

Abstract
Retinal ganglion cells and other central nervous system neurons fail to regenerate after injury. Understanding the obstacles to survival and regeneration, and overcoming them, is key to preserving and restoring function. While comparisons in the cellular changes seen in these non-regenerative cells with those that do have intrinsic regenerative ability has yielded many candidate genes for regenerative therapies, complete visual recovery has not yet been achieved. Insights gained from neurodegenerative diseases, like glaucoma, underscore the importance of axonal transport of organelles, mRNA, and effector proteins in injury and disease. Targeting molecular motor networks, and their cargoes, may be necessary for realizing complete axonal regeneration and vision restoration.
Keywords: CNS regeneration, axon transport, optic nerve, neuroprotection, glaucoma
INTRODUCTION
Like most other mature neurons in the central nervous system (CNS), retinal ganglion cells (RGCs) cannot regenerate their axons in disease and after injury. In the visual system, RGCs carry visual information along their axons down the optic nerve to the superior colliculus (SC) and the lateral geniculate nucleus (LGN) of the thalamus, among other important brain targets. Therapies to protect or restore vision after axon insult must address RGC survival and axon regeneration, and re-integration of the RGC axons into the appropriate visual circuitry. To combat regenerative failure, many strategies have been devised both alone and in combination to allow partial regeneration of injured RGCs to their visual targets. Over the last 30 years, to understand why adult, mammalian RGCs and other CNS neurons do not regenerate after injury, and convert them into neurons that do regenerate, many groups have asked what are the molecular differences between adult, mammalian CNS neurons and 1) “immature” mammalian CNS neurons, which have a higher intrinsic growth capacity; 2) adult peripheral nervous system (PNS) neurons, which do regenerate their axons after injury; and 3) CNS neurons from other species that do show regenerative capacity? Here, we review advances in these parallel strategies, and then discuss a hypothesis that links prior work into a unified model for the role of axon transport in regenerative failure, as well as a new way to target approaches to promote regeneration.
IMMATURE CNS NEURONS HAVE A HIGH REGENERATIVE POTENTIAL
Developing mammalian CNS neurons have a high growth potential that is lost by adulthood. In fact, isolated RGCs from embryonic mice have a far greater in vitro growth potential than RGCs isolated from early postnatal mice (Goldberg et al., 2002a). Manipulating growth regulation pathways in mature neurons may promote regeneration, by increasing intrinsic growth factor-driven signaling pathways such as mTOR, cAMP, suppressors of cytokine signaling (e.g., SOCS3), and mitogen-activated protein kinases (Cai et al., 1999; Zhou et al., 2005; Leaver et al., 2006; Smith et al., 2009; Kurimoto et al., 2010; Park et al., 2010), manipulating responsiveness to extrinsic inhibitory factors such as by blocking Nogo receptor expression or activation (Chen et al., 2000; GrandPré et al., 2000; Fischer et al., 2004), and decreasing transcriptional inhibitors of axon growth such as Krüppel-like family transcription factors (KLFs) (Moore et al., 2011, 2009a). Identifying genes or pathways whose pattern of expression in immature, highly regenerative CNS neurons are drastically altered after maturation may lead to candidate regulators of intrinsic growth potential. In the case of KLFs, where developmental upregulation of KLF9 and KLF4 and downregulation of KLF6 and KLF7 are coincident with the reduction of intrinsic regenerative capacity of RGCs, reversing these expression patterns in adult RGCs or in other CNS neuron pathways after injury allows sprouting or long-distance regeneration (Moore et al., 2009a; Blackmore et al., 2012; Apara et al., 2017; Wang et al., 2017). Exploring the molecular mechanisms of the KLFs further, identification of co-factors such as JNK3 and STAT3, and downstream targets such as serotonin receptors and dual-specificity phosphatase 14 (DUSP14) (Qin et al., 2013; Apara et al., 2017; Trakhtenberg et al., 2017; Galvao et al., 2018) have led to broader understanding of the biology of intrinsic capacity for axon growth. What other molecular targets do KLF family transcription factors and other intrinsic regulators of axon growth affect? Advances in sequencing and mass spectrometry technologies can uncover a more complete understanding of the cellular and molecular changes underlying the developmental loss of regenerative capacity.
Beyond transcriptional regulation, immature and mature CNS neurons have differential axonal transport. In embryonic cortical neurons in vitro, axonal transport included integrins important for axonal growth and elongation; in mature cortical neurons, this transport is lost. While most molecular transport is dependent on motor trafficking, axonal integrin transport has some specificity to a specific kinesin KIF4A (Heintz et al., 2014). It is particularly noteworthy that axonal transport of integrins in mature neurons switches from an anterograde to mainly retrograde transport (Franssen et al., 2015).
These findings present a clear opportunity for future research into the axonal transport changes after optic nerve injury. Does integrin transport change more or less than other proteins after injury? Are all kinesin-transported proteins affected the same by injury, or are there kinesin isoform responses specific to different injuries or insults? While not a direct link, these examples underline how developmental changes in gene expression and axonal transport parallel developmental changes in intrinsic axon growth ability – and a ripe avenue for future regeneration research.
ELECTRICAL STIMULATION AFTER RGC INJURY
An additional element to consider beyond molecular signaling pathways is the functionality of RGCs: electrical activity is critical for transmission of action potentials and visual information, but it turns out it is also beneficial for RGC and other CNS neurons’ responsiveness to survival and growth signals. It has previously been shown that electrical activity regulates mitochondrial localization and motility from the cell body in myelinated axons (Ohno et al., 2011). In addition, electrical modulation through eye-opening, brain-derived neurotrophic factor (BDNF), or tetro-dotoxin administration in vivo during development greatly impact mitochondrial function and trafficking in RGC axons (our unpublished data), demonstrating a direct connection between activity and transport. RGCs extend longer neurites with concurrent electrical stimulation and growth factor administration in vitro (Goldberg et al., 2002b) and in vivo (Lim et al., 2016). Increasing conduction in regenerating axons with a voltage-gated potassium channel blocker enhances visual recovery measured at the level of behavior (Bei et al., 2016). This electrical stimulation pathway acts at least in part through activation of adenylate cyclases, and specifically the calcium-sensitive, soluble adenylate cyclase (sAC) in vitro and in vivo (Corredor et al., 2012; Martinez et al., 2014). Complicating the story, however, is that excessive calcium influx into the axon after injury is a primary step in acute axon degeneration (Knoferle et al., 2010). Pre-loading RGCs with calcium channel blockers before optic nerve crush results in improved survival and regeneration of RGCs (Ribas et al., 2017). How is calcium or downstream cAMP signaling compartmentalized to regulate complex cellular responses? Is there anterograde or retrograde effector transport responsible for long-distance communication? Further work dissecting timing, compartmentalization, and localization of these pathways will be needed to reconcile these data.
PNS NEURONAL REGENERATION INFORMS CNS REGENERATION
Early experiments showing that PNS neuronal grafts can induce CNS axon elongation started a field of comparative work of PNS and CNS neurons, and the glial environments they must regenerate through (Richardson et al., 1980; David and Aguayo, 1981; Benfey and Aguayo, 1982). Exploring differences in the molecular characteristics of the regenerative response after injury between PNS and CNS has uncovered several networks that all could contribute toward inducing RGC regeneration (Smith et al., 2011; Chandran et al., 2016).
