

Abstract
A biological host response to an external stimulus or intervention such as a disease or infection is a dynamic process, which is regulated by an intricate network of many genes and their products. Understanding the dynamics of this gene regulatory network allows us to infer the mechanisms involved in a host response to an external stimulus and hence aids the discovery of biomarkers of phenotype and biological function. In this article, we propose a modeling/analysis pipeline for dynamic gene expression data, called Pipeline4DGEData, which consists of a series of statistical modeling techniques to construct dynamic gene regulatory networks from the large volumes of high-dimensional time course gene expression data that are freely available in the Gene Expression Omnibus (GEO) repository. This pipeline has a consistent and scalable structure that allows it to simultaneously analyze a large number of time course gene expression data sets, and then integrate the results across different studies. We apply the proposed pipeline to influenza infection data from nine studies and demonstrate that interesting biological findings can be discovered with its implementation.
Keywords: Time-course data, Gene Expression Omnibus, Differential Equations, Gene Regulatory Network
Introduction
Gene regulation plays a fundamental role in biological activities, such as cell growth, division, development and response to environmental stimulus. The dynamic gene regulatory network (GRN) specifies the interactions between genes, which in turn determines the expression level of each gene over time. Identifying these networks from genomic big data can shed light on the genetic mechanisms that occur when the cellular processes are impaired, for instance when they are subject to an infection or disease.
It is widely recognized that there may be hundreds or thousands of genes that harbor variations contributing to a specific illness, and there may also be a large genetic variability in host responses to an external stimulus or disease. Thus dynamic GRNs inferred from a single or a few gene expression data sets, may not be sufficient to understand the complexity and heterogeneity of the dynamic interaction process between the host and external stimulus.
Fortunately, in this Big Data era, we have an abundance of high-dimensional time course gene expression data from a variety of biological experiments and conditions. These data sets are generated from microarray, next-generation sequencing and other forms of high-throughput technologies, and are freely available in the Gene Expression Omnibus (GEO) data repository at the National Center for Biotechnology Information (NCBI) (1; 2). As of September 19, 2016, GEO has accumulated gene expression data from approximately 2 million samples from more than 73,000 studies (series) (3). As investigators must submit their genomic data to a data-sharing repository in accordance with grant or journal requirements, the number of data samples in GEO is growing rapidly (e.g., GEO obtained the data from 81,415 new samples between 2015 and 2016 (3)).
While a variety of options exist for reconstructing a gene regulatory network (GRN) based on a single experimental data set (see (4; 5) for an overview). There is limited methodology available to examine a large number of time course data sets from many different experiments to address important biological questions regarding a host genetic response to an external stimulus or disease. Examples of some fundamental questions include: (i) how do we identify genes that have a significant response over time at either population or individual level; (ii) do genes respond differently to the external stimulus or can multiple genes be grouped together based on their behavior over time? (iii) what are the similarities and differences among the genetic time-course patterns for various hosts or experimental conditions? (iv) what network of genetic interactions is formed for the host to respond to the external stimulus? (v) what are the similarities and differences among these networks for various hosts or experimental conditions?
We propose a pipeline for dynamic gene expression data, called Pipeline4DGEData, to address the above questions. This pipeline constructs dynamic gene regulatory networks from the large sets of time course gene expression data available on GEO for different biological experiments under various disease and/or stimulation conditions. We believe that this pipeline will allow us to better understand the pathogenesis of different diseases at both individual and population level. We can examine each host response to a disease or condition at a genetic level so that novel prevention and intervention targets can be discovered and personalized or precision medicine can be achieved (6; 7). We can also integrate the results from a large number of hosts that are responding to various diseases or conditions so that we can gain a better understanding of the heterogeneity that is inherent in the real world population and subsequently identify robust and reproducible signatures of disease phenotypes.
Current approaches for identification of GRNs from gene expression data have pros and cons. For example, information theory models (8; 9) are simple and easy to compute, but are essentially correlation models and do not take into account that multiple genes can co-regulate a target gene. Boolean networks (10; 11) represent the state of a gene using a binary variable, and as such do not account for the continuous nature of gene expressions. Bayesian networks (12; 13; 14) represent conditional dependencies among the genes via a directed graph, but the optimization of the network is computationally expensive, so applications are mostly limited to small-scale systems.
Linear differential equation (LDE) models (15) quantify the rate of change (derivative) of the expression level of one gene in the system as a function of the expression levels of all related genes. Let gi represent the expression level of gene i and Dgi denote the rate of change of this expression level, then for i = 1, …, n, where n denotes the number of genes and βi,j quantifies the regulation effect of gene j on gene i. Some standard statistical methods such as the least squares or maximum likelihood estimation can be used to perform statistical inference for the parameters βi,j from time course gene expression data for small-scale networks (15; 16). However, for LDE models that involve hundreds or even thousands of genes, the standard statistical methods often fail due to the curse-of-dimensionality.
In this article, we propose the Pipeline4DGEData which, consists of a series of cutting-edge statistical analysis and inference techniques to identify dynamic GRNs from high-dimensional time course gene expression data. The pipeline is designed to obtain time-course data from GEO automatically, analyze the data to reconstruct the dynamic GRNs, and output the results into the format of a manuscript for publication. As a consequence, a large number of data sets from GEO can be analyzed and modeled in an effective manner, and the results can be quickly disseminated. We demonstrate the utility of the Pipeline4DGEData by applying it to time course gene expression data from nine studies on viral respiratory infections that are available on GEO.
Dynamic Modelling for Genomic Big Data
The Pipeline for Dynamic Gene Expression Data is implemented in Matlab (R2016a) and is available at https://github.com/j142857z/Pipeline. It obtains GRNs from a large number of time course gene expression data sets. The pipeline has been specifically designed to have a scalable structure that allows it to simultaneously analyze the large volumes of data that are available on GEO. It consists of the following eight steps:
Obtain the time course gene expression data sets from GEO.
Pre-process the probe level data, i.e., background adjustment, normalization, and summarization.
Detect the genes with expression levels that change significantly with respect to time, which we call dynamic response genes (DRGs).
Cluster the DRGs into groups of co-expressed genes, which are referred to as temporal “gene response modules” (GRMs).
Obtain the functional annotation (gene ontology terms, associated pathways and a functional gene-enrichment analysis) of the GRMs.
Construct the high-dimensional gene regulatory networks (GRNs) that determine the interactions between the GRMs using LDE models.
Perform a network feature analysis on the established GRNs
Output the results in the form of a manuscript.
These steps are summarized in the flowchart in Figure (1). Each of these eight steps is detailed in the subsequent subsections.
Figure 1.
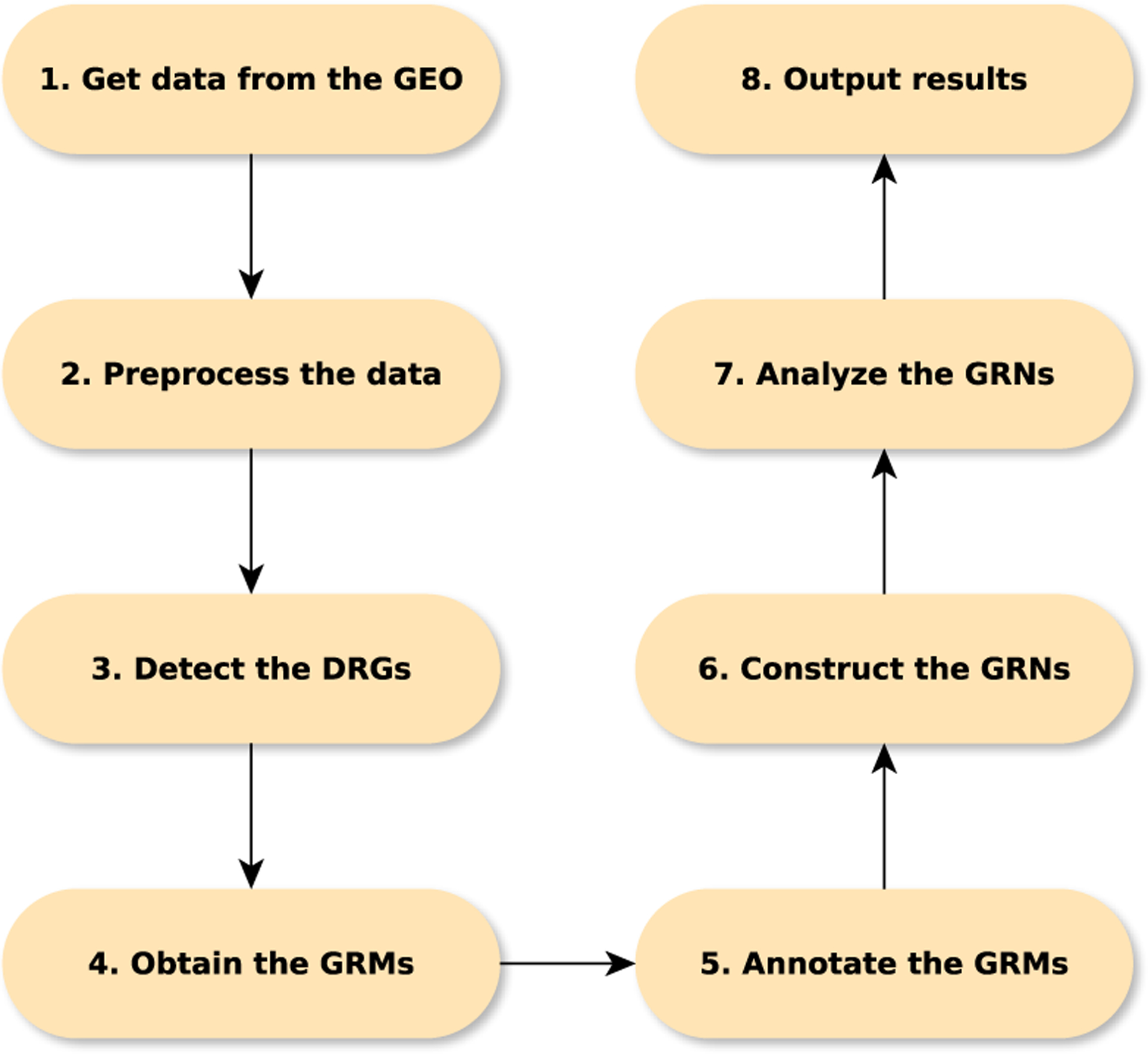
Summary of the eight steps implemented by the Pipeline4DGEData
Step 1. Get the data from GEO
Our pipeline has been developed to model time course gene expression data and is expected to produce reliable results if there are 7 or more time points available. The GEO database is not designed to easily identify the number of time points for the microarray or RNA-Seq experiments. Instead, the experimental design is documented in a text file for each study. We have used text mining, natural language processing and ontology techniques to identify, from the GEO database, potential time course data sets in addition to other characteristics or metadata including the related diseases and cell types from which the data are generated. But these text mining approaches are not the focus of this paper and will be reported elsewhere. The first step of our pipeline is to identify the time course GEO data sets related to a particular keyword, for example, influenza. The resulting GSE numbers, which identify series records in GEO are saved to a file. See (1) and (2) for further details on GEO record types. The pipeline automatically retrieves the data sets and meta information corresponding to these GSE numbers.
Step 2. Pre-process the data
Affymetrix GeneChip arrays are currently among the most widely used high-throughput technologies for the genome-wide measurement of expression profiles. To minimize mis- and cross-hybridization problems, this technology includes both perfect match (PM) and mismatch (MM) probe pairs as well as multiple probes per gene (17). As a result, significant pre-processing is required before an absolute expression level for a specific gene can be accurately assessed. In general, pre-processing probe-level expression data consists of three steps: background adjustment (remove local artefact’s and “noise”), normalization (remove array effects), and summarization at the probe set level (combine probe intensities across arrays to obtain a measure of the expression level of corresponding messenger RNA (mRNA)).
Most datasets in GEO are already pre-processed. By default, the Pipeline for Dynamic Gene Expression Data proceeds with the pre-processing technique used in the original study. However, if the .cell files are available, then the user can select from the following popular pre-processing techniques to reprocess the data if necessary: Microarray Suite 5 (MAS5) (18), Robust Multi-array Average (RMA) (19) and Guanine Cytosine Robust Multi-Array Analysis (GCRMA) (20). Table 1 provides a brief overview of the three pre-processing techniques. See (21; 22) for a detailed comparison.
Table 1.
A brief overview of the pre-processing techniques: Microarray suite 5 (MAS5), Robust Multi-array Average (RMA) and Guanine Cytosine Robust Multi-Array Analysis (GCRMA).
| Technique | Background adjustment | Normalization | Summarization |
|---|---|---|---|
| MAS5 | Ideal (full or partial) MM subtraction | Constant | Turkey biweight |
| RMA | Signal (exponential) and noise (normal) close-form transformation | Quantile | Median polish |
| GCRMA | Optical noise, probe affinity and MM adjustment | Quantile | Median polish |
Step 3. Detect the Dynamic Response Genes (DRGs)
(a) Obtain the estimated gene expression curves
We assume that the centered expression levels of the ith gene, belonging to subject j, denoted here by xi,j, is a smooth function over time t and that the centered gene expression measurement is a discrete observation from this smooth function, which has been distorted by noise, i.e.,
for i = 1, …, n, j = 1, …, N and k = 1, …, Ki,j, where n is the number of genes, N is the number of subjects (or experimental conditions), Ki,j is the number of time points observed for the ith gene, belonging to subject j. The noise ϵi,j is assumed to be an independently identically distributed (i.i.d.) Gaussian random variable with mean 0 and variance σ2.
