


Abstract
Mammalian MutY homologue (MUTYH) is an adenine DNA glycosylase that excises adenine inserted opposite 8-oxoguanine (8-oxoG). The inherited variations in human MUTYH gene are known to cause MUTYH-associated polyposis (MAP), which is associated with colorectal cancer. MUTYH is involved in base excision repair (BER) with proliferating cell nuclear antigen (PCNA) in DNA replication, which is unique and critical for effective mutation-avoidance. It is also reported that MUTYH has a Zn-binding motif in a unique interdomain connector (IDC) region, which interacts with Rad9–Rad1–Hus1 complex (9–1–1) in DNA damage response, and with apurinic/apyrimidinic endonuclease 1 (APE1) in BER. However, the structural basis for the BER pathway by MUTYH and its interacting proteins is unclear. Here, we determined the crystal structures of complexes between mouse MUTYH and DNA, and between the C-terminal domain of mouse MUTYH and human PCNA. The structures elucidated the repair mechanism for the A:8-oxoG mispair including DNA replication-coupled repair process involving MUTYH and PCNA. The Zn-binding motif was revealed to comprise one histidine and three cysteine residues. The IDC, including the Zn-binding motif, is exposed on the MUTYH surface, suggesting its interaction modes with 9–1–1 and APE1, respectively. The structure of MUTYH explains how MAP mutations perturb MUTYH function.
INTRODUCTION
8-Oxoguanine (8-oxoG), a major oxidized form of guanine generated by reactive oxygen species, is highly mutagenic due to its mispairing with adenine. Accumulation of 8-oxoG in DNA causes mutations, eventually leading to cancer and neurodegenerative diseases (1,2). To avoid 8-oxoG accumulation in DNA, mammals have three enzymes: MutT homologue 1 (MTH1), OGG1 (functional counterpart of MutM) and MutY homologue (MUTYH) (1,3). The functions of these enzymes, as well as their bacterial counterparts—MutT, MutM and MutY, have been well characterized by a number of previous studies (4–8). MTH1 hydrolyses mutagenic 8-oxo-dGTP and prevents its misincorporation into DNA (9–13). OGG1 and MUTYH are DNA glycosylases involved in base excision repair (BER) of base pairs containing 8-oxoG (14–17). OGG1 removes the 8-oxoG base paired with cytosine, whereas MUTYH excises a normal adenine base inserted opposite 8-oxoG on the template strand.
Mammalian MUTYH shares 33% amino acid sequence identity with MutY (mouse/human versus B. stearothermophilus, Supplementary Figure S1); further, inherited variations in human MUTYH gene are associated with colorectal polyposis and cancer termed as MUTYH-associated polyposis (MAP) (18–21). MUTYH and MutY have glycosylase activity for normal adenine mispaired with 8-oxoG and are required to excise the adenine misincorporated opposite 8-oxoG on the template strand (the adenine on the nascent strand) during DNA replication because the adenine on the template strand needs to be maintained for preserving the original genetic information. MUTYH interacts with proliferating cell nuclear antigen (PCNA) through the PCNA-interacting protein box (PIP-box) at its C-terminal region (22) (Figure 1A, Supplementary Figure S1), and a mechanism of replication-coupled repair for effective mutation-avoidance has been proposed: MUTYH is recruited to the replication fork and removes adenine on the nascent DNA strand through interaction with PCNA (23,24). Replication-coupled repair by MUTYH is unique among DNA glycosylases related to the 8-oxoG base pairs because the other DNA glycosylases, MutY, MutM and OGG1, seem to search for lesions on DNA by themselves (25–28). Further, it is reported that a unique interdomain connector (IDC) region of MUTYH is involved in its interaction with DNA-associated proteins, such as Rad9–Rad1–Hus1 complex (9–1–1) in DNA damage response and apurinic/apyrimidinic (AP) endonuclease 1 (APE1) in BER (22,29–32) (Figure 1A, Supplementary Figure S1). Previous experiments using inductively coupled plasma-mass spectrometry (ICP-MS) and extended X-ray absorption fine structure (EXAFS) showed that MUTYH has a novel Zn-binding motif composed of at least three cysteines in the IDC (Cys300, Cys307 and Cys310 in mouse), and suggested that this Zn-binding motif in the IDC arranges the N- and C-terminal domains for binding to the A:8-oxoG mispair (33). An additional study using a structural modelling of MUTYH indicates that Cys215 on the [4Fe–4S] domain is a candidate for the fourth ligand of Zn2+ (34).
Figure 1.

Structure of mouse MUTYH. (A) Domain architecture of MUTYH. His56, Cys300, Cys307 and Cys310, which are ligands of the Zn-binding motif, are labelled in red. (B) Overall structure of the MUTYH–DNA complex (Form I). 8-OxoG and the AP site are shown in red. Zn2+ is shown in magenta. N and C indicate the N- and C-terminus of the structure, respectively. (C) Recognition of 8-oxoG. Hydrogen bonds are shown as dashed lines. Van der Waals contacts are shown as dots. A water molecule is shown in red. (D) Recognition of the AP site. (E) Recognition model of the adenine base by MUTYH. The model was generated by superposition of the MutY–DNA complex (gray, PDB ID: 3G0Q) on the MUTYH–DNA complex (white). The superposed adenine nucleoside is shown in pink. Amino acid residues of MutY are shown in parentheses. (F) Possible space for the recognition of 2-oxoadenine. The surface of MUTYH is shown as a mesh.
Biochemical and structural analyses of MutY have proposed a catalytic mechanism for adenine removal following the recognition of A:8-oxoG mispairing (35–38). The crystal structures of MutM and OGG1 have also revealed the repair mechanisms of the 8-oxoG:C pair in DNA (39–41). However, the structural basis for the BER pathway of MUTYH, especially through interactions with DNA-associated proteins, is unclear because only the structure of the N-terminal domain of human MUTYH is available (42). Further, the structure of the Zn-binding motif in the IDC and its role in the BER pathway have not yet been elucidated in the previous studies (33,34). Here, we report the crystal structures of complexes between mouse MUTYH and DNA containing dSpacer (an AP site analogue, See Materials and methods):8-oxoG, and between the C-terminal domain (CTD) with the PIP box of mouse MUTYH and human PCNA. The structures have revealed the repair mechanism for the A:8-oxoG mispair, and proposed the mechanism of replication-coupled repair by MUTYH and PCNA. In addition, the structures showed that the Zn-binding motif is composed of His56 on the [4Fe–4S] domain, Cys300, Cys307 and Cys310 on the IDC. The IDC, including the Zn-binding motif, is exposed on the MUTYH surface, suggesting its interaction modes with 9–1–1 and APE1, respectively. We also examined the structural effects of MAP mutations and discussed how these mutations perturb MUTYH function.
