

Abstract
Despite broad scientific interest in harnessing the power of Earth’s microbiomes, knowledge gaps hinder their efficient use for addressing urgent societal and environmental challenges. We argue that structuring research and technology developments around a design-build-test-learn (DBTL) cycle will advance microbiome engineering and spur new discoveries on the basic scientific principles governing microbiome function. In this Review, we present key elements of an iterative DBTL cycle for microbiome engineering, focusing on generalizable approaches, including top-down and bottom-up design processes, synthetic and self-assembled construction methods, and emerging tools to analyze microbiome function. These approaches can be used to harness microbiomes for broad applications related to medicine, agriculture, energy, and the environment. We also discuss key challenges and opportunities of each approach and synthesize them into best practice guidelines for engineering microbiomes. We anticipate that adoption of a DBTL framework will rapidly advance microbiome-based biotechnologies aimed at improving human and animal health, agriculture, and enabling the bioeconomy.
Keywords: Applied microbiology /631/326/2522, Bacterial techniques and applications /631/326/41/2537, Industrial microbiology /631/326/252, Microbial communities /631/326/2565, Microbiome /631/326/2565/2134, Biomedical engineering /639/166/985
ToC blurb
Microbiome engineering has many potential applications, ranging from agriculture to medicine. In this Review, Lawson, McMahon and colleagues guide us through the design-build-test-learn cycle that has been successful in many disciplines and explain how it applies to microbiome engineering.
Introduction
Microbial communities have seemingly limitless capabilities, driving Earth’s biogeochemical cycles and occupying every environmental niche1,2. Engineers and scientists have tapped into this power for a long time; for example, by manipulating soil microbiomes to increase crop productivity3, by stimulating naturally-occurring or introduced microbiomes to remediate contaminated groundwater4, or by building reactor microbiomes to recover valuable resources from wastewater5. Although these accomplishments highlight the valuable functions of microbiomes, the vast majority of the microbial world’s transformative capabilities have yet to be unlocked and harnessed. Recent insights driven by DNA sequencing have shed light on the high genetic diversity of not-yet-cultured microorganisms and their crucial roles in diverse ecosystems6,7, providing a window on potentially novel biotechnology applications.
In recognition of this unlocked potential, funding agencies and the international science community have called for a global effort to advance microbiome research8,9. These initiatives have recognized the need for microbiome science to move beyond descriptive studies, and embrace a systems approach that generates the mechanistic, predictive, and actionable understanding that enables rational microbiome engineering8. However, achieving this transition is hindered by the lack of tractable experimental systems that permit the detailed functional investigation of microbiomes, the large pool of microbiome gene and metabolite functions that remain unknown10, the many uncharacterized interactions (for example, syntrophy) between microorganisms11, inadequate tools to accurately measure and simulate microbiome functions across time and space, and the limited availability of approaches to precisely manipulate microbiome structure and function.
Integrating basic scientific discovery with engineering can overcome these challenges and develop innovative solutions that support sustainable natural resources management and human and animal health. In particular, engineering approaches can be used to create experimental systems that permit the testing of conceptual knowledge and extraction of new knowledge that advances microbiome research. To accelerate both scientific discovery and translation into innovative solutions, we propose that microbiome engineering adopt an iterative design-build-test-learn (DBTL) cycle to structure research and the technology development process. This cycle involves developing an initial microbiome design or preliminary model system to achieve a defined engineering goal, building the microbiome, testing its function against a set of specified metrics to determine whether the design-build solution(s) produced the design objective (i.e. establish causation), learning what worked, what did not (and why), and incorporating new knowledge into the decision making process of subsequent DBTL cycles (Figure 1). This approach has been used successfully in manufacturing12, metabolic engineering13, and entrepreneurship (‘build, measure, learn’)14, and could rapidly advance our ability to develop much needed tools and design concepts for harnessing microbiomes, delivering innovative solutions and advancing scientific knowledge.
Figure 1.
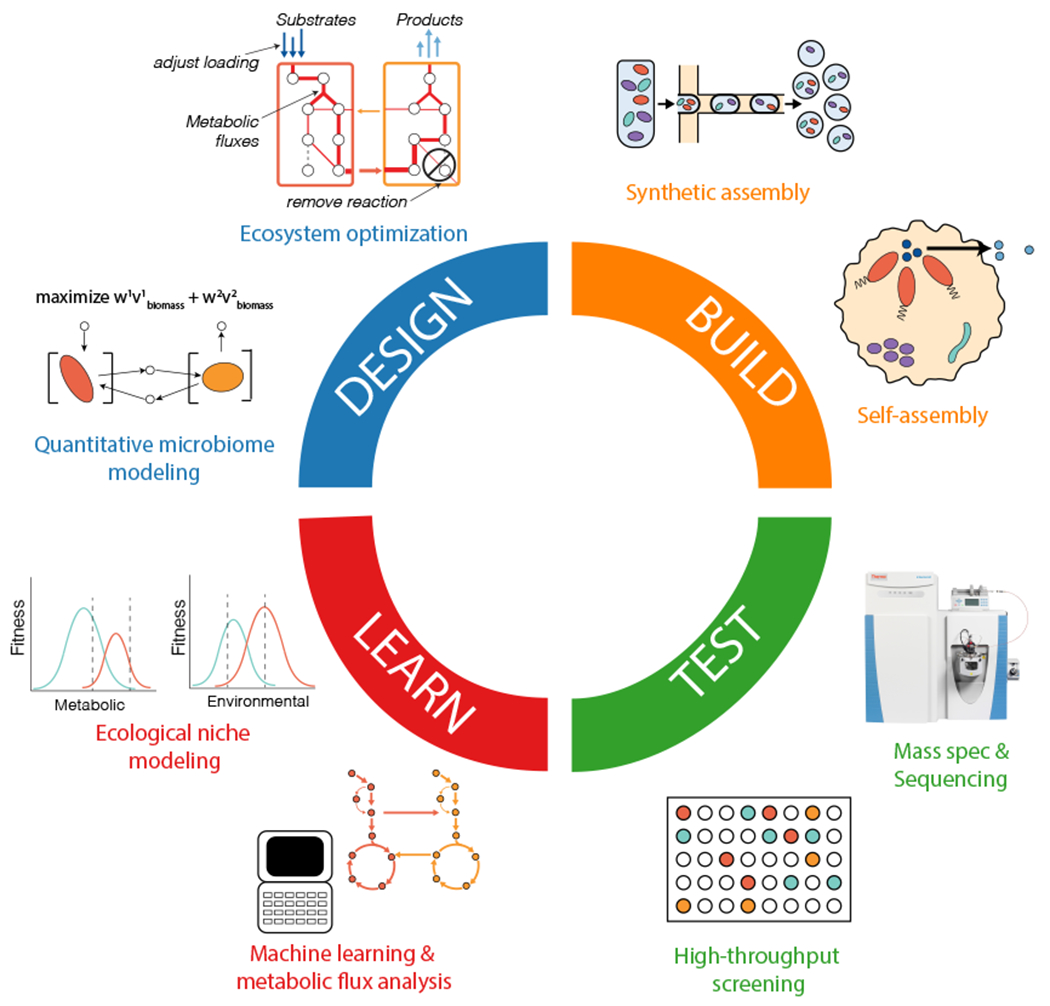
The design-build-test-learn cycle for microbiome engineering. The figure presents key aspects and approaches of each phase of the design-build-test-learn (DBTL) cycle. The cycle starts with a defined engineering objective that determines the design and produces an engineered microbiome that performs the desired function(s).
In this Review, we present key elements of an iterative DBTL approach that can be implemented to advance the rational engineering of microbiomes for functions that benefit society. We review diverse approaches to harness microbiomes in medical, agricultural, energy, and environmental applications, and identify current challenges and opportunities associated with implementing each DBTL phase. Finally, we discuss how the DBTL cycle can be applied to build model systems to establish basic principles of microbial ecosystems and provide an outlook on the frontiers of microbiome engineering.
Designing microbiomes
Because of the high complexity and limited understanding of molecular-scale microbiome processes, microbiome design has conventionally followed a top-down approach. This approach tries to predict how ecosystem-level controls can create a microbiome with desired functions. However, recent advances in multi-omics have provided opportunities to design microbiomes from the bottom-up by predicting how the control of metabolic networks and their interactions can create a microbiome with desired functions. Combined, these approaches offer complementary strategies to design microbiomes for specific engineering goals, ranging from sustainable wastewater treatment to curing microbiome-associated human diseases.
Top-down design.
Rather than deciding which organisms and detailed metabolic pathways to use a priori, the top-down approach uses carefully selected environmental variables (such as certain substrate loading rates, mean-cell retention times, and redox conditions) that force an existing microbiome (naturally occurring or inoculated) through ecological selection to perform the desired biological processes (or ‘metaphenotypes’15) (Figure 2). Here, ‘top’ refers to the ecosystem in which the desired biological process occurs and top-down design denotes the methods used to predict how manipulation of the ecosystem’s physical. chemical. and biological processes (that is. ecosystem processes) obtains the desired function. Predicting how to manipulate an ecosystem is informed by principles of ecological engineering16 (also known as microbial resource management17 or microbial community engineering18). This requires engineers to conceptualize the system as an ecosystem model that captures system inputs and outputs. physicochemical conditions (pH, temperature, redox potential, etc.), known abiotic and biotic processes, and environmental variables, and how their manipulation may promote or inhibit the biological process(es) being optimized19,20. Subsequently. mathematical modeling is used to perform mass balance analysis around chemicals and relevant microorganisms in the system and simulate chemical and biochemical transformation rates. These process-based models capture microbiome functions by representing key physiological or functional guilds of microorganisms (such as methanogens, fermenters, nitrifiers, or phototrophs) with specific stoichiometric (growth and product yields) and kinetic parameters (maximum specific growth rate, substrate uptake rate, and substrate affinity)21,22,23. The models can also integrate equations describing the three-dimensional physical transport processes (diffusion, advection, and dispersion) acting on chemicals and microorganisms, which are especially important in spatially structured systems such as biofilms24,25.
Figure 2.
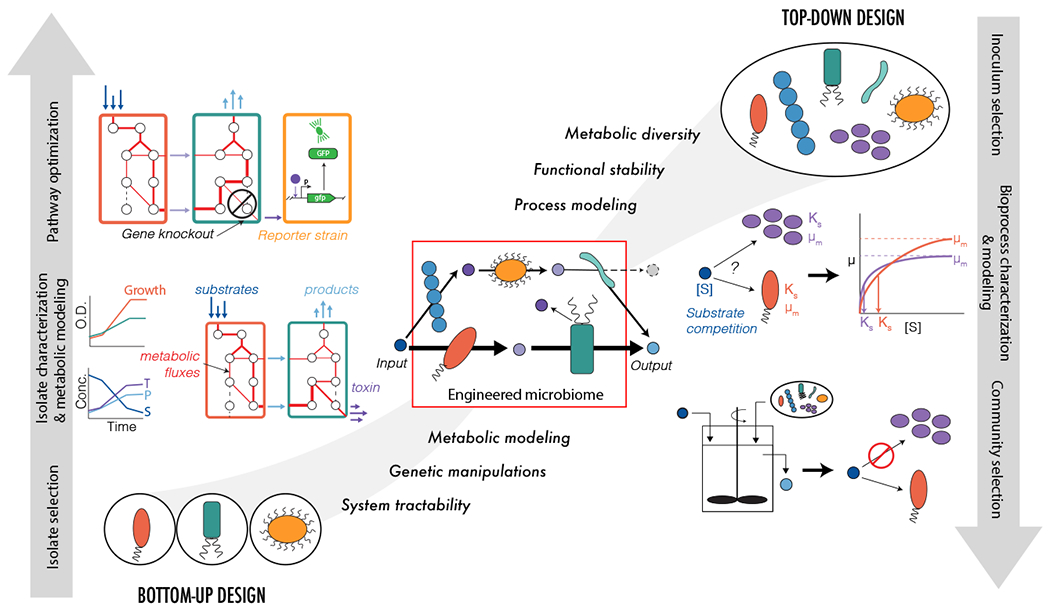
Top-down and bottom-up approaches to design microbiomes. The left panel illustrates a bottom-up design workflow starting from pure isolates. Physiological characterization of individual organisms is performed, and metabolic modeling is used to design consortia for desired function (produce light blue compound from dark blue compound). Genetic engineering and synthetic biology strategies are used to optimize system function (identifying gene editing targets that re-route metabolic flux away from toxin (purple) and towards desired product; designing of toxin reporter strain). The right panel illustrates a top-down design starting with an inoculum containing uncultivated microorganisms from the environment. Community characterization of mixed microbiome is performed, and bioprocess modeling (mass balance analysis including kinetics and microbial growth) is used to develop selection strategies to achieve desired function (produce light blue compound from dark blue compound). Reactor engineering design is used to optimize system function. The middle panel shows an integrated top-down bottom-up design. Combinations of uncultivated consortia and defined cultures are selected to achieve desired functions. Community characterization is performed and microbiome modeling that integrates process-based simulation with metabolic modeling is used to develop selection strategies and analyze microbiome metabolic fluxes. The shapes of the microorganisms represent different isolates or communities selected during design.
