

Abstract
Gastrointestinal cancers encompass a diverse class of tumors arising in the GI tract, including esophagus, stomach, pancreas and colorectum. Collectively, gastrointestinal cancers compose a high fraction of all cancer deaths, highlighting an unmet need for novel and effective therapies. In this context, the transmembrane receptor guanylyl cyclase C (GUCY2C) has emerged as an attractive target for the prevention, detection and treatment of many gastrointestinal tumors. GUCY2C is an intestinally-restricted protein implicated in tumorigenesis that is universally expressed by primary and metastatic colorectal tumors as well as ectopically expressed by esophageal, gastric and pancreatic cancers. This review summarizes the current state of GUCY2C-targeted modalities in the management of gastrointestinal malignancies, with special focus on colorectal cancer, the most incident gastrointestinal malignancy.
Keywords: : biomarker, colorectal cancer, gastrointestinal cancer, GUCY2C, immunotherapy
Cancers derived from the GI tract and its associated organs are among the most prevalent and deadly in the world. Gastrointestinal tumors include esophageal, stomach, pancreatic, small intestinal, liver, colorectal and anal [1]. Among these, colorectal cancer (CRC) represents the highest disease burden and is responsible for >40% of all gastrointestinal tumors [1].
CRC is the third most prevalent cancer and second leading cause of cancer-related death worldwide [2]. Globally, 1.8 million new cases were diagnosed, and 881,000 deaths were attributed, to CRC in 2018 [2]. Despite improvements in CRC screening over the last few decades, a majority of patients still presents with regional or distant disease at the time of diagnosis [3]. Moreover, in patients with distant metastases, the 5-year survival rate is 12%, which has not improved over the past decade [2]. Thus, strategies to prevent and treat CRC represent a substantial unmet clinical need.
An emerging collection of strategies to prevent, identify and treat gastrointestinal malignancies focuses on the intestine-specific cell surface receptor guanylyl cyclase C (GUCY2C). GUCY2C serves as the transmembrane receptor for the diarrheagenic bacterial enterotoxins as well as the intestinal paracrine hormones guanylin and uroguanylin where it functions to regulate cell growth, metabolism and fluid secretion [4,5]. Anatomically, expression of GUCY2C in normal tissues is restricted to intestinal epithelial cells and a subset of hypothalamic neurons [6,7]. In the context of disease, GUCY2C ligand expression is lost while the silenced GUCY2C receptor is maintained throughout CRC carcinogenesis [5,8–10]. Furthermore, GUCY2C receptor is ectopically expressed as part of the genetic reprogramming by a variety of gastrointestinal malignancies [5,8–10]. Collectively, these unique characteristics in health and disease, position GUCY2C as an attractive target for chemoprevention by ligand replacement, a clinical biomarker for detecting CRC metastases in extra-intestinal tissues and a promising immunotherapeutic target in the treatment of gastrointestinal cancers.
The role of GUCY2C in intestinal homeostasis
GUCY2C, originally termed STa receptor, was first identified as the receptor for the heat-stable enterotoxin (STa), the causative agent of traveler’s diarrhea [11,12]. STa is secreted by enterotoxigenic Escherichia coli (ETEC), which is responsible for 222 million global episodes of diarrhea and over 50,000 deaths annually [13,14]. Early studies characterizing STa binding revealed that, in contrast to other diarrheagenic toxins such as cholera toxin, STa resulted in intestinal cyclic GMP (cGMP) accumulation. This observation suggested that STa may signal through a guanylyl cyclase, a family of proteins known to catalyze the conversion of GTP to cGMP and pyrophosphate [15,16]. Thus, using highly conserved sequences from guanylyl cyclase catalytic domains, GUCY2C was first cloned from a rat small intestinal cDNA library and demonstrated to serve as both the STa receptor and source of cGMP accumulation [17]. Human GUCY2C was cloned 1 year later from the CRC tumor cell line T84 [18–20].
Structure
Human GUCY2C protein is a 1050 amino acids protein with a molecular mass of 120 kDa [20]. Its single-transmembrane spanning domain and intracellular domains (a kinase-homology domain, linker domain and guanylyl cyclase domain) bear homology to other membrane-bound guanylyl cyclases, while its unique N-terminal extracellular domain (residue 1–430) defines its ligand specificity [16,21,22]. Within the extracellular domain, STa binds to a microdomain of amino acids close to the transmembrane domain (residues 387–393) [23,24]. The exact mechanisms by which STa binding amplifies the generation of cGMP by GUCY2C remain incompletely defined. GUCY2C is expressed as a preformed homomultimer, and extracellular ligand binding induces intracellular conformational changes that stabilize the catalytically active state of the receptor [25]. The linker domain toggles the activity of the guanylyl cyclase domain after binding of ligand, and without that domain GUCY2C is constitutively active [26]. Furthermore, post-translational modification of GUCY2C greatly alters its activity. GUCY2C is glycosylated at ten different sites, which is necessary for catalytic activity and binding of ligand [27–29]. Phosphorylation of GUCY2C at the kinase homology domain changes GUCY2C activity depending on the mode of stimulation: potentiating ligand-induced cGMP production, but blunting detergent-stimulated cGMP production [30,31]. An added layer of regulation is imposed by the carboxyl-terminal tail, an understudied feature that GUCY2C shares with sensory, but not other, receptor guanylyl cyclases. This 63-residue domain is required for the guanylyl cyclase function of GUCY2C, but also decreases this function through phosphorylation by PKC and association with its unique binding partner, IKEPP (intestinal and kidney-enriched PDZ protein) [32–34]. Clarity on the structure and function of GUCY2C could come from solving its crystal structure, which has yet to be reported.
Molecular mechanisms & physiology
In its canonical role, GUYC2C regulates intestinal fluid secretion through ion channels, fine tuning the osmolality of the 8–9 l of fluid that pass through the human intestine daily [35]. GUCY2C is positioned on the apical membrane on the brush border of intestinal villi, poised to receive luminal signals from its endogenous ligands secreted by the intestinal epithelium [36]. In healthy intestines, these two ligands are guanylin (large intestine) and uroguanylin (small intestine) [37,38]. These endogenous ligands share homology with STa (the exogenous bacterial toxin), highlighting that toxin as an example of molecular mimicry evolved by ETEC [39–41]. Indeed, STa is a superagonist, having a tenfold higher affinity for GUCY2C [42]. These ligands induce GUCY2C to produce the second messenger cGMP. In turn, cGMP binds to membrane-bound cGMP-dependent protein kinase II (PKGII), relieving autoinhibition and activating its catalytic domain [43–45]. Activated PKGII then phosphorylates the cystic fibrosis transmembrane conductance regulator (CFTR), opening the channel that mediates luminal transport of Cl− and HCO3− and the subsequent efflux of water into the GI tract [46–48]. Beyond CFTR, PKGII also phosphorylates sodium hydrogen exchanger 3, inhibiting the absorption of Na+ and decreasing the osmotic influx of water [49,50]. Collectively, these actions result in an increased extracellular electrolyte concentration that drives fluid secretion into the lumen of the intestine, manifesting as diarrhea in the context of overstimulation by STa. While PKGII is the canonical effector of secretory function through GUCY2C, only 50–60% of GUCY2C’s secretory effect is mediated by cGMP/PKGII [45,51]. The cyclic AMP (cAMP) signaling pathway and its effector PKA also phosphorylate CFTR and sodium hydrogen exchanger 3. Complex cyclic nucleotide cross-talk is mediated by a family of enzymes called phosphodiesterases (PDEs). Different PDE isoforms preferentially degrade cAMP or cGMP, and can be activated or repressed by cyclic nucleotides themselves with varying affinity. For example, cGMP binding activates PDE2 and inhibits PDE3. In intestinal epithelial cells, cGMP-mediated inhibition of PDE3 decreases degradation of cAMP, resulting in increased cAMP/PKA-mediated phosphorylation of CFTR [52]. Thus, GUCY2C activity in intestinal enterocytes increases cyclic nucleotides directly through catalysis of GTP to cGMP, and indirectly by increasing the half-life of cAMP, together regulating the activity of these channels, and their trafficking to the membrane surface [53,54].
GUCY2C is conserved across all vertebrates, and recent evidence suggests that evolutionary pressure from ETEC incited a mutational arms race in the receptor and STa, in both primates and bats [55,56]. Human GUCY2C mutations have been characterized by their effects on secretion, with GUCY2C loss-of-function leading to decreased secretion and meconium ileus (neonatal intestinal obstruction), and GUCY2C gain-of-function leading to increased secretion and secretory diarrhea [57,58]. The role of GUCY2C has been confirmed in a genetic mouse model in which the gene for this receptor was disrupted by inserting a neomycin cassette into exon 1, eliminating protein expression [59,60]. These GUCY2C-deficient (GUCY2C−/−) mice are resistant to oral STa but do not suffer from meconium ileus [59,60]. Beyond defects in fluid secretion, GUCY2C−/− mice exhibit aberrations in cell proliferation. Specifically, elimination of GUCY2C in intestinal epithelial cells accelerates cell cycle progression resulting in crypt hyperplasia [61]. Abnormal regulation of intestinal cell division also manifests as increased tumorigenesis. Compared with GUCY2C+/+ mice, GUCY2C−/− mice display higher rates of tumorigenesis in genetic (APCMin/+) and carcinogen-induced models of CRC [62], suggesting that GUCY2C may regulate cell division and tumor initiation.
Activation of the GUCY2C signaling pathway to prevent CRC
Given its roles in intestinal epithelial homeostasis, the GUCY2C/cGMP signaling axis closely interacts with circuits driving intestinal tumorigenesis. The genetic events underlying tumorigenesis have been refined for decades, but a relatively recent observation is the inactivation of GUCY2C/cGMP signaling early in this process. Intestinal transformation is characterized by a step-wise accumulation of genetic mutations, the most common of which have been catalogued by large-scale sequencing studies of colorectal tumors [63]. In that context, >80% of sporadic (nonfamilial) CRCs arise from a two-hit inactivating mutation in the tumor suppressor APC, or an activating mutation in its downstream target β-catenin [64,65]. These ‘gatekeeper’ mutations lift a block on epithelial proliferation, enabling the development of a precancerous polyp (adenoma) [64,65]. Acquisition of subsequent mutations (TP53, KRAS and PIK3CA being the most common after APC), and activation of downstream oncogenic signaling circuits characterizes the transition from adenoma to carcinoma [63]. Recent studies have revealed that the progression through this well-characterized adenoma–carcinoma sequence correlates with disruption of GUCY2C/cGMP signaling. While the mutational events underlying CRC represent irreversible steps, requiring chemotherapy or surgery to remove tumor tissue, the loss of cGMP signaling represents an intriguing reversible step in tumorigenesis, where pharmacologic agents reactivating this signaling axis have the potential to oppose or prevent the disease.