The Ki,j × 1 vector of the estimated centered expression levels evaluated at the points t, for the ith gene, belonging to subject j, is obtained by spline smoothing (23; 24). This approach approximates xi,j by a linear combination of L independent basis functions, , where the Ki,j × L matrix Bi,j denotes the basis functions evaluated at time t and the vector ci,j provides the corresponding coefficients.
The coefficients ci,j can be estimated by minimizing
| (1) |
where the first term defines the squared discrepancy between the observed centered gene expression measurements and the estimated measurements , and the second term contains the L × L matrix Ri,j which is the integral of the squared second derivative of Bi,j. The second term penalizes the curvature of and hence requires it to be sufficiently smooth. The parameter λj controls the trade-off between the fit to the data and the smoothness requirement and hence ensures that has an appropriate amount of regularity. All the genes for each subject are assumed to have the same λj.
We expect that only a small fraction of genes respond to the external stimulus and the majority of the genes have no significant response with relatively flat expression levels over time. Therefore, estimating the parameter λj using the conventional method of minimizing the prediction error with generalized cross validation (GCV), see (25) for details, of all the genes together is not ideal as GCV will tend to select a λj that is large to minimize the prediction error of the majority of unresponsive genes. As we are interested in obtaining an appropriate amount of regularity for the responsive genes, we apply an approach similar to (26) and (27) and choose a subset of the genes that exhibit time course response patterns with relatively smooth trajectories that do not fluctuate widely. Then we rank these genes by their interquartile range and select 200 of the top ranking genes as our estimation subset. The regularity parameter λj is estimated by minimizing the GCV of the responsive genes in our estimation subset, this parameter is then used to smooth the time course data for all the genes.
(b) Perform a hypothesis test to identify the genes with expressions that change significantly over time
Dynamic response genes (DRGs) can be defined as genes with expressions that change significantly over time. In order to determine which genes can be considered DRGs, we use an F-statistic which compares the goodness-of-fit of the null hypothesis versus the alternative hypothesis . The F-statistic is given by,
where dfi,j is the degrees of freedom of the estimated curve , and . are the residual sum of squares under the null and the alternative models for the i-th gene, belonging to subject j. The genes with large F-ratios can be considered as exhibiting notable changes with respect to time. If we wish to have an equal amount of DRGs for each subject, we can rank the F-ratios and select nDRG the number of top ranking dynamic response genes. By default the Pipeline4DGEData sets nDRG to 3000.
As an alternative, the functional principal component analysis (FPCA) approach (27) can also be used to identify the DRGs. The FPCA method needs to borrow information across genes to estimate the covariance and the results will be subject to the estimation accuracy of the covariance matrix.
Step 4. Cluster the DRGs into temporal gene response modules (GRMs)
As many of the DRGs exhibit similar expression patterns over time, we wish to cluster them into co-expressed modules (groups of genes which have similar gene expression patterns over time). This step not only reduces the modeling dimension but also eases the identifiability problem. It is widely recognized that many co-expressed genes may follow similar temporal patterns, but at the same time, some genes may have very few or even no co-expressed genes, and thus may exhibit unique temporal response patterns. Consequently, the GRMs can vary greatly in size, with some being large and containing many genes and others being small or even containing a single gene. To obtain these clusters, we adopt the Iterative Hierarchical Clustering (IHC) method introduced in (28). This approach requires a single parameter that controls the trade-off of the between- and within-cluster correlations. In particular, the average within-cluster correlation will be approximately α, and the between-cluster correlation will be below α. The IHC algorithm is outlined below:
Initialization: Cluster the data for the standardized DRGs using the hierarchical agglomerative clustering approach. Let the distance metric be the Spearman rank correlation with a threshold of α, and the linkage method be the average of the genes in each cluster.
Merge: Treat each of the cluster centers as ‘new genes’ and use the same rule as in the initialization step to merge the centers into new clusters. The cluster centers provide the average time-course pattern of the cluster members.
Prune: Let ci be the center of cluster i. If the correlation between the cluster center and gene j, which will be denoted by ρi,j, is less than α, then remove genej from the cluster i. Let P be the number of genes removed from the existing S clusters. Assign all P genes into single-element clusters. Hence, there is now (S + P) clusters in total.
Repeat Merge-Prune Steps until the index of clusters converges.
Repeat Merge Step until the between-cluster correlations are less than α.
Step 5. Annotate the GRMs
The Pipeline for Dynamic Gene Expression Data outputs an annotation report which extracts biological meaning from the DRGs contained in each GRM. Specifically, it provides the gene ontology terms, associated pathways, and a functional gene enrichment analysis. This is achieved by using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (29; 30).
Step 6. Construct the high-dimensional gene regulatory network (GRN) that determines the interactions between the GRMs
High-dimensional gene regulatory networks map how the change in the expression of any single gene is regulated by its own expression level and other gene expression levels. There is an abundance of literature regarding the use of ordinary differential equation (ODE) modeling to construct a high-dimensional gene regulatory network (GRN) (5; 31; 32). ODEs model gene regulations using rate equations. Here we model the interactions between GRMs using the following ODE
| (2) |
where Dmq,j represents the instantaneous rate of change in qth gene response module, quantifies the regulation effects of the pth gene response module on the rate of change of the qth gene response module Dmq,j. The standard approach for estimating the parameters of differential equations from noisy measurements is nonlinear least squares (NLS) (33; 34). However, this method requires initial estimates of the regulation effects and initial conditions for the expression levels of the gene response modules at time t0. Differential equation models differ from the classical regression models as they capture not only the direct effects that are strong interactions between two gene response modules but they also include indirect effects high correlations that may exist between two gene response modules that are not directly connected but influence each other via a third gene response module they both directly interact with. In general, such indirect interactions may be induced not only by the third gene response module, but equally by the entire collective dynamics of a network.
(a) Initial estimates of the regulation effects
The two-stage smoothing-based estimation method (35; 36) decouples the system of differential equations in (2) and approximates it by a set of pseudo-regression models as in (3). The first step obtains estimates of the average trajectory of the GRMs and their derivative for p, q = 1, …, Q. The estimated trajectories are the average of the smoothed trajectories attained by the spline smoothing approach in part (a) of Step 3, for all the genes contained in the qth GRM. Similarly, is estimated by averaging the derivative of the smoothed trajectories obtained by the spline smoothing approach in part (a) of Step 3, for all the genes contained in the qth GRM. The set of Q pseudo-regression models are then given by
| (3) |
where θp,q,j denotes the direct effects that is the strong relationships between the pth GRM and the rate of change in the qth GRM.
It is widely accepted that gene regulatory networks are sparse, i.e., only a few of the are non-zero. In order to determine which of the regulation effects are significant (i.e., non-zero) our pipeline applies the least absolute shrinkage and selection operator (LASSO) (37) approach or the adaptive LASSO (38) approach (36) to the pseudo-regression model in (3). The LASSO approach requires minimizing
As the penalty term means that the LASSO increases, LASSO sets more coefficients θp,q,j to zero. This means that the LASSO estimator is a smaller model, with fewer predictors. The L1 regularization parameter γ enforces the amount of sparsity in θ and can be chosen by minimizing either the generalized cross validation criterion, the Akaike information criterion or the Bayesian information criterion. As such, LASSO is a model selection and dimensionality reduction technique that determines the significant coefficients θp,q,j or initial estimates of the weighted network edges by maximizing the prediction accuracy (GCV) or the trade-off between the goodness of fit and the complexity (AIC/BIC) of (3). Alternatively, the SCAD approach (31; 39) can also be used in this step. See more details in (31).
(b) Refined estimation of the regulation effects
The parameter estimates from the two-stage method in the above pseudo-regression model are not efficient in terms of estimation accuracy when the model selection is performed simultaneously, and there can be significant approximation error in and its derivatives. The estimation of significant coefficients or network edges can be improved or refined using nonlinear least squares (NLS), maximum likelihood or other more efficient estimation methods once the model selection from part (a) of Step 6 is completed. Our pipeline adopted the NLS approach which minimizes the squared discrepancy between the numerical approximation to the solution of the differential equation (2) and the observations. The non-zero estimates of from part (a) of Step 6 are used as initial estimates for the regulation effects in equation (2). Given the initial estimates, and a set of p initial values, , which are attained by the spline smoothing approach in part (a) of Step 3, an initial numerical approximation of the solution to differential equation (2) can be computed as . Using a linearization of the discrepancy between the data and the numerical solution, the estimated parameters can be refined iteratively. In Lu et al. (31), the mixed-effects modeling approach is used for parameter refinement.
The variability in the GRN is assessed by calculating the confidence intervals of the parameters using the delta method see (40) for details.
Step 7. Perform a network feature analysis on the GRN.
The reconstructed biological networks are significantly different from random networks and often exhibit ubiquitous properties in terms of their structure and organization. Many metrics have been developed to characterize biological networks, see (41) and (42) for an overview.
We use these network metrics to identify influential GRMs and to provide insight on the organization of the GRN describing the host response to the external stimulus or disease. These metrics are divided into two groups: node metrics and network metrics. Node metrics provide information on the modules within each GRN and are useful for identifying GRMs that are likely to be hubs or groups of modules that are strongly interconnected. On the other hand network metrics identify properties or features of each GRN and are useful for comparing different GRNs. The Pipeline for Dynamic Gene Expression Data uses the Matlab SBEToolbox (43) to compute the network metrics for each of the GRNs. This toolbox includes many metrics, some of the most relevant are listed in Table 2. The interaction network between GRMs was represented as as a directed graph with the gene response modules as nodes and interactions as directed edges which are given by the point estimates obtained in step 6. The variability in the network metrics can be estimated by evaluating each metric at the lower endpoint of the 95% confidence interval and the upper endpoint of the 95% confidence interval for .
Table 2.
The node metrics which identify influential modules and the network metrics which provide insight on the organization of the host response to the external stimulus or disease.
| Node Metrics | |
|---|---|
| Degree centrality | is the number of links that a node has with other nodes in the network (44). |
| Closeness centrality | is the average distance from a given node to all other nodes in the network (47). |
| Eccentricity | is the greatest geodesic distance between a node and any other node in the network (48). |
| Bridging centrality | measures the extent to which a node or an edge is located between well-connected regions (50). |
| Clustering Coefficient | measures the degree of interconnectivity in the neighborhood of a node (53). |
| Network Metrics | |
| Density | The proportion of direct ties in a network relative to the total number possible (54). |
| Diameter | is the shortest distance between the two most distant nodes in the network (54). |
Step 8. Output the results: reporting and manuscript drafting.
As GSE numbers can often contain data sets on more than one experimental condition, for example, GSE52428 contains time course gene expression data following intranasal influenza A H1N1 and H3N2 inoculation, for 24 and 17 subjects respectively. The pipeline determines if there is more than one experimental condition (subject/virus strain), and then runs steps 2–8 on each condition/subject. For example, one of the experimental conditions for the GSE52428 study was H1N1 Subject 1. Step 8 creates six folders to store the results from Steps 2–7. The folders are organized in the following hierarchal fashion: GSE number, experimental condition, and steps 2–7. Figure 2 provides a schematic of the folders and Table 3 details the contents of each folder. The folder structure allows for different experimental conditions to be computed in parallel. We have developed and tested a parallel computing version of the pipeline from which the results can be automatically saved on the OneDrive cloud. The experimental condition folder also contains a TEX document (if ran on a Mac, Linux, or Windows machine) and a Word document (if ran on a Windows machine), in the format of a biomedical journal article detailing the results. This article includes: a methods section describing the data and techniques implemented by the Pipeline for Dynamic Gene Expression Data and a draft of the results section which contains tables and figures which are automatically generated by the pipeline. The aim of this document is to facilitate with the drafting of a manuscript for publication, based on the results produced by the pipeline. An example of this article draft is provided in the supplementary material.
Figure 2.