Bottom-up design.
Although the conventional top-down design approach for microbiome engineering offers a framework for macro-scale processes and has been widely successful for wastewater treatment21 and bioremediation4. it often neglects the complex in situ metabolic networks driving microbial and linked chemical transformations26 and ignores processes that depend on intricate interactions between community members; for example. syntrophic interactions through direct interspecies electron27. As a consequence. molecular-scale microbiome processes are often ignored during design. limiting system optimization through molecular-scale mechanistic insight. Recent advances in multi-omics and automation technology (for example, in metagenomics and microfluidics) have enabled researchers to develop bottom-up approaches and focus on engineering the microbiome’s metabolic network and microbial interactions. Here, ‘bottom’ refers to the metabolic networks of individual organisms in the microbiome (expressed from their genomes) and ‘bottom-up design’ denotes the methods used to predict how metabolic flux through these interacting networks obtains the desired function. The general design process is to obtain the genomes of individual members of the microbiome28 (especially keystone species29, when known30), reconstruct their metabolic networks,31,32 and use modeling33 and/or network analysis tools34 to guide design (Figure 2). Existing constraint-based methods such as flux balance analysis (FBA) provide a suitable framework for exploring which combinations of chemical transformations are possible using quantitative models, in which individual populations’ reactions and metabolites can be compartmentalized and metabolic fluxes within and between populations can be simulated using optimality principles35. These models can also simulate steady-state flux distributions over time and space36,37 and can be integrated into process-based and/or individual-based models38 to predict metaphenotypes, self-organizing spatial patterns, and other emergent behaviours. Such bottom-up tools provide the engineer with a computational framework to systematically evaluate the metabolic networks driving biological processes and ecological interactions, and a platform for rationally designing microbiomes with specific properties, such as distributed pathways39,40, modular species interactions41, community resistance and resilience42 and spatiotemporal organization43 that optimize ecosystem function and stability. However, the majority of these bottom-up design examples are based on simple communities with model organisms (such as Escherichia coli and Saccharomyces cerevisiae) that have engineered dependencies. Therefore, extending these designs to systems with non-model organisms of tens to hundreds of different species will require deeper insights into their metabolism and the principles governing their interactions and higher-order behavior.
There are major challenges to implementing this bottom-up approach, including inaccurate and/or incomplete metabolic network reconstructions, unknown functions of many genes, proteins, and metabolites, poorly understood evolutionary pressures driving individual and community-level phenotypes, and limited understanding of gene, metabolic, and ecosystem regulatory schemes (for example, quorum sensing signal-response systems44). These limitations lead to high model uncertainty because key constraints on pathway stoichiometry and enzyme kinetics are either inappropriate or missing, and objective functions fail to capture the true evolutionary drivers of cell behavior45, ultimately leading to poor predictions of in situ phenotypes. As a starting point for bottom-up design, core metabolic models that capture central carbon and energy metabolism can be reconstructed from genome annotations and known physiological information. The predictive power of these models may be limited initially, as they ignore regulatory information, pathway kinetics, secondary metabolism, and evolution. However, when this knowledge is acquired and becomes incorporated into metabolic models through multiple cycles of testing and learning, accurate predictions of system function (for example, metabolic fluxes and metabolite exchange) may emerge. As a complementary approach, data-driven modeling techniques such as ensemble modeling and machine learning may offer more rapid methods to predict microbiome metabolic processes or obtain constraints and parameters required for microbiome modeling, without the need for detailed mechanistic understanding of metabolic regulation46,47. Such modeling frameworks have been used to predict pathway fluxes from proteomic and metabolomic data48, improve metabolite cross-feeding predictions through ensemble modeling-based FBA49, and to obtain key catalytic turnover numbers needed for metabolic models50. Although these approaches are flexible and generalizable enough to be applied to microbial communities, they require substantial amounts of experimental data on the metabolism of individual strains and interacting communities. This information could be leveraged from prior test phases (for example, from high-throughput phenotypic screens and multi-omics) to enable data-driven design.
Integrated design.
Moving forward, we envision that a judiciously balanced blend of top-down and bottom-up approaches will be needed for successful microbiome design, especially when working with complex microbiomes, such as human microbiota or activated sludge (Figure 2). A blended approach could involve selecting both undefined mixtures and defined consortia to achieve desired microbiome functions, merging process-based models with bottom-up metabolic models reconstructed from meta-omic information to simulate ecosystem processes, mass balances, and metabolite fluxes, and using genome-derived information to develop community selection strategies. Capturing higher-order properties in design, such as functional stability and dynamics, will likely also require top-down and bottom-up approaches to converge. In particular, new mathematical modeling approaches that quantify mechanisms of functional degeneracy, niche complementarity, and network buffering51 using a metabolic framework may enable microbiome diversity to be optimized to sustain desired functions in situ. The need for a more comprehensive representation of microbiome metabolism will depend on the specific engineering objective and the degree of ecosystem tractability. For example, a more detailed representation of anaerobic microbiome metabolism is likely required for converting biomass into a specific commodity chemical instead of methane because finer control over metabolism would be needed. In either case, the design phase encompasses defining the engineering problem, developing conceptual and quantitative models, identifying key biological processes to be manipulated, and evaluating multiple candidate design alternatives.
Practical design steps.
There are five key steps when designing microbiomes, in particular complex microbiomes: defining the engineering problem, developing a conceptual ecosystem model, creating an quantitative model, identifying the microbiome process(es) to be engineered, and developing and evaluating candidate design strategies.
To drive the DBTL cycle, a clear definition of the problem with measurable design objectives must be established. These objectives could specify desired outcomes such as product titers, rates and yields, pollutant removal efficiency, crop productivity, or degree of functional stability and robustness. Design objectives should be complemented by techno-economic assessments and/or life cycle analysis to ensure that solutions are economically feasible and have positive environmental and societal impacts52,53.
Conceptual ecosystem models can be used to contextualize the problem. Such models capture system boundaries, inputs and outputs, major pathways of carbon and nutrient flows, key organisms and interspecies interactions responsible for those transformations, and factors influencing their activity (for example, pH, temperature, redox potential, and residence times)19. They provide a concept map that describes current understanding of interactions between the microbiome and physical, chemical, and biological components of the ecosystem, helping to identify important gaps in system understanding and needs for data collection. At this stage, all relevant information should be collected from the literature, existing data (for example, from the Human Microbiome Project54), and online databases (for example, MiDAS (microbial database for activated sludge)55) for ecosystem characterization. This includes reference genomes and physiological information for keystone organisms, previous multi-omic datasets, ecosystem physicochemical properties (such as pH, temperature and chemical concentrations) and processes (such as photochemical reactions and hydrogeological processes), site characteristics (such as nutrient loadings and dynamics, soil profiles and gut anatomy), and all other information needed to characterize the ecosystem. Missing information, such as unknown biochemical pathways and organisms that mediate them, can be targeted during the build-test-learn phases. This conceptual ecosystem model can be used by the scientific community for proposing and testing theories and serves as a roadmap for developing quantitative simulation tools.
Construction of quantitative modeling tools that enable the calculation and simulation of metabolic fluxes, microorganism abundances, mass balances, and ecosystem physicochemical parameters is critical for the systematic design of microbiomes. Several approaches could be used to create such models, including mechanistic metabolic modeling33, process-based modeling21, data-driven modeling (for example, machine learning)48, individual-based modeling38 or their combination. Regardless of the approach, the simulation of complex microbiomes will likely require simplification based on experimentally valid assumptions. Simplification could include reducing the model to a set of core or keystone organisms that represent important functional guilds and control major carbon and energy flows, or reducing the metabolic network size of organisms to central carbon and energy metabolism. Moving forward, it will be important to ensure that models undergo rigorous experimental validation and iteration during build-test-learn cycles to increase their utility and widespread use in microbiome engineering and to identify when modeling efforts fail, revealing gaps in conceptual understanding that can further facilitate model redesign and improvement.
Quantitative microbiome modeling (such as dynamic FBA) helps to identify the core and peripheral biochemical pathways that need to be directly manipulated, added, or removed to achieve the desired engineering objective. Objectives could include increasing butyrate production and non-digestible carbohydrate degradation by fermenting bacteria in the human gut, preventing toxin biosynthesis by cyanobacteria in freshwater ecosystems, or stimulating the degradation of toxic chloroorganics by bioaugmentation with organohalide-respiring bacteria.
Microbiome modeling can predict how environmental (such as substrate loading, pH, and solids retention time) or genetic manipulation (such as gene knockouts, pathway additions, and forced dependencies) could optimize microbiome functions towards the engineering objective. If necessary, synthetic microorganisms could be designed to improve microbiome function. Such synthetic microorganisms will need to be evaluated for their ability to cooperate and compete with existing microbiome members under relevant environmental conditions.
Building microbiomes
The build phase consists of physically assembling the designed microbiome by either top-down manipulation of a natural community (that is, a self-assembled microbiome) or bottom-up assembly using axenic or enrichment cultures of naturally-occurring or engineered microorganisms (that is, a synthetic microbiome). The build phase aims to bring the design specifications and predictions into reality.
Building by self-assembly.
Self-assembled microbiomes may include those built as open mixed cultures using reactor engineering (for example, wastewater treatment bioreactor) or biostimulation (for example, additions to soils, sediments or groundwater aquifers), in which construction creates an environment that promotes the growth and desirable activity of resident microorganisms. Examples include manipulating reactor hydrodynamics to immobilize slow-growing microorganisms into compact granules that enable their retention and proliferation56,57, use of non-human-digestible carbohydrates to stimulate fermentative production of short-chain fatty acids in the gut58, or adding electron donors to drive the metabolism of organohalide-respiring bacteria during bioremediation of toxic chlorinated contaminants4. This approach is powerful when differences in physiological and physicochemical properties between functional guilds can be exploited for assembly through environmental manipulation (for example, differences in growth rates59, main electron donors and acceptors4,60, substrate affinities, cell and/or biofilm densities61, and redox gradients). However, it can be limited when more fine-scale control over microbial metabolism and interactions is necessary (for example, controlling complex competitive interactions62, producing valuable bioproducts at high yields and purity63, or controlling organisms with versatile lifestyles64).
In addition, new strategies for evolutionary engineering have emerged as promising tools to build self-assembled microbiomes. Controlled exposure of an initial microbiome to multiple selection cycles and/or regimes results in the microbiome gaining or optimizing specific functions through adaptation or evolution. For example, successively transferring the microbiomes that maximize plant traits has generated microbiomes that improve plant biomass65 and flowering time66. Response to community-level selection will often be driven by enrichment or adaptation of single species67,68; however, selection for production of community biomass has also been shown to enhance desired species interactions in defined two and three species co-cultures37,69. Re-examining selection experiments to understand when and how mutations and/or adaptations altered microbiome phenotypes could elucidate the mechanisms underlying microbiome fitness optimization and inform design, as has been shown for E. coli in laboratory evolution experiments70,71. As similar evolutionary approaches (for example, adaptive laboratory evolution) have also been successfully applied to optimize strains for metabolic engineering72, extension of experimental and computational protocols already developed for individual microorganisms to microbiomes could streamline the design phase and reduce the time required to complete evolution experiments.
Building synthetic microbiomes.
Direct construction of microbiomes using axenic or enrichment cultures is also promising because of reduced complexity and the use of microorganisms that are genetically tractable and/or well-characterized. This bottom-up approach makes the growing suite of synthetic biology tools accessible for microbiome construction and optimization. An early approach for building microbiomes directly from cultured microorganisms is bioaugmentation. Here, defined laboratory consortia are added back to the environment to enhance the degradation rates of specific contaminants. A successful example has been the addition of consortia containing organohalide-respiring bacteria of the class Dehalococcoidia to contaminated groundwater aquifers and sediments to speed up the degradation of toxic chlorinated solvents. Crucial for the success of this approach was detailed knowledge of the physiology, nutritional requirements, and potential ecological interactions of the keystone dechlorinators with other microorganisms and the geochemical environment4. However, contrary to the success for chlorinated contaminants, bioaugmentation approaches have largely failed for oil spills. Unlike organohalide-respiring Dehalococcoidia members that fill a unique ecological niche and cannot grow without the chlorinated contaminants, organisms capable of degrading oil hydrocarbons (especially aerobic bacteria) are ubiquitous, metabolically versatile, and do not depend on a specific substrate or redox couple for growth64. This metabolic versatility limits their utility for bioaugmentation given their unpredictable in situ activity. Other reasons why bioaugmentation can fail are that unrecognized mutualistic interactions and microorganisms performing critical functions are missing (for example, production of polysaccharide surfactants to increase hydrocarbon bioavailability73), or that consortia selected under laboratory conditions are no longer competitive enough under harsh and/or variable field conditions74,75,76. These examples highlight the need to better understand the interaction networks of synthetic consortia, especially the roles of supporting interactions (secondary functions), and the competitive landscape in situ, which are often difficult to predict in complex ecosystems.
