

Summary
The cholesterol-sensing protein Scap induces cholesterol synthesis by transporting membrane-bound transcription factors called SREBPs from ER to Golgi for proteolytic activation. Transport requires interaction between Scap’s two ER luminal loops (L1 and L7) that flank an intramembrane sterol-sensing domain (SSD). Cholesterol inhibits Scap transport by binding to L1, which triggers Scap’s binding to Insig, an ER retention protein. Here, we used cryo-EM to elucidate two structures of full-length chicken Scap: i) wild-type free of Insigs; and ii) mutant Scap bound to chicken Insig without cholesterol. Strikingly, L1 and L7 intertwine tightly to form a globular domain that acts as a luminal platform connecting the SSD to the rest of Scap. In the presence of Insig, this platform undergoes a large rotation accompanied by rearrangement of Scap’s transmembrane helices. We postulate that this conformational change halts Scap transport of SREBPs and inhibits cholesterol synthesis.
Keywords: cholesterol, Scap, Insig, SREBP, lipid metabolism, membrane homeostasis
Graphical Abstract
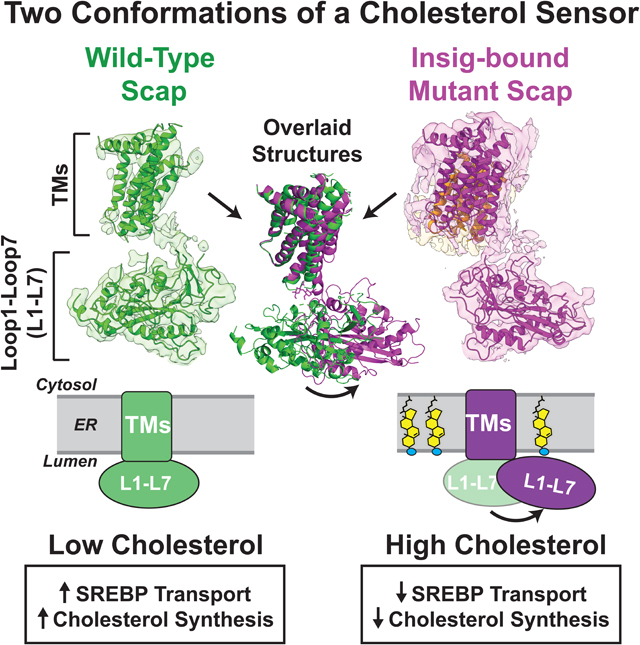
Visualization of Scap, the cholesterol sensor that modulates intracellular localization of SREBP, in complex with Inisg implicates movement of a cholesterol-interacting domain in patrolling proximal membrane regions for sterols.
Introduction
Scap is an integral membrane protein located in the endoplasmic reticulum (ER) that controls the synthesis of cholesterol and fatty acids in animal cells. This control is exerted by the binding of Scap to membrane-bound transcription factors called SREBPs (Sterol Regulatory Element-Binding Proteins) (Brown et al., 2018). When cellular cholesterol levels are low, Scap transports SREBPs to the Golgi, where their transcriptionally active portions are released proteolytically from the membrane to enter the nucleus. Once in the nucleus, SREBPs activate all the genes necessary for cholesterol synthesis and uptake from LDL (Horton et al., 2002). When cholesterol accumulates in ER membranes, it binds to Scap, triggering a conformational change that promotes Scap’s interaction with either one of two ER membrane retention proteins, Insig-1 or Insig-2 (Yang et al., 2002; Yabe et al., 2002a). As a result, transport of the Scap/SREBP complex from ER to Golgi is reduced, and synthesis of cholesterol and uptake of LDL declines. This negative feedback mechanism ensures cholesterol homeostasis (Radhakrishnan et al., 2008).
A detailed biochemical dissection of the hamster ortholog of Scap, carried out over the last 25 years, has led us to advance a working model for its cholesterol-sensing mechanism (Brown and Goldstein, 1997; Goldstein et al., 2006; Brown et al., 2018). As shown in Figure 1, the amino-terminal domain of Scap (amino acids 1–767) contains eight transmembrane (TM) helices separated by seven hydrophilic loops. The soluble, cytoplasmic-facing, carboxy-terminal domain (CTD) of Scap (amino acids 768–1276) binds SREBPs. Scap’s cholesterol-sensing functions are properties of its amino-terminal membrane domain. This domain contains two large luminal loops: Loop 1 (~240 amino acids) and Loop 7 (~175 amino acids). Loop 1 (L1) binds cholesterol with the same affinity and specificity as the entire membrane domain (Motamed et al., 2011; Radhakrishnan et al., 2004; 2007). The segment containing TMs 2–6 is almost entirely buried in the membrane. Three different point mutations in this segment prevent Scap binding to Insig, thus defining this segment as the Insig-binding domain (Yabe et al., 2002). This domain shares sequence homology with at least six other polytopic membrane proteins that play important roles in handling cholesterol, including Niemann-Pick C1 (NPC1) and Patched, and is designated as the sterol-sensing domain (SSD) (Hua et al., 1996a; Kuwabara and Labouesse, 2002). Cytosolic Loop 6 (L6) contains a hexapeptide (MELADL) that serves as a binding site for the COPII complex that clusters ER proteins into coated vesicles for transport to the Golgi (Sun et al., 2005). The second large luminal loop, Loop 7 (L7), exerts a regulatory function that depends on its binding to L1 (Zhang et al., 2016).
Figure 1. Schematic Models for Scap and Insig.
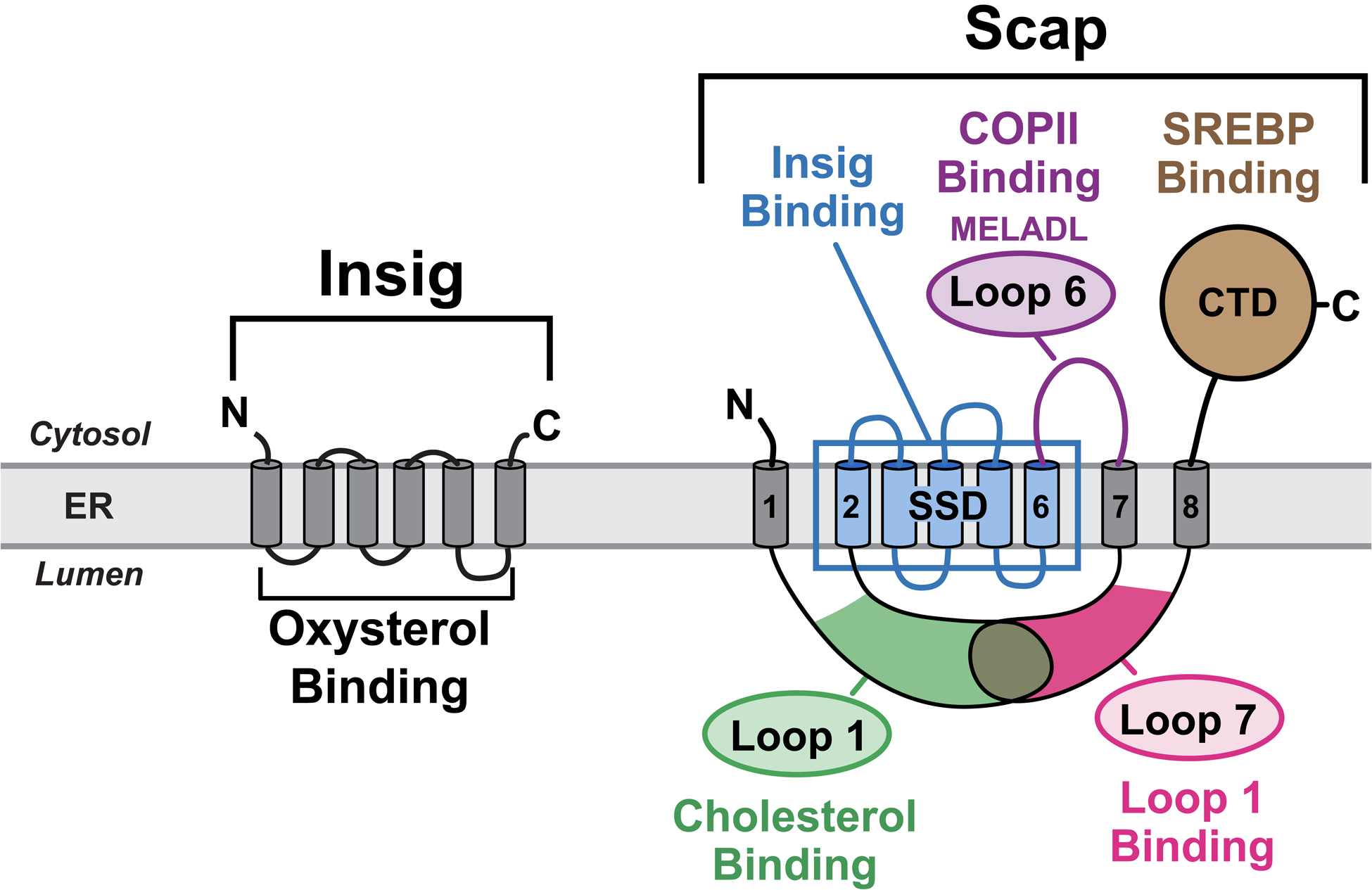
Scap is an ER cholesterol-sensing transmembrane protein with 5 functional subdomains: i) Cholesterol-binding domain (green) localized to luminal Loop 1; ii) Insig-binding domain (blue) localized to TM helices 2–6, which shares homology with other cholesterol regulatory proteins and has thus been defined as the sterol-sensing domain (SSD); iii) COPII-binding domain (purple) localized to cytoplasmic Loop 6 that contains the hexapeptide MELADL, which is recognized by Sec24 proteins for COPII transport to Golgi; iv) Loop1-binding domain localized to luminal Loop 7; and v) SREBP-binding domain localized to the cytoplasmic C-terminal domain (CTD), which contains WD-40 repeats that mediate binding to SREBPs. Insig is an ER oxysterol-sensing transmembrane protein containing six TM helices. Together, Scap and Insig respond to increases in either exogenously-derived cholesterol or endogenously-synthesized oxysterol by forming a Scap/Insig complex, which prevents binding to COPII proteins, blocking the transport of Scap and SREBPs to the Golgi, and reducing SREBP-induced lipid synthesis.
See also Figure S1.
Our previous biochemical studies have provided evidence that L1 binding to L7 places Scap in an active conformation in which the MELADL sequence in L6 is accessible to COPII proteins, allowing Scap to escort SREBPs to the Golgi (Yang et al., 2000; Zhang et al., 2013). Scanning mutagenesis revealed two tyrosine mutations (Y234A in L1 and Y640S in L7) that disrupted the binding of L1 to L7 and blocked the ability of Scap to transport SREBPs to the Golgi (Motamed et al., 2011; Zhang et al., 2013). Moreover, these two loop mutations locked Scap in its cholesterol-bound conformation even in the absence of cholesterol (Gao et al., 2017). We speculated that binding of cholesterol to L1 may disrupt the interaction between L1 and L7, converting Scap to an inactive conformation where it binds Insig, which moves the MELADL sequence such that it is inaccessible to COPII proteins (Brown et al., 2018). Cholesterol binding is not the sole means of converting Scap to an inactive conformation. Oxygenated derivatives of cholesterol such as 25-hydroxycholesterol (25-HC) also convert Scap to an Insig-binding conformation by binding not to Scap, but to Insig (Adams et al., 2004; Radhakrishnan et al., 2007; Sun et al., 2007).
Atomic-level structural insights into the molecular basis of Scap regulation are beginning to emerge. Yan and colleagues (Yan et al., 2021) reported a cryo-EM structure of a truncated, wild-type human Scap (lacking its CTD) bound to human Insig-2. Their model of Scap’s transmembrane region in complex with Insig-2 placed 25-HC at the Scap/Insig interface. The cryo-EM analysis of Yan et al. did not reveal the structure or disposition of L1 and L7.
In the current paper, we report the cryo-EM structure of a full-length, wild-type Scap, free from Insigs or sterols. Our data obtained with the chicken ortholog of Scap (cScap) reveals that L1 and L7 intertwine to form a highly structured domain. We also determined the cryo-EM structure of a complex of chicken Insig-1 with a mutant cScap that binds Insigs in the absence of sterols. These two structures help clarify the role of the L1 and L7 ER luminal loops in controlling the conformation of Scap’s transmembrane helices, thereby regulating transport of SREBPs and assuring cholesterol homeostasis at the level of the cell and the whole organism.
Results
Functional Characterization of cScap
In generating preparations of purified Scap suitable for cryo-EM, we were forced to overcome several obstacles in biochemical purification and cryo-EM imaging. To circumvent the relatively poor behavior of full-length mammalian Scap upon detergent solubilization, we screened 10 vertebrate orthologs and found that full-length cScap had the best biochemical stability after purification. cScap shares high homology with its mammalian counterparts (>75% overall identity, >80% for the membrane domain), including all residues that have been determined to have functional significance (Figure S1A). We tested the function of cScap in multiple assays. We first checked whether cScap could transport human SREBP2 in sterol-depleted Scap-deficient CHO cells, an assay we routinely utilize to characterize Scap constructs. When human SREBP2 was transfected into these cells, its precursor form was barely detectable and no cleaved nuclear form was detected (Figure S1B, lane 2). We have shown previously that SREBPs are rapidly degraded in the absence of Scap (Rawson et al., 1999). Upon co-transfection of wild-type cScap, we observed a marked increase in the amount of the SREBP2 precursor form (Figure S1B, lane 3), indicating that human SREBP2 could bind cScap. Moreover, we also detected a significant amount of the cleaved nuclear form, indicating that cScap transports human SREBP2 for processing. We have shown previously that a point mutation in the intramembrane SSD of hamster Scap (D428A) renders Scap in a configuration that binds Insig in the absence of sterols, and abolishes Scap transport of SREBPs (Feramisco et al., 2005). The analogous residue in cScap is D435. When we mutated D435 to valine, cScap continued to stabilize human SREBP2, but it no longer transported SREBP2 to the Golgi for processing (Figure S1B, lane 4).
To demonstrate directly the binding of cScap to human SREBP2, we used two different assays. In the first assay, we co-transfected HEK293 cells with human SREBP2 and either human Scap or cScap. To enhance detection of the precursor form of human SREBP2, we introduced mutations that disrupt the 3 known cleavage sites on this protein (Hua et al., 1996b; Sakai et al., 1996; Wang et al., 1996). Co-immunoprecipitation showed that FLAG-tagged wildtype and mutant cScaps bound to cleavage-resistant SREBP2 to a degree similar to human Scap (Figure S1C, lanes 3–5). In the second assay, using a system we previously described (Kober et al., 2020), we expressed the CTDs (Figure S1D) of cScap and human SREBP2 in Sf9 insect cells, and showed that these domains co-purified as a stable soluble complex (Figure S1E). Combined, these studies indicate that cScap interacts with human SREBPs, and transports them to the Golgi in a manner similar to mammalian Scap.
Structure of Full-length cScap
Preliminary cryo-EM data on purified cScap did not result in useful 3D reconstructions. To improve resolution, we sought to obtain a Fab fragment that would aid in single particle alignment. We generated and screened over 2,500 hybridoma clones for antibodies that would bind folded cScap but not denatured polypeptide (Figure S2A–F and Methods). We identified a single antibody, IgG 4G10, that aided in cryo-EM analysis. The Fab fragment derived from IgG 4G10 (sequence in Figure S2C), hereafter designated as 4G10Fab, co-purifies with cScap in glyco-diosgenin (GDN) micelles (Figure 2A), and we prepared grids of this complex for cryo-EM imaging (Figure 2A and see Methods). To determine whether 4G10Fab interacted with the luminal or cytoplasmic domain of cScap, we prepared a plasmid encoding a protein representing the ER luminal portion of cScap. This plasmid encoded L1 of cScap followed by L7 of cScap separated by a 15 aa linker, and was preceded by a signal sequence from honey bee mellitin. When overexpressed in Sf9 insect cells, the resulting recombinant protein, hereafter referred to as cScap(L1-L7), was secreted as a soluble protein. An analogous result has been previously observed for a fusion protein combining L1 and L7 of hamster Scap (Zhang et al., 2016), further underscoring the similarities between cScap and mammalian Scap. We demonstrated that 4G10Fab efficiently binds this soluble cScap(L1-L7) fusion protein (Figure 2B), indicating that the antibody fragment interacts with cScap’s luminal loops independent of the presence of the TM helices. Finally, we carried out sterol binding studies as developed previously (Motamed et al., 2011; Radhakrishnan et al., 2004; 2007). Purified cScap in lauryl maltoside neopentyl glycol (LMNG) detergent showed saturable binding to [3H]cholesterol (half-maximal at ~ 75 nM), but no significant binding to [3H]25-HC or [3H]lanosterol (Figure 2C). In competition assays, binding of [3H]cholesterol was reduced by increasing amounts of unlabeled cholesterol, but not by unlabeled 25-HC or lanosterol (Figure 2D). These cScap binding data are consistent with our previous data on mammalian Scap’s sterol binding affinity and specificity (Radhakrishnan et al., 2007; Motamed et al., 2011).
Figure 2. Purification, Sterol Binding, and Cryo-EM Structure of cScap.
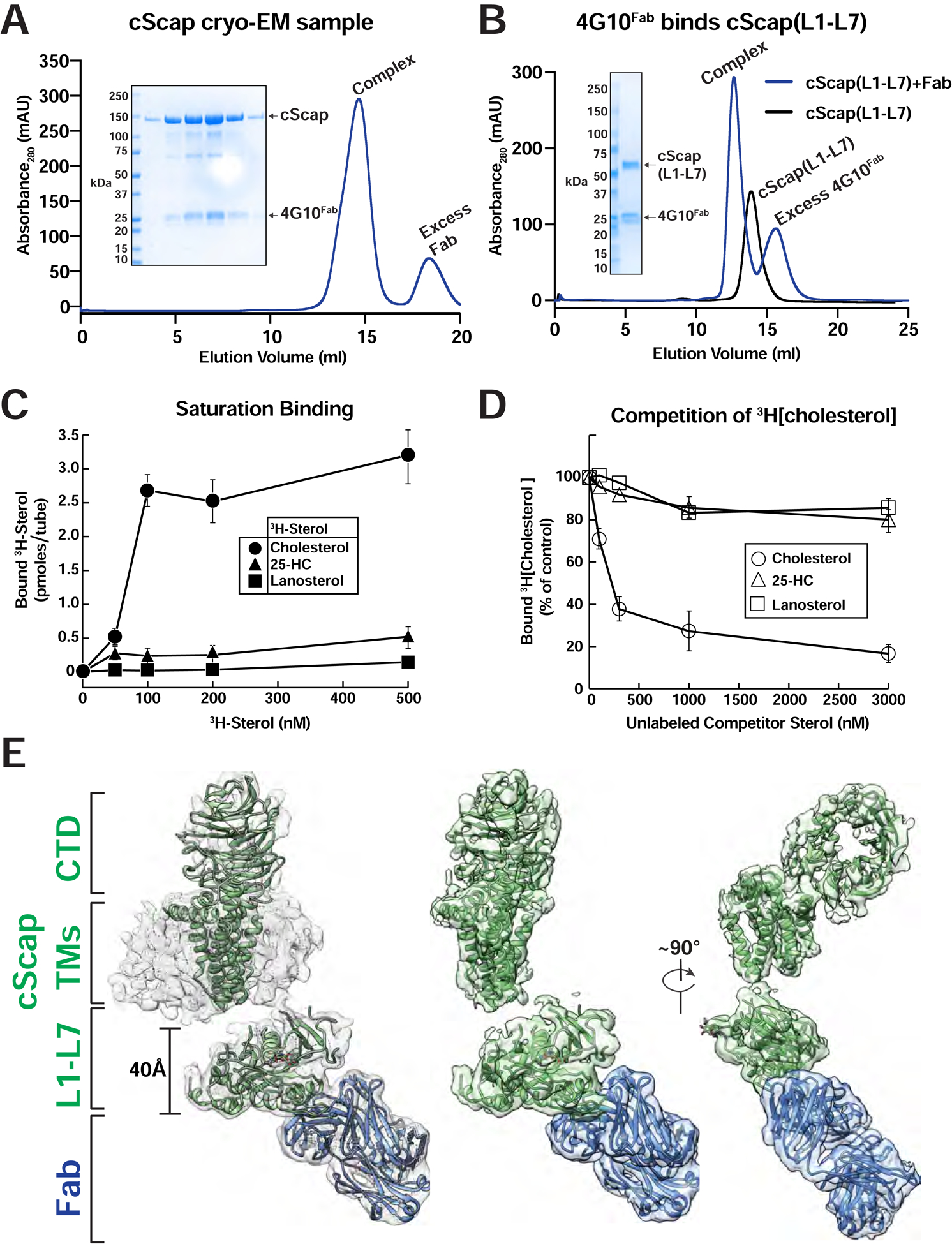
(A) Purification traces for the cryo-EM grid preparation of cScap/4G10Fab complexes. 2 mg of cScap was incubated with 1.3 mg 4G10Fab for 3 hrs on ice. The Scap/4G10Fab complex was then purified by gel filtration (blue trace) as described in Methods. Inset: SDS-PAGE analysis for the Scap/4G10Fab complex used in the cryo-EM grid preparation. The peak eluted at 1 mg/ml in the central fractions. An aliquot of 10 μL of each fraction was subjected to SDS-PAGE on a 4–16% gradient gel. Coomassie staining reveals the presence of stoichiometric cScap and 4G10Fab.