MATERIALS AND METHODS
Protein preparation and mutagenesis
cDNA fragments encoding the core regions of mouse MUTYH (residues 45–487, 35–487 and 45–515) (DDBJ/EMBL/GenBank accession ID: AB117938) (43) were respectively subcloned into the pET48b(+) vector (Novagen). MUTYH with an N-terminal thioredoxin and His tag was expressed using Rosetta-gami2(DE3)pLysS cells. MUTYH was purified using Co-affinity, cation exchange, and heparin columns. The N-terminal tag was removed using the HRV 3C protease before heparin column chromatography. The CTD with the PIP box of MUTYH (331–515) containing an N-terminal GST tag was expressed using the pGEX-6P-1 vector (GE Healthcare) and Rosetta2(DE3)pLysS cells. The CTD was purified using glutathione affinity, cation exchange, and gel filtration columns. The N-terminal GST tag was removed with the HRV 3C protease before cation exchange column chromatography. Human PCNA, which shares 97% amino acid sequence identity with mouse PCNA, was prepared as described previously (44). The CTD–PCNA complex was prepared by mixing the CTD and PCNA solutions at a molar ratio of 1:1 (CTD:monomeric PCNA) and was purified using a gel filtration column. The C300S and F415A/S416A mutants (45–487); the L497A and F500A mutants of CTD (331–515); the ΔC14 mutant of CTD (331–501); the F500A mutant (45–515); and the ΔL mutant (45–515 without 474–486) were purified according to the above-mentioned protocols. Site-directed mutagenesis for all mutants was carried out using the KOD-Plus-Mutagenesis kit (Toyobo).
Crystallization, data collection, and structure determination
MUTYH–DNA complexes were prepared using MUTYH (residues 45–487, 35–487) and DNA annealed with the two oligonucleotides: 5′-ATGAGAC-8OG-GGGACT-3′/5′-TAGTCCC-dS-GTCTCA-3′ (8OG, 8-oxoguanine; dS, dSpacer) (Hokkaido System Science). dSpacer, a tetrahydrofuran derivative, is used as an AP site analogue (45). Hereafter dSpacer is referred to as AP site for simplicity. The data of dynamic light scattering (DLS) of the MUTYH–DNA solution (1 mg/ml) was measured at 298 K using DynaPro NanoStar (Wyatt Technology). Crystals of Form I (residues 35–487 of MUTYH) and Form II (residues 45–487 of MUTYH) were grown using a reservoir solution (0.25 M lithium sulfate or ammonium sulfate, 0.1 M Bis–Tris pH 6.0, 20–22% PEG3350). The expression of MUTYH with 40 μM Zn2+ and crystallization with 100 μM Zn2+ did not affect the quality of electron densities around the Zn-binding motif. We also purified the complex of the D207N mutant of MUTYH (a catalytically-inactive mutant) and DNA containing A:8-oxoG, but could not obtain crystals. The crystals of CTD–PCNA were obtained using a reservoir solution (0.1 M imidazole pH 8.0 and 20% PEG8000). Crystals of MUTYH–DNA and CTD–PCNA were cryoprotected with 15–20% glycerol and frozen at 100 K. X-ray diffraction experiments were performed at 100 K on BL44XU at SPring-8, and on BL17A and AR-NW12A at Photon Factory. The data were processed using HKL-2000 or XDS (46,47). The phases of the MUTYH–DNA data were determined using a combination of molecular replacement with the coordinates of the N-terminal domain of human MUTYH (PDB ID: 3N5N) and single-wavelength anomalous dispersion with iron atoms of the [4Fe–4S] cluster using PHENIX (48). Model building and refinement were carried out using PHENIX and COOT (49). The phases of the CTD–PCNA data were determined by molecular replacement using MOLREP (50) in the CCP4 program suite (51) with the structures of PCNA (PDB ID: 1UL1) and the alanine model of CTD as search models. After several cycles of the refinement of PCNA and CTD, the structure of the PIP box was built and fitted into the electron densities. In the refinement of the CTD–PCNA complex, PCNA and the PIP box were refined with clear electron densities, whereas most of the CTD residues were refined as alanine owing to their weaker electron densities compared to those of PCNA and the PIP box. The data collection and refinement statistics are presented in Supplementary Table S1. All molecular graphics were prepared using PyMOL (Version 2.0 Schrödinger, LLC).
Size-exclusion chromatography
CTDs (wild type, L497A, F500A and ΔC14) and PCNA solutions were mixed at a molar ratio of 1:1 and applied to Superdex 200 10/300 GL (GE Healthcare). The buffer solution for size-exclusion chromatography contained 20 mM Tris–HCl (pH 7.5), 50 mM NaCl and 1 mM β-mercaptoethanol. MUTYH (45–515) (wild type and F500A) and PCNA were mixed at a molar ratio of 1:3, and a buffer solution with 150 mM NaCl was used.
Measurement of glycosylase activity
The glycosylase activities of MUTYH (45–487) (wild type, C300S and F415A/S416A) were measured in a 20 μl reaction buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM DTT and 100 μg/ml BSA. The concentrations of MUTYH were calculated by UV and Bradford methods. The concentrations of MUTYH and DNA were 25 nM and 125 nM, and 100 nM and 20 nM, respectively. The DNA substrate was prepared by annealing the two oligonucleotides: 5′-(FITC)-ATATAGGGAGTCCC-A*-GTCTCAGTCATATT-3′/5′-AATATGACTGAGAC-8OG-GGGACTCCCTATAT-3′ (A* indicates adenine mispaired with 8-oxoG). Reactions were carried out at 37°C and were terminated at various time points by adding 5 μl of 1 M NaOH and heating at 90°C for 10 min. The samples were mixed with 25 μl of formamide, heated at 90°C for 10 min, and electrophoresed on a denaturing 15% (w/v) polyacrylamide gel containing 7 M urea. The ratios of the substrates and products were analysed using a fluorimaging analyser (Typhoon FLA 9000). The data were fitted to the equations in a previous report (52). The glycosylase activity of MUTYH (45–515 and ΔL) in the presence of PCNA was measured using two annealed oligonucleotides: 5′-(FITC)-ATATAGGGAGTCCCAGTCTC-A*-GTCATATT-3′/5′-AATATGAC-8OG-GAGACTGGGACTCCCTATAT-3′. The concentrations of MUTYH, DNA, and PCNA trimer were 100, 20, 50–5000 nM (for the 5-min reaction), and 25, 125, 50–2500 nM (for the 10-min reaction).
