858. [DOI] [PubMed] [Google Scholar]
- 6. Song R, Hennig GW, Wu Q, Jose C, Zheng H, Yan W. Male germ cells express abundant endogenous siRNAs. Proc Natl Acad Sci U S A 2011; 108:13159–13164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, Hodges E, Anger M, Sachidanandam R, Schultz RM, Hannon GJ. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 2008; 453:534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 2006; 442:199–202. [DOI] [PubMed] [Google Scholar]
- 9. Grivna ST, Pyhtila B, Lin H. MIWI associates with translational machinery and PIWI-interacting RNAs (piRNAs) in regulating spermatogenesis. Proc Natl Acad Sci U S A 2006; 103:13415–13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim VN. Small RNAs just got bigger: Piwi-interacting RNAs (piRNAs) in mammalian testes. Genes Dev 2006; 20:1993–1997. [DOI] [PubMed] [Google Scholar]
- 11. Saito K, Nishida KM, Mori T, Kawamura Y, Miyoshi K, Nagami T, Siomi H, Siomi MC. Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the drosophila genome. Genes Dev 2006; 20:2214–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maxwell ES, Fournier MJ. The small nucleolar RNAs. Annu Rev Biochem 1995; 64:897–934. [DOI] [PubMed] [Google Scholar]
- 13. Liao JY, Guo YH, Zheng LL, Li Y, Xu WL, Zhang YC, Zhou H, Lun ZR, Ayala FJ, Qu LH. Both endo-siRNAs and tRNA-derived small RNAs are involved in the differentiation of primitive eukaryote Giardia lamblia. Proc Natl Acad Sci U S A 2014; 111:14159–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 2009; 23:2639–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ro S, Ma HY, Park C, Ortogero N, Song R, Hennig GW, Zheng H, Lin YM, Moro L, Hsieh JT, Yan W. The mitochondrial genome encodes abundant small noncoding RNAs. Cell Res 2013; 23:759–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ambros V. microRNAs: tiny regulators with great potential. Cell 2001; 107:823–826. [DOI] [PubMed] [Google Scholar]
- 17. Axtell MJ. ShortStack: comprehensive annotation and quantification of small RNA genes. RNA 2013; 19:740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hackenberg M, Rodriguez-Ezpeleta N, Aransay AM. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res 2011; 39:W132–W138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 2008; 26:407–415. [DOI] [PubMed] [Google Scholar]
- 20. Wang K, Liang C, Liu J, Xiao H, Huang S, Xu J, Li F. Prediction of piRNAs using transposon interaction and a support vector machine. BMC Bioinformatics 2014; 15:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sai Lakshmi S, Agrawal S. piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res 2008; 36:D173–D177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosenkranz D. piRNA cluster database: a web resource for piRNA producing loci. Nucleic Acids Res 2016; 44:D223–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daub J, Eberhardt RY, Tate JG, Burge SW. Rfam: annotating families of non-coding RNA sequences. Methods Mol Biol 2015; 1269:349–363. [DOI] [PubMed] [Google Scholar]
- 25. Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 2005; 33:D121–D124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res 2003; 31:439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lestrade L, Weber MJ. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res 2006; 34:D158–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics 2008; 24:713–714. [DOI] [PubMed] [Google Scholar]
- 30. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 2009; 37:D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pignatelli M, Vilella AJ, Muffato M, Gordon L, White S, Flicek P, Herrero J. ncRNA orthologies in the vertebrate lineage. Database (Oxford) 2016;2016:bav127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Herrero J, Muffato M, Beal K, Fitzgerald S, Gordon L, Pignatelli M, Vilella AJ, Searle SM, Amode R, Brent S, Spooner W, Kulesha Eet al. . Ensembl comparative genomics resources. Database (Oxford) 2016;2016:bav127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, Girón CG, Gordon Let al. . Ensembl 2016. Nucleic Acids Res 2016; 44:D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zheng LL, Li JH, Wu J, Sun WJ, Liu S, Wang ZL, Zhou H, Yang JH, Qu LH. deepBase v2.0: identification, expression, evolution and function of small RNAs, LncRNAs and circular RNAs from deep-sequencing data. Nucleic Acids Res 2016; 44:D196–D202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 39. An J, Lai J, Lehman ML, Nelson CC. miRDeep*: an integrated application tool for miRNA identification from RNA sequencing data. Nucleic Acids Res 2013; 41:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hackenberg M, Sturm M, Langenberger D, Falcon-Perez JM, Aransay AM. miRanalyzer: a microRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res 2009; 37:W68–W76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schuster A, Tang C, Xie Y, Ortogero N, Yuan S, Yan W. SpermBase – a database for sperm-borne RNA contents. Biol Reprod 2016; In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rice P, Longden I, Bleasby AJ. Tig: EMBOSS: the European molecular biology open software suite. Trends Genet 2000; 16:276–277. [DOI] [PubMed] [Google Scholar]
- 43. Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky NJ. Nar: miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 2012; 40:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li N, You X, Chen T, Mackowiak SD, Friedländer MR, Weigt M, Du H, Gogol-Döring A, Chang Z, Dieterich CJ. Nar: global profiling of miRNAs and the hairpin precursors: insights into miRNA processing and novel miRNA discovery. Nucleic Acids Res 2013; 41:3619–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ziemann M, Kaspi A, El-Osta AJR. Evaluation of microRNA alignment techniques. RNA 2016; 22:1120–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aparicio-Puerta E, Lebrón R, Rueda A, Gómez-Martín C, Giannoukakos S, Jaspez D, Medina JM, Zubkovic A, Jurak I, Fromm BJ. Nar: sRNAbench and sRNAtoolbox 2019: intuitive fast small RNA profiling and differential expression. Nucleic Acids Res 2019; 47:W530–W535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kawano M, Kawaji H, Grandjean V, Kiani J, Rassoulzadegan M. Novel small noncoding RNAs in mouse spermatozoa, zygotes and early embryos. PLoS One 2012; 7:e44542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci 2013; 33:9003–9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Govindaraju A, Uzun A, Robertson L, Atli MO, Kaya A, Topper E, Crate EA, Padbury J, Perkins A, Memili E. Dynamics of microRNAs in bull spermatozoa. Reprod Biol Endocrinol 2012; 10:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Das PJ, McCarthy F, Vishnoi M, Paria N, Gresham C, Li G, Kachroo P, Sudderth AK, Teague S, Love CC, Varner DD, Chowdhary BPet al. . Stallion sperm transcriptome comprises functionally coherent coding and regulatory RNAs as revealed by microarray analysis and RNA-seq. PLoS One 2013; 8:e56535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boerke A, Dieleman SJ, Gadella BM. A possible role for sperm RNA in early embryo development. Theriogenology 2007; 68:S147–S155. [DOI] [PubMed] [Google Scholar]
- 52. Krawetz SA, Kruger A, Lalancette C, Tagett R, Anton E, Draghici S, Diamond MP. A survey of small RNAs in human sperm. Hum Reprod 2011; 26:3401–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sncRNA-Seq datasets have been deposited into the NCBI GEO database with the accession number of GSE81216. Information of AASRA is as follows:
Project name: AASRA.
Source code: https://github.com/biogramming/AASRA.
Scripts used in the manuscript: https://github.com/biogramming/AASRA.
Operating system(s): Linux, Mac.
Programming language: Python.
License: UNR.
Any restrictions to use by non-academics: None.