(B) 4G10Fab binds the cScap(L1-L7) fusion. Secreted cScap(L1-L7) was purified as described in Methods. 860 μg aliquots were mixed with either buffer or with 1760 μg of 4G10Fab and subjected to gel filtration as described in Methods. The binding of 4G10Fab to the cScap(L1-L7) fusion protein results in a left-shift of the elution volume by 0.8 mL relative to cScap(L1-L7) alone. The unbound 4G10Fab elutes after cScap(L1-L7). Inset: SDS-PAGE analysis of the fractions from the complex trace. The wells, from left to right, were loaded with protein standards, input samples for cScap(L1-L7), 4G10Fab, protein standards, and aliquots from the chromatography elution fractions.
(C, D) Sterol binding properties of cScap. Each binding reaction contained 2 μg of His8-tagged cScap and varying concentrations of the indicated 3H-sterol (cholesterol, 106,000 dpm/pmol; 25-HC, 188,000 dpm/pmol; lanosterol, 22,000 dpm/pmol) (C) or 150 nM [3H]cholesterol and varying concentrations of the indicated unlabeled sterol (D). After incubation for 4 hr at 4°C, sterol binding was measured as described in Methods. The 100% of control value in (D), determined in the absence of competitor sterol, was 2.9 pmol/tube. Each data point denotes the average of three assays, and error bars represent ± SEM. When not visible, error bars are smaller than the size of the symbols.
(E) Cryo-EM structure of cScap. Left: cryo-EM density contoured at 0.013 with the model docked in. cScap is colored green and the 4G10Fab is colored blue. The models of the TM domain and CTD of cScap were built using SWISS-MODEL, and rigid-body fitted into the cryo-EM density. Center: Same view as Left, with micelle density masked out. Right: cScap rotated 90° relative to Center.
See also Figures S2, S3, and Table S1.
To elucidate the structure of cScap in its cholesterol-free form in which it transports SREBPs from ER to Golgi, we collected single particle cryo-EM data for cScap/4G10Fab on a Titan Krios microscope (Figure S3). An initial dataset of over 10,000 micrographs and 9 million particles was subjected to extensive 2D and 3D classification to identify subsets for the different reconstructions reported herein (see Methods). Fortunately, the 4G10Fab provides a fiducial marker that drives the alignment of particles during classification (Figure S3). The overall reconstruction of cScap/4G10Fab in GDN detergent micelles shows a clearly delineated Fab fragment (Figure 2E). The maps show that 4G10Fab binds to the ER luminal L1-L7 domain that projects away from the micelle. The global refinement of the best 3D class containing 223K particles resulted in a reconstruction with an overall resolution of 3.0 Å (Figure S3F). The L1-L7 region and the antigen binding region of 4G10Fab have the highest local resolution in the structure (Figure S3C). Focused refinement resulted in a 2.9 Å map that permitted de novo model building (Figure S3D). 3D classification focused on the micellar and CTD regions resulted in a reconstruction at 4.1 Å resolution (Figure S3F). The data revealed density for six TM α-helices and a large feature on the cytosolic face that has a clear WD domain shape identifying it as the CTD (see below). While we could not build a de novo atomic model for these regions, we were able to create a backbone model for TM1, TM3–6, and TM8 of cScap, starting with rigid-body fitting of TM1–6 of the related NPC1 protein (Li et al., 2016b). Cryo-EM data collection and refinement statistics are shown in Table S1.
Luminal Loops, L1 and L7, of cScap Form an Intertwined Globular Structure
The high-resolution focused cryo-EM map for cScap L1-L7 with our refined atomic model is shown in Figure 3A, and representative densities for segments of the protein with different secondary structure are shown in Figure S4A. The loops bind together to form an oblong globular structure that measures 60 Å along its long axis (roughly parallel to the plane of the membrane) and extends ~40 Å away from the surface of the GDN micelle (Figure 2E). Model building and register assignment for L1-L7 was facilitated by clear density for a previously described N-linked glycan modification on an asparagine residue of L7 that is preceded by two tyrosine residues (Nohturfft et al., 1998a; Zhang et al., 2016).
Figure 3. Structure of the L1-L7 Domain of cScap.
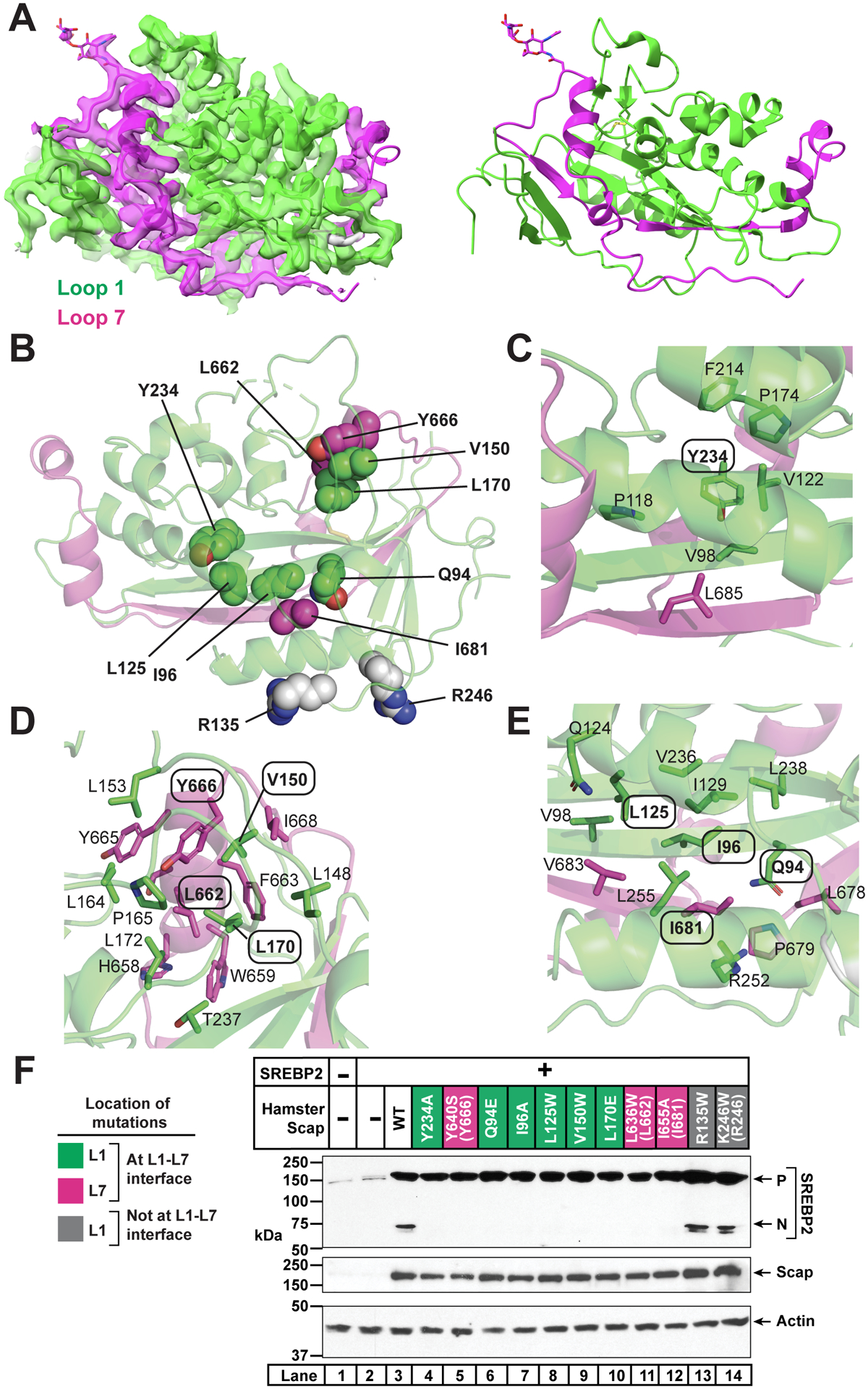
(A) cScap L1-L7 domain. Left: cryo-EM density and model for the cScap L1-L7 domain structure contoured at level 0.0537. The model and the density within 2.5 Å of the coordinates are colored green (L1) or magenta (L7). Image generated using USCF Chimera. Right: Atomic model shown without the cryo-EM density. L1 residues are colored green and L7 residues are colored as magenta. Image generated using Chimera.
(B) Location of key residues in the L1-L7 domain. Domain is rotated 180° towards the reader from the view in A. Side chains of residues interrogated by mutagenesis are depicted as spheres. Green spheres are L1 residues predicted to disrupt the interaction of L1 with L7. Magenta spheres are L7 residues predicted to disrupt the interaction of L7 with L1. Grey spheres are L1 residues in the Fab-binding interface that are solvent-accessible and away from the L1-L7 interface.
(C) Environment of Y234, which is labeled with oval. Side chains within 4 Å of Y234 are shown as sticks and the residue numbers indicated.
(D) Environment of V150, L170, L662, and Y666. These residues are labeled with ovals. Side chains within 4 Å are shown as sticks and the residue numbers indicated.
(E) Environment of Q94, I96, L125, and I681. These residues are labeled with ovals. Side chains within 4 Å of these residues’ side chains are shown as sticks and the residue numbers indicated.
(F) Effect of L1 and L7 mutations on Scap function. Mutations to L1 predicted to disrupt the interaction of L1 with L7 are labeled green. Mutations to L7 predicted to disrupt the interaction of L7 with L1 are labeled magenta. Mutations on the surface of L1 (remote from L7) are labeled grey. Parentheses indicate the analogous residues in cScap when different from hamster Scap. Scap-null hamster SRD-13A cells were set up and transfected as described in Methods with 4 μg pcDNA control plasmid (lane 1), 2 μg pTK-SREBP2 (lane 2), or else 2 μg pTK-SREBP2 and 2 μg WT or mutant pTK-Scap as indicated. The total amount of transfected DNA for all conditions was adjusted to 4 μg with pcDNA control plasmid. The next day, cells were depleted of cholesterol for 1 hr, harvested, and subjected to SDS-PAGE followed by immunoblot analysis of SREBP2 (IgG-22D5), Scap (IgG-4H4), and actin (anti-actin) as described in Methods. P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2.
See also Figures S4, S5, and Table S1.
The folded luminal loop structure contains L1 residues 53–278 and L7 residues 636–704. Compared to typical protein-protein interactions, the interface between the loops is extensive, burying 3760 Å2 of surface area. An N-terminal stretch of L7 residues (ending at the glycosylation site N667) fits into a ridge facing the micelle, and a C-terminal stretch folds back to form part of the central core β-sheet (Figure 3A, magenta). A search using the DALI server (Holm, 2020) showed that the L1-L7 domain of cScap bears structural homology to the middle luminal domain (MLD) of NPC1 (Li et al., 2016a), an integral protein in the lysosomal membrane that transports LDL-derived cholesterol (Infante et al., 2008a; 2008b) (Figure S4B). The topology of the cScap luminal domain is distinctive from the NPC1 MLD in that cScap’s L7 contributes a parallel β-strand to the core β-sheet (see yellow star in Figure S4B) and adds several short α-helices surrounding this core. The result of this topological arrangement is that L1 and L7 are tightly interdigitated.
Inasmuch as our high-resolution cryo-EM reconstructions could be obtained only when the 4G10Fab is bound, it is formally possible that binding of the Fab altered the cScap structure. In our cryo-EM reconstruction of cScap, 4G10Fab binds to a region of L1 away from the micelle, and makes no interactions with L7 (Figure S5A). To test whether the Fab binding affects the activity of cScap, we mutated the cScap residues that contact the Fab, so as to produce a version of cScap that does not bind the Fab. The 4G10Fab interaction is largely mediated by polar contacts, including from residues R135 and D136 to oppositely charged surfaces on the Fab. We produced a charge-swap mutant of these residues (cScapR135E/D136K). This charge-swap mutant lost its ability to interact with 4G10Fab as judged by immunoprecipitation (Figure S5B,C). This alteration of L1 at the Fab binding site did not affect the ability of cScapR135E/D136K to transport SREBP2 in cholesterol-depleted Scap-deficient cells (Figure S5D, lanes 2 and 3). These results suggest that 4G10Fab binds to a site on cScap that is not important for cScap function, and that binding to this site is unlikely to alter cScap structure.
The highly structured nature of the L1-L7 domain agrees with our previous data showing that either co-expression of separate hamster Scap L1 and L7 polypeptides, or alternatively fusion of L7 to the end of L1 (as described above), results in a stable, water-soluble protein (Zhang et al., 2016). The specific interactions seen in our L1-L7 structure agree well with previous alanine scanning mutagenesis studies carried out with hamster Scap, which identified residues Y234 and Y640 (chicken Y666) whose mutation disrupted the interaction between L1 and L7, leading to ER retention of Scap and blocking transport and activation of SREBP2 (Motamed et al., 2011; Zhang et al., 2013) (Figure 3B).
To further interrogate the interaction between L1 and L7, we designed seven additional mutations in hamster Scap (to alanine, glutamate, or tryptophan) that our structure predicted would disrupt the packing between L1 and L7 (Figure 3B–E): Q94E, I96A, L125W, V150W, L170E, L636W (L662 in cScap), and I655A (I681 in cScap). We also made two mutations of surface residues away from the L1-L7 interface (Figure S5A): R135W and K246W (R246 in cScap). We then tested the function of all of these mutant Scaps in SREBP2 cleavage assays. We transfected Scap-deficient cells with human SREBP2 without or with hamster Scap. In the absence of transfected Scap, SREBP2 was barely detected (Figure 3F, lane 2). When hamster Scap was coexpressed, levels of the SREBP2 precursor were greatly increased and the cleaved nuclear form was detected (lane 3). As previously reported (Motamed et al., 2011; Zhang et al., 2013), the Y234A and Y640S mutants stabilized the SREBP2 precursor similar to wild-type, but they did not mediate SREBP2 cleavage (lanes 4 and 5). All seven mutations designed to disrupt the L1-L7 structure stabilized the SREBP2 precursor but did not promote SREBP2 cleavage (lanes 6–12). The two surface mutations that were predicted to maintain the L1-L7 interaction stabilized the SREBP2 precursor and also promoted SREBP2 cleavage similar to wild-type (lanes 13 and 14 compared to lane 3). These new mutagenesis data, together with previous studies (Motamed et al., 2011; Zhang et al., 2013), collectively provide robust functional validation of the L1-L7 structural model.
The structure of L1-L7 revealed that two of L1’s three cysteines, C147 and C169, are engaged in a disulfide bond with clear density in the 2.9 Å map (Figure S5E). To further validate our molecular model for this domain, we assayed a hamster Scap(L1-L7) fusion protein that is secreted from CHO-K1 cells (Figure S5F) as described above (Zhang et al., 2016). This hamster protein contains three cysteines, all in L1 and all of which are conserved in cScap. We mutated each cysteine to alanine or serine. After transfection into CHO-K1 cells, we tested the cell lysate and the medium for non-secreted and secreted forms of L1-L7, respectively. As shown in Figure S5G, wild-type L1-L7 containing all 3 cysteines (CCC) was secreted (lane 7). Replacement of cysteine 147 with alanine (ACC) or cysteine 169 with serine (CSC) abolished secretion (lanes 8 and 9), but replacing cysteine 264 with alanine (CCA) did not affect secretion (lane 10). We then used mPEG-MAL-5000, a high molecular weight thiol-reactive reagent that forms covalent bonds with free cysteines, to determine whether cysteines existed in the reduced state. At least one of the cysteines in wild-type L1-L7 was accessible to mPEG-MAL 5000 (lanes 7 and 11), whereas neither of the two cysteines on mutant L1-L7 (CCA) were accessible to mPEG-MAL-5000 (lanes 10 and 14). After denaturation by heating and disulfide bond reduction with dithiothreitol, one or both of the previously inaccessible cysteines in L1-L7 (CCA) could be modified by mPEG-Mal-5000 (Figure S5H).
If this disulfide bond is also necessary for the function of full-length Scap in cells, we would expect mutation of either of these cysteines to disrupt Scap’s ability to transport SREBP2. To answer this question, we individually mutated the three L1 cysteines to alanine or serine in full-length hamster Scap, and tested these mutants in our SREBP2 cleavage assay. As expected, when transfected into Scap-deficient cells and after sterol depletion, wild-type Scap stabilized the SREBP2 precursor and triggered SREBP2 processing (Figure S5I, lanes 2 and 3). Mutation of either cysteine in the observed disulfide bond (C147A or C169S) did not affect Scap’s ability to stabilize the SREBP2 precursor, but markedly reduced Scap’s ability to trigger SREBP2 processing (lanes 4 and 5). Mutation of the unpaired cysteine (C264A) produced a Scap protein that behaved similarly to wild-type Scap (lane 6). Taken together, these results validate our L1-L7 model and show the requirement of this disulfide bond for Scap’s transport of SREBP2.
Interaction between Scap and Insig
We next sought to characterize how Insig binding changes the conformation of Scap. To determine the structure of an Insig-bound Scap, we used the cScap D435V mutation (described above) that causes Scap to interact with Insig in the absence of sterols. We tested the function of cScapD435V in multiple assays. As shown above, cScapD435V binds to human SREBP2 (Figure S1C, lanes 5) and stabilizes its precursor form to a similar extent as cScap (Figure S1B, lanes 2–4). Unlike with wild-type cScap, we detected no cleaved nuclear form of SREBP2 with cScapD435V, indicating that this mutant was retained in the ER similar to what has been observed for hamster Scap(D428A).
We then tested whether chicken Insig-1 with its first 27 residues deleted (hereafter referred to as cInsig-1) would interact with cScap. In the absence of cInsig-1 and when cScap was overexpressed, cScap mediated transport and cleavage of human SREBP2 even in the presence of sterols (Figure S1F, lane 3). However, when cInsig-1 was overexpressed together with the overexpressed cScap, cleavage of SREBP2 was blocked (Figure S1F, lane 4). These data are consistent with cInsig-1 interacting with cScap under conditions of sterol excess, akin to mammalian Insigs interacting with mammalian Scap (Yang et al., 2002).
Having established the functional similarity of the chicken orthologs of Scap and Insig-1 to their mammalian counterparts, we proceeded to purify a stable complex of FLAG-tagged full-length cScapD435V and untagged cInsig-1 co-expressed in HEK293s GnTI− cells. As with the cScap protein, this complex bound efficiently to 4G10Fab (Figure 4A), and we used this ternary complex for cryo-EM studies (Figure S6). We also measured the sterol binding activity of cScapD435V/cInsig-1 complex in LMNG detergent, using similar assays as described above for cScap. Consistent with the presence of both Scap and Insig in the binding reaction, we detected saturable binding of [3H]cholesterol (half-maximal at ~ 75 nM) and [3H]25-HC (half-maximal at ~ 100 nM) (Figure 4B). [3H]Lanosterol was not bound. We consistently observed that the maximal binding of [3H]cholesterol was approximately two-fold higher than [3H]25-HC. We next conducted separate competition binding assays for bound [3H]cholesterol and bound [3H]25-HC. Binding of [3H]cholesterol was inhibited by unlabeled cholesterol, but not by unlabeled 25-HC or lanosterol (Figure 4C). Binding of [3H]25-HC was inhibited by unlabeled 25-HC, but not by unlabeled cholesterol or lanosterol (Figure 4D). In previous studies, we showed that purified hamster Scap binds cholesterol but not 25-HC (Radhakrishnan et al., 2004), whereas purified human Insig-2 binds 25-HC but not cholesterol (Radhakrishnan et al., 2007). Here we demonstrate that this dual sterol specificity is retained in a purified complex containing both cScap and cInsig-1.
Figure 4. Purification, Sterol Binding, and Cryo-EM Structure of cScapD435V/cInsig-1.