RESULTS AND DISCUSSION
Overall structure of the MUTYH–DNA complex
We determined two forms (Form I and II) of the crystal structures of mouse MUTYH in complex with DNA containing AP:8-oxoG at 2.45- and 1.97-Å resolution, respectively (Figure 1, Supplementary Table S1). Form I showed that MUTYH has a Zn-binding motif composed of His56, Cys300, Cys307 and Cys310 (discussed later), whereas Form II showed swapped Zn-binding with a neighbouring symmetry mate in the crystal (Supplementary Figure S2A, B). DLS analysis showed that the MUTYH–DNA complex (a calculated molecular mass of 58k using the sequence information of MUTYH and DNA) is monodisperse with an estimated molecular mass of 57k (Supplementary Figure S3), indicating that MUTYH is monomeric in solution and the swapped Zn-binding observed in Form II is due to the crystal packing. The structures of Form I and II are very similar except for the Zn-binding motif (r.m.s.d. = 0.5 Å for 387 Cα atoms). Thus, Form I was used for discussing the overall structure of MUTYH, and Form II, with higher resolution, was used for discussing the interactions observed in the MUTYH–DNA complex except for the IDC.
The N-terminal domain (NTD; the six-helix barrel and [4Fe–4S] domains) and the CTD of MUTYH bind to AP:8-oxoG in DNA (Figure 1B). The IDC connecting the NTD and CTD traverses along the major groove of DNA, and MUTYH encircles DNA via interaction between the six-helix barrel domain and the CTD (Figure 1B). The structure of the NTD is similar to that of human MUTYH (PDB ID: 3N5N, residues 65–350 but lacking the Zn-binding motif) except for the IDC and the DNA binding regions (r.m.s.d. = 0.5 Å for 179 Cα atoms) (42) (Supplementary Figure S4A). The overall structure of the MUTYH–DNA complex was similar to that of the B. stearothermophilus MutY–DNA complex (PDB ID: 3G0Q) (37), except for the insertion of IDC and the CTD orientation (Supplementary Figure S4B). The hydrogen bonding network that encloses DNA between the six-helix barrel domain and the CTD of MUTYH differs from that of MutY, which results in different orientations of their CTDs (Supplementary Figures S4B and S5).
Recognition of 8-oxoG, AP site, and adenine
8-OxoG is recognized by Gln110, Thr111, Leu148 and Tyr150 that were inserted into the DNA helix (Figure 1C, Supplementary Figure S6A). Gln110 also forms a hydrogen bond with the phosphate backbone close to the AP site and supports its flip-out structure (Figure 1D, Supplementary Figure S6B). Comparison of the MUTYH–DNA complex and the NTD of human MUTYH showed that structural changes were induced in the DNA binding regions by the insertion of Gln110 and Tyr150 into AP:8-oxoG (Supplementary Figure S4A). N7-H and O8 atoms, which are characteristic features of 8-oxoG, are recognized by hydrogen bonds with Ser416 and van der Waals contacts with Phe415 (Figure 1C). Mutations in Phe415 and Ser416 (F415A/S416A) strongly reduced the glycosylase activity for A:8-oxoG mispairing (Supplementary Figure S7A), supporting that Phe415 and Ser416 are important for 8-oxoG recognition in DNA. This result is consistent with the previous kinetic and DNA binding analyses of MutY, which showed that the corresponding F307A/S308A mutant of MutY has 8-fold lower k2 for A:8-oxoG and 50-fold higher Kd in the binding of DNA containing the A:8-oxoG analogue (53). The binding mode of 8-oxoG and the AP site in MUTYH is similar to that in the complex of MutY and DNA containing A:8-oxoG (PDB ID: 3G0Q), and the recognition of adenine by MUTYH was examined using the structural model of MUTYH complexed with the A:8-oxoG mispair (Figure 1E). The recognition mode of adenine is mostly conserved between MUTYH and MutY, except for Gln251 and Met254 in MUTYH. MUTYH recognizes N6-H of adenine by a hydrogen bond with Oϵ of the polar side chain of Gln251 whereas MutY recognizes the N6-H by a hydrogen bond with the negatively charged side chain of Glu188. Met254 of MUTYH makes van der Waals contacts with adenine instead of Ile191 in MutY. The conformations of the catalytic residues Asp207, Glu105 and Tyr189 in MUTYH are identical to those of Asp144, Glu43 and Tyr126 in MutY, indicating that MutY and MUTYH share similar catalytic mechanisms (35,37,38). MUTYH has glycosylase activity for both 2-oxoadenine (2-oxoA) and adenine (54). There is a space between Arg80 and C2 of adenine, which can accommodate the O2 atom of 2-oxoA (Figure 1F). MUTYH would recognize 2-oxoA through a possible hydrogen bond between O2 of 2-oxoA and Arg80.
Insights into DNA replication-coupled repair by MUTYH and PCNA
It is proposed that MUTYH functions in DNA replication-coupled repair through interaction between its C-terminal PIP box and PCNA (23,24). To investigate replication-coupled repair, the crystal structure of the CTD with the PIP box of mouse MUTYH (residues 331–515) in complex with human PCNA (CTD–PCNA) was determined at 2.7-Å resolution (Supplementary Table S1). There is one CTD–PCNA complex (CTD:monomeric PCNA = 1:1) in the asymmetric unit of the crystal, which comprises two respective layers of PCNA and CTD (Supplementary Figure S8A). PCNA forms a trimer with crystallographic symmetry, which is essentially the same as that reported previously (55). The structure of PCNA and the PIP box could be refined, whereas the electron densities of the CTD were traced with Cα atoms, and most of the CTD residues were refined as alanine (Supplementary Figure S8B). The electron densities of the spacer region between the CTD and the PIP box (470–489) and of the C-terminal tail (504–515) were missing (Figure 1A, Supplementary Figure S1). In the CTD–PCNA complex, specific interactions were observed only between the PIP box of MUTYH and PCNA, which resulted in weaker electron densities of the CTD compared to those of PCNA and the PIP box. We focused on the interactions between the PIP box and PCNA in the CTD–PCNA complex (Figure 2A, B) because the size-exclusion chromatography experiments described later also showed that the C-terminal PIP box is essential for interaction with PCNA.
Figure 2.