For example, cytokines, such as gp130 family members like interleukin-6, are differentially activated after PNS injury as compared to CNS injury (Cafferty et al., 2001). Further studies expanded on the subsequent activation of the JAK/STAT pathway, showing a correlation with enhanced PNS regeneration (Miao et al., 2006). Removing SOCS3, an inhibitor of the JAK/STAT pathway present in high levels following injury in CNS neurons, promotes RGC regeneration (Smith et al., 2009). Surprisingly, STAT3-dependent gene expression is directly inhibited by KLF family member KLF4 after cytokine activation, potentially explaining one mechanism by which KLF4 deletion promotes RGC regeneration (Qin et al., 2013).
Through differential proteomics and bioinformatic network analysis, c-Myc was identified as a hub protein that was downregulated in RGCs but not in dorsal root ganglion PNS neurons after injury (Belin et al., 2015). Furthermore, overexpression of this protein increased survival and regeneration. As another example, the transcription factor SOX11 was first identified as a modulator of regeneration in the PNS (Jankowski et al., 2009). This was extended to the CNS, showing that SOX11 underlies DLK/LZK-mediated cell death, and that overexpression of Sox11 can induce regeneration of some subtypes of RGCs, although also leading to cell death of other RGC subtypes (Norsworthy et al., 2017; Welsbie et al., 2017). This unveiling of factors that differentially promote survival or regeneration depending on the subtype of neuron was also seen with osteopontin and IGF1, which improved survival and regeneration of the alpha-RGCs that preferentially express the relevant receptors (Duan et al., 2015). As more details emerge about not only CNS–PNS differences but also about the heterogeneity of CNS neuron subtypes and their responses to injury, more work will be needed to fine-tune individualized therapies for regeneration.
INTRINSICALLY REGENERATIVE SPECIES
Regenerative failure of the CNS is not a universally conserved phenomenon: in fact, diverse phyla and classes like nematodes (Yanik et al., 2004), fruit-flies (Soares et al., 2014), zebrafish (Cameron, 2000; Sherpa et al., 2008), and reptiles (Lang et al., 1998), demonstrate at least partial neural regeneration after injury. In C. elegans, DLK-1 was first shown to promote and regulate adult axon regeneration, regulating the cells to respond to injury, partially through mRNA stabilization, discussed further below (Hammarlund et al., 2009; Yan et al., 2009). This finding was also seen in peripheral nerve regeneration in mice, with DLK required for retrograde transport of phosphorylated STAT3 to the cell body from the damaged axon (Shin et al., 2012). In one study of Drosophila wing regeneration after injury, transgenic screening highlighted JNK pathway inhibition as pro-growth, a finding conserved in mammalian RGCs after injury (Welsbie et al., 2013; Soares et al., 2014; Apara et al., 2017). In fact, it seems the DLK/JNK pathway underlies cellular responsiveness to injury in intrinsically regenerative species, regenerative PNS neurons, and non-regenerative neurons even in humans, either pro-growth or pro-apoptotic depending on the neuronal context (Le Pichon et al., 2017).
In zebrafish, KLF6a and KLF7a together are necessary for RGC regeneration after optic nerve crush, and similarly promote axon growth and regeneration in rodent RGCs and corticospinal neurons (Veldman et al., 2007; Moore et al., 2009b; Blackmore et al., 2012). However, a downstream target in zebrafish was identified as tuba1a, a key protein for regeneration in fish that has not been found to be relevant for regeneration in mammals (Veldman et al., 2010). Thus exploring conserved and divergent molecular pathways and functions has helped to understand differences in regenerative capacity between mammals and other species and has led to candidate approaches for promoting regeneration.
COMBINING THERAPIES TO ENHANCE REGENERATION
As many of these regenerative therapies target different pathways, the combination of cell intrinsic and cell extrinsic approaches has led to novel insights and improvements in survival and regeneration of ganglion cells. For example, while the deletion of PTEN or SOCS3 independently lead to extensive RGC regeneration, the co-deletion of PTEN and SOCS3 had a synergistic effect for robust, sustained axon regeneration (Sun et al., 2011). Recently, the combination of KLF9 knockdown with zinc chelation by TPEN was shown to lead to more enhanced regeneration and cell survival than either therapy alone (Trakhtenberg et al., 2018). Visual or electrical stimulation also elicits more profound effects in combination with neurotrophic factors or with manipulation of pro-growth signaling pathways in neurons such as RGCs (Goldberg et al., 2002b; Lim et al., 2016). However, despite the best combinations of transcription factors and growth pathways, relatively few RGC axon reach their target regions. It is likely that a cocktail approach manipulating several factors together may enhance regeneration and indeed be necessary for full visual recovery.
Indeed, we must now ask how regenerated RGC synapses compare to those established during development. Do they have adequate transport of pre-synaptic machinery to maintain synaptic connections? Is exogenous expression of guidance molecules necessary for axon targeting? The answers to these questions and more form the next frontier of visual regeneration research. Despite these advances, limited visual recovery has been seen, underscoring the need for a deeper understanding of cellular changes in injury and disease. Many of the regenerative factors discussed above were hypothesized as candidate therapies due to differential expression in regenerative and non-regenerative neurons. Similarly, differential expression of factors in degenerative and non-degenerative neurons can highlight candidates for survival and maintenance of axons, which when combined with regenerative therapy, will lead to enhanced therapeutic response. Indeed, the link between degenerative molecular pathways and those failing to promote regenerative response may be one fertile area to focus on. With that in mind, insights from degenerative changes seen in conditions like glaucoma may suggest new avenues for vision restoration research.
AXON TRANSPORT IN GLAUCOMA AND OTHER NEURODEGENERATIVE DISEASES
Glaucoma is the leading cause of irreversible blindness worldwide and is predicted to affect 80 million people by 2020 (Quigley and Broman, 2006). The biggest risk factor is age; increased intraocular pressure (IOP) is currently the only modifiable risk factor. Vision loss occurs due to dysfunction and death of RGCs and their axons. Furthermore, widespread damage can be seen throughout the visual system, with degenerative changes in the LGN and the visual cortex (Yücel et al., 2000). The molecular pathophysiology of glaucoma is still poorly understood, but increasing evidence implicates interference with axonal transport mechanisms.
Decreased axoplasmic flow between the RGC cell bodies and their axon terminals in the SC or LGN in the face of increased IOP remains one of longest standing hypotheses for pathophysiologic mechanism in this disease. Since the 1970s, studies into axoplasmic transport in glaucomatous degeneration have shown an association between increased IOP and decreased anterograde and retrograde protein transport (Anderson and Hendrickson, 1974, 1977; Minckler et al., 1977; Quigley and Anderson, 1977; Quigley et al., 1979; Crish et al., 2010). Indeed there is a strong link between many neurodegenerative diseases and dysfunctional axon transport (Appel, 1981). Causative mutations in genes that directly or indirectly lead to axon transport deficits may underlie at least a portion of the neuronal death seen in Huntington’s disease (Trushina et al., 2004), amyotrophic lateral sclerosis (ALS) (Pasinelli and Brown, 2006; Nicolas et al., 2018), Parkinson’s disease (Saha et al., 2004), and Alzheimer’s disease (Zhang et al., 2004; Wu et al., 2009). Specifically, decreases in axonal transport precede and possibly contribute to axonal and microtubule, and then somatic, degeneration (Stokin et al., 2005; Morfini et al., 2009).
