

ABSTRACT
Immunotherapy has demonstrated significant activity in a broad range of cancer types, but still the majority of patients receiving it do not maintain durable therapeutic responses. Amino acid metabolism has been proposed to be involved in the regulation of immune response. Here, we investigated in detail the role of arginase 1 (Arg1) in the modulation of antitumor immune response against poorly immunogenic Lewis lung carcinoma. We observed that tumor progression is associated with an incremental increase in the number of Arg1+ myeloid cells that accumulate in the tumor microenvironment and cause systemic depletion of ʟ-arginine. In advanced tumors, the systemic concentrations of ʟ-arginine are decreased to levels that impair the proliferation of antigen-specific T-cells. Systemic or myeloid-specific Arg1 deletion improves antigen-induced proliferation of adoptively transferred T-cells and leads to inhibition of tumor growth. Arginase inhibitor was demonstrated to modestly inhibit tumor growth when used alone, and to potentiate antitumor effects of anti-PD-1 monoclonal antibodies and STING agonist. The effectiveness of the combination immunotherapy was insufficient to induce complete antitumor responses, but was significantly better than treatment with the checkpoint inhibitor alone. Together, these results indicate that arginase inhibition alone is of modest therapeutic benefit in poorly immunogenic tumors; however, in combination with other treatment strategies it may significantly improve survival outcomes.
KEYWORDS: Arginase, tumor microenvironment, immunosuppression, arginase inhibitor, immunotherapy, t-cells response, myeloid cells
Introduction
Cancer immunotherapy has become an effective therapeutic strategy. Many types of solid tumors are being treated with checkpoint inhibitors that inactivate regulatory pathways controlling the immune response. Despite unprecedented antitumor efficacy, checkpoint inhibitors are effective in a minority of cancer patients. The identity of the resistance mechanisms is still poorly understood and has become a priority for cancer researchers. Mounting evidence indicates that the tumor microenvironment alters lymphoid and myeloid cells and converts them into potent immunoregulatory cells.1 Coincidentally, these regulatory pathways impair the development and/or activity of adaptive immune mechanisms that could be involved in the eradication of tumor cells.2 Amino acid metabolism plays a role in the immune response regulation. Increased degradation of several amino acids impairs T-cell activation, proliferation, and effector functions. However, only two groups of enzymes, i.e. those that catabolize ʟ-tryptophan or ʟ-arginine (ʟ-arg) have been reported to be substantially increased in many types of cancers.3–5 Quantification of interstitial fluid metabolites revealed that ʟ-arg is the most strongly depleted amino acid in the microenvironment of murine tumors.6 Moreover, ʟ-arg concentrations in the core regions of solid tumors was shown to be even 5 times lower as compared with tumor periphery and this difference was the highest among all of the amino acids measured.7
Increased expression of arginases (either Arg1 or Arg2) is perceived as a poor prognostic factor in a variety of cancer types including lung cancer,8 head and neck cancer,9 neuroblastoma,10 acute myeloid leukemia,11 pancreatic ductal carcinoma,12 ovarian carcinoma,13 or colorectal cancer.14 Additionally, increased Arg activity was found in skin,15 cervical,16 thyroid follicular,17 thyroid papillary and follicular variant of papillary,17 gastric, bile duct,18 hepatocellular,19 breast20 and esophageal21 cancers, although the clear impact of increased Arg activity on patients’ prognosis has not been reported in these tumor types. There are also ʟ-arg auxotrophic tumors such as melanoma22 and renal carcinoma,23,24 where no correlations between ʟ-arg concentrations and survival have been found.
Although some studies reported that arginases can be produced by tumor cells,10,11,25 it is currently well established that the major source of ʟ-arg-metabolizing cells is found in the tumor stroma. Arg1 is produced by various populations of tumor-infiltrating myeloid cells, such as macrophages, granulocytes, dendritic cells as well as immature precursors of myeloid cells that due to their immunoregulatory functions are referred to as myeloid-derived suppressor cells (MDSCs). Two major subsets of MDSCs, i.e. monocytic (M-MDSC) and granulocytic (G-MDSC also known as polymorphonuclear, PMN-MDSC in humans) are commonly recognized.26 MDSCs express immune checkpoint molecules, deplete essential metabolites, release immunosuppressive adenosine and its metabolites, produce reactive oxygen species, secrete immunoregulatory cytokines, growth-promoting, and proangiogenic factors, contributing to altered immune microenvironment favoring tumor progression.26
ʟ-Arg metabolism plays a role in T-cell-receptor (TCR) signaling. Its depletion by Args downregulates the expression of TCR-associated CD3ζ chain, which is part of the TCR complex involved in signal transduction.27,28 Decreased levels of CD3ζ are associated with attenuated T-cell proliferation triggered by mitogens,28–32 decreased cytokine production,29,30,33 or with impaired acquisition of effector functions by cytotoxic T-cells.34 Arg1-expressing MDSCs were also shown to induce regulatory T-cells (Tregs) in murine tumor models35 as well as in cancer patients.36 All these effects allow tumor cells to escape from T-cell-mediated immune surveillance. Blocking ʟ-arg degradation was therefore proposed to shift the amino acid metabolism to favor lymphocyte proliferation.
Previous studies in mice indicated that treatment with Arg inhibitors such as nor-NOHA or CB-1158 resulted in reduced tumor growth28,37 indicating that Arg is a relevant therapeutic target. In this study, we investigated the role of Arg1 in the tumor microenvironment of lung carcinoma and test whether a novel Arg inhibitor13,38 would be able to modulate antitumor immune response either alone or in combination with other immunotherapeutic approaches. The results obtained in this study confirm previous findings indicating increased infiltration of tumors with Arg1-producing myeloid cells and expand these observations by providing more detailed analysis of cell populations that lead to a systemic decrease in ʟ-arg concentrations. Moreover, we show that a novel arginase inhibitor (OAT-1746) that targets both extracellular and intracellular enzyme exerts antitumor effects. Finally, we confirm that Arg inhibitor can potentiate antitumor effects of checkpoint inhibitor and show for the first time that the antitumor effectiveness can be further improved by the triple combination consisting of Arg inhibitor (OAT-1746), checkpoint inhibitor (anti-PD-1) and STING agonist (DMXAA).
Methods
Mice
All experiments were performed in 8–12-week-old female mice. Wild type (WT) C57BL/6 mice were obtained from the Animal House of the Medical Research Center, Polish Academy of Sciences (Warsaw, Poland). Transgenic mice C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I, stock #003831), B6.129S4-Arg1tm1Lky/J (YARG, stock #015857), B6.Cg-Foxp3tm2Tch/J (Foxp3EGFP, stock #006772), B6(Cg)-Rag2tm1.1Cgn/J (RAG2 knock-out (KO), stock #008449), C57BL/6-Arg1tm1Pmu/J (Arg1fl/fl, stock #008817), B6.129P2-Lyz2tm1(cre)Ifo/J (LysMcre, stock #004781), B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J (R26-CreERT2, stock #008463) were purchased from The Jackson Laboratory. See online supplementary table 1 for a detailed description of the mouse strains used in this study. Animals were housed in controlled environmental conditions in specific-pathogen free (SPF) (transgenic mice) or conventional (WT mice) animal facility of the Medical University of Warsaw with water and food provided ad libitum. The experiments were performed in accordance with the guidelines approved by the 1st Local Ethics Committee in Warsaw (approvals No. 193/2016, 289/2017, and 317/2017), and in accordance with the requirements of EU (Directive 2010/63/EU) and Polish (Dz. U. poz. 266/15.01.2015) legislation.
Generation of myeloid Arg1 KO and tamoxifen-inducible total Arg1 KO mice
Mice with constitutive Arg1 deficiency in the myeloid cells were generated by mating C57BL/6-Arg1tm1Pmu/J (LoxP-flanked (floxed) Arg1 – Arg1fl/fl mice) with the “deleter” strain B6.129P2-Lyz2tm1(cre)Ifo/J (LysMcre mice). The progeny (Arg1fl/fl/LysMcre mice) was genotyped and referred to as myelo Arg1 KO. Mice with tamoxifen-inducible Arg1 deficiency in all tissues were generated by mating C57BL/6-Arg1tm1Pmu/J (Arg1fl/fl mice) with B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J mice (R26-CreERT2 mice). The progeny was genotyped and referred as Arg1 KO. For mice genotyping, DNA was isolated using DNeasy Blood & Tissue Kits (QIAGEN) according to the manufacturer’s instructions. DNA purity and concentration were measured using NanoDrop 2000 c spectrophotometer (Thermo Fisher Scientific). PCR reaction was set up using OneTaq® 2× Master Mix with Standard Buffer (New England Biolabs) and appropriate primers listed in the online supplementary table 2. PCR and agarose electrophoresis conditions were set according to genotyping protocols available on The Jackson Laboratory website (https://www.jax.org). Bands were visualized using ChemiDoc Imaging System (Bio-Rad). Only mice confirmed to have the desired genotype were used in the studies. Mice were bred in the SPF animal facility of the Department of Immunology, Medical University of Warsaw according to the Ministry of Environment Approval No. 69/2018 for the breeding of the genetically modified organisms.
Cell lines
Murine Lewis lung carcinoma cell line (3LL, CRL-1642 also referred to as LL/2 or LLC1), B16 F10 (murine cutaneous melanoma cell line, CRL-6475), K562 (human chronic myeloid leukemia cell line, CRL-3343), EL4 (human T lymphoblast cell line, TIB-39), RAJI (human B-cell lymphoma, CRL-86) were obtained from American Type Culture Collection. PANC02 murine pancreatic cancer cell line PANC 02 murine pancreatic carcinoma cells were kindly obtained from Carsten Ziske (Rheinische Friedrich-Wilhelms-Universität, Bonn, Germany). 3LL, RAJI, K562, and EL4 cells were cultured in RPMI 1640 medium supplemented with heat-inactivated 10% (v/v) fetal bovine serum (FBS, HyClone), 2 mM ʟ-glutamine (Sigma-Aldrich), 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich) at 37°C in an atmosphere of 5% CO2 in the air. B16F10 and PANC02 cells were cultured in DMEM (Sigma-Aldrich) supplemented with aforementioned additives. Cells have been cultured no longer than 3 weeks after thawing and were regularly tested for Mycoplasma contamination using PCR technique and were confirmed to be negative.