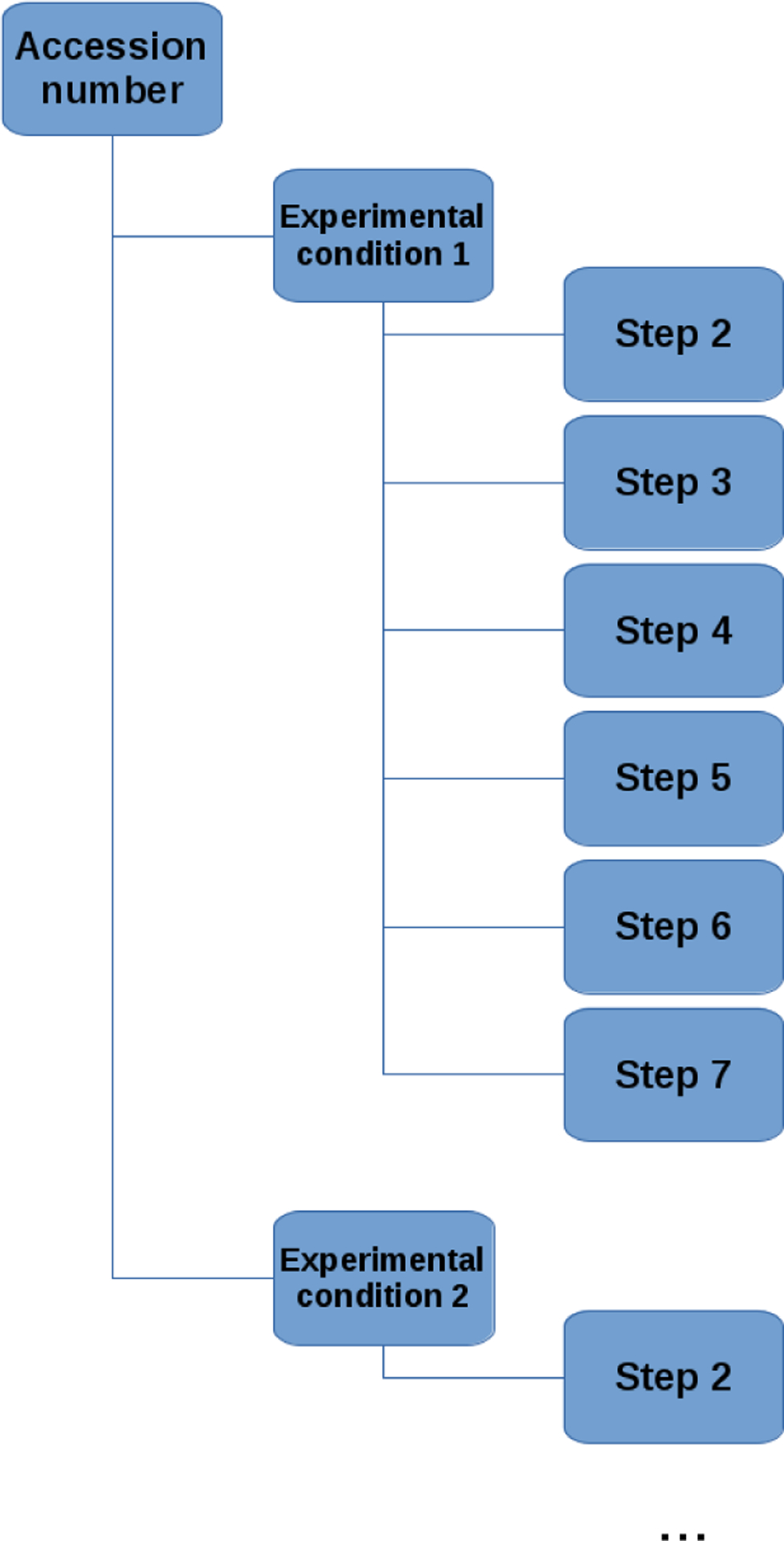
Schematic for the structure of the folders created by the Pipeline for Dynamic Gene Expression Data
Table 3.
The contents of each folder produced by Steps 2–7 of the Pipeline for Dynamic Gene Expression Data
| File name | Description |
|---|---|
| Step 2 | |
| raw_data.mat | A Matlab file containing the raw unprocessed data (if .cell files are available) |
| processed_data.mat | A Matlab file containing the processed data (if .cell files are available) |
| Step 3 | |
| Matlab_Workspace3.mat | A Matlab file containing all the variables that the pipeline has required thus far. |
| Derivative_Fitted_Curves.csv | The first derivative of the estimated smooth trajectories of each gene expression over time. |
| DRG.csv | The probe set ids and the time course genes expression of each of the Dynamic Response Genes. |
| F_value.csv | The F-value for each gene. |
| Fitted_curves.csv | The estimated smooth trajectories of each gene expression over time. |
| Index_Ftest_DRGs.csv | The index of the DRGs ranked by their F-Statistic. |
| Index_Ftest.csv | The index of the probe set ids ranked by their F-Statistic. |
| Probe_set_ID_Ftest_DRG.csv | The probe set ids of the DRGs ranked by their F-Statistic. |
| Step 4 | |
| Matlab_Workspace4.mat | A Matlab file containing all the variables that the pipeline has required thus far. |
| Cluster_IDX.csv | The cluster index for each of the DRGs. |
| Cluster_MT.csv | The probe set ids for each of the DRGs and a number indicating which type of module this probe set belongs to SGM (1), SSM (2), MSM (3) and LSM (4). |
| GRMs.pdf | A file containing a plot of the time-course patterns for all the genes in each GRM. |
| Step 5 | |
| Matlab_Workspace5.mat | A Matlab file containing all the variables that the pipeline has required thus far. |
| M1/Chart_report_of_M1.csv | The annotation and enrichment analysis which highlights the most relevant terms associated with the genes contained in GRM 1. There is a folder and file for each GRM. |
| M1/Table_report_of_M1.csv | The annotation and enrichment analysis which highlights the most relevant terms associated with the genes contained in GRM 1. There is a folder and file for each GRM. |
| Step 6 | |
| Matlab_Workspace6.mat | A Matlab file containing all the variables that the pipeline has required thus far. |
| Coefficients.xls | The estimated regulation effects |
| Dependency_matrix.xls | The GRMs inward and outward connections. |
| Step 7 | |
| Matlab_Workspace7.mat | A Matlab file containing all the variables that the pipeline has required thus far. |
| Adjacency_matrix.xls | The estimated regulation effects |
| Dependency_matrix.xls | The GRMs inward and outward connections. |
| Network_Statistics.xls | The Network metrics for the GRN. |
| Node_metrics.xls | The Network metrics for the GRMs. |
| Network_plot.pdf | A plot of the GRNs. |
The Pipeline for Dynamic Gene Expression Data Applied to Influenza Studies
We demonstrate the utility of the Pipeline for Dynamic Gene Expression Data by applying it to time course gene expression data from nine studies on various influenza viruses. These viruses represent respiratory pathogens that cause either seasonal, endemic infections or unpredictable pandemics. Influenza occurs globally with an annual attack rate estimated at 5% to 10% in adults and 20% to 30% in children (58). Illnesses can result in hospitalization and death mainly among high-risk groups (the very young, elderly or chronically ill). Worldwide, these annual epidemics are estimated to result in about 3 to 5 million cases of severe illness, and about 250,000 to 500,000 deaths (58). We expect that our biological findings on these influenza virus strains, produced by the proposed Pipeline4DGEData, will have a high impact on public health.
The nine studies that we have chosen as examples for the application of the Pipeline for Dynamic Gene Expression Data consist of 58 time course data sets corresponding to different experimental conditions (hosts inoculated with various virus strands) with 1, 554 samples from either blood, epithelial cells, dendritic cells or adenocarcinoma cells (59; 60; 61; 62; 63; 64; 65; 66). Unlike a single cohort experiment study where the goal is to control as many confounding factors as possible, our analysis includes data sets with a broad biological and technical heterogeneity. These data sets include healthy controls, individuals with various types of viral or bacterial infections, and individuals that were vaccinated for influenza. These studies can be grouped into 19 virus subtypes, and Table 4 provides detailed information on these subtypes for each of the nine studies. The majority of the studies (89%) involve human subjects, and their most common (63%) bio-samples are bronchial epithelial cells. The dominating virus subtypes are seasonal influenza virus strains, H1N1 (41% of the data sets) and H3N2 (29% of the data sets). Figure 3 show the distributions of the number of time-points, the number of genes/probes from which expression-level measurements are available and the pre-processing techniques used in the original study. The number of time points range from 7 − 15, with 69% of the data sets containing more than ten time-points. The number of genes or probes range from 11, 961 – 54, 675. Here we choose to analyze the time course gene expression data using the pre-processing techniques employed in the original study. The 58 data sets were pre-processed with one of the following techniques, RMA, GCRMA, MAS5 and LIMMA. The most popular pre-processing technique is RMA (67% of the studies selected this method). The design of the pipeline allows for the systematic integration of the results at various levels. More specifically, results of the pipeline analysis can be examined on an individual by individual basis, e.g., by examining each subject’s DRGs, GRMs and GRN separately. Also, results can be examined across several subjects and experimental conditions (e.g., virus subtypes). This can be achieved by finding the DRGs that are common across groups of subjects, the similarities/differences between the patterns of the GRMs in the groups and the similarities/differences between GRNs across the groups. The following subsections examine the population level similarities/differences between the DRGs, GRMs, and GRN for all 58 time course data sets across the 19 virus subtypes.
Table 4.
The details for each of the 19 virus subtypes for the nine influenza-related studies containing genome-wide time course gene expression data obtained from the GEO database.
| GSE number | Virus Subtype | No. Hosts | Bio Sample | Organism |
|---|---|---|---|---|
| GSE52428 | H1N1/Brisbane | 24 | peripheral blood | Homo sapiens |
| GSE12254 | WE (LCMV-WE) | 1 | liver sample | Macaca mulatta |
| GSE19392 | delNS1 post trypsin | 1 | bronchial epithelial cells | Homo sapiens |
| GSE19392 | PR8 post trypsin | 1 | bronchial epithelial cells | Homo sapiens |
| GSE19392 | vRNA LTX+RNA | 1 | bronchial epithelial cells | Homo sapiens |
| GSE19392 | treated with media alone | 1 | bronchial epithelial cells | Homo sapiens |
| GSE19392 | treated with IFNb IFN | 1 | bronchial epithelial cells | Homo sapiens |
| GSE19392 | treated with LTX transfection reagent | 1 | bronchial epithelial cells | Homo sapiens |
| GSE37571 | A/CA/04/2009 | 1 | Calu-3 lung adenocarcinoma cell line | Homo sapiens |
| GSE37571 | Mock | 1 | Calu-3 lung adenocarcinoma cell line | Homo sapiens |
| GSE30550 | H3N2/Wisconsin | 17 | peripheral blood | Homo sapiens |
| GSE40844 | Mock | 1 | Calu-3 lung adenocarcinoma cell line | Homo sapiens |
| GSE41067 | H1N1 influenza A/New Caledonia/20/1999 | 1 | monocyte-derived dendritic cells | Homo sapiens |
| GSE47960 | dORF6 | 1 | bronchial epithelial cells | Homo sapiens |
| GSE47960 | iSARs | 1 | bronchial epithelial cells | Homo sapiens |
| GSE47960 | Mock | 1 | bronchial epithelial cells | Homo sapiens |
| GSE47961 | BAT | 1 | bronchial epithelial cells | Homo sapiens |
| GSE47961 | dORF6 | 1 | bronchial epithelial cells | Homo sapiens |
| GSE47961 | H1N1 | 1 | bronchial epithelial cells | Homo sapiens |
Figure 3.
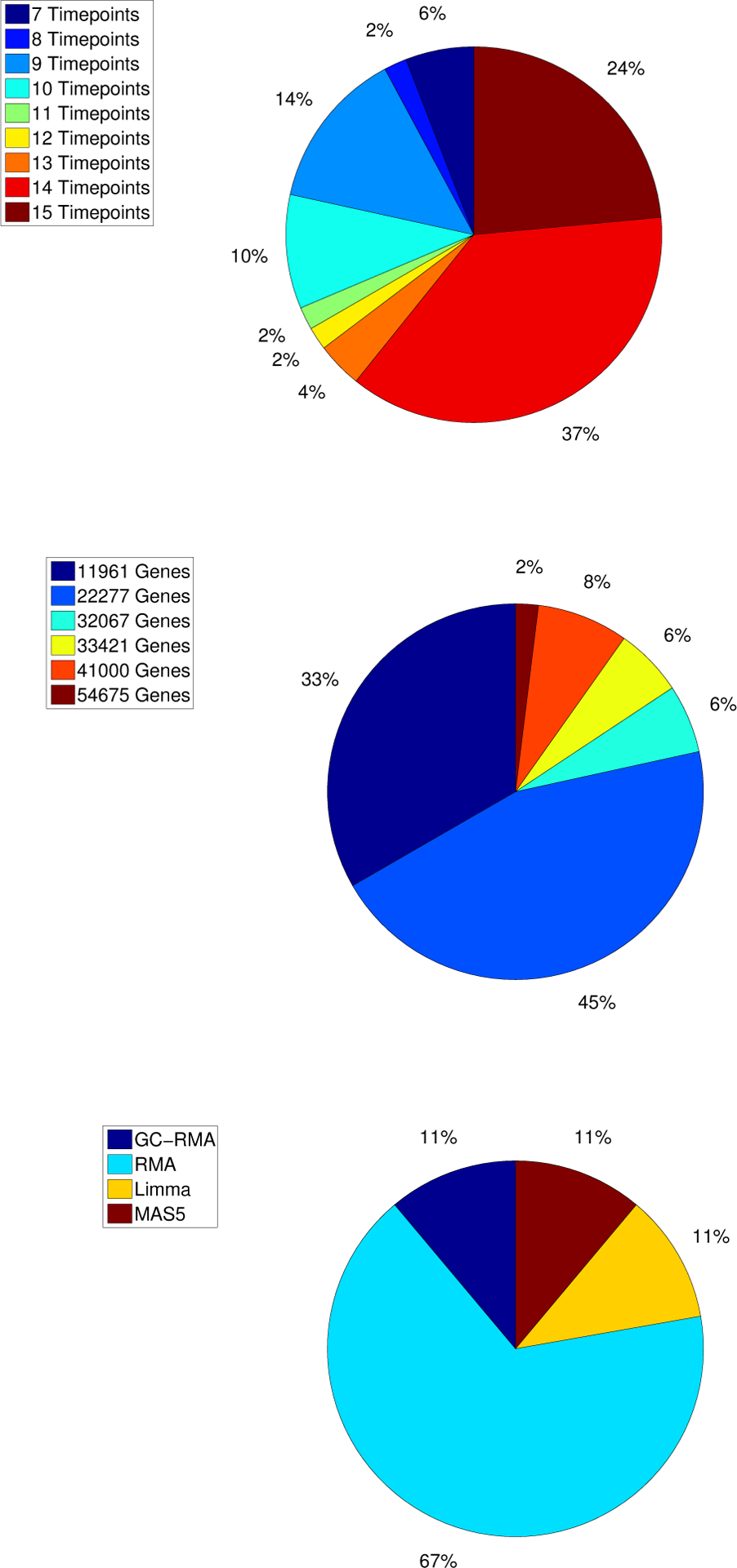
Distribution of the number of time-points and the number of genes/probes
Dynamic Response Genes for Influenza
The F-test statistic in Step 3 of the pipeline was used to identify the top 3, 000 dynamic response genes (DRGs) for each of the 58 datasets. We cut the number of DRGs nDRG = 3, 000 for all experimental conditions to avoid introducing bias due to different sample sizes for different conditions. Alternatively, one could select the number of significant DRGs for each experimental condition using a p-value (with a multiple testing adjustment), but as the number of significant DRGs can vary considerably for different conditions, it can be difficult to draw reliable comparisons between groups of experimental conditions. After careful consideration (e.g., visual inspection of the DRGs for each of the 58 datasets), we felt that 3000 was an appropriate threshold for the number of DRGs. The DRGs are genes that show a strong reaction over time after inoculation with the various virus strands. These genes might translate into clinically valuable biomarkers. The annotation of the DRGs is analysed at both virus subtype and population level in the following subsections.