Loss of GUCY2C signaling in CRC
The loss of homeostatic signaling by the GUCY2C/cGMP axis arises commonly in intestinal transformation. Disruption of several elements of the axis has been reported. Intracellular cGMP accumulation and subcellular localization is tightly regulated by PDEs which hydrolyze the cyclic nucleotides, cGMP and cAMP, to the nucleotides GMP and AMP, respectively. Pathologic upregulation of PDE family members accelerates cyclic nucleotide degradation, reducing signaling by this second messenger. In human colon cancer cell lines, tumor samples and APCmin/+ mouse adenomas, there is increased expression of cGMP-specific PDE5 and PDE10 [66–69]. Pharmacologic inhibition of these overexpressed PDEs decreased tumor cell growth in these studies, suggesting that the restoration of cGMP opposes the oncogenic phenotype. Suppression of the cGMP effector, PKGI, also has been observed in cancer cell line and colon tumor samples [70]. Reconstitution of PKG expression appears to inhibit cancer cell and xenograft growth rates as well.
Interestingly, GUCY2C protein itself seems to be largely spared during tumorigenesis. Retention of GUCY2C expression throughout the adenoma-carcinoma sequence has been widely observed [8,71–75]. Furthermore, The Cancer Genome Atlas reports only 4% (22/537) of colon and rectal tumor specimens harbor mutations in GUCY2C (TCGA-COAD and -READ datasets; https://portal.gdc.cancer.gov/), suggesting that wild-type GUCY2C is retained in the majority of CRCs. Moreover, localization and surface expression of the receptor is also retained in histologic sections from tumors [8,75].
While receptor expression, localization and wild-type sequence are largely retained in transformed tissue, the GUCY2C ligands, guanylin and uroguanylin, are among the most commonly lost gene products in CRC [71,76–79]. Loss of the autocrine and paracrine signaling mediated by these two secreted hormones appears to be a key mechanism of GUCY2C/cGMP signaling dysfunction in cancer. Loss of these hormones occurs early in tumorigenesis, as can be readily observed in histologic sections of human tubular adenomas, the precancerous lesion arising from APC loss of heterozygosity [8,75]. Furthermore, this event is conserved between species, and ligand loss also is a hallmark of intestinal tumorigenesis in mice [8,75]. Blomain et al. recently reported that guanylin loss is part of the altered transcriptional program downstream of mutant APC [8,75]. In mouse models of conditional biallelic Apc deletion, guanylin expression and downstream cGMP signaling disappears within days, and in human colon cancer cells, re-expression of wild-type APC reconstitutes guanylin expression [8,75]. Additionally, mice lacking guanylin through genetic deletion develop crypt hyperplasia, consistent with the phenotype of mice lacking Gucy2c, and consistent with a role for the hormone in epithelial growth control [61]. While it remains unclear if the loss of GUCY2C/cGMP signaling is a bystander effect or a necessary step in tumorigenesis, these observations suggest that the mutations driving intestinal transformation produce a microenvironment of GUCY2C ligand insufficiency, silencing the tumor suppressive properties of this signaling axis. This principal of an otherwise functional, but orphaned, receptor persisting throughout tumorigenesis creates an opportunity to therapeutically restore cGMP signaling through exogenous hormone replacement [80].
Restoring GUCY2C signaling for CRC chemoprevention
In addition to aforementioned observations of GUCY2C/cGMP signaling dysfunction in tumor samples, pathologic GUCY2C ligand loss also arises from diet-induced obesity and intestinal inflammation, conditions which predispose to the development of CRC [6,81–83]. Furthermore, regions of the world with endemic diarrheagenic bacteria (e.g., the bacteria responsible for traveler’s diarrhea), which produce the GUCY2C agonist, STa, have a lower incidence of CRC [84,85]. While these observations are correlative, it is tempting to speculate that pre-existing conditions (obesity, inflammatory syndromes) that reduce intestinal cGMP predispose the epithelium to transformation, while life-long exposure to STs increases GUCY2C activation and opposes epithelial dysplasia. Supporting this concept, in a carcinogen-induced model of tumorigenesis (azoxymethane), mice colonized with ST-secreting E. coli developed fewer tumors than those colonized with control E. coli [86]. Given its tumor-suppressive properties and disruption preceding and during tumorigenesis, the GUCY2C/cGMP signaling axis has emerged as a promising target for CRC prevention.
Several approaches targeting elements of the GUCY2C/cGMP signaling axis have been explored, including targeting cGMP generation through GUCY2C agonists, or cGMP degradation through small-molecule PDE inhibitors. Importantly, several of these pharmacologic agents are already US FDA approved for other purposes. The first FDA-approved synthetic GUCY2C agonist was the ST analog, linaclotide (Ironwood Pharmaceuticals, MA, USA), approved in 2012 for patients with chronic idiopathic constipation and constipation-predominant irritable bowel syndrome [87]. More recently, the uroguanylin analog, plecanatide (Synergy Pharmaceuticals, NY, USA), was approved for the same syndromes and exhibits similar safety and efficacy profiles [88]. Both agents agonize GUCY2C and stimulate cGMP production, taking advantage of canonical cGMP-stimulated fluid secretion to treat symptoms of constipation. Acting downstream of GUCY2C, the PDE5 inhibitor, sildenafil, has been in use for over 20 years to treat erectile dysfunction, and is also approved for the treatment of pulmonary hypertension and benign prostatic hyperplasia [89]. PDE inhibition augments the activity of endogenous GUCY2C ligands by slowing the rate of intracellular cGMP degradation.
Preclinical studies in mice support potential efficacy of cGMP-elevating agents for colon cancer prevention. In the first demonstration of this concept, Shailubhai et al. reported that dietary uroguanylin supplementation reduced tumor burden in mice fed a high-fat diet and genetically predisposed to develop intestinal tumors (APCmin/+) [71]. Over the past 3 years, four additional reports have built upon these initial observations. Much like the initial observations with uroguanylin, oral administration of sildenafil or linaclotide in the drinking water, or plecanatide in the food, reduced tumor multiplicity in the APCmin/+ mice [90,91]. In a different model of tumorigenesis, mediated by the carcinogen azoxymethane and the inflammatory agent, dextran sodium sulfate, sildenafil was also shown to reduce tumor multiplicity [66,92]. Interestingly, the benefit of sildenafil was observed when administered at the earliest stages of transformation, <1 month after the initial insult, suggesting a role for cGMP signaling in the earliest stages of the disease process [66,92].
A body of evidence supports the potential for cGMP-elevating agents to oppose tumorigenesis, but whether these observations will translate to humans remains an open-ended question, and human studies remain in their infancy. The first study to address the question recently examined the incidence of colon cancer in patients taking PDE inhibitors [93]. In a nationwide retrospective cohort study of 36,020 Swedish men diagnosed with a colorectal adenoma, 4849 were prescribed a PDE inhibitor during the study period (2005–2015). Incidence of CRC development was significantly reduced in patients taking the PDE inhibitor (hazard ratio = 0.65; 95% CI: 0.49–0.85). While this represents a correlation between cGMP-agent and lower tumor incidence, the results are encouraging. Given that CRC takes as long as 10 years to progress from polyp to carcinoma, and the new class of GUCY2C synthetic agonists (linaclotide and plecanatide) were only approved in 2012, a correlation between the use of these agents and incidence of cancer has yet to be investigated. Furthermore, these agents are formulated for upper gastrointestinal release, rather than colorectal release. A recent study examined the colorectal bioactivity of linaclotide (formulated for gastric release) in a small human cohort [94]. The agents failed to stimulate colorectal cGMP production at baseline; however, in the context of a bowel prep, which the authors speculate flushed active agent further down the GI tract, linaclotide induced epithelial cGMP and homeostatic signaling, such as phosphorylation of VASP and reduced expression of the epithelial proliferation marker, Ki67. Together these findings suggest that mechanistic insights made in mice are likely conserved in humans. A clinical trial evaluating linaclotide in patients with stage 0–3 CRC is currently ongoing (NCT03796884).
GUCY2C as a biomarker in gastrointestinal malignancies
Seminal studies mapping GUCY2C expression across normal physiologic tissues identified GUCY2C as an intestinally-restricted protein [17]. Subsequent studies examining GUCY2C in gastrointestinal cancers revealed its expression is widely maintained throughout CRC carcinogenesis, and ectopically expressed by other malignancies associated with the GI tract [73,95,96]. Collectively, these data suggested that GUCY2C could be used as a molecular marker for identifying metastatic CRC cells outside of intestinal tissues as well as an indicator of gastrointestinal malignancy.
Colorectal cancer
In CRC, the single most predictive marker for predicting patient outcome is stage of disease at diagnosis [3]. Staging of disease is most commonly based on the tumor node metastasis system which evaluates the primary tumor, extent of lymph node involvement and presence of extra-intestinal metastases [97]. Clinically, patients presenting with ostensibly localized disease undergo surgical resection of the primary tumor as well as the neighboring lymph nodes to define disease staging [98]. Crucially, the presence of lymph node metastases (LNM) is widely considered to be one of the most important factors in CRC staging, as LNM involvement is an indication for more aggressive treatment beyond surgery including neoadjuvant therapy [97,99]. The gold standard for identifying LNM is histopathological assessment by a pathologist [97]. However, histopathological evaluation methods used to identify LNM have limited sensitivity and consequently result in understaging and undertreating CRC. Specifically, up to 30% of patients with pathology-negative nodes exhibit disease recurrence and ultimately die from metastatic disease [100], suggesting the existence of occult metastatic cells missed during histopathological evaluation. Thus, improved techniques for detecting occult metastases are needed.
