

Abstract
The liver is a critical mediator of lipid/glucose homeostasis and is a primary organ involved in dynamic changes during feeding and fasting. Additionally, hepatic-centric pathways are prone to dysregulation during pathophysiological states including metabolic syndrome (MetS) and non-alcoholic fatty liver disease. Omics platforms and GWAS have elucidated genes related to increased risk of developing MetS and related disorders, but mutations in these metabolism-related genes are rare and cannot fully explain the increasing prevalence of MetS-related pathologies worldwide. Complex interactions between diet, lifestyle, environmental factors, and genetic predisposition jointly determine inter-individual variability of disease risk. Given the complexity of these interactions, researchers have focused on master regulators of metabolic responses incorporating and mediating the impact of multiple environmental cues. Transcription factors are DNA binding, terminal executors of signaling pathways that modulate the cellular responses to complex metabolic stimuli and are related to the control of hepatic lipid and glucose homeostasis. Among numerous hepatic transcription factors involved in regulating metabolism, three emerge as key players in transducing nutrient sensing, which are dysregulated in MetS-related perturbations in both clinical and preclinical studies: cAMP Responsive Element Binding Protein 3 Like 3 (CREB3L3), Peroxisome Proliferator Activated Receptor Alpha (PPAR), and Forkhead Box O1 (FOXO1). Additionally, these three transcription factors appear to be amenable to dietary/nutrient-based therapies, being potential targets of nutritional therapy. In this review we aim to describe the activation, regulation, and impact of these transcription factors in the context of metabolic homeostasis. We also summarize their perspectives in MetS and nutritional therapies.
Keywords: CREBH, PPAR, FOXO1, Metabolic syndrome, diet induced obesity, NAFLD
Introduction
As defined by the World Health Organization, Metabolic syndrome (MetS) is a series of disease conditions characterized by “atherogenic dyslipidemia, raised blood pressure, insulin resistance, and pro-thrombotic and pro-inflammatory states” [1]. Prevalence of MetS has become a major health issue in the modern world. According to an estimation based on Centers for Disease Control and Prevention (CDC) data published in 2017, one third of American adults may have diagnosable MetS [2]. Moreover, the spread of western-style-diet and unhealthy lifestyle contributes to prevalent growth of MetS globally. Increases in body-mass index (BMI) and obesity rates have dramatically grown since the 1980s, especially in developing countries [3]. Although the development of MetS is dependent on many lifestyle dependent and independent factors, diet is the key mediator of MetS. Easy access to caloric-rich foods disturbs metabolic homeostasis, which can directly enhance adipose growth and the risk of developing type 2 diabetes, cardiovascular diseases, cancer, and hepatic, respiratory and other MetS-related pathologies [4].
Recently, nutritional scientists and researchers in metabolism-focused disciplines have elucidated key molecular pathways linking sub-optimal diets, especially those of caloric or macronutrient imbalance, to increased risk of MetS. One such regulatory platform utilizes dynamic protein:DNA interactions to allow for both rapid and long-lasting genetic changes to metabolism. Transcription factors (TFs), proteins known to bind DNA sequences, play crucial roles not only in cell differentiation [5], organ development [6], immune response [7], circadian regulation [8], but also in maintaining metabolic homeostasis. Most transcription factors bind specific short sequences on genomic DNA called “motifs/Cis-regulatory elements”, which regulates transcription of target genes positively or negatively [9]. Transcriptional regulation of genes by TFs may vary depending on cell types and physiologic conditions, such as metabolic signals. In the organs majorly related to metabolism including liver, adipose tissue, and skeletal muscle, dysregulation of TF networks impairs the ability to maintain metabolic homeostasis, inducing pathological changes in those organs and the whole body. Therefore, understanding the functions of TFs in metabolic diseases brings insight into intracellular molecular regulations induced by changes in nutrient flux. Generally, about 1600 human TFs have been discovered [9], and most of them involve metabolism to some extent. Due to the sheer number of proteins shown to interact with base pairs of DNA, we have chosen here to focus on three well-studied TFs, cAMP Responsive Element Binding Protein 3 Like 3 (CREB3L3), Peroxisome Proliferator Activated Receptor Alpha (PPARα/PPARA), and Forkhead Box O1 (FOXO1) which are known to be dysregulated in MetS in both clinical and preclinical studies and that have been shown to be modifiable using nutrition-based approaches.
CREB3L3 is known as a common endpoint executor of several metabolic pathways. As a nuclear receptor, PPARα interacts with its multiple ligands to elicit a response in the cell. Activated FOXO1 acts as either activator or repressor for other transcription factors. These TFs involved in metabolic regulation initiate and modify various molecular mechanisms when their transcriptional activation is initiated. However, several upstream pathways and downstream target genes are shared by three TFs. Therefore, the aim of this review is to provide a systematic analysis of the three TFs, with focuses including their protein families, regulating signals, post-translational modifications, genetic variations, gene targets, associated pathological conditions, and clinical applications. In the last section, we will summarize their relationship to mechanisms of nutritional intervention.
Dysregulated glucose and lipid signaling in pathologies related to MetS
Together with obesity, Type 2 diabetes, dyslipidemia, and hypertension, non-alcoholic fatty liver disease (NAFLD) is an additional common feature in MetS. The prevalence of NAFLD in MetS may be expected as the liver is a key metabolic organ that monitors and adjusts glucose and lipid levels in the blood. The liver absorbs blood glucose through glucose transporters and stores it as glycogen under feeding condition. On the other hand, upregulated glycogenolysis during fasting produces glucose from glycogen which in turn can be released to elevate blood glucose or degraded through in situ glycolysis. In addition to glucose signaling, the liver is critical for the homeostasis of lipids through events such as bile acid secretion, fatty acid (FA) uptake and de novo lipogenesis. The anabolism and catabolism of endogenous lipids is a critical metabolic pathway which can be disrupted during MetS where energy is stored as triglycerides (TGs) in lipid droplets and released during lipolysis in conjunction with FA oxidation [10, 11].
It has long been a goal to develop testable biomarkers and to identify critical mediators of MetS and associated disease phenotypes. As MetS is a complex disease state, it may be difficult to identify specific biomarkers to the syndrome that differ considerably from biomarkers of related disease pathologies themselves (e.g., diabetes, NAFLD). For example, insulin resistance, hypertriglyceridemia, and abnormal HDL and uric acid levels are closely associated with NAFLD but also other MetS-related pathologies [12]. Studying NAFLD may be a useful surrogate for studying the hepatic effects of MetS as lipid and glucose signaling are dysregulated in both perturbations. The most widely used biomarker for NAFLD diagnosis is abnormal serum ALT/AST levels [13, 14]. However, serum ALT/AST level alone may lead to high false positive and false negative rates, in spite of the fact that it is the most accurate biomarker currently used [13, 15]. Novel biomarkers of NAFLD and MetS are beginning to be discovered through large scale omics studies. Several studies focusing on plasma lipidomic profiles of patients identified oxidized fatty acids and other lipids as metabolites that may act as biomarkers of disease occurrence or severity (e.g., differentiate NAFLD from more severe nonalcoholic steatohepatitis (NASH) [16–18]. Metabolomics studies [19] have also shown circulating changes in several amino acids and other metabolites in clinical and in vivo models of hepatic dysfunction [20].
NAFLD is currently considered as a disease of multiple lifestyle dependent and independent risk factors and involves multiple organ systems [21]. Dietary choices, gut microbiome structure and function, and genetic predisposition can interact to provoke inflammatory responses between the liver, adipose tissue and the gastrointestinal tract [22]. Due to the complexity of the human NAFLD condition, it is difficult to mimic all characteristics in the progress of metabolic dysfunction in one animal model. Therefore, an ideal model for MetS is selected based on the aim of the individual experiment. Models involving genetic manipulation of genes related to metabolism and/or diet/chemical-induced models are useful when studying NAFLD and other MetS-related pathologies. Rodent models, such as ob/ob and db/db, and diet-induced models including High-Fat Diet (HFD)- or Methionine/Choline Deficient Diet (MCD)-induced mice, or the models combining two methods are common [23, 24]. In general, genetic models quickly recapitulate relatively severe phenotypes of NAFLD; toxic chemical (e.g. CCl4) induced model best mimics fibrosis phenotype in NAFLD; and diet related models (e.g. HFD model) resemble the progress of NAFLD in pathophysiological view [25]. Using these model systems, researchers have identified multiple transcription factors that are dysregulated during times of MetS. Here, we will discuss three of the primary transcription factors that control hepatic glucose and lipid homeostasis and are known to be dysregulated during times of metabolic disruption and stress; CREBH, PPARα, and FOXO1.
CREB3 family transcription factors and MetS
The endoplasmic reticulum localized CREB3 family is a subfamily of the basic leucine zipper (bZIP) transcription factors with five members currently recognized as CREB3, CREB3L1, CREB3L2, CREB3L3, and CREB3L4. CREB3 members are the most studied as related to metabolic disorders and are highly conserved throughout broad species (especially the functional bZIP domain) [26]. CREB3 transcription factors recognize cis-regulatory elements on DNA called cAMP response element (CRE) and B-Box (Figure 1). Mammalian CREB3 TFs are type II transmembrane proteins which are anchored in the ER membrane. Activation of CREB3 TFs requires proteolytic cleavage, which provides another level of regulation other than transcription. Due to tissue distribution differences between CREB3 members, CREB3L3 may be the most critical in relation to metabolism [27–29], but CREB3L2 may also have roles in lipid metabolism which are worthy of further investigation due to the relatively high expression in subcutaneous adipose tissue [26]. CREB3L3, also known as CREB-H, was historically considered to be expressed in a liver-specific manner [30], but recent studies have discovered expression of Creb3l3 in mouse small intestine, another critical organ related to cholesterol absorption/metabolism [31–33]. In humans, according to the GTEx portal [34], CREB3L3 is primarily expressed in the liver, followed by the small intestine with approximately 6-fold lower transcription level on average than in the liver. Regulation of CREB3L3 by metabolic signals demonstrate how CREB3L3 associates with MetS in these metabolic active organs.
Figure 1.
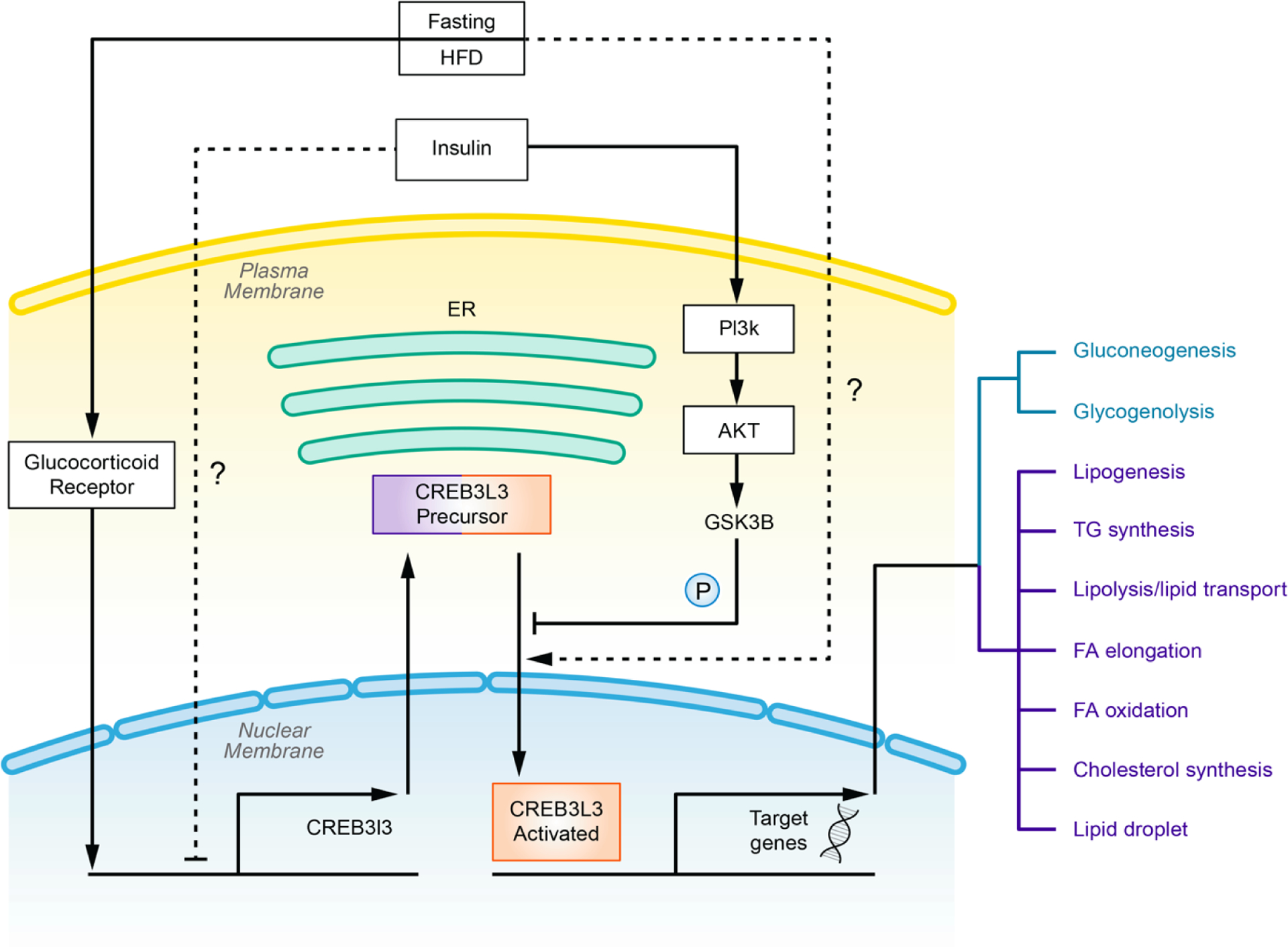
Overview of CREB3L3 regulation and activation. Transcription of Creb3l3 is upregulated by fasting through glucocorticoid receptor. Fasting or HFD treatment may also modulate activation of CREB3L3. Insulin induces phosphorylation of CREB3L3 precursor and prevents its cleavage by activating PI3K pathway. Responding to multiple metabolic signals, CREB3L3 initiates target genes involved in glucose and lipid metabolism.