Virus Subtype-Level Results:
Table 5 provides the annotation for the most significant DRG’s for each of the 19 virus subtypes based on the ranking of the F-test statistic in Step 3 of our pipeline. Many of these genes have been previously identified as been significantly related to influenza infection such as, 2’-5’-oligoadenylate synthetase-like, is part of a system activating RNase L, which is an important unspecific antiviral immune response and is also thought to play a role in the control of cell growth and differentiation (67); DnaJ (Hsp40) homolog, has been implicated in positive regulation of virus replication through co-option by influenza A virus (68); glucan (1,4-alpha-), is related to anti-infectious immunity (69) and N-acyl phosphatidylethanolamine phospholipase D, is an endogenous lipid that modulates pain and inflammation (70).
Table 5.
The most significant DRGs for each of the 19 virus subtypes.
| GSE number | Virus Strand | Most Significant DRG’s |
|---|---|---|
| GSE52428 | H1N1/Brisbane, H3N2/Wisconsin | 2’-5’-oligoadenylate synthetase-like, KH domain containing, RNA binding, signal transduction associated 3 |
| GSE12254 | WE (LCMV-WE) | CHK1 checkpoint homolog, RAD58 homolog (RecA homolog, E. coli), RAD52 homolog |
| GSE19392 | delNS1 post trypsin | DnaJ (Hsp40) homolog, subfamily A, member 1, cyclin A1, inhibitor of DNA binding 2, dominant negative helix-loop-helix protein |
| GSE19392 | PR8 post trypsin | glucan (1,4-alpha-), branching enzyme 1, matrix metallopeptidase 1 (interstitial collagenase), ornithine decarboxylase 1 |
| GSE19392 | vRNA LTX+RNA | STAM binding protein, cysteine and glycine-rich protein 2, polymerase (RNA) III (DNA directed) polypeptide K |
| GSE19392 | treated with media alone | bone marrow stromal cell antigen 2, interferon stimulated exonuclease gene, proteasome (prosome, macropain) subunit |
| GSE19392 | treated with IFNb IFN | N-myc (and STAT) interactor, PHD finger protein 11, interferon-induced protein 44-like, tripartite motif-containing 22 |
| GSE19392 | treated with LTX transfection reagent | interferon induced transmembrane protein 1, interferon stimulated exonuclease gene, radical S-adenosyl methionine domain containing 2 |
| GSE37571 | A/CA/04/2009 | CCAAT/enhancer binding protein (C/EBP), complement factor B, PCSK9, TUBB8, kinesin family member 4A |
| GSE37571 | Mock | major histocompatibility complex, class I, E, complement factor B, major histocompatibility complex, class I, A, HLA-G |
| GSE30550 | H3N2/Wisconsin | Anaphase-promoting complex subunit MND2, Bud site selection protein 22, C-8 sterol isomerase, Vacuolar-sorting protein SNF7 |
| GSE40844 | Mock | tetraspanin 8, kelch-like 24 (Drosophila), hect domain and RLD 5, DEAD (Asp-Glu-Ala-Asp) box polypeptide 60 |
| GSE41067 | H1N1 influenza A/New Caledonia/20/1999 | HLA complex P5, DEXH (Asp-Glu-X-His) box polypeptide 58, interferon, alpha-inducible protein 6 |
| GSE47960 | dORF6 | XIAP associated factor 1, interferon-induced protein 44-like, interferon induced transmembrane protein 1 (9–27) |
| GSE47960 | iSARs | trafficking protein, kinesin binding 1, RAD23 homolog B (S. cerevisiae), chemokine (C-X-C motif) ligand 10 |
| GSE47960 | Mock | tribbles homolog 1 (Drosophila), tumor necrosis factor, alpha-induced protein 2, PR domain containing 1, with ZNF domain |
| GSE47961 | BAT | eyes absent homolog 3, BAI1-associated protein 2, cardiotrophin-like cytokine factor 1, endothelin 2 |
| GSE47961 | dORF6 | N-acyl phosphatidylethanolamine phospholipase D, LIM and senescent cell antigen-like domains 3, zinc finger protein 564 |
| GSE47961 | H1N1 | interferon, alpha-inducible protein 27, nterferon induced transmembrane protein 4 pseudogene, MHC class I polypeptide-related seq B |
Population-Level Results:
As the 58 data sets have a broad biological and technical heterogeneity, we do not expect that the annotation for the 3000 DRGs identified for each data set will contain genes that are common among all 58 data sets. Interestingly, there are 19 data sets out of the 58 that all contain the following three genes in the annotation of the DRG lists: TRADD* which is an apothic pathway activated by TNF and linked to the H1N1 virus (71), ELMO2 which is involved in cytoskeletal rearrangements required for phagocytosis of apoptotic cells and cell motility, and STAT1† which has been shown to be required for ISG induction and resistance to influenza infection (72). As 19 out of the 58 data sets (32% of the population) have identified these genes as having a significant response over time, we can regard them as potential bio-markers for influenza infection response at a population level. Additionally, we found that 17 out of the 58 data sets shared 15 common DRGs (fbx11, Fau, APEH, nadK, Necap1, nsfl1c, RAB6C, YCR015C, CTSS, Cdc25b, COL2A1, CRYAB, SERPING1, RTEL1, POLR2E) that are related to the regulation of cell death, proteins secreted into the cell surroundings and protein binding. Table 6 provides the official gene symbols and the functions of these DRGs. As these represent 29% of the population, they can also be regarded as potential bio-markers for influenza infection response at a population level.
Table 6.
The identified DRGs that are common across 17 or more data sets
| Gene Symbol | Function |
|---|---|
| fbx11 | Substrate recognition component of the a (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complex which mediates the ubiquitination and subsequent proteasomal degradation of target proteins |
| Fau | This protein is synthesized with ribosomal S30 as its C-terminal extension. |
| APEH | This enzyme catalyzes the hydrolysis of the N-terminal peptide bond of an N-acetylated peptide to generate an N-acetylated amino acid and a peptide with a free N-terminus. |
| nadK | Belongs to the NAD kinase family |
| Necap1 | Involved in endocytosis. |
| nsfl1c | Reduces the ATPase activity of VCP. Necessary for the fragmentation of Golgi stacks during mitosis and for VCP-mediated reassembly of Golgi stacks after mitosis. |
| RAB6C | Protein transport. Regulator of membrane traffic from the Golgi apparatus towards the endoplasmic reticulum (ER). |
| YCR015C | Belongs to the UPF0655 family. |
| CTSS | Thiol protease. |
| Cdc25b | Tyrosine protein phosphatase which functions as a dosage-dependent inducer of mitotic progression. |
| COL2A1 | Type II collagen is specific for cartilaginous tissues. It is essential for the normal embryonic development of the skeleton, for linear growth and for the ability of cartilage to resist compressive forces. |
| CRYAB | May contribute to the transparency and refractive index of the lens., mass spectrometry |
| SERPING1 | Activation of the C1 complex is under control of the C1-inhibitor. It forms a proteolytically inactive stoichiometric complex with the C1r or C1s proteases. May play a potentially crucial role in regulating important physiological pathways including complement activation, blood coagulation, fibrinolysis and the generation of kinins. |
| RTEL1 | ATP-dependent DNA helicase required to sup-press inappropriate homologous recombination, thereby playing a central role DNA repair and in the maintenance of genomic stability |
| POLR2E | DNA-dependent RNA polymerase catalyzes the transcription of DNA into RNA using the four ribonucleoside triphosphates as substrates. |
Additionally, the annotation in step 5 provides the functional enrichment of the DRGs for all 58 data sets. The most enriched pathways are: Spliceosome (hsa03040), it is well known that influenza viruses have developed accurate regulation mechanisms to utilize the host spliceosome to enable the expression of specific spliced influenza virus products throughout infection (73); Proteasome (hsa03050) which has been shown to effectively block the influenza virus (74); p53 signaling pathway (hsa04115), the influenza virus infection is known to increases p53 activity (75); and other classical pathways such as cell cycle (hsa04110) and Ribosome (hsa03010) protein synthesis.
Gene Response Modules for Influenza
We use the Pipeline4DGEData Step 4, i.e., the IHC method with a correlation threshold of α = 0.7 to group the 3,000 DRGs for each of the 58 data sets, into between 15 and 476 temporal gene response modules (GRMs).
Virus Subtype-Level Results:
Table 7 shows the number of GRMs for a single data set or the range of the number of GRMs for multiple data sets, for each of the 19 virus subtypes. It also provides a brief description of the time-course patterns of the GRMs. For most virus subtypes, the time-course patterns of the GRMs have either a distinctive up/down regulated feature or multiple features denoting peaks and troughs at various time-points.
Table 7.
GRMs from different studies.
| GSE number | Virus Strand | Number of GRMs | Time Course Pattern |
|---|---|---|---|
| GSE19392 | delNS1 post trypsin | 57 | Two dominating features up/down regulation |
| GSE19392 | PR8 post trypsin | 63 | Two dominating features up/down regulation |
| GSE19392 | vRNA LTX+RNA | 36 | Two dominating features up/down regulation |
| GSE12254 | WE (LCMV-WE) | 57 | Two dominating features up/down regulation |
| GSE37571 | Mock | 18 | Two dominating features up/down regulation |
| GSE40844 | Mock | 17 | Two dominating features up/down regulation |
| GSE41067 | H1N1 influenza A/New Caledonia/20/1999 | 17 | Two dominating features up/down regulation |
| GSE47960 | dORF6 | 47 | Two dominating features up/down regulation |
| GSE47960 | Mock | 83 | Two dominating features up/down regulation |
| GSE47961 | H1N1 | 19 | Two dominating features up/down regulation |
| GSE19392 | treated with media alone | 53 | A few features all denoting peaks and troughs at different timings |
| GSE19392 | treated with LTX transfection reagent | 34 | A few features all denoting peaks and troughs at different timings |
| GSE52428 | H1N1/Brisbane, H3N2/Wisconsin | 200–467 | Many features all denoting peaks and troughs at different timings |
| GSE19392 | treated with IFNb IFN | 56 | Many features all denoting peaks and troughs at different timings |
| GSE37571 | A/CA/04/2009 | 15 | Many features all denoting peaks and troughs at different timings |
| GSE30550 | H3N2/Wisconsin | 291–400 | Many features all denoting peaks and troughs at different timings |
| GSE47960 | iSARs | 75 | Many features all denoting peaks and troughs at different timings |
| GSE47961 | BAT | 36 | Many features all denoting peaks and troughs at different timings |
| GSE47961 | dORF6 | 32 | Many features all denoting peaks and troughs at different timings |
Population-Level Results:
Figure 4 shows the time-course patterns from two different datasets. One with the distinctive up/down regulated time-course patterns (experimental condition Mock from GSE37571) and one with time course patterns that denote peaks and troughs at various time-points (experimental conditions BAT from GSE47961). In this manner, Figure 4 illustrates the different time-course patterns identified in Table 7. These preliminary results indicate that there are two categories of gene response modules for influenza infection. The first is composed of modules of approximately the same size containing genes that have different time course behaviors. The second is composed of modules of a greater size containing genes that have a similar behavior over time (dominating up/down regulated feature).
Figure 4.