The observation that GUCY2C is widely maintained throughout carcinogenesis combined with its restricted expression profile led to the hypothesis that GUCY2C could act as a biomarker to identify occult CRC metastases in lymph nodes [73]. Initially, a retrospective clinical study of 21 stage II CRC patients evaluated pathology-negative nodes for evidence of GUCY2C mRNA. Utilizing reverse-transcription PCR (RT-PCR) to identify GUCY2C mRNA in resected lymph nodes, 11 patients tested negative while ten patients tested positive for GUCY2C. Importantly, detection of GUCY2C in lymph nodes was associated with disease recurrence, suggesting GUCY2C mRNA may reflect the presence of micrometastases and could carry prognostic value [72]. Following this study, a prospective trial verified GUCY2C prognostic value in a multicenter, blinded clinical study. This prospective trial enrolled 257 patients with pathology-negative lymph nodes across nine centers. In total, 2570 resected lymph nodes were assessed by histopathologic evaluation and RT-PCR for GUCY2C mRNA. Following resection, patients were monitored up to 5 years for signs of disease recurrence. The trial identified 225 patients with lymph nodes testing positive for GUCY2C and 32 patients with lymph nodes testing negative for GUCY2C. In this study, patients with lymph nodes testing positive for GUCY2C exhibited a higher risk of recurrence, earlier time to recurrence following resection, and reduced disease-free survival compared with patients testing negative for GUCY2C [101]. Thus, these studies established GUCY2C as a biomarker for identifying occult metastases in regional lymph nodes and assessing patient risk. The utility of GUCY2C RT-PCR has subsequently been independently validated and replicated by several laboratories [102,103].
Recently, a thorough analysis of GUYC2C expression among different CRC types was conducted [8]. CRC is a heterogeneous disease and a prevailing classification system categorizes colorectal carcinomas into three distinct pathways of origin: microsatellite instability (MSI), chromosomal instability (CIN) and the CpG island methylator phenotypes (CIMP) [104,105]. Among these pathways, the majority of CRC tumors (∼75%) arise via the CIN pathway [106]. In this analysis, robust GUCY2C was reported in tumors of CIN and MSI origin, but not in CpG island methylator phenotypes tumors [8]. Absent GUCY2C in serrated adenomas was attributed to loss of cell-derived xenograft 2 (CDX2), a transcription factor required for GUCY2C expression. Collectively, these data suggest that, while the vast majority of CRC tumors exhibit robust GUCY2C expression and are therefore candidates for biomarker detection and GUCY2C-directed therapies, CRC patients with serrated adenomas may not be amenable to these modalities due to GUCY2C loss.
Other gastrointestinal malignancies
While GUCY2C is not expressed in normal esophagus, stomach or pancreas, ectopic GUCY2C expression has been reported in gastrointestinal malignancies derived from these tissues [96]. Ectopic GUCY2C expression by stomach and esophagus is attributed to intestinal metaplasia. During neoplastic transformation, esophageal and stomach adenocarcinomas undergo intestinal metaplasia whereby neoplastic cells acquire molecular and histological characteristics of differentiated intestinal enterocytes. Consequently, these neoplastic cells express intestinal transcription factors, such as CDX2, thereby inducing de novo expression of intestinal-specific genes including GUCY2C [96,107]. Indeed, GUCY2C mRNA and protein is detected in well-differentiated intestinal-type gastric and esophageal tumors, but absent in gastrointestinal tumors not associated with intestinal metaplasia such as poorly differentiated and signet ring carcinomas [108].
A recent comprehensive analysis assessed GUCY2C protein status in 627 gastrointestinal tumors including esophageal, gastric, pancreatic and colorectal [74]. As expected, colorectal tumors exhibited the highest frequency of GUCY2C positivity, with 98% of tumors staining for GUCY2C. Among other gastrointestinal malignancies, the frequency of GCC-positivity was similar with 59% of esophageal, 68% of gastric and 64% of pancreatic tumors staining positive for GUCY2C. Thus, in addition to CRC, the majority of esophageal, gastric and pancreatic tumors may be amenable to GUCY2C detection and targeting.
GUCY2C as an immunotherapeutic target
The concept that the immune system possesses the capacity to recognize and eliminate cancer cells has transformed cancer management. Indeed, widespread success of immune checkpoint inhibitor therapy has demonstrated the potential of immunotherapeutic approaches. However, effective immunotherapeutic options for many gastrointestinal malignancies are limited. For example, in CRC, immune checkpoint inhibitor therapy is only approved for a small minority of patients with MSI tumors [109]. Thus, for the majority of CRC tumors, alternative immunotherapeutic approaches are needed.
Qualities that define GUCY2C as a biomarker of metastatic disease also make it an ideal target for immunotherapy. As described previously, GUCY2C expression in healthy tissues is restricted to intestinal epithelial cells and a small subset of CNS neurons. Thus, GUCY2C detected outside these tissues indicates malignant disease and represents a tumor-specific marker targetable by pharmacologic agents. GUCY2C-directed immunotherapeutics therefore leverage a dichotomous expression profile between normal and cancerous tissues to deliver systemic antitumor responses that eliminate metastases while sparing normal GUCY2C-expressing tissues of the intestinal mucosa and CNS. Initial strategies targeting GUCY2C began with vaccination and expanded into antibody–drug conjugates (ADCs), bispecific T-cell engagers (BiTEs) and chimeric antigen-receptor (CAR)-T cells (CAR T cells) as the field of immunotherapy evolved. A summary of completed and ongoing clinical trials utilizing GUCY2C immunotherapeutics is detailed in Table 1.
Table 1. . Guanylyl cyclase C-targeted immunotherapeutics in clinical trial.
| Type of immunotherapy | Drug name | Phase | Cancer indication | Status | Identifier |
|---|---|---|---|---|---|
| Vaccine | Ad5-GUCY2C-PADRE | I | Colorectal | Completed | NCT01972737 |
| Ad5.F35-GUCY2C-PADRE | IIa | Gastrointestinal malignancies | Initiated | NCT04111172 | |
| Antibody–drug conjugate | TAK-264 | I | Gastrointestinal malignancies | Completed | NCT01577758 |
| II | Pancreatic | Terminated | NCT02202785 | ||
| Bispecific T-cell engager | PF-07062119 | I | Gastrointestinal malignancies | Ongoing | NCT04171141 |
Vaccines
The first immunotherapeutic approach targeting GUCY2C was a vaccine in 2008 [110]. Protein sequence alignment of GUCY2C to the guanylyl cyclase family revealed significant homology across intracellular domains but an antigenically unique extracellular domain. Thus, the extracellular domain of GUCY2C was selected as a vaccine target and cloned into recombinant adenovirus serotype 5 (Ad5) for immunogenicity and CRC tumor challenge studies. Vaccination with Ad5-GUCY2C induced GUCY2C-specific CD8+ T-cell responses but a lack of GUCY2C-specific CD4+ T cell and antibody responses. Moreover, compared with control vaccination, Ad5-GUCY2C significantly reduced tumor burden and increased survival in mouse models of liver and lung metastases (the two most common sites of CRC metastases) [111]. Importantly, Ad5-GUCY2C induced antitumor immunity without generating autoimmunity toward normal GUCY2C-expressing tissues. Induction of GUCY2C-specific antitumor immunity in the absence of colitis is attributed to limited immune cross-talk between systemic and mucosal compartments. Seminal studies using HIV vaccines demonstrated that the route of vaccination profoundly impacts the quality of the immune response. For example, administration of an HIV vaccine orally generates robust mucosal immunity while subcutaneous or intramuscular administration using the same vaccine generates HIV-specific immunity in the spleen but an absence of those responses in mucosal tissues [112–114]. Limited mucosal immunity is attributed to a lack of mucosal homing receptors on T cells primed systemically [115,116]. GUCY2C vaccines therefore exploit immune compartmentalization to constrain GUCY2C immunity to systemic circulation, where metastatic cancer cells may be present, without inducing colitis against GUCY2C on normal mucosal tissues. Similarly, absence of encephalitis following Ad5-GUCY2C vaccination likely reflects limited trafficking and/or access of T cells to the CNS [117,118].
After proof-of-concept studies established GUCY2C as a safe and effective cancer vaccine antigen, additional studies aimed to enhance the therapeutic efficacy of GUCY2C vaccines. Studies comparing Ad5-GUCY2C immunogenicity between GUCY2C+/+ and GUCY2C−/− mice demonstrated that, while Ad5-GUCY2C induces only GUCY2C-specific CD8+ T cells in GUCY2C+/+mice, Ad5-GUCY2C vaccination in GUCY2C−/− mice generates GUCY2C-specific CD4+ T cell and antibody responses [119,120]. Moreover, Ad5-GUCY2C vaccination of GUCY2C−/− mice induces superior antitumor immunity compared with Ad5-GUCY2C vaccination of GUCY2C+/+ mice [120]. Collectively, these data suggest a selective tolerance of GUCY2C-specific CD4+ T cells in GUCY2C+/+ mice, and that GUCY2C immunity in these mice could be enhanced by recruiting CD4+ T cell help. While CD8+ T cells directly lyse tumor cells, helper CD4+ T cells are required for the priming of effector CD8+ T cells with optimal cytolytic function, the development of long-lived memory CD8+ T cells, and the production of antibodies against T-dependent antigens [121,122]. To test this hypothesis, a vector was constructed comprising the GUCY2C extracellular domain fused to an immunogenic CD4+ T-cell epitope from influenza known as site 1 (S1). Vaccination of GUCY2C+/+ mice with Ad5-GUCY2C-S1 induced novel GUCY2C-specific long-lived memory CD8+ T cells and antibodies as well as superior antitumor immunity compared with Ad5-GUCY2C vaccination [120], suggesting inclusion of exogenous CD4+ helper epitopes enhances GUCY2C immunity. Additional studies established that GUCY2C immunity is further improved using a heterologous prime-boost regimen. Indeed, a DNA vaccine against GUCY2C that ‘primes’ GUCY2C immune responses followed by Ad5-GUCY2C-S1 that ‘boosts’ those responses significantly enhanced GUCY2C-specific CD8+ T-cell generation and antitumor immunity over homologous immunization with Ad5 or DNA [123]. Superior antitumor immunity reflected induction of GUCY2C-specific CD8+ T cells with higher T-cell receptor avidity [123].
Recently, a Phase I study evaluated the safety and immunogenicity of a recombinant Ad5 vaccine encoding the extracellular domain of GUCY2C fused to the pan HLA DR-binding epitope (PADRE) in patients with stage I and II CRC [124]. Similar to S1 in mice, PADRE recruits CD4+ helper T cells and enhances GUCY2C-specific CD8+ T-cell responses. After a single intramuscular injection of Ad5-GUCY2C-PADRE, four out of ten patients developed GUCY2C-specific CD8+ responses. Interestingly, induction of GUCY2C-specific antibodies correlated with development of a CD4 response against the helper epitope PADRE, recapitulating observations of selective CD4 tolerance in mice. Ad5-GUCY2C-PADRE vaccination was well tolerated without toxicities in GUCY2C-expressing tissues, or grade 3 or grade 4 toxicities systemically, suggesting that GUCY2C antitumor immunity can be safely induced in patients without autoimmunity. In this trial, pre-existing immunity to Ad5 negatively correlated with development of a GUCY2C-specific immune response following vaccination. Ad5 is a common respiratory pathogen and infection induces antibody responses that neutralize Ad5 upon subsequent exposure. Thus, the GUCY2C-based vaccine was evolved to circumvent limitations associated with pre-existing Ad5-specific immunity. Indeed, a chimeric Ad5.F35 is less susceptible to neutralization than Ad5 in mice and humans with pre-existing Ad5 immunity (Flickinger et al., [125]). Ad5.F35 comprises the Ad5 capsid, but the fiber of the rare Ad35. Natural respiratory infection with Ad5 primarily induces fiber-specific neutralizing antibodies [126], allowing Ad5.F35 to evade that neutralization. A Phase IIA clinical trial (NCT04111172) testing the efficacy of Ad5.F35-hGUCY2C-PADRE in GUCY2C-expressing gastrointestinal malignancies was initiated in 2020.