Intake of dietary factors related to MetS such as lipids and sugars are major regulators of CREB3L3. Long term treatment with well characterized HFD including the atherogenic high fat (AHF) diet [35, 36], Western diet [32], and ketogenic diet [32, 37] have been shown to induce expression and activation of CREB3L3 in mouse models. Moreover, in fasting/re-feeding experiments, starvation also induces activation of CREB3L3. Interestingly, Danno et al. [38] re-fed fasted mice with chow, high-sucrose, or high-fat diet, and they found that the high-fat group showed the least inhibition of fasting-induced Creb3l3 expression and activation. They also found plasma non-esterified fatty acids were correlated with this trend. The molecular mechanisms linking HFD- or fasting to induction of CREB3L3 mRNA expression are not well understood. Activation of glucocorticoid receptor (GR) antagonizes inhibition of CREB3L3 expression by the insulin-PI3K pathway [39], which may partially contribute to the diet-induced CREB3L3 expression. PPARα, an intracellular fatty acids effector [40] (see below), may also regulate Creb3l3 mRNA expression [38], and was considered as the major regulator of CREB3L3 transcription responding to concentration of fatty acids. However, a recent study showed that elevated expression of CREB3L3 by the fatty acid oleate is independent of PPARα [41].
Furthermore, genetic effectors such as Toll Like Receptor 4 (TLR4) [41], Cannabinoid Receptor 1 (CB1R) [42], or hepatocyte nuclear factor 4 alpha (HNF4A) [31] may also contribute to the modifiable expression of CREB3L3. In addition to genetic control, CREB3L3 is post-translationally controlled through regulated intramembrane proteolysis (RIP) [30], in which CREB3L3 precursor is cleaved to an activated form which initiates transcription. However, how this process is related to CREB3L3’s role in metabolism and in MetS is unclear. One possibility is that an unknown mechanism controls transportation of CREB3L3, during HFD/refeeding instances, from ER to Golgi apparatus [43]. Additionally, the protein secretory pathway may play a role, whether by adjusting the ratio of the protein in the anterograde and retrograde transport pathway or by changing the destination from Golgi apparatus to other subcellular organelles, such as lysosome.
Single-nucleotide polymorphisms (SNPs) can affect expression and function of a gene. Therefore, SNPs in CREB3L3 pre-determine genetically activity of CREB3L3, which therefore modulates individual predisposition to MetS. In a meta-analysis of genome-wide association study (GWAS) data, an intronic single nucleotide polymorphism rs2240702 in the CREB3L3 gene was significantly associated with waist-hip ratio in human populations, suggesting a potential genetic linkage of this gene to obesity [44]. Moreover, multiple non-synonymous mutations have been identified on the CREB3L3 exons which are associated with human hypertriglyceridemia [45, 46]. Lee and his colleagues analyzed nonsynonymous substitutions or insertions in CREB3L3 in hypertriglyceridemic group containing 449 individuals, and they found that none of these mutations have been discovered in any of the 238 control individuals. One insertion (245fs), one non-sense mutation (W46X), and multiple point mutations (G105R, P166L, V180M, D182N, E240K) have been discovered in the study. These mutations variously affect transcription ability of CREB3L3: 245fs, W46X, and E240K severely jeopardized transcription of CREB3L3 activity as measured via a luciferase assay while the other above-mentioned mutations have only modest effect on the function of CREB3L3. Interestingly, several synonymous variants have been discovered, which engage similar occurrence in both hypertriglyceridemia and control groups. Taken together with the data from the 1000 Genomes Project in which only G105R mutation has been identified [45, 47], nonsynonymous substitutions, rather than synonymous variations, are rare in the population without hypertriglyceridemia, and they contribute greatly to the hypertriglyceridemic phenotype.
As a transcription factor, CREB3L3 functions majorly through transcribing target genes (Figure 1). Responding to diet-induced metabolic changes, activated CREB3L3 transcribes target genes in the hepatocyte involved in both glucose and lipid metabolism. It has been reported that CREB3L3 promotes expression of phosphoenolpyruvate carboxykinase 1 (PCK1) and glucose-6-phosphatase catalytic subunit (G6PC); genes regulating gluconeogenesis, and liver glycogen phosphorylase (PYGL); a rate-limiting enzyme in glycogenolysis [39, 48]. CREB3L3 adjusts whole body glucose balance, since adenoviral overexpression of CREB3L3 elevates blood glucose and reduces hepatic glycogen [48]. On the other hand, CREB3L3 generates a vast range of effects on lipid metabolism. Zhang et al. [35] categorized dozens of genes related to hepatic lipid regulation in 5 major groups, including lipogenic regulators, TG synthesis, lipolysis/lipid transport, FA elongation, and FA oxidation or cholesterol synthesis. They discovered that expression of these genes was reduced in Creb3l3 knockout (KO) mice, and differences in expression were altered under normal chow or high-fat diet treatment. Moreover, CREB3L3 contributes to expression of FSP27β, a liver isoform of CIDEC which functions in lipid droplet growth [37]. Additionally, AHF-diet fed Creb3l3 KO mice exhibit severe hepatic lipid accumulation and high plasma triglycerides levels [35, 49]. Liver-blood glucose balance is disrupted in Creb3l3 KO mice as evidenced by significantly increased blood glucose levels and concomitant hepatic glycogen level decreases after 12h of fasting [48]. Emerging evidence also implicates CREB3L3 induction with increased expression of the well-studied hepatic secretory hormone, FGF21, under AHF diet [36] or high-fat diet [50] induced metabolic stress. FGF21 exhibits metabolic benefits in both glucose and lipid metabolism (reviewed in [51]). Interestingly, adenoviral overexpression of human FGF21 in the Creb3l3 KO mice rescues ketogenic diet induced hepatic lipid accumulation and inflammation [52].
By regulating hepatic metabolism-related genes, CREB3L3 has become recognized as a critical player in metabolic process and in metabolic diseases. CREB3L3 is involved in regulating both hepatic lipid and glucose metabolism through transcriptional regulation, including lipogenesis, FA and cholesterol metabolism, lipolysis, glycogenolysis, and gluconeogenesis [35, 39, 48]). As a master ranscription regulator in metabolism, CREB3L3 is considered to play a role in MetS through multiple mechanisms. Of all the MetS-related pathologies, CREB3L3 may play the largest role in the development of hepatic lipid dysfunction and NAFLD. As previously mentioned, lipid accumulation is a common phenotype observed in AHF-diet-fed Creb3l3 KO mice [35, 49, 53–55]. These mice display elevated plasma ALT/AST levels and expression of pro-inflammatory cytokines seen in the more severe NASH phenotype as well [35]. Adenoviral over-expression of Creb3l3 significantly ameliorated hepatic lipid accumulation in HFD-fed wildtype rodents [56]. In addition to responding to cellular nutrient cues, CREB3L3 appears to be a metabolic circadian target, regulated majorly by CLOCK/BMAL1/GSK3 pathway [57]. Disruption of routine circadian clock, which is common in the modern life, induces metabolic stress and may play a role in MetS and related pathologies [49]. Although more work needs to be completed in this area, a study indicated that ablation of Creb3l3 not only disturbed circadian regulation of its downstream targets, such as Fgf21, but also interfered with circadian rhythms of serum TG and FFA levels [57]. Moreover, similarly to NAFLD, development of type 2 diabetes (T2D) is associated with dysregulation of CREB3L3, but it is not well established if activation of CREB3L3 is a positive or negative mediator of T2D. For example, CREB3L3, through activation by CB1R, has been reported to deteriorate insulin resistance by increasing the expression of Lipin1 [58]. A separate study however showed that activation of CREB3L3 may be increased by insulin signaling in the liver [35]. Finally, a separate mechanism linking CREB3L3 to diabetes risk is related to iron metabolism which has shown to be associated with the regulation of insulin sensitivity in hepatocytes [59, 60]. Excessive intracellular iron may be caused by a dysregulated induction of the iron-related protein hepcidin, leading to early stages of insulin resistance [61]. Interestingly, hepcidin is a target gene of CREB3L3 in the liver [62].
In summary, the metabolic transcription factor CREB3L3 works to link the metabolic signals from upstream pathways to downstream metabolic effectors. Metabolic stress signals regulate transcription, activation and post-translational modification of CREB3L3, which stimulates transcription of downstream target genes involved in glucose and lipid metabolism. There is evidence that energy storage and consumption conditions (e.g., fed vs. fasted states) trigger activation of CREB3L3 through different pathways. Therefore, it’s been hypothesized that various PTM residues added by those pathways create various affinity towards individual TF partners. Since CREB3L3 works as an endpoint of multiple metabolic pathways, it may be an understudied potential therapeutic target for prevention of MetS.
PPAR family transcription factors and MetS
Peroxisome proliferator-activated receptor (PPARs) family members are vertebrate-specific nutrient sensing nuclear receptors (NRs) [63]. Three members are identified in this family, including PPAR α, β/δ, and γ. Expression and function of the three subtypes of PPAR are unique, as is their tissue distribution. PPARα is highly expressed in liver, skeletal muscle, and brown adipose tissue; PPARγ is mainly expressed in white adipose tissue while PPARβ/δ is ubiquitously expressed. Functions of PPARs are closely associated with energy homeostasis and nutrient sensing (Figure 2). The α and β/δ subtypes are involved in energy utilization and the γ contributes to energy storage in adipose [64]. High sequence homology is shared in the ligand- and DNA-binding domains between PPAR members with a growing list of endogenous and exogenous known ligands[64, 65]. Different from other intracellular messengers, these ligands interact directly with nuclear receptors in the cell after travelling through the cytoplasmic membrane [66]. As transcription factors, PPARs bind to cis-acting elements of the target genes termed as peroxisome proliferators response elements (PPREs), which will either activate or inhibit transcription of the target genes. Similar to other bZIP and basic Helix-Loop-Helix (bHLH) family TFs, PPARs are required to form dimers when they bind to DNA [67]. Different dimer pairs, which are regulated by intracellular abundance and post-translational modifications, change binding preference of cis elements of different target genes. Therefore, PPARs could initiate transcription of a suite of genes under one metabolic state and a unique set of genes when metabolic signals differ. For the past 3 decades, the knowledge about PPARs has been largely expanded, from orphan receptors to critical metabolic regulators, and now to promising treatment targets [68–70]. Here, as before, we focus on the roles that PPARα plays in hepatic metabolism.
Figure 2.
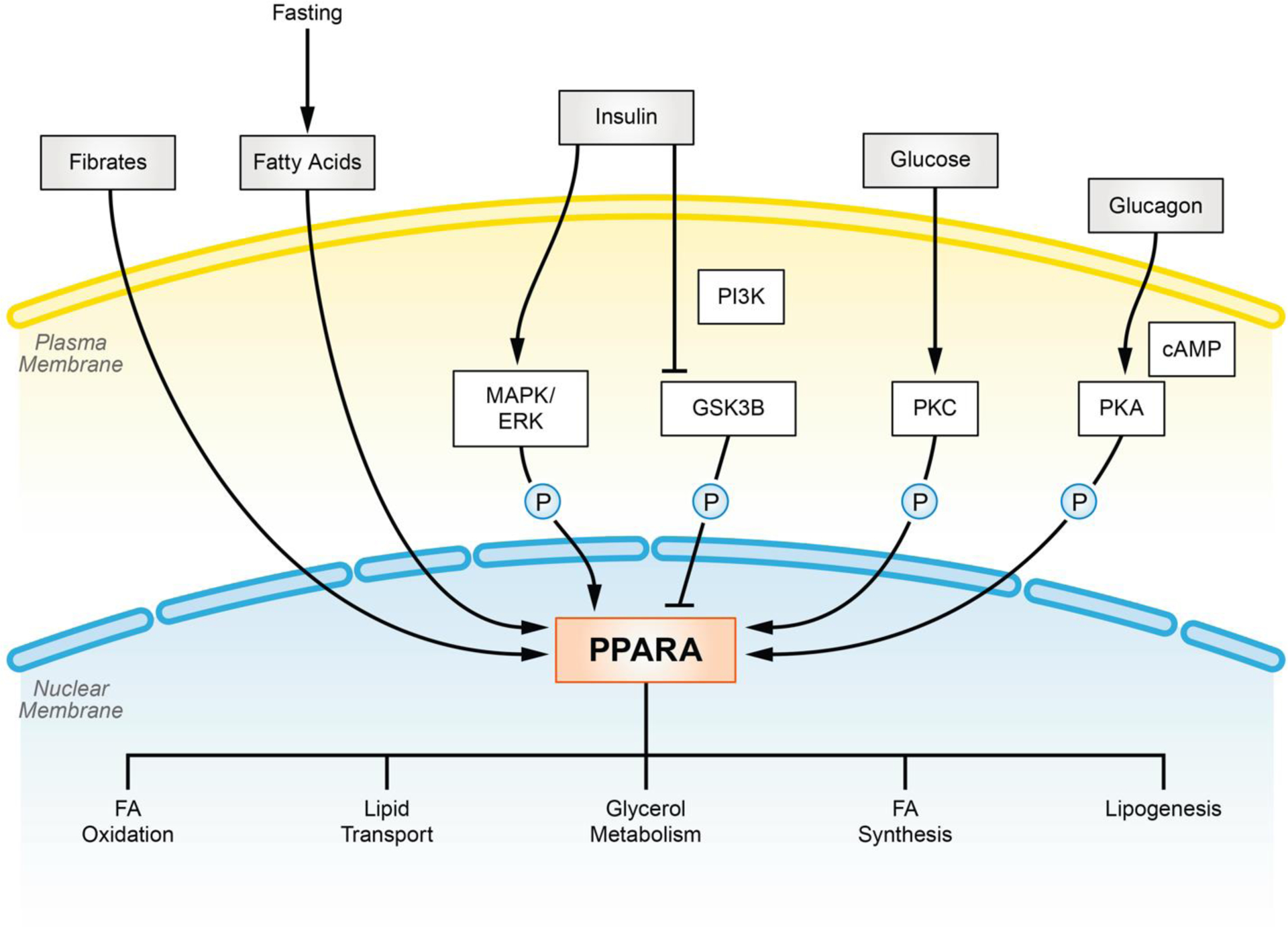
Overview of PPARα activation and regulation. PPARα activity is mediated by intra/extra-cellular metabolic signaling. Fasting induces elevation of fatty acids, the insulin signaling pathway, elevation of blood glucose, and glucagon which may stimulate expression and/or activity of PPARα under physiological metabolic conditions leading to increased lipid consumption.