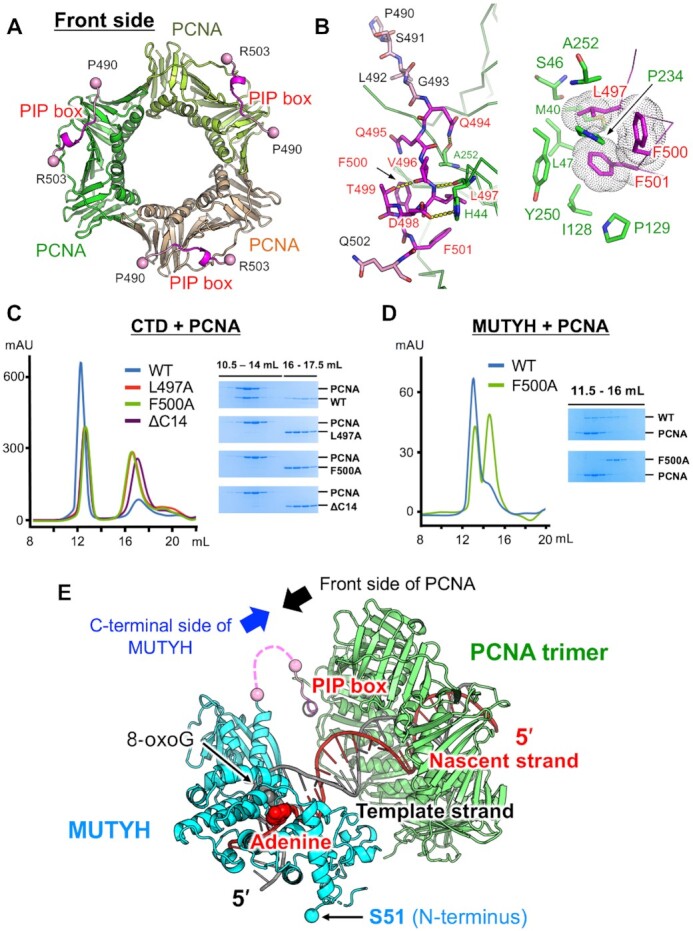
Structure and interactions of the complex between MUTYH and PCNA. (A) Overall structure of the PCNA trimer (green, lightgreen and lightorange) in complex with the PIP box (magenta) of MUTYH. (B) Interactions between the PIP box and PCNA. (C) Size-exclusion chromatography experiments using CTDs in complex with PCNA. Chromatograms of the wild type CTD (blue), L497A (red), F500A (green) and ΔC14 (purple), and SDS-PAGE analyses. WT indicates wild type. (D) Size-exclusion chromatography experiments using MUTYH (45–515) (wild type and F500A) in complex with PCNA. Chromatograms of the wild type (blue) and F500A (green), and SDS-PAGE analyses. (E) Structural model of replication-coupled repair by MUTYH (cyan and pink) and PCNA (green). MUTYH is loaded to DNA in the appropriate direction for the recognition and removal of adenine on the nascent DNA strand (red). The spacer region between the CTD and the PIP box (19 amino acids) is shown as a pink dashed line.
The consensus amino acid sequence of the canonical PIP box is defined by QXXhXXaa (h, hydrophobic; a, aromatic; X, any). The previous study revealed the importance of the PIP box, especially of the conserved Phe518 and Phe519 in human MUTYH (Phe500 and Phe501 in mouse), for the interaction with PCNA (22). The PIP box of MUTYH (QQVLDTFF) binds to the C-terminal side of PCNA (hereafter referred to as the front side) (Figure 2A). The conserved Gln494 in the PIP box forms a hydrogen bond with the main chain of Ala252 in PCNA (Figure 2B left), and the conserved hydrophobic and aromatic residues, Leu497, Phe500 and Phe501, make van der Waals contacts with the hydrophobic pocket of PCNA (Figure 2B right). The non-conserved Asp498 interacts with His44 in PCNA. The hydrogen bond between Thr499 and Val496 in the PIP box maintains its 310 helix structure to fit Leu497, Phe500 and Phe501 into the hydrophobic pocket of PCNA. Size-exclusion chromatography experiments using CTDs (331–515) with mutated PIP box (L497A and F500A) and CTD without the C-terminal 14 residues (ΔC14, 331–501) showed that all mutants lost their binding ability to PCNA (Figure 2C). These results indicate that the PIP box including the C-terminal tail (C-terminal PIP box region) is essential for interaction with PCNA. Although the electron densities of the C-terminal tail (504–515) were missing in the structure of CTD–PCNA, the C-terminal tail of MUTYH would also contribute to the interactions with PCNA, as in FEN1 and translesion DNA polymerases whose main chains at the C-terminal side of the PIP box are involved in interactions with PCNA (44,56). Further, MUTYH (45–515) with the F500A mutation cannot bind to PCNA (Figure 2D), indicating that the C-terminal PIP box region is also required for the interaction between the core region of MUTYH and PCNA. Based on these results, a structural model of replication-coupled repair by MUTYH and PCNA was proposed (Figure 2E). MUTYH is recruited and tethered to the replication fork by binding of its C-terminal PIP box region to PCNA. MUTYH is loaded to the A:8-oxoG mispair on DNA in a direction where the C-terminal side of MUTYH faces the front side of PCNA. MUTYH has a 23-amino acid linker between the CTD and the PIP box (Supplementary Figure S1, a pink dashed line in Figure 2E), and this linker length does not allow recruitment of MUTYH at the back side of PCNA. The binding direction of MUTYH to DNA at the front side of PCNA, which is fixed by tethering between the PIP box and PCNA, is appropriate for MUTYH to remove adenine on the nascent DNA strand, resulting in replication-coupled repair for effective mutation-avoidance. The model also shows that MUTYH cannot access the damaged site while DNA polymerase synthesizes a nascent DNA strand because DNA polymerase also functions at the front side of PCNA (57). In replication-coupled repair, switching from DNA polymerase to MUTYH on PCNA seems to be required, as in the case of FEN1 (57).
We further examined whether PCNA stimulates MUTYH glycosylase activity. Stimulation of MUTYH activity by PCNA was not detected (Supplementary Figure S7B). Similar results were reported with S. solfataricus uracil DNA glycosylase and PCNA (58). We considered that the linker length (23 amino acids) between the CTD and the PIP box may affect stimulation by PCNA because it is longer than that (four amino acids) of FEN1, whose activity is enhanced by PCNA (44). However, the activity of MUTYH with a short linker (ΔL, deletion of 13 amino acids, Lys474–Ser486) was not stimulated by PCNA (Supplementary Figure S7B). Previous experiments using an in vivo repair assay showed that interaction between the PIP box of MUTYH and PCNA is crucial for the efficiency of replication-coupled repair (23). These results indicate that the PIP box of MUTYH has an essential role in the recruitment and tethering of MUTYH to the replication fork for BER. RPA, a single-stranded DNA-binding protein in the replication fork, is another important element for the discussion of the replication-couple repair by MUTYH because MUTYH has an RPA-binding site at its N-terminal region (residues 6–32 in human MUTYH) (22). In the structure of the MUTYH–DNA complex, the N-terminus (Ser51 in the structure) is close to the template strand (Figure 2E). The positions of the N-terminus of MUTYH and the template strand seem to be appropriate for the interaction between MUTYH and RPA which binds to the single-stranded template. These structural insights suggest that PCNA and RPA synergistically act on the recruitment of MUTYH and possibly enhance its activity.