Antibody–drug conjugates
While GUCY2C vaccines rely on mucosal immune compartmentalization and CNS immune privilege to prime systemic antitumor responses, the safety and efficacy of antibody-directed immunotherapies including ADCs, BiTEs and CAR T cells is dependent on architectural differences between normal and cancer cells. In healthy intestinal tissue, membrane-bound GUCY2C is restricted to the luminal surface, and therefore, sits in an anatomically privileged location, separated from the vascular compartment by epithelial tight junctions and consequently inaccessible to circulating GUCY2C ligands [127–129] or GUCY2C-directed immunotherapeutics [130]. However, in CRC, apical-basal polarity is lost [131,132] and barrier permeability is increased [133] resulting in surface GUCY2C that is uniquely accessible to immunotherapeutics in systemic circulation.
ADCs are a class of drug consisting of a recombinant antibody linked to a cytotoxic agent [134,135]. Compared with direct administration of a chemotherapeutic agent alone, ADCs may allow for selective delivery of drugs to tumor cells while reducing exposure of noncancerous tissues [134,135]. To date, at least six ADCs have been FDA approved for use in oncology patients, with the majority of these ADCs targeting hematologic and breast malignancies [136].
Preclinical studies demonstrated proof-of-concept ADC internalization and killing of CRC cells in vitro and in vivo [137]. Subsequently, two clinical trials examined the use of GUCY2C-direted ADCs in patients with gastrointestinal malignancies. Both of these trials utilized an ADC, TAK-264, consisting of an anti-GUCY2C monoclonal antibody conjugated to the antimitotic agent monomethyl auristatin E using a cleavable peptide linker [138]. A Phase I dose escalation clinical study demonstrated TAK-264 safety with no dose-limiting toxicities observed at doses ranging from 0.3–1.5 mg/kg. However, at doses above 1.5 mg/kg, four of 19 patients experienced grade IV toxicities (most commonly neutropenia) [138]. A follow-up Phase II trial assessed TAK-264 efficacy in patients with advanced or metastatic pancreatic adenocarcinoma [139]. In this study, 43 patients received infusion of TAK-264 every 3 weeks, up to six administrations. The overall response rate was 3% with only one patient exhibiting a partial response. The authors concluded that the low efficacy of TAK-264 did not warrant further clinical investigation and the trial was subsequently terminated early due to futility [139]. Limited efficacy was speculated to be attributed to low chemotherapy drug potency, unstable linkers, poor tumor penetration and/or failure of the ADC to internalize, suggesting that further optimization of GUCY2C-targetd ADCs was required [139].
CAR T-cell therapy
Although GUCY2C vaccination generates antitumor immunity with long-lasting memory, homeostatic mechanisms within the T-cell compartment prevent these clones from expanding beyond a fraction of a percent of the T-cell pool [120,140]. Thus, while vaccination has undeniable benefits in prophylaxis and early stage disease models, as tumor burden increases with late-stage metastatic disease alternative approaches are required to increase the number of tumor-directed T cells. One strategy to overcome this limitation and to exponentially expand T-cell numbers is through adoptive cell therapy using engineered T-cell receptors (TCRs). In this therapy, T cells are isolated from a patient, transduced with a TCR clone specific for a patient’s cancer, expanded ex vivo, and administered back to the patient [141]. Tran et al. demonstrated the feasibility and efficacy of this approach to target a G12D mutation in the KRAS gene of a colorectal patient resulting in remission of multiple metastatic lesions [142]. While effective, this TCR-driven approach is highly specific for both a patient’s HLA allele and genetic mutation, thereby limiting widespread use [143].
A more universal approach, termed CAR T-cell therapy, exploits the cytolytic capabilities of T cells, but does so independently of peptide-MHC presentation [143]. This is accomplished using an antibody-derived construct comprised of the variable domains of the antibody heavy and light chains, expressed in a unified molecular format called a single-chain variable fragment (scFv). Binding of scFv to antigen is translated into a cellular response by pairing the extracellular scFv with the intracellular-signaling domains and costimulatory domains of the TCR complex, such as CD3ζ, CD28 and 4-1BB [144–146]. Two CAR T-cell therapeutics, tisagenlecleucel (Kymriah by Novartis, Basel, Switzerland) and axicabtagene ciloleucel (Yescarta by Kite Pharma, CA, USA), employing either 4-1BB or CD28 signaling domains, respectively, recently received FDA approval for leukemias and lymphomas expressing the B-cell antigen, CD19 [147–149]. However, no CAR T cell therapies are currently approved for CRC.
Preliminary studies assessing GUCY2C-specific CAR T-cell safety and efficacy evaluated two CARs with different affinities for mouse GUCY2C. In this study, the high-affinity CAR outperformed the low-affinity construct both in vitro, with cellular cytotoxicity assays and antigen-stimulated cytokine production, and in vivo, in mouse models of colorectal lung metastases [130]. Notably, in tumor challenge studies, the high-affinity CAR T cell was curative to 200 days post tumor inoculation in 25% of animals, whereas none of the animals receiving low-affinity CAR T cells survived beyond 50 days. Of equal importance, no GUCY2C-CAR mediated autoimmunity was observed in animals receiving the high-affinity CAR T cell. To track CAR T cells, the CAR construct included a green fluorescent protein reporter, and while green fluorescent protein-positive cells were observed in both spleen and tumor following injection, no fluorescent signal was detected in either the small intestine or the colon (p < 0.001). Moreover, histopathological scoring of CAR T-cell recipient mice in both the small intestine and colon showed no difference in tissue architecture or inflammatory infiltrates between animals treated with control CAR or GUCY2C-directed CAR T cells [130].
Additional studies employed a CAR design encoding a scFv with specificity for human GUCY2C. Using this CAR design, in a colorectal lung metastases model, dose finding studies demonstrated that a single injection at the highest dose, 107 CAR T cells, was curative in 60% of animals [150]. Repeated administration of CAR T cells at this dose increased curative survival to 80% at 100 days post tumor implantation, suggesting multiple doses may improve efficacy. Moreover, 75% of animals initially treated with GUCY2C CAR T cells survived beyond 80 days following tumor rechallenge, suggesting persistence and functioning of GUCY2C CAR T cells beyond 6 months from the time of initial dosing. Furthermore, in a CDX model of peritoneal metastases, using the human CRC cell line T84, human GUCY2C-directed CAR T cells were curative in 100% of animals tested [150]. These data robustly support the continued investigation of GUCY2C-directed CAR T-cell therapy for clinical implementation in gastrointestinal malignancies.
Bispecific T-cell engagers
BiTEs present an alternate antibody-derived strategy that redirects T-cells residing within tumors to tumor antigens. This is achieved through the fusion of two scFvs into a single molecule. One scFv has specificity for a tumor antigen while the other scFv has specificity for the extracellular domain of CD3ε on T cells [151]. This molecule creates a molecular bridge between the tumor cell and the T cell, promoting the formation of an immunological synapse, degranulation and cell death.
PF-07062119 is a GUCY2C-targeting bispecific molecule developed by Pfizer (NY, USA) for the treatment of gastrointestinal cancers [152]. This molecule is a variation of the BiTE called a dual-affinity retargeting antibody. Rather than conventional scFvs, this construct fuses the tumor antigen-specific antibody light chain to the CD3-specific antibody heavy chain, creating a more compact structure which has demonstrated improved cell killing over conventional BiTEs [153]. The inclusion of an Fc region improves the half-life of their bispecific antibody from 1 h to 1 week. The GUCY2C dual-affinity retargeting antibody-Fc construct controls tumor growth in numerous CRC CDX models, and one patient-derived xenograft model, encompassing varying levels of GUCY2C cell surface expression. In order to study the effect of combinatorial therapies, suboptimal doses of PF-07062119 was administered in combination with anti-PD-1 blockade or anti-VEGF-A antibody. While combination with PD-1 therapy delayed tumor growth by about 10 days, combination with anti-VEGF blockade resulted in tumor ablation. The authors ascribe this to the ability of VEGF-A blockade to promote T-cell infiltration into the tumor; however, higher doses of GUCY2C bispecific monotherapy in early studies was equally curative, suggesting poor T-cell infiltration may not be a barrier to efficacy given a therapeutic dose of the bispecific. A Phase I trial testing the safety of PF-07062119 in combination with anti-PD-1 and anti-VEGF therapies is currently ongoing (NCT04171141).
Of concern is the safety data generated with PF-07062119 in cynomolgus monkeys [152]. The binding affinity of PF-07062119 for human or cynomolgus GUCY2C is similar (human Kd [nM] = 7.47 ± 0.15; cynomolgus Kd [nM] = 3.01 ± 0.03) as well as the binding affinity for CD3 (human Kd [nM] = 23.97 ± 0.97; cynomolgus Kd [nM] = 24.12 ± 0.87). Unlike murine safety studies, cynomolgus monkeys treated with PF-07062119 exhibited villus atrophy in the small intestine, crypt hyperplasia in the small and large intestine, as well as increased immune cell infiltration in both small and large intestine. Interestingly, these latter two observations phenocopy changes imposed by genetically eliminating GUCY2C expression in mice [154] suggesting further evaluation is needed to determine if GUCY2C-expressing intestinal epithelial cells are being affected by this BiTE. The authors noted the absence of intestinal ulceration, erosions and necrosis of these animals suggesting that adverse events may resolve following cessation of therapy. Surveys of other GUCY2C-expressing tissues were not reported.