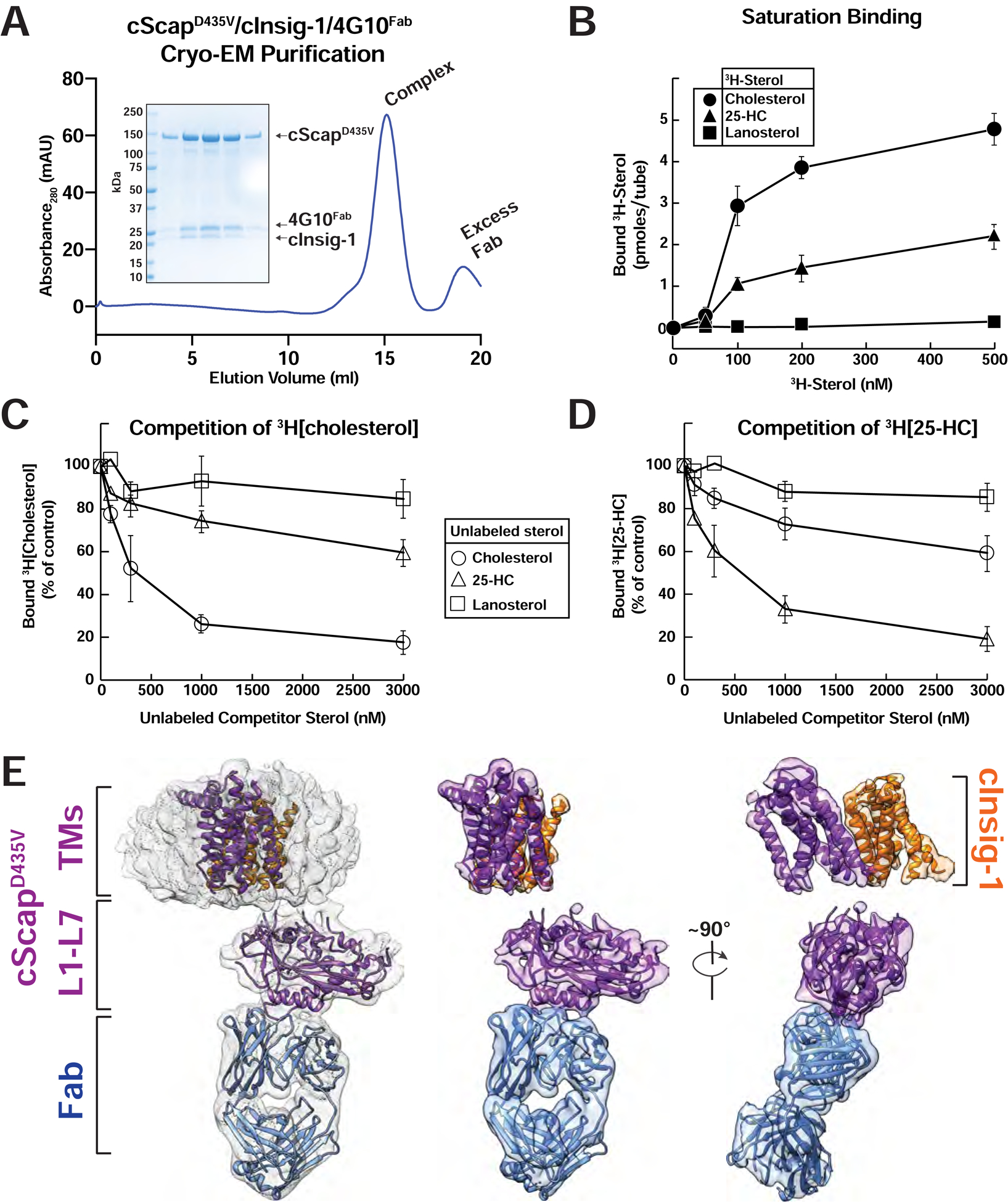
(A) Preparative gel filtration trace of the cScapD435V/cInsig-1/4G10Fab for cryo-EM grid preparation. 1 mg of cScapD435V/cInsig-1 complex was purified and mixed with 0.5 mg 4G10Fab and subjected to gel filtration as described in Methods. Inset: SDS-PAGE analysis for the cScapD435V/cInsig-1/4G10Fab complex used in the cryo-EM grid preparation. The complex eluted at 0.49 mg/mL, and an aliquot of 10 μL of each fraction was subjected to SDS-PAGE on a 4–16% gradient gel. Coomassie staining reveals the presence of stoichiometric cScapD435V, cInsig-1, and 4G10Fab.
(B-D) Sterol binding properties of a complex of cScapD435V/cInsig-1. Each binding reaction contained 2 μg of His8-tagged cScapD435V/cInsig-1 and varying concentrations of the indicated 3H-sterol (cholesterol, 106,000 dpm/pmol; 25-HC, 188,000 dpm/pmol; lanosterol, 22,000 dpm/pmol) (B), 150 nM [3H]cholesterol and varying concentrations of the indicated unlabeled sterol (C), or 150 nM [3H]25-HC and varying concentrations of the indicated unlabeled sterol (D). After incubation for 4 hr at 4°C, sterol binding was measured as described in Methods (n=3). The 100% of control values in (C) and (D), determined in the absence of competitor sterol, were 3.1 and 1.7 pmol/tube, respectively. Each data point denotes the average of three assays, and error bars represent ± SEM. When not visible, error bars are smaller than the size of the symbols.
(E) Cryo-EM structure of cScapD435V/cInsig-1. Left: cryo-EM density contoured at 0.016 with the model docked in. cScapD435V is colored purple, cInsig-1 is colored orange, and the 4G10Fab is colored blue. The models of TM domains for both cScap and cInsig-1 were built using SWISS-MODEL, and rigid-body fitted into the cryo-EM density. Center: Same view as Left, with micelle density masked out. Right: cScapD435V/cInsig-1 rotated 90° relative to Center.
We collected cryo-EM data for the cScapD435V/cInsig-1/4G10Fab complex, which produced a global reconstruction with a resolution of 4.1 Å (Figure S6A–F). As with the cScap sample, 2D and 3D classification relied on the 4G10Fab for accurate particle alignment (Figure S6F). Focused classification on the L1-L7 domain produced a higher-resolution 3.5 Å map (Figure S6D) that permitted de novo model building. 3D classification focused on the TM region resulted in a reconstruction at 4.6 Å resolution that revealed clear density for 4G10Fab, L1-L7, and TM bundles for both cScapD435V and cInsig-1, but lacking density for the CTD (Figure 4E). This map showed clear density for 12 TM α-helices (Figure S6C). Based on this map, we created a backbone model for TM1–6 of cScapD435V and all six TMs of cInsig-1, starting with rigid-body fitting of homologous structures NPC1 TM1–6 (Li et al., 2016b) and mycobacterial Insig (Ren et al., 2015) (Figure 5A). TM7 and TM8 of cScapD435V are not visible in this map.
Figure 5. Structural Basis for the Scap-Insig Interaction.
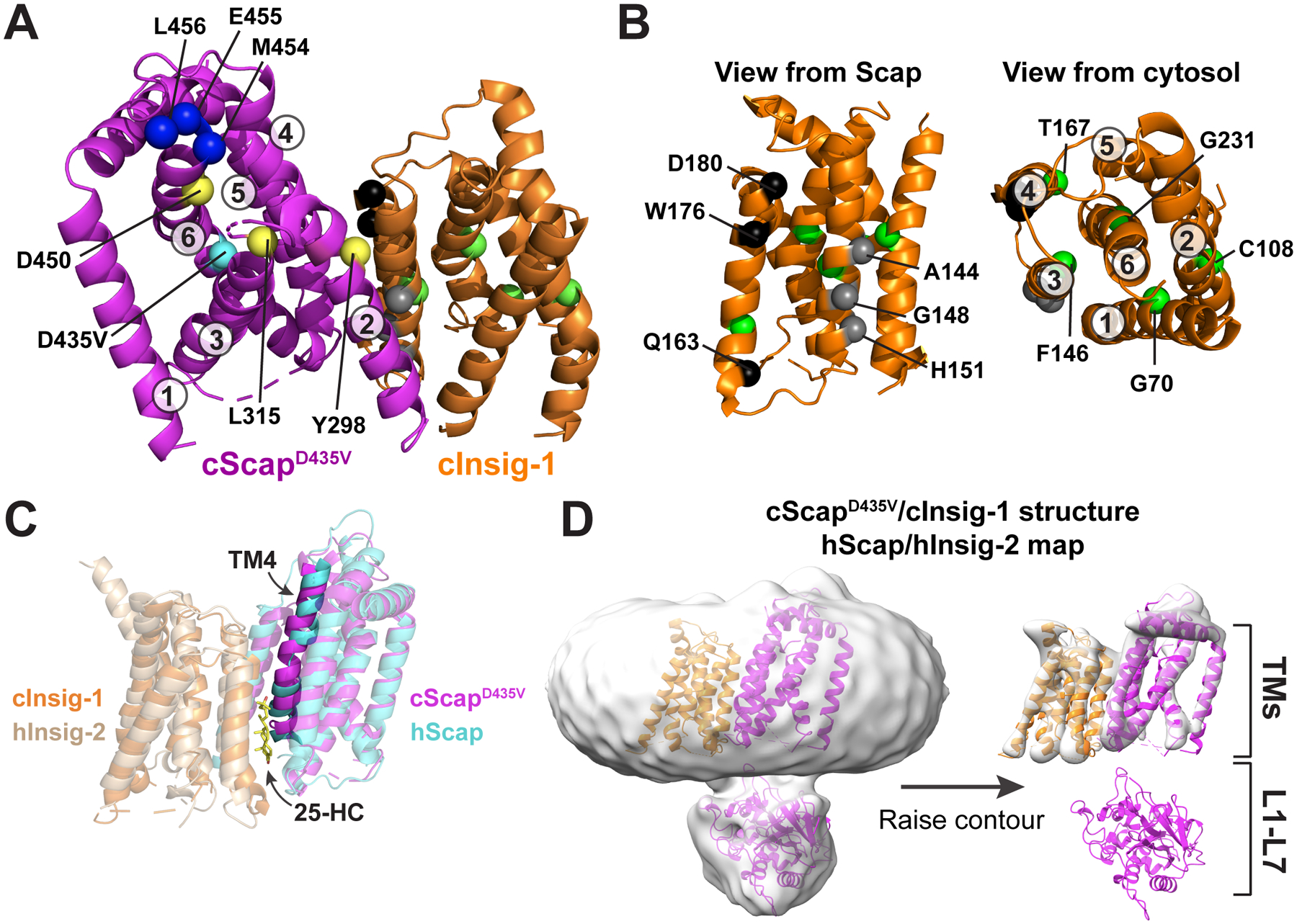
(A) Model of the TM domains of cScapD435V and cInsig-1. cScapD435V is colored magenta and cInsig-1 is colored orange. The Cα atoms of key residues are depicted as spheres. cScap’s TM helices are numbered and the position of key residues are indicated. M454, E455, and L456, shown as blue spheres, are the first visible residues in loop 6 and represent the first 3 of the 6 amino acids of the MELADL motif that is required for COPII binding. Y298, L315, and D450, shown as yellow spheres, render Scap incapable of binding Insig-1 when mutated to the residues indicated in Table S2. The position of the D435V mutation, shown as a cyan sphere, traps Scap in the Insig-binding conformation. See (B) for an explanation of highlighted residues in cInsig-1.
(B): Left: The structure of cInsig-1, viewed from the membrane level as if from cScap. The location of key residues are labeled with black, grey, and green spheres, and are described in Table S3. Black spheres indicate the locations of residues that when mutated as indicated in Table S3 reduce binding to Scap but not to 25-HC. Grey spheres indicate residues that when mutated as indicated in Table S3 lose binding to Scap. Right: cInsig-1 rotated ~90° towards the reader relative to the orientation at bottom left, such that cInsig-1 is viewed from the cytosol. The TM helices are labeled, as are the locations of residues, shown in green spheres, which when mutated as indicated in Table S3 reduce binding to 25-HC.
(C) Comparison of the cScapD435V/cInsig-1 complex with the human Scap/Insig-2/25-HC complex (Yan et al. 2021). Superposition of the transmembrane helices of cScapD435V/cInsig-1 with human Scap/Insig-2/25-HC. Proteins are colored as indicated with transparent cartoons except for TM4. hScap, human Scap; hInsig-2, human Insig-2.
(D) Comparison of the cScapD435V/cInsig-1 structure with the cryo-EM map for the human Scap/Insig-2/25-HC complex. The map (EMD-30074) was gaussian filtered to 3 SD using Chimera, and the cScapD435V/cInsig-1 was docked into the density. The map is contoured at level 0.131 (left) and 0.358 (right). hScap, human Scap; hInsig-2, human Insig-2.
The interface between cScapD435V and cInsig-1 consists of TMs 2, 4, and 5 of cScapD435V, and TMs 3 and 4 of cInsig-1. We sought to interpret previous mutagenesis data for both Scap and Insig (Tables S2 and S3) in light of this structural model. The mutation Y298C in mammalian Scap prevents binding to Insigs and leads to constitutive trafficking of SREBP2 that cannot be blocked by sterols (Nohturfft et al., 1998b). This residue sits at the cytosolic end of TM2, poised for interaction with Insig’s TM3 (Figure 5A). Two other mutations, L315F (Yabe et al., 2002) and D443N (Hua et al., 1996a), also abolish interaction with Insigs. These residues cluster at the cytosolic ends of TM3 and TM6, and may contribute to Insig binding through the proper positioning of TM2 (see below).
Our structure may also explain how mutations in Insig affect binding to Scap (Figure 5B and Table S3). The mutations Q132A, W145A, and D149A in TM4 reduce the interaction of human Insig-2 with Scap without altering its binding to 25-HC (Gong et al., 2006; Radhakrishnan et al., 2007), implying that these mutations do not globally disrupt Insig’s fold. These residues are situated at the cScapD435V/cInsig-1 interface according to our model (Figure 5A, B). Mutations along TM3 (A113W, G117F, and H120F) also block the binding of Insig-2 to Scap (Ren et al., 2015), and are located at the Scap interaction interface in our structure. Five other mutations on multiple TMs of Insig-2 (G39F, C77D, F115A, T136A, G200F) were shown to reduce Scap interaction while also diminishing 25-HC binding (Table S3). In our structural model, all five of these residues face the core of Insig (Figure 5B, green spheres), which supports the idea that oxysterols bind within the interior of the TM bundle.
Superposition of the transmembrane model of the cScapD435V/cInsig-1 complex to that reported by Yan et al. (Yan et al., 2021) for the human Scap/Insig-2 complex showed good agreement (Figure 5C, root mean square deviation 2.8 Å). After docking of our cScapD435V/cInsig-1 model as a rigid body in the map of Yan et al., the TM helices fit well and our L1-L7 domain is situated within an observable, albeit low-resolution, feature that was assigned to the luminal region in the human Scap/Insig-2 reconstruction (Figure 5D). In our de novo model of L1-L7 in the cScapD435V/cInsig-1 complex, we included L1 residues 53–278 and L7 residues 637–704. The L1-L7 domain structure in the complex (Figure 4E) is highly similar to that of cScap (Figure 2E) with rmsd 0.77 Å.
Differences in Conformation Between Scap Alone and Scap Bound to Insig
Superimposing the TM helices in the cScap structure versus cScapD435V/cInsig-1, we observe several changes in conformation involving the SSD (TMs 2–6) and the luminal loops (L1-L7). In the cScap structure, we do not observe any cryo-EM density corresponding to TM2, while this helix is clearly resolved in the complex with cInsig-1 (Figure 6A). However, cScap contains density for a different TM helix that is not resolved in the complex with cInsig-1. Based on the continuity of this TM helix with density for the Scap CTD (Figure 2E), we tentatively assign this density to cScap TM8 (Figure 6A). Viewed from the cytosol (Figure 6B), the putative TM8 in cScap is packed against TM4 and TM5, which blocks the surface on cScap where cInsig-1 would bind. Comparison between the two structures implies that the cInsig-1 interaction with TMs 2, 4, and 5 helps to stabilize the position of TM2 within the SSD. In the absence of cInsig-1, the SSD can bind to cScap’s TM8 intramolecularly, bringing the Scap CTD into close proximity with the SSD TMs and making the CTD resolvable in our cScap cryo-EM map (Figure 2E). The CTD is not resolvable in the cScapD435V/cInsig-1 structure (Figure 4E).
Figure 6. Conformational Differences between cScap and cInsig-1-bound cScapD435V.
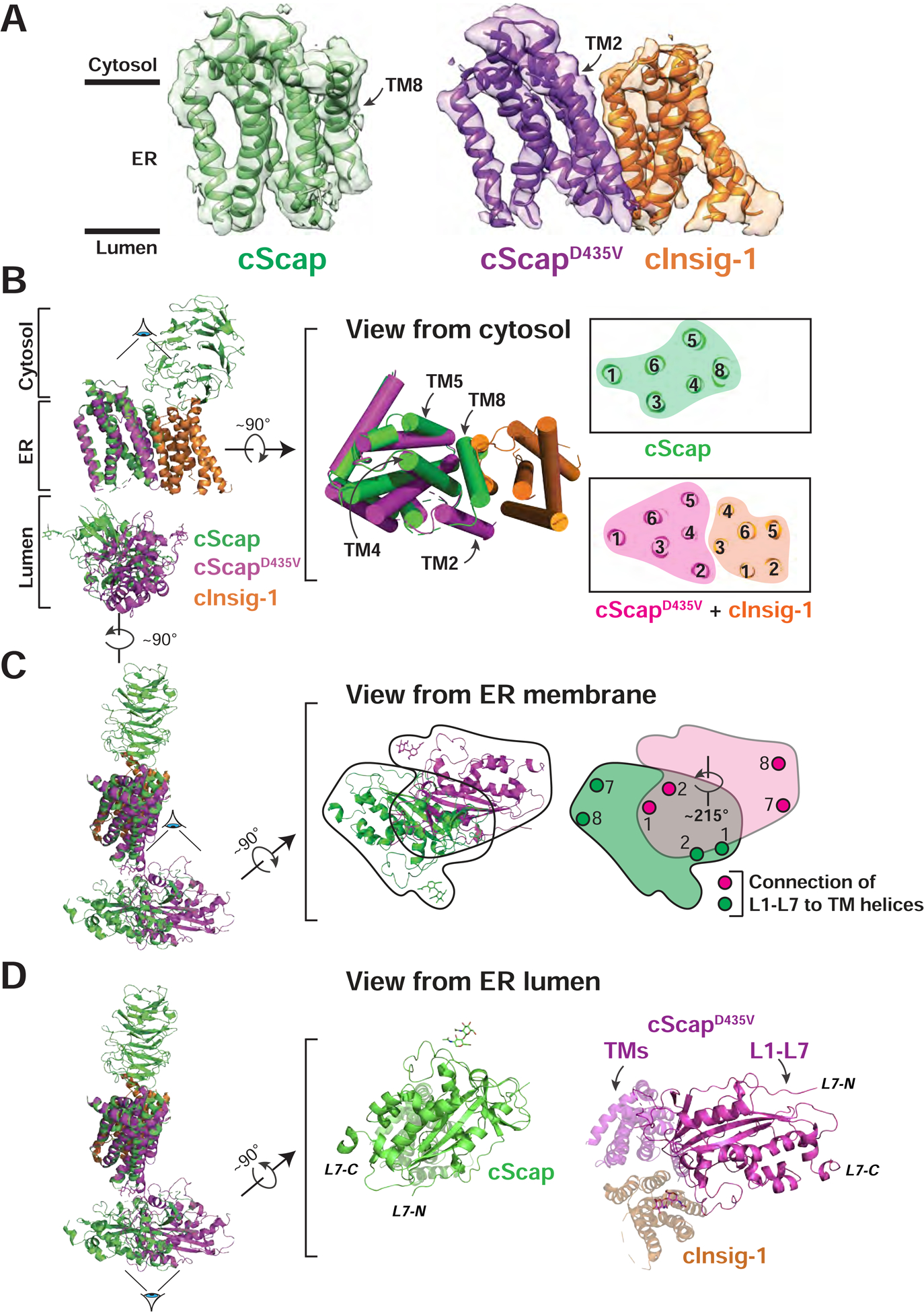
(A) Transmembrane domain models and masked cryo-EM density for the cScap and cScapD435V/cInsig-1 structures. cScap is colored green, cScapD435V is colored purple, and cInsig-1 is colored orange.
(B) Left: cScap and cScapD435V-cInsig-1 superimposed based on the TM helices of cScap. The Scap CTD (resolved only in the cScap structure) and the L1-L7 domains are dimmed to highlight the superposition of the transmembrane domains. cScap is colored green, cScapD435V is colored magenta, and cInsig-1 is colored orange. Middle: The superimposed TM helices, rotated 90° with respect to the view shown at left, as viewed from the cytosol. TM helices are shown as cylinders. Right: Slice of the transmembrane domains from the approximate center of the membrane. Numbers indicate the assigned TM helix. Left and middle panels were generated using Pymol, right panel was generated using UCSF Chimera X.
(C) Left: cScap and cScapD435V/cInsig-1 superimposed based on the TM helices of Scap. The Scap CTD (resolved only in the cScap structure) and the TM helices are dimmed to highlight the different L1-L7 position. cScap is colored green, cScapD435V is colored magenta, and cInsig-1 is colored orange. Middle: The superimposed L1-L7 domains, rotated 90° with respect to the view at left, as viewed from the ER membrane. Black trace outlines the outer boundaries of the L1-L7 domain for each structure. Right: The outlined regions from the Middle panel are colored green or magenta corresponding to the WT and D435V (Insig-bound) forms of cScap, respectively. The grey region denotes the overlap of L1-L7 domains. These two domains are related by a rigid body rotation of ~215° about the membrane normal. Approximate positions where L1 and L7 attach to TM helices (TM1 and TM2 for L1; TM7 and TM8 for L7) are indicated by circles.
(D) Left: Superposition of cScap and cScapD435V/cInsig-1 as in (C). Middle: cScap, rotated 90° with respect to the view at left, as viewed from the ER lumen. cScap is colored green. TM helices extending back from the page are dimmed. Right: cScapD435V/cInsig-1 complex, rotated 90° with respect to the view at left, viewed from the ER lumen. cScapD435V is colored magenta and cInsig-1 is colored orange. All TM helices extending back from the page are dimmed.