Generation of 3LL cells stably expressing murine Arg1
3LL cells stably expressing murine Arg1 (3LL-pLVX-Arg1) were generated by transduction with second-generation lentiviruses. Plasmid encoding murine full-length Arg1 cDNA (pLVX-Arg1-IRES-Puro, Thermo Fisher Scientific) was generated as described previously.13 Transduced 3LL cells were selected with 4.5 μg/ml puromycin (Sigma-Aldrich). 3LL cells transduced with an empty pLVX-IRES-Puro vector served as controls (referred to as 3LL-pLVX).
In vivo tumor models and treatment
3LL cells (1 × 106 or 0.5 × 106 or 0.1 × 106 as stated in each figure caption) were inoculated in 30 µL of phosphate-buffered saline (PBS) subcutaneously into the right thigh of 8–12-week-old female C57BL/6 mice. Arg inhibitor (OAT-1746) provided by OncoArendi Therapeutics, Warsaw, Poland, was dissolved in PBS and administered by intraperitoneal (i.p.) injections twice daily at a dose of 20 mg/kg for the first 14 days post inoculation of tumor cells. Control mice received PBS i.p. Anti-PD-1 (clone: RMP1-14, BioXCell) or rat IgG2a isotype control (clone: 2A3, BioXCell) antibodies were administered i.p. at a dose of 10 mg/kg on days: 6, 9, 12, 15, 18, and 21 after inoculation of tumor cells. DMXAA (5,6-dimethylxanthenone-4-acetic acid, Selleckchem), dissolved in 5% NaHCO3, was given intratumorally on day 8 post inoculation of tumor cells at a dose of 0.5 µg/mouse. Control groups received NaHCO3. To induce total Arg1 KO in mice tamoxifen diluted in peanut oil (both from Sigma-Aldrich) was administered by oral gavage at a dose of 75 mg/kg per mouse on days 7–12 post inoculation of tumor cells. Control groups received peanut oil on the same days. Tumor growth was monitored in three dimensions using a digital caliper starting from days 6–8 post tumor cell inoculation. Tumor volume was calculated according to the formula: V (mm3) = length × width × height × /6 and included thighs in the two measured dimensions. For survival evaluation, humane endpoints were applied including criteria such as severe cachexia or any tumor diameter >15 mm. At the end of the experiment, mice were sacrificed and selected organs such as inguinal lymph nodes, spleens and tumors as well as blood were used for further analysis. Small, intermediate and large tumors were defined as tumors reaching the volumes of ~150, 850 and 3000 mm3, respectively.
Flow cytometry analysis
For flow cytometry analysis, tumors were cut into small pieces and digested for 60 minutes (min) at 37°C with Collagenase type IV (600 U; Sigma-Aldrich) and DNAse I (400 U, Sigma-Aldrich) diluted in RPMI medium. Next, tissues were dissociated using gentleMACS Dissociator (Miltenyi Biotec) and filtered through a 100 µm cell strainer and washed with PBS. Spleens and lymph nodes were mashed through a 70 µm cell strainer and washed with PBS. When necessary, erythrocytes were lysed using red blood cell lysis buffer (155 mmol/L NH4Cl, 10 mmol/L NaH2CO3, and 0.1 mmol/L EDTA, pH 7.3). For detection of cell surface antigens, cells were first stained with Zombie Fixable Viability Kit (BioLegend) according to manufacturer’s instructions, blocked on ice with 5% normal rat serum in FACS buffer (PBS; 1% BSA, 0.01% NaN3) and then incubated for 30 min on ice with fluorochrome-conjugated antibodies (listed in the online supplementary table 3). When necessary, controls for background staining such as isotype or FMO (fluorescence minus one) controls were applied. After washing with FACS buffer, cells were immediately acquired. For intracellular staining, cell surface antigens-stained cells were fixed using Fixation Buffer for 30 min at room temperature (RT), followed by washing in Permeabilization Buffer, and staining with antibody diluted in Permeabilization Buffer for 30 min (RT, Intracellular Fixation & Permeabilization Buffer Set, eBioscience). Flow cytometry analysis was done using FACSCanto II or FACSAria III cytometers (BD Biosciences) operated by FACSDiva v6.0 software. For data analysis Flow Jo v7.6.5 software (Tree Star) was used. Gating strategies are presented in online supplementary figure 1 (immunophenotyping of tumor-infiltrating Arg1-expressing cells) and in online supplementary figure 2 (evaluation of OT-I T-cells proliferation and CD3ζ expression).
In vivo T-cell proliferation assay
Ovalbumin (OVA)-derived peptide 257–264 (SIINFEKL)-specific CD8+ T-cells were isolated from the spleen and lymph nodes of C57BL/6-Tg(TcraTcrb) 1100Mjb/J (OT-I) mice and labeled with Cell Trace Violet (CTV) dye (Thermo Fisher Scientific) for 20 min at 37°C at a final concentration of 2.5 μM. Next, 4–6 × 106 cells in 150 μl of PBS were immediately transferred into the caudal vein of host C57BL/6 mice. The next day, host mice were challenged with 5 μg of full-length OVA protein (grade VII, Sigma-Aldrich) injected subcutaneously in a total volume of 30 μl of PBS into the tumor area (experimental groups) or right thigh (positive control group). The negative control (unstimulated) group did not receive OVA nor was inoculated with tumor cells. Where indicated, OAT-1746 was administered twice daily at a dose of 20 mg/kg intraperitoneally, starting from the day before OT-I T-cells transfer until the end of the experiment. On day 3 post OVA immunization, OVA injection site/tumor-draining inguinal lymph nodes were harvested, mashed through a 70 µm nylon strainer, and cells were incubated with OVA peptide (SIINFEKL)-specific tetramers (iTAg Tetramer/PE H-2Kb OVA, MBL) to detect OT-I CD8+ T-cells, followed by anti-CD3 and anti-CD8 staining. Next, the samples were analyzed for proliferation by flow cytometry. Non-proliferating CTV-stained OT-I T-cells were isolated from OVA-unstimulated mice to setup the proper gating on CTV histogram. All OT-I T-cells with lower CTV fluorescence than non-proliferating T-cells were identified as proliferating cells.
Tissue optical clearing and imaging
Tumor draining and contralateral inguinal lymph nodes (from mice inoculated with 0.5 × 106 of CTV stained CD8+ OT-I T-cells as described above) were dissected and briefly rinsed in 0.01 M PBS (pH 7.4)/heparin (5 IU/ml final concentration) and subjected to 24 hours (hrs) fixation with 4% paraformaldehyde at 4°C. After fixation, lymph nodes were rinsed 3 times for 1 hr with PBS and the residual connective and adipose tissues were gently removed under a stereomicroscope. Next, lymph nodes were prepared using CUBIC-R1-based tissue optical clearing for imaging as described previously.39 Briefly, lymph nodes were immersed in CUBIC-R1 solution for at least 1.5 days (with a longer, up to 7 days incubation time for the enlarged lymph nodes) and subjected to imaging using a Zeiss Lightsheet Z.1 equipped with two 5× objectives (N.A. 0.1) that project two independent co-axial lightsheets onto the lymph nodes from the left and right. Each lightsheet was aligned manually into the same imaging plane, according to the procedure provided by Carl Zeiss AG and the resulting images were automatically fused by ZEN software (Zeiss). Additional detail for tissue optical clearing is provided in supplementary materials.
Statistical analysis
Data are shown as means ± standard deviation (SD). Graphpad Prism 8.3.0 (GraphPad Software) was used to calculate statistical analyses. The normality of data distribution was tested using Shapiro-Wilk test. For statistical analyses of two groups unpaired two-tailed t-test was used. For statistical analyses of three or more groups one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test was applied. Tumor growth curves were analyzed with two-way ANOVA. P < .05 at 95% confidence interval was considered statistically significant. The survival rate was computed using Kaplan-Meier plots and analyzed with log-rank test.
Results
Arg1 is expressed in the tumor microenvironment of murine tumors
To study changes in Arg1 expression levels in tumor-associated immune cells during 3LL tumor progression, we have used genetically engineered YARG mice that co-express yellow fluorescent protein (YFP) and Arg1 under the same Arg1 promoter.40 Tumors were excised, when they reached approximately 5, 10 or 15 mm in the largest diameter and after cell dissociation they were analyzed on flow cytometry (Figure 1a, online supplementary figure 3). We observed that YFP+ cells are nearly exclusively within a CD45+ population of tumor-infiltrating immune cells (online supplementary figure 3). The percentage of YFP+ cells increased with tumor progression, and immune cells infiltrating more advanced tumors expressed more Arg1, as measured by YFP fluorescence intensity (Figure 1a). A more detailed analysis of the phenotypic markers revealed that Arg1-expressing cells are mainly macrophages (CD11b+F4/80+), M-MDSCs (CD11b+Ly6C+), and to a lesser degree G-MDSCs (CD11b+Ly6G+) (Figure 1b and c). In two other tumor models (PANC02 and B16F10 melanoma) syngeneic with C57BL/6 mice we have also observed increased accumulation of YFP+CD45+ cells in the tumor microenvironment, although the percentages of immune cells with Arg1 were lower as compared with 3LL tumors (online supplementary figure 4). Next, we sought to investigate whether tumor progression is associated with changes in ʟ-arg metabolism. To address this, we measured plasma ʟ-arg and ʟ-ornithine concentrations, when the tumors reached sizes corresponding to large, intermediate and small tumors, respectively (see materials and methods). As shown in Figure 2, the concentrations of ʟ-arg were the lowest in the plasma of mice with the most advanced tumors (reaching 26.15 ± 9.34 µM in mice with the largest tumors), and this was associated with increased plasma ʟ-ornithine concentrations. Altogether, these results indicate that tumor progression is associated with a gradual accumulation of Arg1+ myeloid cells that leads to systemic ʟ-arg depletion.