Conclusion & future perspective
Gastrointestinal cancers, and specifically CRC, account for a large fraction of all cancer deaths, suggesting a need for the development of new pharmacologic agents. In this context, the transmembrane protein GUCY2C has emerged as a promising target for the prevention, detection and treatment of gastrointestinal malignancies. GUCY2C exhibits a limited expression profile across healthy tissues and is normally confined to the apical surface of intestinal tissues and select CNS neurons. Detection of GUCY2C outside of these tissues represents malignant disease and GUCY2C is an established marker for identifying occult CRC metastases in pathology-negative lymph nodes. Moreover, beyond use as a prognostic biomarker in CRC, differences in GUCY2C expression, signaling and cellular localization between normal tissues and tumors permits safe and selective delivery of therapeutics to gastrointestinal cancers using GUCY2C as a target.
The applicability of GUCY2C-directed therapies covers the spectrum of disease and extends throughout the process of CRC carcinogenesis (Figure 1). In healthy patients who are at high risk for developing CRC, GUCY2C ligands may be effective as chemopreventive agents that halt disease before it occurs. In patients who present with localized tumors, GUCY2C vaccines may induce long-term immunity that actively opposes recurrence or new metastases. Finally, in patients who present with advanced disease, immunotherapeutics including ADCs, CAR T cells or BiTEs may be effective in eliminating established tumors. Beyond CRC, ectopic GUCY2C expression by esophageal, gastric and pancreatic malignancies suggest GUCY2C-directed therapies possess widespread utility. Indeed, it is estimated that approximately 25% of all cancer-related deaths in the USA derive from GUCY2C-expressing gastrointestinal malignancies and may therefore be susceptible to GUCY2C-targeted therapies [124,155].
Figure 1. . Guanylyl cyclase C-targeted therapies in colorectal cancer.
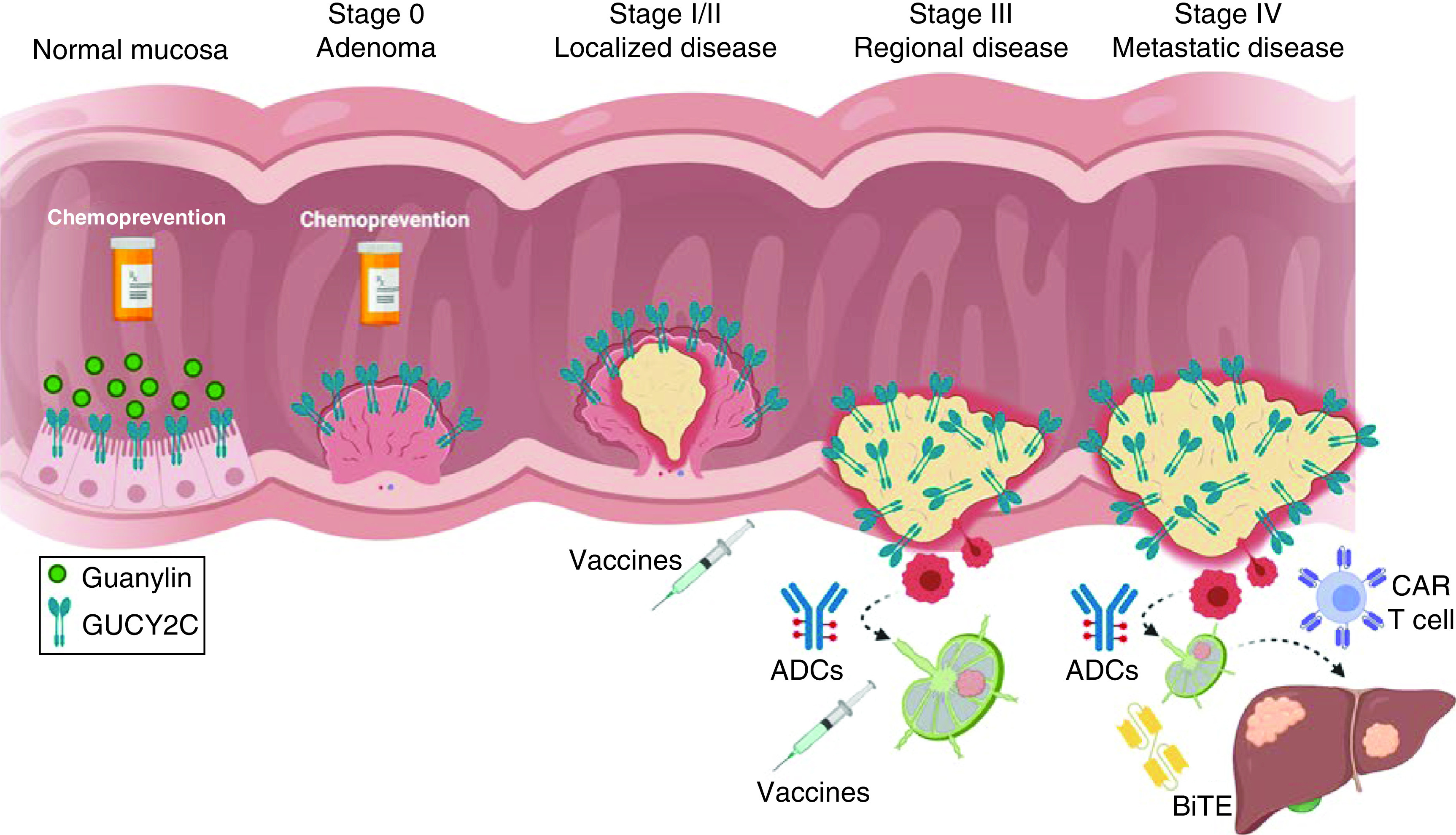
GUCY2C is an intestinal receptor that binds to its endogenous ligand, guanylin, in the colon. In healthy mucosa, the guanylin-GUCY2C signaling axis regulates homeostatic mechanisms including cell proliferation. During early stages of carcinogenesis, guanylin is silenced resulting in disruption of the guanylin-GUCY2C signaling axis and creation of a protumorigenic niche. Oral administration of GUCY2C ligands may restore the guanylin-GUCY2C signaling axis and prevent the development of colorectal cancer. As tumorigenesis progresses, GUCY2C-expressing colorectal cancer cells spread from the intestinal mucosa to neighboring lymph nodes and eventually to distant organs including the liver and lung. Administration of GUCY2C-targeted vaccines in patients with localized and regional disease may generate life-long GUCY2C immunity actively preventing colorectal cancer seeding to neighboring lymph nodes and distant organs respectively. In late-stage disease, GUCY2C-targeted ADCs, CAR T cells or BiTEs may be effective immunotherapeutics in eradicating established metastatic tumors.
ADC: Antibody–drug conjugate; BiTE: Bispecific T-cell engager; CAR: Chimeric-antigen receptor; GUCY2C: Guanylyl cyclase C.
GUCY2C-directed therapies represent a nascent field with broad potential. Preclinical studies have demonstrated antitumor efficacy across multiple models. Studies to date have focused on GUCY2C-targeted therapies in CRC, the malignancy canonically associated with GUCY2C expression. While presumably effective across other GUCY2C-positive gastrointestinal malignancies, future studies are required to demonstrate the translatability of this approach. Beyond these studies, clinical testing is necessary to advance GUCY2C-directed therapies into patient populations. Currently, clinical trials assessing GUCY2C-targeted chemoprevention, vaccines and BiTEs are underway, and it is expected that trials testing CAR T cells against GUCY2C will be initiated in the coming years. Data from these trials are forthcoming and will be highly informative in assessing the ability of GUCY2C-targeted therapies to prevent and/or treat gastrointestinal malignancies.
Executive summary.
In healthy people, guanylyl cyclase C (GUCY2C) is an intestinally-restricted transmembrane receptor that regulates fluid secretion, cell proliferation and intestinal homeostasis.
The endogenous GUCY2C ligand, guanylin, is silenced in colorectal cancer (CRC) tumorigenesis and GUCY2C ligand replacement in preclinical models prevents tumor development.
Across gastrointestinal cancers, GUCY2C is near universally expressed by primary and metastatic CRC tumors and ectopically expressed by a majority of esophageal, gastric and pancreatic tumors.
Detection of GUCY2C outside of intestinal tissues represents malignant disease and carries prognostic value in CRC patients.
Numerous GUCY2C-targeted immunotherapeutics including vaccines, antibody–drug conjugates, chimeric antigen-receptor-T cells and bispecific T-cell engagers have demonstrated safety and antitumor efficacy in preclinical models.
Acknowledgments
Figure 1 was created using BioRender.com.
Footnotes
Author contributions
JC Flickinger Jr, AE Snook and SA Waldman conceived the review. JC Flickinger Jr, JA Rappaport, JR Barton and TR Baybutt wrote the manuscript. AM Pattison constructed the figure. AE Snook and SA Waldman revised the manuscript.