In addition to this difference in TM architecture, we observe a large conformational difference in the relative position and orientation of the oblong L1-L7 domain in the two structures (Figure 6C). Compared to cScap, when cInsig-1 is bound in the cScapD435V/cInsig-1 complex, L1-L7 has rotated by ~215° horizontally in a plane parallel to the membrane (right panel). A consequence of this rotation is a dramatic change in the positioning of the L1-L7 domain relative to the SSD (Figure 6D). In cScap, L1-L7 is situated under the SSD (middle panel). In contrast, in the cScapD435V/cInsig-1 complex, L1-L7 is no longer fully positioned under the SSD (right panel). In this conformation, most of L1 (~80%) extends away from the SSD where it would reside under the lipid bilayer. L7 is anchored to the membrane by TM7 and TM8, which are displaced the most from the SSD. The multiple structural changes described above are summarized in schematic form in Figures 7A and 7B and elaborated on in the Discussion.
Figure 7. Model for cScap Conformational Changes.
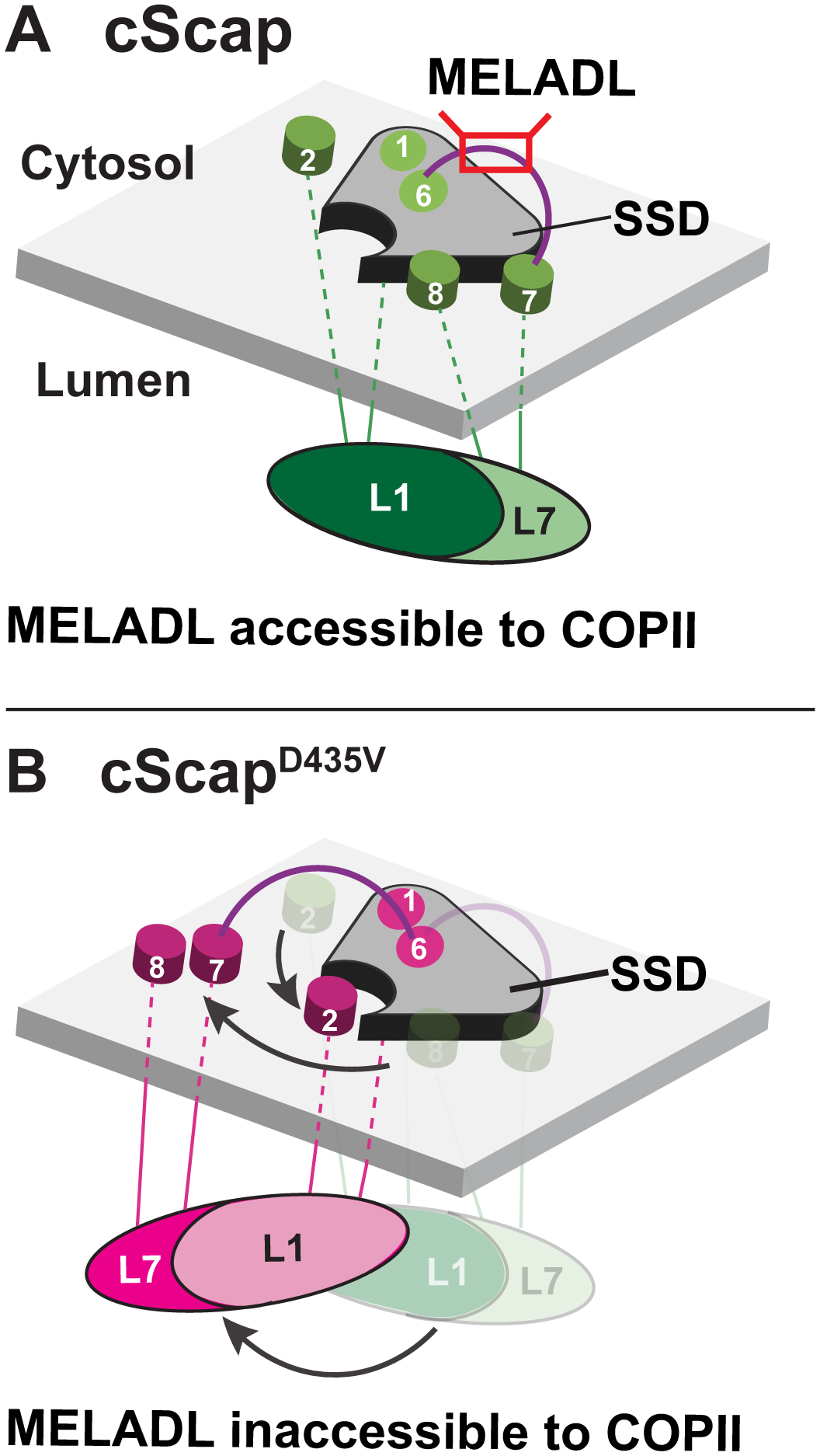
(A) When cScap is free from cInsig-1, the L1-L7 domain is positioned directly underneath the SSD module and the TM7-TM8 module is bound to the SSD. In this conformation, the MELADL sequence on the cytoplasmic loop connecting TM6 and TM7 is accessible to COPII proteins.
(B) In the cInsig-1 bound form triggered by mutation of cScap’s D435 to V, the L1-L7 domain rotates away from the cScap SSD module. For clarity, cInsig-1 is not shown. The rotation of the L1-L7 domain moves TM2 into position to create a binding interface for Insig-1 as shown in Figure 6B. Moreover, the TM7-TM8 module is moved away from the SSD to provide Insig-1 access to this interface. These conformational changes on the luminal side may cause a concerted change in MELADL-containing L6 on the cytoplasmic side to prevent interactions with COPII proteins.
See also Figure S7.
Figure S7 compares the architecture of Scap in its two conformations with four other SSD-containing proteins: NPC1, Patched, Dispatched, and NPC1-L1. In both cScap structures, the L1-L7 domain adopts an orientation with its long axis roughly parallel to the membrane. In contrast, in the other related SSD-containing proteins, the middle luminal domain (MLD)-related subunits have their long axis pointing down from the membrane. The freedom of the cScap L1-L7 structure to adopt such different relative orientations may arise from the fact that Scap has a single transmembrane SSD, whereas these other proteins all have two pseudo-symmetrically interacting ‘SSD-MLD’ units on the same polypeptide, which may restrict possible MLD motions. The variation in SSD-MLD disposition also highlights the adaptability of these subunits for different sterol-related functions.
Discussion
Extensive studies of Scap have led to a working model for how it regulates cellular cholesterol levels. In this model, the interaction between Scap’s L1 and L7 is necessary for the transport of SREBPs from ER to Golgi, leading to an increase in cholesterol synthesis (Brown et al., 2018; Zhang et al., 2016). Feedback regulation is initiated by binding of cholesterol to L1, which changes L1’s interaction with L7 on the luminal side of the ER membrane. The cholesterol signal is then propagated through the membrane-embedded SSD and culminates with movement of the cytosolic MELADL sequence, thus preventing binding to COPII proteins, halting SREBP transport, and decreasing cholesterol synthesis (Brown et al., 2018). Our current structures highlight the modular nature of Scap in which TMs 2–6 of the SSD and TM7–8 are linked in two ways: i) through the MELADL-containing L6 on the cytosolic side; and ii) through a luminal ‘platform’ created by the intertwining of L1 and L7 (Figures 3 and 7).
The significance of this modular architecture was revealed by comparison of our cScap structure to our cScapD435V/cInsig-1 structure (Figure 6). When cInsig-1 binds cScapD435V, three major changes occur. First, although TM2 is not visible in the cScap structure, it becomes ordered and resolvable in cScapD435V at the cInsig-1 interface (Figure 6A). Second, the cInsig-1 TM bundle displaces cScap’s TM8 (Figure 6B). Third, the entire L1-L7 luminal platform rotates, positioning the TM7–8 module away from the rest of the SSD (Figure 6C–D). We hypothesize that cholesterol can change cScap’s conformation in a manner similar to that which we observe for the cScapD435V/cInsig-1 complex. A consequence of this large conformational change could be to alter the accessibility of the MELADL sequence on L6 (which is linked to TM7) to the COPII transport machinery (Figure 7). In this model, cInsig-1 and cholesterol could act cooperatively to displace cScap’s TM8 from binding to the Scap SSD (Figure 6B), which may explain why both cholesterol and Insigs are required to fully curb ER-to-Golgi transport of Scap (Lee et al., 2005).
The cScapD435V/cInsig-1 complex transmembrane region in our structure is generally similar to that reported in the human Scap/Insig-2 complex (Figure 5C, D). An exception is that the human Scap/Insig-2 interface is bridged by a putative 25-HC molecule in a cavity facing the ER lumen, created by a distortion in TM4 of Scap (Yan et al., 2021). While a similar cavity is not apparent in the structure of our cScapD435V/cInsig-1 complex, we still observe binding to 25-HC (Figure 4B, D), suggesting that the proposed 25-HC site in Yan et al. may not be the sole binding site for oxysterols. Notably, in both structures, the residues in Insig whose mutations affect high-affinity 25-HC binding as well as Scap binding are localized to the core of Insig, whereas residues whose mutations do not alter 25-HC binding but still affect Scap binding are at the interface (Figure 5 A, B). Moreover, Insig-2 can bind 25-HC with high affinity in the absence of Scap (Radhakrishnan et al., 2007), and Insig binding to Scap can occur in the absence of 25-HC, when triggered by cholesterol binding to Scap (Adams et al., 2004). Additional studies will be needed to elucidate the mechanism by which oxysterols promote the interaction between Scap and Insigs.
Cholesterol and 25-HC both trigger the binding of Scap to Insigs. In this work, we have characterized the structures of cScap and cScapD435V/cInsig-1 in the absence of either cholesterol or 25-HC. Although our purified chicken proteins bind these sterols with the expected affinity and specificity (Figures 2C–D, 4B–D), we have not yet obtained structures with bound cholesterol. As such, we cannot say how cholesterol interacts with the luminal loops, nor how the L1 and L7 elements undergo conformational changes or dissociation in the presence of cholesterol. However, our atomic model of L1-L7 does provide insights into how cholesterol could change the conformation of Scap. This insight comes from the location of residues that, when mutated, lock Scap into a conformation that binds Insigs in the absence of cholesterol, and prevents it from transporting SREBP2 (Figure 3). For example, the previously characterized Y234A mutation on L1 will have a disruptive effect on this domain through destabilization of the core β-sheet, and the Y666S mutation on L7 (Y640S in hamster) will remove stabilizing hydrophobic packing interactions between L1 and L7 (Figure 3B). Such destabilization will disfavor the conformation observed for cScap alone by preventing the proper positioning of the L1-L7 luminal platform. Consistent with this hypothesis, targeted mutagenesis predicted to destabilize L1-L7 based on our structures also prevented Scap’s transport of SREBP2 (Figure 3F). Understanding the precise manner in which the tightly associated L1 and L7 undergo conformational changes and/or dissociation in the presence of cholesterol will require the structural elucidation of cholesterol-bound cScap.
In our previous working model, we speculated that cholesterol binding to L1 causes L1 to dissociate from L7 and that this dissociation triggers a conformational change that promotes Insig binding, thereby blocking movement of the Scap/SREBP complex to the Golgi (Brown et al., 2018). The structural information in the current paper leads us to consider the alternate model shown in Figure 7. In the new model, the L1-L7 complex does not dissociate but is in equilibrium between two positions: i) residing directly underneath the SSD, and ii) extending away from the SSD, allowing more contact with cholesterol in the membrane. If L1-L7 encounters cholesterol in the membrane, it remains trapped in the extended state. In this state, the position of the TM7–8 module is altered, precluding exposure of the MELADL sequence at the cytosolic end of Loop 6 and blocking its access to COPII proteins. In the cholesterol-bound conformation, TM8 would move away from the SSD, allowing Insig to bind. The association of Insig would stabilize the cholesterol-bound conformation of Scap and thereby assure that Scap cannot move to the Golgi. A corollary of this model is that in the absence of membrane cholesterol, the L1-L7 complex may move continuously between its two positions, allowing Loop1 to scan the membrane for cholesterol. Distinguishing between our two working models will require determination of the structure of Scap bound to cholesterol. Attempts to determine this structure are underway.
Limitations of the Study
We have used the structure of the cInsig1-bound cScapD435V as a proxy for cholesterol-bound Scap that is retained in the ER. A structure of Scap bound to cholesterol, either in detergent or in a nanodisc membrane, is needed to draw definitive conclusions regarding the mechanism of cholesterol sensing. Unlike the high resolution of the luminal L1-L7 region in our cryo-EM structures, the local resolution is low in the transmembrane and cytosolic regions. Achieving higher resolution throughout the protein will be necessary to determine precisely how rotation of the luminal L1-L7 domain changes the accessibility of the cytosolic MELADL sequence, which mediates ER-to-Golgi transport of SREBPs.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to Daniel Rosenbaum (dan.rosenbaum@utsouthwestern.edu), Department of Biophysics, UT Southwestern Medical Center, Dallas, TX 75390.
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Structural data have been deposited in the Protein Data Bank (PDB) and the Electron Microscopy Data Bank (EMDB). The atomic models of the cScap L1-L7 domain/4G10Fab complex and the cScapD435V L1-L7 domain/4G10Fab complex have been deposited in the PDB with accession codes 7LKF and 7LKH, respectively. The cryo-EM maps for the global cScap/4G10Fab reconstruction and the refinement focused on the cScap L1-L7 domain have been deposited in the EMDB with accession codes EMD-23406 and EMD-23405, respectively. The cryo-EM maps for the global cScapD435V/cInsig-1/4G10Fab reconstruction and the refinement focused on the cScapD435V L1-L7 domain have been deposited in the EMDB with accession codes EMD-23407 and EMD-23408, respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Animal experiments to generate antibodies were performed at UT Southwestern with the approval of the Institutional Animal Care and Research Advisory Committee (#0745–07-01–1 for IgG-17H1; #2017–102391 for IgG-4G10). Mice were housed at 72°F and under a 12 hr light cycle (6AM on, 6PM off).
Cell Lines
HEK 293s GnTI−cells
HEK 293s GnTI− cells were obtained from ATCC (CRL-3022). Cells were thawed in Dulbecco’s modified Eagle’s medium (DMEM) (high glucose) medium supplemented with 10% v/v fetal calf serum (FCS, Sigma-Aldrich Cat# F2442), 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate and passaged in monolayer culture at 37°C in 5% CO2. After expanding to ten 10-cm dishes, cells were sloughed off and transferred to suspension culture in FreeStyle 293 medium (Thermo Scientific) supplemented with 2% v/v FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. Suspension cells were grown with orbital shaking at 130 rpm in baffled flasks at 37°C, 8% CO2, and 70% humidity. Cells were passaged to maintain a suspension density within the range of 0.4 × 106 cells/ml to 2.5 × 106 cells/ml.
HEK293T cells
HEK293T cells were maintained in monolayer culture at 37°C and 5% CO2 in DMEM (high glucose) supplemented with 5% v/v FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate.
CHO-K1 cells
CHO-K1 cells were maintained in monolayer culture at 37 °C and 8–9% CO2 in a 1:1 mixture of Ham’s F-12 and DMEM supplemented with 5% v/v FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate.
SRD-13A cells
Scap-deficient CHO cells (SRD-13A) (Rawson et al., 1999) were maintained in monolayer culture at 37°C and 8–9% CO2 in a 1:1 mixture of Ham’s F-12 and DMEM supplemented with 5% v/v FCS, 1 mM sodium mevalonate, 20 μM sodium oleate, 5 μg/ml cholesterol, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate.
Sf9 cells
Spdoptera frugiperda (Sf9) cells were cultured in SF900 II SFM media (Gibco) at 27°C with orbital shaking at 120 rpm.
Cell line validation
To guard against potential genomic instability, an aliquot of each cell line was passaged for only 4–6 weeks, after which a fresh batch of cells was thawed and propagated. All cell lines were confirmed to be free of mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza).
METHOD DETAILS
Plasmids
Constructs for mammalian cell protein expression and purification
The expression plasmid pEZT-cScapD435V-3xFLAG encodes, in order from its NH2-terminus, chicken Scap (XP_025003137.1, residues 1–1323) containing a point mutation (D435V) followed by 3 copies of the FLAG motif DYKDDDDKGSDYKDDDDKGSDYKDDDDK. This construct is expressed downstream of the strong cytomegalovirus (CMV) promoter. The coding sequence of this construct was codon-optimized for expression in human cells and produced as synthetic dsDNA pieces (gBlocks, IDT). These dsDNA pieces were assembled into a XhoI- and KpnI-linearized pEZT vector (Morales-Perez et al., 2016) using the NEBuilder HiFi DNA assembly kit according to the manufacturer’s instructions. A tagless pEZT-cInsig-1 (Uniprot Q5ZMT9, residues 28–252) was similarly designed, produced, and assembled into a pEZT vector that had been linearized with NheI and NotI restriction enzymes. The pEZT-cScapD435V-3xFLAG construct was used as a template for site-directed mutagenesis to generate pEZT-cScap-3xFLAG. For sterol-binding assays, we generated His-tagged Scap proteins where a His8 tag was introduced at the NH2-terminus of the pEZT-cScap-3xFLAG constructs to generate the plasmids pEZT-His8-cScap-3xFLAG and pEZT-His8-cScapD435V-3xFLAG.
For large-scale expression of the Scap-Insig complex for immunizations, Fab characterization, and cryo-EM studies, we generated a pEZT plasmid modified to express both cInsig-1 and cScapD435V separated by a 2A “cleavage” peptide (Liu et al., 2017). This construct encodes, in order from its NH2-terminus, the residues MTG, cInsig-1 (residues 28–252), a 30-amino acid spacer SRENLYFQGGSGATNFSLLKQAGDVEENPĜPA containing the 2A peptide from Porcine teschovirus (P2A), and the cScapD435V-3xFLAG described above. The ^ denotes the break in the peptide chain caused by the 2A sequence leading to the production of cInsig-1 and cScap as two separate peptides. This plasmid is designated as pEZT-cInsig-1–2A-cScapD435V-3xFLAG.
Constructs for insect cell protein expression and purification
The expression plasmid pFastBac-His10-hamster Scap(L1-L7) fusion has been previously described (Zhang et al., 2016). Point mutations were introduced into this construct using site-directed mutagenesis. The expression plasmid encoding FLAG-human SREBP2 CTD has been previously described (Kober et al., 2020). The expression plasmid pFastBac-His10-cScap CTD encodes, in order from its NH2-terminus, a His10 tag followed by cScap residues 779–1323. This construct was generated by subcloning from the pEZT-cScap-3xFLAG construct described above. The expression plasmid pFastBac-cScap(L1-L7) fusion encodes, in order from its NH2 terminus, a honeybee melittin signal peptide, cScap residues 46–284, a 15-amino acid linker containing a TEV recognition site, SENLYFQGSSGSSGS, cScap residues 542–738, and a His10 tag. This fusion was generated by subcloning from the pEZT-cScap-3xFLAG plasmid described above.
Constructs for SREBP2 processing assays
For these assays, we generated plasmids that express proteins under the control of the weak Thymidine Kinase (TK) promotor. The pTK-HSV-SREBP2 plasmid (Hua et al., 1996b), pTK-Scap plasmid encoding hamster Scap (Hua et al., 1996a), and the pEZT-myc-SREBP2 (WT) plasmid (Kober et al., 2020) were described previously in the indicated references. pTK-cScap-3xFLAG and pTK-cScapD435V-3xFLAG plasmids were subcloned from the pEZT constructs described above using Gibson assembly. Mutations were introduced into the above parental plasmids by site-directed mutagenesis.
Constructs for Scap-SREBP2 co-immunoprecipitation assays
The pEZT-myc-SREBP2 plasmid described previously (Kober et al., 2020) served as a template for the SREBP2 plasmid used here. This construct was modified by site-directed mutagenesis to eliminate the recognition sites for Site-1 Protease (R519A) (Hua et al., 1996b), Site-2 Protease (S480A, R481S) (Sakai et al., 1996), and Caspase-2 (D466A) (Wang et al., 1996). The resulting SREBP2 cleavage-resistant construct is termed SREBP2(CR). pEZT-human Scap-3xFLAG (Accession sequence NP_036367.2) was generated from synthetic DNA blocks in the same way as described above for chicken Scap.
Constructs for Scap(L1-L7) fusion protein secretion assays
The previously described pCMV-hamster Scap(L1-L7) plasmid (Zhang et al., 2016) encodes, in order from the NH2-terminus, the 21-aa signal sequence from honeybee mellitin, an epitope tag consisting of 12 histidines, L1 of hamster Scap (aa 46–284), a 15-aa linker that includes a TEV protease cleavage site (SENLYFQGSSGSSGS), and L7 of hamster Scap (aa 535–710). Mutation of each of the three cysteines in L1 to alanine or serine for disulfide bond characterization (Figure S5) was carried out by site-directed mutagenesis.