Figure 1.
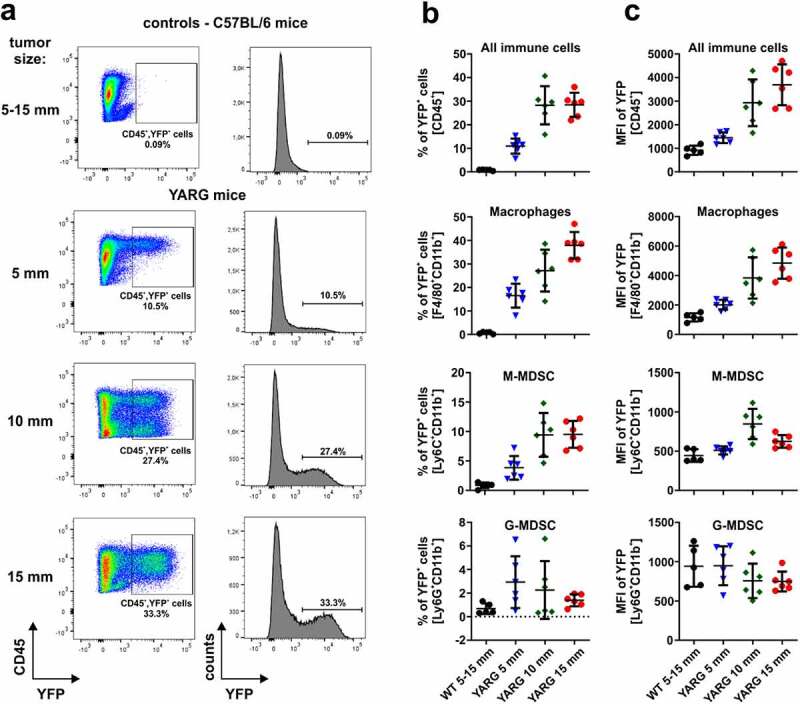
Dynamics of 3LL tumors infiltration with Arg1-expressing immune cells. WT or YARG mice were inoculated subcutaneously with 1 × 106 of 3LL cells and tumors were harvested when reached approximately 5, 10 or 15 mm in the largest diameter. Tumor samples were digested to single-cell suspension, stained with fluorochrome-coupled antibodies and analyzed in flow cytometry. A. Representative dot plots and histograms of WT and YARG mice showing gating for YFP+ cells. B. Percentages of YFP+ (Arg1+) cells within the populations of the total immune cells (CD45+), macrophages (CD11b+F4/80+), M-MDSC (CD11b+Ly6C+), and G-MDSC (CD11b+Ly6G+) infiltrating 3LL tumors at different stages of the disease progression. Data show means ± SD; n = 5–6. C. Mean fluorescence intensity (MFI) of YFP within the defined populations. Data show means ± SD; n = 5–6
Figure 2.
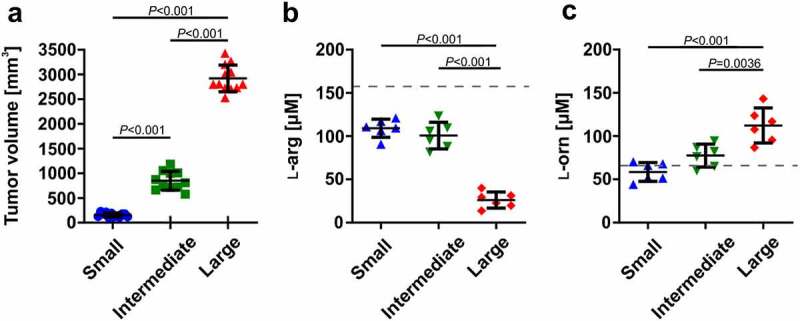
Tumor progression is associated with decreased ʟ-arg and increased ʟ-ornithine plasma concentrations. A. WT mice were inoculated subcutaneously with 0.5 × 106 of 3LL tumor cells on days 0, 7, and 14 of the experiment in order to generate large, intermediate, and small tumors, respectively. Data show means ± SD; n = 11–12 (the results are pooled from two independent experiments); P values were calculated with one-way ANOVA with Tukey’s multiple comparisons test. B. ʟ-arginine (ʟ-arg) and C. ʟ-ornithine (ʟ-orn) concentrations in plasma samples assessed by mass spectrometry. Data show means ± SD; n = 6 (measurements were done in the second experiment only); P values were calculated with one-way ANOVA with Tukey’s multiple comparisons test. The dashed lines represent ʟ-arg and ʟ-orn concentrations in healthy C57BL/6 female mice (151 ± 5 µM and 59 ± 5 µM, respectively41)
Arg1 deficiency delays tumor growth
3LL cells do not express Arg1 (online supplementary figure 5). Considering that both hematological malignancies, as well as solid tumors, have been reported to produce arginases,10,13,25,42 we sought to investigate whether forced overexpression of Arg1 in 3LL cells induced by lentiviral transduction could affect tumor progression. As shown in Figure 3a, we have observed that 3LL tumors overexpressing Arg1 (3LL-pLVX-Arg1) grow faster in mice than mock-transfected (3LL-pLVX) or unmodified (3LL-WT) tumors. The in vivo tumor growth-promoting effects of Arg1 overexpression were abrogated in immunodeficient RAG2 KO mice indicating their association with an impaired immune response (Figure 3b).
Figure 3.
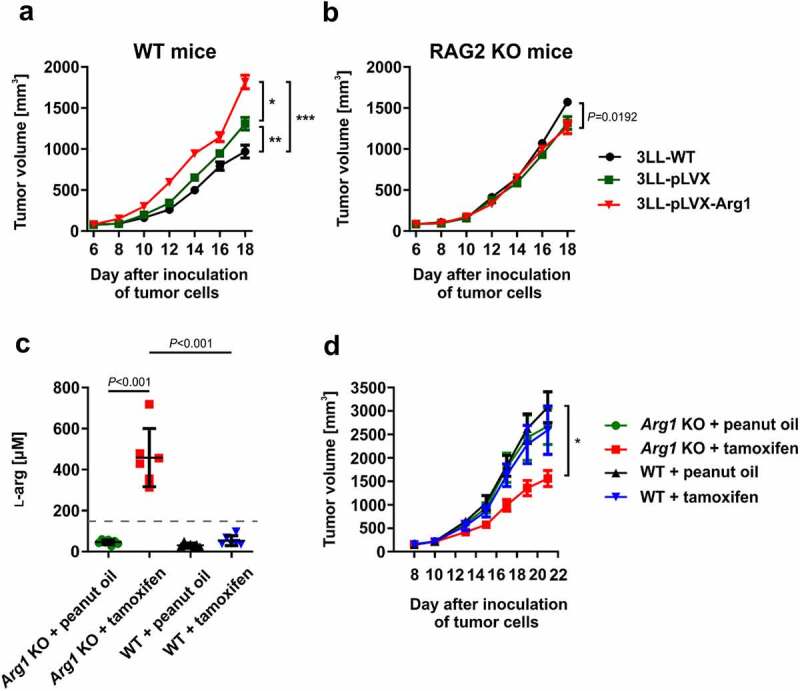
Tumor progression is modulated by Arg1. A, B. Mice were inoculated subcutaneously with 0.5 × 106 of Arg1-overexpressing (3LL-pLVX-Arg1) or control tumor cells (3LL-WT and 3LL-pLVX – 3LL transduced with empty vector) in WT [A] and RAG2 KO [B] mice. Data show mean tumor volumes ± SD; n = 7–8. P values were calculated with two-way ANOVA. *P = .002; **P = .0161; ***P < .0001. Individual growth curves are presented in the Supplementary Fig. 11 and 12. C, D. Inducible total Arg1 KO and WT mice were inoculated subcutaneously with 0.5 × 106 of 3LL cells on day 0. Next, from day 7 until day 12 mice received tamoxifen (75 mg/kg p.o.) or peanut oil (solvent). C. ʟ-arg concentrations in plasma collected on day 21 and assessed by mass spectrometry. Data show means ± SD; n = 6–7. P values were calculated with one-way ANOVA with Tukey post-hoc test. The dashed line represents ʟ-arg concentration in healthy C57BL/6 female mice (151 ± 5 µM41). D. Tumor volume in time. Data show mean tumor volumes ± SD; n = 6–7. P values were calculated with two-way ANOVA. *P < .0001. Individual growth curves are presented in the Supplementary Fig. 13
Next, we sought to investigate whether a lack of Arg1 in mice would affect tumor progression. To address this, we have crossed Arg1fl/fl mice with R26-CreERT2 mice to obtain a strain of mice allowing for the whole body tamoxifen-inducible Arg1 deletion (hereafter referred to as Arg1 KO mice). Tamoxifen administration to tumor-bearing Arg1 KO mice led to a significant increase in the plasma ʟ-arg concentration (Figure 3c). Tumor growth was significantly inhibited after tamoxifen administration as compared with three control groups (Arg1 KO mice treated with peanut oil used as tamoxifen solvent, WT mice treated with peanut oil or WT mice treated with tamoxifen) (Figure 3d). Altogether, these results indicate that the increased Arg1 in the tumor environment is associated with accelerated tumor progression that might be related to impaired development of the immune response.
