

Abstract
Treatment failure of antibiotic therapy due to insufficient efficacy or occurrence of toxicity is a major clinical challenge, and is expected to become even more urgent with the global rise of antibiotic resistance. Strategies to optimize treatment in individual patients are therefore of crucial importance. Currently, therapeutic drug monitoring plays an important role in optimizing antibiotic exposure to reduce treatment failure and toxicity. Biomarker‐based strategies may be a powerful tool to further quantify and monitor antibiotic treatment response, and reduce variation in treatment response between patients. Host response biomarkers, such as CRP, procalcitonin, IL‐6, and presepsin, could potentially carry significant information to be utilized for treatment individualization. To achieve this, the complex interactions among immune system, pathogen, drug, and biomarker need to be better understood and characterized. The purpose of this tutorial is to discuss the use and evidence of currently available biomarker‐based approaches to inform antibiotic treatment. To this end, we also included a discussion on how treatment response biomarker data from preclinical, healthy volunteer, and patient‐based studies can be further characterized using pharmacometric and system pharmacology based modeling approaches. As an illustrative example of how such modeling strategies can be used, we describe a case study in which we quantitatively characterize procalcitonin dynamics in relation to antibiotic treatments in patients with sepsis.
Antibiotic treatment of severe infections and sepsis is driven by empirical treatment protocols.1 Treatment failure of antibiotic therapy due to insufficient efficacy, or occurrence of toxicity is associated with significant morbidity and mortality.2 The emergence of antibiotic resistance is increasingly complicating effective antibiotic treatment3 and may be further promoted by suboptimal antibiotic treatment. Strategies to optimize antibiotic treatment for optimal efficacy and minimized risks for toxicity and resistance are therefore needed.
Therapeutic drug monitoring (TDM) represents the best‐established strategy toward individualizing antibiotic drug treatment to reach predefined pharmacokinetic (PK) targets associated with efficacy or toxicity. Nonetheless, even after the use of TDM‐based dose individualization, substantial unpredictable variation in antibiotic treatment response remains, as can be concluded from suboptimal outcomes for major classes of severe infections and sepsis.2 The sources of the variation leading to treatment failure may be related to pharmacodynamic (PD) pathogen characteristics, the patient‐specific immune response, sensitivity to develop drug‐induced organ damage or other adverse reactions, or effects of inflammation on PKs. Strategies to further predict interindividual variation (IIV) in treatment response are thus warranted.
Biomarker‐based strategies that characterize patient‐specific response to antibiotic therapy are of interest to further optimize antibiotic treatment with respect to efficacy, toxicity, and antibiotic resistance risk (Figure 1). Biomarkers hold the potential to inform treatment strategies and aid in decisions throughout all phases of an infection. However, the use of biomarkers in patient care remains limited and is primarily based on diagnosis of pathogens.3 In particular, knowledge gaps in quantitative understanding of the relationships among drug exposure, pathogen dynamics, and biomarker dynamics hinder clinical use of treatment response biomarkers.
Figure 1.
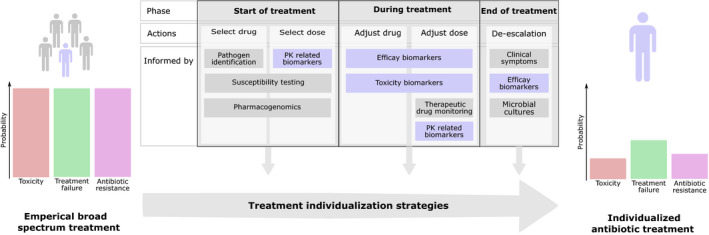
Overview of the use of biomarker‐informed treatment individualization strategies. Current empirical antibiotic treatments are associated with significant risk of toxicity (red), treatment failure (green), and antibiotic resistance development (purple). These risks could be reduced by optimizing antibiotic treatments at an individual level. Specifically, treatment individualization strategies informed by biomarkers (blue) could play an important part. Such biomarkers can inform on pharmacokinetics (PKs), efficacy, and toxicity, and guide the treatment throughout all phases of infection.
This tutorial provides an overview of biomarker‐based strategies to predict antibiotic treatment response, with a particular focus on PD biomarkers. We will discuss three classes of patient‐associated biomarkers: (i) immune response‐associated treatment efficacy biomarkers, (ii) antibiotic‐induced toxicity biomarkers, and (iii) biomarkers to predict variation in (target site) PKs. We then discuss how quantitative pharmacological modeling approaches can be used to address key data analytical challenges, as further illustrated using a case study focusing on the quantification of antibiotic treatment response based on procalcitonin dynamics in patients with sepsis. Overall, this tutorial will thus provide a comprehensive primer with respect to both the current state‐of‐the‐art as well as concrete directions for the analysis of complex infection‐associated and antibiotics‐associated biomarker datasets.
TREATMENT EFFICACY BIOMARKERS
Immune response biomarkers are of interest to quantify antibiotic treatment efficacy because direct quantification of bacterial disease load during clinical infection is typically not possible, and because the effects of infection‐induced inflammation contribute to ultimately observed efficacy.4 The immune response triggered by pathogen associated molecular patterns (PAMPs) present on bacteria results in the production of several immune response biomarkers that involve interactions between multiple cell types and tissues (Figure 2). Several host immune response biomarkers have been shown to be associated with clinical outcomes, such as mortality, duration of hospitalization, and antibiotic treatment duration, with the majority of studies focusing on critically ill patients with sepsis and serious respiratory tract infections (RTIs) (Table 1). Importantly, approaches to minimize and/or optimize the use of antibiotics by such biomarkers could also reduce the risk for resistance development.5 We provide an overview of the biological characteristics and level of evidence to predict clinical outcomes for the most important host immune response biomarkers for severe infections and sepsis. Emerging biomarkers soluble triggering receptor expressed on myeloid cells‐1 (sTREM‐1),6, 7 proadrenomedullin (proADM),8, 9, 10, 11 and pentraxin‐312 were not included due to currently insufficient evidence for their clinical relevance.
Figure 2.