Structure of a Zn-binding motif
The previous study showed that mouse MUTYH has a novel Zn-binding motif composed of at least three cysteines, Cys300, Cys307 and Cys310 (33). Further, a structural modelling of MUTYH identified Cys215 as a candidate for the fourth ligand of Zn2+ (34). Our crystal structures showed that the fourth ligand of the Zn-binding motif is His56 on the N-terminal [4Fe–4S] domain, and that the Zn-binding motif is formed by His56, Cys300, Cys307 and Cys310 (Figure 3). Cys215, which is also conserved in human, forms a hydrophobic core close to the Zn-binding motif, with Phe326 and Pro327 on the IDC as well as Leu211, Ile220, Pro224 and Leu233 on the [4Fe–4S] domain (Supplementary Figure S2C). The activity measurement results showing that the C300S mutant is less active than the wild type, were consistent with previous experiments (33) (Supplementary Figure S7A). In Form I, amino acids from Arg285 to Ile297 and from Leu302 to Gln306 in the IDC were disordered (Figure 3A). MUTYH was prepared and crystallized in the presence or absence of Zn2+, which did not affect the quality of electron densities around the Zn-binding motif (as mentioned in Materials and methods). The IDC in Form I has higher B-factors and the IDC in Form II shows swapped Zn-binding in the crystal, indicating that a part of the IDC is flexible even in the complex form with DNA (Supplementary Figure S2A, B). The flexible nature of the IDC would be necessary for its binding to different partners such as 9–1–1 and APE1 (discussed later). The IDC covers the [4Fe–4S] domain and traverses along the major groove of DNA (Figure 3A). The conserved amino acid residues between mice and humans, such as Leu311, Trp317, Asn325 and Phe326 on the IDC as well as Ala219 and Tyr55 on the [4Fe–4S] domain make van der Waals contacts, and the Zn-binding motif is located on the protein surface (Figure 3B, C).
Figure 3.
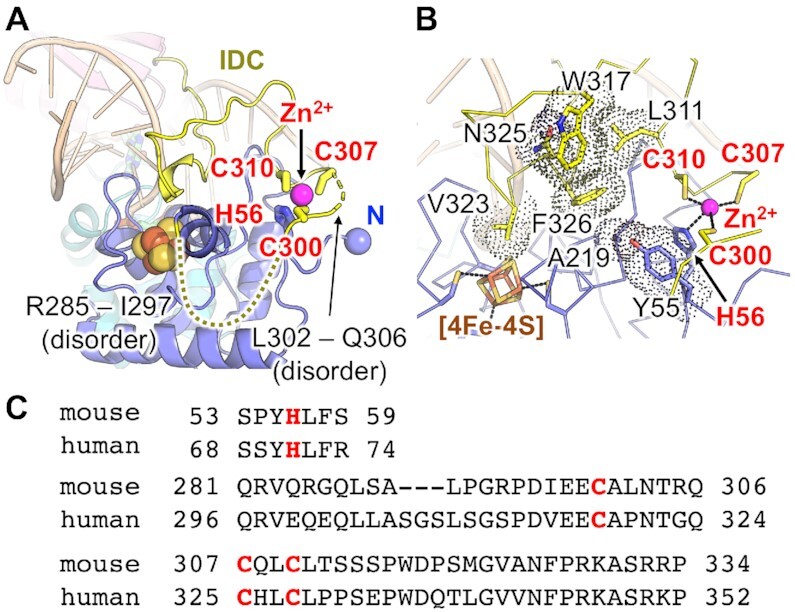
IDC and Zn-binding motif. (A) Structure of the IDC and the Zn-binding motif. The amino acid residues Arg285–Ile297 and Leu302–Gln306 are disordered. (B) Van der Waals contacts observed in the IDC and [4Fe–4S] domain. (C) Amino acid sequences around the Zn-binding motif.
Structural insights into the functions of MUTYH coupled with 9–1–1 and APE1 through IDC were examined using the structures of MUTYH, 9–1–1 and APE1 as well as the biochemical data for their interactions (Figure 4). 9–1–1, which is involved in the DNA damage checkpoint pathway, is a DNA damage-specific clamp for the recruitment of the proteins in checkpoint signalling and DNA repair (59). Recent study has described that MUTYH and 9–1–1 are recruited to oxidatively damaged telomeres through SIRT6, an NAD+-dependent histone deacetylase (60). It is reported that 9–1–1 interacts with MUTYH through the loop region (Pro134–Asp155) in its Hus1 subunit (30). Structural studies on 9–1–1 indicate that 9–1–1 is a double-faced DNA clamp that can recruit proteins at both the front and back sides (61–64). FEN1 is indicated to bind to the front side of 9–1–1 (63), whereas RHINO, which is involved in DNA damage response through CHK1 activation, interacts with the edge close to the back side of 9–1–1 (64). The location of the binding interface between the IDC of MUTYH and the loop region in Hus1 of 9–1–1 suggests that MUTYH functions at the front side of 9–1–1 which is loaded on the damaged DNA (29,30) (Figure 4A). FEN1 also seems to be recruited at the front side of 9–1–1, but in a different way, through the interaction between its PIP box and 9–1–1 (63).
Figure 4.

Suggested binding modes of MUTYH with 9–1–1 and APE1. (A) Binding mode between MUTYH and 9–1–1 (green, pink and cyan, PDB ID: 3A1J) in the cartoon and surface style. The IDC of MUTYH and the binding surface for MUTYH on 9–1–1 are shown in yellow. (B) Binding mode between MUTYH and APE1 (cyan, PDB ID: 1DEW) (66) in the cartoon and surface style (left), and in the cartoon style (right). The binding surface for MUTYH on APE1 is shown in yellow. The AP site is shown with red spheres.
During BER, MUTYH is reported to protect its own product, the generated AP site opposite 8-oxoG, from other enzymes such as APE1 (in the presence of 5-fold molar excess) and OGG1, as MUTYH has a high affinity for its own product (65). On the other hand, another report found that APE1 stimulates the glycosylase activity of MUTYH in excessive amounts of APE1 (10-fold to 100-fold molar excess) (31). These results indicate that increasing the frequency of physical interaction between MUTYH and APE1 promotes the appropriate transfer of the generated AP site from MUTYH to APE1. The AP site transfer is believed to be an essential process in the BER pathway, as the AP site is cytotoxic when left unprocessed. A previous NMR study showed that APE1 interacts with the IDC of MUTYH through two regions, its catalytic site and the back site distal to the catalytic site (32). The binding interface between the IDC of MUTYH and only the back site of APE1 (His116–Gln117 and Arg136–Cys138) suggests the interaction for the AP site transfer from MUTYH to APE1 (Figure 4B). Through interaction with the IDC of MUTYH, APE1 is located at the 3′ side of the AP site generated by MUTYH. APE1 accepts the AP site from MUTYH without interfering with the interaction between MUTYH and PCNA because APE1 interacts with MUTYH at the opposite side of the interface between MUTYH and PCNA. In this model, the binding of APE1 to the MUTYH–DNA complex induces local structural changes of MUTYH and DNA, which promote the release of the AP site from MUTYH and the AP site transfer to APE1 via sliding of DNA (Figure 4B).