What mechanistic insights can be derived from such consistent, distal-to-proximal cellular neurodegeneration? Identifying the molecular cargoes of bidirectional cellular transport mechanisms and ensuring adequate transport of these molecules to their targets may be a key component to achieving long-distance regeneration and re-innervation of RGCs to the brain.
DENDRITIC AND SYNAPTIC DEGENERATION AND TRANSPORT IN GLAUCOMA
RGCs require functional connections with pre-synaptic neurons in the retina and post-synaptic neurons in the brain to maintain transmission of visual information, and re-establishing and maintaining synaptic communication is vital to survival of RGCs (Della Santina et al., 2013). In different glaucoma models, the DBA/2J mouse and a microbead injection-induced IOP model of glaucoma, axon transport fails early, with synaptic transmission and axon and dendritic dysfunction preceding the eventual RGC death, implicating axon transport in disease pathology (Buckingham et al., 2008; Sappington et al., 2010; Ou et al., 2016; Ward et al., 2014). Additional work has also highlighted early dendritic field reorganization in different RGC subtypes, before measurable axonal degeneration, and well before cell death (Della Santina et al., 2013; El-Danaf and Huberman, 2015). Thus failure of long-distance transport down axonal or dendritic neurites may underlie early phases of degeneration.
MOLECULAR MOTORS UNDERLYING TRANSPORT ARE LINKED TO NEURODEGENERATIVE DISEASE
In neurons, microtubule motor proteins, dyneins, and kinesins, drive organelle and molecular axonal transport (Vale et al., 1985; Hirokawa, 1998; Teng et al., 2005; Hirokawa et al., 2009). Given the importance of axon transport in the homeostatic maintenance of neuronal survival, disruptions to these motor proteins underly a variety of neurological diseases. For example, Charcot–Marie–Tooth disease type 2A can be caused by mutations in KIF1B1, and congenital fibrosis of the extraocular muscles (which is a neuropathy, not a myopathy) can be caused by mutations in KIF21A (Zhao et al., 2001; Yamada et al., 2003). Kinesins can form axonal aggregates in some neurodegenerative conditions like Alzheimer’s disease, especially in cases with certain amyloid precursor protein mutants, with blockages occurring before the characteristic amyloid plaque accumulations (Stokin et al., 2005). A loss-of-function mutation in KIF5A results in hereditary spastic paraplegia, characterized by a progressive loss of function and degeneration of upper motor neurons, starting with synaptic degradation (Reid, 2003; Xia et al., 2003; Morfini et al., 2009). Different loss-of-function mutations in KIF5A, all affecting the cargo binding domain, are causative in some cases of ALS (Nicolas et al., 2018). Charcot–Marie–Tooth syndromes can include optic atrophy among the peripheral neuropathies that define the disease, supporting the premise of shared axon transport-related pathophysiologies among axonopathies. The vital role of molecular transport in injury and disease, and their corresponding variety of mRNA, protein, and mitochondrial cargoes, highlight the necessity of addressing axonal transport when attempting to regenerate and re-innervate CNS neurons.
Does neurodegeneration follow a general decline in transport, or is the transport of specific, key cargoes causative in these different diseases. In the last few decades, increasing evidence has shown that kinesin subtypes and adaptor proteins have at least partial cargo specificity (Chevalier-Larsen and Holzbaur, 2006). For example, KIF1A and KIF1B of the Kinesin 3 family transport synaptic vesicle precursors synaptophysin and synaptotagmin, but do not transport syntaxin 1A or SNAP25 (Okada et al., 1995), whereas KIF5 motors do transport syntaxin 1A and SNAP25, and also transport synaptotagmin (Toda et al., 2008). The physiologic relevance of this partial specificity and partial redundancy is not fully elucidated but could reflect compensatory mechanisms for vital cargo to maintain cellular function and survival.
Regulation of these motors’ active state and specific cargoes also depends on cell signaling cascades and post-translational modifications such as phosphorylation and changes in adaptor proteins. For KIF5 motors, protein kinase A phosphorylation inhibits the binding of synaptic vesicles and glycogen synthase kinase-3 phosphorylation inhibits the binding of membrane organelles (Sato-Yoshitake et al., 1992; Morfini et al., 2002). In mitochondrial trafficking, the adaptor proteins Milton and Miro bridge KIF5 motors to the mitochondria in a calcium dependent manner (Glater et al., 2006; MacAskill et al., 2009). The wide heterogeneity of these identified cargoes leads directly to the question of how specificity is effected, if they form functional groups, and how they change after injury or during regeneration. What are the key molecules and organelles transported in axons? And, which are affected in RGC axons in optic neuropathies like glaucoma with associated axon transport loss?
AXONAL TRANSPORT OF MITOCHONDRIA
Mitochondria are perhaps the most studied organelle being shuttled up and down the axon by motor proteins. Mitochondria are responsible for ATP generation by oxidative phosphorylation, generation of reactive oxygen species, calcium buffering, among many other functions (Werth and Thayer, 1994). Dysregulation of mitochondria and mitochondrial distribution can lead to apoptotic cell death. Mitochondrial trafficking is essential for neurite outgrowth in vitro (Morris and Hollenbeck, 1993), and in vivo transport of mitochondria after injury has recently been more appreciated. In vivo imaging of mitochondria in the retina has shown a general decrease in motility and transport in aged mice compared to adult mice, as well as a reduced number of transported mitochondria in a glaucoma model (Takihara et al., 2015). Interestingly, aged mice are more susceptible to the mitochondrial transport disruption of glaucoma compared to younger adult mice, correlating with the increased incidence of glaucoma as human age. Mitochondria traffic to injured axons in C. elegans is required for normal regeneration (Han et al., 2016). Similarly, after optic nerve crush in mice, the mitochondrial protein Armcx1 is upregulated during injury in a regenerative condition, and further overexpression enhances both survival and regeneration of RGCs. This effect is hypothesized to be due to an increased in mobilization of mitochondria after injury, consistent with the results seen in C. elegans and in the mammalian sciatic nerve (Cartoni et al., 2016; Han et al., 2016; Zhou et al., 2016). Thus promoting increased mitochondrial transport promotes regenerative responses in the mammalian optic nerve, although it is not yet understood how regulation of this mitochondrial redistribution and energetics modulation contributes to survival and regeneration.