PPARα is considered to be a master hepatic transcriptional regulator in regard to metabolic function. Structurally, PPARα is divided into 6 domains from A to F [71, 72]. Domain C directly binds to PPRE motif on promoter sequences of DNA whereas A/B and E/F domain act as activating functional domain which renders specificity to target genes and transcription partners. PPARα can bind dietary factors including FAs and their derivatives, leading to structural protein modifications, post-transcriptional changes, and increased binding affinity to co-factors and PPRE motifs [73–75]. During fasting, a time of elevated free FAs, PPARα is activated to adjust hepatic metabolism accordingly during this time of overall nutrient shortage [76] [77]. Currently, it is not well understood if PPARα is a primary sensor of both hepatic and circulating plasma levels of FAs [78], or if another PPAR family member such as PPARβ/δ is primarily responsible for responding to FA levels in circulation [79]. De novo synthesized fatty acids are the major contributor to activation of PPARα [78].
In addition to changes in fatty acid concentrations, several other molecular pathways interact with PPARα under fasting conditions to rapidly balance energy deficits (Figure 2). Of the many levels of PPAR regulation, post-translational modifications (PTMs), such as phosphorylation may be some of the most critical. Various PPARα phosphorylation sites have been identified by in silico discovery, and some of these phosphorylation sites have been verified in vivo [80]. One well studied mechanism involves the insulin signal effector, MAPK/ERK, which can directly phosphorylate PPARα on Ser12/Ser21 residues [81]. This phosphorylation event stabilizes PPARα against ubiquitination-mediated degradation ultimately promoting activation [82]. Another example, involving PKA, has been reported to similarly increase stability and activity of PPARα by phosphorylating Thr129/Ser163 residues [83]. Additionally, two in vitro studies discovered a series of serine and threonine residues on the C and D domain of PPARα which can be phosphorylated by PKC [84, 85]. Phosphorylation at these sites may alter interactions of PPARα with other transcription partners and target genes. Although many phosphorylation events appear to lead to increased stability or activation, phosphorylation of Ser73 by GSK3β has been shown to increase degradation and reduce activity of PPARα [86]. These PPARα regulating kinases are the downstream executors of metabolic pathways, such as insulin and glucagon signaling pathway. Therefore, changes in dietary energy supply could adjust abundance and activity of PPARα indirectly by PTMs. Furthermore, expression level of PPARα is fluctuated in response to dietary factors. Binding motifs of various nutrient-regulated transcription factors and nuclear receptors, including LXR, PXR, HNF4, CREB3L3 and even PPARα itself [87–90] have been reported on the PPARα promoter region, and the mRNA expression of PPARα is under the regulation thereof. Finally, although it appears phosphorylation events may be critical mediators of PPAR activity, other PTMs such as SUMOylation may also play a role in regulation of PPARα activation [91, 92].
PTMs lead to structural changes in TFs, which leads to oligomerization of a group of TFs prior to their induction/modulation of transcription. PPARα belongs to the NR1C branch of the nuclear receptor superfamily, of which a dimer is required to promote transcription [67]. Due to high similarity in the sequence of DNA binding motifs, other nuclear receptors dimerize with PPARα to bind the tandem hexad motif 5’-AGGTCA-3′. Genome-wide profiling indicated DNA binding sites of the nuclear receptors, including LXR, RXR, and PPARs are highly overlapped [93]. A prime example of this heterodimer formation involves retinoid X receptors (RXRs) [94], which are also called NR2B under the NR superfamily context. Upon heterodimerization, PPARα:RXR can bind a conserved DNA binding element named PPRE direct repeat 1 (DR1) found within the promoter region of target genes [95, 96]. Although RXR was considered as the default transcription factor pair for PPARα, new heterodimer partners have been identified recently. Interactions between PPARα and LXRα have been reported, presumably regulating expression of genes such as Apoa1 under metabolic challenging states [97]. Also, as briefly described above, PPARα can also heterodimerize with proteins outside of the nuclear receptor family such as CREBH. The importance of this interaction between CREBH and PPARα during metabolic flux is illustrated within the context of the hepatic secretory hormone FGF21 which is highly induced in fasted or AHF-diet treated mice. Studies show that hepatic expression of Fgf21 relies on expression both of PPARα [98, 99] and of CREBH [36, 52]. Co-immunoprecipitation, Chromatin immunoprecipitation (ChIP), and electrophoretic mobility shift assay (EMSA) experiments further validate the interaction between PPARα and CREBH, and promoter region of Fgf21 [36]. After the heterodimer locates the binding motif within FGF21, cofactors are recruited as co-activators or co-repressors which regulate transactivation activity [100] of this critical hormone. Following the dimerization, homo-/hetero-dimers of PPARα will locate PPAR response element (PPRE) in the promoter of target genes to allow initiation of transcription.
Functionally, PPARα works as a bridge across upstream metabolic signals and downstream effector proteins. Therefore, PPARα target genes play critical roles in regulating metabolism. As before, we are focusing on hepatic expression of PPARα, which is enriched in the liver and is where its major functions arise. Rakhshandehroo et al. discovered 1847 genes expression changes in livers of PPARα knockout mice in the fasted state using microarray-based global analysis. Many of these genes can be classified functionally in FA oxidation, lipid transport, glycerol metabolism, FA synthesis, and lipogenesis [101], though not all of these candidate genes are regulated directly through transcription initiated by PPARα. Experimentally validated PPARα target genes have been summarized elsewhere [102]. Interestingly, PPARα has been shown to promote transcription of not only metabolic effectors (e.g. FGF21, FADS, FASN, PCK1), but also metabolic-regulated transcription factors such as LXR, NR1D1, and CREBH. Finally, it is important to note that there are diverse expression patterns resulting from PPARα between mice and humans [103]. These organismal discrepancies are important to understand and have also been observed in clinical situations in that humans react differently to many PPARα agonists compared to established mouse models.
Studies in rodent models have been an important driving force in our understanding of PPARα as a major metabolic regulator in the liver. Activation of PPARα has generally been shown to improve the symptoms of metabolic syndrome, and many of the beneficial effects have been observed through activated PPARα leading to the upregulation of genes concerned with fatty acid oxidation. Using the New Zealand Obese (NZO) mouse model, which mimics a polygenic syndrome closely resembling the complexity of human metabolic syndrome, studies have identified PPARα-mediated pathways and targets involved in fatty acid and cholesterol metabolism were the most significantly altered from healthy mice [104, 105]. Insulin resistance, one of the major causes of the metabolic syndrome, often leads to late complications such as type 2 diabetes. Mouse models utilized in diabetes research have highlighted the utility of PPARα in improving disease progression. In both a high fat diet-induced and a genetic mouse model of insulin resistance, activation of PPARα with PPARα-selective fibrates resulted in reduced insulin resistance and increased expression of enzymes involved in lipid oxidation [106–108]. In a mouse model of Alström Syndrome, marked by obesity, hyperinsulinemia, and T2D, activation of PPARα via the agonist WY14643 led to improved insulin sensitivity and reduced steatosis [109]. Interestingly, PPARα-null mice fed a high fat diet were shown to be protected from developing insulin resistance compared to control mice [110, 111]. However, these studies were largely conducted in the fasted state. When similar experiments were conducted in the non-fasted state, PPARα-null mice were not shown to be protected from insulin resistance [112, 113]. Since PPARα-null mice have an impaired response to fasting that leads to reduced fatty acid oxidation, it is possible that glycogen stores are instead preferentially used, leading to the perceived insulin resistance [77, 113]. PPARα has also been closely associated with the progression of the metabolic syndrome into the development of NAFLD [114, 115]. Studies using PPARα-null mice have shown that lack of PPARα leads to decreased expression of fatty acid-metabolizing enzymes, resulting in a fatty liver phenotype [116, 117]. In the non-fasted state, PPARα deficiency causes mild fatty liver, while fasting results in much more severe steatosis and steatohepatitis [118, 119]. In models using a high fat diet, PPARα-null mice demonstrated significantly increased inflammation, hepatic triglyceride levels, and higher NAFLD activity scores [120, 121]. In mice fed an MCD diet to induce NASH, PPARα-null mice also demonstrated more severe steatohepatitis [122]. Furthermore, treatment of MCD diet-fed wild-type mice with the PPARα agonist Wy-14643 has been shown to prevent and even reverse steatohepatitis [123]. Other PPARα agonists, notably fibrates, have also been shown to decrease hepatic steatosis in high fat- and high fructose-induced rodent models of NASH [109, 124, 125]. Taken together, research conducted over the past several decades has elucidated PPARα as a critical metabolic regulator that’s activation has generally been found to improve conditions of metabolic syndrome and NAFLD in rodent models.
However, it should be kept in mind that species variations are the leading cause of failure when the results from animal experiment are translated to pharmacological interventions for humans . Much of what we know regarding differences between mouse and humans in regard to PPARα agonists has been determined by the broad use of fibrates clinically. Fibrates are commonly prescribed pharmaceuticals known to activate PPARα effectively lowering circulating triglycerides and decreasing cardiovascular disease risk. Fibrates have been used in practice of hyperlipidemia therapy for more than half a century [126]. High safety of fibrates has been observed in clinical trials except for minor risk for increasing incidence of gallstones, blood clots and muscle-related diseases [127]. Currently, 6 fibrates have been approved in hyperlipidemia therapy, all of which targets PPARα or works as the pan-PPAR agonist, i.e. Clofibrate, Fenofibrate, Bezafibrate, Gemfibrozil, Ciprofibrate, and Pemafibrate [128]. However, as the first fibrate developed to regulate lipid metabolism, Clofibrate was discontinued due to increasing in non-cardiovascular mortality [129]. To achieve higher therapeutic efficiency and less adverse effects, multiple fibrate-like chemicals have been tested in clinical trials, such as Elafibranor [130], Icosabutate [131], and lanifibranor [132]. More potent PPARα agonists, including WY14643, GW9578, and GW7647, have never been applied clinically, but have been widely used in the research [133]. However, administration of fibrates to mice have side-effects not clearly observed in human patients. One example involves severe carcinogenic effects observed in mice and rats treated chronically with fibrates [134]. Overexpression of hepatic PPARα in rodents may lead to hepatic peroxisome proliferation and expression of peroxisomal acyl-CoA oxidases (ACOX1), which can elevate oxidative stress levels, perhaps leading to increased carcinogenesis risk in mice [135]. More generally, hepatic cellular over-proliferation is another common observation in rodents treated with PPARα, which has not been observed in human and nonhuman primate studies as of yet [135]. To accommodate these species differences and to better extrapolate observations in rodent models to humans, humanized PPARα mouse models have been developed [136], in which human PPARα is expressed in a mouse PPARα-null background. In the humanized model, both peroxisome and hepatocyte proliferation and peroxisomal ACO level are not elevated as observed in the WT mice [136]. Differences in the effect on lipid levels induced by the fibrate gemfibrozil between human PPARα transgenic mice and wild-type mice have been reported that gemfibrozil significantly increased HDL level in the transgenic mice but not in the WT cohorts, while hepatocyte hypertrophy is less severe in the transgenic, gemfibrozil-treated group compared with the WT cohorts [137]. Further studies in this area are needed to better understand the potential utility of human PPARα transgenic mice in metabolic syndrome and NAFLD models. Although the humanized PPARα mouse may not replicate the complete human molecular environment, it may work as a more appropriate preclinical animal model for anti- hypertriglyceridemia drug development as well as in studies investigating the importance of PPARα in pathologies of metabolism. For the past decades, our knowledge of the metabolic roles of PPARα and its role in MetS has been expanded on by animal experiments and clinical observations. PPARα expression and activity is modulated by various metabolic pathways and through a variety of PTMs which lead to different dimer combinations and target gene activation. Therefore, a more complete understanding of the intricate intracellular networks related to PPARα, may lead to more effective MetS drugs with more advantageous pharmacodynamic parameters and reduced adverse effects.