Financial & competing interests disclosure
This work was supported in part by the National Institutes of Health (R01 CA204881, R01 CA206026 and P30 CA56036), the Defense Congressionally Directed Medical Research Program W81XWH-17-PRCRP-TTSA and Targeted Diagnostic & Therapeutics, Inc., to SA Waldman. AE Snook received a Research Starter Grant in Translational Medicine and Therapeutics from the PhRMA Foundation and was supported by the Defense Congressionally Directed Medical Research Programs (#W81XWH-17-1-0299, #W81XWH-19-1-0263 and #W81XWH-19-1-0067). SA Waldman and AE Snook also were supported by a grant from The Courtney Ann Diacont Memorial Foundation. JC Flickinger Jr and JR Barton are supported by a PhRMA Foundation Predoctoral Fellowship In Pharmacology/Toxicology. JA Rappaport is supported by an NIH Ruth Kirschstein Individual Predoctoral MD/PhD Fellowship (F30 CA232469). AM Pattison is supported by a Ruth Kirschstein Individual Research Fellowship Award (F31 CA225123). SA Waldman is the Samuel M.V. Hamilton Professor of Thomas Jefferson University. The authors have no other relevant affiliations or financialinvolvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Ashktorab H, Kupfer SS, Brim H, Carethers JM. Racial disparity in gastrointestinal cancer risk. Gastroenterology 153(4), 910–923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 14(2), 89–103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Goding Sauer Aet al. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 70(3), 145–164 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Pattison AM, Merlino DJ, Blomain ES, Waldman SA. Guanylyl cyclase C signaling axis and colon cancer prevention. World J. Gastroenterol. 22(36), 8070–8077 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rappaport JA, Waldman SA. The guanylate cyclase C-cGMP signaling axis opposes intestinal epithelial injury and neoplasia. Front. Oncol. 8, 299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valentino MA, Lin JE, Snook AEet al. A uroguanylin-GUCY2C endocrine axis regulates feeding in mice. J. Clin. Invest. 121(9), 3578–3588 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merlino DJ, Barton JR, Charsar BAet al. Two distinct GUCY2C circuits with PMV (hypothalamic) and SN/VTA (midbrain) origin. Brain Struct. Funct. 224(8), 2983–2999 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bashir B, Merlino DJ, Rappaport JAet al. Silencing the GUCA2A-GUCY2C tumor suppressor axis in CIN, serrated, and MSI colorectal neoplasia. Hum. Pathol. 87, 103–114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aka AA, Rappaport JA, Pattison AM, Sato T, Snook AE, Waldman SA. Guanylate cyclase C as a target for prevention, detection, and therapy in colorectal cancer. Expert Rev. Clin. Pharmacol. 10(5), 549–557 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baybutt TR, Aka AA, Snook AE. The heat-stable enterotoxin receptor, guanylyl cyclase C, as a pharmacological target in colorectal cancer immunotherapy: a bench-to-bedside current report. Toxins (Basel) 9(9), (2017). https://www.mdpi.com/2072-6651/9/9/282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gardner P, Chao AC, De Sauvage F. STa receptors: physiological and pathophysiological regulation of intestinal secretion by 5′-cyclic guanosine monophosphate. Gastroenterology 109(1), 325–327 (1995). [DOI] [PubMed] [Google Scholar]
- 12.de Sauvage FJ, Horuk R, Bennett G, Quan C, Burnier JP, Goeddel DV. Characterization of the recombinant human receptor for Escherichia coli heat-stable enterotoxin. J. Biol. Chem. 267(10), 6479–6482 (1992). [PubMed] [Google Scholar]
- 13.GBD 2015 LRI Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect. Dis. 17(11), 1133–1161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosangadi D, Smith PG, Kaslow DC, Giersing BK. WHO ETEC & Shigella Vaccine Consultation Expert Group. WHO consultation on ETEC and shigella burden of disease, Geneva, 6–7th April 2017: meeting report. Vaccine 37(50), 7381–7390 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Kuhn M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 96(2), 751–804 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Basu N, Arshad N, Visweswariah SS. Receptor guanylyl cyclase C (GC-C): regulation and signal transduction. Mol. Cell. Biochem. 334(1–2), 67–80 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Schulz S, Green CK, Yuen PS, Garbers DL. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell 63(5), 941–948 (1990). [DOI] [PubMed] [Google Scholar]
- 18.Singh S, Singh G, Heim JM, Gerzer R. Isolation and expression of a guanylate cyclase-coupled heat stable enterotoxin receptor cDNA from a human colonic cell line. Biochem. Biophys. Res. Commun. 179(3), 1455–1463 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Murakami H, Masui H. Hormonal control of human colon carcinoma cell growth in serum-free medium. Proc. Natl Acad. Sci. USA 77(6), 3464–3468 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Sauvage FJ, Camerato TR, Goeddel DV. Primary structure and functional expression of the human receptor for Escherichia coli heat-stable enterotoxin. J. Biol. Chem. 266(27), 17912–17918 (1991). [PubMed] [Google Scholar]
- 21.Biswas KH, Shenoy AR, Dutta A, Visweswariah SS. The evolution of guanylyl cyclases as multidomain proteins: conserved features of kinase-cyclase domain fusions. J. Mol. Evol. 68(6), 587–602 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Vaandrager AB. Structure and function of the heat-stable enterotoxin receptor/guanylyl cyclase C. Mol. Cell. Biochem. 230(1–2), 73–83 (2002). [PubMed] [Google Scholar]
- 23.Wada A, Hirayama T, Kitaura Het al. Identification of ligand recognition sites in heat-stable enterotoxin receptor, membrane-associated guanylyl cyclase C by site-directed mutational analysis. Infect. Immun. 64(12), 5144–5150 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasegawa M, Hidaka Y, Matsumoto Y, Sanni T, Shimonishi Y. Determination of the binding site on the extracellular domain of guanylyl cyclase C to heat-stable enterotoxin. J. Biol. Chem. 274(44), 31713–31718 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Lucas KA, Pitari GM, Kazerounian Set al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 52(3), 375–414 (2000). [PubMed] [Google Scholar]
- 26.Saha S, Biswas KH, Kondapalli C, Isloor N, Visweswariah SS. The linker region in receptor guanylyl cyclases is a key regulatory module: mutational analysis of guanylyl cyclase C. J. Biol. Chem. 284(40), 27135–27145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arshad N, Ballal S, Visweswariah SS. Site-specific N-linked glycosylation of receptor guanylyl cyclase C regulates ligand binding, ligand-mediated activation and interaction with vesicular integral membrane protein 36, VIP36. J. Biol. Chem. 288(6), 3907–3917 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arshad N, Visweswariah SS. Glycosylation of human receptor guanylyl cyclase C. BMC Pharmacol. 9(S1), (2009). [Google Scholar]
- 29.Ghanekar Y, Chandrashaker A, Tatu U, Visweswariah SS. Glycosylation of the receptor guanylate cyclase C: role in ligand binding and catalytic activity. Biochem. J. 379(Pt. 3), 653–663 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaleel M, Saha S, Shenoy AR, Visweswariah SS. The kinase homology domain of receptor guanylyl cyclase C: ATP binding and identification of an adenine nucleotide sensitive site. Biochemistry 45(6), 1888–1898 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Bhandari R, Suguna K, Visweswariah SS. Guanylyl cyclase C receptor: regulation of catalytic activity by ATP. Biosci. Rep. 19(3), 179–188 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Scott RO, Thelin WR, Milgram SL. A novel PDZ protein regulates the activity of guanylyl cyclase C, the heat-stable enterotoxin receptor. J. Biol. Chem. 277(25), 22934–22941 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Crane JK, Shanks KL. Phosphorylation and activation of the intestinal guanylyl cyclase receptor for Escherichia coli heat-stable toxin by protein kinase C. Mol. Cell. Biochem. 165(2), 111–120 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Wada A, Hasegawa M, Matsumoto Ket al. The significance of Ser1029 of the heat-stable enterotoxin receptor (STaR): relation of STa-mediated guanylyl cyclase activation and signaling by phorbol myristate acetate. FEBS Lett. 384(1), 75–77 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Leiper JB. Fate of ingested fluids: factors affecting gastric emptying and intestinal absorption of beverages in humans. Nutr. Rev. 73(Suppl. 2), 57–72 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Krause WJ, Cullingford GL, Freeman RHet al. Distribution of heat-stable enterotoxin/guanylin receptors in the intestinal tract of man and other mammals. J. Anat. 184(Pt. 2), 407–417 (1994). [PMC free article] [PubMed] [Google Scholar]
- 37.Currie MG, Fok KF, Kato Jet al. Guanylin: an endogenous activator of intestinal guanylate cyclase. Proc. Natl Acad. Sci. USA 89(3), 947–951 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamra FK, Forte LR, Eber SLet al. Uroguanylin: structure and activity of a second endogenous peptide that stimulates intestinal guanylate cyclase. Proc. Natl Acad. Sci. USA 90(22), 10464–10468 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taxt A, Aasland R, Sommerfelt H, Nataro J, Puntervoll P. Heat-stable enterotoxin of enterotoxigenic Escherichia coli as a vaccine target. Infect. Immun. 78(5), 1824–1831 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fasano A. Toxins and the gut: role in human disease. Gut 50(Suppl. 3), III9–14 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozaki H, Sato T, Kubota H, Hata Y, Katsube Y, Shimonishi Y. Molecular structure of the toxin domain of heat-stable enterotoxin produced by a pathogenic strain of Escherichia coli. A putative binding site for a binding protein on rat intestinal epithelial cell membranes. J. Biol. Chem. 266(9), 5934–5941 (1991). [PubMed] [Google Scholar]
- 42.Hamra FK, Eber SL, Chin DT, Currie MG, Forte LR. Regulation of intestinal uroguanylin/guanylin receptor-mediated responses by mucosal acidity. Proc. Natl Acad. Sci. USA 94(6), 2705–2710 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wall ME, Francis SH, Corbin JDet al. Mechanisms associated with cGMP binding and activation of cGMP-dependent protein kinase. Proc. Natl Acad. Sci. USA 100(5), 2380–2385 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaandrager AB, Hogema BM, de Jonge HR. Molecular properties and biological functions of cGMP-dependent protein kinase II. Front. Biosci. 10(1–3), 2150–2164 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Bijvelds MJC, Tresadern G, Hellemans Aet al. Selective inhibition of intestinal guanosine 3′,5′-cyclic monophosphate signaling by small-molecule protein kinase inhibitors. J. Biol. Chem. 293(21), 8173–8181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Markert T, Vaandrager AB, Gambaryan Set al. Endogenous expression of type II cGMP-dependent protein kinase mRNA and protein in rat intestine. Implications for cystic fibrosis transmembrane conductance regulator. J. Clin. Invest. 96(2), 822–830 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cuthbert AW, Hickman ME, MacVinish LJet al. Chloride secretion in response to guanylin in colonic epithelial from normal and transgenic cystic fibrosis mice. Br. J. Pharmacol. 112(1), 31–36 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaandrager AB, Smolenski A, Tilly BCet al. Membrane targeting of cGMP-dependent protein kinase is required for cystic fibrosis transmembrane conductance regulator Cl- channel activation. Proc. Natl Acad. Sci. USA 95(4), 1466–1471 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaandrager AB, Bot AG, Ruth P, Pfeifer A, Hofmann F, De Jonge HR. Differential role of cyclic GMP-dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology 118(1), 108–114 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Chen T, Kocinsky HS, Cha Bet al. Cyclic GMP kinase II (cGKII) inhibits NHE3 by altering its trafficking and phosphorylating NHE3 at three required sites: identification of a multifunctional phosphorylation site. J. Biol. Chem. 290(4), 1952–1965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y, Tan Q, Riederer Bet al. Deciphering ion transporters, kinases and PDZ-adaptor molecules that mediate guanylate cyclase C agonist-dependent intestinal fluid loss in vivo. Biochem. Pharmacol. 178, 114040 (2020). [DOI] [PubMed] [Google Scholar]