Despite the appeal of building microbiomes bottom-up and the growing collection of cultured microorganisms from specific habitats77,78, the majority of microorganisms relevant for human health, agriculture, and environmental applications remain uncultured, poorly characterized, genetically intractable, and difficult to maintain, making the construction of synthetic microbiomes challenging. To capture this uncharacterized metabolic diversity, innovative isolation and controlled microbiome assembly techniques are needed, such as singlecell sorting79 coupled to high-throughput culturing (culturomics)80,81 and phenotyping82,83 across multiple conditions in parallel. Microfluidics84,85, that is, creation and manipulation of microliter droplets, can facilitate this approach. Microfluidic chips can enable automated assembly and analysis of microbial communities from axenic or enrichment cultures through droplet combination86, elimination of specific species87, sequencing, and multi-omics phenotyping of individual cells88,89. Combined with new gene editing techniques, such as CRISPR-based genomic tools90 that improve the efficiency of homologous recombination-based gene editing91,92, microfluidics could also automate synthetic biology techniques for the engineering of cells and microbiomes with novel capabilities93.
Another challenge with synthetic microbiomes is maintaining their functional stability in the laboratory or in open systems (for example, human gut, soil, and wastewater treatment plants), which are susceptible to invasion by naturally-occurring microorganisms and dynamic heterogeneous environments. As mentioned above, the major reason for the success of bioaugmentation with organohalide-respiring Dehalococcoidia members is their highly specialized lifestyle that enables them to occupy an open ecological niche using chlorinated electron acceptors. However, the functional stability of organisms with versatile lifestyles in open systems is much less predictable. Few studies have examined the functional stability of synthetic consortia in open systems and the knowledge required to rationally engineer stable ecological interactions is limited. However, engineered bacteria have been successfully deployed as diagnostic sensors in the mammalian gut for up to 200 days maintaining robust function94,95. This feat, together with the bioaugmentation example of Dehalococcoidia4, demonstrates that synthetic consortia can form stable microbiomes with previously established community members, provided key players can compete with resident microorganisms.
Observations from self-assembled microbiomes suggest that building communities with spatiotemporal organization will be important for achieving stable and multi-functional synthetic microbiomes. Highly diverse microbial communities, such as human microbiota or those used for wastewater treatment, self-assemble as biofilms, flocs, or granules comprised of multiple single-species microcolonies attached together via species-specific extracellular polymeric substances (including polysaccharides, proteins, and DNA) and other poorly defined macromolecules (such as humics)96,97. These self-organizing microbial assemblages create diverse microenvironments and ecological niches that support the combination of seemingly incompatible functions (for example, both aerobic and anaerobic processes98,99) and functionally diverse population structures that can compensate for disturbances, such as changes in nutrients, physicochemical condition, or predation100,101. Although building such fine-scale and sophisticated architectures into synthetic microbiomes is nascent, microfluidic-based systems have been used to assemble simple communities with improved functional stability by controlling spatial structure and chemical communication102. Additionally, 3D bioprinting platforms could enable the construction of spatially organized systems, in which populations can be physically separated while remaining chemically interactive103,104. How to scale these spatially defined structures from experimental laboratory systems to real-world applications remains to be resolved, although knowledge gained from test and learn phases with model systems (such as synthetic polysaccharide particles 105,106) should provide more insights. Until then, existing approaches based on top-down assembly and/or engineered biofilm carrier media107 could be used to build self-organized synthetic microbiomes with better stability and functionality.
Designing synthetic genetic circuits in engineered hosts that can robustly perform sense-compute-respond programs in complex environments also remains a major challenge108. Therefore, it will be important to examine the molecular mechanisms that determine microbiome stability and adaptation to environmental perturbation in natural and engineered ecosystems, in order to extract design principles that can be used for rationally engineering robust functions. Given the potential utility of genetically engineered microorganisms and microbiomes in diverse open environments, safeguards such as biocontainment systems (such as two-layered gene circuits and essential synthetic auxotrophies109) will also require further development and will be needed as integral components of constructed synthetic microbiomes that use genetically modified organisms in the future.
Integrating approaches.
The ultimate goal for rational microbiome design is to develop tools that enable engineers to directly add, remove, or modify specific functions and phenotypes in situ over a range of desirable operational conditions. One emerging technique with promise to achieve such flexibility is in situ metagenomic engineering110,111, which involves delivery of engineered mobile genetic elements to resident microorganisms. For example, donor strains engineered with integrative and conjugative elements have transferred DNA carrying a reporter and antibiotic resistance genes or multi-gene pathways (for example, nitrogen fixation (nif) gene cluster112) to bacteria in highly heterogeneous and diverse environments, such as soil112 and the mammalian gut111. Further development of such tools in combination with existing CRISPR-Cas gene editing techniques would enable the precise manipulation of the microbiome’s metabolic network in situ, effectively combining self-assembled and synthetic microbiomes (Figure 3; Box 1)
Figure 3.
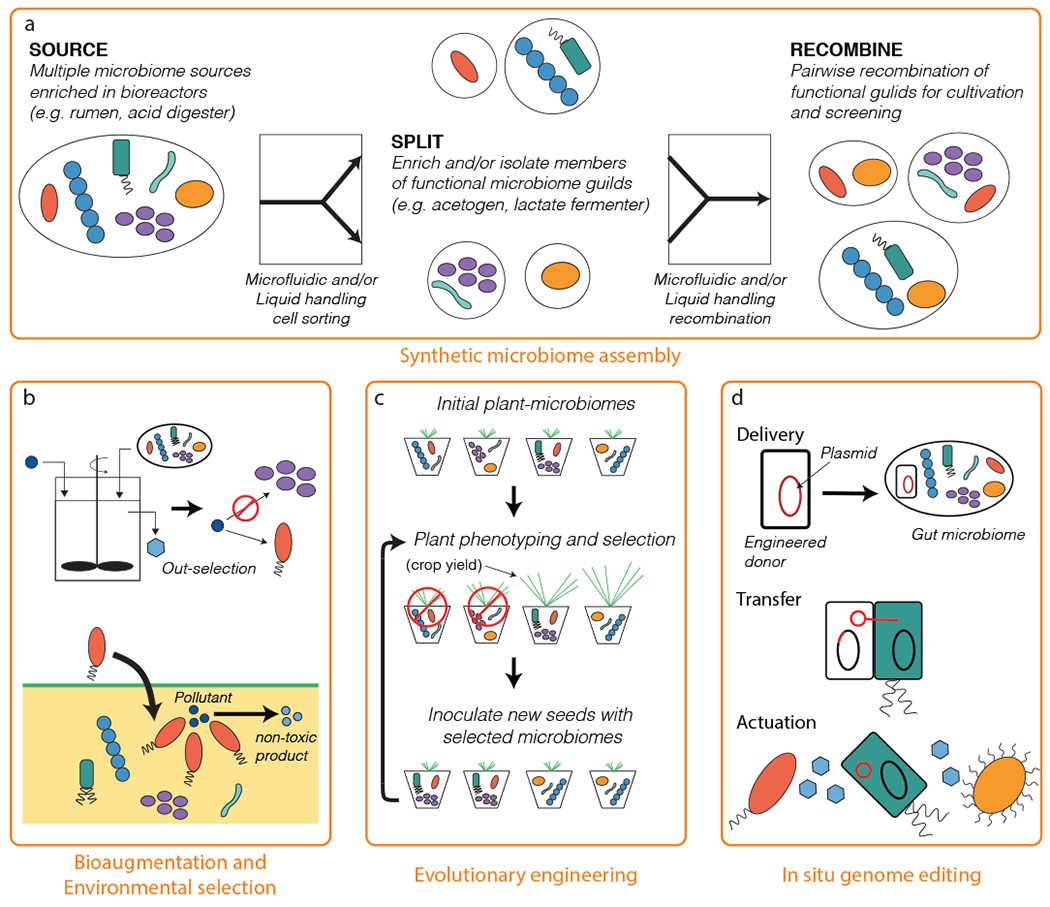
Building self-assembled and synthetic microbiomes. (a) This example shows a protocol for assembling synthetic microbiomes from multiple microbiome sources. Complex microbiomes can be taken apart into key functional members using automated microfluidic cell sorting techniques. Isolated or enriched members can then be recombined into synthetic consortia using liquid handling robotics for downstream screening and/or cultivation. (b) Microbiome assembly can also be achieved through environmental selection via bioreactor manipulation or biostimulation (top) or using bioaugmentation with defined cultures (bottom). (c) Another option is microbiome assembly through directed adaptation and/or evolution of the microbiome to acquire or optimize a desired function. (d) In situ microbiome engineering can be used to add new functions to microbiomes residing in the environment.
Box 1 - A DBTL cycle to create synthetic microbiomes with desired functions.
Here, we present a generalized DBTL cycle for creating synthetic microbiomes with desired functions, integrating both top-down and bottom-up approaches. We briefly describe two iterations of the cycle and identify opportunities for incorporating high-throughput approaches and automation to increase speed and reproducibility.
[b1] Top-down approach
[b2] Design: identify biological process(es)
An example of a process to harness or replicate is anaerobic conversion of complex lignocellulosic biomass into valuable commodity chemicals. The initial design step includes selection of different innocula that may contain microorganisms with desired functions (for example, acid phase anaerobic digester, herbivore rumen, or others). Conceptual ecosystem models that include environmental parameters (pH, temperature, nutrients, etc.) and expected functional guilds (hydrolytic bacteria, fermenting bacteria, methanogens, etc.) are used to select enrichment variables.
[b2] Build: enrich microbiomes from multiple sources
Source innocula are cultivated under different environmental conditions to select for desired function using real (for example, lignocellulosic hydrolysate or rumen fluid) and synthetic media. Modulation of environmental conditions and medium composition are done to improve desired function. For complex environments (such as soil) model laboratory ecosystems could be ideal platforms for microbiome enrichment146.
[b2] Test: evaluate performance
Performance of enriched microbiomes are tested on real and synthetic media using high-throughput phenotypic screens. High-throughput screens could be developed using microfluidic or automated microbioreactor experiments. Deeper multi-omic measurements (such as metagenomics, metatranscriptomics, and metaproteomics) are collected from high performing microbiomes.
[b2] Learn: identify key functional roles of microbiome members
Besides key functions, bottlenecks for the desired function are identified using metabolic reconstruction and multi-omic analysis. This understanding helps to refine conceptual models of microbiome function and create quantitative models.
Bottom-up approach
[b2] Design: screen for new potential microbial partners
In silico metabolic modeling is used to screen for interacting microorganisms from high performing microbiome enrichments. Metagenome-assembled genomes (MAGs) can be used to reconstruct metabolic models of key microbiome members. Automated computational workflows (together with manual curation) will accelerate model building. FBA is used to predict each microorganism’s requirements for optimal growth and activity, and unify individual metabolic models into a microbiome model to identify new potential partners that improve the design objective (for example, higher titers, rates, or yields of valuable product).
[b2] Build: recombine key microorganisms into new synthetic consortia
Following their isolation or enrichment, key microorganisms are assembled into new synthetic consortia based on in silico predictions at various ratios (for example, 1:1, 1:10). Microfluidic devices and/or liquid handling robotics could be used for high-throughput isolation and recombination.
[b2] Test: test function and stability of consortia
High-throughput phenotypic screening coupled to multi-omic measurements can be used for testing. This step should also include validation of predicted metabolisms of individual isolates or enrichments.
[b2] Learn: identify microbial interactions that control function
Analyzing the metabolism of microorganisms growing in consortia versus in isolation using metabolic flux analysis (MFA) can identify important mechanisms and interactions. This understanding can be used to propose how microbiome function and stability could be optimized by environmental manipulation and/or in situ genome-engineering.