FOXO family transcription factors and MetS
A third transcription factor associated with MetS, FOXO1, belongs to a large TF family called the Forkhead box proteins (FOXs) family. The FOXs are multifunctional transcription factors that include more than 50 members discovered in humans; classified into 16 subclasses from FOXA to FOXS [138, 139]. All FOX proteins possess a highly conserved DNA binding domain, FOX-DBD, which interacts with conserved motif 5′-(G/A)(T/C)(A/C)AA(C/T)A-3′ on the promoter of target genes [139]. In mammalian cells, the type O class (FOXO) includes 4 members: FOXO1, FOXO3, FOXO4, and FOXO6. The FOXO members are characterized by DNA binding to a consensus motif sequence 5′-TTGTTTAC-3′. As with the other transcription factors discussed thus far, tissue specificity and PTM state can determine overall FOXO activation as well as which downstream effectors are induced [140, 141]. FOXO1, FOXO3, and FOXO4 are expressed ubiquitously at varying levels among multiple tissues while FOXO6 is expressed primarily in the brain and liver [142, 143]. FOXO proteins undergo multiple PTMs including phosphorylation, acetylation and ubiquitination [144]. These PTMs determines transcriptional activity, subcellular localization and turnover time of FOXO proteins. A growing body of evidence implicates FOXO proteins as critical mediators of metabolism and are highly incorporated into the regulation of energy metabolism, proliferation, cellular differentiation, the alleviation of cellular stress, and ageing [141, 145]. Initial study of FOXO proteins using C. elegens, determined that the worm homolog, DAF-16, was a crucial regulator of life-span [146] and insulin signaling [147].
In this review article, we focus on metabolic roles of a representative member of the FOXO subfamily, FOXO1. FOXO1 (previously known as FKHR) is critical to organismal development, and global deletion of FOXO1, as opposed to other FOXO members, is embryonic lethal [148, 149]. In this review, we will focus on the role FOXO1 plays in diet-regulated hepatic metabolic changes and MetS (Figure 3). Three types of PTMs have been identified for FOXO1; phosphorylation, acetylation, and ubiquitination. FOXO1 is inactivated in the basal state largely due to PTMs regulated through the PI3K pathway [150]. FOXO1 levels are controlled through a complex regulatory cascade that involves indirect sensing of insulin or growth factors (Figure 3). Specifically, PI3K phosphorylates AKT through second messengers which allows AKT to translocate to the nucleus and phosphorylate FOXO1 at three residues (T24, S256, and S319). An additional phosphorylation event of S319 promotes sequential phosphorylations at S322 and S325 by growth factor-induced activation of CK1 [151]. These two phosphorylation events help facilitate binding to RAN-XPO1-mediated nuclear exporting machinery which allow the FOXO1 complex to translocate to the cytosol for degradation [141]. This cascade occurs primarily when glucose/insulin levels are high.
Figure 3.
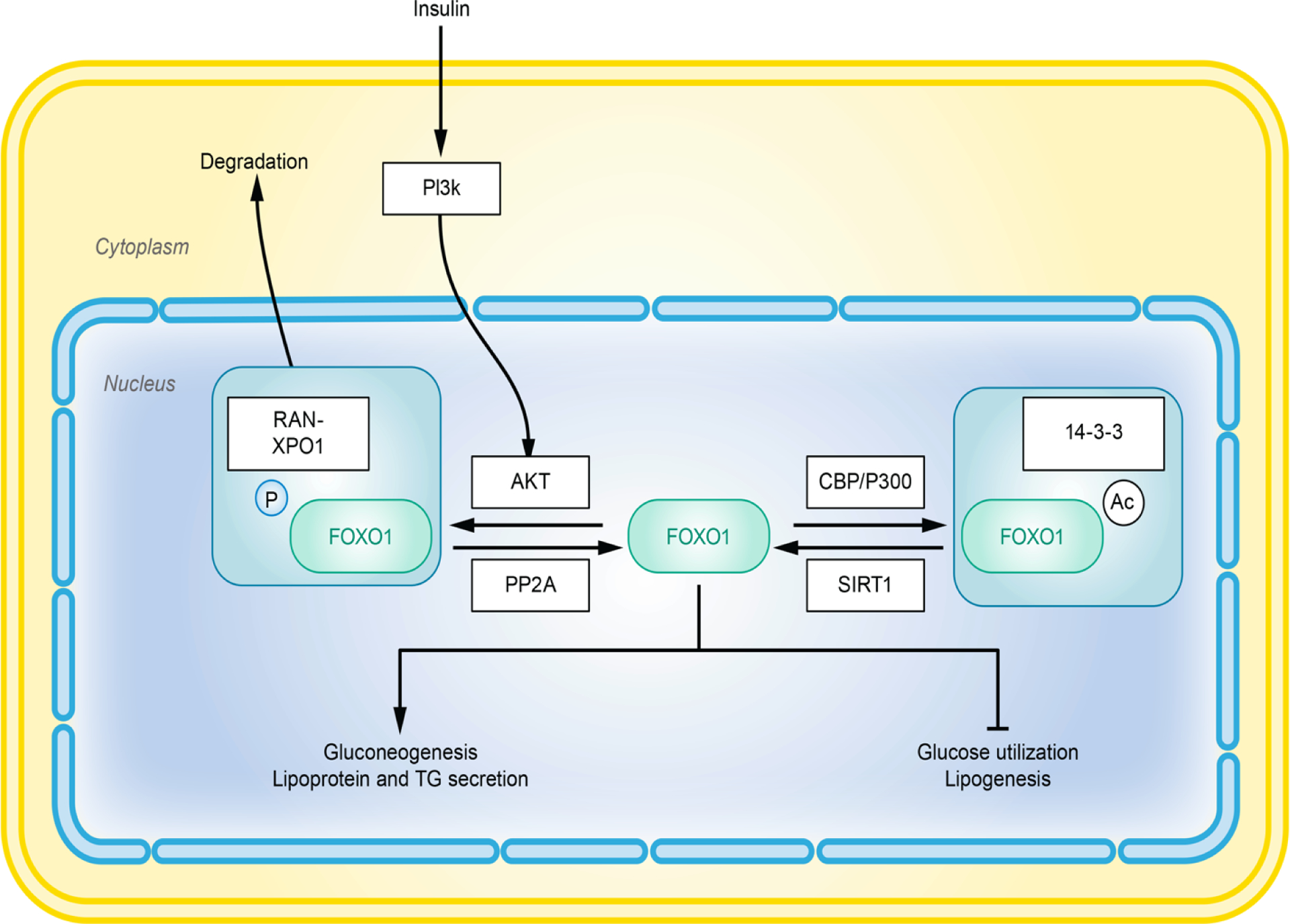
Overview of FOXO1 activation and regulation. Post translational modifications determine the activity and half-life of FOXO1. Phosphorylation of FOXO1 facilitates its binding with RAN-XPO1 complex, which mediates nuclear export and degradation of FOXO1. Acetylation increases affiliation of FOXO1 with the 14-3-3 proteins, leading to inhibition.
On the other hand, additional regulatory phosphorylation events initiated by cyclin-dependent kinase-2 (CDK2) (S249), [152] and JNK can inhibit or promote FOXO1 translocation respectively [153]. Acetylation of FOXO1 is increased by histone acetyltransferases (HATs), such as CREBBP, and is decreased by histone deacetylases (HDACs), such as SIRT1. The roles of these acetylation or deacetylation events in FOXO1 activity is complicated and not completely understood. Currently, the majority of studies support the paradigm that acetylation by the CREBBP/EP300 complex reduces DNA binding, by increasing FOXO1’s affinity to inhibitory 14-3-3 protein. Alternatively, SIRT1 is a protein that can promote the removal of acetyl groups on lysine residues, which acts to activate FOXO1 [141, 154–156]. SIRT1-induced deacetylation may in fact override AKT-mediated phosphorylation of FOXO1 [155, 157]. Additional regulatory platforms such as methylation of mRNA and microRNAs may also regulate abundance of FOXO1 [138, 158], but will not be described here.
PTMs determine stability and function of FOXO1 in various tissues. FOXO1 is highly expressed in all classic tissues regulating energy homeostasis, including liver, pancreas, skeletal muscle, adipose tissue and hypothalamus [159]. FOXO1 is a critical functional regulator in the liver, especially in hepatic glucose and lipid metabolism. As other TFs, FOXO1 requires the formation of a complex with another TF or other coactivators to initiate transcription. Interestingly, FOXO1 may act as a transcriptional repressor in some scenarios. This provides FOXO1 versatility in regulation under different metabolic context in order to maintain metabolic homeostasis. For example, FOXO1 may promote or inhibit different target genes when being acetylated or deacetylated [160]. FOXO1 facilitates gluconeogenesis, lipoprotein and TG secretion but inhibits expression of genes involved in glucose utilization and lipogenesis in the liver [158]. During the feeding state, high concentrations of insulin circulates throughout the blood and stimulates PI3K pathway leading to inhibition of FOXO1 by AKT-induced phosphorylation. Alternatively, fasting may increase both the expression and activity of FOXO1. The mRNA level of FOXO1 dramatically increases during prolonged fasting and is reduced during refeeding [161]. The transcription of gluconeogenesis-related genes such as Pck1 and G6pc are induced by FOXO1 during fasting [162], which may be facilitated by the transcription co-activator PGC1α [163] and transcription factor C/EBPα [164]. On the other hand, FOXO1 negatively regulates transcription of glucose utilization genes including Gck during fasting [165]. The SIN3A/HDAC repressor complex may play a major role in this inhibitory process [166, 167]. Although it is clear that FOXO1 plays major roles in hepatic control of glucose, FOXO1 also has important roles in lipid homeostasis as well. Interestingly, overexpression of FOXO1 during fasting upregulated plasma TG levels [168]. It is believed that FOXO1 can impact on the expression of two genes directly related to TG levels, MTTP and APOC3. MTTP functions as a rate-limiting enzyme in hepatic VLDL assembly, while APOC3 inhibits hydrolysis and uptake TG content from VLDL by binding to lipoprotein lipase (LPL) in the circulation. Both proteins are regulated by FOXO1 at the level of the hepatocyte[169, 170]. Therefore, during fasting, expression of Mttp and Apoc3 are suppressed which can alter the hepatic lipid profile.
Dysregulation of FOXO1 and target genes is observed during MetS. During MetS and diet-induced obesity, FOXO1 may play a critical role in insulin resistance. In the high-fat-diet-induced obesity model, treatment with FOXO1 antisense oligonucleotides reduced hepatic expression of Pck1 and G6pc, ultimately improving both insulin sensitivity and hepatic triglyceride levels [171]. FOXO1 haploinsufficiency can also improve insulin sensitivity in the adipose tissue [5, 172]. Interestingly, other models of MetS including high- fructose diets have also been shown to induce insulin resistance and diabetic dyslipidemia through mechanisms involving FOXO1 [169]. As evidence increases implicating dysregulated FOXO1 signaling in multiple chronic disease pathologies, some efforts have been made to design molecules that can target the pathway. However, since the DNA recognition motif of FOXO1 is short, the number of potential FOXO1 target genes is large, which may lead to unintended effects. Moreover, activity of FOXO1 is controlled by multiple PTMs [141], which differ depending on the metabolic state of the cell. Therefore, to design a molecule that works directly on FOXO1 aimed at achieving a single specific effect on metabolism seems less plausible than one targeting upstream controllers of FOXO1, such as PI3K pathway factors or SIRT1. A high-throughput screening study published in 2010 identified a group of compounds bound to FOXO1 [173]. AS1708727 is one of them which potently inhibits activity of FOXO1. Apart from the pre-clinical studies in cell culture [174] and in animal models [175], there is however no published clinical follow-up related to AS1708727. Additionally, Langlet et al. reported in 2017 that corepressor SIN3A provided specificity to target selecting of FOXO1 [167]. They took advantage of this property to develop a series of molecules regulating FOXO1 which selectively affect transcription of G6pc rather than Gck or vice versa. They claimed that due to pharmacokinetic issues they failed to utilize the chemicals in an in vivo experiment. In addition, multiple natural compounds may effectively manipulate activity of FOXO1, and these are reviewed here [176].
At the molecular level, FOXO1 is a transcriptional regulator of gluconeogenesis, is regulated by insulin, and impacts on hepatic lipid metabolism [177]. One of FOXO1’s critical roles is to regulate the insulin response during fasting and feeding states, and the liver is one of FOXO1’s critical sites of action. During feeding, insulin mediates an increase of glucose uptake into hepatocytes, suppression of gluconeogenesis and glycogenolysis, and upregulation of glycogen synthesis. During times of fasting, the limited insulin stimulation results in increases of gluconeogenesis through an upregulation of PCK1 and G6PC largely mediated through interplay between AKT and FOXO1. When FOXO1 is constitutively expressed in the liver, fasting blood glucose rises [178]. During fasting conditions, FOXO1 is dephosphorylated at AKT sites, localized in the nucleus, and activated leading to the transcriptional induction of two critical gluconeogenic enzymes, G6PC and PCK1 [179] and increased hepatic glucose production. In the fed state, insulin signaling activates PI3K and the subsequent production of PIP3 activates AKT. AKT phosphorylates FOXO1 at Thr24, Ser253 and Ser316 leading to its nuclear exportation and inactivation [180] with subsequent suppression of gluconeogenesis. NAFLD is characterized by hepatic insulin resistance [181], proatherogenic dyslipidemia, and vascular damage [182]. In approximately 30 percent of NAFLD cases, fatty liver may progress to NASH, which is thought to be triggered by lipid peroxidation and mitochondrial dysfunction leading to oxidative stress and systemic inflammation [183]. The progression of NAFLD to more severe and permanent disorders including NASH, cirrhosis, or liver failure has been shown to be linked to the activation of glucose and lipogenesis-regulating transcription factor, FOXO1 [184]. For example, although NAFLD is characterized by hyperinsulinemia, under oxidative stress conditions, as in those observed in NASH [183], FOXO1 becomes unresponsive to insulin because of interaction with the deacetylase sirtuin 1, resulting in induction of glucogenic genes [157, 185]. In summary, FOXO1 is an essential gene and functions as a MetS related transcription factor. It binds to its heterodimer partners, which triggers activation or repression of the multiple other TFs and metabolic related genes. This dual regulation by FOXO1 highlights its role in the glucose and lipid metabolism, indicating a therapeutic potential in the MetS.