MUTYH-associated polyposis mutations
The common missense mutations in patients with MAP are Y179C and G396D (Y150C and G365D in mice) (18,19). In addition to Y179C and G396D, a number of MAP-related mutations were identified, and their mutational effects on glycosylase activity or DNA binding were examined (21). The missense mutations were mapped to the structure of the mouse MUTYH–DNA complex (Figure 5A). All residues mapped in Figure 5A were conserved between humans and mice. Therefore, the amino acid numbering of mouse MUTYH is used hereafter. Y150C, G365D and five other missense mutations (R153H, R156W, R212W, M254V and G257E) are located in the DNA-binding region (Figure 5B). These residues contribute to the interactions with A:8-oxoG or the DNA backbone, and their mutations would perturb recognition of the A:8-oxoG mispair. In the common missense mutations, the phenotypic effects of Y150C are relatively severe compared to those of G365D (19). The data agrees with the MUTYH–DNA structure in which Tyr150 is involved in a direct interaction with 8-oxoG whereas Gly365 interacts with the DNA backbone close to the 8-oxoG. The mutations R216H/L, V217F and P266L, observed in the [4Fe–4S] domain, would disrupt the structure of the [4Fe–4S] cluster (Figure 5C). The R245Q mutation distal to the [4Fe–4S] cluster loses hydrogen bonds with Asp76 and Asp248. The remaining three mutations (I194V, L357P and P374L) seem to destabilize the hydrophobic cores in the MUTYH structure (Figure 5D).
Figure 5.

MUTYH-associated polyposis (MAP) mutations. (A) Mapping of the MAP-related mutations. Amino acid residues of mouse MUTYH corresponding to the MAP-related mutations are labelled. The amino acid numbering of human MUTYH is shown in parentheses. The amino acid residues for DNA binding and for the stabilization of protein folding are shown in red and blue, respectively. (B–D) Interactions observed in the amino acid residues involved in MAP.
CONCLUSION
Our structural and biochemical studies have elucidated the repair mechanism for the A:8-oxoG mispair and proposed the DNA replication-coupled repair process involving MUTYH, wherein MUTYH is loaded onto DNA by PCNA in a fixed direction for the recognition and removal of adenine on the nascent DNA strand. Further, the structure of a Zn-binding motif and the structural effects on MAP mutations were revealed. The Zn-binding motif at the IDC comprises His56, Cys300, Cys307 and Cys310, and is exposed to the protein surface. The structures of MUTYH, 9–1–1 and APE1, including their respective interacting regions, suggest testable models for examining the BER mechanism of MUTYH coupled with 9–1–1 and APE1, respectively.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession number (7EF8, MUTYH–DNA Form I; 7EF9, MUTYH–DNA Form II; 7EFA, CTD–PCNA).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the beamline staff at SPring-8 and Photon Factory for their help in the data collection. Synchrotron experiments were performed with the approval of the Cooperative Research Program of Institute for Protein Research, Osaka University (2017A6761), and the Photon Factory Program Advisory Committee (2017G167 and 2019G027). We also thank M.A. Suico for critical reading of the manuscript.
Contributor Information
Teruya Nakamura, Graduate School of Pharmaceutical Sciences, Kumamoto University, 5-1 Oehonmachi, Chuo-ku, Kumamoto, 862-0973 Kumamoto, Japan; Priority Organization for Innovation and Excellence, Kumamoto University, 5-1 Oehonmachi, Chuo-ku, Kumamoto, 862-0973 Kumamoto, Japan.
Kohtaro Okabe, Graduate School of Pharmaceutical Sciences, Kumamoto University, 5-1 Oehonmachi, Chuo-ku, Kumamoto, 862-0973 Kumamoto, Japan.
Shogo Hirayama, Graduate School of Pharmaceutical Sciences, Kumamoto University, 5-1 Oehonmachi, Chuo-ku, Kumamoto, 862-0973 Kumamoto, Japan.
Mami Chirifu, Graduate School of Pharmaceutical Sciences, Kumamoto University, 5-1 Oehonmachi, Chuo-ku, Kumamoto, 862-0973 Kumamoto, Japan.
Shinji Ikemizu, Graduate School of Pharmaceutical Sciences, Kumamoto University, 5-1 Oehonmachi, Chuo-ku, Kumamoto, 862-0973 Kumamoto, Japan.
Hiroshi Morioka, Graduate School of Pharmaceutical Sciences, Kumamoto University, 5-1 Oehonmachi, Chuo-ku, Kumamoto, 862-0973 Kumamoto, Japan.
Yusaku Nakabeppu, Division of Neurofunctional Genomics, Department of Immunobiology and Neuroscience, Medical Institute of Bioregulation, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan.