Testing microbiome function
The test phase involves measuring microbiome-associated phenotypes and properties to determine the efficacy of the design-build solution. The measurements should determine whether the design outcomes were achieved (for example, measuring the titer-rate-yield of a bioproduct, pollutant removal efficiency, or crop productivity) and whether the design-build solution was responsible for the observed outcome (establishing cause and effect). This typically requires readouts of ecosystem physicochemical properties (such as pH, temperature, and chemical concentrations), as well as the stoichiometry and kinetics of key ecosystem processes and microbiome functions (such as biomass growth, chemical transformations, nutrient assimilation, and metabolic fluxes). For example, acetate degradation rates and pathways to methane in an anaerobic digester microbiome could be tested using 13C-labelled acetate and online biogas analysis that measures the flux through acetoclastic methanogenesis versus syntrophic acetate oxidation coupled to hydrogenotrophic methanogenesis113. While the level of microbiome granularity measured during testing will depend on the specific design objectives and ecosystem complexity, the ability to quantify molecular microbial processes (for example, metabolic pathway rates and routes, enzyme activities, and individual organism growth rates) goes beyond bulk activity measurements and enables testing the specific mechanisms responsible for the observed microbiome functions. The challenge will be to develop tools that are high-throughput, quantitative, affordable, and easy to use, such that routine analyses of the microbiome over time, space, and under dynamic conditions can be accomplished.
Towards this goal, we envision a test phase comprised of high-throughput phenotypic screening of microbiome design-build solutions, followed by deeper investigation of promising solutions using multi-omic and metabolic flux analyses to obtain greater insights on underlying mechanisms (Figure 4). High-throughput phenotypic testing of constructed microbiomes could be achieved using droplet microfluidics, as has recently been demonstrated for screening ~100,000 synthetic communities114. Fully automated microbioreactor platforms that combine liquid handling and advanced sensing with microtiter plate or scaled-down bioreactor cultivation could also be used82,83. Combined with emerging methods to measure metabolic network activity and metabolic processes in heterogeneous environments (Box 2), rich information will be obtained to facilitate learning.
Figure 4.
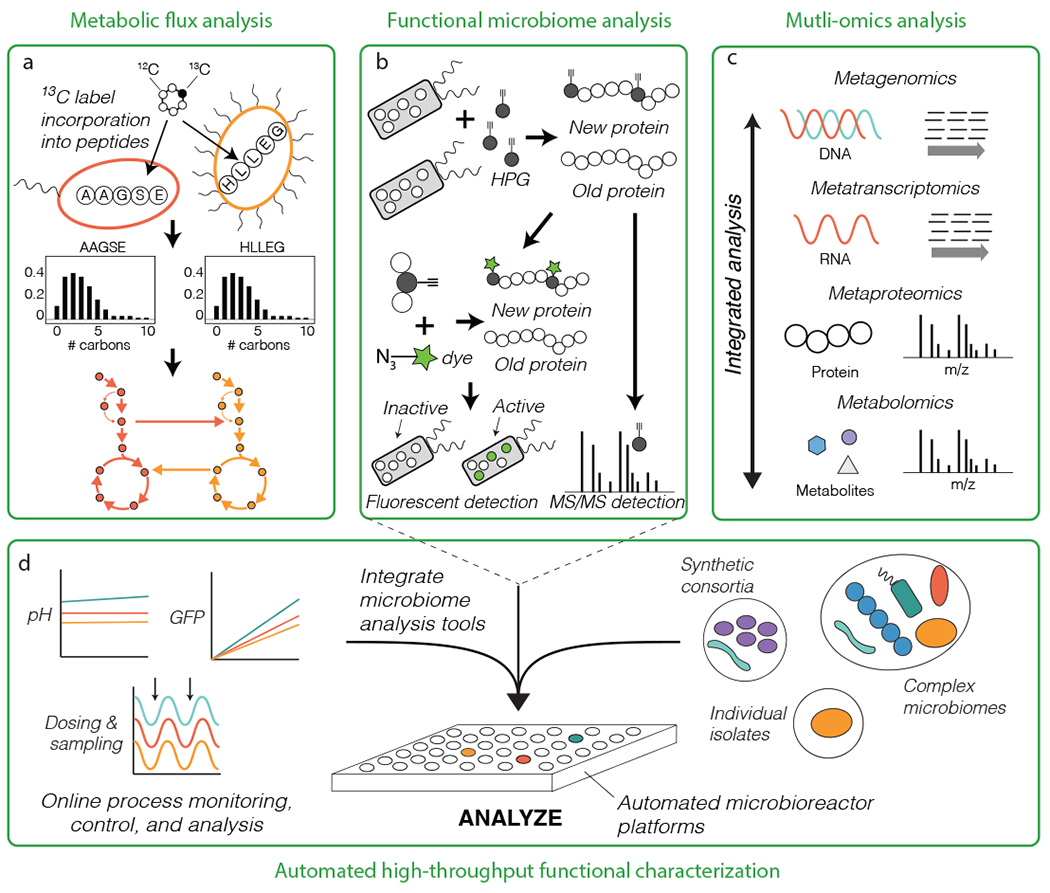
Testing microbiome function. (a) Isotopic tracers combined with metaproteome can also be used to measure microbiome metabolic flux by analyzing isotopic labelling patterns of short peptides rather than amino acids (metabolome). (b) Biorthogonal non-canonical amino acid tagging (BONCAT) is a method for rapid profiling of the anabolic processes (growth) in situ using either fluorescent detection or metaproteomics. (c) Metagenomics, metatranscriptomics, metaproteomics, and metabolomics can be integrated to reconstruct and analysis metabolic network expression in microbiomes. (d) An automated microbioreactor platform enables high-throughput analysis of microbiome processes across diverse conditions (for example, with changing environmental or physiological variables). The platform can integrate tools for detailed functional analysis of individual microbiome members to complex communities. HPG: the amino acid homopropargylglycine.
Box 2 - A toolbox for measuring microbiome function.
[b2] Multi-omics integration.
The ability to assemble genomes from metagenomic data28 has enabled the genome-resolved analysis of individual transcriptomes63 and proteomes118 from diverse communities and greatly increased the interpretive power of multi-omic datasets. A key challenge moving forward will be the integration of metabolomic information163, both intracellular and extracellular, which cannot be readily assigned to individual members of the microbiome such as DNA, RNA, and proteins can be. The large amount of unknown or poorly characterized genes, enzymes and metabolites currently limits the interpretive power of multi-omic information. It does, however, create novel targets for further biochemical studies. Advances in bioinformatic tools, such as data-driven approaches (for example, statistical or machine learning methods) and knowledge-based approaches (for example, interaction networks or genome-scale metabolic modeling)164,165, will be key to the success of systematic measurements of microbiome function through coherent multi-omics data integration.
[b2] Isotopic tracers.
Isotopic tracers have a long history in functional analysis in both pure cultures and communities, and have been combined with DNA166, RNA167, and protein116 measurements to link individual populations to specific in situ functions. Moving forward, more efforts to incorporate isotopic tracers with multi-omics (especially metaproteomics and metabolomics) are needed for illuminating the complex metabolic networks within microbiomes. The combination of these techniques should also pave the way for measurement of intracellular and extracellular reaction rates (‘metafluxomics’)124,125, which has been one of the most powerful tools for elucidating in vivo phenotypes, pathway constraints, and metabolic regulation in pure cultures used for engineering purposes.
[b2] Mass spectrometry imaging.
Mass spectrometry imaging (MSI) techniques visualize the distribution of elements and their isotopes as well as biomolecules within complex samples. MSI is well suited for the analysis of spatially structured microbiomes and for the investigation of cellular interactions. When combined with FISH, MSI also enables the linking of microbiome structure with function168,169. The chemical coverage, spatial resolution, and sample preparation that can be obtained with different MSI techniques depends on the selected ionization method132. Although nanoscale secondary ion mass spectrometry (nanoSIMS) has superior lateral resolution compared to matrix-assisted laser desorption-ionization (MALDI) or desorption electrospray ionization (DESI; 50 nm, 3-50 mm and 100 mm, respectively), its relative chemical versatility is very low (elements and isotopes versus peptides, lipids, metabolites, and other molecules). Therefore, nanoSIMS has generally been applied to study substrate use of single cells, whereas MALDI has been used to visualize chemical interactions between populations132. Although MALDI-MSI and DESI-MSI are more accessible than nanoSIMS170 and could be well positioned to visualize the broad range of chemical interactions within microbiomes, they have very low throughput and their lateral resolution and sensitivity currently prohibit single-cell metabolic profiling132. A technique that combines the best of these two methods is nanostructure-initiator mass spectrometry (NIMS). NIMS is a matrix-free desorption-ionization technique that depends on initiator molecules trapped in 30 nm large pores to achieve the ionization of small molecules adsorbed to the pore surface. NIMS offers a lateral resolution of ~150 nm and is particularly well suited for the analyses of peptides and metabolites171. So far, NIMS has only seen limited application in microbiology172,173. We expect advances that improve these issues will make MSI a useful and more widely applied tool for functional analysis of microbiomes in the near future174.
[b2] Bioorthogonal chemistry.
Metabolic labeling techniques, such as bioorthogonal non-canonical amino acid tagging (BONCAT), offer additional approaches to measure microbiome anabolic activity in situ. BONCAT is based on the in vivo translational incorporation of a non-canonical amino acid (for example, L-azidohomoalanine, a L-methionine surrogate), followed by fluorescent labelling of tagged cellular proteins by azide-alkyne click chemistry175. The technique can be used together with rRNA-targeted FISH to directly link taxonomy with in situ activity175. BONCAT has also been combined with FACS to separate active cells from complex samples and further characterize them by DNA sequencing133. In addition, tagged proteins can be selectively enriched through bead-capture and subjected to proteomic analysis176. The combined application of these methods could enable the high-throughput tracking of newly synthesized proteins from uncultivated microorganisms under different physicochemical conditions. Although BONCAT can be limited due to differences in cellular amino acid uptake and metabolic perturbation, the technique offers a flexible tool for the comparatively simple, inexpensive, and high-throughput analysis of in situ activity on a single-cell level.
[b2] Microfluidics.
Devices that enable the high-throughput analyses of microorganisms at single-cell resolution will be important for the rapid cultivation and functional analysis of microbiomes. Microfabricated devices such microfluidic ‘lab-on-chip’ technology could offer multiple applications, including isolation of individual cells and populations from complex microbiomes177, creation of in vitro cell-based models that facilitate assembly of synthetic microbiomes and experimentation under heterogenous microenvironmental conditions178, and online diagnostics for rapid monitoring and detection of desired phenotypes. These applications are still in early stages of development and several challenges remain, including reliable detection of microorganisms in droplets, precise control of gas concentrations, cross contamination, and technology accessibility177,179.
[b2] Automation.
To increase the reproducibility, throughput, efficiency, and standardization of microbiome engineering, advances in automation will be necessary. This includes incorporating liquid handling robotics, microfluidic devices, automated cultivation systems, online physicochemical measurement sensors, and software into data generation and analysis workflows. Emerging examples include the use of liquid handling robotics coupled to automated microfermentation platforms for high-throughput cultivation82, or microfluidics to automate the analysis of thousands of droplet experiments that probe microbial community interactions180,114. Such automated platforms could also integrate several functional tools (for example, single-cell analyses and multi-omics), resulting in rich reproducible data sets that could be leveraged for machine learning and other big data analytics.
Microbiome metabolic network activity.
To test predictions of microbiome function at a systems-level, measurement of the microbiome’s in situ metabolic network structure and activity is critical. Multi-omic approaches (metagenomics, metatranscriptomics, metaproteomics, metabolomics) combined with bioinformatic tools have enabled the genome-centric analysis of individual species (or even strains115) within microbiomes and global measurement of sequences, proteins, and metabolites116,117,118. These tools measure the microbiome’s components on a spectrum from functional potential (for example, gene abundance) to expressed products (for example, protein and metabolite abundance), and through their combined activity produce microbiome metaphenotypes that drive system function. Currently, multi-omic approaches used to infer microbiome function have focused on correlating gene abundances or gene expression data across time and space with ecosystem geochemical data or process rates. This has included measurements of key functional genes and transcripts using qPCR assays (for example, ammonia monoxygenase119), microarrays (for example, GeoChip120), or untargeted high-throughput approaches (metatranscriptome and/or metaproteome). Although useful for overall system characterization and discovery, these approaches focus on measuring the components or “parts list” of the system, which are often limited predictors of emergent phenotypes due to metabolic network complexity, interactions, and regulation121,122. Therefore, new approaches and tools are needed to measure the in situ stoichiometry and fluxes of microbiome metabolic networks to permit the direct testing of design predictions and offer mechanistic insights into metabolic regulation.