Transcription factors as targets of nutritional interventions
As has been discussed throughout this review, signaling pathways involving transcription factors such as CREB3, PPARA, and FOXO1 are critical mediators of nutrient sensing and disease states related to MetS including NAFLD. As these pathways are primed to respond to specific bioactive nutrient stimuli as well as overall energy availability, it is not a surprise that these three transcription factors have been shown to be modulated by multiple nutrient-based therapies that have been shown effective at ameliorating MetS and related disorders. For example, caloric restriction (CR) is an effective remedy that applies a lower-calorie but nutrition-balanced diet for metabolic disorders [186, 187]. Studies on beneficial effects of CR application have sustained for decades in varies animal models, from yeast, C. elegans, rodents to primates [186, 188]. CR has been applied to genetically modified obesity models such as ob/ob mice, to evaluate its effects in energy metabolism and body composition in NAFLD [187, 189, 190]. In the well-established db/db obesity model, CR modulated hepatic lipidomic profiling, with that more than 100 lipid species differently identified after CR treatment [191]. CR treatment also influences the regulation of TG synthesis and lipogenesis through more than one molecular mechanism, especially through regulating TFs. For example, hepatic SIRT1 has been downregulated in the db/db mouse but it was reversed under CR. As above mentioned, SIRT1, an activator of FOXO1, can indirectly involve in regulation of hepatic gluconeogenesis. On the other hand, increased expression of PPARA in db/db mice fails to be reversed by CR treatment. However, whether or not CREB3L3 plays some roles in the beneficial metabolic effects of CR is currently unclear. CREB3L3 is upregulated during short-term fasting and this effect is observed in the zebra fish model being fasted as long as 3 weeks [192]. On the contrary, evidence has shown that, as the major target of CREB3L3 during metabolic challenge, FGF21 has no role in CR induced metabolic changes [193]. Therefore, it’s uncertain if CREB3L3 extends its roles in short-term fasting to long term CR, and which targets does CREB3L3 regulate during CR. Moreover, short term CR treatment could alleviate obesity associated unfolded protein response (UPR) in the liver of ob/ob mouse model [190]. In the human studies, CR has been suggested to be beneficial for reducing hepatic lipid level either in the non-NAFLD obesity individuals [189] or in the NAFLD patients [187].
Additionally, the ingestion of plant-based polyphenols has been shown to regulate metabolism and MetS-related pathologies through mechanisms involving the model transcription factors described herein. Polyphenolic compounds constitute more than 4000 members with many inducing beneficial effects on metabolic health. In the final section of this review we will discuss how three model polyphenols, Epigallocatechin gallate (EGCG), curcumin, and resveratrol interact with the transcription factors discussed thus far to perhaps impact on hepatic metabolism and metabolic diseases such as NAFLD.
EGCG
Flavonoids are a large subgroup of polyphenols found in fruits and vegetables and have been shown to have multiple bioactive properties. It has been shown that isoflavones, one subset of flavonoids, regulate PPARα through aldose reductase [194]or directly as a ligand [195]. Epigallocatechin gallate, a tea enriched flavanol, has been shown to activate PPARA using cell-based assays. Specifically, tea extracts (green tea, black tea,oolong tea and doongule tea) and tea components (epigallocatechin gallate, epigallocatechin, epicatechin gallate, epicatechin and gallic acid), were shown to activate mouse cloned Ppara to differeing degrees with green tea and black tea extracts, and epigallocatechin gallate, increasing the activation of Ppara 1.5–2 times compared with the control [196]. Similar effects have been observed using feeding studies from a variety of species including dogs [197] and rats [198][199]. For example, rats with metabolic syndrome were fed green extracts for 9 weeks and fasting blood glucose and triglycerides, were significantly lower due to EGCG supplementation. Ppara gene expression was significantly higher in EGCG fed mice [199]. In addition to activation of Ppara, tea catechins may exert protective effects through FOXO1. In hyperlipidemic rats, EGCG was shown to decrease liver injury and oxidative stress by activating SIRT1 and increasing FOXO1 protein. Interestingly, these changes were not observed in rats deficient in Srebp2 [200]. In a separate cell culture study, treatment of 10 μM EGCG decreased hepatic glucose production, repressed both gluconeogenesis and glycogenolysis, blocked phosphorylation of FOXO1 at S273, and suppressed FOXO1 translocation [201]. EGCG has also been shown to counteract the PI3K/AKT/FOXO1 pathway to reduce hepatic injury in rats exhibiting NAFLD [202]. Finally, the impact of EGCG on CREB3L3 has not been readily reported. There is one report that EGCG can induce CREBZF expression through FOXO1, and CREBZF is known to interact with CREBH to inhibit its transcriptional activity [203] [204].
Curcumin
Additionally, curcumin, a flavonoid found in turmeric has also been shown to have wide-ranging effects on metabolism. Misra et al. [204] discovered that curcumin increased expression of transmembrane O-mannosyltransferase targeting cadherins 3 (TMTC3) through the AMPK pathway. TMTC3 may compete with PGC1α, a transcriptional co-activator of CREBH, resulting in diminished expression of the target genes of CREBH. Curcumin has been shown to protect against streptozotocin-induced diabetic phenotypes perhaps through the enhanced phosphorylation of AKT and inhibited acetylation of FOXO1 [205]. Additionally, tetrahydrocurcumin, a metabolite of curcumin, has been shown in hepatocyte cell models to impact on glucose signaling through modulation of phosphorylation of the insulin receptor substrate 1 (IRS1)/PI3K/AKT and FOXO1 [206]. Curcumin also increases expression of FOXO1 in white adipose tissue perhaps impacting on lipid metabolism and storage [207]. A large body of work has implicated the importance of PPAR signaling in regard to the protective effects observed with curcumin supplementation. Although PPARG has received an abundance of attention [208], a growing body of evidence shows that curcumin may also impact on PPARA expression and transcriptional activity. For example, in fish fed a high fat diet, enrichment with 0.04 % curcumin for 10 weeks resulted in higher hepatic expression of ppara, cpt1, and acox1 and reduced hepatic lipid deposition [209]. In rats, curcumin supplementation, has been shown to lead to decreased concentrations of serum insulin and glucose and total hepatic cholesterol; PPARA expression was also increased in these rats [210]. Interestingly, curcumin may impact on PPAR expression through epigenetic mechanisms not yet fully appreciated [211].Similarly, in a mouse model of hyperlipidemia, curcumin treatment can lead to upregulation of hepatic Ppara expression, which may ameliorate hepatic cholesterol metabolism in LDLR knockout mice [212].
Resveratrol
Finally, resveratrol, a stilbenoid phenol found in red wine and studied intensively for its possible antiaging and other beneficial effects on metabolic disorders, has been shown to decrease both mRNA level and activated form of CREBH in HFD treated mouse liver, likely through the regulation of ATF6/SIRT1 [213]. Interestingly, SIRT1 is a possible deacetylase that could modulate the acetylation states of CREB3l3 under fasting conditions [214]. Additionally, Vaticanol B, a resveratrol tetramer has been shown to impact on endoplasmic reticulum stress response; which CREB3L3 can play a role [215]. Regarding PPARA, multiple experiments have shown that resveratrol directly binds PPARA and the 4′-hydroxyl group of resveratrol may be critical for the direct activation of PPARA [216, 217]. Hepatic expression of PPARα is elevated by treatment of resveratrol, majorly through PKA/AMPK/PPARA pathway and downregulation of PPARA levels may impair hepatic protective effect of resveratrol [218, 219]. As with curcumin, there may be understudied mechanisms related to epigenetics as well [220]. Finally, resveratrol may also work through the FOXO1 pathway, as decreased expression of SREBP1 has been observed after resveratrol treatment in cells exposed to media containing high palmitate and glucose [221]. Resveratrol may impact on gluconeogenic genes (e.g., PCK1 and G6PC) through mechanisms related to decreased phosphorylation of AKT and FOXO1. Additionally, resveratrol increases nuclear localization of FOXO1 where it can be deacetylated by SIRT1 [222]. Another mechanism that Resveratrol may impact on hepatic metabolism is through the nutrient sensor, AMPK. AMPK was recently shown to phosphorylate FOXO1 at T649. This phosphorylation is critical for FOXO1 stability and nuclear localization [223]. The importance of FOXO1, AMPK, and SIRT1 in the beneficial effects of resveratrol were also observed in a 12-week supplementation study in mice. At study conclusion, circulating triglycerides were decreased due to Resveratrol supplementation and expression levels of hepatic SIRT1, and phosphorylated AMPK and FOXO1 were increased [224].
Conclusion
Transcription factors are the terminal executors of signaling pathways and play a major role in determining the type and extent of response towards metabolic stimuli. Among the many environmental stimuli regulating cellular responses, diet and nutrient sensing is the predominate signal upstream of TFs that are known to maintain hepatic metabolic homeostasis. Dysregulation of TF networks are highly associated with a series of metabolic disorders including MetS and related pathologies such as NAFLD and T2D. Using dietary and lifestyle interventions such as CR or flavonoid enriched diets may relieve MetS and help to reset dysregulation of TF networks [225]. In this review, we have evaluated three TFs that are major contributors to hepatic metabolism; CREB3L3, PPARα, and FOXO1, with a focus on describing how they are regulated at the molecular level, how their downstream targets modulate hepatic and lipid homeostasis, and how they are modulated during times of metabolic stress and disease. With a better understanding of the complex interactions between transcription factors and the genes that they regulate, more effective treatment strategies especially nutrition-based manipulation may be developed to mitigate or prevent MetS in the future.
Highlights.
CREB3L3 responds to multiple metabolic signals to regulate target genes involved in glucose and lipid metabolism.
PPARα is activated by multiple intra/extra-cellular metabolic signals leading to modulation of lipid homeostasis.
FOXO1 is a transcriptional regulator of gluconeogenesis and is activated by multiple metabolic signals.
Transcription factors play a critical role in metabolic dysfunction and may be amenable to nutrient-based interventions.
Funding:
This work was supported by the National Institute of Environmental Health Sciences [P30 ES020957, R00ES028734] and the National Institute of Diabetes and Digestive and Kidney [R01DK106540] at the National Institutes of Health and the Office of the Vice President for Research at Wayne State University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviation
- ACOX1
Acyl-CoA oxidase 1
- AHF
Atherogenic high fat diet
- AKT
RAC-alpha serine/threonine-protein kinase
- ALT/AST
Aspartate transaminase/alanine transaminase ratio
- AMPK
AMP-activated protein kinase
- APOA1
Apolipoprotein A1
- APOC3
Apolipoprotein C-III
- ATF6
Activating transcription factor 6
- bHLH
basic Helix-Loop-Helix
- BMAL1
Aryl Hydrocarbon Receptor Nuclear Translocator Like (ARNTL)
- bZIP
Basic leucine zipper
- C/EBP
CCAAT-enhancer-binding protein
- CB1R
Cannabinoid Receptor 1
- CCl4
Carbon tetrachloride
- CDC
Centers for Disease Control and Prevention
- CDK2
Cyclin-dependent kinase-2
- ChIP
Chromatin immunoprecipitation
- CIDEC/FSP27
Cell Death Inducing DFFA Like Effector C
- CLOCK
Clock Circadian Regulator
- CPT1
Carnitine Palmitoyltransferase 1
- CR
Caloric restriction
- CRE
cAMP response element
- CREB3L3
cAMP Responsive Element Binding Protein 3 Like 3
- CREBBP
CREB-binding protein
- CREBZF
CREB/ATF BZIP Transcription Factor
- DR1
PPRE direct repeat 1
- EMSA
Electrophoretic mobility shift assay
- EP300
E1A Binding Protein P300
- ER
Endoplasmic reticulum
- FA
Fatty acid
- FADS
Flavin Adenine Dinucleotide Synthetase 1 (FLAD1)
- FASN
Fatty Acid Synthase
- FGF21
Fibroblast growth factor 21
- FOXO1
Forkhead Box O1
- G6PC
Glucose-6-phosphatase catalytic subunit
- GCK
Glucokinase
- GSK3
Glycogen Synthase Kinase 3
- GWAS
Genome-wide association study
- HAT
Histone acetyltransferase
- HDAC
Histone deacetylase
- HFD
High-fat diet
- HNF4
Hepatocyte Nuclear Factor 4
- HNF4A
Hepatocyte nuclear factor 4 alpha
- IRS1
Insulin receptor substrate 1
- KO
Knockout
- LDLR
Low density lipoprotein receptor
- LPL
Lipoprotein lipase
- LXR
Liver X receptor
- MAPK/ERK
Mitogen-Activated Protein Kinase
- MCD
Methionine/choline deficient diet
- MetS
Metabolic syndrome
- MTTP
Microsomal triglyceride transfer protein
- NAFLD
Non-alcoholic fatty liver disease
- NASH
Nonalcoholic steatohepatitis
- NR
Nuclear receptor
- NR1D1
Nuclear Receptor Subfamily 1 Group D Member 1
- NZO
New Zealand Obese mouse model
- PCK1
Phosphoenolpyruvate carboxykinase 1
- PGC1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PI3K
Phosphoinositide 3-kinase
- PKA
Protein kinase A
- PKC
Protein kinase C
- PPAR
Peroxisome proliferator-activated receptor
- PPREs
Peroxisome proliferators response elements
- PTMs
Post-translational modifications
- PXR
Pregnane X receptor
- PYGL
Liver glycogen phosphorylase
- RAN
RAN, Member RAS Oncogene Family
- RXR
Retinoid X receptor
- SIN3A
SIN3 Transcription Regulator Family Member A
- SIRT1
Sirtuin 1
- SREBP1
Sterol Regulatory Element Binding Transcription Factor 1
- T2D
Type 2 diabetes
- TF
Transcription factor
- TG
triglyceride
- TLR4
Toll Like Receptor 4
- TMTC3
Transmembrane O-mannosyltransferase targeting cadherins 3
- UPR
Unfolded protein response
- VLDL
Very low-density lipoprotein
- WT
Wildtype
- XPO1
Exportin 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that there are no competing financial interests.