Yuriko Yamagata, Graduate School of Pharmaceutical Sciences, Kumamoto University, 5-1 Oehonmachi, Chuo-ku, Kumamoto, 862-0973 Kumamoto, Japan; Shokei University and Shokei University Junior College, 2-6-78, Kuhonji, Chuo-ku, Kumamoto, 862-8678 Kumamoto, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Leading Initiative for Excellent Young Researchers (to T.N.); Grants-in-Aid for Scientific Research [25460038 to T.N.]; Grant-in-Aid for Scientific Research on Innovative Areas [22131007 to Y.Y.]; Grant for Basic Science Research Projects from The Sumitomo Foundation (to T.N.); Takeda Science Foundation (to T.N.); Kato Memorial Bioscience Foundation (to T.N.); KUMAYAKU Alumni Research Foundation (to T.N.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Nakabeppu Y., Sakumi K., Sakamoto K., Tsuchimoto D., Tsuzuki T., Nakatsu Y.. Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol. Chem. 2006; 387:373–379. [DOI] [PubMed] [Google Scholar]
- 2. Nakabeppu Y. Wilson D.M. III Neurodegeneration caused by accumulation of an oxidized base lesion, 8-oxoguanine, in nuclear and mitochondrial DNA: from animal models to human diseases. The Base Excision Repair Pathway: Molecular Mechanisms and Role in Disease Development and Therapeutic Design. 2017; World Scientific; 523–556. [Google Scholar]
- 3. Tsuzuki T., Nakatsu Y., Nakabeppu Y.. Significance of error-avoiding mechanisms for oxidative DNA damage in carcinogenesis. Cancer Sci. 2007; 98:465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maki H., Sekiguchi M.. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature. 1992; 355:273–275. [DOI] [PubMed] [Google Scholar]
- 5. Tchou J., Kasai H., Shibutani S., Chung M.H., Laval J., Grollman A.P., Nishimura S.. 8-oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Michaels M.L., Tchou J., Grollman A.P., Miller J.H.. A repair system for 8-Oxo-7, 8-dihydrodeoxyguanine. Biochemistry. 1992; 31:10964–10968. [DOI] [PubMed] [Google Scholar]
- 7. Tajiri T., Maki H., Sekiguchi M.. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res. 1995; 336:257–267. [DOI] [PubMed] [Google Scholar]
- 8. Nakamura T., Meshitsuka S., Kitagawa S., Abe N., Yamada J., Ishino T., Nakano H., Tsuzuki T., Doi T., Kobayashi Y.et al.. Structural and dynamic features of the MutT protein in the recognition of nucleotides with the mutagenic 8-oxoguanine base. J. Biol. Chem. 2010; 285:444–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mo J.Y., Maki H., Sekiguchi M.. Hydrolytic elimination of a mutagenic nucleotide, 8-oxodGTP, by human 18-kilodalton protein: sanitization of nucleotide pool. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:11021–11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sakumi K., Furuichi M., Tsuzuki T., Kakuma T., Kawabata S., Maki H., Sekiguchi M.. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J. Biol. Chem. 1993; 268:23524–23530. [PubMed] [Google Scholar]
- 11. Svensson L.M., Jemth A.-S., Desroses M., Loseva O., Helleday T., Högbom M., Stenmark P.. Crystal structure of human MTH1 and the 8-oxo-dGMP product complex. FEBS Lett. 2011; 585:2617–2621. [DOI] [PubMed] [Google Scholar]
- 12. Waz S., Nakamura T., Hirata K., Koga-Ogawa Y., Chirifu M., Arimori T., Tamada T., Ikemizu S., Nakabeppu Y., Yamagata Y.. Structural and kinetic studies of the human nudix hydrolase MTH1 reveal the mechanism for its broad substrate specificity. J. Biol. Chem. 2017; 292:2785–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakamura T., Hirata K., Fujimiya K., Chirifu M., Arimori T., Tamada T., Ikemizu S., Yamagata Y.. X-ray structure analysis of human oxidized nucleotide hydrolase MTH1 using crystals obtained under microgravity. Int. J. Micrograv. Sci. Appl. 2019; 36:360103. [Google Scholar]
- 14. Aburatani H., Hippo Y., Ishida T., Takashima R., Matsuba C., Kodama T., Takao M., Yasui A., Yamamoto K., Asano M.et al.. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res. 1997; 57:2151–2156. [PubMed] [Google Scholar]
- 15. Nishioka K., Ohtsubo T., Oda H., Fujiwara T., Kang D., Sugimachi K., Nakabeppu Y.. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell. 1999; 10:1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Slupska M.M., Baikalov C., Luther W.M., Chiang J.H., Wei Y.F., Miller J.H.. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol. 1996; 178:3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohtsubo T., Nishioka K., Imaiso Y., Iwai S., Shimokawa H., Oda H., Fujiwara T., Nakabeppu Y.. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000; 28:1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Al-Tassan N., Chmiel N.H., Maynard J., Fleming N., Livingston A.L., Williams G.T., Hodges A.K., Davies D.R., David S.S., Sampson J.R.et al.. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 2002; 30:227–232. [DOI] [PubMed] [Google Scholar]
- 19. Nielsen M., Joerink - van de Beld M.C., Jones N., Vogt S., Tops C.M., Vasen H.F.A., Sampson J.R., Aretz S., Hes F.J.. Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology. 2009; 136:471–476. [DOI] [PubMed] [Google Scholar]
- 20. Oka S., Nakabeppu Y.. DNA glycosylase encoded by MUTYH functions as a molecular switch for programmed cell death under oxidative stress to suppress tumorigenesis. Cancer Sci. 2011; 102:677–682. [DOI] [PubMed] [Google Scholar]
- 21. Mazzei F., Viel A., Bignami M.. Role of MUTYH in human cancer. Mutat. Res. 2013; 743–744:33–43. [DOI] [PubMed] [Google Scholar]
- 22. Parker A., Gu Y., Mahoney W., Lee S.H., Singh K.K., Lu A.L.. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem. 2001; 276:5547–5555. [DOI] [PubMed] [Google Scholar]
- 23. Hayashi H., Tominaga Y., Hirano S., McKenna A.E., Nakabeppu Y., Matsumoto Y.. Replication-associated repair of adenine:8-oxoguanine mispairs by MYH. Curr. Biol. 2002; 12:335–339. [DOI] [PubMed] [Google Scholar]
- 24. van Loon B., Hübscher U.. An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase lambda. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:18201–18206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Banerjee A., Yang W., Karplus M., Verdine G.L.. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature. 2005; 434:612–618. [DOI] [PubMed] [Google Scholar]
- 26. Banerjee A., Santos W.L., Verdine G.L.. Structure of a DNA glycosylase searching for lesions. Science. 2006; 311:1153–1157. [DOI] [PubMed] [Google Scholar]
- 27. Blainey P.C., Van Oijen A.M., Banerjee A., Verdine G.L., Xie X.S.. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang L., Chakravarthy S., Verdine G.L.. Structural basis for the lesion-scanning mechanism of the MutY DNA glycosylase. J. Biol. Chem. 2017; 292:5007–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi G., Chang D.Y., Cheng C.C., Guan X., Venclovas Č., Lu A.L.. Physical and functional interactions between MutY glycosylase homologue (MYH) and checkpoint proteins Rad9-Rad1-Hus1. Biochem. J. 2006; 400:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hwang B.J., Jin J., Gunther R., Madabushi A., Shi G., Wilson G.M., Lu A.L.. Association of the Rad9-Rad1-Hus1 checkpoint clamp with MYH DNA glycosylase and DNA. DNA Repair. 2015; 31:80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang H., Clendenin W.M., Wong D., Demple B., Slupska M.M., Chiang J.H., Miller J.H.. Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res. 2001; 29:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luncsford P.J., Manvilla B.A., Patterson D.N., Malik S.S., Jin J., Hwang B.J., Gunther R., Kalvakolanu S., Lipinski L.J., Yuan W.et al.. Coordination of MYH DNA glycosylase and APE1 endonuclease activities via physical interactions. DNA Repair. 2013; 12:1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Engstrom L.M., Brinkmeyer M.K., Ha Y., Raetz A.G., Hedman B., Hodgson K.O., Solomon E.I., David S.S.. A zinc linchpin motif in the MUTYH glycosylase interdomain connector is required for efficient repair of DNA damage. J. Am. Chem. Soc. 2014; 136:7829–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nuñez N.N., Khuu C., Babu C.S., Bertolani S.J., Rajavel A.N., Spear J.E., Armas J.A., Wright J.D., Siegel J.B., Lim C.et al.. The zinc linchpin motif in the DNA repair glycosylase MUTYH: identifying the Zn2+ ligands and roles in damage recognition and repair. J. Am. Chem. Soc. 2018; 140:13260–13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guan Y., Manuel R.C., Arvai A.S., Parikh S.S., Mol C.D., Miller J.H., Lloyd R.S., Tainer J.A.. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat. Struct. Biol. 1998; 5:1058–1064. [DOI] [PubMed] [Google Scholar]