AXONAL TRANSPORT OF MRNA
Effectors from the cell body arrive at pre-synaptic terminals, growth cones, dendrites, and sites of injury by two methods: local axonal translation after transport of mRNA, and direct long-distance transport of proteins. Before the detection of mRNA transport into mammalian axons, local translation was predicted based on an efficiency hypothesis: over long distances, transport of few mRNA molecules that could be translated many times over at the desired location conserves energy over synthesizing these proteins at the cell body and transporting them (Spaulding and Burgess, 2017). Evidence for local translation in PNS neurons has first been shown through radioactive protein synthesis labeling in vitro and in vivo, followed by microscopic evidence of ribosomes in vivo (Koenig, 1991; Eng et al., 1999; Bleher and Martin, 2001). The presence of ribosomes and mRNA in the axons of mature CNS neurons is a pre-requisite for local translation. PolyA and rRNA are seen in developing hippocampal neurons in vitro, and axonal protein synthesis contributes to growth cone stabilization in isolated, regenerating DRG neurons in vivo (Kleiman et al., 1994; Zheng et al., 2001). Furthermore, specific mRNA molecules whose transport is increased after injury have been seen in both PNS and CNS axons in vivo. (Hanz et al., 2003; Willis et al., 2011). RNA-binding proteins and ribosomes for local translation have been found in peripheral neurons (Zheng et al., 2001; Spillane et al., 2013), and bound to mitochondria in RGC axons in vitro (our unpublished data). Isolated mRNA from purified axons of cortical neurons using a specialized microfluidic chamber also revealed many transcripts related to RNA translation machinery and transport (Taylor et al., 2009). Indeed even with these data in CNS neurons, having less translation machinery in CNS axons than PNS axons (Verma et al., 2005) may contribute to the differential regenerative capacities between these two populations. Is a relative lack of mRNA transport and/or locally translated effector proteins a fundamental reason for regenerative failure? While further work in this field is necessary to determine if increased translation after injury in RGC axons can improve regeneration, progress has been made in identifying and targeting specific mRNA transport pathways.
Are there links between mRNA transport and molecular pathways implicated in neuroprotection or regeneration? One strong example involves the dual leucine zipper kinase (DLK-1) pathway, which was first shown to lead to cebp-1 mRNA stabilization and local translation in C. elegans (Yan et al., 2009), and later tied to RGC survival in mice (Watkins et al., 2013; Welsbie et al., 2013). In a high-throughput siRNA assay, DLK inhibition was seen to be pro-survival in primary RGCs given an axonal injury. Similarly, inhibition of leucine zipper kinase (LZK), whose C. elegans homolog is DLK-1, in conjunction with DLK knockdown, more completely prevents RGC death both in vitro and in vivo (Welsbie et al., 2017). Downstream effectors of this pathway have been identified, including SOX11, MEF2A, JUN, and ATF2, and a number of these also affect RGC survival and optic nerve regeneration. It is not known, however, whether DLK or these DLK/LZK pathway effectors lead to mRNA stabilization and local translation or transport changes in mammalian axons, similar to DLK’s mechanism of action in C. elegans.
Even more broadly, how can we discover the identities of these pools of mRNA that are actively being translated in RGC axons? Ribotrap techniques to isolate actively translating RNA in the visual system, and identifying them with RNA-Seq, takes this exploration of in vivo axonal translation a step further (Shigeoka et al., 2016). Briefly, affinity-tagged ribosomes are expressed in a cell-specific manner, cross-linked, and isolated. The bound mRNA to these ribosomes, the “translatome,” gives insight into actively translating mRNA in a specific cell type. In neurons with spatial separation of compartments, such as RGCs, optic nerve, and synaptic terminals can be isolated to identify locally translating axonal proteins. Quantifying changes of intra-axonal protein synthesis in the normal, injured, and regenerating optic nerves as compared to intrinsically regenerating axons will identify aspects of the translatome most relevant to neuro-regeneration.
AXONAL TRANSPORT OF PROTEINS
Directly transported proteins have been identified and studied in glaucoma and acute optic nerve injury, with an experimental focus on strong candidates for involvement in neurodegeneration, such as the transport of BDNF (Pease et al., 2000). To truly appreciate the complement of proteins transported normally or disrupted in glaucoma or other insults, unbiased methods are needed for broad identification. Mass spectrometry for proteins and lipids continue to show the most promise for tackling such questions. As these technologies continue to advance, methods for subdividing these pools, including time resolution for synthesis and degradation of these molecules and compartmentalized sequencing, will provide a clearer picture of molecular interactions.
Historically, studies in goldfish (Benowitz et al., 1981; Perry et al., 1985), tadpoles (Szaro et al., 1984), toads (Skene and Willard, 1981), and mammals (McKerracher et al., 1990) using radiolabeled amino acids have shown that there is a global loss of axonal transport following nerve injury, with selective increases in proteins of certain molecular weights. In non-regenerating mammalian RGCs, there is a preferential loss of slow compared to fast axonal transport, which may underlie some of the differences in regenerative potential between species. Studies that try to dissect the identities of axonal proteins and protein changes are confounded by proteins originating from non-axonal sources, such as glia (Perry et al., 1985). Approaches to separate axonal from glial proteins have included isolating axoplasm, e.g., from ligated sciatic nerves with or without injury; however, biological variability limited attempts to quantify protein differences even when using clustering methods to correlate and group transport machinery to a set of proteins (Michaelevski et al., 2010). Nonetheless, there is evidence of increased anterograde transport of structural components of translation machinery and mRNAs in PNS neurons that may not occur in CNS neurons after injury or in degenerative disease. Together these findings paint a picture of a coordinated cellular response to injury that relies on transporting both formed proteins and the machinery to synthesize new proteins (See Fig. 1).
Figure 1.