References
- 1.National Cholesterol Education Program Expert Panel on Detection, E. and A. Treatment of High Blood Cholesterol in, Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation, 2002. 106(25): p. 3143–421. [PubMed] [Google Scholar]
- 2.Saklayen MG, The Global Epidemic of the Metabolic Syndrome. Curr Hypertens Rep, 2018. 20(2): p. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng M, et al. , Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet, 2014. 384(9945): p. 766–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonomini F, Rodella LF, and Rezzani R, Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis, 2015. 6(2): p. 109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakae J, et al. , The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell, 2003. 4(1): p. 119–29. [DOI] [PubMed] [Google Scholar]
- 6.Kietzmann T, Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol, 2017. 11: p. 622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh H, Khan AA, and Dinner AR, Gene regulatory networks in the immune system. Trends Immunol, 2014. 35(5): p. 211–8. [DOI] [PubMed] [Google Scholar]
- 8.Kawai M and Rosen CJ, PPARgamma: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol, 2010. 6(11): p. 629–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lambert SA, et al. , The Human Transcription Factors. Cell, 2018. 172(4): p. 650–665. [DOI] [PubMed] [Google Scholar]
- 10.Rui L, Energy metabolism in the liver. Compr Physiol, 2014. 4(1): p. 177–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bechmann LP, et al. , The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol, 2012. 56(4): p. 952–64. [DOI] [PubMed] [Google Scholar]
- 12.Paschos P and Paletas K, Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia, 2009. 13(1): p. 9–19. [PMC free article] [PubMed] [Google Scholar]
- 13.Kotronen A, et al. , Prediction of non-alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology, 2009. 137(3): p. 865–72. [DOI] [PubMed] [Google Scholar]
- 14.Yki-Jarvinen H, Diagnosis of nonalcoholic fatty liver disease (NAFLD). Duodecim, 2016. 132(22): p. 2099–106. [PubMed] [Google Scholar]
- 15.Friedman SL, et al. , Mechanisms of NAFLD development and therapeutic strategies. Nat Med, 2018. 24(7): p. 908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puri P, et al. , The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology, 2009. 50(6): p. 1827–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feldstein AE, et al. , Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Lipid Res, 2010. 51(10): p. 3046–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loomba R, et al. , Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis. J Lipid Res, 2015. 56(1): p. 185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, et al. , Noninvasive Detection of Nonalcoholic Steatohepatitis Using Clinical Markers and Circulating Levels of Lipids and Metabolites. Clin Gastroenterol Hepatol, 2016. 14(10): p. 1463–1472 e6. [DOI] [PubMed] [Google Scholar]
- 20.Alonso C, et al. , Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology, 2017. 152(6): p. 1449–1461 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tilg H and Moschen AR, Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology, 2010. 52(5): p. 1836–46. [DOI] [PubMed] [Google Scholar]
- 22.Tilg H, Adolph TE, and Moschen AR, Multiple Parallel Hits Hypothesis in NAFLD - Revisited After a Decade. Hepatology, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi Y, Soejima Y, and Fukusato T, Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol, 2012. 18(19): p. 2300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jahn D, et al. , Animal models of NAFLD from a hepatologist’s point of view. Biochim Biophys Acta Mol Basis Dis, 2019. 1865(5): p. 943–953. [DOI] [PubMed] [Google Scholar]
- 25.Van Herck MA, Vonghia L, and Francque SM, Animal Models of Nonalcoholic Fatty Liver Disease-A Starter’s Guide. Nutrients, 2017. 9(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khan HA and Margulies CE, The Role of Mammalian Creb3-Like Transcription Factors in Response to Nutrients. Front Genet, 2019. 10: p. 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang M, et al. , Human leucine zipper protein promotes hepatic steatosis via induction of apolipoprotein A-IV. FASEB J, 2017. 31(6): p. 2548–2561. [DOI] [PubMed] [Google Scholar]
- 28.Kim TH, et al. , Identification of Creb3l4 as an essential negative regulator of adipogenesis. Cell Death Dis, 2014. 5: p. e1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim TH, et al. , Effects of low-fat diet and aging on metabolic profiles of Creb3l4 knockout mice. Nutr Diabetes, 2015. 5: p. e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang K, et al. , Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell, 2006. 124(3): p. 587–99. [DOI] [PubMed] [Google Scholar]
- 31.Luebke-Wheeler J, et al. , Hepatocyte nuclear factor 4alpha is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology, 2008. 48(4): p. 1242–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X, et al. , Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J Lipid Res, 2014. 55(5): p. 850–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kikuchi T, et al. , Intestinal CREBH overexpression prevents high-cholesterol diet-induced hypercholesterolemia by reducing Npc1l1 expression. Mol Metab, 2016. 5(11): p. 1092–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The Broad Institute of MIT and Harvard. GTEx portal. 2019; Available from: https://www.gtexportal.org/home/.
- 35.Zhang C, et al. , Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology, 2012. 55(4): p. 1070–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim H, et al. , Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor alpha to regulate metabolic hormone FGF21. Endocrinology, 2014. 155(3): p. 769–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu X, et al. , Transcriptional activation of Fsp27 by the liver-enriched transcription factor CREBH promotes lipid droplet growth and hepatic steatosis. Hepatology, 2015. 61(3): p. 857–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Danno H, et al. , The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARalpha. Biochem Biophys Res Commun, 2010. 391(2): p. 1222–7. [DOI] [PubMed] [Google Scholar]
- 39.Lee MW, et al. , Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab, 2010. 11(4): p. 331–9. [DOI] [PubMed] [Google Scholar]
- 40.Hihi AK, Michalik L, and Wahli W, PPARs: transcriptional effectors of fatty acids and their derivatives. Cell Mol Life Sci, 2002. 59(5): p. 790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gentile CL, et al. , Fatty acids regulate CREBh via transcriptional mechanisms that are dependent on proteasome activity and insulin. Mol Cell Biochem, 2010. 344(1–2): p. 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chanda D, et al. , Cannabinoid receptor type 1 (CB1R) signaling regulates hepatic gluconeogenesis via induction of endoplasmic reticulum-bound transcription factor cAMP-responsive element-binding protein H (CREBH) in primary hepatocytes. J Biol Chem, 2011. 286(32): p. 27971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun LP, et al. , Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem, 2005. 280(28): p. 26483–90. [DOI] [PubMed] [Google Scholar]
- 44.Pulit SL, et al. , Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet, 2019. 28(1): p. 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JH, et al. , The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med, 2011. 17(7): p. 812–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johansen CT, Kathiresan S, and Hegele RA, Genetic determinants of plasma triglycerides. J Lipid Res, 2011. 52(2): p. 189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Genomes Project C, et al. , A map of human genome variation from population-scale sequencing. Nature, 2010. 467(7319): p. 1061–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim H, et al. , CREBH Maintains Circadian Glucose Homeostasis by Regulating Hepatic Glycogenolysis and Gluconeogenesis. Mol Cell Biol, 2017. 37(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Z, et al. , Interaction between stress responses and circadian metabolism in metabolic disease. Liver Res, 2017. 1(3): p. 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei J, et al. , HRD1-ERAD controls production of the hepatokine FGF21 through CREBH polyubiquitination. EMBO J, 2018. 37(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fisher FM and Maratos-Flier E, Understanding the Physiology of FGF21. Annu Rev Physiol, 2016. 78: p. 223–41. [DOI] [PubMed] [Google Scholar]
- 52.Park JG, et al. , CREBH-FGF21 axis improves hepatic steatosis by suppressing adipose tissue lipolysis. Sci Rep, 2016. 6: p. 27938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brunt EM, et al. , Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol, 1999. 94(9): p. 2467–74. [DOI] [PubMed] [Google Scholar]
- 54.Wang M, Zhao S, and Tan M, bZIP transmembrane transcription factor CREBH: Potential role in non-alcoholic fatty liver disease (Review). Mol Med Rep, 2016. 13(2): p. 1455–62. [DOI] [PubMed] [Google Scholar]
- 55.Kleiner DE, et al. , Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology, 2005. 41(6): p. 1313–21. [DOI] [PubMed] [Google Scholar]
- 56.Min AK, et al. , cAMP response element binding protein H mediates fenofibrate-induced suppression of hepatic lipogenesis. Diabetologia, 2013. 56(2): p. 412–22. [DOI] [PubMed] [Google Scholar]
- 57.Zheng Z, et al. , CREBH Couples Circadian Clock with Hepatic Lipid Metabolism. Diabetes, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chanda D, et al. , Activation of cannabinoid receptor type 1 (Cb1r) disrupts hepatic insulin receptor signaling via cyclic AMP-response element-binding protein H (Crebh)-mediated induction of Lipin1 gene. J Biol Chem, 2012. 287(45): p. 38041–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chitturi S and George J, Interaction of iron, insulin resistance, and nonalcoholic steatohepatitis. Curr Gastroenterol Rep, 2003. 5(1): p. 18–25. [DOI] [PubMed] [Google Scholar]
- 60.Dongiovanni P, et al. , Iron in fatty liver and in the metabolic syndrome: a promising therapeutic target. J Hepatol, 2011. 55(4): p. 920–32. [DOI] [PubMed] [Google Scholar]
- 61.Tsuchiya H, et al. , High-fat, high-fructose diet induces hepatic iron overload via a hepcidin-independent mechanism prior to the onset of liver steatosis and insulin resistance in mice. Metabolism, 2013. 62(1): p. 62–9. [DOI] [PubMed] [Google Scholar]
- 62.Vecchi C, et al. , ER stress controls iron metabolism through induction of hepcidin. Science, 2009. 325(5942): p. 877–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Palanker L, et al. , Drosophila HNF4 regulates lipid mobilization and beta-oxidation. Cell Metab, 2009. 9(3): p. 228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grygiel-Gorniak B, Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications--a review. Nutr J, 2014. 13: p. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chinetti-Gbaguidi G, Fruchart JC, and Staels B, Role of the PPAR family of nuclear receptors in the regulation of metabolic and cardiovascular homeostasis: new approaches to therapy. Curr Opin Pharmacol, 2005. 5(2): p. 177–83. [DOI] [PubMed] [Google Scholar]
- 66.Sever R and Glass CK, Signaling by nuclear receptors. Cold Spring Harb Perspect Biol, 2013. 5(3): p. a016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amoutzias GD, et al. , Choose your partners: dimerization in eukaryotic transcription factors. Trends Biochem Sci, 2008. 33(5): p. 220–9. [DOI] [PubMed] [Google Scholar]
- 68.Willson TM, et al. , The PPARs: from orphan receptors to drug discovery. J Med Chem, 2000. 43(4): p. 527–50. [DOI] [PubMed] [Google Scholar]
- 69.Berger J and Moller DE, The mechanisms of action of PPARs. Annu Rev Med, 2002. 53: p. 409–35. [DOI] [PubMed] [Google Scholar]
- 70.Dubois V, et al. , Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J Clin Invest, 2017. 127(4): p. 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pawlak M, Lefebvre P, and Staels B, Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol, 2015. 62(3): p. 720–33. [DOI] [PubMed] [Google Scholar]
- 72.Diradourian C, Girard J, and Pegorier JP, Phosphorylation of PPARs: from molecular characterization to physiological relevance. Biochimie, 2005. 87(1): p. 33–8. [DOI] [PubMed] [Google Scholar]
- 73.Georgiadi A and Kersten S, Mechanisms of gene regulation by fatty acids. Adv Nutr, 2012. 3(2): p. 127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu HE, et al. , Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell, 1999. 3(3): p. 397–403. [DOI] [PubMed] [Google Scholar]
- 75.Poulsen L, Siersbaek M, and Mandrup S, PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol, 2012. 23(6): p. 631–9. [DOI] [PubMed] [Google Scholar]
- 76.Montagner A, et al. , Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut, 2016. 65(7): p. 1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kersten S, et al. , Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest, 1999. 103(11): p. 1489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chakravarthy MV, et al. , “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab, 2005. 1(5): p. 309–22. [DOI] [PubMed] [Google Scholar]
- 79.Sanderson LM, et al. , Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) but not PPARalpha serves as a plasma free fatty acid sensor in liver. Mol Cell Biol, 2009. 29(23): p. 6257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burns KA and Vanden Heuvel JP, Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta, 2007. 1771(8): p. 952–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Juge-Aubry CE, et al. , Regulation of the transcriptional activity of the peroxisome proliferator-activated receptor alpha by phosphorylation of a ligand-independent trans-activating domain. J Biol Chem, 1999. 274(15): p. 10505–10. [DOI] [PubMed] [Google Scholar]