- 36. Fromme J.C., Banerjee A., Huang S.J., Verdine G.L.. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004; 427:652–656. [DOI] [PubMed] [Google Scholar]
- 37. Lee S., Verdine G.L.. Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:18497–18502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Woods R.D., O’Shea V.L., Chu A., Cao S., Richards J.L., Horvath M.P., David S.S.. Structure and stereochemistry of the base excision repair glycosylase MutY reveal a mechanism similar to retaining glycosidases. Nucleic Acids Res. 2016; 44:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fromme J.C., Verdine G.L.. Structural insights into lesion recognition and repair by the bacterial 8-oxoguanine DNA glycosylase MutM. Nat. Struct. Biol. 2002; 9:544–552. [DOI] [PubMed] [Google Scholar]
- 40. Fromme J.C., Verdine G.L.. DNA lesion recognition by the bacterial repair enzyme MutM. J. Biol. Chem. 2003; 278:51543–51548. [DOI] [PubMed] [Google Scholar]
- 41. Bruner S.D., Norman D.P., Verdine G.L.. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000; 403:859–866. [DOI] [PubMed] [Google Scholar]
- 42. Luncsford P.J., Chang D.-Y., Shi G., Bernstein J., Madabushi A., Patterson D.N., Lu A.-L., Toth E.A.. A structural hinge in eukaryotic MutY homologues mediates catalytic activity and Rad9-Rad1-Hus1 checkpoint complex interactions. J. Mol. Biol. 2010; 403:351–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ichinoe A., Behmanesh M., Tominaga Y., Ushijima Y., Hirano S., Sakai Y., Tsuchimoto D., Sakumi K., Wake N., Nakabeppu Y.. Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res. 2004; 32:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sakurai S., Kitano K., Yamaguchi H., Hamada K., Okada K., Fukuda K., Uchida M., Ohtsuka E., Morioka H., Hakoshima T.. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005; 24:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takeshita M., Chang C.N., Johnson F., Will S., Grollman A.P.. Oligodeoxynucleotides containing synthetic abasic sites. Model substrates for DNA polymerases and apurinic/apyrimidinic endonucleases. J. Biol. Chem. 1987; 262:10171–10179. [PubMed] [Google Scholar]
- 46. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 47. Kabsch W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adams P.D., Afonine P. V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W.et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 50. Vagin A., Teplyakov A.. Molecular replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010; 66:22–25. [DOI] [PubMed] [Google Scholar]
- 51. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A.et al.. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kundu S., Brinkmeyer M.K., Livingston A.L., David S.S.. Adenine removal activity and bacterial complementation with the human MutY homologue (MUTYH) and Y165C, G382D, P391L and Q324R variants associated with colorectal cancer. DNA Repair. 2009; 8:1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Russelburg L.P., O’Shea Murray V.L., Demir M., Knutsen K.R., Sehgal S.L., Cao S., David S.S., Horvath M.P. Structural basis for finding OG lesions and avoiding undamaged G by the DNA glycosylase MutY. ACS Chem. Biol. 2020; 15:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ushijima Y., Tominaga Y., Miura T., Tsuchimoto D., Sakumi K., Nakabeppu Y.. A functional analysis of the DNA glycosylase activity of mouse MUTYH protein excising 2-hydroxyadenine opposite guanine in DNA. Nucleic Acids Res. 2005; 33:672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gulbis J.M., Kelman Z., Hurwitz J., O’Donnell M., Kuriyan J.. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell. 1996; 87:297–306. [DOI] [PubMed] [Google Scholar]
- 56. Hishiki A., Hashimoto H., Hanafusa T., Kamei K., Ohashi E., Shimizu T., Ohmori H., Sato M.. Structural basis for novel interactions between human translesion synthesis polymerases and proliferating cell nuclear antigen. J. Biol. Chem. 2009; 284:10552–10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lancey C., Tehseen M., Raducanu V.S., Rashid F., Merino N., Ragan T.J., Savva C.G., Zaher M.S., Shirbini A., Blanco F.J.et al.. Structure of the processive human Pol δ holoenzyme. Nat. Commun. 2020; 11:1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dionne I., Bell S.D.. Characterization of an archaeal family 4 uracil DNA glycosylase and its interaction with PCNA and chromatin proteins. Biochem. J. 2005; 387:859–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Helt C.E., Wang W., Keng P.C., Bambara R.A.. Evidence that DNA damage detection machinery participates in DNA repair. Cell Cycle. 2005; 4:529–532. [DOI] [PubMed] [Google Scholar]
- 60. Tan J., Wang X., Hwang B.J., Gonzales R., Konen O., Lan L., Lu A.L.. An ordered assembly of MYH glycosylase, SIRT6 protein deacetylase, and Rad9-Rad1-Hus1 checkpoint clamp at oxidatively damaged telomeres. Aging (Albany NY). 2020; 12:17761–17785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Doré A.S., Kilkenny M.L., Rzechorzek N.J., Pearl L.H.. Crystal structure of the rad9-rad1-hus1 DNA damage checkpoint complex–implications for clamp loading and regulation. Mol. Cell. 2009; 34:735–745. [DOI] [PubMed] [Google Scholar]
- 62. Sohn S.Y., Cho Y.. Crystal structure of the human Rad9-Hus1-Rad1 clamp. J. Mol. Biol. 2009; 390:490–502. [DOI] [PubMed] [Google Scholar]
- 63. Xu M., Bai L., Gong Y., Xie W., Hang H., Jiang T.. Structure and functional implications of the human Rad9-Hus1-Rad1 cell cycle checkpoint complex. J. Biol. Chem. 2009; 284:20457–20461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hara K., Iida N., Tamafune R., Ohashi E., Sakurai H., Ishikawa Y., Hishiki A., Hashimoto H.. Structure of the RAD9-RAD1-HUS1 checkpoint clamp bound to RHINO sheds light on the other side of the DNA clamp. J. Biol. Chem. 2020; 295:899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tominaga Y., Ushijima Y., Tsuchimoto D., Mishima M., Shirakawa M., Hirano S., Sakumi K., Nakabeppu Y.. MUTYH prevents OGG1 or APEX1 from inappropriately processing its substrate or reaction product with its C-terminal domain. Nucleic Acids Res. 2004; 32:3198–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mol C.D., Izumi T., Mitra S., Tainer J.A.. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000; 403:451–456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession number (7EF8, MUTYH–DNA Form I; 7EF9, MUTYH–DNA Form II; 7EFA, CTD–PCNA).