

Abstract
Motivation
DNA methylation can be measured at the single CpG level using sodium bisulfite conversion of genomic DNA followed by sequencing or array hybridization. Many analytic tools have been developed, yet there is still a high demand for a comprehensive and multifaceted tool suite to analyze, annotate, QC and visualize the DNA methylation data.
Results
We developed the CpGtools package to analyze DNA methylation data generated from bisulfite sequencing or Illumina methylation arrays. The CpGtools package consists of three types of modules: (i) ‘CpG position modules’ focus on analyzing the genomic positions of CpGs, including associating other genomic and epigenomic features to a given list of CpGs and generating the DNA motif logo enriched in the genomic contexts of a given list of CpGs; (ii) ‘CpG signal modules’ are designed to analyze DNA methylation values, such as performing the PCA or t-SNE analyses, using Bayesian Gaussian mixture modeling to classify CpG sites into fully methylated, partially methylated and unmethylated groups, profiling the average DNA methylation level over user-specified genomics regions and generating the bean/violin plots and (iii) ‘differential CpG analysis modules’ focus on identifying differentially methylated CpGs between groups using different statistical methods including Fisher’s Exact Test, Student’s t-test, ANOVA, non-parametric tests, linear regression, logistic regression, beta-binomial regression and Bayesian estimation.
Availability and implementation
CpGtools is written in Python under the open-source GPL license. The source code and documentation are freely available at https://github.com/liguowang/cpgtools.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
DNA methylation is one of the most extensively studied epigenetic modifications that is involved in many cellular processes, such as transcription regulation, genomic imprinting and chromatin remodeling. The Illumina DNA methylation microarrays, including the HumanMethylation450 BeadChip (i.e. 450 K array) and Infinium MethylationEPIC BeadChip (i.e. 850 K array), as well as whole-genome and reduced representation bisulfite sequencing are the most widely used techniques to interrogate DNA methylation at single-nucleotide level. Many tools have been developed to analyze these data (Supplementary Table S1). Most of these tools are dedicated to one data type or focus on certain aspects of DNA methylation analyses, such as bisulfite reads mapping or DMR (differentially methylated region) calling. The goal of this study was to develop a comprehensive Python package to perform DNA methylation data QC, conversion, dimensionality reduction, annotation, statistical comparison and visualization.
2 Features and methods
Programs in CpGtools package can be broadly divided into three categories (Supplementary Table S2).
(1) CpG position modules can be used to analyze, annotate and visualize CpGs by their genomic locations. For example, we developed modules to calculate the distribution of given CpGs on chromosomes (Supplementary Fig. S1); on gene-centered genomic regions (Supplementary Fig. S2) and on user-specified genomic regions (Supplementary Fig. S3). To better annotate a given list of CpGs with epigenomic features, we built several annotation files using data generated from the ENCODE and other resources (Supplementary Table S3). In addition, we developed CpG_logo.py to visualize the local genomic context of a given list of CpGs. To demonstrate the utility of this module, we applied it to 2932 CHG or CHH (H = A, C or T) sites extracted from the Illumina 450K probes, and a novel DNA motif was identified as shown in Figure 1A. We also implemented CpG_to_gene.py to assign given CpGs to their putative targets using the algorithms developed by GREAT (McLean et al., 2010).
(2) CpG signal modules focus on the analysis and visualization of methylation signals. We developed modules to visualize the distributions of beta values for each sample using the jitter plot overlaid by violin plot (Fig. 1B) and stacked bar plot (Supplementary Fig. S4), the mean methylation profiles over gene-centered genomic regions (Fig. 1C) and user-specified genomic regions (Supplementary Fig. S5). We implemented the PCA (principal component analysis) and t-Distributed Stochastic Neighbor Embedding (t-SNE) algorithms to perform dimensionality reduction and to visualize the local and global structures within the data. For example, we observed a clear separation between tumor and normal samples when applying PCA and t-SNE algorithms to 450K array data generated from the TCGA-LIHC cohort (Fig. 1D, Supplementary Fig. S6). In addition, we also developed a module to perform methylation status calling using Bayesian Gaussian mixture modeling. This module is able to classify each CpG into one of the three discrete states: methylated, partially methylated, or unmethylated (Fig. 1E).