Arg1 impairs proliferation of antigen-specific T-cells in mice
It has previously been shown that Arg activity or low ʟ-arg concentrations suppress the proliferation of T-cells,43,44 and that these suppressive effects are associated with decreased levels of CD3ζ chain, a critical component of TCR that transmits activation signals in T lymphocytes.45 We have confirmed these findings in ex vivo cultures of human CD4+ and CD8+ T-cells triggered to proliferate by αCD3/CD28-coupled beads (online supplementary figure 6). To further investigate whether the increased numbers of Arg1-expressing myeloid cells in the tumor microenvironment might be associated with the impaired proliferation of antigen-specific T-cells, we adoptively transferred OT-I T-cells into mice with tumors at different stages of progression and subcutaneously immunized mice with OVA protein in the tumor area, according to the scheme depicted in Figure 4a. Flow cytometric analysis of tumor-draining lymph nodes revealed that the proliferation of adoptively transferred OT-I T-cells was impaired in mice with the largest tumors (Figure 4b). Concomitantly, in mice with the largest tumors, CD3ζ levels in OT-I T-cells were significantly decreased (Figure 4c). Similarly, direct counting of T-cells in tumor-draining lymph nodes revealed that in OVA-immunized mice with large tumors the number of adoptively transferred OT-I T-cells was significantly lower as compared with mice bearing small or intermediate tumors (Figure 4d, online supplementary figure 7, online supplementary video 1–2 https://immunologia.wum.edu.pl/node/236). Moreover, adoptive transfer of OT-I T-cells into tumor-bearing Arg1 KO mice treated with tamoxifen resulted in a nearly two-fold increase of tumor-infiltrating cells as compared with controls (inducible total Arg1 KO mice treated with peanut oil) (Figure 5a-c). We then compared in vivo proliferation of OT-I T-cells adoptively transferred into tumor-bearing WT mice or myelo Arg1 KO mice with constitutive Arg1 KO in the myeloid lineage driven by Cre recombinase under the control of Lyz2 promoter. Tumor volumes in myelo Arg1 KO mice were significantly smaller as compared with WT mice (Figure 5d). Also in these experiments, the proliferation of OT-I T-cells was inhibited in WT mice with large tumors as compared with mice with small tumor burden (Figure 5e), and these findings correlated with decreased ʟ-arg (figure 5f) and increased ʟ-ornithine (Figure 5g) concentrations in the plasma. In myelo Arg1 KO mice, however, the OT-I T-cell proliferation rate, as well as plasma ʟ-arg concentrations, were comparable in mice with small or large tumors. ʟ-Orn concentrations were lower in mice bearing large tumors as compared to mice with small tumors, however still within the normal range.
Figure 4.
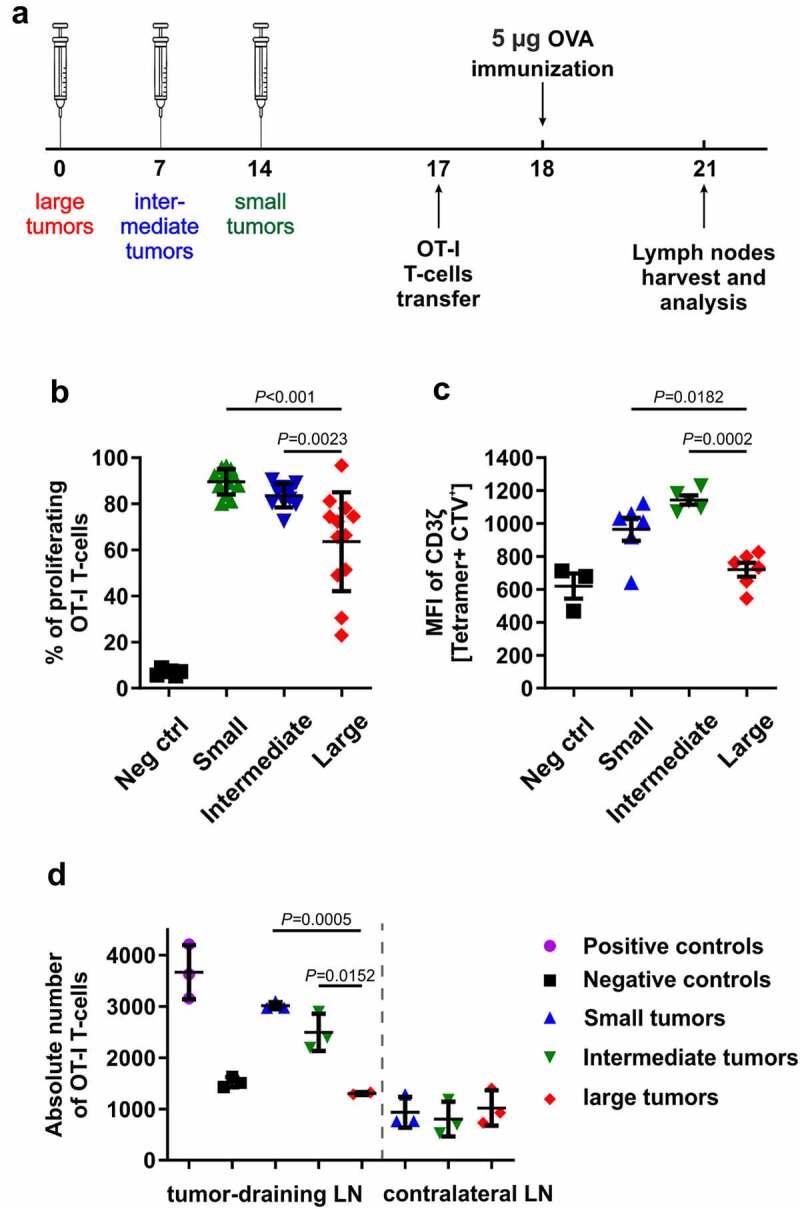
Arg1 impairs proliferation of antigen-specific T-cells in 3LL-bearing mice. Mice were injected subcutaneously with 0.5 × 106 of 3LL cells. A. Schematic presentation of the experimental setting. B. Percentage of proliferating OT-I T cells. Data show means ± SD; n = 11–12. P values were calculated with one-way ANOVA with Tukey post-hoc test. Tumor-free mice transferred with OT-I T-cells but not immunized with OVA served as negative control (Neg ctrl). C. Mean fluorescence intensity (MFI) of CD3ζ staining in OT-I T-cells. Data show means ± SD; n = 3–6. P values were calculated with one-way ANOVA with Tukey post-hoc test; negative controls (Neg ctrl) are WT mice 72 hrs post OT-I T-cells transfer without tumor and OVA protein immunization. D. Numbers of OT-I T-cells in tumor-draining inguinal and contralateral lymph nodes counted after tissue optical clearing after the adoptive transfer of the 0.5 × 106 CTV-stained cells. Tumor-free mice transferred with OT-I T-cells but not immunized with OVA served as negative control (Neg ctrl) while tumor-free mice transferred with OT-I T-cells and immunized with OVA served as positive control (Pos ctrl). Data show means ± SD; n = 2–3. P values were calculated with one-way ANOVA with Tukey post-hoc test
Figure 5.
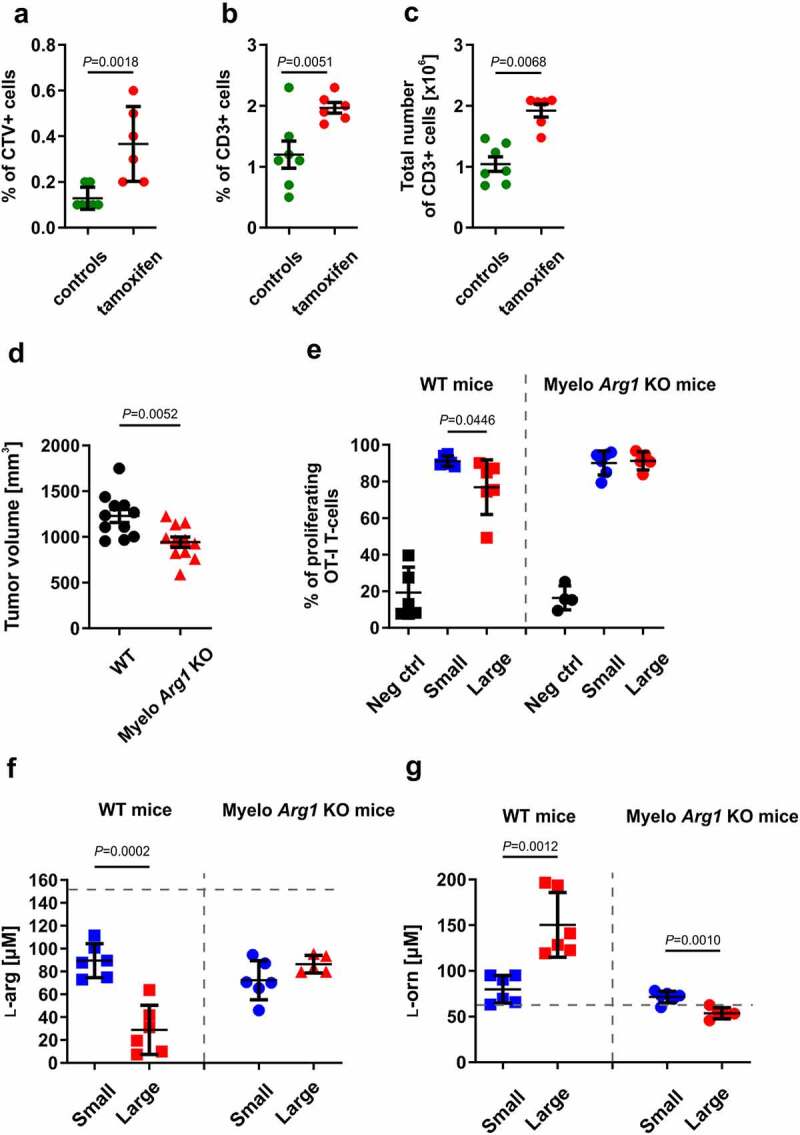
Total or myeloid-specific Arg1 deficiency improves antigen-specific T-cells proliferation in 3LL-tumor-bearing mice. A-C. Inducible total Arg1 KO mice were inoculated subcutaneously with 0.5 × 106 3LL cells on day 0. Next, from day 7 until day 12 mice received tamoxifen (75 mg/kg p.o.) or peanut oil (solvent). On day 17 each mouse was inoculated i.v. with 4–6 × 106 of CTV-stained OT-I T-cells. Antigen-specific proliferation was triggered with 5 µg of OVA protein injected subcutaneously in the tumor area on day 18. On day 21 tumor-draining lymph nodes were isolated for subsequent analysis. A. Percentages of adoptively transferred CTV+ OT-I T-cells in inguinal tumor-draining lymph nodes. Data show means ± SD; n = 6–7. P values were calculated with unpaired t-test. B. Percentage and C. absolute numbers of CD3+ tumor-infiltrating lymphocytes (TILs) analyzed in flow cytometry. Data show means ± SD; n = 6–7. P values were calculated with unpaired t-test. D-G. WT or myelo Arg1 KO mice were subcutaneously inoculated with 0.5 × 106 of 3LL cells. D. Tumor volumes in WT or myelo Arg1 KO mice on day 17 after the inoculation of tumor cells. Data show means ± SD; n = 11. P values were calculated with unpaired t-test. E. Mice with small and large tumors were inoculated i.v. with 4–6 × 106 of CTV-stained OT-I T-cells. Antigen-specific proliferation was triggered with 5 µg of OVA protein injected subcutaneously in the tumor area on the following day. Tumor-draining inguinal lymph nodes were isolated to determine percentages of proliferating OT-I T-cells 72 hours post immunization. Data show means ± SD; n = 4–6. P values were calculated with unpaired t-test; negative control (Neg ctrl) – WT mice 72 hrs post OT-I T-cells transfer without tumor and OVA protein immunization. F. ʟ-arginine (ʟ-arg) and G. ʟ-ornithine (ʟ-orn) concentrations in the plasma samples of WT or myelo Arg1 KO mice subcutaneously inoculated with 0.5 × 106 of 3LL cells, assessed by mass spectrometry. Data show means ± SD; n = 5–6. P values were calculated with unpaired t-test. Dashed lines represent ʟ-arg and ʟ-orn concentrations in healthy C57BL/6 female mice (151 ± 5 µM and 59 ± 5 µM, respectively41)
Collectively, these data suggest that high Arg1 activity in mice with advanced tumors negatively affects the development of antigen-specific immune response in local secondary lymphoid organs and leads to reduced numbers of effector T-cells in the tumor microenvironment.