Construct validation
All plasmid open reading frames were verified by Sanger sequencing. Gene block sequences and primers for plasmid construction are available upon request.
Assays
SREBP2 processing (Low sterol conditions)
Scap-deficient hamster SRD-13A cells were set up on day 0 at a density of 700,000 cells per 10-cm dish. On day 1, cells were switched into fresh medium consisting of a 1:1 mixture of Ham’s F-12 and DMEM supplemented with 5% v/v FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. Cells were transfected with DNA complexed with Xtreme-GENE HP using a 3:1 μl reagent: μg DNA ratio in Opti-MEM according to the manufacturer’s instructions. On day 2, cells were washed with phosphate-buffered saline (PBS) and switched into cholesterol-depleting medium consisting of a 1:1 mixture of Ham’s F-12 and DMEM supplemented with 5% v/v newborn calf lipoprotein-deficient serum (LPDS), 50 μM sodium compactin, 50 μM sodium mevalonate, 1% w/v 2 hydroxypropyl-β-cyclodextrin (HPCD), 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate for 1 hr. LPDS (Goldstein et al., 1983) and solutions of sodium compactin and sodium mevalonate (Brown et al., 1978) were prepared as described in the indicated reference. Following this treatment, cells were washed twice in PBS, solubilized in sodium dodecyl sulfate (SDS) lysis buffer (10 mM Tris-HCl (pH 6.8), 100 mM NaCl, 1% w/v SDS, 1 mM EDTA, 1 mM EGTA, 200 μg/ml PMSF, 20 μg/ml ALLN, 25 mU/μl benzonase, and protease inhibitors) and subjected to 10% SDS-PAGE and immunoblot analysis.
SREBP2 processing (High sterol conditions)
Scap-deficient SRD-13A cells were set up on day 0 at a density of 700,000 cells per 10-cm dish. On day 1, cells were switched into fresh medium consisting of a 1:1 mixture of Ham’s F-12 and DMEM supplemented with 5% v/v FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. Cells were transfected with DNA complexed with FuGENE6 (Promega) using a 3:1 μl reagent: μg DNA ratio in Opti-MEM according to the manufacturer’s instructions. On day 2, cells were washed with PBS and switched into cholesterol-depleting medium consisting of a 1:1 mixture of Ham’s F-12 and DMEM supplemented with 5% v/v LPDS, 50 μM sodium compactin, 50 μM sodium mevalonate, 1% w/v 2 hydroxypropyl-β-cyclodextrin (HPCD),100 units/ml penicillin, and 100 μg/ml streptomycin sulfate, for 1 hr. Following cholesterol depletion, cells were washed with PBS and switched into oxysterol-replete medium consisting of a 1:1 mixture of Ham’s F-12 and DMEM supplemented with 5% v/v LPDS, 50 μM sodium compactin, 50 μM sodium mevalonate, 2.5 μM 25-hydroxycholesterol (25-HC), 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate, for 3 hours. Following this treatment, cells were washed twice in PBS, solubilized in SDS lysis buffer and subjected to 10% SDS-PAGE and immunoblot analysis.
Scap-SREBP2 co-immunoprecipitation
HEK 293T cells were set up on day 0 at a density of 700,000 cells per 10-cm dish. On day 1, cells were switched to fresh culture medium consisting of DMEM (high glucose) supplemented with 5% v/v FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. Cells were transfected with DNA complexed with FuGENE6 using a 3:1 μl reagent: μg DNA ratio in Opti-MEM according to the manufacturer’s instructions. On day 2, the cells were harvested, washed in PBS, and the cells from two dishes were pooled and resuspended in 1 ml of solubilization buffer (500 mM NaCl, 50 mM HEPES-NaOH (pH 7.5), 1% w/v lauryl maltose neopentyl glycol (LMNG) detergent, 0.2 % w/v cholesteryl hemisuccinate (CHS), 0.2% w/v sodium cholate, 20% v/v glycerol, 1mM EDTA, 0.5 mM TCEP, 25 mU/μl benzonase, and protease inhibitors (160 μg/ml benzamidine, 1 mM PMSF, 1 mM E64, and 2.5 μg/ml leupeptin)). Cells were lysed by passing 10 times through a 22-gauge needle and the cell membranes were solubilized by rotating for 1 hr at 4°C. Next, the samples were centrifuged at 100,000 × g for 30 min at 4°C to pellet insoluble material. At this point, 50 μl (5%) of the sample was retained for immunoblot analysis of the input material. Supernatants were incubated for 1 hr with 20 μl anti-FLAG M2 affinity resin that had been equilibrated in solubilization buffer. The resin was washed three times by rotating for 30 min at 4°C in 1 ml wash buffer (50 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, 2% v/v glycerol, 0.1% w/v LMNG, 0.02% w/v CHS, 0.02% w/v sodium cholate, and 0.5 mM TCEP). Bound proteins were eluted with 200 μL of wash buffer supplemented with 400 μg/ml 3xFLAG peptide (APExBio). Eluted samples were subjected to 10% SDS-PAGE and immunoblot analysis.
Secretion of Scap L1-L7 and modification of cysteines
On day 0, CHO-K1 cells were set up for experiments in 3 ml of medium A (1:1 mixture of DMEM and Ham’s F12 supplemented with 5% v/v FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate) at a density of 6 × 105 cells/60-mm dish. On day 1, cells were switched to 3 ml of medium A containing 1% v/v insulin-transferrin-selenium (ITS) and transfected with wildtype and mutant versions of hamster Scap(L1-L7) using FuGENE 6 as the transfection agent. On day 3, media was collected from each group (two dishes/condition) and pooled (6 ml total), and the cells from each group were harvested, pooled, and resuspended in PBS (1 ml total). Equivalent fractions (by volume) of cells and media were then processed and subjected to immunoblot analysis. In some experiments, the medium from transfected cells containing secreted proteins was incubated with 6 mM mPEG-MAL-5000 (methoxy poly-(ethylene glycol) maleimide) for 30 min at room temperature, followed by immunoblot analysis. In some of these modification assays, the medium was subjected to heating and incubation for 15 min with reducing agents before treatment with mPEG-MAL-5000.
Immunoblot analysis
Following SDS-PAGE, proteins were transferred to nitrocellulose membranes using the Trans-Blot Turbo Transfer System (BioRad Laboratories, Hercules, CA). Membranes were probed with the following primary antibodies: monoclonal Anti-SREBP2 IgG-22D5 (McFarlane et al., 2015) (used at 5 μg/ml), monoclonal anti-Scap L7 IgG-9D5 (Sakai et al., 1997) (used at 5 μg/ml), monoclonal anti-Scap L1 IgG-7G5 (Zhang et al., 2016) (used at 5 μg/ml), monoclonal anti-Insig 17H1 (used at 3 μg/ml), monoclonal anti-FLAG M2 clone (Sigma-Aldrich) (used at 1 μg/ml), and monoclonal anti-Actin (Sigma-Aldrich) (used at 1 μg/ml). Bound antibodies were detected by chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, Thermo Scientific, Waltham, MA) using a 1:5000 dilution of anti-mouse IgG or anti-rabbit IgG conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Membranes were exposed to Blue X-ray Film (Phoenix Research Products, Pleasanton CA) for 1–30 s.
3H-Sterol binding
Binding assays were carried out as previously described (Motamed et al., 2011). Each reaction, in a final volume of 100 μl of the LMNG-containing “binding buffer” (see section below titled “cScap and cScapD435V/cInsig-1 complexes for sterol binding assays”), contained 2 μg of either His8-cScap-3xFLAG (13.4 pmol) or His8-cScapD435V-3xFLAG/Insig-1 complex (11.5 pmol of a putative 1:1 complex) and varying concentrations of the indicated 3H-sterol (delivered in 3 μl of ethanol) in the absence or presence of the indicated competitor unlabeled sterol (delivered in 2 μl of ethanol). After incubation for 4 hr at 4°C, each reaction was loaded onto a column packed with 0.3 ml of Ni2+-NTA-agarose beads pre-equilibrated with 2 ml of binding buffer. The columns were washed with 3 ml of binding buffer and then eluted with 1 ml of the same buffer containing 250 mM imidazole. Aliquots of the eluate (0.5 ml) were assayed for radioactivity in a liquid scintillation counter (Beckman Coulter LS 6500).
Analytical Fab binding
Binding between 4G10Fab and cScapD435V/cInsig-1 was tested as follows: 500 μg of the cScapD435V/cInsig-1 complex purified in glyco-diosgenin (GDN) as described below was incubated without or with 320 μg 4G10Fab for 30 min and then subjected to gel filtration at a flow rate of 0.7 ml/min over a Superose 6 Increase 10/300 column that had been equilibrated in a buffer consisting of 20 mM HEPES-NaOH, 150 mM NaCl, 0.02% w/v GDN, and 0.5 mM TCEP.
Binding between cScap(L1-L7) fusion and 4G10Fab was tested as follows: 860 μg of the cScap(L1-L7) fusion protein was incubated without or with 1,760 μg 4G10Fab for 1 hr and then subjected to analytical gel filtration at a flow rate of 0.7 ml/min over a Superdex 200 Increase 10/300 column (GE) equilibrated in 50 mM HEPES-NaOH (pH 7.5) and 150 mM NaCl.
Immunoprecipitation of cScap by 4G10Fab
HEK 293T cells were set up on day 0 at a density of 700,000 cells per 10-cm dish. On day 1, cells were switched to fresh culture medium consisting of DMEM (high glucose) supplemented with 5% v/v FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. Cells were transfected with DNA complexed with FuGENE6 using a 3:1 μl reagent:μg DNA ratio in Opti-MEM according to the manufacturer’s instructions. On day 2, the cells were harvested, washed in PBS, and the cells from five dishes were pooled and resuspended in 3 ml of solubilization buffer (150 mM NaCl, 50 mM HEPES-NaOH (pH 7.5), 1% w/v GDN, 1 mM EDTA, 0.5 mM TCEP, and protease inhibitors, including 160 μg/ml benzamidine, 1 mM PMSF, 1 mM E64, and 2.5 μg/ml leupeptin). Cells were lysed by passing 10 times through a 22-gauge needle and the cell membranes were solubilized while rotating for 2 hr at 4°C. Next, the samples were centrifuged at 100,000 × g for 30 min at 4°C to pellet insoluble material. Supernatants were incubated for 1 hr with 20 μl anti-FLAG M2 affinity resin that had been equilibrated in solubilization buffer. The resin was washed three times by rotating for 30 min at 4°C in 1 ml wash buffer (50 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, 0.05% w/v GDN and 0.5 mM TCEP). Bound proteins were eluted with 1 ml of wash buffer supplemented with 400 μg/ml 3xFLAG peptide (APExBio). At this point, 50 μl (5%) of the sample was retained for immunoblot analysis. Eluted samples were then incubated for 1 hr with 20 μl protein L agarose which had been complexed with 40 μg 4G10Fab and then equilibrated with wash buffer to remove unbound Fab. Following the 1 hr incubation, the resin was washed three times while rotating for 1 hr at 4°C in 1 ml wash buffer. Bound proteins were eluted in 200 μl SDS lysis buffer supplemented with 100 mM glycine-HCl (pH 3.0). Eluted samples were subjected to 10% SDS-PAGE and immunoblot analysis with HRP-conjugated M2 anti-FLAG antibody.
Reproducibility of assays
All cellular assays were carried out at least 3 times, using different batches of cells set up on different days. All 3H-sterol binding assays were carried out in triplicate using two different batches of purified proteins.
Antibody Selection and Production
To produce the mouse anti-Scap monoclonal antibody IgG-4G10, BALB/c mice were immunized with a mixture consisting of 50 μg chicken cScapD435V/cInsig-1 complex purified in GDN detergent as described below and Ribi Adjuvant System (Sigma-Aldrich) according to the manufacturer’s instructions. This initial immunization was followed by 11 boosts, each consisting of 50 μg protein. Over 2500 hybridoma cells were created by fusing SP2-mIL6 mouse myeloma cells with splenic B lymphocytes derived from the immunized animal. Hybridoma culture supernatants were screened by ELISA for reactivity. ELISA screening was carried out in detergent-free conditions using biotinylated Scap-Insig complex that was reconstituted into lipid nanodiscs as described below and captured on streptavidin-coated plates. 504 ELISA-positive clones were counter-screened by SDS-PAGE immunoblot against 10 ng purified Scap-Insig complex to select ELISA-positive but immunoblot-negative clones. 24 ELISA-positive, immunoblot-negative hybridomas were subcloned by serial dilution three times and their antibodies were purified by affinity chromatography using Protein G Sepharose 4 Fast Flow columns. The eluted antibodies were dialyzed overnight against mAb dialysis buffer (10 mM NaPO4 (pH 7.5) and 50 mM NaCl) and assayed for binding to detergent-solubilized cScapD435V-cInsig-1 by gel filtration. Three antibodies exhibited binding to Scap, but two of these were not useful in subsequent cryo-EM studies. Fortunately, the remaining antibody, designated IgG-4G10 (subclass 1, κ), proved to be useful as described below.
The Fab region of IgG-4G10 was sequenced as follows: RNA was extracted from the 4G10 hybridoma cells using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Single strand DNA (ssDNA) was generated from this RNA using the Superscript III First Strand kit (Invitrogen) with both random hexamers and oligo dT primers according to the manufacturer’s instructions. The resulting ssDNA was used to prepare double strand DNA (dsDNA) using the GoTaq Polymerase kit (Promega) according to the manufacturer’s instructions. The resulting dsDNA was probed with a series of primers designed to amplify different possible variable regions resulting from recombination (primers listed in Table S4). PCR products corresponding to the expected size of ~500 bp were gel-extracted and sequenced. The sequences were analyzed using the IMGT database (Giudicelli et al., 2011) to identify possible complementarity-determining regions (CDRs). The 4G10 hybridoma generated a single nucleotide sequence for both a heavy IgG and light IgK. To confirm this sequence, the 4G10 CDR was further subcloned into a shuttle vector and sequenced. The resulting coding sequence of the Fab domain is provided in Figure S2C. To generate 4G10 Fab, IgG-4G10 was digested with 1:250 w/w papain for 2 hr at 37°C in mAb dialysis buffer (see above) supplemented with 1mM EDTA, 10 mM L-Cysteine hydrochloride, and 50 mM NaPO4 (pH 7.5). Following digestion, the papain was inactivated with 30 mM iodoacetamide for 15 min on ice and the digested Fab was purified by gel filtration at a flow rate of 0.7 ml/min over a Superdex 200 Increase 10/300 column equilibrated in 50 mM HEPES-NaOH (pH 7.5) and 150 mM NaCl. Purified Fab was used immediately for cScapD435V/Insig-1 complex binding experiments, including cryo-EM grid preparation, or else supplemented with 20% v/v glycerol and stored at −80°C for use in cScap(L1-L7) fusion binding experiments.
To produce the monoclonal antibody IgG-17H1 against Insig-1, BALB/c mice were immunized with recombinant full-length human Insig-1 containing a His10 epitope tag at its NH2-terminus and a FLAG epitope tag at its COOH-terminus that was purified using the same procedures as described previously for human Insig-2 (Radhakrishnan, 2007). The purified protein was combined with Sigma Adjuvant System (Sigma-Aldrich Cat. S6322) (100 μg primary injection, followed by three 100 μg boosts, and ending with a final boost of 50 μg). Hybridoma cells were generated by fusing the splenic B lymphocytes with SP2-mIL6 mouse myeloma cells. After ELISA screening of hybridoma culture supernatants, one positive hybridoma, designated IgG-17H1 (subclass 2a, κ), was subcloned by serial dilution three times and purified by affinity chromatography using protein G-Sepharose4 Fast Flow columns.
Production of Baculoviruses for Protein Overexpression
Baculoviruses for Sf9 expression were produced using the Bac-to-Bac expression system according to the manufacturer’s instructions. BacMam baculoviruses for mammalian protein expression from pEZT constructs were produced using the Bac-to-Bac expression system and the GFP-expressing viruses were titered by serial dilution in Sf9 cells as previously described (Morales-Perez et al., 2016). Viruses were concentrated by centrifugation at 66,700 × g for 1 hr at 4°C to achieve titers necessary to infect HEK GnTI− cells at a multiplicity of infection (MOI) of 1–3 viruses per cell while using no more than 10% total volume of virus in the final culture.
Protein Expression and Purification
Saposin
Saposin A was expressed in Rosetta Gami-2 (DE3) cells. Cells were grown in LB at 37°C on an orbital shaker (225 rpm) to a density of OD600 = 0.8, at which point Saposin expression was induced with 1 mM IPTG for 4 hours. Cells were pelleted by centrifugation, washed in PBS, and stored at −80°C until purification. Cells were lysed by Dounce homogenization in lysis buffer (50 mM HEPES-NaOH (pH 7.5), 300 mM NaCl, 1% v/v Triton X-100) supplemented with 1 mg/ml lysozyme and 25 mU/μl benzonase. Lysates were incubated while rotating for 1 hr at 4°C and then centrifuged at 10,000 × g for 20 min at 4°C. The resulting supernatant was heated for 10 min at 85°C and precipitated proteins were eliminated by centrifugation at 50,000 × g for 30 min at 4°C. The clarified supernatant was incubated with Ni2+-NTA resin for 2 hrs at 4°C. The resin was collected by centrifugation at 1000 × g for 5 min at 4°C and then transferred to a gravity column. The resin was washed first with 15 column volumes (CV) of lysis buffer, then with 15 CV of buffer consisting of 50 mM HEPES-NaOH (pH 7.5), 300 mM NaCl, 20 mM imidazole (pH 7.5), and 50 mM sodium cholate, and then finally with 15 CV of buffer consisting of 50 mM HEPES-NaOH (pH 7.5), 300 mM NaCl, 50 mM imidazole (pH 7.5). Saposin was eluted in the final wash buffer supplemented with imidazole to a final concentration of 400 mM. Eluted protein was mixed with 5% w/w TEV protease and dialyzed overnight against a buffer consisting of 20 mM HEPES-NaOH (pH 7.5) and 150 mM NaCl. The following day, the protein was concentrated using a 3,000 MWCO Amicon centrifugal filter and subjected to gel filtration at a flow rate of 0.7 ml/min over a Superdex 200 Increase 10/300 column (GE Healthcare, Chicago, IL) equilibrated in 20 mM HEPES-NaOH (pH 7.5) and 150 mM NaCl. Purified saposin was flash-frozen in liquid N2 and stored at −80°C until use.
cScap CTD-human SREBP2 CTD complex
On day 0, Sf9 cells were set up at a density of 1 × 106 cells/ml, infected with 20 ml of high titer baculovirus per liter of culture, and cultured for 72 hrs. Cells were then harvested by centrifugation at 4,000 × g for 10 min at 4°C, washed with PBS, and the pellets were stored at −80°C until purification.
Purification of the CTD complex was conducted as described previously (Kober et al., 2020). All steps were conducted on ice or at 4°C. Cell pellets from 12 L cultures were resuspended in 500 ml of buffer consisting of 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mg/ml iodoacetamide, 1 mM EDTA, 0.5 mM TCEP, and protease inhibitors (160 μg/ml benzamidine, 1 mM PMSF, 1 mM E64, and 2.5 μg/ml leupeptin). Cells were lysed by Dounce homogenization. Insoluble material was removed by centrifugation at 20,000 × g for 30 min. The clarified supernatant was incubated with 15 ml Ni2+-NTA beads for 1 hr to bind the His10-tagged cScap CTD. Following incubation, the resin was pelleted by centrifugation at 1,000 × g for 5 min and transferred to an empty column. The resin was washed with 10 CV of wash buffer (20 mM Tris-HCl (pH 7.5) and 150 mM NaCl) that was supplemented with 50 mM imidazole (pH 7.5) and bound proteins were eluted in wash buffer supplemented with 300 mM imidazole directly onto a column containing 5 ml anti-FLAG M2 affinity resin in order to bind the 3xFLAG-tagged human SREBP2 CTD. After washing with 3 CV wash buffer, bound complexes were eluted in wash buffer supplemented with 100 μg/ml 3xFLAG peptide. Eluted proteins were concentrated to 1 ml using a 50,000 MWCO Amicon centrifugal filter and subjected to gel filtration chromatography at a flow rate of 0.5 ml/min over a Superdex 200 Increase 10/300 column that had been equilibrated in buffer consisting of 20 mM HEPES-NaOH (pH 7.5) and 150 mM NaCl.
cScap(L1-L7) fusion
On day 0, 6 L cultures of Sf9 cells were set up at a density of 1 × 106 cells/ml, infected with 20 ml of high titer baculovirus per liter of culture, and cultured for 72 hrs. After pelleting the cells by centrifugation at 4,000 × g for 10 min at 4°C, the supernatants were immediately used for purification using procedures similar to that described previously (Zhang et al., 2016). Supernatants were clarified by filtration through 0.2-micron filters and then passed through a column containing 10 ml Ni2+-NTA resin. The resin was washed with 10 CV of wash buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl) supplemented with 50 mM imidazole and bound proteins were eluted in wash buffer supplemented with 300 mM imidazole. The eluate was concentrated to 1 ml using a 30,000 MWCO Amicon centrifugal filter and subjected to gel filtration chromatography at a flow rate of 0.7 ml/min over a Superdex 200 Increase 10/300 column that had been equilibrated in buffer consisting of 50 mM HEPES-NaOH (pH 7.5) and 150 mM NaCl.
cScap and cScapD435V/cInsig-1 complex for cryo-EM and sterol binding studies
For cryo-EM samples, 6 L cultures of HEK 293s GnTI− cells at a density of 2.5 million cells/ml were transduced with a single BacMam baculovirus (either pEZT-cScap-3xFLAG or pEZT-cInsig-1–2A-cScapD435V-3xFLAG) at an MOI of 3 virions per cell. For cholesterol binding studies, either a single BacMam virus (pEZT-His8-cScap-3xFLAG at MOI of 3) or two BacMam viruses (pEZT-cInsig-1 at MOI of 3 and pEZT-His8-cScapD435V-3xFLAG at MOI of 1) was used to infect cells. The cultures were supplemented with 2.5 mM sodium butyrate at the time of transduction in order to increase expression. Transduced cells were cultured for 60–72 hrs with orbital shaking at 130 rpm in an incubator maintained at 30°C, 8% CO2, and 70% humidity. Cells were harvested by centrifugation at 4,000 × g for 10 min at 4°C, washed with PBS supplemented with protease inhibitors (160 g/ml benzamidine, 1 mM PMSF, 1 mM E64, and 2.5 μg/ml leupeptin), and stored at −80°C until purification.