(3) Differential CpG analysis modules aim to identify differentially methylated cytosines using various statistic approaches (Supplementary Table S4). To analyze beta values generated from Illumina BeadChip array, CpGtools offer methods including t-test (automatically switches to ANOVA if more than two groups are provided), generalized linear mode and Bayesian estimation (Kruschke, 2013). Non-parametric analyses using the Mann–Whitney U test (two-group comparisons) and the Kruskal–Wallis H-test (multiple-groups comparisons) are also provided. Despite the different statistical methods employed, we found the resulting P-values are highly concordant (Supplementary Fig. S7). To analyze count-based proportion values generated from RRBS/WGBS experiments, CpGtools provide Fisher’s exact test, logistic regression and beta-binomial regression. Fisher’s exact test is less commonly used as most experiments have biological replicates. We found the resulting P-values from logistic regression model (with the quasi-binomial family to deal with over-dispersion) and beta-binomial regression are highly correlated with each other as well as with the published methods, such as DSS (Wu et al., 2015) (Supplementary Fig. S8).
Fig. 1.
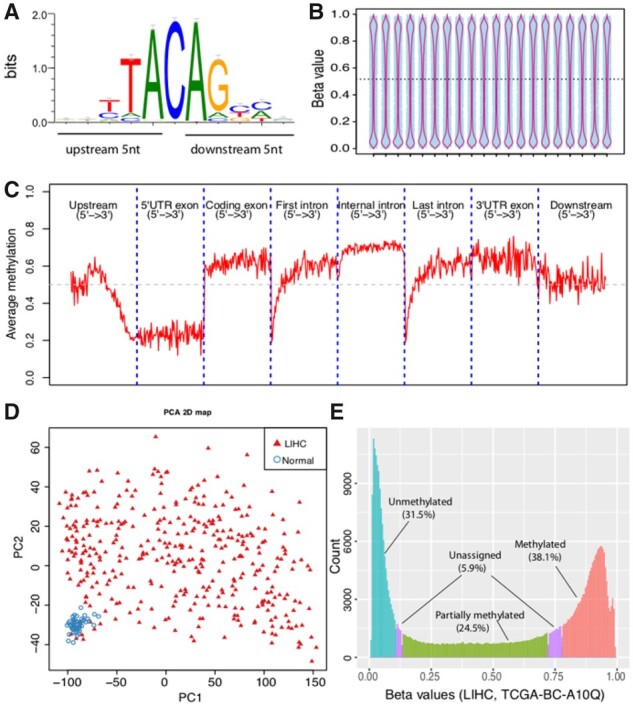
Example output from CpGtools. (A) The consensus DNA motif logo calculated from CHG and CHH context contained in 450 K. (B) Jitter and violin plots generated from 450 K data of 20 TCGA samples. (C) Mean methylation profiles across genomic regions centered on RefSeq genes. All genomic regions were scaled into the same length. (D) Two-dimensional scatter plot showing the PCA analysis of 429 TCGA samples, including 50 normals (blue circles) and 379 LIHC samples (red triangles). (E) Methylation status calling from one TCGA LIHC patient. The frequencies (y-axis) of ‘Methylated’, ‘partially methylated’, ‘unmethylated’ and ‘unassigned’ CpGs are indicated by red, green, blue and purple bars, respectively. LIHC, Liver hepatocellular carcinoma
3 Conclusion
The CpGtools package provides a number of modules that can analyze and visualize CpGs by their genomic positions and methylation signals, as well as perform statistical comparisons. Written in the Python genera-purpose programming language, it is easy to use and capable of handling large-scale DNA methylation data.
Funding
This work was supported in part by the NIH grants [R01CA224917 and R01AA27179 to K.R.].
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Ting Wei, Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN 55905, USA.
Jinfu Nie, Anhui Province Key Laboratory of Medical Physics and Technology, Center of Medical Physics and Technology, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei 230031, Anhui, China.
Nicholas B Larson, Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN 55905, USA.
Zhenqing Ye, Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN 55905, USA.
Jeanette E Eckel-Passow, Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN 55905, USA.
Keith D Robertson, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905, USA.
Jean-Pierre A Kocher, Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN 55905, USA.
Liguo Wang, Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN 55905, USA.
References
- Kruschke J.K. (2013) Bayesian estimation supersedes the t test. J. Exp. Psychol. Gen., 142, 573–603. [DOI] [PubMed] [Google Scholar]
- McLean C.Y. et al. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H. et al. (2015) Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res., 43, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.