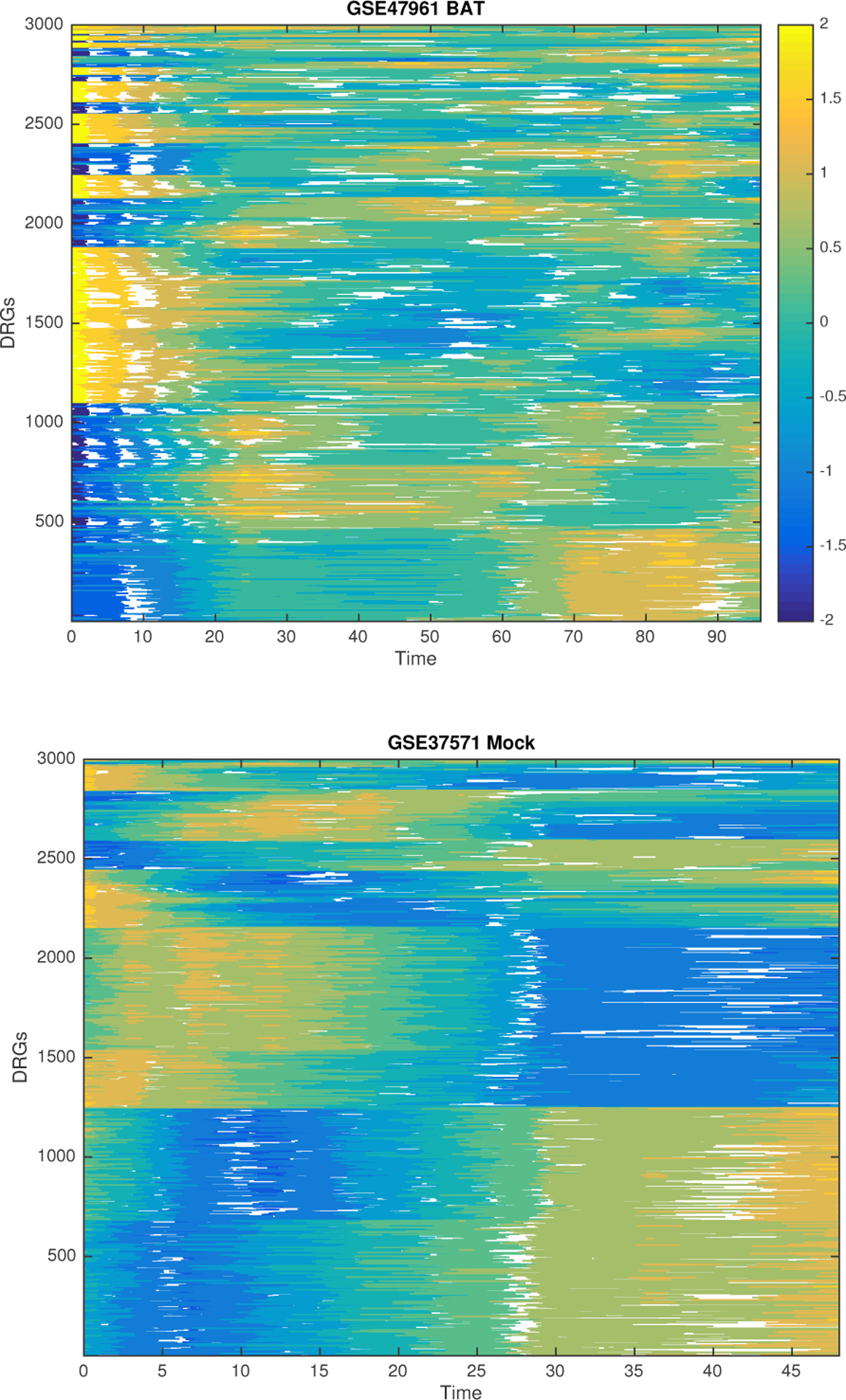
Typical Time-Course Patterns of the GRMs
Gene Regulatory Networks for Influenza
Steps 6 and 7 of our pipeline are applied to the GRMs attained from each data set contained in the 9 influenza-related studies, the gene response networks for these data sets are established and their network features are analyzed. In this section, we analyze the network features of the 58 gene regulatory networks. The nodes of these networks are the GRMs, and the edges are the regulation effects β. The results are summarized as follows.
Virus Subtype-Level Results:
Here we use the network centrality measures to compare the features of the GRNs for each of the 19 virus subtypes. Figure 5 illustrates the virus subtypes with GRNs that have relatively large centrality measures by showing the standardized clustering coefficient, density, bridging centrality and closeness centrality of the GRNs grouped by their virus subtype. For virus subtypes with multiple GRNs, we obtain the average of the metric for all experimental conditions pertaining to that particular virus subtype.
Figure 5.
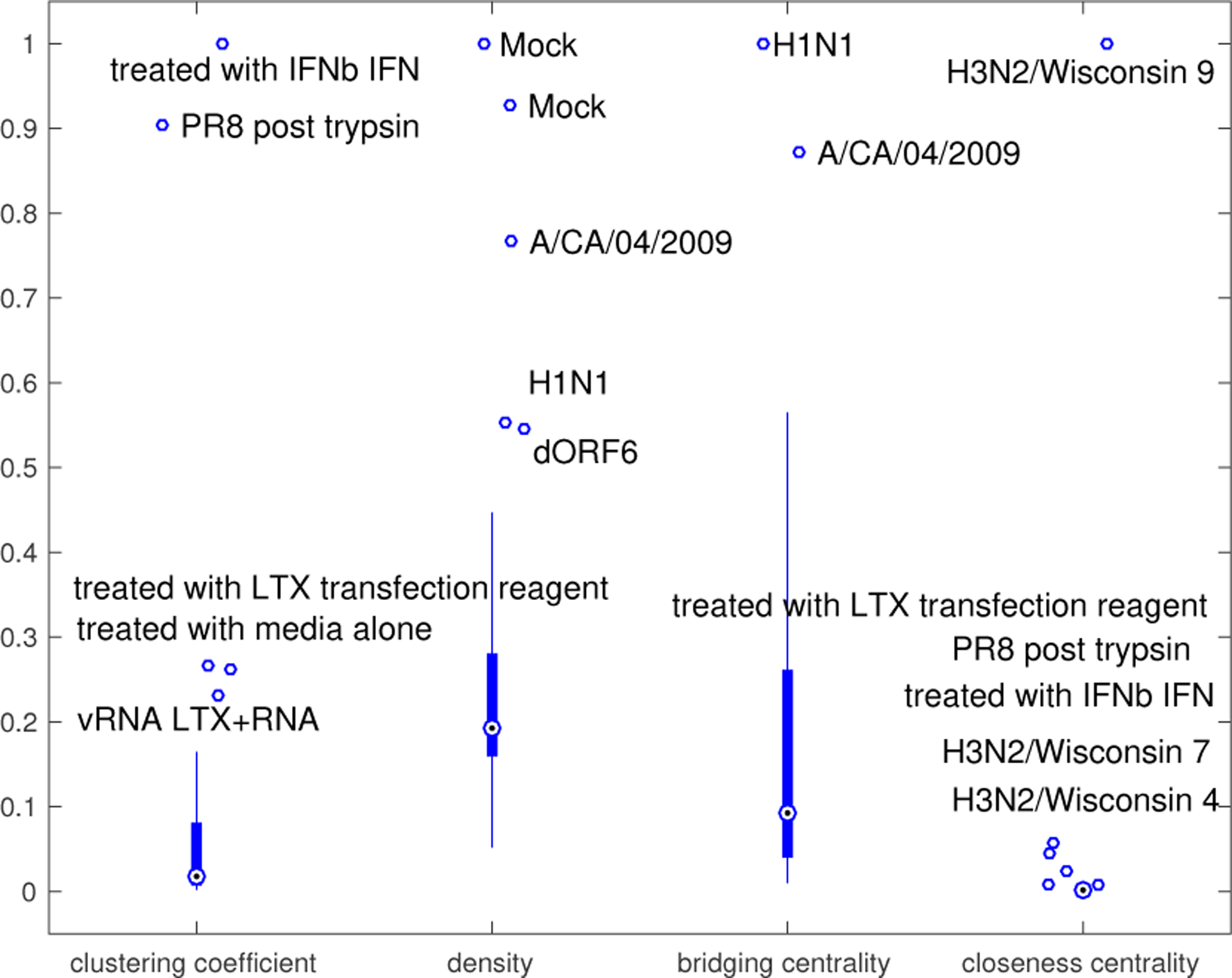
The standardized clustering coefficient, density, bridging centrality and closeness centrality of the GRNs for the 19 virus subtypes.
The virus subtypes with relatively high clustering coefficients are: In the GSE19392 study, cells treated with IFNb, treated with media alone, treated with LTX transfection reagent, vRNA LTX+RNA, and PR8 post trypsin. This suggests that there is a strong interconnectivity between the modules in the GRNs for these studies. Figure 6 illustrates the GRN for the cells treated with interferon beta (IFNb), which has a relatively high clustering coefficient (top), and the GRN for the cells infected with PR8 virus lacking the NS1 gene (DNS1), which has a relatively low clustering coefficient (bottom). The size of the nodes indicates the number of genes contained in the modules, (i.e., the larger the node, the greater the number of genes contained in that module). It is clear that both GRNs only have two very large modules containing many genes and that these modules are not closely related to each other as they have a large distance between one another. The GRN for the cells treated with interferon beta (IFNb) has a closer-knit community of modules than the cells infected with the PR8 virus lacking the NS1 gene (DNS1), which has a dispersed community of modules.
Figure 6.
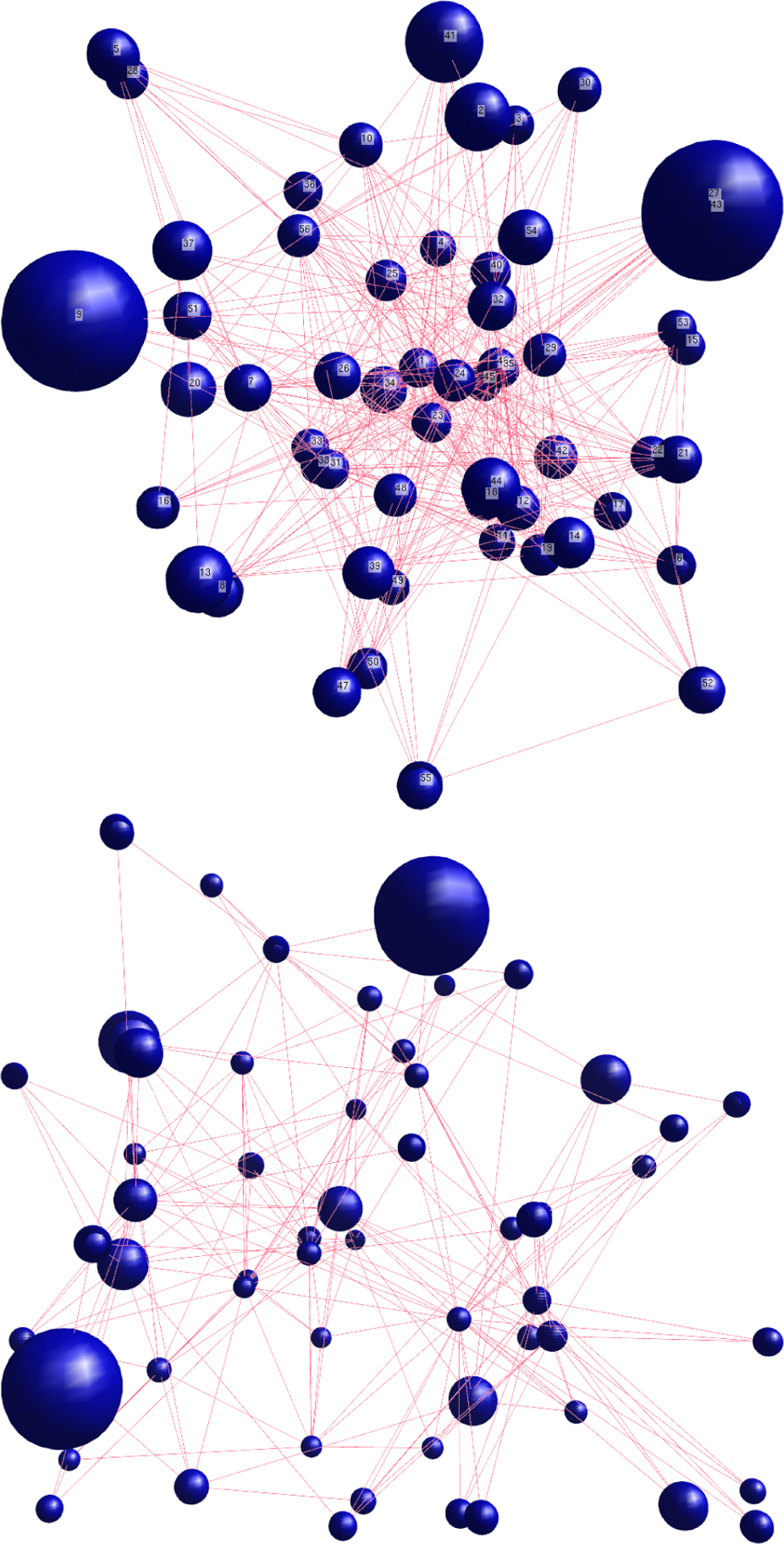
The GRNs for two experimental conditions in the study of GSE19392: cells treated with IFNb (top) and PR8 virus lacking the NS1 gene (bottom)
The studies with relatively high densities are GSE37572 Mock, GSE40844 Mock, GSE37571 A/CA/04/2009, GSE47961 H1N1 and dORF6. As Table 2 describes, this metric provides a measure of the general interdependence between all nodes in the network. GRNs with higher densities are those where the GRMs tend to be closely dependent on each other. In such networks, the expression of each module tends to be affected by many or all of the other modules. Figure 7 illustrates the GRN for GSE37571 A/CA/04/2009, which has a relatively high density (top), and the GRN for GSE52428 H3N2 Subject 9, which has a relatively low density (bottom). The GRN for GSE37571 A/CA/04/2009 contains a few GRMs, but these modules are highly connected to one another. While the GRN for GSE52428 H3N2 Subject 9 has many GRMs, but each module only regulates a relatively small proportion of the other modules.