All purification steps were conducted on ice or at 4°C. Cell pellets were thawed, loosened by Dounce homogenization, and lysed in hypotonic buffer (10 mM HEPES-NaOH (pH 7.5), 2.5 mM EDTA, 0.5 mM TCEP, and protease inhibitors (160 μg/ml benzamidine, 1 mM PMSF, 1 mM E64, and 2.5 μg/ml leupeptin) while stirring for 30 min. Membranes were pelleted by centrifugation at 21,000 × g for 30 min. Membranes were solubilized by Dounce homogenization in buffer containing a final concentration of 50 mM HEPES-NaOH (pH 7.5), 500 mM NaCl, 20% v/v glycerol, 1% w/v LMNG, 0.2% w/v CHS, 0.2 % w/v sodium cholate, 0.5 mM TCEP, 1 mU/μl benzonase, and protease inhibitors. After stirring for 1 hr, insoluble material was pelleted by centrifugation for 1 hour at 100,000 × g. The supernatant was supplemented with additional protease inhibitors and incubated with 10 ml anti-FLAG M2 affinity resin for 4 hr. After the incubation, the resin was collected by centrifugation at 1,000 × g for 5 min at 4°C, transferred to a gravity column, and washed with 10 CV M2 wash buffer (50 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, 0.1% w/v GDN, 0.001% w/v CHS, 10% v/v glycerol, and 0.5 mM TCEP) supplemented with protease inhibitors, followed by a final wash with 5 CV of M2 wash buffer without protease inhibitors. Bound proteins were eluted with M2 wash buffer supplemented with 400 μg/ml FLAG peptide (DYKDDDDA, Biomatik). Eluted proteins were concentrated to 1 ml using a 100,000 MWCO Amicon centrifugal filter and subjected to gel filtration at a flow rate of 0.7 ml/min over a Superose 6 Increase 10/300 column (GE Healthcare) equilibrated in 20 mM HEPES-NaOH, 150 mM NaCl, 0.02% w/v GDN, 0.002% w/v CHS, and 0.5 mM TCEP. A typical purification from a 6 L culture yielded 2–3 mg of Scap or Scap-Insig complex.
cScap/4G10Fab and cScapD435V/cInsig-1/4G10Fab complexes
To prepare samples for cryo-EM grids, the purified cScap and cScapD435V/cInsig-1 complexes were incubated for 2 hr on ice with 1.5 molar excess of 4G10Fab. The mixtures were concentrated to 1 ml using a 100,000 MWCO Amicon centrifugal filter, and then subjected to gel filtration at a flow rate of 0.7 ml/min over a Superose 6 Increase 10/300 column equilibrated in 20 mM HEPES, 150 mM NaCl, 0.004% w/v GDN, 0.0004% w/v CHS, and 0.5 mM TCEP. Fractions from the center of the gel filtration peak were concentrated to 5 mg/ml using a Sartorius 100 kDa centrifuge concentrator. The final sample was centrifuged at 20,000 × g for 5 min at 4°C to pellet any debris and then the sample was vitrified as as quickly as possible as described below.
cScapD435V/cInsig-1 complexes for immunizations, ELISA assays, and Fab binding studies
For immunizations, cScapD435V/cInsig-1 complexes were expressed as described above except that the transduced cultures were further supplemented with 300 nM 25-HC. Protein complexes were purified as described above for cryo-EM studies except that the M2 wash buffer consisted of 50 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, 0.02% w/v GDN, 600 nM 25-HC, 10% v/v glycerol, and 0.5 mM TCEP, and the gel filtration buffer consisted of 20 mM HEPES-NaOH, 150 mM NaCl, 0.02% w/v GDN, 600 nM 25-HC, and 0.5 mM TCEP.
For ELISA assays on hybridoma clones, the detergent-solubilized cScapD435V/cInsig-1 complex was reconstituted into detergent-free saposin nanodiscs. For these studies, cScapD435V/cInsig-1 was purified as described above for cryo-EM studies except that the M2 wash buffer consisted of 50 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, 0.05% w/v LMNG, 0.01% w/v cholate, 0.01% w/v CHS, 600 nM 25-HC, 10% v/v glycerol, and 0.5 mM TCEP, and the gel filtration buffer consisted of 20 mM HEPES-NaOH, 150 mM NaCl, 0.01% w/v LMNG, 600 nM 25-HC, 0.002% w/v CHS, 0.002% w/v cholate, and 0.5 mM TCEP. Purified complexes were biotinylated with a 5-fold molar excess of EZ-link Biotin NHS ester for 30 min at room temperature. Unreacted biotin was quenched by addition of Tris-HCl (pH 8.0) to a final concentration of 50 mM. The lipid mixture for reconstitution was prepared by combining 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), and cholesterol in a molar ratio of 3:1:0.3 POPC:POPE:cholesterol, solubilizing the powder mixture in chloroform in glass vials, and then drying the lipid mixtures under Argon to form a thin film on the glass surface. The lipid film was hydrated in a buffer consisting of 20 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, and 1% w/v cholate to yield a final concentration of 10 mM lipids, and then subjected to bath sonication for further solubilization. To initiate reconstitution, the biotinylated cScapD435V/cInsig-1 complex was mixed with a 60-fold molar excess of the solubilized lipids for 10 min on ice. After this incubation, saposin was added at a 3-fold molar excess of cScapD435V/cInsig-1 and the mixture was rotated for 1 hr at 4°C. Detergent was removed by incubating the sample with ~50 mg Bio-Beads SM-2 Absorbent Media (BioRad) for 1 hr while rotating, after which the Bio-Beads were allowed to settle and the sample supernatant was transferred to a fresh tube containing ~50 mg Bio-Beads. This process was repeated 4 additional times over the course of 12 hours. The final detergent-free sample was subjected to gel filtration at a flow rate of 0.7 ml/min over a Superose 6 Increase 10/300 column equilibrated in buffer consisting of 20 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, 600 nM 25-HC, and 0.5 mM TCEP.
For 4G10Fab binding studies (Figure S2E, F), cScapD435V/cInsig-1 complexes were purified as for immunizations.
cScap and cScapD435V/cInsig-1 complexes for sterol binding assays
His8-cScap and His8-cScap/cInsig-1 complexes were purified by virtue of their FLAG tag, as described above for cryo-EM studies, with the exception that LMNG detergent was used instead of GDN. The M2 wash buffer consisted of 50 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, 0.05% w/v LMNG, 0.01% w/v cholate, 0.01% w/v CHS, 10% v/v glycerol, and 0.5 mM TCEP. Following elution from the anti-FLAG M2 affinity resin with M2 wash buffer supplemented with 400 μg/ml FLAG peptide, the proteins were subjected to gel filtration at a flow rate of 0.7 ml/min over a Superose 6 Increase 10/300 column equilibrated in 20 mM HEPES-NaOH, 150 mM NaCl, 0.01% w/v LMNG, 0.002% w/v CHS, 0.002% w/v cholate, and 0.5 mM TCEP. This gel filtration buffer was used as the “binding buffer” for all steps of the 3H-sterol binding assays described above.
Cryo-electron Microscopy
Grid preparation
Quantifoil Au 300 mesh R 1.2/1.3 grids were plasma cleaned with air at 30 mA for 80 seconds using a Pelco easiglow machine. 3 μl of cScap/4G10Fab or cScapD435V/cInsig-1/4G10Fab samples were applied to grids at 4°C in 100% humidity and blotted for 4.5 sec followed by freezing in liquid ethane cooled by liquid N2, using a Vitrobot Mark IV. Grids were stored in liquid N2 until imaging.
Data collection
Micrographs were acquired on a Titan Krios G2 microscope (Thermos Fisher) with a K3 Summit direct electron detector (Gatan) in the super-resolution CDS counting mode, operated at 300 kV using SerialEM software. The calibrated magnification is 46,296. 9 images were collected for each stage movement using the beam-tilt imaging method. The slit width of the GIF-Quantum energy filter was set to 20 eV. Micrographs were dose-fractioned into 36 frames at the dose rate of ~1.6 e−/Å2/frame.
Image processing
Movie frames of cScap/4G10Fab micrographs were motion-corrected and binned two-fold, resulting in a pixel size of 1.08 Å, and dose-weighted using MotionCor2(Zheng et al., 2017). CTF correction was performed using GCTF (Zhang, 2016). The rest of the image processing steps were carried out using RELION 3.1 (Zivanov et al., 2018). Particles were first roughly picked by using the Laplacian-of-Gaussian blob method, and then subjected to 2D classification. Class averages representing projections of the cScap/4G10Fab in different orientations were used as templates for reference-based particle picking. A total of 9,052,025 particles were picked from 10,641 micrographs. Particles were extracted and binned by three times (leading to 3.24 Å/pixel) and subjected to another round of 2D classification. Particles in good 2D classes were chosen (7,476,803 in total) for 3D classification using an initial model generated from a subset of the particles in RELION. Particles from one of the 3D classes showing good secondary structural features were selected and re-extracted into the original pixel size of 1.08 Å. Subsequently, we performed finer 3D classification by using local search in combination with small angular sampling. One resulting class showed improved density for the entire protein, and had better refinement statistics (resolution, alignment accuracy) than all other individual classes or combinations thereof. Therefore, this single class was selected for further processing. Refinement of the 223,673 particles in this class produced a reconstruction at 3.0 Å resolution after CTF refinement and particle polishing. To improve the resolution for L1-L7, we performed focused 3D refinement, along with CTF refinement and particle polishing, by using a soft mask around L1-L7 and part of 4G10Fab. The final reconstruction of L1-L7 was resolved at 2.9 Å resolution. To improve the resolution for the TM region and CTD, we performed another round of 3D classification by using local angular search. One of the resulting classes shows improved density for the TM regions and visible density for CTD. 3D refinement of this class along with subtraction of density for the detergent micelle resulted in 3D reconstruction to an overall resolution of 4.1 Å after CTF refinement and particle polishing.
A total of 11,542,708 particles were picked from 14,526 micrographs of cScapD435V/cInsig-1/4G10Fab. 9,521,887 particles were selected by 2D classification. Subsequently two rounds of 3D classification with 7.5 degree angular sampling together with one round of 3D classification with 3.75 degree angular sampling revealed one good class showing good density for L1-L7 and TMD. Refinement of the 155,187 particles in this class produced a reconstruction at 4.1 Å resolution after CTF refinement and particle polishing. All other classes, which had poorer refinement statistics (resolution, alignment accuracy) and displayed poorer density in both L1-L7 and TMD, were not selected for further processing. Additional 3D refinement was focused on L1-L7 and part of 4G10Fab, leading to a reconstruction of L1-L7 at 3.5 Å resolution after CTF refinement and particle polishing. To improve the density of TM region, we performed a focused 3D classification with density subtraction (Bai et al., 2015). In this approach, the density corresponding to L1-L7 and 4G10Fab as well as the detergent micelle density was subtracted from the original particles. The subsequent 3D classification on the modified particles was carried out by applying a mask around the TMD and having all of the orientations fixed at the value determined in the 3D refinement. This round of 3D classification yielded 5 classes of particles, among which one major class showed good density for the TM region. 3D refinement of this class yielded a structure at 4.6 Å resolution after CTF refinement and particle polishing.
Model Building
Luminal domain
To begin de novo building of the L1-L7 domain, two continuous chains of poly-alanine were built into the density de novo in Coot (Emsley et al., 2010). One chain was identified as L7 by the unambiguous density for the glycan at N667, which is preceded by two well-ordered tyrosine residues. The other chain was identified as L1 and the register assignment was validated by the presence of the disulfide bond between C147 and C169 (see Fig. S5E). The Fab domains were built from templates created by docking the individual Ig domains from the heavy chain of PDB 6SVL (Ebenhoch et al., 2019) and the light chain of PDB 5E94 (Hennen et al., 2016) into the density. The full L1-L7/4G10Fab model was built in Coot and subjected to rounds of iterative real-space refinement in Phenix (Liebschner et al., 2019) followed by manual corrections in Coot and ISOLDE (Croll, 2018). The final cScap L1-L7 domain model includes L1 residues 53–278 and L7 residues 636–704. Residues 70–79 and 201–204 were omitted. The cScapD435V L1-L7 domain model includes L1 residues 53–278 and L7 residues 637–704. Residues 73–80 were omitted.
Transmembrane region and CTD
The density of the TM regions for both samples is not sufficiently resolved for de novo model building. The models of cScap and cInsig-1 TM regions were first built using the SWISS-MODEL homology-modelling server (Waterhouse et al., 2018) with the structures of NPC1 (Li et al., 2016b) and mycobacterial Insig (Ren et al., 2015) as templates. The generated models were manually inspected and flexible regions were removed. These models were rigid-body fitted into the cryo-EM density of cScap/4G10Fab or cScapD435V/cInsig-1/4G10Fab in Chimera (Pettersen et al., 2004) with good agreement. To reduce ambiguity in model building, all side chains were removed from the coordinates. The resulting backbone model was subsequently refined against the map using strong secondary structure restraints in Phenix (Liebschner et al., 2019). The model of the CTD was built by using SWISS-MODEL sequence-based homology-modelling approach (Waterhouse et al., 2018). This model was rigid-body fitted into the cryo-EM density of cScap/4G10Fab, showing good agreement in shape and size. However, the precise orientation of the CTD relative to the TM domain cannot be determined unambiguously, due to the lack of resolved secondary structural features. Model geometries were assessed using Molprobity as part of the Phenix validation tools.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cryo-EM data collection, analysis, and refinement statistics are shown in Table S1. For 3H-sterol binding studies (Figure 2C, D and Figure 4B–D), error bars indicate standard error of the mean of triplicate assays.
Supplementary Material
Figure S1. Alignment of Scap Orthologs and Functional Analysis of Chicken Scap and Chicken Insig-1, Related to Figure 1
(A) Alignment of chicken Scap (Uniprot A0A3Q3ANV4, residues 1–758), human Scap (Uniprot Q12770, residues 1–729) and hamster Scap (Uniprot P97260, residues 1–729). Sequences aligned with Clustal Omega and visualized with ESPript 3.0 web server. Identical residues indicated with bold text and grey shading. Key residues labeled with colors to match main text figures. TM helix boundaries are indicated with boxes. Boundaries for the L1 and L7 structures are indicated with green and magenta lines, respectively. Red line denotes disulfide bond between C147 and C169, also indicated in red type. Green residues denote functionally important amino acids in L1. Yellow residues denote amino acids important for interaction with Insigs. Blue residue denotes an aspartic acid that when mutated to alanine or valine traps Scap in an Insig-binding conformation. Purple box denotes MELADL motif required for COPII binding. Magenta residues denote functionally important amino acids in L7. Glycan denotes N-linked sugar visible in reconstruction.
(B) Ability of cScap constructs to stabilize and transport human SREBP2. Scap-null hamster SRD-13A cells were set up and transfected as described in Methods with 4 μg pcDNA control plasmid (lane 1) or 2 μg pTK-SREBP2 and 2 μg WT or mutant pTK-cScap-3xFLAG as indicated. The total amount of transfected DNA for all conditions was adjusted to 4 μg with pcDNA control plasmid. The next day, cells were depleted of cholesterol for 1 hr, harvested, and subjected to SDS-PAGE followed by immunoblot analysis of SREBP2 (IgG-22D5), cScap (anti-FLAG), and actin (anti-actin) as described in Methods. P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2; hSREBP2, human SREBP2.
(C) Co-immunoprecipitation of human SREBP2 with human Scap and cScap. HEK 293T cells were set up and transfected as described in Methods with 6 μg pcDNA control plasmid (lane 1) or 3 μg pEZT-Myc-SREBP2(cleavage resistant) and 3 μg of the indicated species of pEZT-Scap-3xFLAG, as indicated. The total amount of transfected DNA in all dishes was adjusted to 6 μg with pcDNA control plasmid. The following day, FLAG-tagged Scap was precipitated using M2 anti-FLAG resin and eluted with 3xFLAG peptide as described in Methods. A portion of the solubilized input sample (5%) was withheld for immunoblot analysis. Input samples (0.75% of total) and eluate samples (7.5% of total) were subjected to SDS-PAGE followed by immunoblot of Scap (anti-FLAG) and SREBP2 (IgG-22D5). CR, cleavage resistant; hScap, human Scap.
(D) Schematic of SREBP2 and Scap highlighting the interaction between their CTDs.
(E) His-tagged cScap CTD (residues 779–1323) and 3xFLAG-tagged human SREBP2 CTD (residues 793–1068) were co-expressed in Sf9 cells and purified as described in Methods. A representative gel filtration purification trace is shown. Bracket indicates the elution volume of the purified complex, the homogeneity of which was assessed by Coomassie staining after 10% SDS-PAGE (inset).
(F) Ability of cInsig-1 to regulate transport of cScap. Scap-null hamster SRD-13A cells were set up and transfected as described in Methods with 5 μg pcDNA control plasmid (lane 1) or 1 μg pEZT-human SREBP2, 2 μg pEZT-cInsig-1 and 2 μg WT or mutant pEZT-cScap-3xFLAG as indicated. The total amount of transfected DNA for all conditions was adjusted to 5 μg with pcDNA control plasmid. The next day, cells were depleted of cholesterol for 1 hr and then switched into media with 2.5 μM 25-HC for three hours, as described in Methods. Following this incubation, cells were harvested and subjected to SDS-PAGE followed by immunoblot analysis of SREBP2 (IgG-22D5), cScap (anti-FLAG), cInsig-1 (IgG-17H1) and actin (anti-actin) as described in Methods. P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2. hSREBP2, human SREBP2.
Figure S2. Generation and Characterization of a Scap-binding Antibody (4G10), Related to Figure 2
(A) Representative purification of cScapD435V/cInsig-1 complex in GDN micelles for animal immunizations. cScapD435V/cInsig-1 complexes were expressed in HEK GnTI− cells, solubilized in LMNG micelles, exchanged into GDN micelles, affinity-purified, and subjected to gel filtration over a Superose 6 Increase 10/300 column as described in Methods. The cScapD435V/cInsig-1 complex elutes as a symmetric peak with a slight left shoulder. Inset: SDS-PAGE sample demonstrating biochemical purity of the sample.
(B) Representative purification of biotinylated cScapD435V/cInsig-1 complex reconstituted in Saposin-lipid nanodiscs for ELISA screening. 1.9 mg of cScapD435V/cInsig-1 complex purified in LMNG micelles was biotinylated and subjected to reconstitution into detergent-free lipid nanodiscs as described in Methods. Inset: SDS-PAGE sample demonstrating biochemical purity of cScapD435V and cInsig-1 in the nanodiscs.
(C) Amino acid sequences obtained for the Fab domain of IgG-4G10.
(D) Immunoblot analysis of the mAb IgG-4G10. 100 ng of purified cScapD435V/cInsig-1 complex was denatured with SDS loading buffer and subjected to SDS-PAGE followed by immunoblot analysis using 1 μg/mL of either M2 anti-FLAG antibody or the IgG 4G10 antibody. In contrast to anti-FLAG, IgG 4G10 does not detect denatured cScapD435V.
(E) 4G10Fab binds cScapD435V/cInsig-1 complex. cScapD435V/cInsig-1 complexes were purified as described in Methods. 500 μg of this purified protein was mixed with 320 μg 4G10Fab for 30 min on ice and then subjected to analytical gel filtration over a Superose 6 Increase 10/300 as described in Methods. Binding to the Fab results in a left-shift of about 0.5 mL relative to cScapD435V/Insig-1 complex alone. Unbound 4G10Fab elutes after the cScapD435V/cInsig-1 complex. Bracket indicates fractions selected for SDS-PAGE analysis in (F).
(F) Coomassie staining of gel filtration fractions for the cScapD435V/cInsig-1/4G10Fab trace in (E) after 4–16% gradient SDS-PAGE.