Arg inhibitor restores Arg1-mediated suppression of T-cells and exerts antitumor effects in mice
We then investigated whether OAT-1746, an arginase inhibitor (online supplementary figure 8), could reverse enzyme-mediated suppression of T-cell proliferation in vitro. We confirmed that recombinant human ARG1 (rhARG1) suppressed the proliferation of human CD4+ and CD8+ T-cells triggered by αCD3/CD28 beads and reduced expression of CD3ζ and CD3ε in these cells (online supplementary figure 6). We observed that OAT-1746 dose-dependently restored CD4+ and CD8+ T-cell proliferation in response to αCD3/CD28 beads, where T-cells were co-incubated with rhARG1 (online supplementary figure 6). OAT-1746 also restored CD3ε and CD3ζ expression that had been reduced by ARG1 (online supplementary figure 6). Intriguingly, rhARG1 did not suppress the cytotoxic effects of primed OT-I T-cells nor spontaneous or antibody-dependent cellular cytotoxicity (ADCC) of NK cells (online supplementary figure 9). The latter express activating receptors that use CD3ζ.46
Having established potential immunoregulatory effects of OAT-1746 in ex vivo proliferation assays with T-cells, we sought whether this Arg inhibitor would be able to exert antitumor effects in mice. To this end, we have inoculated 3LL cells into mice and one day later we started administration of OAT-1746 or its diluent twice daily for 14 days. We observed a slight, but significant inhibition of tumor growth (Figure 6a) as well as prolonged survival of tumor-bearing mice (Figure 6b). Similar effects were observed in mice inoculated with Arg1-overexpressing tumors (Figure 6c). To elucidate whether OAT-1746 administration is associated with improved T-cell proliferation we adoptively transferred OT-I T-cells into mice with small and large tumors and measured their proliferation after subcutaneous OVA priming in the tumor area. We observed that OT-I proliferation was significantly suppressed in mice with large tumors as compared with mice with small tumors (Figure 4b) and that OAT-1746 restored T-cell proliferation (Figure 6d). Additionally, using transgenic FoxP3EGFPmice that co-express green fluorescence protein (EGFP) and Treg-specific transcription factor FoxP3 we observed that OAT-1746 administration significantly reduced the percentage of Tregs (Figure 6e), while the percentage of non-Treg T-cells among the tumor-infiltrating lymphocytes was concomitantly increased (figure 6f).
Figure 6.
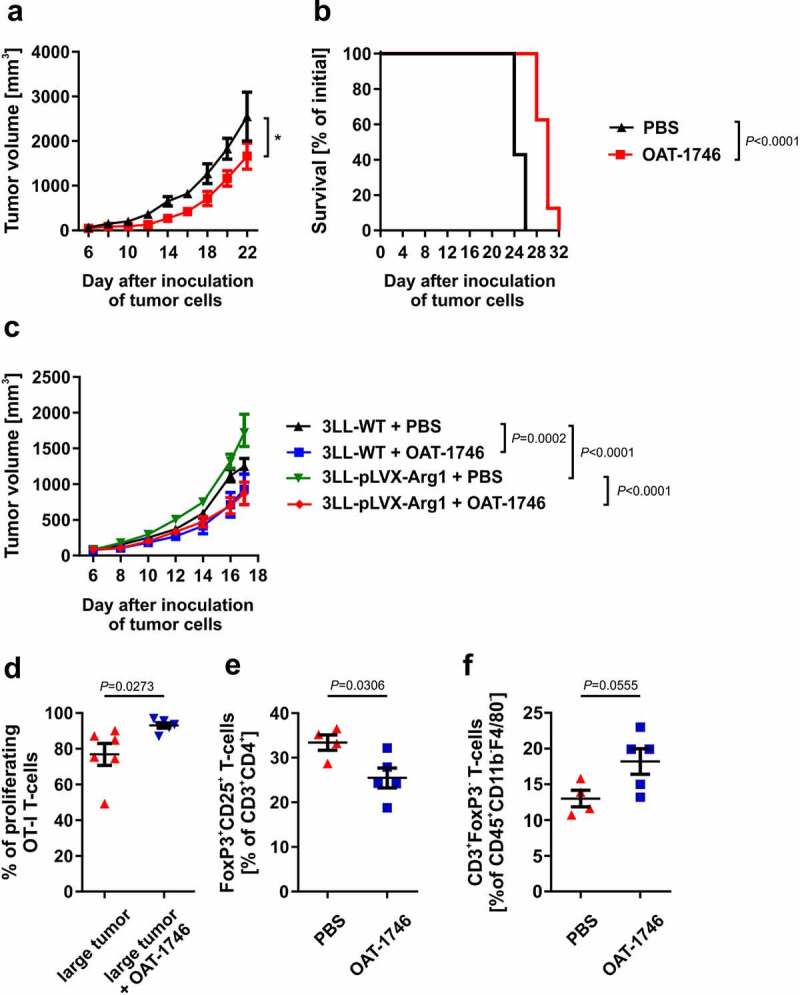
Arginase inhibitor inhibits 3LL tumors growth and restores T-cells proliferation in vivo. C57BL/6 mice were inoculated subcutaneously with 0.1 × 106 of 3LL tumor cells. OAT-1746 was administered twice daily by an intraperitoneal route at a dose of 20 mg/kg for the first 14 days. A. Tumor volume in time. Data show means ± SD; n = 7–8. P values were calculated with two-way ANOVA. Individual growth curves are presented in the Supplementary Fig. 14. B. Animal survival curve. P values were calculated with log-rank test. C. C57BL/6 mice were inoculated subcutaneously with 0.5 × 106 of control 3LL WT or Arg1-overexpressing 3LL-pLVX-Arg1 cells. OAT-1746 was administered twice daily by an intraperitoneal route at a dose of 20 mg/kg for the subsequent 14 days starting from day 1 after inoculation. The graph presents tumor volumes in time. Data show means ± SD; n = 7–8. P values were calculated with two-way ANOVA. Individual growth curves are presented in the Supplementary Fig. 15. D. CTV stained OT-I CD8+ T-cells were transferred to 3LL small or large tumor-bearing mice. The next day mice were immunized s.c. in the tumor area with OVA. On day 3 post-immunization proliferation of OT-I T-cells was evaluated in tumor-draining inguinal lymph nodes in flow cytometry. Where indicated, mice were treated i.p. with 20 mg/kg OAT-1746 twice daily on days −1 until +3 after OT-I T-cells transfer. Data show means ± SD; n = 4–6. P values were calculated with unpaired t-test. E-F. FoxP3EGFP mice were inoculated on day 0 with 1 × 106 3LL cells and beginning from day 1 post inoculation with tumor cells were treated i.p. with 20 mg/kg OAT-1746 administered twice daily. On day 18 the percentages of tumor-infiltrating Tregs (CD3+CD4+CD25+FoxP3+ cells) (e) as well as non-Treg T-cells (CD45+CD3+FoxP3− cells) (f) were measured in flow cytometry. Data are presented as means ± SD; n = 4–5, P values were calculated with unpaired t-test
OAT-1746 potentiates antitumor effects of immunotherapies
Considering that OAT-1746 alone exerts rather modest antitumor efficacy in 3LL model, we decided to investigate its effects in combination with anti-PD-1 checkpoint inhibitors. Again OAT-1746 revealed modest antitumor effects that were stronger than treatment with anti-PD-1 monoclonal antibodies (mAbs) alone (Figure 7a). Nonetheless, this combination treatment failed to significantly prolong mouse survival (Figure 7b). Thus, we hypothesized that in addition to treatment with two inhibitors of negative regulatory pathways, the effective immunotherapy could benefit from the activation of immunostimulatory pathway. To this end, we combined OAT-1746, anti-PD-1 mAbs, and STING agonist, DMXAA. We observed that such triple combination is not only more effective than administration of single immunomodulators or double combinations, but also leads to a significant prolongation of mice survival (Figure 7c-d, online supplementary figure 10).