- 52.De Jonge HR, Tilly BC, Hogema BMet al. cGMP inhibition of type 3 phosphodiesterase is the major mechanism by which C-type natriuretic peptide activates CFTR in the shark rectal gland. Am. J. Physiol. Cell Physiol. 306(4), C343–C353 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chao AC, de Sauvage FJ, Dong YJ, Wagner JA, Goeddel DV, Gardner P. Activation of intestinal CFTR Cl- channel by heat-stable enterotoxin and guanylin via cAMP-dependent protein kinase. EMBO J. 13(5), 1065–1072 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beltrán AR, Carraro-Lacroix LR, Bezerra CNAet al. Escherichia coli heat-stable enterotoxin mediates Na+/H+ exchanger 4 inhibition involving cAMP in T84 human intestinal epithelial cells. PLoS ONE 10(12), e0146042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carey CM, Apple SE, Hilbert ZA, Kay MS, Elde NC. Conflicts with diarrheal pathogens trigger rapid evolution of an intestinal signaling axis. BioRxiv (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fitzpatrick DA, O'Halloran DM, Burnell AM. Multiple lineage specific expansions within the guanylyl cyclase gene family. BMC Evol. Biol. 6, 26 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Müller T, Rasool I, Heinz-Erian Pet al. Congenital secretory diarrhoea caused by activating germline mutations in GUCY2C. Gut 65(8), 1306–1313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith A, Bulman DE, Goldsmith Cet al. Meconium ileus in a Lebanese family secondary to mutations in the GUCY2C gene. Eur. J. Hum. Genet. 23(7), 990–992 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schulz S, Lopez MJ, Kuhn M, Garbers DL. Disruption of the guanylyl cyclase-C gene leads to a paradoxical phenotype of viable but heat-stable enterotoxin-resistant mice. J. Clin. Invest. 100(6), 1590–1595 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mann EA, Jump ML, Wu J, Yee E, Giannella RA. Mice lacking the guanylyl cyclase C receptor are resistant to STa-induced intestinal secretion. Biochem. Biophys. Res. Commun. 239(2), 463–466 (1997). [DOI] [PubMed] [Google Scholar]
- 61.Steinbrecher KA, Wowk SA, Rudolph JA, Witte DP, Cohen MB. Targeted inactivation of the mouse guanylin gene results in altered dynamics of colonic epithelial proliferation. Am. J. Pathol. 161(6), 2169–2178 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li P, Schulz S, Bombonati Aet al. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology 133(2), 599–607 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487(7407), 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rowan AJ, Lamlum H, Ilyas Met al. APC mutations in sporadic colorectal tumors: a mutational “hotspot” and interdependence of the “two hits”. Proc. Natl Acad. Sci. USA 97(7), 3352–3357 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kwong LN, Dove WF. APC and its modifiers in colon cancer. Adv. Exp. Med. Biol. 656, 85–106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin S, Wang J, Wang Let al. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am. J. Cancer Res. 7(1), 41–52 (2017). [PMC free article] [PubMed] [Google Scholar]
- 67.Lee K, Piazza G A. The interaction between the Wnt/β-catenin signaling cascade and PKG activation in cancer. J. Biomed. Res. 31(3), 189–196 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li N, Lee K, Xi Yet al. Phosphodiesterase 10A: a novel target for selective inhibition of colon tumor cell growth and β-catenin-dependent TCF transcriptional activity. Oncogene 34(12), 1499–1509 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Whitt JD, Li N, Tinsley HNet al. A novel sulindac derivative that potently suppresses colon tumor cell growth by inhibiting cGMP phosphodiesterase and β-catenin transcriptional activity. Cancer Prev. Res. (Phila.) 5(6), 822–833 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kwon I-K, Schoenlein PV, Delk Jet al. Expression of cyclic guanosine monophosphate-dependent protein kinase in metastatic colon carcinoma cells blocks tumor angiogenesis. Cancer 112(7), 1462–1470 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Shailubhai K, Yu HH, Karunanandaa Ket al. Uroguanylin treatment suppresses polyp formation in the Apc(Min/+) mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer Res. 60(18), 5151–5157 (2000). [PubMed] [Google Scholar]; •• The first report demonstrating guanylyl cyclase C (GUCY2C) ligand replacement prevents the development of polyps in mouse intestinal cancer models.
- 72.Cagir B, Gelmann A, Park Jet al. Guanylyl cyclase C messenger RNA is a biomarker for recurrent stage II colorectal cancer. Ann. Intern. Med. 131(11), 805–812 (1999). [DOI] [PubMed] [Google Scholar]
- 73.Carrithers SL, Barber MT, Biswas Set al. Guanylyl cyclase C is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proc. Natl Acad. Sci. USA 93(25), 14827–14832 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Danaee H, Kalebic T, Wyant Tet al. Consistent expression of guanylyl cyclase-C in primary and metastatic gastrointestinal cancers. PLoS ONE 12(12), e0189953 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; • A comprehensive analysis detailing GUCY2C protein status across hundreds of gastrointestinal tumors.
- 75.Blomain ES, Rappaport JA, Pattison AMet al. APC-β-catenin-TCF signaling silences the intestinal guanylin-GUCY2C tumor suppressor axis. Cancer Biol. Ther. 21(5), 441–451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilson C, Lin JE, Li Pet al. The paracrine hormone for the GUCY2C tumor suppressor, guanylin, is universally lost in colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 23(11), 2328–2337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates the universal loss of guanylin in human primary colorectal tumors.
- 77.Steinbrecher KA, Tuohy TM, Heppner Goss Ket al. Expression of guanylin is downregulated in mouse and human intestinal adenomas. Biochem. Biophys. Res. Commun. 273(1), 225–230 (2000). [DOI] [PubMed] [Google Scholar]
- 78.Zhang L, Zhou W, Velculescu VEet al. Gene expression profiles in normal and cancer cells. Science 276(5316), 1268–1272 (1997). [DOI] [PubMed] [Google Scholar]
- 79.Cohen MB, Hawkins JA, Witte DP. Guanylin mRNA expression in human intestine and colorectal adenocarcinoma. Lab. Invest. 78(1), 101–108 (1998). [PubMed] [Google Scholar]
- 80.Blomain ES, Pattison AM, Waldman SA. GUCY2C ligand replacement to prevent colorectal cancer. Cancer Biol. Ther. 17(7), 713–718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harmel-Laws E, Mann EA, Cohen MB, Steinbrecher KA. Guanylate cyclase C deficiency causes severe inflammation in a murine model of spontaneous colitis. PLoS ONE 8(11), e79180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Di Guglielmo MD, Perdue L, Adeyemi A, van Golen KL, Corao DU. Immunohistochemical staining for uroguanylin, a satiety hormone, is decreased in intestinal tissue specimens from female adolescents with obesity. Pediatr. Dev. Pathol. 21(3), 285–295 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin JE, Colon-Gonzalez F, Blomain Eet al. Obesity-induced colorectal cancer is driven by caloric silencing of the guanylin-GUCY2C paracrine signaling axis. Cancer Res. 76(2), 339–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pitari GM, Di Guglielmo MD, Park J, Schulz S, Waldman SA. Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc. Natl Acad. Sci. USA 98(14), 7846–7851 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pitari GM, Zingman LV, Hodgson DMet al. Bacterial enterotoxins are associated with resistance to colon cancer. Proc. Natl Acad. Sci. USA 100(5), 2695–2699 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li P, Lin JE, Snook AE, Waldman SA. ST-producing E. coli oppose carcinogen-induced colorectal tumorigenesis in mice. Toxins (Basel) 9(9), (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schoenfeld P, Lacy BE, Chey WDet al. Low-dose linaclotide (72 μg) for chronic idiopathic constipation: a 12-week, randomized, double-blind, placebo-controlled trial. Am. J. Gastroenterol. 113(1), 105–114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shah ED, Kim HM, Schoenfeld P. Efficacy and tolerability of guanylate cyclase-C agonists for irritable bowel syndrome with constipation and chronic idiopathic constipation: a systematic review and meta-analysis. Am. J. Gastroenterol. 113(3), 329–338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Andersson KE. PDE5 inhibitors - pharmacology and clinical applications 20 years after sildenafil discovery. Br. J. Pharmacol. 175(13), 2554–2565 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sharman SK, Islam BN, Hou Yet al. Cyclic-GMP-elevating agents suppress polyposis in APCmin mice by targeting the preneoplastic epithelium. Cancer Prev. Res. (Phila.) 11(2), 81–92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chang W-CL, Masih S, Thadi Aet al. Plecanatide-mediated activation of guanylate cyclase-C suppresses inflammation-induced colorectal carcinogenesis in Apc+/Min-FCCC mice. World J. Gastrointest. Pharmacol. Ther. 8(1), 47–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Islam BN, Sharman SK, Hou Yet al. Sildenafil suppresses inflammation-driven colorectal cancer in mice. Cancer Prev. Res. (Phila.) 10(7), 377–388 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang W, Sundquist J, Sundquist K, Ji J. Use of phosphodiesterase 5 inhibitors is associated with lower risk of colorectal cancer in men with benign colorectal neoplasms. Gastroenterology 157(3), 672–681.e4 (2019). [DOI] [PubMed] [Google Scholar]
- 94.Weinberg DS, Lin JE, Foster NRet al. Bioactivity of oral linaclotide in human colorectum for cancer chemoprevention. Cancer Prev. Res. (Phila.) 10(6), 345–354 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carrithers SL, Parkinson SJ, Goldstein S, Park P, Robertson DC, Waldman SA. Escherichia coli heat-stable toxin receptors in human colonic tumors. Gastroenterology 107(6), 1653–1661 (1994). [DOI] [PubMed] [Google Scholar]
- 96.Park J, Schulz S, Haaf J, Kairys JC, Waldman SA. Ectopic expression of guanylyl cyclase C in adenocarcinomas of the esophagus and stomach. Cancer Epidemiol. Biomarkers Prev. 11(8), 739–744 (2002). [PubMed] [Google Scholar]
- 97.Ong MLH, Schofield JB. Assessment of lymph node involvement in colorectal cancer. World J. Gastrointest. Surg. 8(3), 179–192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Van Cutsem E, Oliveira J. ESMO Guidelines Working Group. Primary colon cancer: ESMO clinical recommendations for diagnosis, adjuvant treatment and follow-up. Ann. Oncol. 20(Suppl. 4), 49–50 (2009). [DOI] [PubMed] [Google Scholar]
- 99.Nagtegaal ID, Schmoll H-J. Colorectal cancer: what is the role of lymph node metastases in the progression of colorectal cancer? Nat. Rev. Gastroenterol. Hepatol. 14(11), 633–634 (2017). [DOI] [PubMed] [Google Scholar]
- 100.Rahbari NN, Bork U, Motschall Eet al. Molecular detection of tumor cells in regional lymph nodes is associated with disease recurrence and poor survival in node-negative colorectal cancer: a systematic review and meta-analysis. J. Clin. Oncol. 30(1), 60–70 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Waldman SA, Hyslop T, Schulz Set al. Association of GUCY2C expression in lymph nodes with time to recurrence and disease-free survival in pN0 colorectal cancer. JAMA 301(7), 745–752 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The first prospective clinical study defining GUCY2C as a prognostic biomarker in colorectal cancer patients with histopathology-negative lymph nodes.