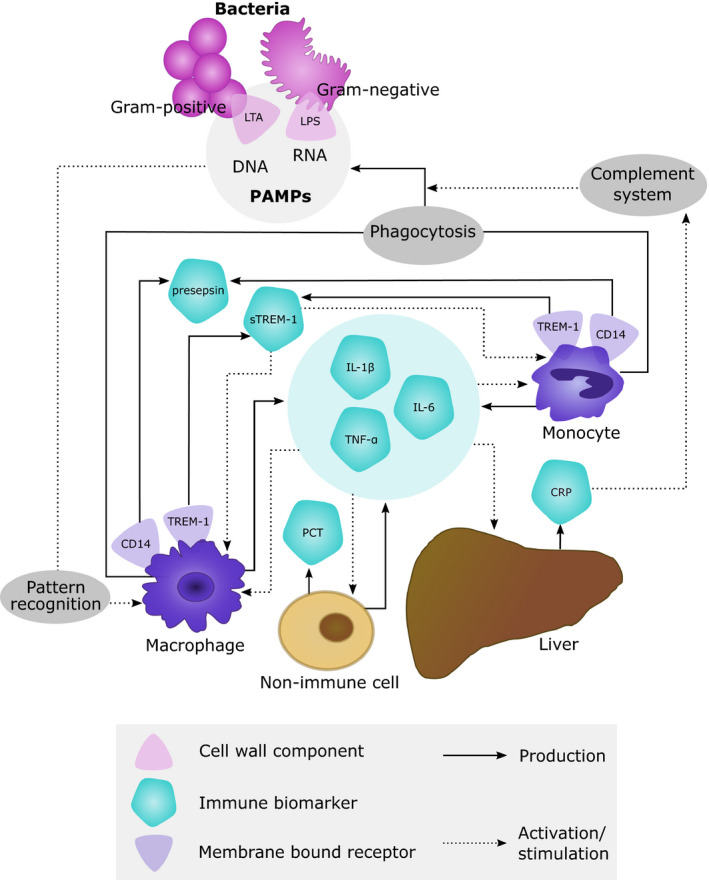
Biological basis of immune response biomarkers produced by host cells after exposure to bacterial pathogens. CRP, C‐reactive protein; IL, interleukin; LPS, lipopolysaccharide; LTA, lipoteichoic acid; PAMPs, pathogen associated molecular patterns; PCT, procalcitonin; (s)TREM‐1, (soluble) triggering receptor expressed on myeloid cells 1; TNF‐α, tumor necrosis factor‐α.
Table 1.
Biomarker‐clinical outcome relationships
| Infection type | Study type | Patients | Antibiotic discontinuation | Reduction of antibiotic treatment duration | Reduction of hospital stay | Reduction of short‐term mortalitya |
|---|---|---|---|---|---|---|
| Procalcitonin | ||||||
| RTIs | Interventional15, 16 | 6,708 | NA | ✓ | ? | ✓ |
| Interventional17 | 337 | NA | ✓ | X | ? | |
| Interventional18 | 101 | < 0.5 μg/L or < 20% BL | ✓ | X | X | |
| Sepsis | Interventional84 | 3,489 | ≤ 0.5 μg/L or < 10% BL | ✓ | X | ? |
| Interventional85 | 68 |
BL PCT ≥ 1 µg/L ≤ 0.25 µg/L or < 10% BL |
✓ | X | X | |
|
BL < 1 µg/L PCT < 0.1 µg/L | ||||||
| Interventional22 | 110 | < 1 µg/L or < 35% BL | ✓ | X | ? | |
| Interventional20 | 1,575 | ≤ 0.25 μg/L or 20% BL | ✓ | ? | ✓ | |
| Interventional21 | 394 | ≤ 0.25 μg/L or < 10% BL | X | ? | X | |
| Interventional86 | 621 | < 0.5 µg/L or < 20% BL | ✓ | X | ? | |
| Observational10 | 1,089 | NA | ? | ? | ✓ | |
| PCT < 0.1 μg/L or < 10% BL | ||||||
| CRP | ||||||
| RTIs | Observational11 | 37 | NA | ? | ? | ✓ |
| Sepsis | Observational23 | 891 | NA | ? | ? | ✓ |
| ProADM | ||||||
| RTIs | Observational11 | 37 | NA | ? | ? | X |
| Observational8 | 19 | NA | ? | ? | X | |
| Septic shock | Observational10 | 1,089 | NA | ? | ? | ✓ |
| sTREM‐1 | ||||||
| Sepsis | Observational6 | 50 | NA | ? | ? | ✓ |
| Observational7 | 90 | NA | ? | ? | ✓ | |
| Presepsin | ||||||
| Sepsis | Observational34 | 109 | NA | ? | ? | ✓ |
| IL‐6 | ||||||
| Sepsis | Observational6 | 50 | NA | ? | ? | ✓ |
| Observational7 | 90 | NA | ? | ? | ✓ | |
✓, biomarker is predictive of outcome; ?, biomarker has not been related to the outcome; %BL, percent of baseline biomarker concentration; IL‐6, interleukin 6; NA, not available; PCT, procalcitonin; proADM, mid‐regional proadrenomedullin; RTI, respiratory tract infection; sTREM‐1, soluble triggering receptor expressed on myeloid cells‐1; X, biomarker is unpredictive of outcome.
Short‐term mortality refers to both in‐hospital and 28‐day mortality.
Procalcitonin
Procalcitonin (PCT) is a precursor to the hormone calcitonin, and is, under normal conditions, produced only intracellularly by parafollicular cells in thyroidal tissues. During microbial infections and severe systemic inflammation, PCT production is induced throughout the body where it is thought to be associated with immune modulatory properties.13 PCT production is inhibited by interferon‐γ, which therefore leads to only limited elevation of PCT during viral infections.14 PCT‐guided antibiotic treatment termination can lead to a significant reduction of antibiotic exposure in sepsis and RTIs.15, 16, 17, 18, 19 Studies in sepsis and RTIs show varying results for the correlation of PCT with mortality.15, 16, 18, 19, 20 The relative change in PCT has been successfully used to guide antibiotic treatment duration,21, 22 illustrating the relevance of considering the dynamics of PCT.