MFA is the most authoritative method for measuring in vivo fluxes. This method calculates fluxes from metabolite stable isotope measurements obtained during isotopic labelling experiments using metabolic network modeling123. Although MFA has been used to measure fluxes in co-cultures124, flux analysis in communities is challenging because metabolite pools cannot be easily assigned to individual cells and the number of possible reactions in a microbiome greatly exceed those of an individual organism. Nonetheless, isotopic tracers combined with exometabolomics and/or off-gas analysis have been used to determine process fluxes driving important microbiome functions, such as syntrophic acetate oxidation and methanogenesis during anaerobic digestion116. To circumvent the challenges with metabolite measurements, a method analyzing labelling patterns from short peptides instead of amino acids for MFA was proposed125. Peptides can be assigned to individual species in a microbiome using high-throughput metaproteomic approaches, which opens the door to determining fluxes in microbial communities (that is, to ‘metafluxomics’). Given that fluxes represent the final outcome of cellular regulation across all levels126, further development and demonstration of metafluxomics will be essential for advancing microbiome engineering efforts and our understanding of metabolic regulation in microbiomes. This will also require new software packages for associated computational analyses, similar to existing 13C-MFA software127. Such data may also allow metabolic modelers to infer, rather than assume, community and individual-level objective functions and to identify new constraints, enabling the accurate prediction and measurement of reaction rates driving microbiome function.
Measuring function in spatially heterogeneous environments.
Most natural microbiomes, such as those associated with plants (for example, rhizosphere), humans (for example, oral microbiome), and industrial processes (for example, acid mine drainage), display highly-organized spatial organization across micro-scale physicochemical gradients that directly influences microbiome function. For example, the spatial proximity of microorganisms can control whether they interact through diffusible substrates or direct transfer128, whereas variations in colony size can dramatically influence apparent substrate affinity constants and substrate competition between biofilm microorganisms129. Therefore, one of the biggest challenges will be to create tools that measure and report on microbiome spatial structure and function across all relevant scales (from μm to km). Current methods to measure structure-function relationships have focused on the μm to mm scale using approaches such as fluorescence in situ hybridization (FISH) combined with stable isotope labeling (SIP)130, chemical fingerprinting131, mass spectrometry imaging132, and/or fluorescence-based biorthogonal non-canonical amino acid tagging (BONCAT)133 (Box 2). Although these techniques have successfully identified the substrate use and activity patterns of spatially distributed microorganisms in microbiomes, they are limited by throughput and can only examine and/or differentiate a limited number of organisms. The integrated application of labelling techniques (for example, SIP and BONCAT) with metaproteomics and cell sorting (for example, fluorescence-activated cell sorting (FACS)133) could be used to measure the metabolic activity of microorganisms in high throughput with spatial resolution. Combined with microsensor devices that profile microenvironmental chemical properties, for example, through microelectrodes134 or engineered biosensors95, microbiome structure, function, and ecosystem physicochemical parameters could be monitored in real-time.
Learning microbiome design principles
Progressing through the design-build-test phases of microbiome engineering presents a unique opportunity to learn from previous failures and successes, and to incorporate new knowledge into subsequent cycles. Indeed, the learn phase of the DBTL cycle is critical for success and for improving microbiome engineering efficacy. To date there are no general strategies, techniques, or approaches that guarantee success in translating information obtained from the test phase into new knowledge that informs the next design phase. Therefore, we stress the importance of devoting enough emphasis and resources to the learn phase early on, so as to avoid, for example, the difficulties encountered in metabolic engineering due to a relative lack of investment in the learn step13. Further development of computational methods to formalize the learn phase will be needed, including machine learning algorithms48,135,136, metabolic flux analysis and constraint- based analysis36,124,125,137, ecosystem modeling approaches138, and regulatory network analysis139. Together, these analyses could isolate the principal drivers of microbiome interactions and function from large datasets to inform microbiome design. For example, generalized Lotka-Volterra equations could infer interacting species from temporal population dynamics data that become the starting point for bottom-up design140 or constraint-based analysis could be applied to identify key metabolite exchange reactions from 13C-metabolomic data that improve flux simulation accuracy and design of anaerobic consortia137.
More broadly, we envision the learn phase to focus on translating data into generalizable principles for microbiome engineering, through the continuous refinement of conceptual knowledge and proposed theory (for example, from traditional macroecology141,142,143,144,51) with each DBTL cycle. We propose that model laboratory ecosystems should be utilized to drive microbiome engineering inquiry and learning. Model laboratory ecosystems are experimental platforms that can replicate the physicochemical conditions of a complex environment (natural or engineered) in a simplified and controlled manner and contain model microbial communities (for example, the model rhizosphere microbiome THOR145) that can be used as testing grounds for learning how to design, construct, and optimize engineered microbiomes. These ecosystems have reduced complexity, are accessible for experimentation, and can be established in a reproducible manner, which is often not possible when working in natural environments.
Recently, model laboratory ecosystems have been developed for studying plant-soil microbiome interactions146. These fabricated ecosystem (EcoFAB) use 3D printing, sensing, and analytical and imagining technologies to create an experimental device that replicates the native soil ecosystem, in which microorganism and host phenotypes can be monitored in response to changing variables, enabling the systematic dissection of microbial interactions and metabolite exchanges influencing plant health146,147. EcoFABs offer a middle ground between model organisms and complex natural microbiomes, and can be established collaboratively between expert investigators to create standardized and reproducible devices and protocols for dissemination to the broader research community. Such model systems offer the ability to experimentally develop engineered microbiomes with desired functions in a tractable manner, and permit results to be compared with results from natural settings. This cross-examination between model and natural ecosystems will be a valuable and necessary approach for learning engineering principles and practices that are relevant to real-world systems (not laboratory artifacts), and for acquiring knowledge on scaling-up lab-based engineering strategies to full-scale applications (Figure 5). For example, microfluidic-based in vitro models of the human gut microbiome that contain co-cultures of human cells with different bacterial consortia are already producing physiological (including epithelial cell monolayer formation, cell growth and viability, cytokine levels, and metabolomic profiles) and environmental (including oxygen gradients and laminar flow) variables that are comparable to in vivo variables148.
Figure 5.
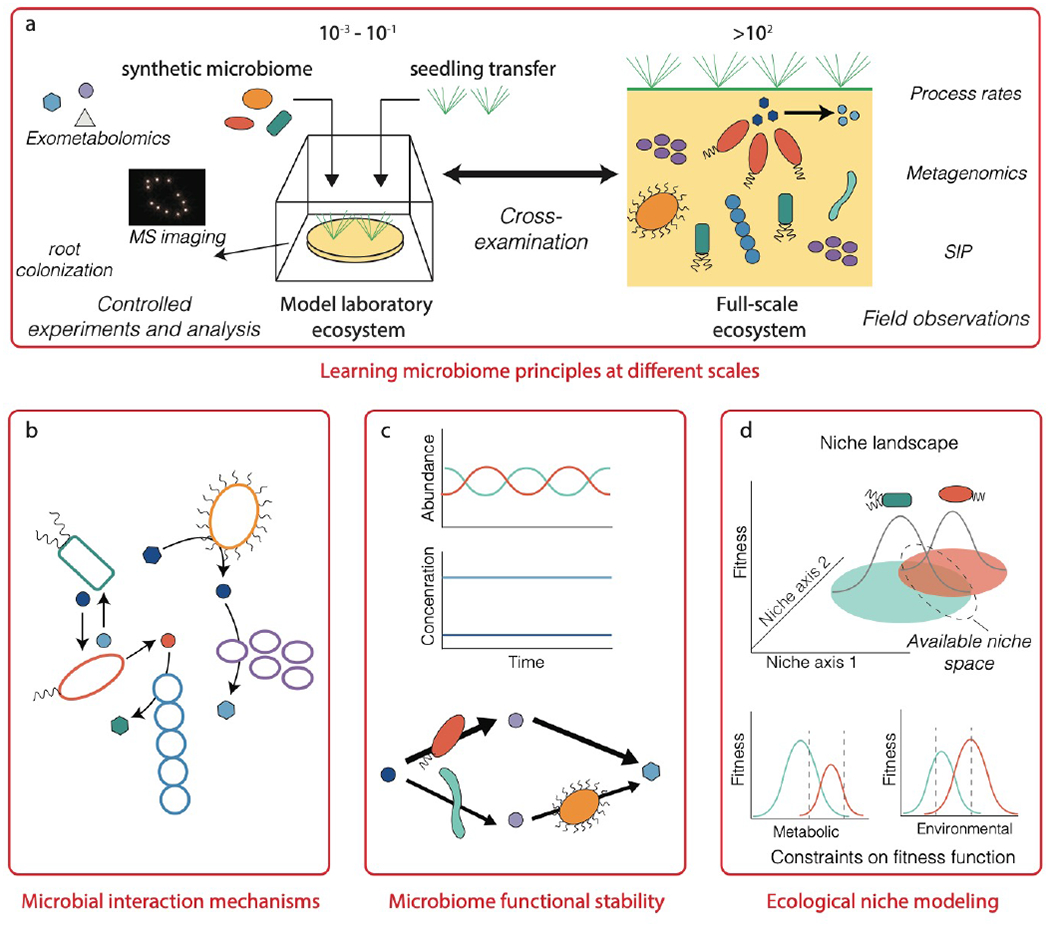
Learning fundamental principles for microbiome engineering. (a) Model laboratory ecosystems can be used for controlled experiments with simplified microbiomes and environmental properties, representing an in-between of pure lab conditions (such as test tubes or flasks) and complex natural environments (such as soil or the ocean). Continuous cross-examination between laboratory-scale models and natural complex ecosystems will be needed for developing engineering principles and practices that are robust in real systems, while also tractable in the lab. This will require close collaboration between multiple stakeholders, including researchers and end-users (such as hospitals or treatment plants) that have expertise and experience with issues specific to each scale. Key principles that need to be learned to enable systematic microbiome engineering are microbial interaction mechanisms, mechanisms governing functional stability and degeneracy, and frameworks for quantitatively mapping and simulating ecological niches in complex ecosystems.
The combination of model ecosystems with the DBTL cycle may be particularly fruitful for understanding the mechanisms governing microbial interactions and functional stability. Substantial knowledge is available on specific microorganisms that co-aggregate and exchange metabolites, such as bacteria involved in nitrogen cycling2, consortia of methane-oxidizing archaea and sulphate-reducing bacteria149,150,128, and syntrophic bacteria partnered with hydrogenotrophic methanogens151,152. However, we are only beginning to understand the complex mechanisms (such as quorum sensing and secondary metabolites) involved in regulating the behavior, interactions, and kin discrimination of microorganisms in communities153. Although studies have established links between microbiome functional redundancy, diversity, and stability154, a framework to predict or engineer functionally stable microbiomes has not been attained. Through the use of model laboratory ecosystems together with existing knowledge of microbial ecology and engineering design, it may be possible to decipher the chemical language of microbiomes and discover mechanisms of other important processes (including evolution, selection, dispersal limitation, and neutral processes155) that enable robust and stable microbiome function. Translating this theory into engineering design practice will require a quantitative framework that links these mechanisms to metabolic interaction networks, and new approaches that enable ecological properties to emerge from metabolic models (Box 3).
Box 3 - Emerging principles for microbiome engineering: a case for niche modeling.
Ecological niche modeling could be used to systematically design higher-order properties such as functional stability and robustness into engineered microbiomes. However, to develop such a framework, mechanistic understanding on how diversity is maintained within microbiomes and how it imparts properties such as functional stability is needed. Here we propose that this understanding could come from applying the DBTL cycle to answer key questions:
[b1] Does functional degeneracy lead to productivity and functional stability?
Diversity has been correlated with productivity and functional stability in communities of macroorganisms143,181, yet the role that diversity has in improving microbiome function and functional stability remains open. For microbiome engineering, we propose that diversity be viewed, discussed, and defined through the lens of functional redundancy (as described previously154), or more specifically, functional degeneracy. This is the degree to which a set of organisms perform an identical role in ecosystem functionality (for example, methane oxidation, nitrogen fixation, or polymer hydrolysis), but exhibit degeneracy with respect to other physiological traits (for example, pH optima or biofilm formation), which enables them to achieve realized niche space and coexistence51. The DBTL cycle offers an excellent opportunity to understand the molecular basis of functional degeneracy and to examine how emergent community-level properties, such as resilience to perturbation or susceptibility to invasion by another species, are predictable from quantifying the fundamental and realized niche space in microbiomes. We propose that ecological niche modeling could be a particularly useful framework to achieve this goal.
[b1] How is diversity maintained in microbial ecosystems?