Figure 7.
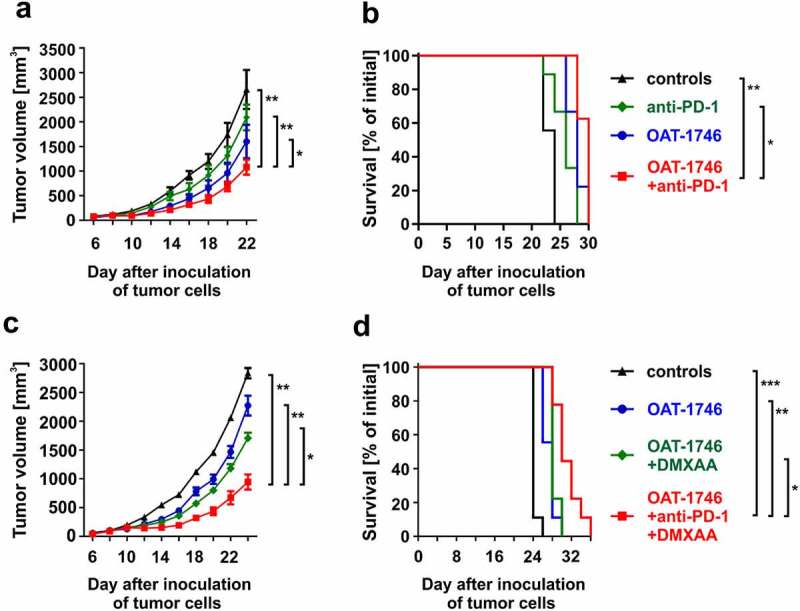
Arginase inhibitor potentiates antitumor activity of immunotherapies. A-B. C57BL/6 mice were inoculated with 0.1 × 106 of 3LL tumor cells. OAT-1746 was administered twice daily by an intraperitoneal route at a dose of 20 mg/kg for the first 14 days, and anti-PD-1 or isotype control antibodies i.p. at the dose of 10 mg/kg on days 6, 9, 12, 15, and 18. Number of mice in the experimental groups: ctrl n = 9, anti-PD-1 n = 9, OAT-1746 n = 9, OAT-1746+ anti-PD-1 n = 8. A. The graph shows tumor volumes in time. Data show means ± SD; n = 8–10. P values were calculated with two-way ANOVA. *P = .0151; **P < .0001. Individual growth curves are presented in the Supplementary Fig. 16. B. Animal survival curve. P values were calculated with log-rank test. *P = .0011; **P < .0001. C-D. C57BL/6 mice were inoculated with 0.1 × 106 of 3LL tumor cells. OAT-1746 was administered twice daily by an intraperitoneal route at a dose of 20 mg/kg for the first 14 days, anti-PD-1 or isotype control antibodies i.p. at the dose of 10 mg/kg on days 6, 9, 12, 15, and 18 and DMXAA (or NaHCO3 used as DMXAA diluent) by intratumoral injection at a dose of 0.5 µg/mouse on day 8. Number of mice in the experimental groups: ctrl n = 9, OAT-1746 n = 9, OAT-1746+ DMXAA n = 9, OAT-1746+ anti-PD-1+ DMXAA n = 9. C. The graph presents tumor volumes in time. Data show means ± SD; n = 9–10. P values were calculated with two-way ANOVA. *P = .0002; **P < .0001. Individual growth curves are presented in the Supplementary Fig. 17. D. Animal survival curve. P values were calculated with log-rank test. *P = .0078; **P = .0017; ***P < .0001. Additional experimental groups for C and D are shown in Supplementary Figure 12
Discussion
Using genetically engineered reporter mice co-expressing eYFP with Arg1, we have confirmed previous findings indicating that Arg1-positive immune cells infiltrate murine 3LL tumors.28,47 Our studies expand these observations by showing kinetic data, i.e. a progressive accumulation of Arg1+ myeloid cells with increasing expression levels of this enzyme, and that not only mature (CD11b+F4/80+) macrophages, but also immature M-MDSCs (CD11b+Ly6C+) express Arg1. Using the same Arg1-eYFP reporter mice Arlauckas et al. have also observed that F4/80+ macrophages are the predominant source of Arg1 in a syngeneic MC38 colon carcinoma model.48 The mechanisms involved in the recruitment of Arg+ myeloid cells to the tumor or in the upregulation of Arg1 in these cells are currently unknown. Previous studies indicate that Arg1 induction can be mediated by hypoxia-inducible factor 1α induced by tumor-derived lactic acid,47 prostaglandins generated by cyclooxygenase-2,49 or type 2 cytokines in STAT3-dependent mechanisms.50 Moreover, we show that the presence of Arg+ myeloid cells in more advanced tumors (Figure 1) is associated with a gradual systemic loss of ʟ-arg (Figure 2). Decreased ʟ-arg concentrations have also been observed in cancer patients with various types of malignancies37,51,52 as well as with lung cancer53 indicating that the murine model to some extent corresponds to clinical data. However, it should be noted that in the majority of studies involving cancer patients’ Arg1+ cells were reported to be of granulocytic lineage.5,54 A limitation of our studies is that the 3LL model is based on ectopically growing transplantable tumors. Despite their barriers, transplantable tumor models allowed development of important therapeutic principles in immuno-oncology.55
To better understand the immunoregulatory role of Arg1 we have stably transduced 3LL cells with lentiviral particles encoding this enzyme. Although immunoblotting and enzymatic assays revealed that 3LL-pLVX-Arg1 cells produced active Arg1 (online supplementary figure 5), we have no data on whether the enzyme can be released from tumor cells. Despite the limitation of this artificial model, we have clearly observed that increased Arg1 production by tumor cells is associated with accelerated tumor progression, which is abrogated in immunodeficient mice or in mice treated with Arg inhibitor (Figure 3a,b and 5c).
Normal T-cells survive at low ʟ-arg concentrations; however, their activation and proliferation are strongly inhibited, with complete cell cycle arrest observed at ʟ-arg concentrations below 23 µM.56 Serum ʟ-arg concentrations in mice with large tumors reached the values that were reported to suppress T-cell proliferation in in vitro conditions (Figure 2). Accordingly, the proliferation of OVA-specific T-cells adoptively transferred to mice with large 3LL tumors was suppressed as compared with mice having small or intermediate size tumors (Figure 4). Decreased T-cell proliferation might possibly be related to higher OVA dilution when inoculated to mice with large tumors. However, the suppression of T-cell proliferation by large tumors was not observed, when the adoptive transfer of OT-I T-cells was done into mice with tamoxifen-induced Arg1 deletion or into mice with a constitutive knock-out of Arg1 in the myeloid lineage (Figure 5a,e). Tamoxifen-induced systemic deletion of Arg1 resulted in a significant increase in the serum ʟ-arg concentrations of mice with large tumors (Figure 3c) indicating that systemic drop in this amino acid results from the accumulation of Arg1+ immune cells.
Furthermore, an increase in Arg1 induced by lentiviral transduction of tumor cells further accelerated tumor progression in WT mice, but this effect was not observed in immunodeficient mice, indicating that ʟ-arg degradation might lead to suppression of the endogenous antitumor immune response (Figure 3). Similar findings were reported with recombinant pegylated-Arg1 administered into tumor-bearing mice, where suppressed T-cell responses were associated with accelerated progression of 3LL tumors in mice.57 Also in other lung cancer models, Arg1 has been shown to cause T-cell dysfunction. For example, in KRASG12D genetically engineered mice that develop lung tumors resembling NSCLC, G-MDSCs were observed to cause T-cell suppression by ʟ-arg depletion. Arg inhibitor has not only restored T-cell function, but caused significant regressions of tumors in these mice.8
In contrast to malignant T-cells that undergo cell cycle arrest and apoptosis at low ʟ-arg concentrations,58 normal T-cells resume responsiveness to mitogens as soon as normal ʟ-arg concentration is restored.30 Although dietary supplementation of ʟ-arginine was shown to improve T-cell functions in mice,44 a moderate-to-poor oral ʟ-arginine bioavailability (~20%) as well as its rapid renal clearance (T½ of 1–2 h)59 prompted development of Arg inhibitors as a potential approach to mitigate the negative outcomes of increased ʟ-arg degradation. Here, we observed that OAT-1746 restores T-cell activities that were suppressed by Arg1 activity, including proliferation triggered by anti-CD3/CD28 beads as well as expression of CD3ε and CD3ζ (online supplementary figure 6). Intriguingly, cytotoxic effects of T-cells were unaffected by Arg1, despite the fact that CD3ζ and CD3ε were downregulated, and thus it may be expected that TCR signal transduction should be inhibited. In in vivo studies, we observed that OAT-1746 not only improved OVA-induced proliferation of adoptively transferred OT-I T-cells, but also decreased the percentage of intratumor Tregs (Figure 6d-e). FoxP3+ Tregs were recently shown to induce Arg1 in dendritic cells, thereby increasing amino acid consumption in the local microenvironment and activating mTOR signaling that favors the development of additional Tregs.60 Additionally, Arg2 is expressed at higher levels in Tregs from metastatic melanomas as compared with normal skin, and Arg2 in Tregs was demonstrated to attenuate mTOR activity and conferred Tregs with enhanced suppressive activity.61
Abrogation of Arg1 activity by either systemic knock-out or specific Arg1 deletion in myeloid cells only was associated with inhibition of tumor growth (figures 3d, 5d) and improvement of antigen-specific proliferative response of adoptively transferred T-cells (Figure 5a, e). These observations indicate that Arg1 inhibition might be an effective therapeutic approach to improve priming conditions for antigen-recognizing T-cells. Several Arg inhibitors have been described so far, including both natural and synthetic compounds.38,62,63 Almost all developed as drug candidates are competitive inhibitors of both isoenzymes (Arg1 and Arg2). Finding an isoform-specific Arg inhibitor is challenging due to extensive homology in the active site of the enzymes. The first group of Arg inhibitors such as N-hydroxy-nor-ʟ-arginine (nor-NOHA) were reversible Arg1 and Arg2 inhibitors with rather poor pharmacokinetic properties or insufficient penetration through the plasma membrane. The second generation of Arg inhibitors, i.e. boronic acid analogs of ʟ-arginine such as ABH had improved pharmacokinetic and pharmacodynamic properties and are extensively reviewed.38,63 Due to potential toxicity in humans further analogs have been developed for oncology studies, and one (CB-1158) is under evaluation in clinical trials. CB-1158 effectively blocks extracellular Arg, but penetrates plasma membrane rather poorly. Here, we have used a novel Arg inhibitor (OAT-1746) that blocks both extracellular and intracellular Arg activity and has pharmacokinetic properties allowing for administration twice daily.38,64 We have observed that OAT-1746 completely restores T-cell proliferation suppressed by recombinant Arg1 added to the culture media. Arg1 suppressed T-cell proliferation (online supplementary figure 6), but did not suppress cytotoxicity effector functions of T-cells and NK cells (online supplementary figure 9). Thus, it seems that increased Arg activity observed in tumors negatively regulates mainly the initial phases of the immune response, but effector functions are largely unhampered. More detailed analyses of the immunoregulatory effects of arginases are clearly needed.