C‐reactive protein
C‐reactive protein (CRP) is a hepatic acute phase protein playing a crucial role in the innate host defense by activating the complement system, promoting phagocytosis of pathogens. Admission levels of CRP do not predict outcome in sepsis and RTIs,11, 23, 24 but CRP levels in patients showing a clinical response to antibiotics decreased faster11, 23 or lower levels after therapy.24, 25, 26
Interleukin‐6
Interleukin‐6 (IL‐6) is a cytokine produced by immune cells and stromal cells and is involved in inflammation. IL‐6 plays a pivotal role in orchestrating the immune response to infection. This pleiotropic cytokine has both pro‐inflammatory and anti‐inflammatory effects, and is involved in neutrophil infiltration, activation, and proliferation of T‐cells and B‐cells, and acute phase response.27 IL‐6 is of potential interest as a biomarker in sepsis to reflect disease severity.28 Increased IL‐6 levels have been associated with increased mortality in patients with sepsis.29, 30
Presepsin
Presepsin is a cleavage product of the CD14 membrane coreceptor involved in pathogen recognition and initiation of the innate immune response,31 and is involved in regulation of bacterial phagocytosis.32 Presepsin is a rapidly responding biomarker for bacterial and fungal infections. Baseline presepsin levels have been shown to predict mortality33, 34 and correlate with sepsis disease severity.33 Furthermore, the dynamic time course of presepsin also predicts survival.34
Biomarker dynamics
The majority of clinically used immune response biomarker studies have considered primarily static threshold concentrations to inform clinical decision making. Less attention has been given to the full dynamics of immune response biomarkers for evaluation of treatment efficacy. To guide and optimize antibiotic treatment in individual patients, immune response biomarkers should ideally reflect underlying current pathogen load (Figure 3a), which is typically not feasible to measure directly. Consequently, characterization of the onset and decline of biomarkers in response to infectious challenges is an important step toward their interpretation in context of antibiotic treatment response. Biomarkers with a delayed onset or prolonged half‐life therefore reflect poorly the current state of infection. Substantial differences exist between the kinetic properties of biomarkers (Table 2), and kinetic properties should be considered when they are investigated as antibiotic treatment response biomarkers to evaluate treatment response (Figure 3b).
Figure 3.
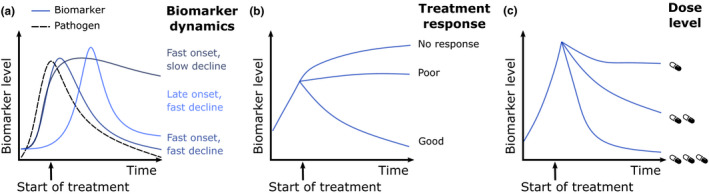
Key characteristics of treatment response biomarkers. To enable the assessment of treatment response biomarker should (a) be rapidly induced and have a relatively short half‐life to satisfactory follow the course of infection, (b) be able to stratify treatment response, and (c) have a characterized drug exposure‐response relationship.
Table 2.
Kinetics characteristics of immune‐response biomarkers
| Biomarker | Tincrease, hours | Peak, hours | T50%, hours | Reference |
|---|---|---|---|---|
| Procalcitonin | 3–4b | 6–24b | 24a | 43, 87 |
| C‐reactive protein | 6b | 24–48b | 19b | 88, 89 |
| Presepsin | 2c | 3c | 4–5c | 90 |
| Soluble triggering receptor expressed on myeloid cells‐1 | 2b | 6b | NA | 91 |
| Proadrenomedullin | 2b | 4b | 2d | 92, 93 |
T50%, biomarker (elimination) half‐life; T increase, time to detectable biomarker increase (after lipopolysaccharide injection).
Clinically observed.
Healthy volunteer study.
In vivo.
In vitro.
Antibiotic exposure‐biomarker response
The relationship between antibiotic drug exposure and changes in biomarker dynamics to quantify treatment response can be considered as another important, but currently also still poorly characterized step toward treatment response biomarkers to enable dose optimization (Figure 3c). This relationship reflects antibiotic‐induced killing of pathogens, which leads to a change in biomarker levels, in particular when such a biomarker has kinetic properties closely associating with the underlying infection.
Currently, only for CRP, such a relationship has been reported for infections treated with teicoplanin and vancomycin in pediatric and adult patients, respectively. Through the use of a PK‐PD model, PK models were associated with logistic growth models accounting for pathogen‐induced CRP increase. The subsequent reduction of CRP growth kinetics was then captured by antibiotic concentration‐effect (Emax) models.35, 36 For teicoplanin, individual CRP‐derived PD area under the curve (AUC):half‐maximal effective concentration (EC50) targets could be derived, which aim for CRP normalization. Nonetheless, these studies should be considered as important first steps toward characterization of antibiotic exposure‐response relationships for treatment response biomarkers.
Pharmacological characterization of efficacy biomarkers
Addressing the current knowledge gaps with respect to the pharmacological properties of treatment efficacy biomarkers (i.e., their kinetic and exposure‐response properties), is an important step to develop actionable biomarkers to optimize antibiotic treatments. Because studies in patients with severe infections and sepsis are often associated with significant IIV, there is a need for complementary strategies, including preclinical in vivo models and healthy volunteer challenge models.
In vivo infection and challenge models
Preclinical animal infection models can contribute key insights into host immune response biomarker dynamics that may be challenging to obtain from human studies. For several clinically relevant biomarkers, including PCT, CRP, and IL‐6, in vivo infection models have been described. Examples of such studies that performed such characterization with a focus on host immune response biomarkers include bacteremia models in pigs,37 mice studies, and in vivo sepsis models in baboons and hamsters.38, 39
Preclinical in vivo experiments involving immunogenic PAMPs further allow to study biomarker dynamics for biomarkers in a controlled setting. The PAMP lipopolysaccharide (LPS) derived from Gram‐negative bacteria is commonly used for such in vivo challenge studies, which are relevant to study biomarker dynamic profiles. Particularly in combination with PK‐PD modeling, these studies can lead to important insights. Held et al. described a dose ranging study with LPS in rats measuring TNF‐α time profiles, which were subsequently modeled using a kinetic‐PD modeling approach to characterize the time course and dose‐response of this biomarker, identifying a peak‐shift in TNF‐α response at increasing LPS doses.40 Another relevant example was the development of a pharmacometric model‐based analysis for a porcine LPS challenge model that was recently described in which the LPS exposure was also considered in relation to dynamics of TNF‐α and IL‐6.41 The explicit consideration of LPS exposure can help to further bridge these findings to patients.
These preclinical infection and related PAMP‐challenge models can be of significant value to under controlled settings to characterize key dynamics and pathogen exposure‐response of relevant immune response biomarkers to help further interpret such markers in the clinical setting, although it must be acknowledged that interspecies immune system differences exist and must be taken into account.
Healthy volunteer challenge studies
The use of healthy volunteer challenge studies based on administration of pathogen‐isolates PAMPs, heat‐killed, or low‐dose bacteria forms a relevant intermediate step to characterize the dynamics of host immune response biomarkers in relation to bacterial pathogen load in humans. In particular, LPS challenge studies form a relevant clinical model to study host biomarker response under controlled conditions, and have already contributed to further quantitatively characterize biomarker dynamic profiles.