To create a framework for ecological niche modeling, it will be important to understand how diversity is maintained. Competitive exclusion suggests that two species with identical resource requirements cannot coexist in the same ecological niche144. Therefore, we need to understand the mechanisms that create niche space and enable diversity to develop and be maintained. For example, what role do the processes of spatiotemporal variability, dormancy, predation, nutrient loading, secondary metabolite production and resistance, cell motility, and biofilm formation have in niche differentiation? And how can these processes be manipulated to achieve and maintain a desired level of functional degeneracy in a microbiome? Answers to these questions will offer microbiome engineering mechanisms to design and control ecological niche space for desired microbiome properties.
[b1] How does ecological niche modeling underlie microbiome engineering?
To enable the systematic engineering of desirable higher-order microbiome properties, we propose that microbiome engineering develops a framework for ecological niche modeling. The goal of this framework would be to quantify community and individual fundamental niche and realized niche space by integrating multi-omic data, physiological information, nutrient availability, and environmental parameters, and use them to develop strategies for controlling cooperation and competition in microbiomes. To achieve this goal, new mathematical representations of the fundamental and realized niche of an organism or guild will need to be defined, together with fitness functions that describe responses to environmental variables. When incorporated into microbiome modeling, this framework will enable the ecological forecasting of higher-order properties, as well as quantification of cooperative and competitive microbiome landscapes. Moreover, such frameworks will help guide important unresolved microbiome design questions, such as the trade-off between functional redundancy and minimal diversity.
Outlook
True advancement in microbiome engineering will need multiple rounds of DBTL to capture the necessary ecological principles to manipulate microbiomes in a precise manner with predictable outcomes (Figure 1). For example, incorporating direct interspecies electron transfer discovered during previous DBTL cycles into metabolic models and bioreactor construction (for example, by adding conductive materials) could optimize the efficiency of biogas production from waste27; or designing engineered E. coli to control levels of previously discovered autoinducers could tailor gut microbiota under conditions of dysbiosis towards a healthier state156. However, developing new knowledge and tools with fast turnaround will require next-generation infrastructure for data collection, data sharing, and knowledge integration. To accelerate progress, developing the predictive capabilities needed for the learn phase is a priority. Model laboratory ecosystems combined with advances in automation, such as liquid-handling robots, microfluidics, and data analysis pipelines157,158, will offer a starting point for the testing of multiple designs in a rigorous and reproducible manner. Capturing new knowledge from this process and integrating information into subsequent DBTL cycles will accelerate microbiome engineering developments, creating innovative biotechnologies and practices for the management of microbiomes across medicine, agriculture, manufacturing, and environmental stewardship. Examples that show particular promise for advancing microbiome engineering across these fields include illuminating the roles that phages and metabolite cross-feeding have in controlling ruminal carbon turnover159, harnessing untapped anaerobic fungal-bacterial consortia to improve biomass conversion to valuable bioproducts160,161, creating microfluidic cell sorting techniques to automatically sort stable isotope-labelled cells from high diversity samples for subsequent multi-omic analysis or cultivation162, and developing in situ metagenomic engineering tools to introduce new functions into microbiomes in their native environment111.
To move the DBTL approach forward, interdisciplinary research teams with expertise in experimentation (for example, in culturing, molecular genetics, or biochemistry) computation (for example, metabolic modeling, machine learning, or bioinformatics), automation (for example, robotics, or microfluidics), and practice (for example, professional engineers, or medical doctors) are essential. The road ahead for microbiome engineering seems long, given our nascent understanding of microbial ecology; however, structuring research and technology developments around the DBTL cycle offers a promising approach for advancing microbiome engineering and providing innovative solutions for addressing pressing societal and environmental problems.
Acknowledgements
The authors would like to acknowledge the College of Engineering at the University of Wisconsin-Madison that provided financial support for a workshop during the Madison Microbiome Meeting on April 27th, 2018 in which all authors attended and participated in discussions that led to the creation of this article. CEL is supported by a Postgraduate Scholarship-Doctoral (PGS-D) award from the National Sciences and Engineering Research Council of Canada (NSERC) and a Wisconsin Distinguished Graduate Fellowship. KDM and DRN acknowledge support from the National Science Foundation (CBET-1803055 and MCB-1518130) and from the University of Wisconsin-Madison Wisconsin Alumni Research Foundation via the Microbiome Initiative. DRN and BFP acknowledge support from the U.S. Department of Energy (DOE) Great Lakes Bioenergy Research Center grants (DOE Office of Science BER DE-SC0018409). BFP acknowledges support from the National Science Foundation (CBET-1703504; MCB-1716594). MAO and HGM are funded by the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley Laboratory and the U.S. Department of Energy. HGM is also funded by the DOE Agile BioFoundry (http://agilebiofoundry.org), supported by the U.S. Department of Energy, Energy Efficiency and Renewable Energy, Bioenergy Technologies Office, through the same contract DE-AC02-05CH11231. HGM is also supported by the Basque Government through the BERC 2018-2021 program and by Spanish Ministry of Economy and Competitiveness MINECO through BCAM Severo Ochoa excellence accreditation SEV-2017-071. FEL acknowledges support by DOD’s Strategic Environmental Research and Development Program (SERDP) and the Governor’s Chair program through the University of Tennessee and Oak Ridge National Laboratory.
Glossary
- Microbiome science
discovery and testing of fundamental principles governing microbiome function and assembly
- Microbiome engineering
leveraging fundamental scientific principles and quantitative design to create microbiomes that perform desired functions
- Metaphenotypes
sets of emergent functions of a microbiome resulting from the interactions between individual microbial genomes (metagenome) and their interaction with the environment
- Ecological engineering
the process of designing and operating bioreactors and other engineered systems to foster the development of specific microbial communities that can perform desired functional processes
- Exometabolomics
an analytical technique to quantify extracellular small molecule metabolites from environmental and/or biological samples typically through gas/liquid chromatography-mass spectrometry or nuclear magnetic resonance
- Functional guilds
groups organisms that use similar resources (for example, electron donors, electron acceptors, or carbon source) and occupy a similar ecological niche
- Fundamental niche
the entire set of environmental conditions in which an organism can survive and reproduce (that is, an organism’s niche in the absence of interspecific competition)
- Generalized Lotka-Volterra equation
A set of ordinary differential equations used to represent population dynamics based on experimentally inferred species interaction parameters
- Off-gas analysis
the monitoring of gas flow rate and chemical composition (e.g. carbon dioxide, hydrogen, methane) produced from a biological system
- Realized niche
the set of environmental conditions used by a species after considering interspecific competition (competition, predation, and others).
- Keystone species
An organism that has a disproportionately large effect on maintaining the microbiome’s function and microbial interactions (both between micoorganisms and with the environment).
- Flux balance analysis
a constraint-based mathematical modeling technique for simulating metabolic fluxes through a metabolic network reconstructed from genomic information
- Ensemble modeling
Use of multiple models to address uncertainty by simulating a set of possibilities and selecting those consistent with measured data.
- Machine learning
A technique used to build predictive models through patterns and inferences obtained from sample data, rather than explicit or mechanistic relationships
- Self-assembled microbiome
a microbiome built through environmental manipulation that selects for desired functions
- Synthetic microbiome
a microbiome built using pre-defined axenic or enrichment cultures to achieve a desired function
- Syntrophy
an obligately mutualistic process that is mediated by metabolite cross-feeding between two or more organisms that cannot be catalyzed by one organism alone
- Techno-economic assessment
A tool used to evaluate the technical and economic viability of an integrated process through a combination of process design, modeling, and economic evaluation
- Life cycle analysis
a tool used to evaluate the environmental impacts associated with all stages of a product or processes life, such as energy and water consumption, and air pollutant and greenhouse gas emissions
- Integrative and conjugative elements (ICEs)
ICEs are mobile genetic elements able to integrate into DNA sites via site-specific recombination that carry genes encoding the machinery necessary for conjugation
- Structure-function relationships
the influence of the microbiomes three-dimensional spatial organization on its function
Footnotes
Competing interests
The authors declare no competing interests.
Reference
- 1.Falkowski PG, Fenchel T & Delong EF The microbial engines that drive earth ’s biogeochemical cycles. Science. 320, 1034–1039 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Kuypers MMM, Marchant HK & Kartal B The microbial nitrogen-cycling network. Nat. Rev. Microbiol 16, 263–276 (2018). [DOI] [PubMed] [Google Scholar]
- 3.O’Connell KP, Goodman RM & Handelsman J Engineering the rhizosphere: Expressing a bias. Trends Biotechnol. 14, 83–88 (1996). [Google Scholar]
- 4.Löffler FE & Edwards EA Harnessing microbial activities for environmental cleanup. Curr. Opin. Biotechnol 17, 274–284 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Mccarty PL, Bae J & Kim J Domestic wastewater treatment as a net energy producer à can this be achieved ? Environ. Sci. Technol 45, 7100–7106 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Rinke C et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–7 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Anantharaman K et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun 7, 1–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alivisatos AP et al. A unified initiative to harness Earth’s microbiomes. Science. 350, 507–508 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Dubilier N, McFall-Ngai M & Zhao L Create a global microbiome effort. Nature 526, 631–634 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Price MN et al. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 557, 503–509 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Zengler K & Zaramela LS The social network of microorganisms — how auxotrophies shape complex communities. Nat. Rev. Microbiol 16, 383–390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheelwright SC & Clark KB Revolutionizing Product Development: Quantum Leaps in Speed, Efficiency, and quality. (The Free Press, 1992). [Google Scholar]
- 13.Nielsen J & Keasling JD Engineering cellular metabolism. Cell 164, 1185–1197 (2016). [DOI] [PubMed] [Google Scholar]; This review highlights experiences, success stories, and challenges associated with implementing the DBTL cycle for metabolic engineering.
- 14.Blank S & Dorf B The Startup Owner’s Manual: The Step-by-Step Guide for Building a Great Company. 1, (K&S Ranch, Inc., 2012). [Google Scholar]
- 15.Jansson JK & Hofmockel KS The soil microbiome — from metagenomics to metaphenomics. Curr. Opin. Microbiol 43, 162–168 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Briones A & Raskin L Diversity and dynamics of microbial communities in engineered environments and their implications for process stability. Curr. Opin. Biotechnol 14, 270–276 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Verstraete W et al. Microbial resource management: the road to go for environmental biotechnology. Eng. Life Sci 7, 117–126 (2007). [Google Scholar]
- 18.Moralejo-Gárate H, Mar’Atusalihat E, Kleerebezem R & Van Loosdrecht MCM Microbial community engineering for biopolymer production from glycerol. Appl. Microbiol. Biotechnol 92, 631–639 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Nielsen PH et al. A conceptual ecosystem model of microbial communities in enhanced biological phosphorus removal plants. Water Res. 44, 5070–5088 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Winkler M-KH et al. An integrative review of granular sludge for the biological removal of nutrients and recalcitrant organic matter from wastewater. Chem. Eng. J 336, 489–502 (2018). [Google Scholar]
- 21.Henze M, Gujer W, Mino T & Van Loosdrecht MCM Activated sludge models ASM1, ASM2, ASM2d and ASM3. (IWA Publishing, 2000). [Google Scholar]
- 22.Batstone DJ, Puyol D & Rodri XFJ Mathematical modelling of anaerobic digestion processes : applications and future needs. 595–613 (2015). [Google Scholar]
- 23.Muñoz-Tamayo R, Giger-Reverdin S & Sauvant D Mechanistic modelling of in vitro fermentation and methane production by rumen microbiota. Anim. Feed Sci. Technol 220, 1–21 (2016). [Google Scholar]
- 24.Picioreanu C, Kreft J & Loosdrecht M. C. M. Van. Particle-based multidimensional multispecies biofilm model. Appl. Environ. Microbiol 70, 3024–3040 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Yunjie, Carlos Domingo-Félez Benedek Gy. Plósz, and B. F. S. Intermittent aeration suppresses nitrite-oxidizing bacteria in membrane-aerated biofilms : A Model-Based Explanation. Environ. Sci. Technol 51, 6146–6155 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Nobu MK et al. Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor. ISME J. 9, 1710–1722 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rotaru AE et al. A new model for electron flow during anaerobic digestion: Direct interspecies electron transfer to Methanosaeta for the reduction of carbon dioxide to methane. Energy Environ. Sci 7, 408–415 (2014). [Google Scholar]
- 28.Albertsen M et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol 31, 533–538 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Banerjee S, Schlaeppi K & Heijden MGA Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol 16, 567–576 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Röttjers L & Faust K Can we predict keystones? Nat. Rev. Microbiol 17, 193 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Thiele I & Palsson BØ A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc 5, 93–121 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Dash S, Ng CY & Maranas CD A review of computational tools for design and reconstruction of metabolic pathways. Synth. Syst. Biotechnol 2, 243–252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zomorrodi AR & Segre D Synthetic ecology of microbes: mathematical models and applications. j. mol. biol 428, 837–86 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borenstein E, Kupiec M, Feldman MW & Ruppin E Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc. Natl. Acad. Sci 105, 14482–14487 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orth JD, Thiele I & Palsson BØ What is flux balance analysis? Nat. Biotechnol 28, 245–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhuang K et al. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 5, 305–316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study integrates multiple genome-scale models for dynamic flux balance analysis of a microbial community.