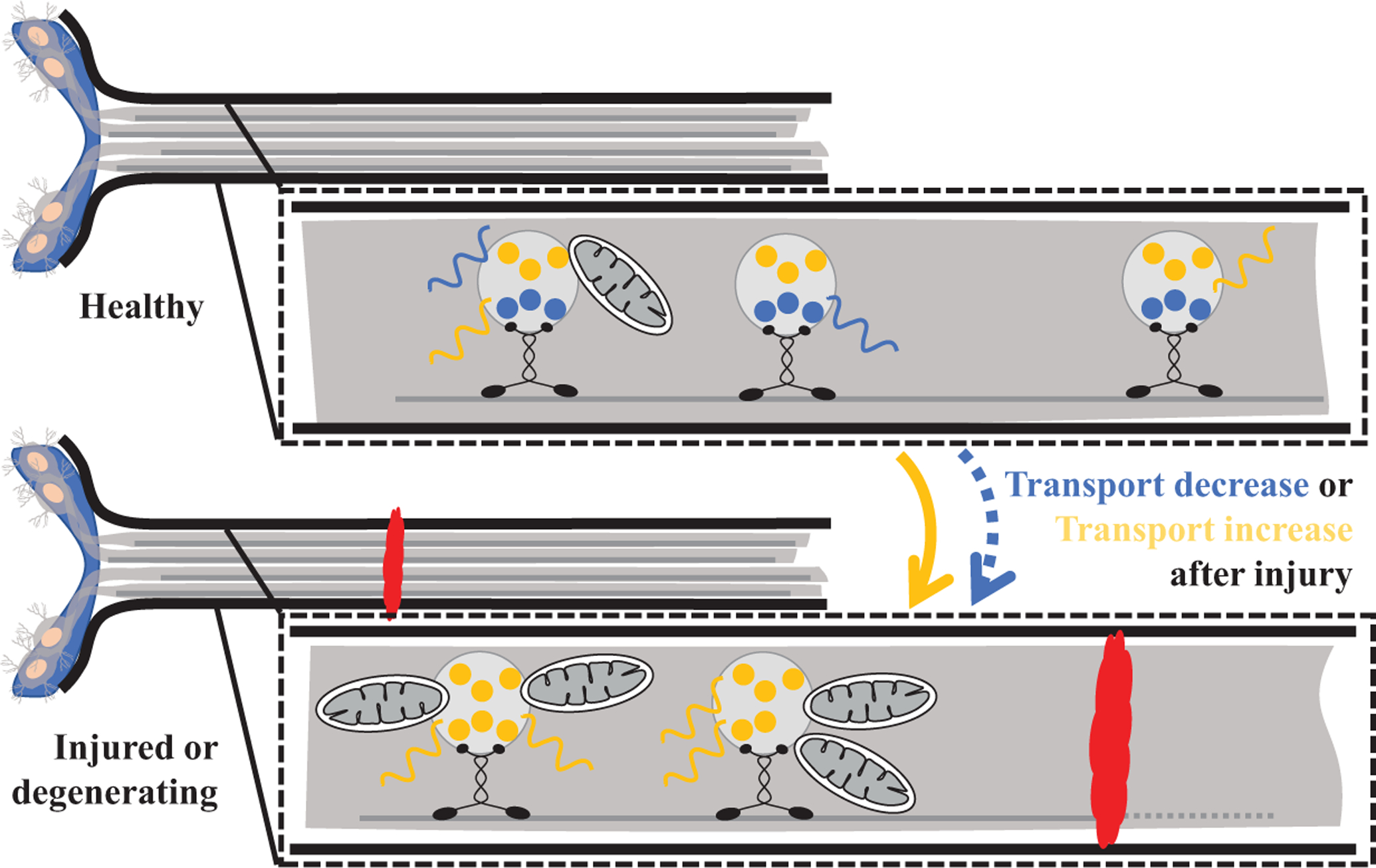
After axonal injury, anterograde transport of mitochondria, mRNA, and proteins are all affected. Certain proteins and mRNA decrease in transport, while the transport of other specific transcripts, proteins, and mitochondria toward the injury site may increase.
Even better than such indirect, bioinformatics-based approaches to identify and separate axonally transported proteins from glial proteins after injury would be direct detection of anterogradely or retrogradely transported proteins in vivo. This has been challenging, primarily due to the low proportion of transported axonal proteins compared to those from the surrounding white matter milieu. Are regenerated RGCs able to adequately transport synaptic proteins and mRNA for local translation from the cell body? In fact, what proteins make up the pre-synaptic compartment in RGCs, and do these differ by RGC subtype or target region? While synaptosomal proteomics have improved with novel compartment labeling techniques (Ting et al., 2016), these have for the most part been restricted to cell culture.
Recent advances in mass spectrometry-compatible signal detection in vivo have allowed a re-evaluation of this open question in transport biology (Schiapparelli et al., 2014), to the point that we can now directly detect changes in CNS axoplasmic protein transport in injury and disease. Labeling a group of proteins with an affinity tag, such as biotin, is a common method of separating proteins of interest from the background. In such paradigms, these labeled proteins are enriched with streptavidin pull-down, and once isolated, trypsinized, and identified with mass spectrometry. This technique has mostly been limited to situations where labeled proteins are a large portion of total proteins, as can be controlled in cell culture, but is a challenge when labeled proteins are only a small fraction of total proteins. In vivo, the percentage of proteins transported from a cell body to the axon is low compared to all the proteins found in the optic nerve, resulting in a high false-positive rate of contaminating unlabeled proteins. To overcome this limitation, reversing the order of the technique, trypsinizing all protein, and then pulling down and searching by mass spec for only the biotinylated peptides can now allow direct detection and high specificity even in rare samples (Schiapparelli et al., 2014). A second technical improvement is the ability to multiplex tags with slightly different molecular weight biotin groups, similar to tandem mass tagging, allowing for quantitative differences in protein abundance between conditions (Thompson et al., 2003). We have been exploring these approaches in optic nerve injury and regeneration, and our early findings suggest feasibility of the technique and identification of promising candidates.
COULD AXON TRANSPORT BE ONE UNIVERSAL EFFECTOR REGULATING REGENERATIVE FAILURE OR SUCCESS?
Re-examining recent advances in regeneration with a renewed and technically improved focus on axonal transport underscores that many of these strategies may depend or be enhanced by functioning molecular motor networks. For example, KLF4 has been directly linked to mitochondrial biogenesis and autophagy in non-neuronal cells, and proper translocation of mitochondrial to the site of injury is critical for axon growth (Jang and Arany, 2015). While KLF4 deletion promotes partial RGC regeneration, could negative regulation of mitochondrial translocation limit the effectiveness of this therapy? Dissection of the KLF9–Dusp14 link in RGC regeneration may potentially unveil a direct link between nuclear gene expression changes and the transport of signals sent to the RGC axon. As discussed above, the DLK/LZK pathway, which ties in upstream to the role of Sox11 in RGC survival and regeneration, is also known to affect mRNA stabilization and translation in other species. Could transport failure after injury be a cause of incomplete regeneration with visual recovery seen in each of these studies?
As mentioned previously, functional axonal transport depends on multiple kinesins and dynein. How are these motors, and their respective cargoes, differentially expressed and regulated after injury? What is the redundancy and specificity of cargo transport between motors? In uninjured neurons, loss of KIF4a reduces integrin trafficking, but overexpression is not able to increase integrin transport to the axonal compartment (Heintz et al., 2014). Approaching this problem from the opposite side, how do known regenerative or survival therapies affect kinesin expression, and protein translocation? Identifying the upstream regulators of transport through comparisons between regenerative and non-regenerative neurons, and specifically targeting them, may unmask the intrinsic growth capacities of adult CNS neurons.
In summary, recent large-scale òmics studies, both transcriptomic and proteomic, have opened up the ability to quantitatively probe all changes in the neuronal cell body – which may generate a deluge of differentially expressed candidates – and now changes in transport to other compartments, like the axon, which may narrow such molecular candidates to those most relevant for axonal degeneration and regeneration. Indeed probing these together may suggest a mechanistic link between gene transcription, protein expression, and molecular transport to affected axons. We hypothesize that understanding such mechanistic links will not only impact axon regeneration approaches, but also address myelination in regenerating fibers, electrophysiology and axon conductance of action potentials, and formation of synapses, all key to restoring the diversity of visual responses and behavior.
ACKNOWLEDGMENTS
Supported by NEI R01EY026766 and Research to Prevent Blindness (J.L.G.).
Contract grant sponsor: Research to Prevent Blindness; contract grant number: NEI R01EY026766.