Several healthy volunteer LPS challenge studies have been conducted for which the induction of clinically relevant immune response markers, such as PCT, TNF‐a, IL‐1β, and IL‐10.42, 43 A current knowledge gap concerns the relationship between LPS concentrations, which often very rapidly decline, and biomarker dynamics. Such explicit consideration would allow to deconvolute the rapid decline of systems LPS concentrations and related biomarker levels.
Age‐dependent between‐patient variability in biomarker levels
Biomarker concentration in the absence of acute inflammation or infection might be dependent on a number of factors, including the age of the patient. For example, plasma levels of IL‐6, IL‐1, and TNF‐α have been observed to be elevated in elderly subjects, which may be associated with frailty and an underlying overactivation of the immune system.44, 45 This subinflammatory status has earlier been described as inflamm‐aging46 and may need to be considered for biomarker‐guided treatment optimization strategies. Similarly, preterm neonates are still in the midst of the development of their innate immune response,47 and significantly reduced pro‐inflammatory cytokine (e.g., IL‐1β, IL‐6, and TNF‐α) responses to endotoxin stimulation have been observed in this population.48 In addition, higher levels of CRP and PCT have been described in the first 2–3 days after birth in preterm as well as term neonates, most likely associated with the stress related to being born.
ANTIBIOTIC EXPOSURE BIOMARKERS
Optimization of antibiotic‐drug exposure is one key component toward optimizing antibiotic efficacy, minimizing toxicity, and reducing the risk of antibiotic resistance. In this context, population PK models are commonly used to identify patient‐specific predictors for IIV in PK parameters, and typically include static covariates, such as body weight, age, and glomerular filtration rate, to predict variation in drug clearance of distribution volume. However, particularly in patients with severe infections and sepsis, substantial unexplained IIV in PK remains after the consideration of such commonly included covariates,49 likely due to infection‐induced inflammation effects on absorption, distribution, metabolism, and elimination.50 To this end, immune response biomarkers that capture infection‐induced inflammation effects, and predict IIV on systematic clearance, systemic distribution, and target site concentrations at the site of infection, are of significant relevance for further individualization of antibiotic treatments.
Systemic clearance
Biomarkers to predict inflammation association IIV in systemic clearance are likely associated with inflammation effects on either renal or hepatic clearance. For renal clearance, CRP and IL‐6 have been shown to be associated with renal function.51, 52 Potential explanations could be a reduced clearance of CRP or IL‐6 due to reduced kidney function, or vascular inflammation causing reduced kidney function.52 CRP or IL‐6 may therefore be a relevant biomarker to predict the effect of inflammation on kidney function and therefore renal drug clearance. Hepatic drug clearance may be affected by infection‐associated inflammation through modulation of drug metabolizing enzyme activity, resulting in decreased drug metabolism.53, 54 This could be supported by observations in patients with rheumatoid arthritis, known to have elevated levels of IL‐6, that often show increased exposure of CYP metabolized drugs, such as simvastatin. Such drug exposure has been shown to reduce in these patients when treated with an IL‐6 inhibitor.55 However, no quantitative data on the extent of reduction of CYP activity for specific CYP enzymes are available so far.50 Nevertheless, the available evidence has already led to using infection/inflammation biomarkers as a predictor for changes observed in PK. For instance, IL‐6 and CRP have been shown to predict hepatic clearance.53, 56
Systemic distribution volume
Inflammation can influence volume of distribution due to capillary leakage, vasodilatation, edema formation, and administration of intravenous fluids. For example, for β‐lactams, the volume of distribution can be increased up to fivefold in patients with sepsis.57 Potential biomarkers to reflect these changes have not been thoroughly studied to our knowledge. A relevant example includes a reported shift of albumin from plasma to interstitial fluid, as observed in severe infections, was found to increase apparent volume of distribution by up to 150%.58
Target site pharmacokinetics
Consideration of target site concentrations (i.e., at the site of infection), is well‐accepted to be of considerable importance for successful antibiotic treatment. Such target concentrations can deviate substantially from plasma concentrations,59 and may lead to persisting infections,60 and potentially and resistant development.57
Tissue concentrations can be measured using various approaches, including tissue homogenates, cantharis‐induced skin blister fluid,59 microdialysis,61 or bronchoalveolar lavage for RTIs.62 For routine patient care, the implementation of such techniques are often not feasible as they are invasive and costly. However, tissue concentrations can be predicted using physiologically‐based PK (PBPK) models. Such PBPK models integrate drug‐specific and system‐specific parameters to obtain tissue‐specific PK predictions and can be informed with patient‐specific characteristics. Due to the physiological basis of PBPK models, they allow for alteration of system parameters to capture disease‐specific changes of PK. Such an approach was used to describe the plasma PK of vancomycin in patients with sepsis.50
During severe infections and sepsis, inflammation may induce alterations in drug penetration into specific tissue compartments.63 Larger variation in target site concentrations is often seen in patients with infections compared with healthy volunteer studies that investigate target site PKs.64 The use of systemically measurable immune response biomarkers to evaluate relationships of the extent of inflammation and the rate and extent of tissue penetration is thus relevant to further optimize antibiotic drug exposure at the target site in individual patients. Currently, to our knowledge, such studies in which extent of inflammation and target site concentrations are studied are lacking.
TOXICITY BIOMARKERS
Antibiotic‐induced toxicities are a major challenge for multiple classes of antibiotics, several of which have narrow therapeutic windows. Explicitly considering toxicity guided by toxicity biomarkers could lead to safer treatments reducing morbidity and mortality. To date, TDM is still the most important clinical strategy to prevent occurrence of drug‐induced toxicities, but these approaches rely on static antibiotic exposure/concentration cutoffs. Such PK‐driven strategies may not be sufficient to predict the individual development of antibiotic‐induced toxicities, stressing the need for toxicodynamic (or PD) biomarkers to quantify and predict antibiotic‐induced toxicity.
Antibiotic‐induced toxicities can be either categorized as acute dose‐limiting toxicities or long‐term toxicological effects. Acute dose‐limiting toxicities can impact antibiotic treatment efficacy, thus potentially threaten efficient eradication of pathogens, and subsequently lead to increased risk of emergence of resistance. Examples of such toxicities include acute kidney injury caused by polymyxins, erythromycin‐induced hepatotoxicity, and β‐lactam‐induced mitochondrial toxicity.65 Acute toxicity is often reversible or alleviated by the termination of antibiotic treatment. Long‐term toxicity can have lasting and potentially permanent impact on patients, such as aminoglycoside‐associated ototoxicity. A summary of relevant examples of antibiotic‐related toxicities can be found in Table 3.