- 37.Harcombe WR et al. Ecosystem interactions and spatial dynamics. Cell Rep. 7, 1104–1115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hellweger FL, Clegg RJ, Clark JR, Plugge CM & Kreft J Advancing microbial sciences by individual-based modelling. Nat. Rev. Microbiol 14, 461–471 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Zhou K, Qiao K, Edgar S & Stephanopoulos G Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat. Biotechnol 33, 377–383 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that designing distributed metabolic pathways over multiple microbial taxa can optimize a desired function.
- 40.Lilja EE & Johnson DR Segregating metabolic processes into different microbial cells accelerates the consumption of inhibitory substrates. ISME J. 10, 1568–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mee MT, Collins JJ, Church GM & Wang HH Syntrophic exchange in synthetic microbial communities. Proc. Natl. Acad. Sci. U. S. A 111, E2149–E2156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shade A et al. Fundamentals of microbial community resistance and resilience. Front. Microbiol 3, 417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balagaddé FK et al. A synthetic Escherichia coli predator-prey ecosystem. Mol. Syst. Biol. 4, 1–8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Papenfort K & Bassler BL Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol 14, 576–588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feist AM & Palsson BO What do cells actually want? Genome Biol. 17, 110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oyetunde T, Bao FS, Chen JW, Martin HG & Tang YJ Leveraging knowledge engineering and machine learning for microbial bio-manufacturing. Biotechnol. Adv 36, 1308–1315 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Tran LM, Rizk ML & Liao JC Ensemble modeling of metabolic networks. Biophys. J 95, 5606–5617 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Costello Z & Martin HG A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data. npj syst. Biol. Appl 4, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Medlock GL et al. Inferring metabolic mechanisms of interaction within a defined gut microbiota. Cell Syst. 7, 245–257.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heckmann D et al. Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models. Nat. Commun 9, 5252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Konopka A, Lindemann S & Fredrickson J Dynamics in microbial communities: unraveling mechanisms to identify principles. ISME J. 9, 1488 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith AL et al. Navigating wastewater energy recovery strategies: A life cycle comparison of anaerobic membrane bioreactor and conventional treatment systems with anaerobic digestion. Environ. Sci. Technol 48, 5972–5981 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Balakrishnan M et al. Novel pathways for fuels and lubricants from biomass optimized using life-cycle greenhouse gas assessment. Proc. Natl. Acad. Sci 112, 7645–7649 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turnbaugh PJ et al. The human microbiome project. Nature 449, 804–810 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McIlroy SJ et al. MiDAS: The field guide to the microbes of activated sludge. Database 2015, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arne Alphenaar P, Visser A & Lettinga G The effect of liquid upward velocity and hydraulic retention time on granulation in UASB reactors treating wastewater with a high sulphate content. Bioresour. Technol 43, 249–258 (1993). [Google Scholar]
- 57.Liu Y & Tay J-H The essential role of hydrodynamic shear force in the formation of biofilm and granular sludge. Water Res. 36, 1653–1665 (2002). [DOI] [PubMed] [Google Scholar]
- 58.L. Z et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 359, 1151–1156 (2018). [DOI] [PubMed] [Google Scholar]; This study shows how biostimulation strategies can be used to create microbiomes with desired functions, such as enhanced short chain fatty acid production.
- 59.Van Dongen U, Jetten MSM & Van Loosdrecht MCM The SHARON®-Anammox® process for treatment of ammonium rich wastewater. Water Sci. Technol 44, 153–160 (2001). [PubMed] [Google Scholar]
- 60.Mueller UG & Sachs JL Engineering microbiomes to improve plant and animal health. Trends Microbiol. 23, 606–617 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Winkler MKH, Kleerebezem R, Kuenen JG, Yang J & LoosdrechtVan MCM. Segregation of biomass in cyclic anaerobic / aerobic granular sludge allows the enrichment of anaerobic ammonium oxidizing bacteria at low temperatures. Environ. Sci. Technol 45, 7330–7337 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Laureni M et al. Biomass segregation between bio film and flocs improves the control of nitrite-oxidizing bacteria in mainstream partial nitritation and anammox processes. Water Res. 154, 104–116 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Scarborough MJ, Lawson CE, Hamilton JJ, Donohue TJ & Noguera DR Metatranscriptomic and thermodynamic insights into medium-chain fatty acid production using an anaerobic microbiome. mSystems 3, 1–21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides a detailed reconstruction of the metabolism and interactions of an anaerobic microbiome using multi-omic and thermodynamic analyses.
- 64.Head IM, Jones DM & Röling WFM Marine microorganisms make a meal of oil. Nat. Rev. Microbiol 4, 173–82 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Swenson W, Wilson DS & Elias R Artificial ecosystem selection. Proc. Natl. Acad. Sci 97, 9110 – 9114 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Panke-buisse K, Poole AC, Goodrich JK, Ley RE & Kao-kniffin J Selection on soil microbiomes reveals reproducible impacts on plant function. 9, 980–989 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams HTP & Lenton TM Artificial selection of simulated microbial ecosystems. 104, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.King KC et al. Rapid evolution of microbe-mediated protection against pathogens in a worm host. ISME J. 10, 1915–1924 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hillesland KL & Stahl DA Rapid evolution of stability and productivity at the origin of a microbial mutualism. 107, 1–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barrick JE et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461, 1243–1247 (2009). [DOI] [PubMed] [Google Scholar]
- 71.Utrilla J et al. Global rebalancing of cellular resources by pleiotropic point mutations illustrates a multi-scale mechanism of adaptive evolution. Cell Syst. 2, 260–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.LaCroix RA, Palsson BO & Feist AM A model for designing adaptive laboratory evolution experiments. Appl. Environ. Microbiol 83, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Iwabuchi N et al. Extracellular polysaccharides of rhodococcus rhodochrous s-2 stimulate the degradation of aromatic components in crude oil by indigenous marine bacteria. Appl. Environ. Microbiol 68, 2337–2343 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Palková Z Multicellular microorganisms: laboratory versus nature. EMBO Rep. 5, 470–476 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eydallin G, Ryall B, Maharjan R & Ferenci T The nature of laboratory domestication changes in freshly isolated Escherichia coli strains. Environ. Microbiol 16, 813–828 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Steensels J, Gallone B, Voordeckers K & Verstrepen KJ Domestication of industrial microbes. Curr. Biol 29, R381–R393 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Seshadri R et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat. Biotechnol 36, 359 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Forster SC et al. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol 37, 186–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang C-Y et al. High-throughput single-cell cultivation on microfluidic streak plates. Appl. Environ. Microbiol 82, 2210–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lagier JC et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol 1, (2016). [DOI] [PubMed] [Google Scholar]; This study, together with Browne et al.81, describe approaches for high-throughput culturing of microorganisms that can be integrated with metagenomics and enable genome-sequencing, archiving, and phenotypic analysis.
- 81.Browne HP et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huber R et al. Robo-Lector - A novel platform for automated high-throughput cultivations in microtiter plates with high information content. Microb. Cell Fact 8, 1–15 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Clark C et al. Characterization of TAP Ambr 250 disposable bioreactors, as a reliable scale-down model for biologics process development. Biotechnol. Prog 33, 478–489 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Gach PC et al. A droplet microfluidic platform for automating genetic engineering. ACS Synth. Biol 5, 426–433 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Prakadan SM, Shalek AK & Weitz DA Scaling by shrinking: Empowering single-cell ‘omics’ with microfluidic devices. Nat. Rev. Genet 18, 345–361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cole RH et al. Printed droplet microfluidics for on demand dispensing of picoliter droplets and cells. Proc. Natl. Acad. Sci 114, 8728–8733 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ando H, Lemire S, Pires DP & Lu TK Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 1, 187–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lan F, Demaree B, Ahmed N & Abate AR Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat. Biotechnol 35, 640–646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heinemann J et al. On-chip integration of droplet microfluidics and nanostructure-initiator mass spectrometry for enzyme screening. Lab Chip 17, 323–331 (2017). [DOI] [PubMed] [Google Scholar]
- 90.Shapiro RS, Chavez A & Collins JJ CRISPR-based genomic tools for the manipulation of genetically intractable microorganisms. Nat. Rev. Microbiol 16, 333–339 (2018). [DOI] [PubMed] [Google Scholar]
- 91.Cobb RE, Wang Y & Zhao H High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth. Biol 4, 723–728 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nayak DD & Metcalf WW Cas9-mediated genome editing in the methanogenic archaeon Methanosarcina acetivorans. Proc. Natl. Acad. Sci. 114, 2976–2981 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shih SCC et al. A versatile microfluidic device for automating synthetic biology. ACS Synth. Biol. 4, 1151–1164 (2015). [DOI] [PubMed] [Google Scholar]
- 94.Kotula JW et al. Programmable bacteria detect and record an environmental signal in the mammalian gut Jonathan. Proc. Natl. Acad. Sci 111, 4838–4843 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Riglar DT et al. Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nat. Biotechnol 35, 653–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nielsen PH, Saunders AM, Hansen AA, Larsen P & Nielsen JL Microbial communities involved in enhanced biological phosphorus removal from wastewater — a model system in environmental biotechnology. Curr. Opin. Biotechnol 23, 452–459 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Nadell CD, Drescher K & Foster KR Spatial structure, cooperation and competition in biofilms. Nat. Rev. Microbiol 14, 589 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Vlaeminck SE, Cloetens LFF, Carballa M, Boon N & Verstraete W Granular biomass capable of partial nitritation and anammox. Water Sci. Technol 58, 1113–1120 (2008). [DOI] [PubMed] [Google Scholar]
- 99.Mark Welch JL, Rossetti BJ, Rieken CW, Dewhirst FE & Borisy GG Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci 113, E791 LP–E800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Werner JJ et al. Bacterial community structures are unique and resilient in full-scale bioenergy systems. Proc. Natl. Acad. Sci. U. S. A 108, 4158–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gruber-Dorninger C et al. Functionally relevant diversity of closely related Nitrospira in activated sludge. ISME J. 9, 643–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim HJ, Boedicker JQ, Choi JW & Ismagilov RF Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc. Natl. Acad. Sci 105, 18188 – 18193 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Connell JL, Ritschdorff ET, Whiteley M & Shear JB 3D printing of microscopic bacterial communities. Proc. Natl. Acad. Sci 110, 18380 – 18385 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schaffner M, Rühs PA, Coulter F, Kilcher S & Studart AR 3D printing of bacteria into functional complex materials. Sci. Adv 3, eaao6804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Datta MS, Sliwerska E, Gore J, Polz M & Cordero OX Microbial interactions lead to rapid micro-scale successions on model marine particles. Nat. Commun 7, 11965 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows how synthetic polysaccharide particles can be used as a model system to study ecological processes and microbe-microbe interactions.
- 106.Enke TN et al. Modular assembly of polysaccharide-degrading marine microbial communities. Curr. Biol 29, 1528–1535.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 107.Rusten B, Eikebrokk B, Ulgenes Y & Lygren E Design and operations of the Kaldnes moving bed biofilm reactors. Aquac. Eng 34, 322–331 (2006). [Google Scholar]
- 108.Venturelli OS, Egbert RG & Arkin AP Towards engineering biological systems in a broader context. J. Mol. Biol 428, 928–944 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Lee JW, Chan CTY, Slomovic S & Collins JJ Next-generation biocontainment systems for engineered organisms. Nat. Chem. Biol 14, 530–537 (2018). [DOI] [PubMed] [Google Scholar]
- 110.Sheth RU, Cabral V, Chen SP & Wang HH Manipulating bacterial communities by in situ microbiome engineering. Trends Genet. 32, 189–200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ronda C, Chen SP, Cabral V, Yaung SJ & Wang HH Metagenomic engineering of the mammalian gut microbiome in situ. Nat. Methods 16, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study, together with Brophy et al.112, provide new techniques to transfer engineered mobile genetic elements into microorganisms living in their native environment.