Pharmacologic inhibition of arginase led to a slight, but significant inhibition of tumor growth. Also, other Arg inhibitors were previously shown to exert modest antitumor effects.8,28,37,47,48,65 These data indicate that inhibition of Arg activity is too meager to trigger a sufficiently strong antitumor response that would lead to tumor eradication. CB-1158 was recently demonstrated to significantly potentiate the antitumor effects of anti-PD-L1 antibodies in a strongly immunogenic CT-26 colon adenocarcinoma model.37 However, in a less immunogenic 4T1 breast cancer model, the combination of CB-1158 with anti-PD-L1 or anti-CTLA4 antibodies was not effective. In another poorly immunogenic mouse model of 3LL lung carcinoma, we observed a rather modest antitumor activity of Arg inhibitor combined with anti-PD1 antibodies (Figure 7). Interestingly, potentiated antitumor efficacy of similar combination was not observed in MC38 colon carcinoma model. The discrepancy can be explained by a different tumor mode used as well as a different arginase inhibitor (ABH vs OAT-1746). ABH is poorly inhibiting intracellular arginases. It will be important in future studies to address potential toxic effects, especially hepatotoxicity, of intracellularly active Arg1 inhibitors. Checkpoint inhibitors and Arg inhibitors inactivate negative immunoregulatory pathways. Therefore, we sought to investigate the antitumor effectiveness of double and triple combinations with a potent activator of the immunostimulatory pathway associated with innate immune response. The results of these experiments revealed that although it is possible to further improve antitumor efficacy and prolong the survival of tumor-bearing mice (Figure 7), the therapeutic effects are still insufficient to induce complete responses in mice. A limitation of these studies is that Arg inhibitor administration was started soon after inoculation of tumor cells. It will be important in future studies to see whether the treatment is also effective in a more therapeutically relevant setting once the tumor is fully established. Altogether, the existing preclinical data seem to indicate that therapeutic targeting of the immunomodulatory Arg1 might be a useful addition to other immunotherapeutic strategies rather than being an effective single target treatment.
Supplementary Material
Acknowledgments
We would like to thank Dr. Anna Kowalczyk and Ms. Sylwia Figurska for their exceptional help with animal breeding and genotyping as well as Mrs. Elzbieta Gutowska, Mrs. Ewa Chmiel, Mrs. Ewa Pieta and Mrs. Karolina Siudakowska for their outstanding technical support.
Funding Statement
This work was supported by the following grants: Narodowe Centrum Badań i Rozwoju [265503/3/NCBIR/15 (STRATEGMED2)]; Ministerstwo Nauki i Szkolnictwa Wyższego [iONCO (Regionalna Inicjatywa Doskonalosci)]; Narodowe Centrum Nauki [2016/23/B/NZ6/03463].
Disclosure Statement
M.G, P.S. and R.B. are employees in OncoArendi Therapeutics, Warsaw, Poland. All remaining authors have nothing to disclose.
Authors’ contribution
AS performed in vitro and in vivo experiments, analyzed data and participated in writing the manuscript. JC-T, ZP, TMG, ZR, OS, and TPR participated in experiments in animal models. PM, AG, AW performed light sheet microscopy on transparent lymph nodes. MG, PS carried out enzymatic assays. RB designed and synthesized ARG1 inhibitor. DN and JG designed and supervised the study, analyzed data, secured funding, and wrote the manuscript. All authors read and approved the final manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Veglia F, Perego M, Gabrilovich D.. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108–15. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shalapour S, Karin M.. Pas de Deux: control of anti-tumor immunity by cancer-associated inflammation. Immunity. 2019;51:15–26. doi: 10.1016/j.immuni.2019.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray PJ. Amino acid auxotrophy as a system of immunological control nodes. Nature Immunol. 2016;17:132–139. doi: 10.1038/ni.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemos H, Huang L, Prendergast GC, Mellor AL. Immune control by amino acid catabolism during tumorigenesis and therapy. Nature Rev Cancer. 2019;19(3):162–175. doi: 10.1038/s41568-019-0106-z. [DOI] [PubMed] [Google Scholar]
- 5.Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D, Golab J. Myeloid cell-derived arginase in cancer immune response. Front Immunol. 2020;11:938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, Muir A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife. 2019;8. doi: 10.7554/eLife.44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez-Davies JE, Rosales KK, Li H, et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18:1090–1101. doi: 10.1038/ncb3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miret JJ, Kirschmeier P, Koyama S, Zhu M, Li YY, Naito Y, Wu M, Malladi VS, Huang W, Walker W, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti-tumor immunity. J Immunother Cancer. 2019;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bron L, Jandus C, Andrejevic-Blant S, Speiser DE, Monnier P, Romero P, Rivals J-P, et al. Prognostic value of arginase-II expression and regulatory T-cell infiltration in head and neck squamous cell carcinoma. Int J Cancer. 2013;132:E85–E93. [DOI] [PubMed] [Google Scholar]
- 10.Mussai F, Egan S, Hunter S, Webber H, Fisher J, Wheat R, McConville C, Sbirkov Y, Wheeler K, Bendle G, et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. 2015;75(15):3043–3053. doi: 10.1158/0008-5472.CAN-14-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, Qureshi A, Dazzi F, Vyas P, Cerundolo V, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. 2013;122(5):749–758. doi: 10.1182/blood-2013-01-480129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ino Y, Yamazaki-Itoh R, Oguro S, Shimada K, Kosuge T, Zavada J, Kanai Y, Hiraoka N. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS One. 2013;8(2):e55146. doi: 10.1371/journal.pone.0055146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czystowska-Kuzmicz M, Sosnowska A, Nowis D, Ramji K, Szajnik M, Chlebowska-Tuz J, Wolinska E, Gaj P, Grazul M, Pilch Z, et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat Commun. 2019;10(1):3000. doi: 10.1038/s41467-019-10979-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Z, Lian J, Yang M, Wuyang J, Zhao C, Chen W, Liu C, Zhao Q, Lou C, Han J, Zhang Y. Overexpression of Arginase-1 is an indicator of poor prognosis in patients with colorectal cancer. Pathol Res Pract. 2019;215:152383. doi: 10.1016/j.prp.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 15.Gokmen SS, Aygit AC, Ayhan MS, Yormulaz F, Gulen S, et al. Significance of arginase and ornithine in malignant tumors of the human skin. J Lab Clin Med. 2001;137:340–344. doi: 10.1067/mlc.2001.114543. [DOI] [PubMed] [Google Scholar]
- 16.Bedoya AM, Tate DJ, Baena A, Córdoba CM, Borrero M, Pareja R, Rojas F, Patterson JR, Herrero R, Zea AH, et al. Immunosuppression in cervical cancer with special reference to arginase activity. Gynecol Oncol. 2014;135(1):74–80. doi: 10.1016/j.ygyno.2014.07.096. [DOI] [PubMed] [Google Scholar]
- 17.Cerutti JM, Delcelo R, Amadei MJ, Nakabashi C, Maciel RMB, Peterson B, Shoemaker J, Riggins GJ. A preoperative diagnostic test that distinguishes benign from malignant thyroid carcinoma based on gene expression. J Clin Invest. 2004;113(8):1234–1242. doi: 10.1172/JCI19617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akiba J, Nakashima O, Hattori S, Naito Y, Kusano H, Kondo R, Nakayama M, Tanikawa K, Todoroki K, Umeno Y, et al. The expression of arginase-1, keratin (K) 8 and K18 in combined hepatocellular-cholangiocarcinoma, subtypes with stem-cell features, intermediate-cell type. J Clin Pathol. 2016;69(10):846–851. doi: 10.1136/jclinpath-2015-203491. [DOI] [PubMed] [Google Scholar]
- 19.Obiorah IE, Chahine J, Ko K, Park BU, deGuzman J, Kallakury B. Prognostic implications of arginase and cytokeratin 19 expression in hepatocellular carcinoma after curative hepatectomy: correlation with recurrence-free survival. Gastroenterology Res. 2019;12(2):78–87. doi: 10.14740/gr1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porembska Z, Luboinski G, Chrzanowska A, Mielczarek M, Magnuska J, Barańczyk-Kuźma A. Arginase in patients with breast cancer. Clin Chim Acta. 2003;328(1–2):105–111. doi: 10.1016/S0009-8981(02)00391-1. [DOI] [PubMed] [Google Scholar]
- 21.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60(10):1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ascierto PA, Scala S, Castello G, Daponte A, Simeone E, Ottaiano A, Beneduce G, De Rosa V, Izzo F, Melucci MT, et al. Pegylated arginine deiminase treatment of patients with metastatic melanoma: results from phase I and II studies. J Clin Oncol. 2005;23(30):7660–7668. doi: 10.1200/JCO.2005.02.0933. [DOI] [PubMed] [Google Scholar]