Table 3.
Overview of antibiotic related toxicities and potential toxicity biomarkers
| Antibiotic induced toxicity | Site or organ damage | Biomarkers | Ref |
|---|---|---|---|
| Nephrotoxicity | |||
|
Antibiotic (classes) involved: Aminoglycosides β‐lactams Sulfonamides |
Glomerulus | Total protein, albumin, cystatin C, MCP‐1, β2‐microglobulin, osteopontin | 72 |
| Proximal tubule | β2‐microglobulin, clusterin, KIM‐1, L‐FABP, MCP‐1, NAG, NGAL, osteopontin | 66, 67 | |
| Distal tubule | Clusterin, NGAL, osteopontin | 67 | |
| Hepatotoxicity | |||
|
Antibiotic (classes) involved: Tetracyclines Erythromycin Sulfonamides Rifampicin |
General | ALT, AST, ALP, bilirubin | 94 |
| Liver‐specific mitochondrial damage | GLDH, OCT | 76, 95 | |
| Necrosis and/or apoptosis | GLDH, HMGB‐1, K18 | 96 | |
| Inflammation | HMGB‐1 | 96 | |
| Liver‐specific damage | miRNA‐122, miRNA‐192 | 66, 96 | |
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GLDH, glutamate dehydrogenase; HMGB‐1, high‐mobility group box 1; K18, Keratin‐18; KIM‐1, kidney injury molecule‐1; L‐FABP, liver‐type fatty acid‐binding protein; MCP‐1, monocyte chemotactic protein‐1; NAG, N‐acetyl‐β‐glucosaminidase; NGAL, neutrophil gelatinase‐associated lipocalin; OCT, ornithine carbamoyltransferase.
Direct quantification of toxicity, such as drug‐induced tissue damage quantified by histology, is generally unfeasible in patients. For that reason, biomarkers relating to such toxicity can be of great clinical value to assess toxicity levels. The current clinical practice uses standard biomarkers, such as serum creatinine and liver enzymes, to monitor kidney and liver function, respectively. These markers are general indicators of organ function and do not specifically/exclusively reflect antibiotic toxicity. Although many potential toxicity biomarkers have been identified, they have yet to make it into clinical practice.
Biomarkers for toxicity can provide insight on current state of toxicity but also identify patients at high risk of developing toxicity. Identifying patients at high risk of toxicity related to specific antibiotics can aid in selection of appropriate drug therapy on an individual level, which may result in minimized risk of toxicity. The development of toxicity during a treatment can be assessed by measuring biomarkers relating to early toxicity signs, allowing for timely adjustment of treatment to prevent severe toxicities. Biomarkers can also play a role in confirming the occurrence, as well as quantifying the extent of toxicity. Quantitative knowledge about the state of toxicity can inform on appropriate treatment measures. The combination of such biomarkers into multiplex panels have been suggested to provide an even better estimation of the state of toxicity.66
Characterizing exposure‐toxicity relationships
Understanding of exposure‐toxicity relationships (i.e., toxicodynamics), is a crucial step in order to derive guidelines for treatment individualization. Relating toxicity, such as organ damage, to easily measurable biomarkers is imperative to facilitate such individualization in the clinic. A noncomprehensive summary of the currently most ubiquitously used toxicity biomarkers can be found in Table 3. However, the kinetic properties of several (potential) biomarkers for toxicity remain poorly characterized. Such characterization is challenging due to the complex mechanisms that govern biomarker kinetics, which include interactions with underlying disease and inflammation, but also indirect toxicity‐induced organ function effects that influence the kinetics of toxicity biomarkers.
Preclinical models can play an important role in the quantitative characterization of antibiotic toxicity by generating exposure‐response data as well as correlating toxicity to biomarkers. Such efforts have been made in several studies, including studies relating antibiotic exposure to histopathological changes of kidney tissue and associated urinary biomarker dynamics.67, 68 To enable the clinical use of toxicity‐related biomarkers, exposure‐toxicity‐biomarker dynamics need to be fully characterized. A quantitative characterization of such relationships would aid in the clinical interpretations of biomarker levels.
Hematological toxicity
Severe antibiotic‐related hematological toxicities include leukopenia, thrombocytopenia, and coagulation dysfunction, and are caused by several antibiotics from different classes. The severity of such toxicities is directly measurable in the blood of patients through quantification of these blood cell types, and therefore feasible to directly quantify in the clinic. However, prognostic and predictive biomarkers can play an important role to prevent and reduce hematological toxicities. An example is that baseline platelet concentration has been shown to be a predictive biomarker of hematological toxicity induced by linezolid within 7 days of treatment.69 This early emerging toxicity was predictive of 30‐day mortality, thus highlighting the possibility to reduce mortality by reducing toxicity. In order to develop biomarker‐guided dose adjustment strategies for hematological toxicities, consideration of the specific mode of action of antibiotics is important, and can be complex due to maturation delays of hematopoietic precursor cells and homeostatic feedback mechanisms. The use of pharmacometric models has been shown to address these challenges.70
Nephrotoxicity
Nephrotoxicity is associated with a number of antibiotics (Table 3). Currently, serum creatinine and blood urea nitrogen are the most commonly used biomarkers for monitoring acute kidney injury, however, they are nonspecific and the serum levels of these markers will rise only after a considerable loss kidney function.71
Preclinical studies have been performed to investigate potential biomarkers of antibiotic‐related nephrotoxicity. In antibiotic treated rats, serum cystatin C has been found to outperform the standard serum kidney injury markers in regard to specificity and selectivity, and using a panel of novel urinary biomarkers, including kidney injury molecule‐1, the onset of toxicity, as well as the reversion after discontinuation of treatment, could be identified.72 It has shown correlation with histopathological and proximal tubule damage in rats treated with several different nephrotoxic antibiotics and can be detected shortly after initiation of antibiotic therapy.68 Such biomarkers of early nephrotoxicity could potentially be utilized for treatment monitoring and prevent emergence of severe toxicity.66
Hepatotoxicity
Antibiotic‐induced hepatotoxicity can lead to severe, possibly fatal, hepatic damage. Several antibiotics are known to be associated with hepatotoxicity (Table 3). Current treatment monitoring relies on increasing concentrations of the liver enzymes alanine aminotransferase and aspartate aminotransferase, with circulation half‐lives of 47 and 17 hours, respectively.73 Although these enzymes are markers for liver damage, they are not predictive of overall liver function nor correlate well with observed hepatic pathology.74 Alternative biomarkers for hepatotoxicity include glutamate dehydrogenase (clinically observed half‐life of 11–18 hours75), outperforming standard enzyme biomarkers to predict acute toxicity,76 and also microRNA‐based biomarkers have gained acceptance as markers of liver damage.74 Mechanistic models can play an important role in increase the understanding of such complex antibiotic exposure‐toxicity‐biomarker relationships. For instance, for hepatotoxicity, several mechanistic system models have been published, which allows to derive relationships among drug exposure, time‐dependent cell death, and associated biomarker dynamics.77
MODEL‐BASED ANALYSIS OF BIOMARKERS
The use of mathematical and statistical modeling approaches for analysis of biomarker data for either toxicity or efficacy is of pivotal importance because of (i) extensive variability and covariates that must be considered and corrected for the analysis of clinical datasets, and (ii) the possibility to effectively integrate knowledge about biomarker dynamics in relation to pathogen exposure and outcomes. An overview table of different modeling techniques and their applications can be seen in Table 4.