- 112.Brophy JAN et al. Engineered integrative and conjugative elements for efficient and inducible DNA transfer to undomesticated bacteria. Nat. Microbiol 3, 1043–1053 (2018). [DOI] [PubMed] [Google Scholar]
- 113.Mulat DG et al. Quantifying contribution of synthrophic acetate oxidation to methane production in thermophilic anaerobic reactors by membrane inlet mass spectrometry. Environ. Sci. Technol 8, 2505–2511 (2014). [DOI] [PubMed] [Google Scholar]
- 114.Kehe J et al. Massively parallel screening of synthetic microbial communities. Proc. Natl. Acad. Sci 116, 12804–12809 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study develops a high-throughput phenotypic screen using droplet-based microfluidics that can analyze ~100,000 multi-species synthetic communities per day against any optically assayable function.
- 115.Truong DT, Tett A, Pasolli E, Huttenhower C & Segata N Microbial strain-level population structure and genetic diversity diversity from metagenomes. Nat. Methods 12, 626–638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mosbaek F et al. Identification of syntrophic acetate-oxidizing bacteria in anaerobic digesters by combined protein-based stable isotope probing and metagenomics. ISME J. 10, 2405–2418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lawson CE et al. Metabolic network analysis reveals microbial community interactions in anammox granules. Nat. Commun 8, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hawley AK, Brewer HM, Norbeck AD, Pasa-Toli L & Hallam SJ Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc. Natl. Acad. Sci 111, 11395–11400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bowen JL, Babbin AR, Kearns PJ & Ward BB Connecting the dots : linking nitrogen cycle gene expression to nitrogen fluxes in marine sediment mesocosms. Front. Microbiol 5, 1–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.He Z et al. GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J. 1, 67–77 (2007). [DOI] [PubMed] [Google Scholar]
- 121.Hellerstein MK In vivo measurement of fluxes through metabolic pathways: The missing link in functional genomics and pharmaceutical research. Annu. Rev. Nutr 23, 379–402 (2003). [DOI] [PubMed] [Google Scholar]
- 122.Sauer U Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol 2, 62 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Antoniewicz MR, Kelleher JK & Stephanopoulos G Elementary Metabolite Units (EMU): a novel framework for modeling isotopic distributions. Metab. Eng 9, 68–86 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gebreselassie NA & Antoniewicz MR (13)C-metabolic flux analysis of co-cultures: A novel approach. Metab. Eng 31, 132–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ghosh A et al. A peptide-based method for 13C metabolic flux analysis in microbial communities. PLoS Comput. Biol 10, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study develops a novel method for calculating metabolic fluxes in microbial communities using 13C-labelled peptides.
- 126.Nielsen J It Is All about Metabolic Fluxes. J. Bacteriol 185, 7031–7035 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Beyß M, Azzouzi S, Weitzel M, Wiechert W & Nöh K The design of FluxML: A universal modeling language for 13C metabolic flux analysis. Front. Microbiol 10, 1022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.McGlynn SE, Chadwick GL, Kempes CP & Orphan VJ Single cell activity reveals direct electron transfer in methanotrophic consortia. Nature 526, 531–535 (2015). [DOI] [PubMed] [Google Scholar]
- 129.Picioreanu C, Pérez J & van Loosdrecht MCM Impact of cell cluster size on apparent half-saturation coefficients for oxygen in nitrifying sludge and biofilms. Water Res. 106, 371–382 (2016). [DOI] [PubMed] [Google Scholar]
- 130.Nielsen JL & Nielsen PH Advances in microscopy: Microautoradiography of single cells. Methods Enzymol. 397, 237–256 (2005). [DOI] [PubMed] [Google Scholar]
- 131.Huang WE et al. Raman-FISH: Combining stable-isotope Raman spectroscopy and fluorescence in situ hybridization for the single cell analysis of identity and function. Environ. Microbiol 9, 1878–1889 (2007). [DOI] [PubMed] [Google Scholar]
- 132.Dunham SJB, Ellis JF, Li B & Sweedler JV Mass spectrometry imaging of complex microbial communities. Acc. Chem. Res 50, 96–104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hatzenpichler R et al. Visualizing in situ translational activity for identifying and sorting slow-growing archaeal–bacterial consortia. Proc. Natl. Acad. Sci 113, E4069–E4078 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study develops a high-throughput approach for visualizing protein synthesis in individual cells within microbiomes by combining bioorthogonal non-canonical amino acid tagging with fluorescence-activated cell sorting.
- 134.Okabe S, Satoh H & Watanabe Y Analysis of microbial structure and function of nitrifying biofilms. Methods Ecol. Evol 337, 213–224 (2001). [DOI] [PubMed] [Google Scholar]
- 135.DiMucci D, Kon M & Segrè D Machine learning reveals missing edges and putative interaction mechanisms in microbial ecosystem networks. mSystems 3, e00181–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Qu K, Guo F, Liu X, Lin Y & Zou Q Application of machine learning in microbiology. Front. Microbiol 10, 827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang P-H et al. An interspecies malate–pyruvate shuttle reconciles redox imbalance in an anaerobic microbial community. ISME J. 13, 1042–1055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study combines metabolic modeling with 13C metabolomic experiments to resolve poorly understood metabolite exchange reactions driving ecosystem function in anaerobic microbiomes.
- 138.Faust K & Raes J Microbial interactions: from networks to models. Nat. Rev. Microbiol 10, 538 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Imam S, Noguera DR & Donohue TJ An integrated approach to reconstructing genome-scale transcriptional regulatory networks. PLoS Comput. Biol 11, 1–35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Venturelli OS et al. Deciphering microbial interactions in synthetic human gut microbiome communities. Mol. Syst. Biol 14, e8157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.MacArthur R Fluctuations of animal populations and a measure of community stability. Ecology 36, 533–536 (1955). [Google Scholar]
- 142.Martin HG & Goldenfeld N On the origin and robustness of power-law species – area relationships in ecology. Proc. Natl. Acad. Sci 103, 10310–10315 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tilman D Biodiversity : Population versus ecosystem stability. Ecology 77, 350–363 (1996). [Google Scholar]
- 144.Hardin Garrett. The competitive exclusion principle. Science. 131, 1292–1297 (1960). [DOI] [PubMed] [Google Scholar]
- 145.Lozano GL et al. Introducing THOR, a model microbiome for genetic dissection of community behavior. MBio 10, e02846–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zengler K et al. EcoFABs: advancing microbiome science through standardized fabricated ecosystems. Nat. Methods 16, 567–571 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the construction and use of standardized fabricated ecosystems for the development of theory and predictive models for microbiomes.
- 147.Zhalnina K, Zengler K, Newman D & Northen TR Need for laboratory ecosystems to unravel the structures. MBio 9, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shah P et al. A microfluidics-based in vitro model of the gastrointestinal human–microbe interface. Nat. Commun 7, 11535 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wegener G, Krukenberg V, Riedel D, Tegetmeyer HE & Boetius A Intercellular wiring enables electron transfer between methanotrophic archaea and bacteria. Nature 526, 587–590 (2015). [DOI] [PubMed] [Google Scholar]
- 150.Scheller S, Yu H, Chadwick GL, McGlynn SE & Orphan VJ Artificial electron acceptors decouple archaeal methane oxidation from sulfate reduction. Science. 351, 703–707 (2016). [DOI] [PubMed] [Google Scholar]
- 151.Schink B Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev 61, 262–280 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jackson BE & McInerney MJ Anaerobic microbial metabolism can proceed close to thermodynamic limits. Nature 415, 454–456 (2002). [DOI] [PubMed] [Google Scholar]
- 153.Phelan VV, Liu WT, Pogliano K & Dorrestein PC Microbial metabolic exchange--the chemotype-to-phenotype link. Nat. Chem. Biol 8, 26–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Louca S et al. Function and functional redundancy in microbial systems. Nat. Ecol. Evol 2, 936–943 (2018). [DOI] [PubMed] [Google Scholar]
- 155.Ladau J & Eloe-Fadrosh EA Spatial, temporal, and phylogenetic scales of microbial ecology. Trends Microbiol. 27, 662–669 (2019). [DOI] [PubMed] [Google Scholar]
- 156.Thompson JA, Oliveira RA, Djukovic A, Ubeda C & Xavier KB Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep. 10, 1861–1871 (2015). [DOI] [PubMed] [Google Scholar]
- 157.Arkin AP et al. KBase: The united states department of energy systems biology knowledgebase. Nat. Biotechnol 36, 566–569 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Morrell WC et al. The experiment data depot: A web-based software tool for biological experimental data storage, sharing, and visualization. ACS Synth. Biol 6, 2248–2259 (2017). [DOI] [PubMed] [Google Scholar]
- 159.Solden LM et al. Interspecies cross-feeding orchestrates carbon degradation in the rumen ecosystem. Nat. Microbiol 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Podolsky IA et al. Harnessing nature’s anaerobes for biotechnology and bioprocessing. Annu. Rev. Chem. Biomol. Eng 10, 105–128 (2019). [DOI] [PubMed] [Google Scholar]
- 161.Swift CL, Brown JL, Seppälä S & O’Malley MA Co-cultivation of the anaerobic fungus Anaeromyces robustus with Methanobacterium bryantii enhances transcription of carbohydrate active enzymes. J. Ind. Microbiol. Biotechnol (2019). doi: 10.1007/s10295-019-02188-0 [DOI] [PubMed] [Google Scholar]
- 162.Lee KS et al. An automated Raman-based platform for the sorting of live cells by functional properties. Nat. Microbiol 4, 1035–1048 (2019). [DOI] [PubMed] [Google Scholar]
- 163.Swenson TL, Karaoz U, Swenson JM, Bowen BP & Northen TR Linking soil biology and chemistry in biological soil crust using isolate exometabolomics. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Noor E, Cherkaoui S & Sauer U Biological insights through omics data integration. Curr. Opin. Syst. Biol 15, 39–47 (2019). [Google Scholar]
- 165.Zampieri Guido, Vijayakumar Supreeta, Yaneske Elisabeth, A C. Machine and deep learning meet genome-scale metabolic modelling. PLoS Comput. Biol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ziels RM, Sousa DZ, Stensel HD & Beck DAC DNA-SIP based genome-centric metagenomics identifies key long-chain fatty acid-degrading populations in anaerobic digesters with different feeding frequencies. ISME J. 12, 112–123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fortunato CS & Huber JA Coupled RNA-SIP and metatranscriptomics of active chemolithoautotrophic communities at a deep-sea hydrothermal vent. ISME J. 10, 1925–1938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Orphan VJ, Orphan VJ, House CH & Hinrichs K Methane-consuming archaea revealed by directly coupled isotopic and phylogenetic analysis. 484, 484–488 (2013). [DOI] [PubMed] [Google Scholar]
- 169.Kaltenpoth M, Strupat K & Svatoš A Linking metabolite production to taxonomic identity in environmental samples by (MA)LDI-FISH. ISME J. 10, 527–531 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nuñez J, Renslow R, Cliff JB & Anderton CR NanoSIMS for biological applications: Current practices and analyses. Biointerphases 13, 03B301 (2018). [DOI] [PubMed] [Google Scholar]
- 171.Northen TR et al. Clathrate nanostructures for mass spectrometry. Nature 449, 1033 (2007). [DOI] [PubMed] [Google Scholar]
- 172.Louie KB et al. “Replica-extraction-transfer” nanostructure-initiator mass spectrometry imaging of acoustically printed bacteria. Anal. Chem 85, 10856–10862 (2013). [DOI] [PubMed] [Google Scholar]
- 173.Johnson CH et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 21, 891–897 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gilmore IS, Heiles S & Pieterse CL Metabolic imaging at the single-cell scale: recent advances in mass spectrometry imaging. Annu. Rev. Anal. Chem 12, 201–224 (2019). [DOI] [PubMed] [Google Scholar]
- 175.Hatzenpichler R et al. In situ visualization of newly synthesized proteins in environmental microbes using amino acid tagging and click chemistry. Environ. Microbiol 16, 2568–2590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ma Y & Yates JR Proteomics and pulse azidohomoalanine labeling of newly synthesized proteins: what are the potential applications? Expert Rev. Proteomics 15, 545–554 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kaminski TS, Scheler O & Garstecki P Droplet microfluidics for microbiology: Techniques, applications and challenges. Lab Chip 16, 2168–2187 (2016). [DOI] [PubMed] [Google Scholar]
- 178.Bein A et al. Microfluidic organ-on-a-chip models of human intestine. Cmgh 5, 659–668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Aleklett K et al. Build your own soil: Exploring microfluidics to create microbial habitat structures. ISME J. 12, 312–319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hsu RH, Clark RL, Tan JW, Romero PA & Venturelli OS Rapid microbial interaction network inference in microfluidic droplets. bioRxiv 521823 (2019). doi: 10.1101/521823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tilman D et al. Diversity and productivity in a long-term grassland experiment. Science. 294, 843–846 (2001). [DOI] [PubMed] [Google Scholar]