REFERENCES
- Anderson DR and Hendrickson A (1974) Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Investigative Ophthalmology & Visual Science, 13, 771–783. [PubMed] [Google Scholar]
- Anderson DR and Hendrickson AE (1977) Failure of increased intracranial pressure to affect rapid axonal transport at the optic nerve head. Investigative Ophthalmology & Visual Science, 16, 423–426. [PubMed] [Google Scholar]
- Apara A, Galvao J, Wang Y, Blackmore M, Trillo A, Iwao K, et al. (2017) KLF9 and JNK3 interact to suppress axon regeneration in the adult CNS. The Journal of Neuroscience, 37, 9632–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SH (1981) A unifying hypothesis for the cause of amyotrophic lateral sclerosis, parkinsonism, and Alzheimer disease. Annals of Neurology, 10, 499–505. [DOI] [PubMed] [Google Scholar]
- Bei F, Lee HHC, Liu X, Gunner G, Jin H, Ma L, et al. (2016) Restoration of visual function by enhancing conduction in regenerated axons. Cell, 164, 219–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin S, Nawabi H, Wang C, Tang S, Latremoliere A, Warren P, et al. (2015) Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron, 86, 1000–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfey M and Aguayo AJ (1982) Extensive elongation of axons from rat brain into peripheral nerve grafts. Nature, 296, 150–152. [DOI] [PubMed] [Google Scholar]
- Benowitz LI, Shashoua VE and Yoon MG (1981) Specific changes in rapidly transported proteins during regeneration of the goldfish optic nerve. The Journal of Neuroscience, 1, 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore MG, Wang Z, Lerch JK, Motti D, Zhang YP, Shields CB, et al. (2012) Kruppel-like Factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proceedings of the National Academy of Sciences, 109, 7517–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleher R and Martin R (2001) Ribosomes in the squid giant axon. Neuroscience, 107, 527–534. [DOI] [PubMed] [Google Scholar]
- Buckingham BP, Inman DM, Lambert W, Oglesby E, Calkins DJ, Steele MR, et al. (2008) Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. Journal of Neuroscience, 28, 2735–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WBJ, Gardiner NJ, Gavazzi I, Powell J, Mcmahon SB, Heath JK, et al. (2001) Leukemia inhibitory factor determines the growth status of injured adult sensory neurons. The Journal of Neuroscience, 21, 7161–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Yingjing S, De Bellard ME, Tang S and Filbin MT (1999) Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron, 22, 89–101. [DOI] [PubMed] [Google Scholar]
- Cameron DA (2000) Cellular proliferation and neurogenesis in the injured retina of adult zebrafish. Visual Neuroscience, 17, 789–797. [DOI] [PubMed] [Google Scholar]
- Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, et al. (2016) The mammalian-specific protein Armcx1 regulates mitochondrial transport during axon regeneration (Neuron (2016) 92(6) (1294–1307) (S0896627316308340) (10.1016/j.neuron.2016.10.060)). Neuron, 94, 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Coppola G, Nawabi H, Omura T, Versano R, Huebner EA, et al. (2016) A systems-level analysis of the peripheral nerve intrinsic axonal growth program. Neuron, 89, 956–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MS, Huber AB, Van Der Haar MED, Frank M, Schnell L, Spillmann AA, et al. (2000) Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature, 403, 434–439. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen E and Holzbaur ELF (2006) Axonal transport and neurodegenerative disease. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease, 1762, 1094–1108. [DOI] [PubMed] [Google Scholar]
- Corredor RG, Trakhtenberg EF, Pita-Thomas W, Jin X, Hu Y and Goldberg JL (2012) Soluble adenylyl cyclase activity is necessary for retinal ganglion cell survival and axon growth. Journal of Neuroscience, 32, 7734–7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crish SD, Sappington RM, Inman DM, Horner PJ and Calkins DJ (2010) Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proceedings of the National Academy of Sciences, 107, 5196–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S and Aguayo AJ (1981) Axonal elongation into peripheral nervous system “Bridges” after central nervous system injury in adult rats. Science, 214, 931–933. [DOI] [PubMed] [Google Scholar]
- Della Santina L, Inman DM, Lupien CB, Horner PJ and Wong ROL (2013) Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. The Journal of Neuroscience, 33, 17444–17457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Qiao M, Bei F, Kim IJ, He Z and Sanes JR (2015) Subtype-Specific regeneration of retinal ganglion cells following axotomy: effects of osteopontin and mTOR signaling. Neuron, 85, 1244–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Danaf RN and Huberman AD (2015) Characteristic patterns of dendritic remodeling in early-stage glaucoma: evidence from genetically identified retinal ganglion cell types. Journal of Neuroscience, 35, 2329–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng H, Lund K and Campenot RB (1999) Synthesis of B-tubulin, actin, and other proteins in axons of sympathetic neurons in compartmented cultures. Journal of Neuroscience, 19, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, He Z and Benowitz LI (2004) Counteracting the Nogo receptor enhances optic nerve regeneration if retinal ganglion cells are in an active growth state. Journal of Neuroscience, 24, 1646–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen EHP, Zhao RR, Koseki H, Kanamarlapudi V, Hoogenraad CC, Eva R, et al. (2015) Exclusion of integrins from CNS axons is regulated by Arf6 activation and the AIS. Journal of Neuroscience, 35, 8359–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvao J, Iwao K, Apara A, Wang Y, Ashouri M, Shah N, et al. (2018) The Krüppel-like factor gene target Dusp14 regulates axon growth and regeneration. Investigative Ophthalmology & Visual Science, 59, 2736–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS and Schwarz TL (2006) Axonal transport of mitochondria requires Milton to recruit kinesin heavy chain and is light chain independent. The Journal of Cell Biology, 173, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL, Klassen MP, Hua Y and Barres BA (2002a) Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science, 296, 1860–1865. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Espinosa JS, Xu Y, Davidson N, Kovacs GT, Barres BA (2002b) Retinal ganglion cells do not extend axons by default. Neuron, 33, 689–702. [DOI] [PubMed] [Google Scholar]
- GrandPré T, Nakamura F, Vartanlan T and Strittmatter SM (2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature, 403, 439–444. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM and Bastiani M (2009) Axon regeneration requires a conserved MAP kinase pathway. Science, 323, 802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SM, Baig HS and Hammarlund M (2016) Mitochondria localize to injured axons to support regeneration. Neuron, 92, 1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, et al. (2003) Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron, 40, 1095–1104. [DOI] [PubMed] [Google Scholar]
- Heintz TG, Heller JP, Zhao R, Caceres A, Eva R and Fawcett JW (2014) Kinesin KIF4A transports integrin β1 in developing axons of cortical neurons. Molecular and Cellular Neuroscience, 63, 60–71. [DOI] [PubMed] [Google Scholar]
- Hirokawa N (1998) Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science, 279, 519–526. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y and Niwa S (2009) Kinesin superfamily motor proteins and intracellular transport. Nature Reviews Molecular Cell Biology, 10, 682–696. [DOI] [PubMed] [Google Scholar]
- Jang C and Arany Z (2015) Mitochondria Cripple without Krüppel. Trends in Endocrinology & Metabolism, 26, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski MP, McIlwrath SL, Jing X, Cornuet PK, Salerno KM, Koerber HR, et al. (2009) Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Research, 1256, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman R, Banker G and Steward O (1994) Development of subcellular mRNA compartmentation in hippocampal neurons in culture. The Journal of Neuroscience, 14, 1130–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, et al. (2010) Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proceedings of the National Academy of Sciences, 107, 6064–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig E (1991) Evaluation of local synthesis of axonal proteins in the goldfish mauthner cell axon and axons of dorsal and ventral roots of the rat in vitro. Molecular and Cellular Neuroscience, 394, 384–394. [DOI] [PubMed] [Google Scholar]