Figure 7.
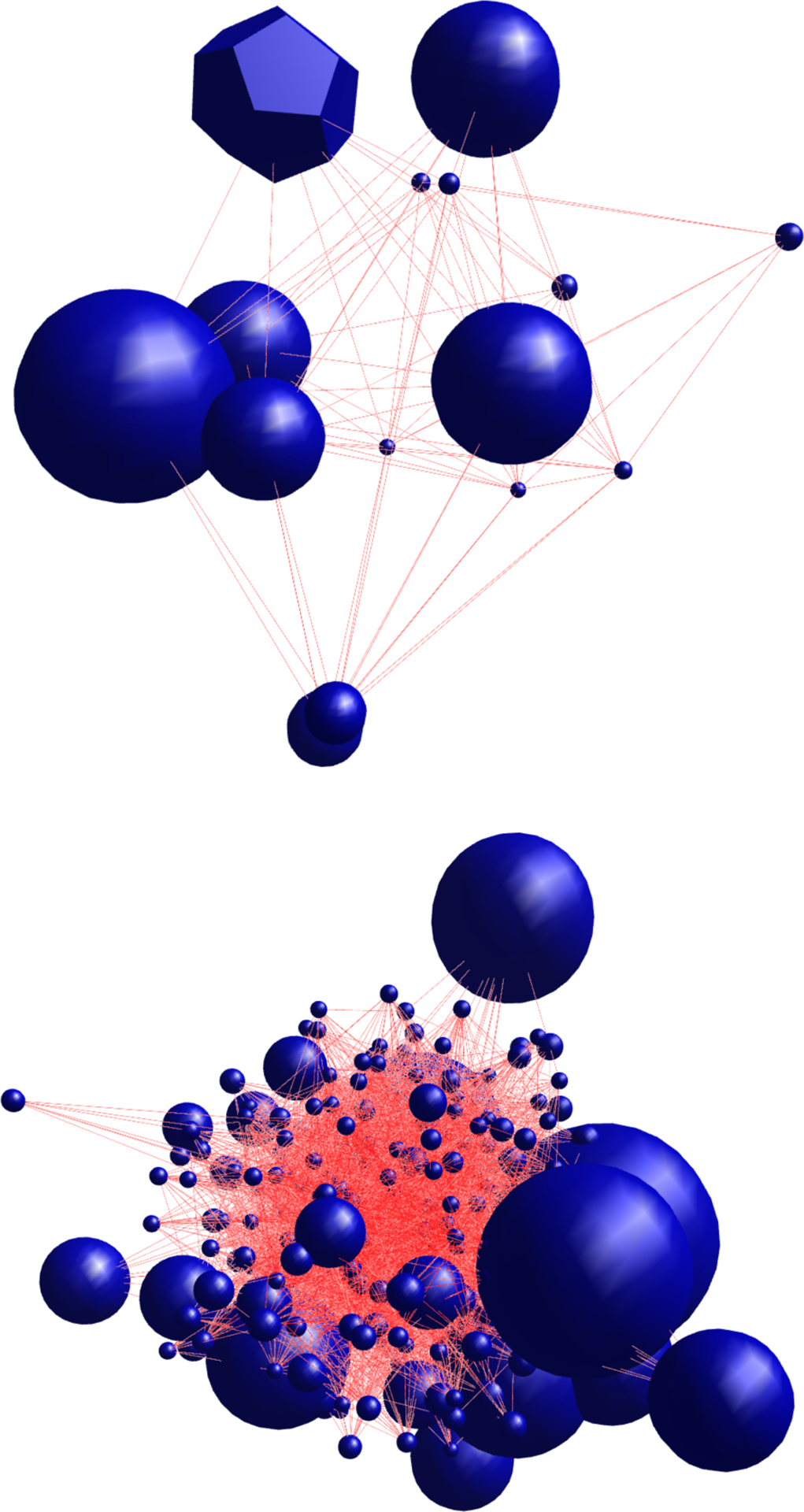
The GRNs for GSE37571 A/CA/04/2009 (top) and GSE52428 H3N2 Subject 9 (bottom)
The studies with relatively high bridging centralities are GSE19392 treated with LTX transfection reagent, GSE37572 A/CA/04/2009, and GSE47961 H1N1. This suggests that these studies have modules that regulate highly-connected groups of GRMs. Figure 6 shows the GRN for GSE37571 A/CA/04/2009. The GRM that is shaped like a dodecahedron is the module with the largest bridging centrality, from Figure 6, it is clear that this module connects different groups of highly connected GRMs. We analyzed the functional enrichment of the DRGs in this GRM. The most enriched pathways are Pathogenic Escherichia coli infection (hsa05830), Gap junction (hsa04540) and Phagosome (hsa04666), in particular, these all reference the tubulin protein superfamily as being significantly related to these DRGs. This supports recent research showing that influenza A virus induces the disruption of the microtubule network β with and α-tubulin (76; 77)
The studies with relatively high closeness centralities are GSE52428 H3N2/Wisconsin Subject 7, 4 and 9, GSE19392 study, cells treated with IFNb, treated with LTX transfection reagent, and PR8 post trypsin. Figure 7 shows the GRNs for GSE37571 A/CA/04/2009 and GSE52428 H3N2/Wisconsin Subject 9. The GRN for GSE37571 A/CA/04/2009 has a relatively low closeness centrality metric with a few modules that are relatively far apart or dispersed, while the GRN for GSE52428 H3N2/Wisconsin Subject 9 has a high closeness centrality metric with many of the modules in close proximity to one another.
Population-Level Results:
Clustering is an important property of any network as it measures the interconnectivity of the network. Our results suggest that a lower clustering coefficient may indicate an expanded response to the influenza infection. The evidence to support this claim comes from 22 data sets in the GSE19392 and GSE30550 studies which account for 43% of the population. The data sets in GSE19392 concern cells infected by the influenza virus but under five different conditions, each highlighting a distinct aspect of the response: i) cells were infected with the wild-type PR8 influenza virus that can mount a complete replicative cycle (PR8 post trypsin); ii) cells were transfected with viral RNA isolated from influenza particles. This does not result in the production of viral proteins or particles (vRNA LTX+RNA); iii) cells were treated with interferon beta, to distinguish the portion of the response which is mediated through Type I IFNs (treated with IFNb IFN); iv) cells were infected with a PR8 virus lacking the NS1 gene (delNS1 post trypsin); v) cells were treated with media alone as a control. All the conditions except for condition (iv) have an unusually high clustering coefficient (this is also true for diameter and mean distance, two alternative measures of inter-connectivity). The NS1 protein normally inhibits vRNA- or IFNb-induced pathways, and its deletion can reveal an expanded response to infection (61). We suspect that a lower clustering coefficient is an indication of an increase in a host response to influenza infection. This is further supported by the data sets in the GSE30550 study. These data sets are divided into two groups according to the clinical symptom chart based on the standardized symptom scoring (78): symptomatic (Sx) group with nine subjects (subjects 1,5,6,7,8,10,12,13,15) and asymptomatic (Asx) group with eight subjects (subjects 2,3,4,9,11,14,16,17). Interestingly, the majority of the Asx group (subjects 2,3,4,9,11,14,16), which we would expect to have a high response to influenza infection as they are not experiencing symptoms, have the lowest average clustering coefficients among all the studies.
In summary, we have briefly discussed some of the results from our analysis of several virus infection studies using Pipeline4DGEData. The primary goal of this article is to propose the methodology for the Pipeline4DGEData, a tool for the construction of gene regulatory networks from large amounts of high-dimensional time course gene expression data. Here we demonstrate the pipeline’s ability to identify potential bio-markers for influenza infection and to discern interesting characteristics of the dynamic interaction process between the host and influenza virus. More detailed biological findings from these viral infection studies will be reported elsewhere in the near future.
The Pipeline for Dynamic Gene Expression Data Applied to Simulated Data Sets
The accuracy of the results provided by Pipeline4DGEData was evaluated by examining its performance on simulated expression data. Simulation 1 is derived from previously discovered GRMs, i.e., the estimated GRMs were used to simulate the underlying time course data. The pipeline analysis was run on the simulated data in order to confirm if the new results are consistent with the original results. Simulation 2 adds various levels of random noise to the the time course gene expression data set GSE52428, subject 1. The pipeline analysis was run on the simulated data in order to confirm if the new results are consistent with the original results.
Simulation 1
The simulated data was obtained as follows. Let ci be the average standardized expression level over time of the genes in the ith cluster attained from Step 4 of the Pipeline. The simulated time course data for the jth dynamic response gene in the ith cluster can be generated as follows:
for j = 1, …, Qi, where Qi is the number of genes in the ith cluster, also provided by the step 4, ϵi,j(t) is normally distributed random variable with mean 0 and variance 1 whereas σ quantifies the magnitude of the measurement error. This produces simulated time course measurements for the 3000 DRGs. The remaining non-responsive genes (NRG) were generated by
The simulated data sets for the DRGs and the non-responsive genes are combined to form the test data set. The simulated data were generated for three measurement error levels: σ = 0.1, σ = 0.2 and σ = 0.3 and each simulation was performed 1000 times.
The method described above was used to simulate time course data based on the pipeline results from the data set GSE52428, subject 1. This data set contains time course measurements for 22, 277 genes. Step 3 of the pipeline identifies 3000 of these genes as DRGs and the remaining 19, 277 genes are considered non-responsive genes (NRGs). Step 4 subsequently groups these DRGs into 128 clusters. Using the method described above, simulated expression data of the 22, 277 genes were obtained from the 128 modules. The results obtained from the application of the pipeline to this simulated data is described as follows.
Dynamic Response Genes:
The F-test statistic in Step 3 of the pipeline was used to identify the top 3,000 dynamic response genes (DRGs) for each of the simulated datasets. We computed a binary vector that is one if the gene has been selected as a DRG and zero otherwise. Then we computed the difference between this binary vector and a binary vector corresponding to the true DRG assignment. If the difference is negative then the gene has been identified as DRGs but in fact is a non-responsive gene (false positive) and if its is positive then the gene has been identified as a non-responsive gene but is in fact a DRG (false positive). Table 8 shows the average percentage of false positives and percentage of false negatives. As is evidenced in Table (8) Step 3 provides an accurate identification of the top 3,000 dynamic response genes with an error which ranges from 3.74% for σ = 0.1 to 5.24% for σ = 0.3.
Table 8.
Accuracy of the DRG Identification
| Error Level | False Positives | False Negatives |
|---|---|---|
| Simulation 1 | ||
| 0.10 | 1.87% | 1.87% |
| 0.20 | 2.19% | 2.19% |
| 0.30 | 2.62% | 2.62% |
| Simulation 2 | ||
| 0.10 | 1.00% | 1.00% |
| 0.20 | 2.05% | 2.05% |
| 0.30 | 2.84% | 2.84% |
Gene Response Modules:
The IHC clustering method in Step 4 of the pipeline was used to group the top 3,000 DRGs into gene response modules for each of the simulated datasets. The adjusted rand index (ARI) (79) provides an overall similarity measure between two clustering assignments taking into account that the agreement between partitions could arise by chance alone. Thus, the ARI provides a measure of the similarity of two clustering methods. This index has an expected value of zero for independent clusterings and maximum value 1 for identical clusterings. Table 9 shows the adjusted rand index for the true cluster assignment and the cluster assignment identified by Step 4 of the pipeline. Overall, step 4 provides clusters that are very similar to the true cluster assignment with an similarity which ranges from 82% for σ = 0.1 to 75% for σ = 0.3.
Table 9.
Accuracy of the GRM identification.
| Error Level | Adjusted rand index (ARI) |
|---|---|
| Simulation 1 | |
| 0.10 | 0.82 |
| 0.20 | 0.79 |
| 0.30 | 0.75 |
| Simulation 2 | |
| 0.10 | 0.90 |
| 0.20 | 0.81 |
| 0.30 | 0.74 |
Gene Response Network:
Step 6 attains the gene response networks for each simulation data set. As these networks are likely to have differing number of nodes as different simulations can generate a different number of clusters, we cannot compare these networks directly. Instead we compare the edge weights in terms of the DRGs as opposed to the clusters by computing a 3000 × 3000 matrix which assigns the edge of each cluster to each of the DRGs in that cluster. Table 9 uses one minus the Hamming distance to compare the percentage of coordinates (i.e., nodes) that are the same. Step 6 provides networks that are very similar to the true networks with a similarity which ranges from 79% for σ = 0.1 to 73% for σ = 0.3.