Figure S3. Cryo-EM Analysis of cScap/4G10Fab, Related to Figure 2
(A) Representative electron micrograph and 2D class averages of cScap/4G10Fab, showing clear densities for Fab as well as L1-L7, TM domain, and CTD of cScap.
(B) Gold-standard Fourier shell correlation (FSC) curve for the cryo-EM maps shown in Figure 2E and 3A. FSC curve for the full cScap/4G10Fab reconstruction is blue, and FSC curve for the L1-L7/4G10Fab focused refinement is red.
(C) Cryo-EM map of entire cScap/4G10Fab complex colored by local resolution.
(D) Focused cryo-EM map of L1-L7 domain of cScap and part of 4G10Fab colored by local resolution.
(E) Euler angle distribution of particles used in the final 3D reconstructions.
(F) Flowchart of cryo-EM data processing.
Figure S4. L1-L7 Density and Model Building, Related to Figure 3
(A) Representative EM densities for the indicated segments of cScap L1 and L7. The density contour of the map within 2.5 Å of the selected residues of the 2.9Å WT L1-L7 map was set at level 0.0684 in Chimera. L1 residues are colored green, L7 residues are colored purple. Residues labeled with ovals were subjected to mutagenesis studies (Figure 3F).
(B) Left: Secondary structure diagram of cScap L1-L7 domain depicted using Topdraw. The conserved MLD fold is highlighted with red box. L1 is depicted in green, L7 is depicted in magenta. Brown line indicates the disulfide bridge. Yellow star indicates β-strand of L7 that contributes to the core β-sheet. Right: Secondary structure diagram of MLD domain of NPC1 (residues 400–607) depicted using Topdraw. The conserved structural elements between L1-L7 and the NPC1 MLD are highlighted with a red box. Brown lines indicate the two disulfide bridges. Secondary structure assignment is based on PDB 5KWY.
Figure S5. Biochemical Characterization of 4G10Fab/L1-L7 Interaction and Disulfide bond in L1, Related to Figures 2 and 3
(A) Structural details of the L1-L7/4G10Fab interaction. L1-L7 domain is shown as cartoon with L1 in green and L7 in magenta. The solvent-accessible electrostatic surface of 4G10Fab (generated using the APSB Pymol plugin) is shown colored with a range of −3 kT/e (red) to +3 kT/e (blue). Inset: closeup of the 4G10Fab/L1-L7 interaction. Residues in L1 with sidechains that make interactions with the 4G10Fab (within 5Å) are shown as sticks.
(B) Schematic for the immunoprecipitation of cScap by 4G10Fab.
(C) Immunoprecipitation of wild-type and mutant cScap by 4G10Fab. HEK 293T cells were set up and transfected as described in Methods with 6 μg pEZT-cScap-3xFLAG WT or pEZT-cScap-3xFLAG R135E D136K plasmids. The next day, the cells were harvested and solubilized in GDN detergent as described in Methods. Supernatants were subjected to immunoprecipitation by anti-FLAG M2 resin and eluted cScap proteins were then subjected to immunoprecipitation by 4G10Fab bound to protein L resin as described in Methods. The final eluates were subjected to 10% SDS-PAGE followed by immunoblot analysis of cScap (M2 anti-FLAG-HRP).
(D) SREBP2 cleavage assay. Scap-deficient SRD-13A cells were set up and transfected as described in Methods with 6 μg pcDNA control plasmid (lane 1) or else 2 μg pEZT-Myc-SREBP2 together with 4μg of either pEZT-cScap-3xFLAG WT or pEZT-cScap-3xFLAG R135E D136K. The next day, cells were depleted of cholesterol for 1 hr, harvested, and subjected to SDS-PAGE followed by immunoblot analysis of SREBP2 (IgG-22D5), cScap (anti-FLAG), and actin (anti-actin) as described in Methods. P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2.
(E) Structure of cScap L1-L7 showing the location of the disulfide bond between C147 and C169 of L1. Inset: Continuous density for residues C147 and C169 showing the disulfide bond. Density within 2 Å of the two residues was contoured at sigma level 5 in Pymol.
(F) Schematic of recombinant hamster Scap(L1-L7) fusion that is secreted by CHO-K1 cells. Starting at the NH2-terminus, the construct contains the honeybee melittin signal sequence (brown), followed by a His12 epitope tag (blue), hamster Scap-Loop 1 (aa 46–284) containing three cysteines at the indicated locations, a 15 amino acid linker (orange), and hamster Scap-Loop 7 (aa 535–710). L7 does not contain any cysteines.
(G) Secretion of wild-type (WT) and cysteine mutants of hamster Scap(L1-L7). CHO-K1 cells were set up and transfected as described in Methods with 4 μg of either wildtype pCMV-Scap(L1-L7) plasmid containing all three cysteines (CCC) or mutant versions where cysteines had been individually mutated – C147A (ACC), C169S (CSC), C264A (CCA). 48 hr after transfection, cells were harvested, and media was collected. Aliquots of media (1.5% of total) were treated with 6 mM mPEG-MAL-5000 to modify free and exposed cysteines as described in Methods. Equivalent fractions of cells and media were then subjected to immunoblot analysis with 5 μg/ml anti-L7 (IgG 9D5).
(H) Analysis of cysteine-modified forms of hamster Scap(L1-L7) (C264A). Media from cells transfected with mutant pCMV-Scap(L1-L7) (C264A) in (C) was collected and incubated at the indicated temperatures in the absence or presence of 1 mM DTT for 15 min. The media was then treated without or with 6 mM mPEG-MAL-5000 as indicated to modify free and exposed cysteines as described in Methods, after which the samples were subjected to immunoblot analysis with 5 μg/ml anti-L7 (IgG 9D5). RT, room temperature.
(I) The C147-C169 disulfide bond is essential for Scap transport of SREBP2. Scap-deficient SRD-13A cells were set up and transfected as described in Methods with 4 μg pcDNA control plasmid (lane 1) or 2 μg pTK-SREBP2 and 2 μg WT or mutant pTK-Scap as indicated. The total amount of transfected DNA for all conditions was adjusted to 4 μg with pcDNA control plasmid. The next day, cells were depleted of cholesterol for 1 hr, harvested, and subjected to 10% SDS-PAGE followed by immunoblot analysis of SREBP2 (IgG-22D5), Scap (IgG-4H4), and actin (anti-actin) as described in Methods. P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2.
Figure S6 – Cryo-EM Analysis of cScapD435V/cInsig-1/4G10Fab, Related to Figure 4
(A) Representative electron micrograph and 2D class averages of cScapD435V/cInsig-1/4G10Fab, showing clear densities for Fab, L1-L7 domain of cScap, and TM domains of both cScap and cInsig-1.
(B) Gold-standard Fourier shell correlation curve for the cryo-EM map shown in Figure 4E and Figure S6D. FSC curve for the full cScapD435V/cInsig-1/4G10Fab reconstruction is blue, and FSC curve for the L1-L7/4G10Fab focused refinement is red.
(C) Cryo-EM map of entire cScapD435V/cInsig-1/4G10Fab complex colored by local resolution.
(D) Focused cryo-EM map of L1-L7 domain of cScapD435V and part of 4G10Fab colored by local resolution.
(E) Euler angle distribution of particles used in the final 3D reconstructions.
(F) Flowchart of cryo-EM data processing.
Figure S7. Orientation of Loop Structures in cScap and Other SSD-containing Proteins, Related to Figure 7
The SSDs of cScap (green for WT, magenta for D435V) were aligned with the SSDs of NPC1 (yellow, PDB 6V3H), Patched (orange, PDB 6OEU), Dispatched (cyan, PDB 6TBU), and NPC1-L1 (tan, PDB 6W5U). For clarity, the NTD-TM1 regions of NPC1 (residues 1–291) and NPC1-L1 (residues 1–311) were omitted. In both forms of cScap, the L1-L7 is rotated such that the β-sheet is roughly parallel to the membrane. In the other SSD-containing proteins, the middle luminal domain (MLD)-related subunits are angled downward such that their core β-sheets are roughly perpendicular to the membrane. Grey cartoons denote simplified orientations of SSDs and structures related to NPC1’s MLD. Other than Scap, these SSD-containing proteins all have two pseudo-symmetrically interacting ‘SSD-MLD’ units on the same polypeptide.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal Anti-FLAG M2 clone | Sigma-Aldrich | F1804; RRID:AB_262044 |
| Mouse monoclonal Anti-FLAG M2-Peroxidase | Sigma-Aldrich | A8592; RRID:AB_439702 |
| Rabbit monoclonal Anti-Actin | Sigma-Aldrich | A2066; RRID:AB_476693 |
| Rabbit monoclonal Anti-SREBP2 | (McFarlane et al., 2015) | IgG-22D5 |
| Mouse monoclonal Anti-Scap L7 | (Sakai et al., 1997) | IgG-9D5 |
| Mouse monoclonal Anti-Insig | This paper | IgG-17H1 |
| Bacterial and Virus Strains | ||
| XL10-Gold Ultracompetent cells | Agilent | Cat# 200317 |
| MAX Efficiency DH10Bac Competent cells | ThermoFisher | Cat# 10361012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) | Avanti | Cat# 850457 |
| 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) | Avanti | Cat# 850757P |
| 2-Hydroxypropyl-β-cyclodextrin (HPCD) | Cyclodextrin Technologies Development | Cat# THPB-P |
| 25-Hydroxycholesterol | Steraloids | Cat# C6550–000 |
| [3H]Cholesterol | American Radiolabeled Chemicals | Cat# ART 2133 |
| [3H]25-hydroxycholesterol | American Radiolabeled Chemicals | Cat# ART 0766 |
| [3H]Lanosterol | American Radiolabeled Chemicals | Cat# ART 0546A |
| 3x-FLAG peptide | APExBio | Cat# A6001 |
| 5-Cholesten-3ß-OL Hemmisuccinate (CHS) | Steraloids | Cat# C6823–000; CAS 1510-21-0 |
| Anti-FLAG M2 affinity resin | Sigma-Aldrich | Cat# A2220 |
| Benzamidine hydrochloride monohydrate | Goldbio | Cat# B-050–250; CAS 1670-14-0 |
| Benzonase | EMDMillipore | Cat# 70746–3 |
| Bio-Beads SM-2 Absorbent Media | BIO-RAD | Cat# 1523920 |
| Cholesterol | Sigma | Cat# C8667–5g |
| E64 | Goldbio | Cat# E-064–100; CAS 66701-25-5 |
| EZ-Link NHS-LC-LC-Biotin | ThermoFisher | Cat# 21343 |
| Fetal Calf Serum | Sigma-Aldrich | F2442–500ML |
| FLAG peptide | Biomatik | Custom order |
| FuGENE6 transfection reagent | Promega | Cat# E2691 |
| Glyco-diosgenin (GDN) | Anatrace | Cat# GDN101; CAS 1402423-29-3 |
| Iodoacetamide | Sigma-Aldrich | Cat# I1149 |
| Lanosterol | Sigma Aldrich | Cat# L5768 |
| Lauryl Maltose Neopentyl Glycol (LMNG) | Anatrace | Cat# NG310; CAS 1257852-96-2 |
| Leupeptin hemisulfate | Goldbio | Cat# L-010–100; CAS 103476-89-7 |
| mPEG maleimide, MW 5000 | Nanocs Inc. | Cat.# PG1-ML-5k-1 |
| NEBuilder HiFI DNA Assembly | NEB | Cat# E2621L |
| Newborn calf lipoprotein-deficient serum (LPDS) | (Goldstein et al., 1983) | N/A |
| Ni-NTA agarose | Qiagen | Cat# 30250 |
| Papain | Worthington | Cat# LS003119 |
| Phenylmethylsulfonylfluoride (PMSF) | Goldbio | Cat# P-470–25; CAS 329-98-6 |
| Protease Inhibitor Cocktail Set III, EDTA-Free | Calbiochem | Cat# 539134 |
| Protein L agarose | Pierce | Cat# 20510 |
| Sodium butyrate | Sigma-Aldrich | Cat# 303410; CAS 156-54-7 |
| Sodium cholate | Sigma-Aldrich | Cat# C1254; CAS 206986-87-0 |
| Sodium compactin | (Brown et al., 1978) | N/A |
| Sodium mevalonate | (Brown et al., 1978) | N/A |
| Sodium oleate | (Goldstein et al., 1983) | N/A |
| Tris (2-Carboxyethyl) phosphine Hydrochloride (TCEP- HCl) | Goldbio | CAT# TCEP1 CAS 51805- 45-9 |
| X-tremeGENE HP DNA transfection reagent | Millipore Sigma | Cat# XTGHP-RO |
| Critical Commercial Assays | ||
| RNEAsy Mini kit | Qiagen | Cat# 74106 |
| Superscript III First Strand kit | Invitrogen | Cat# 18080–51 |
| GoTaq Polymerase kit | Promega | M3005 |
| Deposited Data | ||
| PDB model of cScap L1-L7/4G10Fab | This paper | PDB 7LKF |
| PDB model of cScapD435V L1-L7/4G10Fab | This paper | PDB 7LKH |
| Focused refinement map of cScap L1-L7/4G10Fab | This paper | EMD-23405 |
| Focused refinement map of cScapD435V L1-L7/4G10Fab | This paper | EMD-23408 |
| Global map of cScap L1-L7/4G10Fab | This paper | EMD-23406 |
| Global map of cScap L1-L7/4G10Fab | This paper | EMD-23407 |
| PDB for starting template of Fab | (Ebenhoch et al., 2019) | PDB 6SVL |
| PDB for starting template of Fab | (Hennen et al., 2016) | PDB 5E94 |
| PDB template for Scap TM homology model | (Li et al., 2016b) | PDB 5I31 |
| PDB template for Insig homology model | (Ren et al., 2015) | PDB 4XU4 |
| Experimental Models: Cell Lines | ||
| Human: HEK 293s GnTI− cells | ATCC | CRL-3022 |
| Human: HEK 293T cells | ATCC | CRL-3216 |
| Hamster: Scap-deficient SRD-13A cells | (Rawson et al., 1999) | N/A |
| Hamster: CHO K1 | ATCC | CCL-61 |
| Spodoptera frugiperda: Sf9 cells | Invitrogen | Cat# 11496015 |
| Oligonucleotides | ||
| Primers for Fab sequencing are listed in Table S3 | This paper | N/A |
| Recombinant DNA Plasmids | ||
| pEZT-Chicken Scap (D435V)-3xFLAG | This paper | N/A |
| pEZT-Chicken Scap (WT)-3xFLAG | This paper | N/A |
| pEZT-Chicken Scap (R135E D136K)-3xFLAG | This paper | N/A |
| pEZT-His8-Chicken Scap-3xFLAG D435V | This paper | N/A |
| pEZT-His8-Chicken Scap-3xFLAG WT | This paper | N/A |
| pEZT-Chicken Insig-1 | This paper | N/A |
| pEZT-Chicken Insig-1–2A-Scap (D435V)-3xFLAG | This paper | N/A |
| pEZT-Human Scap WT | This paper | N/A |
| pEZT-Myc-Human SREBP2 (CR) (D468A, S480A, R481S, and R519A) | This paper | N/A |
| pTK-HSV-Human SREBP2 | (Hua et al., 1996b) | N/A |
| pTK-Chicken Scap (D435V)-3xFLAG | This paper | N/A |
| pTK-Chicken Scap (WT)-3xFLAG | This paper | N/A |
| pTK-Hamster Scap | (Hua et al., 1996a) | N/A |
| pTK-Hamster Scap Y234A | (Motamed et al., 2011) | N/A |
| pTK-Hamster Scap Y640S | (Zhang et al., 2013) | N/A |
| pTK-Hamster Scap C147A | This paper | N/A |
| pTK-Hamster Scap C169S | This paper | N/A |
| pTK-Hamster Scap C264A | This paper | N/A |
| pTK-Hamster Scap Q94E | This paper | N/A |
| pTK-Hamster Scap I96A | This paper | N/A |
| pTK-Hamster Scap L125W | This paper | N/A |
| pTK-Hamster Scap R135W | This paper | N/A |
| pTK-Hamster Scap V150W | This paper | N/A |
| pTK-Hamster Scap K246W | This paper | N/A |
| pTK-Hamster Scap L636W | This paper | N/A |
| pTK-Hamster Scap I655A | This paper | N/A |
| pFastBac His10 Hamster(L1-L7) | (Zhang et al., 2016) | N/A |
| pFastBac His10 Hamster(L1-L7) C147A | This paper | N/A |
| pFastBac His10 Hamster(L1-L7) C169S | This paper | N/A |
| pFastBac His10 Hamster(L1-L7) C246A | This paper | N/A |
| pFastBac-Chicken (L1-L7)-His10 | This paper | N/A |
| pFastBac-His10-Chicken Scap CTD | This paper | N/A |
| pFastBac-3xFLAG-Human SREBP2 CTD | (Kober et al., 2020) | N/A |
| Software and Algorithms | ||
| PyMOL (2.3.5) | Schrödinger | https://pymol.org/2/ |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| ISOLDE | (Croll, 2018) | https://isolde.cimr.cam.ac.uk/ |
| SWISS-MODEL | (Waterhouse et al., 2018) | https://swissmodel.expasy.org/ |
| Relion 3.1 | (Zivanov et al., 2018) | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| Phenix (1.18.2–3874) | (Liebschner et al., 2019) | https://www.phenix-online.org/download/ |
| DALI server | (Holm, 2020) | http://ekhidna2.biocenter.helsinki.fi/dali/ |
| PROMALS3D server | (Pei et al., 2008) | http://prodata.swmed.edu/promals3d/promals3d.php |
| Coot | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| IMGT database | (Giudicelli et al., 2011) | http://www.imgt.org/ |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
Cryo-EM structures of wild-type Scap and a mutant Scap bound to Insig
Scap’s two ER luminal loops L1 and L7 intertwine tightly to form a globular domain
Scap’s binding to Insig triggers a large rotation of the luminal L1-L7 domain
L1-L7 rotation suggests how cholesterol blocks Scap’s transport of SREBPs
Acknowledgements
We are grateful to Linda Donnelly and Angela Carroll for antibody production; Lisa Beatty, Karen Chapman, Ije Dukes, Bilkish Bajaj, and Camille Harry for assistance with cell culture and baculovirus production; Yinxin Zhang and Kwang-min Lee for characterization of the disulfide bond in Scap; Shimeng Xu for assistance with transfection experiments. This project was supported by the National Institutes of Health (R35GM116387 to D.M.R.; P01HL20948 to A.R., M.S.B., and J.L.G.; R01GM136976 to X.B), the Welch Foundation (I-1770 to D.M.R.; I-1793 to A.R.; I-1944 to X.B), Mallinckrodt Foundation Scholar Award (to D.M.R.), Fondation Leducq (19CVD04 to A.R.) and CPRIT (RR160082 to X.B.). D.L.K. is a recipient of a postdoctoral fellowship from the American Heart Association (18POST34080141). Cryo-EM data were collected at the University of Texas Southwestern Medical Center Cryo-EM Facility, which is funded by the CPRIT Core Facility Support Award RP170644.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, and Goldstein JL (2004). Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem 279, 52772–52780. [DOI] [PubMed] [Google Scholar]
- Bai X-C, Rajendra E, Yang G, Shi Y, and Scheres SHW (2015). Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife 4, 1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Faust JR, Goldstein JL, Kaneko I, and Endo A (1978). Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J Biol Chem 253, 1121–1128. [PubMed] [Google Scholar]
- Brown MS, Radhakrishnan A, and Goldstein JL (2018). Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem 87, 783–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll TI (2018). ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D Struct Biol 74, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebenhoch R, Akhdar A, Reboll MR, Korf-Klingebiel M, Gupta P, Armstrong J, Huang Y, Frego L, Rybina I, Miglietta J, Pekcec A, Wollert KC, Nar H (2019). Crystal structure and receptor-interacting residues of MYDGF - a protein mediating ischemic tissue repair. Nat Commun 10, 5379–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhou Y, Goldstein JL, Brown MS, and Radhakrishnan A (2017). Cholesterol-induced conformational changes in the sterol-sensing domain of the Scap protein suggest feedback mechanism to control cholesterol synthesis. Journal of Biological Chemistry 292, 8729–8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli V, Brochet X, and Lefranc M-P (2011). IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc 2011, 695–715. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Basu SK, and Brown MS (1983). Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Meth Enzymol 98, 241–260. [DOI] [PubMed] [Google Scholar]
- Gong Y, Lee JN, Brown MS, Goldstein JL, and Ye J (2006). Juxtamembranous aspartic acid in Insig-1 and Insig-2 is required for cholesterol homeostasis. Proc Natl Acad Sci USA 103, 6154–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennen S, Kodra JT, Soroka V, Krogh BO, Wu X, Kaastrup P, Ørskov C, Rønn SG, Schluckebier G, Barbateskovic S, Gandhi PS, Reedtz-Runge S (2016). Structural insight into antibody-mediated antagonism of the Glucagon-like peptide-1 Receptor. Sci Rep 6, 26236–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L (2020). DALI and the persistence of protein shape. Protein Sci. 29, 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, and Brown MS (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Nohturfft A, Goldstein JL, and Brown MS (1996a). Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell 87, 415–426. [DOI] [PubMed] [Google Scholar]
- Hua X, Sakai J, Brown MS, and Goldstein JL (1996b). Regulated cleavage of sterol regulatory element binding proteins requires sequences on both sides of the endoplasmic reticulum membrane. J Biol Chem 271, 10379–10384. [DOI] [PubMed] [Google Scholar]
- Infante RE, Abi-Mosleh L, Radhakrishnan A, Dale JD, Brown MS, and Goldstein JL (2008a). Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J Biol Chem 283, 1052–1063. [DOI] [PubMed] [Google Scholar]
- Infante RE, Radhakrishnan A, Abi-Mosleh L, Kinch LN, Wang ML, Grishin NV, Goldstein JL, and Brown MS (2008b). Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J Biol Chem 283, 1064–1075. [DOI] [PubMed] [Google Scholar]
- Kober DL, Xu S, Li S, Bajaj B, Liang G, Rosenbaum DM, and Radhakrishnan A (2020). Identification of a degradation signal at the carboxy terminus of SREBP2: A new role for this domain in cholesterol homeostasis. Proceedings of the National Academy of Sciences 117, 28080–28091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara PE, and Labouesse M (2002). The sterol-sensing domain: multiple families, a unique role? Trends Genet 18, 193–201. [DOI] [PubMed] [Google Scholar]