- Kurimoto T, Yin Y, Omura K, Gilbert H, Kim D, Cen L, et al. (2010) Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. The Journal of Neuroscience, 30, 15654–15663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang DM, Monzón-Mayor M, Bandtlow CE and Stuermer CAO (1998) Retinal axon regeneration in the lizard Gallotia galloti in the presence of CNS myelin and oligodendrocytes. Glia, 23, 61–74. [DOI] [PubMed] [Google Scholar]
- Le Pichon CE, Meilandt WJ, Dominguez S, Solanoy H, Lin H, Ngu H, et al. (2017) Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Science Translational Medicine, 9, eaag0394. [DOI] [PubMed] [Google Scholar]
- Leaver SG, Cui Q, Plant GW, Arulpragasam A, Hisheh S, Verhaagen J, et al. (2006) AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Therapy, 13, 1328–1341. [DOI] [PubMed] [Google Scholar]
- Lim J-HA, Stafford BK, Nguyen PL, Lien BV, Wang C, Zukor K, et al. (2016) Neural activity promotes long-distance, target-specific regeneration of adult retinal axons. Nature Neuroscience, 19, 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, et al. (2009) Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron, 61, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Stessin AM, Campana A, Hou J, Nikulina E, Buck J, et al. (2014) Soluble adenylyl cyclase is necessary and sufficient to overcome the block of axonal growth by myelin-associated factors. The Journal of Neuroscience, 34, 9281–9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerracher L, Vidal-Sanz M, Essagian C and Aguayo AJ (1990) Selective impairment of slow axonal transport after optic nerve injury in adult rats. The Journal of Neuroscience, 10, 2834–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao T, Wu D, Zhang Y, Bo X, Subang MC, Wang P, et al. (2006) Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. Journal of Neuroscience, 26, 9512–9519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelevski I, Medzihradszky KF, Lynn A, Burlingame AL and Fainzilber M (2010) Axonal transport proteomics reveals mobilization of translation machinery to the lesion site in injured sciatic nerve. Molecular & Cellular Proteomics, 9, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minckler DS, Bunt AH and Johanson GW (1977) Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Investigative Ophthalmology & Visual Science, 16, 426–441. [PubMed] [Google Scholar]
- Moore DL, Apara A and Goldberg JL (2011) Kruppel-like transcription factors in the nervous system: novel players in neurite outgrowth and axon regeneration. Molecular and Cellular Neuroscience, 47, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, et al. (2009a) KLF family members regulate intrinsic axon regeneration ability. Science, 326, 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, et al. (2009b) KLF family members regulate intrinsic axon regeneration ability. Science, 1007, 2007–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, et al. (2009) Axonal transport defects in neurodegenerative diseases. The Journal of Neuroscience, 29, 12776–12786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J and Brady ST (2002) Fast axonal transport misregulation and Alzheimer’s disease. NeuroMolecular Medicine, 2, 89–99. [DOI] [PubMed] [Google Scholar]
- Morris RL and Hollenbeck PJ (1993) The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. Journal of Cell Science 104 (Pt 3), 917–927. [DOI] [PubMed] [Google Scholar]
- Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, et al. (2018) Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron, 97, 1268–1283.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norsworthy MW, Bei F, Kawaguchi R, Wang Q, Tran NM, Li Y, et al. (2017) Sox11 expression promotes regeneration of some retinal ganglion cell types but kills others. Neuron, 94, 1112–1120.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N, Kidd GJ, Mahad D, Kiryu-seo S, Avishai A, Komuro H, et al. (2011) Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of ranvier. Journal of Neuroscience, 31, 7249–7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y and Hirokawa N (1995) The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell, 81, 769–780. [DOI] [PubMed] [Google Scholar]
- Ou Y, Jo RE, Ullian EM, Wong ROL, and Della Santina L (2016). Selective Vulnerability of Specific Retinal Ganglion Cell Types and Synapses after Transient Ocular Hypertension. Journal of Neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Kanter JL and He Z (2010) PTEN/mTOR and axon regeneration. Experimental Neurology, 223, 45–50. [DOI] [PubMed] [Google Scholar]
- Pasinelli P and Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nature Reviews Neuroscience, 7, 710–723. [DOI] [PubMed] [Google Scholar]
- Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA and Zack DJ (2000) Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Investigative Opthalmology & Visual Science, 41, 764–774. [PubMed] [Google Scholar]
- Perry GW, Burmeister DW and Grafstein B (1985) Changes in protein content of goldfish optic nerve during degeneration and regeneration following nerve crush. Journal of Neurochemistry, 44, 1142–1151. [DOI] [PubMed] [Google Scholar]
- Qin S, Zou Y and Zhang CL (2013) Cross-talk between klf4 and stat3 regulates axon regeneration. Nature Communications, 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA and Anderson DR (1977) Distribution of axonal transport blockade by acute intraocular pressure elevation in the primate optic nerve head. Investigative Opthalmology & Visual Science, 16, 640–644. [PubMed] [Google Scholar]
- Quigley HA and Broman AT (2006) The number of people with glaucoma worldwide in 2010 and 2020. British Journal of Ophthalmology, 90, 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Guy J and Anderson DR (1979) Blockade of rapid axonal transport: effect of intraocular pressure elevation in primate optic nerve. Archives of Ophthalmology, 97, 525–531. [DOI] [PubMed] [Google Scholar]
- Reid E (2003) Science in motion: common molecular pathological themes emerge in the hereditary spastic paraplegias. Journal of Medical Genetics, 40, 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas VT, Koch JC, Michel U, Bähr M and Lingor P (2017) Attenuation of axonal degeneration by calcium channel inhibitors improves retinal ganglion cell survival and regeneration after optic nerve crush. Molecular Neurobiology, 54, 72–86. [DOI] [PubMed] [Google Scholar]
- Richardson PM, McGuinness UM and Aguayo AJ (1980) Axons from CNS neurones regenerate into PNS grafts. Nature, 284, 264–265. [DOI] [PubMed] [Google Scholar]
- Saha AR, Hill J, Utton MA, Asuni AA, Ackerley S, Grierson AJ, et al. (2004) Parkinson’s disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. Journal of Cell Science, 117, 1017–1024. [DOI] [PubMed] [Google Scholar]
- Sappington RM, Carlson BJ, Crish SD and Calkins DJ (2010) The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Investigative Opthalmology & Visual Science, 51, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato-Yoshitake R, Yorifuji H, Inagaki M and Hirokawa N (1992) The phosphorylation of kinesin regulates its binding to synaptic vesicles. The Journal of Biological Chemistry, 267, 23930–23936. [PubMed] [Google Scholar]