Table 4.
Overview of modeling techniques with application in individualization of antibiotic treatments
| Model | General purpose | Antibiotic treatment and biomarker applications | Approach | Input (example) | Output | Model type | NLME | ||
|---|---|---|---|---|---|---|---|---|---|
| Drug | System | Effect data | |||||||
| PK | Characterize relationship between dosing regimen and plasma PK profiles, quantify variability and identify covariates predictive of IIV. | Design and evaluation of clinical PK studies. Clinical dose selection, TDM, and treatment optimization. | Data driven | Dose information, longitudinal concentration data | Patient specific covariates (sex, age, biomarker concentration) | NA | Plasma PK profiles | ODE modela | Yes |
| PBPK | Predict plasma and tissue‐specific drug concentrations and investigate the impact of patient‐specific or disease‐specific physiological changes. | Predict infection site PK. Perform between‐species or ‐population PK translation. | Mechanistic | Dose information, drug‐specific properties (Mw, LogP) | System‐specific properties (blood flows, volumes) | NA | Plasma and tissue PK profiles | ODE model | No |
| PD | Characterize concentration‐effect relationships, typically in experimental settings that do not include PK. | Quantify drug effect in static in vitro bacterial time‐kill experiments. Characterize ex vivo biomarker dose‐response to LPS. | Data driven | Static drug concentration or dose | NA | Bacterial density, biomarker concentration | Drug concentration‐PD response relationships | ODE model | Yes |
| Kinetic‐PD |
Characterize dynamic concentration‐effect relationships in patients when dosing information is available but PK data is lacking. |
Characterize biomarker response to drug and/or immune trigger (e.g., pathogen, LPS) in vivo, for which quantification is lacking. | Data driven | Dose information | Patient specific covariates (sex, age) | Bacterial density, biomarker concentration | Dose concentration‐PD response relationships | ODE model | Yes |
| PK‐PD | Characterize relationship between dosing regimen, drug concentration and effect, quantify PK and PD variability, and identify significant covariates. |
Design and evaluation of clinician studies based on efficacy and toxicity. Biomarker guided treatment optimization. |
Data driven | Dose information, longitudinal concentration data | Patient specific covariates (sex, age) | Bacterial density, biomarker concentration | Plasma PK profiles and related PD response | ODE model | Yes |
| Time‐to‐event | Characterize probability of events over time and risk‐based population stratification. | Identify high‐risk subpopulations. Characterize treatment effect on outcome (e.g., time‐to‐negative bacterial culture) when linked to PK‐PD model. | Data driven | Dose information | Patient specific covariates (sex, age, biomarker concentration) | Time of event (death or toxicity) | Survival curves | Parametric or non‐parametric survival model | Possible |
| Quantitative systems pharmacology | Characterizes or predict system behavior by integration of different level data from multiple sources. Capturing over time behavior of a whole system under perturbation. | Target identification. Quantitatively characterize complex interactions drug‐pathogen‐host. Relating biomarker dynamics to treatment efficacy and toxicity. | Mechanistic | Dose information, drug specific data (receptor binding) | System specific knowledge (immune response network) | Multi‐layered data (biomarker concentrations, cell densities, gene expression levels) | System level PD response | ODE model | No |
IIV, interindividual variation; LPS, lipopolysaccharide; NA, not available; NLME, nonlinear mixed effect; ODE, ordinary differential equation; PBPK, physiologically‐based pharmacokinetic; PD, pharmacodynamic; PK, pharmacokinetic; TDM, therapeutic drug monitoring.
Or analytical expression.
Pharmacometric models typically utilize a combination of nonlinear mixed effect models and differential equation models. Such models are of relevance to quantify biomarker onset and decline characteristics, relationships with PK or antibiotic exposure, and quantification of specific sources of interindividual variability and patient‐associated predictors thereof. Pharmacometric PK‐PD models for the glycopeptides teicoplanin and vancomycin establishing antibiotic exposure‐response relationships for CRP represent relevant examples.35, 36
The use of pharmacometric models for biomarkers—clinical outcome relationships—is another important potential use, as demonstrated for other disciplines.78 For instance, data on time to negative culture, which may reflect successful antibiotic therapy,79 could be readily captured with time‐to‐event models and associated with dynamic models capturing biomarker dynamics.
Developing quantitative understanding of biomarker and pathogen dynamics, and drug exposure‐pathogen‐biomarker relationships is complex and likely requires integration of data from preclinical, healthy volunteer, and patient studies. Model‐based approaches, and, in particular, quantitative systems pharmacology‐based models, can play an important role to reach this aim, as these models readily allow such mechanism‐based integration. Important framework models have been already established (i.e., such as for the immune system),80 and can be further expanded to include biomarkers of clinical interest.
A clear example how quantitative systems pharmacology‐type modeling can provide insight into pathogenesis of bacterial pneumonia in relation to multiple immunological biomarkers was described by Diep et al.,81 who integrated bacterial growth dynamics of Acetobacter baumannii in context of a preclinical in vivo infection model. Model components included neutrophils counts, TNF‐α, IL‐1β, and cytokine‐induced neutrophil chemoattractant‐1. The model allowed to further characterize the relationship between bacterial burden and immune biomarkers. This example demonstrated how modeling of preclinical infection studies can lead to further insights into the specific role of biomarkers in reflecting underlying bacterial pathogenesis and the potential effect of antibiotic treatments.