- 82.Blanquart C, et al. , Peroxisome proliferator-activated receptor alpha (PPARalpha) turnover by the ubiquitin-proteasome system controls the ligand-induced expression level of its target genes. J Biol Chem, 2002. 277(40): p. 37254–9. [DOI] [PubMed] [Google Scholar]
- 83.Kou XH, et al. , Bilobetin ameliorates insulin resistance by PKA-mediated phosphorylation of PPARalpha in rats fed a high-fat diet. Br J Pharmacol, 2012. 165(8): p. 2692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blanquart C, et al. , The protein kinase C signaling pathway regulates a molecular switch between transactivation and transrepression activity of the peroxisome proliferator-activated receptor alpha. Mol Endocrinol, 2004. 18(8): p. 1906–18. [DOI] [PubMed] [Google Scholar]
- 85.Gray JP, et al. , Regulation of peroxisome proliferator-activated receptor alpha by protein kinase C. Biochemistry, 2005. 44(30): p. 10313–21. [DOI] [PubMed] [Google Scholar]
- 86.Hinds TD Jr., et al. , Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3beta Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) alpha. J Biol Chem, 2016. 291(48): p. 25179–25191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Inoue J, et al. , PPARalpha gene expression is up-regulated by LXR and PXR activators in the small intestine. Biochem Biophys Res Commun, 2008. 371(4): p. 675–8. [DOI] [PubMed] [Google Scholar]
- 88.Hayhurst GP, et al. , Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol, 2001. 21(4): p. 1393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pineda Torra I, et al. , Characterization of the human PPARalpha promoter: identification of a functional nuclear receptor response element. Mol Endocrinol, 2002. 16(5): p. 1013–28. [DOI] [PubMed] [Google Scholar]
- 90.Nakagawa Y, et al. , Hepatic CREB3L3 controls whole-body energy homeostasis and improves obesity and diabetes. Endocrinology, 2014. 155(12): p. 4706–19. [DOI] [PubMed] [Google Scholar]
- 91.Leuenberger N, Pradervand S, and Wahli W, Sumoylated PPARalpha mediates sex-specific gene repression and protects the liver from estrogen-induced toxicity in mice. J Clin Invest, 2009. 119(10): p. 3138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pourcet B, et al. , SUMOylation of human peroxisome proliferator-activated receptor alpha inhibits its trans-activity through the recruitment of the nuclear corepressor NCoR. J Biol Chem, 2010. 285(9): p. 5983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boergesen M, et al. , Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor alpha in mouse liver reveals extensive sharing of binding sites. Mol Cell Biol, 2012. 32(4): p. 852–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Castelein H, Declercq PE, and Baes M, DNA binding preferences of PPAR alpha/RXR alpha heterodimers. Biochem Biophys Res Commun, 1997. 233(1): p. 91–5. [DOI] [PubMed] [Google Scholar]
- 95.DiRenzo J, et al. , Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Mol Cell Biol, 1997. 17(4): p. 2166–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tzeng J, et al. , An Ideal PPAR Response Element Bound to and Activated by PPARalpha. PLoS One, 2015. 10(8): p. e0134996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bedi S, Identification of Novel Ligands and Structural Requirements for Heterodimerization of the Liver X Receptor Alpha. Browse all Theses and Dissertations, 2017. 1743. [Google Scholar]
- 98.Badman MK, et al. , Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab, 2007. 5(6): p. 426–37. [DOI] [PubMed] [Google Scholar]
- 99.Inagaki T, et al. , Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab, 2007. 5(6): p. 415–25. [DOI] [PubMed] [Google Scholar]
- 100.Viswakarma N, et al. , Coactivators in PPAR-Regulated Gene Expression. PPAR Res, 2010. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rakhshandehroo M, et al. , Comprehensive analysis of PPARalpha-dependent regulation of hepatic lipid metabolism by expression profiling. PPAR Res, 2007. 2007: p. 26839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bougarne N, et al. , Molecular Actions of PPARalpha in Lipid Metabolism and Inflammation. Endocr Rev, 2018. 39(5): p. 760–802. [DOI] [PubMed] [Google Scholar]
- 103.Rakhshandehroo M, et al. , Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One, 2009. 4(8): p. e6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Herberg L and Coleman DL, Laboratory animals exhibiting obesity and diabetes syndromes. Metabolism, 1977. 26(1): p. 59–99. [DOI] [PubMed] [Google Scholar]
- 105.Knebel B, et al. , Alteration of Liver Peroxisomal and Mitochondrial Functionality in the NZO Mouse Model of Metabolic Syndrome. Proteomics Clin Appl, 2018. 12(1). [DOI] [PubMed] [Google Scholar]
- 106.Guerre-Millo M, et al. , Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem, 2000. 275(22): p. 16638–42. [DOI] [PubMed] [Google Scholar]
- 107.Kim H, et al. , Peroxisome proliferator-activated receptor-alpha agonist treatment in a transgenic model of type 2 diabetes reverses the lipotoxic state and improves glucose homeostasis. Diabetes, 2003. 52(7): p. 1770–8. [DOI] [PubMed] [Google Scholar]
- 108.Haluzik MM, et al. , Improvement of insulin sensitivity after peroxisome proliferator-activated receptor-alpha agonist treatment is accompanied by paradoxical increase of circulating resistin levels. Endocrinology, 2006. 147(9): p. 4517–24. [DOI] [PubMed] [Google Scholar]
- 109.Larter CZ, et al. , Peroxisome proliferator-activated receptor-alpha agonist, Wy 14,643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. J Gastroenterol Hepatol, 2012. 27(2): p. 341–50. [DOI] [PubMed] [Google Scholar]
- 110.Guerre-Millo M, et al. , PPAR-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes, 2001. 50(12): p. 2809–14. [DOI] [PubMed] [Google Scholar]
- 111.Tordjman K, et al. , PPARalpha deficiency reduces insulin resistance and atherosclerosis in apoE-null mice. J Clin Invest, 2001. 107(8): p. 1025–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Haluzik M, Gavrilova O, and LeRoith D, Peroxisome proliferator-activated receptor-alpha deficiency does not alter insulin sensitivity in mice maintained on regular or high-fat diet: hyperinsulinemic-euglycemic clamp studies. Endocrinology, 2004. 145(4): p. 1662–7. [DOI] [PubMed] [Google Scholar]
- 113.Haluzik MM and Haluzik M, PPAR-alpha and insulin sensitivity. Physiol Res, 2006. 55(2): p. 115–22. [DOI] [PubMed] [Google Scholar]
- 114.Liss KH and Finck BN, PPARs and Nonalcoholic Fatty Liver Disease. Biochimie, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tailleux A, Wouters K, and Staels B, Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta, 2012. 1821(5): p. 809–18. [DOI] [PubMed] [Google Scholar]
- 116.Aoyama T, et al. , Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem, 1998. 273(10): p. 5678–84. [DOI] [PubMed] [Google Scholar]
- 117.Akiyama TE, et al. , Peroxisome proliferator-activated receptor-alpha regulates lipid homeostasis, but is not associated with obesity: studies with congenic mouse lines. J Biol Chem, 2001. 276(42): p. 39088–93. [DOI] [PubMed] [Google Scholar]
- 118.Leone TC, Weinheimer CJ, and Kelly DP, A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci U S A, 1999. 96(13): p. 7473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sugden MC, et al. , Peroxisome-proliferator-activated receptor-alpha (PPARalpha) deficiency leads to dysregulation of hepatic lipid and carbohydrate metabolism by fatty acids and insulin. Biochem J, 2002. 364(Pt 2): p. 361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abdelmegeed MA, et al. , PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr, 2011. 141(4): p. 603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Patsouris D, et al. , Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology, 2006. 147(3): p. 1508–16. [DOI] [PubMed] [Google Scholar]
- 122.Ip E, et al. , Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology, 2003. 38(1): p. 123–32. [DOI] [PubMed] [Google Scholar]
- 123.Ip E, et al. , Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology, 2004. 39(5): p. 1286–96. [DOI] [PubMed] [Google Scholar]
- 124.Shiri-Sverdlov R, et al. , Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J Hepatol, 2006. 44(4): p. 732–41. [DOI] [PubMed] [Google Scholar]
- 125.Abd El-Haleim EA, Bahgat AK, and Saleh S, Resveratrol and fenofibrate ameliorate fructose-induced nonalcoholic steatohepatitis by modulation of genes expression. World J Gastroenterol, 2016. 22(10): p. 2931–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lalloyer F and Staels B, Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol, 2010. 30(5): p. 894–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Davidson MH, et al. , Safety considerations with fibrate therapy. Am J Cardiol, 2007. 99(6A): p. 3C–18C. [DOI] [PubMed] [Google Scholar]
- 128.Hong F, Xu P, and Zhai Y, The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int J Mol Sci, 2018. 19(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.W.H.O. cooperative trial on primary prevention of ischaemic heart disease using clofibrate to lower serum cholesterol: mortality follow-up. Report of the Committee of Principal Investigators. Lancet, 1980. 2(8191): p. 379–85. [PubMed] [Google Scholar]
- 130.Westerouen Van Meeteren MJ, Drenth JPH, and Tjwa E, Elafibranor: a potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin Investig Drugs, 2020. 29(2): p. 117–123. [DOI] [PubMed] [Google Scholar]
- 131.Bays HE, et al. , Icosabutate for the treatment of very high triglycerides: A placebo-controlled, randomized, double-blind, 12-week clinical trial. J Clin Lipidol, 2016. 10(1): p. 181–91 e1–2. [DOI] [PubMed] [Google Scholar]
- 132.Wettstein G, et al. , The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol Commun, 2017. 1(6): p. 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brown PJ, et al. , Identification of a subtype selective human PPARalpha agonist through parallel-array synthesis. Bioorg Med Chem Lett, 2001. 11(9): p. 1225–7. [DOI] [PubMed] [Google Scholar]
- 134.Klaunig JE, et al. , PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol, 2003. 33(6): p. 655–780. [DOI] [PubMed] [Google Scholar]
- 135.Peters JM, Cheung C, and Gonzalez FJ, Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? J Mol Med (Berl), 2005. 83(10): p. 774–85. [DOI] [PubMed] [Google Scholar]
- 136.Cheung C, et al. , Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res, 2004. 64(11): p. 3849–54. [DOI] [PubMed] [Google Scholar]
- 137.Li Y, et al. , Application research on PPARalpha-transgenic mice in preclinical safety evaluation of gemfibrozil. Toxicol Res (Camb), 2017. 6(1): p. 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li Y, et al. , A global perspective on FOXO1 in lipid metabolism and lipid-related diseases. Prog Lipid Res, 2017. 66: p. 42–49. [DOI] [PubMed] [Google Scholar]
- 139.Laissue P, The forkhead-box family of transcription factors: key molecular players in colorectal cancer pathogenesis. Mol Cancer, 2019. 18(1): p. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Furuyama T, et al. , Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J, 2000. 349(Pt 2): p. 629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.van der Horst A and Burgering BM, Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol, 2007. 8(6): p. 440–50. [DOI] [PubMed] [Google Scholar]
- 142.van der Vos KE and Coffer PJ, The extending network of FOXO transcriptional target genes. Antioxid Redox Signal, 2011. 14(4): p. 579–92. [DOI] [PubMed] [Google Scholar]
- 143.Kim DH, et al. , FoxO6 integrates insulin signaling with gluconeogenesis in the liver. Diabetes, 2011. 60(11): p. 2763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vogt PK, Jiang H, and Aoki M, Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle, 2005. 4(7): p. 908–13. [DOI] [PubMed] [Google Scholar]
- 145.Accili D and Arden KC, FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell, 2004. 117(4): p. 421–6. [DOI] [PubMed] [Google Scholar]
- 146.Lin K, et al. , daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science, 1997. 278(5341): p. 1319–22. [DOI] [PubMed] [Google Scholar]
- 147.Ogg S, et al. , The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature, 1997. 389(6654): p. 994–9. [DOI] [PubMed] [Google Scholar]
- 148.Hosaka T, et al. , Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A, 2004. 101(9): p. 2975–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Salih DA, et al. , FoxO6 regulates memory consolidation and synaptic function. Genes Dev, 2012. 26(24): p. 2780–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Eijkelenboom A and Burgering BM, FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol, 2013. 14(2): p. 83–97. [DOI] [PubMed] [Google Scholar]
- 151.Rena G, et al. , Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J, 2002. 21(9): p. 2263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Huang H, et al. , CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science, 2006. 314(5797): p. 294–7. [DOI] [PubMed] [Google Scholar]
- 153.Weng Q, et al. , Oxidative Stress Induces Mouse Follicular Granulosa Cells Apoptosis via JNK/FoxO1 Pathway. PLoS One, 2016. 11(12): p. e0167869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Motta MC, et al. , Mammalian SIRT1 represses forkhead transcription factors. Cell, 2004. 116(4): p. 551–63. [DOI] [PubMed] [Google Scholar]