- 102.Beaulieu M, Desaulniers M, Bertrand Net al. Analytical performance of a qRT-PCR assay to detect guanylyl cyclase C in FFPE lymph nodes of patients with colon cancer. Diagn. Mol. Pathol. 19(1), 20–27 (2010). [DOI] [PubMed] [Google Scholar]
- 103.Haince J-F, Houde M, Beaudry Get al. Comparison of histopathology and RT-qPCR amplification of guanylyl cyclase C for detection of colon cancer metastases in lymph nodes. J. Clin. Pathol. 63(6), 530–537 (2010). [DOI] [PubMed] [Google Scholar]
- 104.Wang W, Kandimalla R, Huang Het al. Molecular subtyping of colorectal cancer: recent progress, new challenges and emerging opportunities. Semin. Cancer Biol. 55, 37–52 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bae JM, Kim JH, Kang GH. Molecular subtypes of colorectal cancer and their clinicopathologic features, with an emphasis on the serrated neoplasia pathway. Arch. Pathol. Lab. Med. 140(5), 406–412 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Marisa L, de Reyniès A, Duval Aet al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 10(5), e1001453 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Park J, Schulz S, Waldman SA. Intestine-specific activity of the human guanylyl cyclase C promoter is regulated by Cdx2. Gastroenterology 119(1), 89–96 (2000). [DOI] [PubMed] [Google Scholar]
- 108.Birbe R, Palazzo JP, Walters R, Weinberg D, Schulz S, Waldman SA. Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum. Pathol. 36(2), 170–179 (2005). [DOI] [PubMed] [Google Scholar]
- 109.Ganesh K, Stadler ZK, Cercek Aet al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 16(6), 361–375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Snook AE, Stafford BJ, Li Pet al. Guanylyl cyclase C-induced immunotherapeutic responses opposing tumor metastases without autoimmunity. J. Natl Cancer Inst. 100(13), 950–961 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Riihimäki M, Hemminki A, Sundquist J, Hemminki K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 6, 29765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Belyakov IM, Ahlers JD, Brandwein BYet al. The importance of local mucosal HIV-specific CD8(+) cytotoxic T lymphocytes for resistance to mucosal viral transmission in mice and enhancement of resistance by local administration of IL-12. J. Clin. Invest. 102(12), 2072–2081 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cromwell MA, Veazey RS, Altman JDet al. Induction of mucosal homing virus-specific CD8(+) T lymphocytes by attenuated simian immunodeficiency virus. J. Virol. 74(18), 8762–8766 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Belyakov IM, Derby MA, Ahlers JDet al. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc. Natl Acad. Sci. USA 95(4), 1709–1714 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lycke N. Recent progress in mucosal vaccine development: potential and limitations. Nat. Rev. Immunol. 12(8), 592–605 (2012). [DOI] [PubMed] [Google Scholar]
- 116.Chang S-Y, Ko H-J, Kweon M-N. Mucosal dendritic cells shape mucosal immunity. Exp. Mol. Med. 46, e84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 18(2), 123–131 (2017). [DOI] [PubMed] [Google Scholar]
- 118.Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J. Clin. Invest. 120(5), 1368–1379 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Snook AE, Li P, Stafford BJet al. Lineage-specific T-cell responses to cancer mucosa antigen oppose systemic metastases without mucosal inflammatory disease. Cancer Res. 69(8), 3537–3544 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Snook AE, Magee MS, Schulz S, Waldman SA. Selective antigen-specific CD4(+) T-cell, but not CD8(+) T- or B-cell, tolerance corrupts cancer immunotherapy. Eur. J. Immunol. 44(7), 1956–1966 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 18(10), 635–647 (2018). [DOI] [PubMed] [Google Scholar]
- 122.Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 16(2), 102–111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xiang B, Baybutt TR, Berman-Booty Let al. Prime-boost immunization eliminates metastatic colorectal cancer by producing high-avidity effector CD8+ T cells. J. Immunol. 198(9), 3507–3514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Snook AE, Baybutt TR, Xiang Bet al. Split tolerance permits safe Ad5-GUCY2C-PADRE vaccine-induced T-cell responses in colon cancer patients. J. Immunother. Cancer 7(1), 104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The first clinical trial detailing the safety and immunogenicity of a GUCY2C-targeted vaccine in humans.
- 125.Flickinger JC, Singh J, Carlson Ret al. Chimeric Ad5.F35 vector evades anti-adenovirus serotype 5 neutralization opposing GUCY2C-targeted antitumor immunity. J. Immunother. Cancer 8(2), e001046 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cheng C, Gall JGD, Nason Met al. Differential specificity and immunogenicity of adenovirus type 5 neutralizing antibodies elicited by natural infection or immunization. J. Virol. 84(1), 630–638 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wolfe HR, Mendizabal M, Lleong Eet al. In vivo imaging of human colon cancer xenografts in immunodeficient mice using a guanylyl cyclase C--specific ligand. J. Nucl. Med. 43(3), 392–399 (2002). [PubMed] [Google Scholar]
- 128.Guarino A, Cohen MB, Overmann G, Thompson MR, Giannella RA. Binding of E. coli heat-stable enterotoxin to rat intestinal brush borders and to basolateral membranes. Dig. Dis. Sci. 32(9), 1017–1026 (1987). [DOI] [PubMed] [Google Scholar]
- 129.Carrithers SL, Parkinson SJ, Goldstein SD, Park PK, Urbanski RW, Waldman SA. Escherichia coli heat-stable enterotoxin receptors. A novel marker for colorectal tumors. Dis. Colon Rectum 39(2), 171–181 (1996). [DOI] [PubMed] [Google Scholar]
- 130.Magee MS, Kraft CL, Abraham TSet al. GUCY2C-directed CAR T cells oppose colorectal cancer metastases without autoimmunity. Oncoimmunology 5(10), e1227897 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Halaoui R, McCaffrey L. Rewiring cell polarity signaling in cancer. Oncogene 34(8), 939–950 (2015). [DOI] [PubMed] [Google Scholar]
- 132.Darido C, Buchert M, Pannequin Jet al. Defective claudin-7 regulation by Tcf-4 and Sox-9 disrupts the polarity and increases the tumorigenicity of colorectal cancer cells. Cancer Res. 68(11), 4258–4268 (2008). [DOI] [PubMed] [Google Scholar]
- 133.Soler AP, Miller RD, Laughlin KV, Carp NZ, Klurfeld DM, Mullin JM. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis 20(8), 1425–1431 (1999). [DOI] [PubMed] [Google Scholar]
- 134.Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 16(5), 315–337 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Chau CH, Steeg PS, Figg WD. Antibody–drug conjugates for cancer. Lancet 394(10200), 793–804 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Leung D, Wurst JM, Liu T, Martinez RM, Datta-Mannan A, Feng Y. Antibody conjugates-recent advances and future innovations. Antibodies (Basel) 9(1), (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Marszalowicz GP, Snook AE, Magee MS, Merlino D, Berman-Booty LD, Waldman SA. GUCY2C lysosomotropic endocytosis delivers immunotoxin therapy to metastatic colorectal cancer. Oncotarget 5(19), 9460–9471 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Almhanna K, Kalebic T, Cruz Cet al. Phase I study of the investigational anti-guanylyl cyclase antibody–drug conjugate TAK-264 (MLN0264) in adult patients with advanced gastrointestinal malignancies. Clin. Cancer Res. 22(20), 5049–5057 (2016). [DOI] [PubMed] [Google Scholar]
- 139.Almhanna K, Wright D, Mercade TMet al. A Phase II study of antibody–drug conjugate, TAK-264 (MLN0264) in previously treated patients with advanced or metastatic pancreatic adenocarcinoma expressing guanylyl cyclase C. Invest. New Drugs 35(5), 634–641 (2017). [DOI] [PubMed] [Google Scholar]
- 140.Alanio C, Lemaitre F, Law HKW, Hasan M, Albert ML. Enumeration of human antigen-specific naïve CD8+ T cells reveals conserved precursor frequencies. Blood 115(18), 3718–3725 (2010). [DOI] [PubMed] [Google Scholar]
- 141.Zhang J, Wang L. The emerging world of TCR-T cell trials against cancer: a systematic review. Technol. Cancer Res. Treat. 18, (2019) (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tran E, Robbins PF, Lu Y-Cet al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 375(23), 2255–2262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhao L, Cao YJ. Engineered T cell therapy for cancer in the clinic. Front. Immunol. 10, 2250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T-cell receptor with antibody type-specificity. Transplant. Proc. 21(1 Pt. 1), 127–130 (1989). [PubMed] [Google Scholar]
- 145.Brentjens RJ, Santos E, Nikhamin Yet al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 13(18 Pt. 1), 5426–5435 (2007). [DOI] [PubMed] [Google Scholar]
- 146.Milone MC, Fish JD, Carpenito Cet al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 17(8), 1453–1464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Maude SL, Laetsch TW, Buechner Jet al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378(5), 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schuster SJ, Bishop MR, Tam CSet al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 380(1), 45–56 (2019). [DOI] [PubMed] [Google Scholar]
- 149.Neelapu SS, Locke FL, Bartlett NLet al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377(26), 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Magee MS, Abraham TS, Baybutt TRet al. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol. Res. 6(5), 509–516 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates the efficacy of GUCY2C-targeted chimeric antigen-receptor-T cells in eradicating human colorectal tumors.
- 151.Suurs FV, Lub-de Hooge MN, de Vries EGE, de Groot DJA. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 201, 103–119 (2019). [DOI] [PubMed] [Google Scholar]
- 152.Mathur D, Root AR, Bugaj-Gaweda Bet al. A novel GUCY2C-CD3 T-cell engaging bispecific construct (PF-07062119) for the treatment of gastrointestinal cancers. Clin. Cancer Res. 26(9), 2188–2202 (2020). [DOI] [PubMed] [Google Scholar]
- 153.Moore PA, Zhang W, Rainey GJet al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 117(17), 4542–4551 (2011). [DOI] [PubMed] [Google Scholar]
- 154.Li P, Lin JE, Chervoneva I, Schulz S, Waldman SA, Pitari GM. Homeostatic control of the crypt-villus axis by the bacterial enterotoxin receptor guanylyl cyclase C restricts the proliferating compartment in intestine. Am. J. Pathol. 171(6), 1847–1858 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J. Clin. 67(1), 7–30 (2017). [DOI] [PubMed] [Google Scholar]