CASE STUDY: PROCALCITONIN TO QUANTIFY ANTIBIOTIC TREATMENT RESPONSE IN SEPSIS
Biomarkers that capture pathogen load or infection severity are of significant interest for evaluation of drug‐treatment response in patients. PCT is one important host immune response biomarker to support this goal. As discussed, several studies support a quantitative relationship between PCT and pathogen load and/or infection severity. However, extensive variation in observed biomarker dynamics remains a major challenge limiting further use of PCT as treatment response biomarker in patients with severe infections and sepsis. Such variability is associated with differences in underlying microbial etiologies, comorbidities,82 and antibiotic treatments. All together, these factors make it more challenging to interpret and evaluate PCT biomarker time course data. To this end, the use of quantitative nonlinear mixed effect modeling of PCT dynamics measured in individual patients is of relevance to extract relevant drug‐treatment response information and further understand the onset and decline of PCT levels in relation to disease severity. The aim of the current case study is to demonstrate how nonlinear mixed effect dynamic modeling can be used for analysis of PCT time course data in order to quantitate antibiotic treatment effects in patients with sepsis.
In this case study, we utilize a large historical study dataset (Figure 4a) in patients diagnosed with sepsis for which daily PCT measurements were collected in a prospective setting.83 Based on the previously discussed quantitative relationship between PCT and pathogen load, we make an important and plausible assumption that the rate of PCT change can be used as metric to quantify treatment response in patients. A decline in PCT thus indicates an efficacious treatment response and the rate of decline quantifies the treatment‐specific efficacy. Enrolled patients received a diverse range of empirical antibiotic treatments, often in combination to ensure sufficient coverage. Patients could switch between antibiotic treatments as guided by clinical symptoms or microbiological testing, representing a common challenge in the analysis of patient‐based biomarker data. For this analysis, we focused on patients who only received antibiotic therapy but no other antimicrobial agents. We furthermore excluded rare antibiotic combination treatments that were present with insufficient frequency, yielding a dataset of 587 patients who received 23 different combination treatments that contained 1 to 3 antibiotics, with a median number of 2 treatments per patient. A total of 3,484 PCT samples were available.
Figure 4.
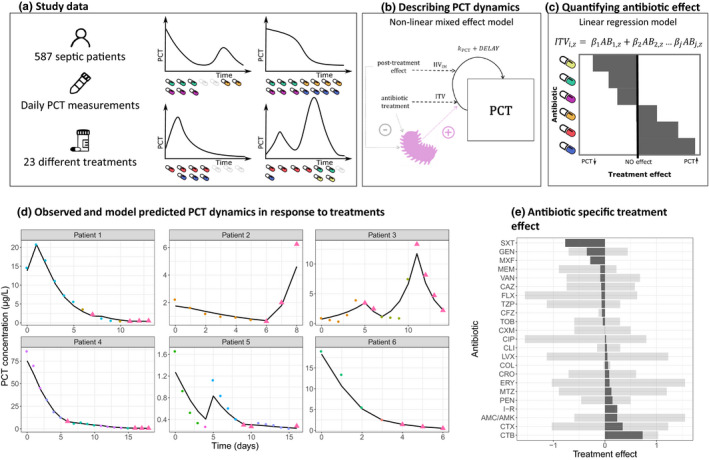
Procalcitonin (PCT) biomarker case study workflow. (a) Overview of study data and examples of biomarker time course data in relation to antibiotic therapy. (b) Pharmacodynamic model to capture PCT dynamics and antibiotic drug effects, where kpct represents a first‐order infection induced production or degradation rate of PCT, delay a time‐dependent delay of antibiotic effect, interindividual variation (IIV)IM post‐treatment immune response, and intertreatment variation (ITV). (c) Quantification of individual antibiotic effects using a linear regression analysis. (d) A selection of observed PCT profiles (points) and model predictions (solid lines), illustrating the diversity of dynamics and treatments. Colored points indicate different treatments, consisting up to three antibiotics, whereas triangles indicate observation without any treatment. (e) The mean (dark gray area) and the range (shaded area) of the treatment response related to pairwise combinations and mono‐treatments per individual antibiotic. A negative treatment response value is associated with decrease of PCT while a positive is associated with an increase.
We used a quantitative modeling approach to quantify expected drug treatment response as a surrogate for treatment efficacy from available PCT time course profiles. Due to the extensive variation observed between and within patients, the use of a nonlinear mixed effect modeling framework is essential. A compartment differential equation model was used to model PCT dynamics (Figure 4b) during and after discontinuation of antibiotic treatment. The model included the following parameters: (i) baseline PCT (PCT0) accounting for PCT level at start of treatment; (ii) time delay (PCTdelay) factor to quantify delay in drug‐induced decline in PCT; and (iii) first‐order production or degradation rate of PCT (kPCT) as induced by the infection. Random effects were added to quantify IIV in PCT0 and PCTdelay. IIV in kPCT as induced during and after drug treatment was estimated using two random effect parameters intertreatment variation (ITV) and post‐treatment immune response (IM). ITV was related to variation between unique treatments within a patient.
The developed model adequately captured the observed PCT time course profiles, which are illustrated by selected examples of observed and model‐predicted individual PCT time course profiles (Figure 4c) and in the Supplementary Materials Figures S1 and S2. Model parameters were estimated with sufficient precision of estimated parameters (Table S1). The derived parameters and associated individual and treatment‐specific parameters shed light on ITV underlying PCT time profiles, such as between‐treatment differences in drug‐induced changes in PCT, delays in drug‐induced PCT changes, and post‐treatment changes in PCT.
We then utilized the model‐estimated ITV values reflecting treatment effects on PCT to quantify antibiotic treatment response values for each treatment and each individual. A linear regression analysis of ITV values and their corresponding antibiotic (combination) treatments was used to estimate antibiotic‐specific treatment effects, under the assumption of additivity. The mean and the range of regression estimates per antibiotic are shown in Figure 4d, with variation being associated with the magnitude of variation observed for each antibiotic.