- Schiapparelli LM, McClatchy DB, Liu H-H, Sharma P, Yates JR, et al. (2014) Direct Detection of Biotinylated Proteins by Mass spectrometry. Journal of Proteome Research, 13, 3966–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa T, Fimbel SM, Mallory DE, Maaswinkel H, Spritzer SD, Sand JA, Li L, Hyde DR and Stenkamp DL (2008), Ganglion cell regeneration following whole-retina destruction in zebrafish. Devel Neurobio, 68: 166–181. doi: 10.1002/dneu.20568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, et al. (2016) Dynamic axonal translation in developing and mature visual circuits. Cell, 166, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V and Diantonio A (2012) Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron, 74, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene JHP and Willard M (1981) Changes in axonally transported proteins during axon regeneration in toad retinal ganglion cells. The Journal of Cell Biology, 89, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Sun F, Park KK, Cai B, Wang C, Kuwako K, et al. (2009) SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron, 64, 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RP, Lerch-Haner JK, Pardinas JR, Buchser WJ, Bixby JL and Lemmon VP (2011) Transcriptional profiling of intrinsic PNS factors in the postnatal mouse. Molecular and Cellular Neuroscience, 46, 32–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares L, Parisi M and Bonini NM (2014) Axon injury and regeneration in the adult drosophila. Scientific Reports 6, 6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding EL and Burgess RW (2017) Accumulating evidence for axonal translation in neuronal homeostasis. Frontiers in Neuroscience, 11, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillane M, Ketschek A, Merianda TT, Twiss JL and Gallo G (2013) Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Reports, 5, 1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, et al. (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science, 307, 1282–1288. [DOI] [PubMed] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, et al. (2011) Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature, 480, 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaro BG, Faulkner LA, Kevin Hunt R and Peng Loh Y (1984) Axonal transport of [35S]methionine labeled proteins in xenopus optic nerve: phases of transport and the effects of nerve crush on protein patterns. Brain Research, 297, 337–355. [DOI] [PubMed] [Google Scholar]
- Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, et al. (2015) In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proceedings of the National Academy of Sciences, 112, 10515–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Berchtold NC, Perreau VM, Tu CH, Jeon NL and Cotman CW (2009) Axonal mRNA in uninjured and regenerating cortical mammalian axons. The Journal of Neuroscience, 29, 4697–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng J, Rai T, Tanaka Y, Takei Y, Nakata T, Hirasawa M, et al. (2005) The KIF3 motor transports N-cadherin and organizes the developing neuroepithelium. Nature Cell Biology, 7, 474–482. [DOI] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, et al. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical Chemistry, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- Ting AY, Stawski PS, Draycott AS, Udeshi ND, Lehrman EK, Wilton DK, et al. (2016) Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell, 166, 1295–1307.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda H, Mochizuki H, Flores R, Josowitz R, Krasieva TB, Lamorte VJ, et al. (2008) UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes & Development, 22, 3292–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakhtenberg EF, Li Y, Feng Q, Tso J, Rosenberg PA, Goldberg JL, et al. (2018) Zinc chelation and Klf9 knockdown cooperatively promote axon regeneration after optic nerve injury. Experimental Neurology, 300, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakhtenberg EF, Pita-Thomas W, Fernandez SG, Patel KH, Venugopalan P, Shechter JM, et al. (2017) Serotonin receptor 2C regulates neurite growth and is necessary for normal retinal processing of visual information. Developmental Neurobiology, 77, 419–437. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, Ure D, Eide L, Tran DD, et al. (2004) Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Molecular and Cellular Biology, 24, 8195–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Reese TS and Sheetz MP (1985) Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell, 42, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman MB, Bemben MA and Goldman D (2010) Tuba1a gene expression is regulated by KLF6/7 and is necessary for CNS development and regeneration in zebrafish. Molecular and Cellular Neuroscience, 43, 370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman MB, Bemben MA, Thompson RC and Goldman D (2007) Gene expression analysis of zebrafish retinal ganglion cells during optic nerve regeneration identifies KLF6a and KLF7a as important regulators of axon regeneration. Developmental Biology, 312, 596–612. [DOI] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell D, Meyer R, Holt CE, et al. (2005) Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. The Journal of Neuroscience, 25, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Winsor K, Nienhaus C, Hess E and Blackmore MG (2017) Combined chondroitinase and KLF7 expression reduce net retraction of sensory and CST axons from sites of spinal injury. Neurobiology of Disease, 99, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NJ, Ho KW, Lambert WS, Weitlauf C and Calkins DJ (2014) Absence of transient receptor potential vanilloid-1 accelerates stress-induced axonopathy in the optic projection. The Journal of Neuroscience, 34, 3161–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, et al. (2013) DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proceedings of the National Academy of Sciences, 110, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Mitchell KL, Jaskula-Ranga V, Sluch VM, Yang Z, Kim J, et al. (2017) Enhanced functional genomic screening identifies novel mediators of dual leucine zipper kinase-dependent injury signaling in neurons. Neuron, 94, 1142–1154.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, et al. (2013) Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proceedings of the National Academy of Sciences, 110, 4045–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth JL and Thayer SA (1994) Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. The Journal of Neuroscience, 14, 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis DE, Xu M, Donnelly CJ, Tep C, Kendall M, Erenstheyn M, et al. (2011) Axonal localization of transgene mRNA in mature PNS and CNS neurons. The Journal of Neuroscience, 31, 14481–14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Cui B, He L, Chen L and Mobley WC (2009) The coming of age of axonal neurotrophin signaling endosomes. Journal of Proteomics, 72, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C-H, Roberts E, Her L-S, Liu X, Williams DS, Cleveland DW, Goldstein LSB (2003) Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. The Journal of Cell Biology, 161, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Andrews C, Chan W-M, McKeown CA, Magli A, de Berardinis T, et al. (2003) Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nature Genetics, 35, 318–321. [DOI] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD and Jin Y (2009) The DLK-1 Kinase Promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell, 138, 1005–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanik MF, Cinar H, Cinar HN, Chisholm AD, Jin Y and Ben-Yakar A (2004) Functional regeneration after laser axotomy. Nature, 432, 822. [DOI] [PubMed] [Google Scholar]
- Yücel YH, Zhang Q, Gupta N, Kaufman PL and Weinreb RN (2000) Loss of neurons in magnocellular and parvocellular layers of the lateral geniculate nucleus in glaucoma. Archives of Ophthalmology, 118, 378–384. [DOI] [PubMed] [Google Scholar]
- Zhang B, Higuchi M, Yoshiyama Y, Ishihara T, Forman MS, Martinez D, et al. (2004) Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. The Journal of Neuroscience, 24, 4657–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, et al. (2001) Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell, 105, 587–597. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Kelly TK, Chang B, Ryazantsev S, Rajasekaran AK, Martin KC, Twiss JL (2001) A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. The Journal of Neuroscience, 21, 9291–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Pernet V, Hauswirth WW and Di Polo A (2005) Activation of the extracellular signal-regulated kinase 1/2 pathway by AAV gene transfer protects retinal ganglion cells in glaucoma. Molecular Therapy, 12, 402–412. [DOI] [PubMed] [Google Scholar]
- Zhou B, Yu P, Lin MY, Sun T, Chen Y and Sheng ZH (2016) Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. The Journal of Cell Biology, 214, 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]