- Lee PCW, Sever N, and DeBose-Boyd RA (2005). Isolation of sterol-resistant Chinese hamster ovary cells with genetic deficiencies in both Insig-1 and Insig-2. J Biol Chem 280, 25242–25249. [DOI] [PubMed] [Google Scholar]
- Li X, Saha P, Li J, Blobel G, and Pfeffer SR (2016a). Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proceedings of the National Academy of Sciences 113, 10079–10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang J, Coutavas E, Shi H, Hao Q, and Blobel G (2016b). Structure of human Niemann-Pick C1 protein. Proceedings of the National Academy of Sciences 113, 8212–8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung L-W, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, Wang L, Vaseghi HR, Qian L, and Liu J (2017). Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci Rep 7, 2193–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane MR, Cantoria MJ, Linden AG, January BA, Liang G, and Engelking LJ (2015). Scap is required for sterol synthesis and crypt growth in intestinal mucosa. J Lipid Res 56, 1560–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Perez CL, Noviello CM, and Hibbs RE (2016). Manipulation of Subunit Stoichiometry in Heteromeric Membrane Proteins. Structure 24, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motamed M, Zhang Y, Wang ML, Seemann J, Kwon HJ, Goldstein JL, and Brown MS (2011). Identification of luminal loop 1 of scap as the sterol sensor that maintains cholesterol homeostasis. Journal of Biological Chemistry 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohturfft A, Brown MS, and Goldstein JL (1998a). Topology of SREBP cleavage-activating protein, a polytopic membrane protein with a sterol-sensing domain. J Biol Chem 273, 17243–17250. [DOI] [PubMed] [Google Scholar]
- Nohturfft A, Brown MS, and Goldstein JL (1998b). Sterols regulate processing of carbohydrate chains of wild-type SREBP cleavage-activating protein (SCAP), but not sterol-resistant mutants Y298C or D443N. Proc Natl Acad Sci USA 95, 12848–12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A, Goldstein JL, McDonald JG, and Brown MS (2008). Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab 8, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, and Goldstein JL (2007). Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci USA 104, 6511–6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Sun L-P, Kwon HJ, Brown MS, and Goldstein JL (2004). Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol Cell 15, 259–268. [DOI] [PubMed] [Google Scholar]
- Rawson RB, DeBose-Boyd R, Goldstein JL, and Brown MS (1999). Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J Biol Chem 274, 28549–28556. [DOI] [PubMed] [Google Scholar]
- Ren R, Zhou X, He Y, Ke M, Wu J, Liu X, Yan C, Wu Y, Gong X, Lei X, et al. (2015). Crystal structure of a mycobacterial Insig homolog provides insight into how these sensors monitor sterol levels. Science 349, 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, and Goldstein JL (1996). Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Sakai J, Nohturfft A, Cheng D, Ho YK, Brown MS, and Goldstein JL (1997). Identification of complexes between the COOH-terminal domains of sterol regulatory element-binding proteins (SREBPs) and SREBP cleavage-activating protein. J Biol Chem 272, 20213–20221. [DOI] [PubMed] [Google Scholar]
- Sun L-P, Li L, Goldstein JL, and Brown MS (2005). Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem 280, 26483–26490. [DOI] [PubMed] [Google Scholar]
- Sun L-P, Seemann J, Goldstein JL, and Brown MS (2007). Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci USA 104, 6519–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zelenski NG, Yang J, Sakai J, Brown MS, and Goldstein JL (1996). Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. Embo J 15, 1012–1020. [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe D, Xia Z-P, Adams CM, and Rawson RB (2002). Three mutations in sterol-sensing domain of SCAP block interaction with insig and render SREBP cleavage insensitive to sterols. Proc Natl Acad Sci USA 99, 16672–16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Cao P, Song W, Qian H, Du X, Coates HW, Zhao X, Li Y, Gao S, Gong X, et al. (2021). A structure of human Scap bound to Insig-2 suggests how their interaction is regulated by sterols. Science 263, eabb2224. [DOI] [PubMed] [Google Scholar]
- Yang T, Goldstein JL, and Brown MS (2000). Overexpression of membrane domain of SCAP prevents sterols from inhibiting SCAP.SREBP exit from endoplasmic reticulum. J Biol Chem 275, 29881–29886. [DOI] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, and Brown MS (2002). Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500. [DOI] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lee KM, Kinch LN, Clark L, Grishin NV, Rosenbaum DM, Brown MS, Goldstein JL, and Radhakrishnan A (2016). Direct demonstration that Loop1 of Scap binds to Loop7: A crucial event in cholesterol homeostasis. Journal of Biological Chemistry 291, 12888–12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Motamed M, Seemann J, Brown MS, and Goldstein JL (2013). Point mutation in luminal loop 7 of Scap protein blocks interaction with loop 1 and abolishes movement to Golgi. Journal of Biological Chemistry 288, 14059–14067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Meth 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Alignment of Scap Orthologs and Functional Analysis of Chicken Scap and Chicken Insig-1, Related to Figure 1
(A) Alignment of chicken Scap (Uniprot A0A3Q3ANV4, residues 1–758), human Scap (Uniprot Q12770, residues 1–729) and hamster Scap (Uniprot P97260, residues 1–729). Sequences aligned with Clustal Omega and visualized with ESPript 3.0 web server. Identical residues indicated with bold text and grey shading. Key residues labeled with colors to match main text figures. TM helix boundaries are indicated with boxes. Boundaries for the L1 and L7 structures are indicated with green and magenta lines, respectively. Red line denotes disulfide bond between C147 and C169, also indicated in red type. Green residues denote functionally important amino acids in L1. Yellow residues denote amino acids important for interaction with Insigs. Blue residue denotes an aspartic acid that when mutated to alanine or valine traps Scap in an Insig-binding conformation. Purple box denotes MELADL motif required for COPII binding. Magenta residues denote functionally important amino acids in L7. Glycan denotes N-linked sugar visible in reconstruction.
(B) Ability of cScap constructs to stabilize and transport human SREBP2. Scap-null hamster SRD-13A cells were set up and transfected as described in Methods with 4 μg pcDNA control plasmid (lane 1) or 2 μg pTK-SREBP2 and 2 μg WT or mutant pTK-cScap-3xFLAG as indicated. The total amount of transfected DNA for all conditions was adjusted to 4 μg with pcDNA control plasmid. The next day, cells were depleted of cholesterol for 1 hr, harvested, and subjected to SDS-PAGE followed by immunoblot analysis of SREBP2 (IgG-22D5), cScap (anti-FLAG), and actin (anti-actin) as described in Methods. P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2; hSREBP2, human SREBP2.
(C) Co-immunoprecipitation of human SREBP2 with human Scap and cScap. HEK 293T cells were set up and transfected as described in Methods with 6 μg pcDNA control plasmid (lane 1) or 3 μg pEZT-Myc-SREBP2(cleavage resistant) and 3 μg of the indicated species of pEZT-Scap-3xFLAG, as indicated. The total amount of transfected DNA in all dishes was adjusted to 6 μg with pcDNA control plasmid. The following day, FLAG-tagged Scap was precipitated using M2 anti-FLAG resin and eluted with 3xFLAG peptide as described in Methods. A portion of the solubilized input sample (5%) was withheld for immunoblot analysis. Input samples (0.75% of total) and eluate samples (7.5% of total) were subjected to SDS-PAGE followed by immunoblot of Scap (anti-FLAG) and SREBP2 (IgG-22D5). CR, cleavage resistant; hScap, human Scap.
(D) Schematic of SREBP2 and Scap highlighting the interaction between their CTDs.
(E) His-tagged cScap CTD (residues 779–1323) and 3xFLAG-tagged human SREBP2 CTD (residues 793–1068) were co-expressed in Sf9 cells and purified as described in Methods. A representative gel filtration purification trace is shown. Bracket indicates the elution volume of the purified complex, the homogeneity of which was assessed by Coomassie staining after 10% SDS-PAGE (inset).
(F) Ability of cInsig-1 to regulate transport of cScap. Scap-null hamster SRD-13A cells were set up and transfected as described in Methods with 5 μg pcDNA control plasmid (lane 1) or 1 μg pEZT-human SREBP2, 2 μg pEZT-cInsig-1 and 2 μg WT or mutant pEZT-cScap-3xFLAG as indicated. The total amount of transfected DNA for all conditions was adjusted to 5 μg with pcDNA control plasmid. The next day, cells were depleted of cholesterol for 1 hr and then switched into media with 2.5 μM 25-HC for three hours, as described in Methods. Following this incubation, cells were harvested and subjected to SDS-PAGE followed by immunoblot analysis of SREBP2 (IgG-22D5), cScap (anti-FLAG), cInsig-1 (IgG-17H1) and actin (anti-actin) as described in Methods. P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2. hSREBP2, human SREBP2.
Figure S2. Generation and Characterization of a Scap-binding Antibody (4G10), Related to Figure 2
(A) Representative purification of cScapD435V/cInsig-1 complex in GDN micelles for animal immunizations. cScapD435V/cInsig-1 complexes were expressed in HEK GnTI− cells, solubilized in LMNG micelles, exchanged into GDN micelles, affinity-purified, and subjected to gel filtration over a Superose 6 Increase 10/300 column as described in Methods. The cScapD435V/cInsig-1 complex elutes as a symmetric peak with a slight left shoulder. Inset: SDS-PAGE sample demonstrating biochemical purity of the sample.
(B) Representative purification of biotinylated cScapD435V/cInsig-1 complex reconstituted in Saposin-lipid nanodiscs for ELISA screening. 1.9 mg of cScapD435V/cInsig-1 complex purified in LMNG micelles was biotinylated and subjected to reconstitution into detergent-free lipid nanodiscs as described in Methods. Inset: SDS-PAGE sample demonstrating biochemical purity of cScapD435V and cInsig-1 in the nanodiscs.
(C) Amino acid sequences obtained for the Fab domain of IgG-4G10.
(D) Immunoblot analysis of the mAb IgG-4G10. 100 ng of purified cScapD435V/cInsig-1 complex was denatured with SDS loading buffer and subjected to SDS-PAGE followed by immunoblot analysis using 1 μg/mL of either M2 anti-FLAG antibody or the IgG 4G10 antibody. In contrast to anti-FLAG, IgG 4G10 does not detect denatured cScapD435V.
(E) 4G10Fab binds cScapD435V/cInsig-1 complex. cScapD435V/cInsig-1 complexes were purified as described in Methods. 500 μg of this purified protein was mixed with 320 μg 4G10Fab for 30 min on ice and then subjected to analytical gel filtration over a Superose 6 Increase 10/300 as described in Methods. Binding to the Fab results in a left-shift of about 0.5 mL relative to cScapD435V/Insig-1 complex alone. Unbound 4G10Fab elutes after the cScapD435V/cInsig-1 complex. Bracket indicates fractions selected for SDS-PAGE analysis in (F).
(F) Coomassie staining of gel filtration fractions for the cScapD435V/cInsig-1/4G10Fab trace in (E) after 4–16% gradient SDS-PAGE.
Figure S3. Cryo-EM Analysis of cScap/4G10Fab, Related to Figure 2
(A) Representative electron micrograph and 2D class averages of cScap/4G10Fab, showing clear densities for Fab as well as L1-L7, TM domain, and CTD of cScap.
(B) Gold-standard Fourier shell correlation (FSC) curve for the cryo-EM maps shown in Figure 2E and 3A. FSC curve for the full cScap/4G10Fab reconstruction is blue, and FSC curve for the L1-L7/4G10Fab focused refinement is red.
(C) Cryo-EM map of entire cScap/4G10Fab complex colored by local resolution.
(D) Focused cryo-EM map of L1-L7 domain of cScap and part of 4G10Fab colored by local resolution.
(E) Euler angle distribution of particles used in the final 3D reconstructions.
(F) Flowchart of cryo-EM data processing.
Figure S4. L1-L7 Density and Model Building, Related to Figure 3
(A) Representative EM densities for the indicated segments of cScap L1 and L7. The density contour of the map within 2.5 Å of the selected residues of the 2.9Å WT L1-L7 map was set at level 0.0684 in Chimera. L1 residues are colored green, L7 residues are colored purple. Residues labeled with ovals were subjected to mutagenesis studies (Figure 3F).
(B) Left: Secondary structure diagram of cScap L1-L7 domain depicted using Topdraw. The conserved MLD fold is highlighted with red box. L1 is depicted in green, L7 is depicted in magenta. Brown line indicates the disulfide bridge. Yellow star indicates β-strand of L7 that contributes to the core β-sheet. Right: Secondary structure diagram of MLD domain of NPC1 (residues 400–607) depicted using Topdraw. The conserved structural elements between L1-L7 and the NPC1 MLD are highlighted with a red box. Brown lines indicate the two disulfide bridges. Secondary structure assignment is based on PDB 5KWY.
Figure S5. Biochemical Characterization of 4G10Fab/L1-L7 Interaction and Disulfide bond in L1, Related to Figures 2 and 3
(A) Structural details of the L1-L7/4G10Fab interaction. L1-L7 domain is shown as cartoon with L1 in green and L7 in magenta. The solvent-accessible electrostatic surface of 4G10Fab (generated using the APSB Pymol plugin) is shown colored with a range of −3 kT/e (red) to +3 kT/e (blue). Inset: closeup of the 4G10Fab/L1-L7 interaction. Residues in L1 with sidechains that make interactions with the 4G10Fab (within 5Å) are shown as sticks.
(B) Schematic for the immunoprecipitation of cScap by 4G10Fab.
(C) Immunoprecipitation of wild-type and mutant cScap by 4G10Fab. HEK 293T cells were set up and transfected as described in Methods with 6 μg pEZT-cScap-3xFLAG WT or pEZT-cScap-3xFLAG R135E D136K plasmids. The next day, the cells were harvested and solubilized in GDN detergent as described in Methods. Supernatants were subjected to immunoprecipitation by anti-FLAG M2 resin and eluted cScap proteins were then subjected to immunoprecipitation by 4G10Fab bound to protein L resin as described in Methods. The final eluates were subjected to 10% SDS-PAGE followed by immunoblot analysis of cScap (M2 anti-FLAG-HRP).
(D) SREBP2 cleavage assay. Scap-deficient SRD-13A cells were set up and transfected as described in Methods with 6 μg pcDNA control plasmid (lane 1) or else 2 μg pEZT-Myc-SREBP2 together with 4μg of either pEZT-cScap-3xFLAG WT or pEZT-cScap-3xFLAG R135E D136K. The next day, cells were depleted of cholesterol for 1 hr, harvested, and subjected to SDS-PAGE followed by immunoblot analysis of SREBP2 (IgG-22D5), cScap (anti-FLAG), and actin (anti-actin) as described in Methods. P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2.
(E) Structure of cScap L1-L7 showing the location of the disulfide bond between C147 and C169 of L1. Inset: Continuous density for residues C147 and C169 showing the disulfide bond. Density within 2 Å of the two residues was contoured at sigma level 5 in Pymol.
(F) Schematic of recombinant hamster Scap(L1-L7) fusion that is secreted by CHO-K1 cells. Starting at the NH2-terminus, the construct contains the honeybee melittin signal sequence (brown), followed by a His12 epitope tag (blue), hamster Scap-Loop 1 (aa 46–284) containing three cysteines at the indicated locations, a 15 amino acid linker (orange), and hamster Scap-Loop 7 (aa 535–710). L7 does not contain any cysteines.
(G) Secretion of wild-type (WT) and cysteine mutants of hamster Scap(L1-L7). CHO-K1 cells were set up and transfected as described in Methods with 4 μg of either wildtype pCMV-Scap(L1-L7) plasmid containing all three cysteines (CCC) or mutant versions where cysteines had been individually mutated – C147A (ACC), C169S (CSC), C264A (CCA). 48 hr after transfection, cells were harvested, and media was collected. Aliquots of media (1.5% of total) were treated with 6 mM mPEG-MAL-5000 to modify free and exposed cysteines as described in Methods. Equivalent fractions of cells and media were then subjected to immunoblot analysis with 5 μg/ml anti-L7 (IgG 9D5).
(H) Analysis of cysteine-modified forms of hamster Scap(L1-L7) (C264A). Media from cells transfected with mutant pCMV-Scap(L1-L7) (C264A) in (C) was collected and incubated at the indicated temperatures in the absence or presence of 1 mM DTT for 15 min. The media was then treated without or with 6 mM mPEG-MAL-5000 as indicated to modify free and exposed cysteines as described in Methods, after which the samples were subjected to immunoblot analysis with 5 μg/ml anti-L7 (IgG 9D5). RT, room temperature.
(I) The C147-C169 disulfide bond is essential for Scap transport of SREBP2. Scap-deficient SRD-13A cells were set up and transfected as described in Methods with 4 μg pcDNA control plasmid (lane 1) or 2 μg pTK-SREBP2 and 2 μg WT or mutant pTK-Scap as indicated. The total amount of transfected DNA for all conditions was adjusted to 4 μg with pcDNA control plasmid. The next day, cells were depleted of cholesterol for 1 hr, harvested, and subjected to 10% SDS-PAGE followed by immunoblot analysis of SREBP2 (IgG-22D5), Scap (IgG-4H4), and actin (anti-actin) as described in Methods. P, precursor form of SREBP2; N, cleaved nuclear form of SREBP2.
Figure S6 – Cryo-EM Analysis of cScapD435V/cInsig-1/4G10Fab, Related to Figure 4
(A) Representative electron micrograph and 2D class averages of cScapD435V/cInsig-1/4G10Fab, showing clear densities for Fab, L1-L7 domain of cScap, and TM domains of both cScap and cInsig-1.
(B) Gold-standard Fourier shell correlation curve for the cryo-EM map shown in Figure 4E and Figure S6D. FSC curve for the full cScapD435V/cInsig-1/4G10Fab reconstruction is blue, and FSC curve for the L1-L7/4G10Fab focused refinement is red.
(C) Cryo-EM map of entire cScapD435V/cInsig-1/4G10Fab complex colored by local resolution.
(D) Focused cryo-EM map of L1-L7 domain of cScapD435V and part of 4G10Fab colored by local resolution.
(E) Euler angle distribution of particles used in the final 3D reconstructions.
(F) Flowchart of cryo-EM data processing.
Figure S7. Orientation of Loop Structures in cScap and Other SSD-containing Proteins, Related to Figure 7
The SSDs of cScap (green for WT, magenta for D435V) were aligned with the SSDs of NPC1 (yellow, PDB 6V3H), Patched (orange, PDB 6OEU), Dispatched (cyan, PDB 6TBU), and NPC1-L1 (tan, PDB 6W5U). For clarity, the NTD-TM1 regions of NPC1 (residues 1–291) and NPC1-L1 (residues 1–311) were omitted. In both forms of cScap, the L1-L7 is rotated such that the β-sheet is roughly parallel to the membrane. In the other SSD-containing proteins, the middle luminal domain (MLD)-related subunits are angled downward such that their core β-sheets are roughly perpendicular to the membrane. Grey cartoons denote simplified orientations of SSDs and structures related to NPC1’s MLD. Other than Scap, these SSD-containing proteins all have two pseudo-symmetrically interacting ‘SSD-MLD’ units on the same polypeptide.
Data Availability Statement
Structural data have been deposited in the Protein Data Bank (PDB) and the Electron Microscopy Data Bank (EMDB). The atomic models of the cScap L1-L7 domain/4G10Fab complex and the cScapD435V L1-L7 domain/4G10Fab complex have been deposited in the PDB with accession codes 7LKF and 7LKH, respectively. The cryo-EM maps for the global cScap/4G10Fab reconstruction and the refinement focused on the cScap L1-L7 domain have been deposited in the EMDB with accession codes EMD-23406 and EMD-23405, respectively. The cryo-EM maps for the global cScapD435V/cInsig-1/4G10Fab reconstruction and the refinement focused on the cScapD435V L1-L7 domain have been deposited in the EMDB with accession codes EMD-23407 and EMD-23408, respectively.