Simulation 2
The simulated data was obtained as follows. Let η be a matrix size 22, 277 × 15 containing the time course gene expression data set GSE52428, subject 1. The simulated time course data can be generated as follows:
for j = 1, …, 22, 277, indexing each gene and i = 1, …, 15, indexing each time point, ϵj(ti) is normally distributed random variable with mean 0 and variance 1 whereas σj quantifies the magnitude of the measurement error. The simulated data were generated for three measurement error levels: σj = 0.1κj, σj = 0.2κj and σj = 0.3κj, where κj represents the sample standard deviation of the jth gene and each simulation was performed 1000 times. The results obtained from the application of the pipeline to this simulated data is described as follows.
Dynamic Response Genes:
The F-test statistic in Step 3 of the pipeline was used to identify the top 3,000 dynamic response genes (DRGs) for each of the simulated datasets. Table 8 shows the average percentage of genes that were identified as DRGs but were in fact non-responsive genes (false positives) and average percentage of genes that were identified as non-responsive genes but were in fact DRGs (false negatives). As is evidenced in Table (8) Step 3 provides an accurate identification of the top 3,000 dynamic response genes with an error which ranges from 2% for σ = 0.1 to 5.68% for σ = 0.3.
Gene Response Modules:
The IHC clustering method in Step 4 of the pipeline was used to group the top 3,000 DRGs into gene response modules for each of the simulated datasets. The ARI provides a measure of the similarity of two clustering methods. This index has an expected value of zero for independent clusterings and maximum value 1 for identical clusterings. Table 9 shows the adjusted rand index for the true cluster assignment and the cluster assignment identified by Step 4 of the pipeline. Overall, step 4 provides clusters that are very similar to the true cluster assignment with an similarity which ranges from 90% for σ = 0.1 to 74% for σ = 0.3.
Gene Response Network:
Step 6 attains the gene response networks for each simulation data set. Table 9 uses one minus the Hamming distance to compare the percentage of coordinates (i.e., nodes) that are the same. Step 6 provides networks that are very similar to the true networks with a similarity which ranges from 81% for σ = 0.1 to 80% for σ = 0.3.
These results indicate that there is consistency in the output returned by the pipeline. In other words, given a set of genomic time course data with underlying behavioural patterns shared by groups of dynamically-responsive genes, the pipeline is able to identify these groups of genes as DRGs, cluster them into GRMs based on their expression patterns and determine the GRN that describes the interactions between these modules. This consistency is manifest in the objective correspondence between the results (DRGs, GRMs, and GRN) obtained from the original data and those obtained from the simulated data sets. Table 8 shows that rates of false positives and negatives between DRGs from real and simulated are low. In other words, if a group of genes within the dataset are dynamically responsive, then the pipeline will correctly class them, on average, as DRGs. Similarly, the high rand indices (close to 1) observed when comparing real and simulated modules, and displayed in Table 9, show that the clustering method used by the pipeline performs well. Finally, correspondence was observed between the GRNs from real and simulated data, as shown in Table 11. The coherence between the results from real and simulated data serves as strong evidence that the output returned by the pipeline is truly representative of the dynamic response patterns hidden in a large data set, such as those provided by the GEO.
Table 11.
Sensitivity of the GRN Identification
| Step 3 | |||
| SS vrs SS | FPCA vrs FPCA | SS vrs FPCA | |
| % of DRGs that differ | |||
| nDRG : 2000 vs 3000 | 4.48% | 4.48% | 11.76% |
| nDRG : 4000 vs 3000 | 4.48% | 4.48% | 15.76% |
| nDRG : 5000 vs 3000 | 8.97% | 8.97% | 18.59% |
| Step 3 | |||
| Adjusted Rand Index (ARI) | |||
| α : 0.60 vs 0.7 | 0.99 | 0.99 | 0.39 |
| α : 0.80 vs 0.7 | 0.99 | 0.99 | 0.39 |
| α : 0.90 vs 0.7 | 0.99 | 0.99 | 0.39 |
| Step 6 | |||
| LASSO | |||
| γ : AIC vs GCV | 0.87 | 0.64 | 0.90 |
| γ : BIC vs GCV | 0.88 | 0.77 | 0.91 |
| γ : AIC vs BIC | 0.87 | 0.64 | 0.90 |
| SCAD | |||
| γ : AIC vs GCV | 0.87 | 0.64 | 0.87 |
| γ : BIC vs GCV | 0.87 | 0.77 | 0.88 |
| Y : AIC vs BIC | 0.88 | 0.64 | 0.87 |
| LASSO vs SCAD | |||
| γ = GCV | 0.87 | 0.35 | |
The Pipeline for Dynamic Gene Expression Data Sensitivity Analysis
Various steps of the Pipeline for Dynamic Gene Expression Data have tuning parameters that must be provided by the user or set to default for instance: step 3 either uses spline smoothing (default) or functional principal component analysis to estimate the trajectory of the genes and subsequently selects a fixed number of dynamic response genes, nDRG (default is 3000); step 4 selects the average within cluster correlations and the higher bound for the within cluster corelations (default α = 0.7); step 6 uses either LASSO (default) or SCAD to do variable selection and selects the corresponding regulation parameter γ using either GCV, AIC or BIC (default approach selects γ by minimizing GCV). In this section, we examine the sensitivity of the results to each of these parameters.
Dynamic Response Genes:
The F-test statistic in Step 3 of the pipeline was used to identify the dynamic response genes (DRGs) for the time course gene expression data set GSE52428, subject 1. We compare three different numbers of the top ranking dynamic response genes nDRG set to 2000, 4000 or 5000 to the default for that approach namely, nDRG = 3000 for both spline smoothing (SS) and functional principal components analysis (FPCA). We also compare the difference between the different approaches SS vrs FPCA for nDRG set to 2000, 4000 or 5000. Table 8 shows the sum of the percentage of genes that were identified as DRGs but were in fact non-responsive genes (false positives) and the percentage of genes that were identified as non-responsive genes but were in fact DRGs (false negatives) for each scenario. As is evidenced in Table (8) Step 3 provides an stable identification of the top dynamic response genes with a variation which ranges from 4.48% for nDRG = 2000 to 8.97% for nDRG = 5000 for SS and FPCA. As expected there are some differences between the DRGS identified by the SS and FPCA approaches with an agreement regarding DRG selection of 90.40% for nDRG = 2000 and 79.66% for nDRG = 5000.
Gene Response Modules:
The IHC clustering method in Step 4 of the pipeline was used to group the top 3,000 DRGs into gene response modules for the time course gene expression data set GSE52428, subject 1. Table 9 shows the adjusted rand index for the cluster assignment identified by Step 4 of the pipeline with α = 0.7 and the cluster assignment identified by Step 4 of the pipeline with α set to 0.6, 0.8 or 0.9 for both the SS and FPCA approaches. Overall, step 4 provides clusters that are robust to the selection of the parameter α as the ARI of the clusters is 99% for all levels of α for both the SS and FPCA approaches. As expected theres is not much similarity between clusters identified by the SS and FPCA approches with an agreement regarding cluster assignment of 39% for all levels of α.. The FPCA approach borrows information across genes to attain an estimate the trajectories of the genes, thus the estimated trajectories will be more similar to one another for this approach than SS approach where the trajectories for each gene are attained from smoothing the observations from each gene individually. In light of this the FPCA is likely to produce less clusters containing more genes.
Gene Response Network:
Step 6 attains the gene response networks for each simulation data set. Here we use one minus the Hamming distance to compare the percentage of coordinates (i.e., nodes) that are the same. Table 9 shows the percentage of common coordinates for the various approaches for selecting the regulation parameter γ, namely, GCV, AIC and BIC. For the SS approach Step 6 provides networks that are robust to the technique used to select the regulation γ parameter with an average agreement of 0.87. However, for the FPCA approach there is a considerable variability in the variable selection for various values of γ with an average agreement of 0.68. We also compare the two proposed approaches LASSO vs SCAD for the SS and FPCA. For the SS approach Step 6 provides networks that are robust to the model selection technique used with an average agreement of 0.87. However, for the FPCA approach there is a considerable variability in networks wit respect to the model selection technique used with an average agreement of 0.35.
Conclusions
In this article, we propose the Pipeline4DGEData for the statistical modeling and analysis of the time course gene expression data sets that are available on GEO. Pipeline4DGEData identifies dynamic gene regulatory networks that determine the host response to an external stimulus or disease. This pipeline incorporates the following eight steps: (i) obtain the data from GEO; (ii) pre-process the data; (iii) detect the dynamic response genes; (iv) cluster the dynamic response genes into gene response modules; (v) annotate these gene response modules; (vi) construct a gene regulatory network that describes the interactions between these gene response modules using differential equations; (vii) perform a network feature analysis to identify influential GRMs and to characterize various properties of the network and (viii) output the results in a manner that make the publication process more efficient, i.e., provide a manuscript file for a biological journal that contains all tables and figures automatically generated by the pipeline and a template for the descriptions of the studies analyzed and methodologies implemented. Furthermore, the pipeline has a consistent and scalable structure that facilitates comparative analysis across large groups of data sets.
We demonstrate the utility of the Pipeline for Dynamic Gene Expression Data by using it to analyze time course gene expression data from nine studies on various influenza viruses. The goal of our analysis is to simultaneously analyze multiple heterogeneous sources of time course gene expression data on various influenza viruses to (i) identify robust bio-markers for influenza infection and (ii) determine virus-specific bio-markers. These two goals facilitate the diagnosis of virus strains and the identification of vaccine responders at a population level.
We detect the most significant dynamic response genes for each of the 19 virus subtypes (see Table 5). Furthermore, we establish the following potential bio-markers for influenza infection response at population level (37% of the data sets reporting these genes as having a significant response over time): TRADD, an adapter molecule for TNFRSF1A/TNFR1 that specifically associates with the cytoplasmic domain of activated TNFRSF1A/TNFR1 mediating its interaction with FADD; ELMO2 involved in cytoskeletal rearrangements required for phagocytosis of apoptotic cells and cell motility; and STAT1 A signal transducer and activator of transcription that mediates signaling by interferons IFNs.
Additionally, we determine that there are two types of gene response modules for influenza infection: (i) there are approximately the same number of genes in each module and each module’s time course behavior is different (modules with many features); (ii) there are a lot of genes in each module and these have a similar behavior over time (dominating up/down regulated feature).
The network feature analysis can help identify key gene response modules in the gene regulatory network while also providing insight into the structure of the network allowing us to make comparisons across various virus subtypes. We find that a lower clustering coefficient may indicate an expanded response to the influenza infection with 43% of the data sets supporting this claim.
While the Pipeline for Dynamic Gene Expression Data is shown to produce interesting biological results, this approach does have some limitations which may warrant more research in the future. In the network construction step (Step 6), the linear ODE model is used, but for some applications, nonlinear ODE models or other network models may be more appropriate. The two-stage approach for linear ODE model selection is highly dependent on the accuracy of the estimated time course gene expression trajectories and their derivatives and can produce inaccurate results if these estimates are poor. As a result, a more robust approach to model selection would be favorable. Finally, we also need to develop more comparative tools to allow us to produce more in-depth across-study analyses.
In summary, the Pipeline for Dynamic Gene Expression Data allows us to take advantage of the vast amounts of publicly available data sets and investigate gene regulatory networks for different biological conditions efficiently. We can examine the dynamic interaction processes between many hosts and external stimulus across multiple conditions. We believe that these types of analyses will lead to a better understanding of the heterogeneity of the real world population. This approach also provides an examination of each host response to a disease or experimental condition at a genetic level, so that novel prevention and intervention targets can be discovered at an individual level and personalized or precision medicine can be achieved.
Supplementary Material
Table 10.
Accuracy of the GRN Identification
| Error Level | 1-Hamming Distance |
|---|---|
| Simulation 1 | |
| 0.10 | 0.79 |
| 0.20 | 0.78 |
| 0.30 | 0.73 |
| Simulation 2 | |
| 0.10 | 0.81 |
| 0.20 | 0.81 |
| 0.30 | 0.80 |
Acknowledgements
This research was partially supported by the NIH grants HHSN272201000055C and R01 AI087135. The authors are grateful to the students, postdoctoral fellows and faculty members who are participating in the GEO-BigData project at the University of Texas Health Science Center at Houston. The authors would like to thank Canglin Wu for providing us with the list of GSE numbers and Dr. Nan Deng for helpful discussions.
Footnotes
adapter molecule for TNFRSF1A/TNFR1 that specifically associates with the cytoplasmic domain of activated TNFRSF1A/TNFR1 mediating its interaction with FADD
signal transducer and activator of transcription that mediates signaling by interferons IFNs
References
- [1].Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research. 2002;30(1):207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data setsupdate. Nucleic acids research. 2013;41(D1):D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].NCBI. GEO Summary; 2016. Available from: http://www.ncbi.nlm.nih.gov/geo/summary/.