- 23.Yoon CY, Shim YJ, Kim EH, Lee J-H, Won N-H, Kim J-H, Park I-S, Yoon D-K, Min B-H. Renal cell carcinoma does not express argininosuccinate synthetase and is highly sensitive to arginine deprivation via arginine deiminase. Int J Cancer. 2007;120(4):897–905. doi: 10.1002/ijc.22322. [DOI] [PubMed] [Google Scholar]
- 24.Ochocki JD, Khare S, Hess M, Ackerman D, Qiu B, Daisak JI, Worth AJ, Lin N, Lee P, Xie H, et al. Arginase 2 suppresses renal carcinoma progression via biosynthetic cofactor pyridoxal phosphate depletion and increased polyamine toxicity. Cell Metab. 2018;27(1263–80.e6):1263–1280.e6. doi: 10.1016/j.cmet.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gannon PO, Godin-Ethier J, Hassler M, Delvoye N, Aversa M, Poisson AO, Péant B, Alam Fahmy M, Saad F, Lapointe R, et al. Androgen-regulated expression of arginase 1, arginase 2 and interleukin-8 in human prostate cancer. PLoS One. 2010;5(8):e12107. doi: 10.1371/journal.pone.0012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nature Comm. 2016;7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim EM, Miyake B, Aggarwal M, VoeTlause R, Griffith M, Zavazava N. Embryonic stem cell-derived haematopoietic progenitor cells down-regulate the CD3 xi chain on T cells, abrogating alloreactive T cells. Immunology. 2014;142:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. [DOI] [PubMed] [Google Scholar]
- 29.Munder M, Engelhardt M, Knies D, Medenhoff S, Wabnitz G, Luckner-Minden C, Feldmeyer N, Voss RH, Kropf P, Müller I, et al. Cytotoxicity of tumor antigen specific human T cells is unimpaired by arginine depletion. PLoS One. 2013;8:e63521. doi: 10.1371/journal.pone.0063521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munder M, Schneider H, Luckner C, Giese T, Langhans C-D, Fuentes JM, Kropf P, Mueller I, Kolb A, Modolell M, et al. Suppression of T-cell functions by human granulocyte arginase. Blood. 2006;108:1627–1634. doi: 10.1182/blood-2006-11-010389. [DOI] [PubMed] [Google Scholar]
- 31.Chuang JC, Yu CL, Wang SR. Modulation of lymphocyte proliferation by enzymes that degrade amino acids. Clin Exp Immunol. 1990;82:469–472. doi: 10.1111/j.1365-2249.1990.tb05473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, Allen PM. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res. 2009;69:3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu H-R, Tsai C-C, Chang L-S, Huang HC, Cheng HH, Wang JY, Sheen JM, Kuo HC, Hsieh KS, Huang YH, et al. l-Arginine-dependent epigenetic regulation of interleukin-10, but not transforming growth factor-β, production by neonatal regulatory T lymphocytes. Front Immunol. 2017;8:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Van Ginderachter JA, Brys L, De Baetselier P, Raes G, Geldhof AB. Nitric oxide-independent CTL suppression during tumor progression: association with arginase-producing (M2) myeloid cells. J Immunol. 2003;170:5064. [DOI] [PubMed] [Google Scholar]
- 35.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68:5439–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, Greten TF, Korangy F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135:234–243. [DOI] [PubMed] [Google Scholar]
- 37.Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, Li W, MacKinnon AL, Makkouk A, Marguier G, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borek B, Gajda T, Golebiowski A, Blaszczyk R. Boronic acid-based arginase inhibitors in cancer immunotherapy. Bioorg Med Chem. 2020;28(18):115658. doi: 10.1016/j.bmc.2020.115658. [DOI] [PubMed] [Google Scholar]
- 39.Matryba P, Sosnowska A, Wolny A, Bozycki L, Greig A, Grzybowski J, Stefaniuk M, Nowis D, Gołąb J. Systematic evaluation of chemically distinct tissue optical clearing techniques in murine lymph nodes. J Immunol. 2020;204(5):1395–1407. doi: 10.4049/jimmunol.1900847. [DOI] [PubMed] [Google Scholar]
- 40.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447(7140):92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luiking YC, Hallemeesch MM, Vissers YL, Vissers YL, Lamers WH, Deutz NE. In vivo whole body and organ arginine metabolism during endotoxemia (sepsis) is dependent on mouse strain and gender. J Nutr. discussion 96S-97S 2004;134:2768S–74S. doi: 10.1093/jn/134.10.2768S. [DOI] [PubMed] [Google Scholar]
- 42.Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, Qureshi A, Dazzi F, Vyas P, Cerundolo V, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. 2013;122(5):749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109:1568–1573. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meer F, Mann M, et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(829–42):e13. doi: 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, Park H-J, Zabaleta J, Ochoa AC. L-Arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell Immunol. 2004;232(1–2):21–31. doi: 10.1016/j.cellimm.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Chiesa S, Mingueneau M, Fuseri N, Malissen B, Raulet DH, Malissen M, Vivier E, Tomasello E. Multiplicity and plasticity of natural killer cell signaling pathways. Blood. 2006;107(6):2364–2372. doi: 10.1182/blood-2005-08-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, Weissleder R. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics. 2018;8(21):5842–5854. doi: 10.7150/thno.26888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodriguez PC, Hernandez CP, Quiceno D, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–939. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, Blosser RL, Tam AJ, Bruno T, Zhang H, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. 2013;123:1580–1589. doi: 10.1172/JCI60083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vissers YL, Dejong CH, Luiking YC, Fearon KC, von Meyenfeldt MF, Deutz NE. Plasma arginine concentrations are reduced in cancer patients: evidence for arginine deficiency? Am J Clin Nutr. 2005;81(5):1142–1146. doi: 10.1093/ajcn/81.5.1142. [DOI] [PubMed] [Google Scholar]
- 52.Kaplan I, Aydin Y, Bilen Y, Geenc F, Keles MS, Eroglu A. The evaluation of plasma arginine, arginase, and nitric oxide levels in patients with esophageal cancer. Turkish J Med Sci. 2012;42:403–409. [Google Scholar]
- 53.Naini AB, Dickerson JW, Brown MM. Preoperative and postoperative levels of plasma protein and amino acid in esophageal and lung cancer patients. Cancer. 1988;62:355–360. doi:. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, Ochoa AC. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69:1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016;16(12):759–773. doi: 10.1038/nrc.2016.91. [DOI] [PubMed] [Google Scholar]
- 56.Van de Velde L-A, Murray PJ. Proliferating helper T cells require Rictor/mTORC2 complex to integrate signals from limiting environmental amino acids. J Biol Chem. 2016;291:25815–25822. doi: 10.1074/jbc.C116.763623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, Sanchez-Pino D, Hernandez C, Wyczechowska DD, Ochoa AC. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015;75(2):275–283. doi: 10.1158/0008-5472.CAN-14-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morrow K, Hernandez CP, Raber P, Del Valle L, Wilk AM, Majumdar S, Wyczechowska D, Reiss K, Rodriguez PC. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia. 2013;27(3):569–577. doi: 10.1038/leu.2012.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tangphao O, Grossmann M, Chalon S, Hoffman BB, Blaschke TF. Pharmacokinetics of intravenous and oral L-arginine in normal volunteers. Br J Clin Pharmacol. 1999;47:261–266. doi: 10.1046/j.1365-2125.1999.00883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, Fairchild PJ, Mellor AL, Ron D, Waldmann H. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci U S A. 2009;106:12055–12060. doi: 10.1073/pnas.0903919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lowe MM, Boothby I, Clancy S, Ahn RS, Liao W, Nguyen DN, Schumann K, Marson A, Mahuron KM, Kingsbury GA, et al. Regulatory T cells use arginase 2 to enhance their metabolic fitness in tissues. JCI Insight. 2019;4(24). doi: 10.1172/jci.insight.129756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pudlo M, Demougeot C, Girard-Thernier C. Arginase inhibitors: a rational approach over one century. Med Res Rev. 2017;37:475–513. doi: 10.1002/med.21419. [DOI] [PubMed] [Google Scholar]
- 63.Pham TN, Liagre B, Girard-Thernier C, Demougeot C. Research of novel anticancer agents targeting arginase inhibition. Drug Discov Today. 2018;23(4):871–878. doi: 10.1016/j.drudis.2018.01.046. [DOI] [PubMed] [Google Scholar]
- 64.Blaszczyk R, Brzezinska J, Dymek B, Stanczak PS, Mazurkiewicz M, Olczak J, Nowicka J, Dzwonek K, Zagozdzon A, Golab J, et al. Discovery and pharmacokinetics of sulfamides and guanidines as potent human arginase 1 inhibitors. ACS Med Chem Lett. 2020;11(4):433–438. doi: 10.1021/acsmedchemlett.9b00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Secondini C, Coquoz O, Spagnuolo L, Spinetti T, Peyvandi S, Ciarloni L, Botta F, Bourquin C, Rüegg C. Arginase inhibition suppresses lung metastasis in the 4T1 breast cancer model independently of the immunomodulatory and anti-metastatic effects of VEGFR-2 blockade. Oncoimmunology. 2017;6(6):e1316437. doi: 10.1080/2162402X.2017.1316437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.