- 155.Kodani N and Nakae J, Tissue-Specific Metabolic Regulation of FOXO-Binding Protein: FOXO Does Not Act Alone. Cells, 2020. 9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hatta M, Liu F, and Cirillo LA, Acetylation curtails nucleosome binding, not stable nucleosome remodeling, by FoxO1. Biochem Biophys Res Commun, 2009. 379(4): p. 1005–8. [DOI] [PubMed] [Google Scholar]
- 157.Frescas D, Valenti L, and Accili D, Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem, 2005. 280(21): p. 20589–95. [DOI] [PubMed] [Google Scholar]
- 158.Peng S, et al. , A Review of FoxO1-Regulated Metabolic Diseases and Related Drug Discoveries. Cells, 2020. 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kousteni S, FoxO1, the transcriptional chief of staff of energy metabolism. Bone, 2012. 50(2): p. 437–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Brunet A, et al. , Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science, 2004. 303(5666): p. 2011–5. [DOI] [PubMed] [Google Scholar]
- 161.Imae M, et al. , Nutritional and hormonal factors control the gene expression of FoxOs, the mammalian homologues of DAF-16. J Mol Endocrinol, 2003. 30(2): p. 253–62. [DOI] [PubMed] [Google Scholar]
- 162.Postic C, Dentin R, and Girard J, Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab, 2004. 30(5): p. 398–408. [DOI] [PubMed] [Google Scholar]
- 163.Puigserver P, et al. , Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature, 2003. 423(6939): p. 550–5. [DOI] [PubMed] [Google Scholar]
- 164.Sekine K, et al. , Foxo1 links insulin signaling to C/EBPalpha and regulates gluconeogenesis during liver development. EMBO J, 2007. 26(15): p. 3607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Matsumoto M, et al. , Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab, 2007. 6(3): p. 208–16. [DOI] [PubMed] [Google Scholar]
- 166.Haeusler RA, et al. , Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat Commun, 2014. 5: p. 5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Langlet F, et al. , Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell, 2017. 171(4): p. 824–835 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Altomonte J, et al. , Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J Clin Invest, 2004. 114(10): p. 1493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Qu S, et al. , PPAR{alpha} mediates the hypolipidemic action of fibrates by antagonizing FoxO1. Am J Physiol Endocrinol Metab, 2007. 292(2): p. E421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kamagate A, et al. , FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest, 2008. 118(6): p. 2347–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Samuel VT, et al. , Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes, 2006. 55(7): p. 2042–50. [DOI] [PubMed] [Google Scholar]
- 172.Kim JJ, et al. , FoxO1 haploinsufficiency protects against high-fat diet-induced insulin resistance with enhanced peroxisome proliferator-activated receptor gamma activation in adipose tissue. Diabetes, 2009. 58(6): p. 1275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nagashima T, et al. , Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice. Mol Pharmacol, 2010. 78(5): p. 961–70. [DOI] [PubMed] [Google Scholar]
- 174.Zou P, et al. , Targeting FoxO1 with AS1842856 suppresses adipogenesis. Cell Cycle, 2014. 13(23): p. 3759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tanaka H, et al. , Effects of the novel Foxo1 inhibitor AS1708727 on plasma glucose and triglyceride levels in diabetic db/db mice. Eur J Pharmacol, 2010. 645(1–3): p. 185–91. [DOI] [PubMed] [Google Scholar]
- 176.Kim CG, et al. , Role of Forkhead Box Class O proteins in cancer progression and metastasis. Semin Cancer Biol, 2018. 50: p. 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tikhanovich I, Cox J, and Weinman SA, Forkhead box class O transcription factors in liver function and disease. J Gastroenterol Hepatol, 2013. 28Suppl 1: p. 125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Matsumoto M, et al. , Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest, 2006. 116(9): p. 2464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhang W, et al. , FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem, 2006. 281(15): p. 10105–17. [DOI] [PubMed] [Google Scholar]
- 180.Lu M, et al. , Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med, 2012. 18(3): p. 388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Marchesini G, et al. , Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes, 2001. 50(8): p. 1844–50. [DOI] [PubMed] [Google Scholar]
- 182.Targher G, et al. , Relations between carotid artery wall thickness and liver histology in subjects with nonalcoholic fatty liver disease. Diabetes Care, 2006. 29(6): p. 1325–30. [DOI] [PubMed] [Google Scholar]
- 183.Day CP, From fat to inflammation. Gastroenterology, 2006. 130(1): p. 207–10. [DOI] [PubMed] [Google Scholar]
- 184.Dong XC, FOXO transcription factors in non-alcoholic fatty liver disease. Liver Res, 2017. 1(3): p. 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nakae J, et al. , The LXXLL motif of murine forkhead transcription factor FoxO1 mediates Sirt1-dependent transcriptional activity. J Clin Invest, 2006. 116(9): p. 2473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Redman LM and Ravussin E, Caloric restriction in humans: impact on physiological, psychological, and behavioral outcomes. Antioxid Redox Signal, 2011. 14(2): p. 275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Yu H, Jia W, and Guo Z, Reducing Liver Fat by Low Carbohydrate Caloric Restriction Targets Hepatic Glucose Production in Non-Diabetic Obese Adults with Non-Alcoholic Fatty Liver Disease. J Clin Med, 2014. 3(3): p. 1050–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Heilbronn LK and Ravussin E, Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr, 2003. 78(3): p. 361–9. [DOI] [PubMed] [Google Scholar]
- 189.Larson-Meyer DE, et al. , Effect of 6-month calorie restriction and exercise on serum and liver lipids and markers of liver function. Obesity (Silver Spring), 2008. 16(6): p. 1355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tsutsumi A, et al. , Caloric restriction decreases ER stress in liver and adipose tissue in ob/ob mice. Biochem Biophys Res Commun, 2011. 404(1): p. 339–44. [DOI] [PubMed] [Google Scholar]
- 191.Kim KE, et al. , Caloric restriction of db/db mice reverts hepatic steatosis and body weight with divergent hepatic metabolism. Sci Rep, 2016. 6: p. 30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Craig PM and Moon TW, Fasted zebrafish mimic genetic and physiological responses in mammals: a model for obesity and diabetes? Zebrafish, 2011. 8(3): p. 109–17. [DOI] [PubMed] [Google Scholar]
- 193.Kim KH and Lee MS, FGF21 as a Stress Hormone: The Roles of FGF21 in Stress Adaptation and the Treatment of Metabolic Diseases. Diabetes Metab J, 2014. 38(4): p. 245–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Qiu LX and Chen T, Novel insights into the mechanisms whereby isoflavones protect against fatty liver disease. World J Gastroenterol, 2015. 21(4): p. 1099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kim S, et al. , Genistein enhances expression of genes involved in fatty acid catabolism through activation of PPARalpha. Mol Cell Endocrinol, 2004. 220(1–2): p. 51–8. [DOI] [PubMed] [Google Scholar]
- 196.Lee K, Transactivation of peroxisome proliferator-activated receptor alpha by green tea extracts. J Vet Sci, 2004. 5(4): p. 325–30. [PubMed] [Google Scholar]
- 197.Serisier S, et al. , Effects of green tea on insulin sensitivity, lipid profile and expression of PPARalpha and PPARgamma and their target genes in obese dogs. Br J Nutr, 2008. 99(6): p. 1208–16. [DOI] [PubMed] [Google Scholar]
- 198.Lukitasari M, et al. , Beneficial effects of green coffee and green tea extract combination on metabolic syndrome improvement by affecting AMPK and PPAR-alpha gene expression. J Adv Pharm Technol Res, 2020. 11(2): p. 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lukitasari M, Nugroho DA, and Rohman MS, 28 GREEN TEA EXTRACT ADMINISTRATION HAD A BENEFICIAL EFFECT ON PPAR ALPHA AND PPAR GAMMA GENE EXPRESSION IN METABOLIC SYNDROME RAT MODEL. Journal of Hypertension, 2018. 36: p. e9. [Google Scholar]
- 200.Li Y and Wu S, Epigallocatechin gallate suppresses hepatic cholesterol synthesis by targeting SREBP-2 through SIRT1/FOXO1 signaling pathway. Mol Cell Biochem, 2018. 448(1–2): p. 175–185. [DOI] [PubMed] [Google Scholar]
- 201.Li X, et al. , Epigallocatechin Gallate Inhibits Hepatic Glucose Production in Primary Hepatocytes via Downregulating PKA Signaling Pathways and Transcriptional Factor FoxO1. J Agric Food Chem, 2019. 67(13): p. 3651–3661. [DOI] [PubMed] [Google Scholar]
- 202.Xiao J, et al. , Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1, and NF-kappa B pathways. Eur J Nutr, 2014. 53(1): p. 187–99. [DOI] [PubMed] [Google Scholar]
- 203.Kim YJ, et al. , Epigallocatechin-3-Gallate (EGCG)-Inducible SMILE Inhibits STAT3-Mediated Hepcidin Gene Expression. Antioxidants (Basel), 2020. 9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Misra J, et al. , Curcumin differentially regulates endoplasmic reticulum stress through transcriptional corepressor SMILE (small heterodimer partner-interacting leucine zipper protein)-mediated inhibition of CREBH (cAMP responsive element-binding protein H). J Biol Chem, 2011. 286(49): p. 41972–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ren BC, et al. , Curcumin alleviates oxidative stress and inhibits apoptosis in diabetic cardiomyopathy via Sirt1-Foxo1 and PI3K-Akt signalling pathways. J Cell Mol Med, 2020. 24(21): p. 12355–12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chen JW, et al. , Tetrahydrocurcumin ameliorates free fatty acid-induced hepatic steatosis and improves insulin resistance in HepG2 cells. J Food Drug Anal, 2018. 26(3): p. 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Weisberg SP, Leibel R, and Tortoriello DV, Dietary curcumin significantly improves obesity-associated inflammation and diabetes in mouse models of diabesity. Endocrinology, 2008. 149(7): p. 3549–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mazidi M, et al. , Potential effects of curcumin on peroxisome proliferator-activated receptor-gamma in vitro and in vivo. World J Methodol, 2016. 6(1): p. 112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ji R, et al. , Effects of dietary curcumin on growth, antioxidant capacity, fatty acid composition and expression of lipid metabolism-related genes of large yellow croaker fed a high-fat diet. Br J Nutr, 2020: p. 1–10. [DOI] [PubMed] [Google Scholar]
- 210.Niu Y, et al. , Curcumin attenuates insulin resistance and hepatic lipid accumulation in a rat model of intra-uterine growth restriction through insulin signalling pathway and sterol regulatory element binding proteins. Br J Nutr, 2019. 122(6): p. 616–624. [DOI] [PubMed] [Google Scholar]
- 211.Li YY, et al. , Fatty liver mediated by peroxisome proliferator-activated receptor-alpha DNA methylation can be reversed by a methylation inhibitor and curcumin. J Dig Dis, 2018. 19(7): p. 421–430. [DOI] [PubMed] [Google Scholar]
- 212.Shin SK, et al. , Long-term curcumin administration protects against atherosclerosis via hepatic regulation of lipoprotein cholesterol metabolism. Mol Nutr Food Res, 2011. 55(12): p. 1829–40. [DOI] [PubMed] [Google Scholar]
- 213.Zhou R, et al. , Resveratrol Ameliorates Lipid Droplet Accumulation in Liver Through a SIRT1/ATF6-Dependent Mechanism. Cell Physiol Biochem, 2018. 51(5): p. 2397–2420. [DOI] [PubMed] [Google Scholar]
- 214.Kim H, et al. , Lysine Acetylation of CREBH Regulates Fasting-Induced Hepatic Lipid Metabolism. Mol Cell Biol, 2015. 35(24): p. 4121–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Tabata Y, et al. , Vaticanol B, a resveratrol tetramer, regulates endoplasmic reticulum stress and inflammation. Am J Physiol Cell Physiol, 2007. 293(1): p. C411–8. [DOI] [PubMed] [Google Scholar]
- 216.Calleri E, et al. , Resveratrol and its metabolites bind to PPARs. Chembiochem, 2014. 15(8): p. 1154–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Rigano D, Sirignano C, and Taglialatela-Scafati O, The potential of natural products for targeting PPARalpha. Acta Pharm Sin B, 2017. 7(4): p. 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jiang L, et al. , Resveratrol prevents hepatic steatosis induced by hepatitis C virus core protein. Biotechnol Lett, 2012. 34(12): p. 2205–12. [DOI] [PubMed] [Google Scholar]
- 219.Huang Y, et al. , Resveratrol protects against nonalcoholic fatty liver disease by improving lipid metabolism and redox homeostasis via the PPARalpha pathway. Appl Physiol Nutr Metab, 2020. 45(3): p. 227–239. [DOI] [PubMed] [Google Scholar]
- 220.Wu J, et al. , Resveratrol Attenuates High-Fat Diet Induced Hepatic Lipid Homeostasis Disorder and Decreases m6A RNA Methylation. 2020, Research Square. [DOI] [PMC free article] [PubMed]
- 221.Wang GL, et al. , Resveratrol inhibits the expression of SREBP1 in cell model of steatosis via Sirt1-FOXO1 signaling pathway. Biochem Biophys Res Commun, 2009. 380(3): p. 644–9. [DOI] [PubMed] [Google Scholar]
- 222.Park JM, et al. , Role of resveratrol in FOXO1-mediated gluconeogenic gene expression in the liver. Biochem Biophys Res Commun, 2010. 403(3–4): p. 329–34. [DOI] [PubMed] [Google Scholar]
- 223.Yun H, et al. , AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J, 2014. 281(19): p. 4421–38. [DOI] [PubMed] [Google Scholar]
- 224.Zhu W, et al. , Effects and mechanisms of resveratrol on the amelioration of oxidative stress and hepatic steatosis in KKAy mice. Nutr Metab (Lond), 2014. 11: p. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.de la Iglesia R, et al. , Dietary Strategies Implicated in the Prevention and Treatment of Metabolic Syndrome. Int J Mol Sci, 2016. 17(11). [DOI] [PMC free article] [PubMed] [Google Scholar]