- [4].De Jong H Modeling and simulation of genetic regulatory systems: a literature review. Journal of computational biology. 2002;9(1):67–103. [DOI] [PubMed] [Google Scholar]
- [5].Hecker M, Lambeck S, Toepfer S, Van Someren E, Guthke R. Gene regulatory network inference: data integration in dynamic modelsa review. Biosystems. 2009;96(1):86–103. [DOI] [PubMed] [Google Scholar]
- [6].Hood L, Friend SH. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nature Reviews Clinical Oncology. 2011;8(3):184–187. [DOI] [PubMed] [Google Scholar]
- [7].Khoury MJ, Gwinn ML, Glasgow RE, Kramer BS. A population approach to precision medicine. American journal of preventive medicine. 2012;42(6):639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Steuer R, Kurths J, Daub CO, Weise J, Selbig J. The mutual information: detecting and evaluating dependencies between variables. Bioinformatics. 2002;18(suppl 2):S231–S240. [DOI] [PubMed] [Google Scholar]
- [9].Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. science. 2003;302(5643):249–255. [DOI] [PubMed] [Google Scholar]
- [10].Thomas R Boolean formalization of genetic control circuits. Journal of theoretical biology. 1973;42(3):563–585. [DOI] [PubMed] [Google Scholar]
- [11].Shmulevich I, Dougherty ER, Kim S, Zhang W. Probabilistic Boolean networks: a rule-based uncertainty model for gene regulatory networks. Bioinformatics. 2002;18(2):261–274. [DOI] [PubMed] [Google Scholar]
- [12].Friedman N, Linial M, Nachman I, Pe’er D. Using Bayesian networks to analyze expression data. Journal of computational biology. 2000;7(3–4):601–620. [DOI] [PubMed] [Google Scholar]
- [13].Kim SY, Imoto S, Miyano S. Inferring gene networks from time series microarray data using dynamic Bayesian networks. Briefings in bioinformatics. 2003;4(3):228–235. [DOI] [PubMed] [Google Scholar]
- [14].Perrin BE, Ralaivola L, Mazurie A, Bottani S, Mallet J, dAlche Buc F. Gene networks inference using dynamic Bayesian networks. Bioinformatics. 2003;19(suppl 2):ii138–ii148. [DOI] [PubMed] [Google Scholar]
- [15].Holter NS, Maritan A, Cieplak M, Fedoroff NV, Banavar JR. Dynamic modeling of gene expression data. Proceedings of the National Academy of Sciences. 2001;98(4):1693–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sakamoto E, Iba H. Inferring a system of differential equations for a gene regulatory network by using genetic programming. In: Evolutionary Computation, 2001. Proceedings of the 2001 Congress on vol. 1. IEEE; 2001. p. 720–726. [Google Scholar]
- [17].Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nature genetics. 1999;22(3):231–238. [DOI] [PubMed] [Google Scholar]
- [18].Affymetririx. Statistical Algorithms Description Document. Affymetrix white papers. 2002;. [Google Scholar]
- [19].Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. [DOI] [PubMed] [Google Scholar]
- [20].Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. Journal of the American statistical Association. 2004;99(468):909–917. [Google Scholar]
- [21].Millenaar FF, Okyere J, May ST, van Zanten M, Voesenek LA, Peeters AJ. How to decide? Different methods of calculating gene expression from short oligonucleotide array data will give different results. BMC bioinformatics. 2006;7(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lim WK, Wang K, Lefebvre C, Califano A. Comparative analysis of microarray normalization procedures: effects on reverse engineering gene networks. Bioinformatics. 2007;23(13):i282–i288. [DOI] [PubMed] [Google Scholar]
- [23].Green PJ, Silverman BW. Nonparametric regression and generalized linear models: a roughness penalty approach. CRC Press; 1993. [Google Scholar]
- [24].Silverman B, Ramsay J. Functional Data Analysis. Springer; 2005. [Google Scholar]
- [25].Golub GH, Heath M, Wahba G. Generalized cross-validation as a method for choosing a good ridge parameter. Technometrics. 1979;21(2):215–223. [Google Scholar]
- [26].Yao F, Müller HG, Wang JL. Functional data analysis for sparse longitudinal data. Journal of the American Statistical Association. 2005;100(470):577–590. [Google Scholar]
- [27].Wu S, Wu H. More powerful significant testing for time course gene expression data using functional principal component analysis approaches. BMC bioinformatics. 2013;14(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Carey M, Wu S, Gan G, Wu H. Correlation-based iterative clustering methods for time course data: the identification of temporal gene response modules for influenza infection in humans. Infectious Disease Modelling, in press, http://dxdoiorg/101016/jidm201607001. 2016;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- [30].Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lu T, Liang H, Li H, Wu H. High-dimensional ODEs coupled with mixed-effects modeling techniques for dynamic gene regulatory network identification. Journal of the American Statistical Association. 2011;106(496). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wu S, Liu ZP, Qiu X, Wu H. High-Dimensional Ordinary Differential Equation Models for Reconstructing Genome-Wide Dynamic Regulatory Networks. In: Topics in Applied Statistics. Springer; 2013. p. 173–190. [Google Scholar]
- [33].Hemker P Numerical methods for differential equations in system simulation and in parameter estimation. Analysis and Simulation of biochemical systems. 1972;p. 59–80. [Google Scholar]
- [34].Bard Y, Bard Y. Nonlinear parameter estimation; 1974.
- [35].Voit EO, Almeida J. Decoupling dynamical systems for pathway identification from metabolic profiles. Bioinformatics. 2004;20(11):1670–1681. [DOI] [PubMed] [Google Scholar]
- [36].Liang H, Wu H. Parameter estimation for differential equation models using a framework of measurement error in regression models. Journal of the American Statistical Association. 2008;103(484). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tibshirani R Regression shrinkage and selection via the lasso. Journal of the Royal Statistical Society Series B (Methodological). 1996;p. 267–288. [Google Scholar]
- [38].Zou H The Adaptive Lasso and Its Oracle Properties. Journal of the American Statistical Association. 2006;101(476):1418–1429. [Google Scholar]
- [39].Fan J, Li R. Variable Selection via Nonconcave Penalized Likelihood and its Oracle Properties. Journal of the American Statistical Association. 2001;96(456):1348–1360. [Google Scholar]
- [40].Bates DM, Watts DG. Nonlinear regression: iterative estimation and linear approximations. Nonlinear regression analysis and its applications. 1988;p. 32–66. [Google Scholar]
- [41].Huber W, Carey VJ, Long L, Falcon S, Gentleman R. Graphs in molecular biology. BMC bioinformatics. 2007;8(6):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P. Coexpression analysis of human genes across many microarray data sets. Genome research. 2004;14(6):1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Konganti K, Wang G, Yang E, Cai JJ. SBEToolbox: a Matlab toolbox for biological network analysis. Evolutionary Bioinformatics. 2013;9:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nieminen J On the centrality in a graph. Scandinavian journal of psychology. 1974;15(1):332–336. [DOI] [PubMed] [Google Scholar]
- [45].Jeong H, Mason SP, Barabási AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411(6833):41–42. [DOI] [PubMed] [Google Scholar]
- [46].Koschützki D, Schreiber F. Centrality analysis methods for biological networks and their application to gene regulatory networks. Gene regulation and systems biology. 2008;2:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bavelas A Communication patterns in task-oriented groups. The Journal of the Acoustical Society of America. 1950;22(6):725–730. [Google Scholar]
- [48].Hage P, Harary F. Eccentricity and centrality in networks. Social networks. 1995;17(1):57–63. [Google Scholar]
- [49].Barrenas F, Chavali S, Holme P, Mobini R, Benson M. Network properties of complex human disease genes identified through genome-wide association studies. PloS one. 2009;4(11):e8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hwang W, Cho Yr, Zhang A, Ramanathan M. Bridging centrality: identifying bridging nodes in scale-free networks. In: Proceedings of the 12th ACM SIGKDD international conference on Knowledge discovery and data mining; 2006. p. 20–23. [Google Scholar]
- [51].Hallinan JS. Cluster analysis of the p53 genetic regulatory network: Topology and biology. In: Computational Intelligence in Bioinformatics and Computational Biology, 2004. CIBCB’04. Proceedings of the 2004 IEEE Symposium on IEEE; 2004. p. 1–8. [Google Scholar]
- [52].Palumbo MC, Colosimo A, Giuliani A, Farina L. Functional essentiality from topology features in metabolic networks: a case study in yeast. FEBS letters. 2005;579(21):4642–4646. [DOI] [PubMed] [Google Scholar]
- [53].Watts DJ, Strogatz SH. Collective dynamics of small-worldnetworks. nature. 1998;393(6684):440–442. [DOI] [PubMed] [Google Scholar]
- [54].Wasserman S, Faust K. Social network analysis: Methods and applications. vol. 8. Cambridge university press; 1994. [Google Scholar]
- [55].Cai JJ, Borenstein E, Petrov DA. Broker genes in human disease. Genome biology and evolution. 2010;2:815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Huang CH, Chen TH, Ng KL. Graph theory and stability analysis of protein complex interaction networks. IET systems biology. 2016;10(2):64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Grewal N, Singh S, Chand T. Effect of Aggregation Operators on Network-based Disease Gene Prioritization: A Case Study on Blood Disorders. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2016;. [DOI] [PubMed] [Google Scholar]
- [58].(WHO) WHO. Influenza (Seasonal) fact-sheet; 2016. Available from: http://www.who.int/mediacentre/factsheets/fs211/en/.
- [59].Woods CW, McClain MT, Chen M, Zaas AK, Nicholson BP, Varkey J, et al. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS One. 2013;8(1):e52198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Djavani M, Crasta OR, Zhang Y, Zapata JC, Sobral B, Lechner MG, et al. Gene expression in primate liver during viral hemorrhagic fever. Virology Journal. 2009;6(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139(7):1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jin S, Li Y, Pan R, Zou X. Characterizing and controlling the inflammatory network during influenza A virus infection. Scientific reports. 2014;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Huang Y, Zaas AK, Rao A, Dobigeon N, Woolf PJ, Veldman T, et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet. 2011;7(8):e1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Thakar J, Hartmann BM, Marjanovic N, Sealfon SC, Kleinstein SH. Comparative analysis of anti-viral transcriptomics reveals novel effects of influenza immune antagonism. BMC immunology. 2015;16(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Niu Z, Chasman D, Eisfeld AJ, Kawaoka Y, Roy S. Multi-task Consensus Clustering of Genome-wide Transcriptomes from Related Biological Conditions. Bioinformatics. 2016;p. btw007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mitchell HD, Eisfeld AJ, Sims AC, McDermott JE, Matzke MM, Webb-Robertson BJM, et al. A network integration approach to predict conserved regulators related to pathogenicity of influenza and SARS-CoV respiratory viruses. PloS one. 2013;8(7):e69374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Itkes A. Oligoadenylate and cyclic AMP: interrelation and mutual regulation. In: Biological Response ModifiersInterferons, Double-Stranded RNA and 2, 5-Oligoadenylates. Springer; 1994. p. 209–221. [DOI] [PubMed] [Google Scholar]
- [68].Cao M, Wei C, Zhao L, Wang J, Jia Q, Wang X, et al. DnaJA1/Hsp40 is co-opted by influenza A virus to enhance its viral RNA polymerase activity. Journal of virology. 2014;88(24):14078–14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bohn JA, BeMiller JN. (1 3)-β-D-Glucans as biological response modifiers: a review of structure-functional activity relationships. Carbohydrate polymers. 1995;28(1):3–14. [Google Scholar]
- [70].LoVerme J, La Rana G, Russo R, Calignano A, Piomelli D. The search for the palmitoylethanolamide receptor. Life sciences. 2005;77(14):1685–1698. [DOI] [PubMed] [Google Scholar]
- [71].Yuan S Drugs to cure avian influenza infection–multiple ways to prevent cell death. Cell death & disease. 2013;4(10):e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon αβ in acute influenza infection. Nature communications. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Dubois J, Terrier O, Rosa-Calatrava M. Influenza viruses and mRNA splicing: doing more with less. MBio. 2014;5(3):e00070–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dudek SE, Luig C, Pauli EK, Schubert U, Ludwig S. The clinically approved proteasome inhibitor PS-341 efficiently blocks influenza A virus and vesicular stomatitis virus propagation by establishing an antiviral state. Journal of virology. 2010;84(18):9439–9451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Turpin E, Luke K, Jones J, Tumpey T, Konan K, Schultz-Cherry S. Influenza virus infection increases p53 activity: role of p53 in cell death and viral replication. Journal of virology. 2005;79(14):8802–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Husain M, Harrod KS. Enhanced acetylation of alpha-tubulin in influenza A virus infected epithelial cells. FEBS letters. 2011;585(1):128–132. [DOI] [PubMed] [Google Scholar]
- [77].Han X, Li Z, Chen H, Wang H, Mei L, Wu S, et al. Influenza virus A/Beijing/501/2009 (H1N1) NS1 interacts with β-tubulin and induces disruption of the microtubule network and apoptosis on A549 cells. PLoS One. 2012;7(11):e48340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].JACKSON GG, DOWLING HF, SPIESMAN IG, BOAND AV. Transmission of the common cold to volunteers under controlled conditions: I. The common cold as a clinical entity. AMA Archives of Internal Medicine. 1958;101(2):267–278. [DOI] [PubMed] [Google Scholar]
- [79].Hubert L, Arabie P. Comparing partitions. Journal of classification. 1985;2(1):193–218. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.