With this case example we show how model‐based analysis of PCT biomarker data can be used to extract pharmacological knowledge from notoriously challenging clinical biomarker data in sepsis. The model files used for this analysis are available as Supplementary Material S1, S2. The described characterization of PCT dynamics in relation to changes in treatment is a first step to better understand how this biomarker can be used to individualize treatment response monitoring and further guide antibiotic therapy. Nonetheless, we can conclude that significant variation in PCT‐inferred response to antibiotics occur (Figure 4e), which can be affected by a variety of covariates not included in this case study. The application of similar analysis strategies in patient cohorts suffering from severe infections but not sepsis with potentially greater uniformity underlying pathogens and without the systematic inflammation component in sepsis could potentially yield to more directly actionable drug‐biomarker exposure relationships to guide treatment. Another step represents the association of biomarker dynamic profiles with readouts reflecting the underlying infection (e.g., using readouts such as time to negative blood or sputum culture). In summary, this case example demonstrated the under‐used utility of pharmacometric model‐based analysis to further enable the use of treatment response biomarkers to guide antibiotic treatment.
CONCLUSION
In this tutorial, we have discussed two main applications of biomarker‐guided treatment optimization. In drug development, biomarkers can be used to guide dose selection and further treatment optimization, taking into account factors that lead to IIV. Through the use of quantitative (systems) modeling integration of biomarker data from different species and experiments can be effectively integrated.
In patient care, biomarkers can be used to further develop individualized treatment strategies. At this point, data clearly quantifying the added value of host biomarkers in comparison to existing minimum inhibitory concentration (MIC) and TDM‐based approaches are lacking. Nonetheless, biomarkers have the potential to enhance efficacy and reduce toxicity of antibiotic therapy, as they may hold information on both PK and PD treatment response on a patient‐specific level. Such biomarker‐based treatment individualization should be used in conjunction with current MIC and TDM‐based strategies. The combined strategies could lead to improved therapy, as they would simultaneously consider patient‐specific immune response, pathogen, and infection site.
In order to be able to fully benefit from the use of biomarkers, we first need to gain a better understanding of the complex relationship underlying host‐pathogen‐drug interactions. Specifically, developing further understanding in biomarker response dynamics in relation to specific pathogens, the pathogen burden, and the contribution of other noninfectious inflammatory processes contribute to this picture, represents a crucial next step. Data from healthy volunteers, in vitro experiments, and animal studies can help in quantifying different aspects of these relationships under controlled conditions. This allows for disentanglement of the complex system, leading to an opportunity to study specific independent interactions (Figure 5). For example, in vitro time‐kill studies can help us to increase our understanding of the relationship between drug concentration and pathogen. In vivo studies performed in immunocompetent animals provide insight in the dynamics of immune biomarkers in response to infection. LPS challenges in healthy volunteers can aid in characterizing human immune biomarker dynamics in response to pathogens. Although studying specific interactions in different systems has many advantages, such approach will generate large quantities of data originating from many different sources that need to be integrated. Modeling and simulation techniques, like systems modeling, PBPK modeling, and population modeling, can be powerful tools to help us bring all of the information together. In the future, these tools can facilitate the effective use of biomarkers to increase treatment efficacy and decrease the risk of toxicity and antibiotic resistance development (Figure 5).
Figure 5.
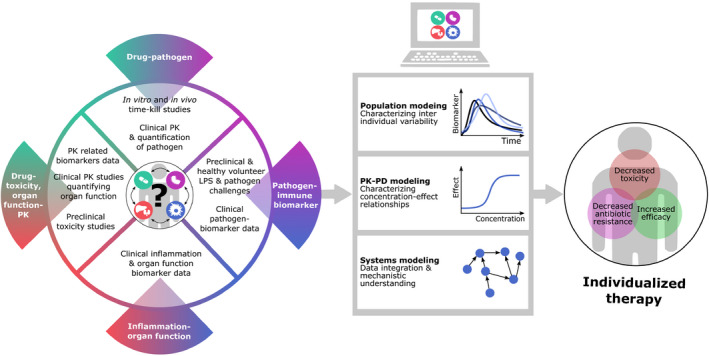
Strategies to develop biomarker‐based strategies to individualize antibiotic therapy. The success of treatment of bacterial infections is affected by several interacting factors. These relationships need to be specifically characterized to enable individualized antibiotic treatments. Such characterization requires data form multiple sources, such as preclinical experiments, healthy volunteer studies, and clinical data, each contributing with unique information. The analysis and integration of such datasets require advanced modeling techniques, including population, pharmacokinetic‐pharmacodynamic (PK‐PD), and systems modeling. From these models we can obtain valuable insights aiding treatment optimization.
FUNDING
This study was funded by ZonMW ABR (Project 541001007).
CONFLICT OF INTEREST
The authors report no conflict of interest. As Editor‐in‐Chief for Clinical Pharmacology and Therapeutics, Piet H. van der Graaf was not involved in the peer review and editorial decision of this manuscript.
Supporting information
Table S1
Fig S1
Fig S2
Supinfo S1‐S2
References
- 1.Garnacho‐Montero, J.et al. Impact of adequate empirical antibiotic therapy on the outcome of patients admitted to the intensive care unit with sepsis. Crit. Care Med. 31, 2742–2751 (2003). [DOI] [PubMed] [Google Scholar]
- 2.De Waele, J.J.et al. Risk factors for target non‐attainment during empirical treatment with β‐lactam antibiotics in critically ill patients. Intensive Care Med. 40, 1340–1351 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Belgrader, P.et al. Rapid pathogen detection using a microchip PCR array instrument. Clin. Chem. 44, 2191–2194 (1998). [PubMed] [Google Scholar]
- 4.Thorsted, A., Nielsen, E.I. & Friberg, L.E. Pharmacodynamics of immune response biomarkers of interest for evaluation of treatment effects in bacterial infections. Int. J. Antimicrob. Agents 56, 106059 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Llewelyn, M.J.et al. The antibiotic course has had its day. BMJ 3418, j3418 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Ríos‐Toro, J.J.et al. Soluble membrane receptors, interleukin 6, procalcitonin and C reactive protein as prognostic markers in patients with severe sepsis and septic shock. PLoS One 12, e0175254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giamarellos‐Bourboulis, E.J.et al. Soluble triggering receptor expressed on myeloid cells 1 as an anti‐inflammatory mediator in sepsis. Intensive Care Med. 32, 237–243 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Pereira, J.M.et al. Mid‐regional proadrenomedullin: an early marker of response in critically ill patients with severe community‐acquired pneumonia? Rev. Port. Pneumol. 22, 308–314 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Angeletti, S.et al. Diagnostic and prognostic role of procalcitonin (PCT) and MR‐pro‐Adrenomedullin (MR‐proADM) in bacterial infections. APMIS 123, 740–748 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Elke, G.et al. The use of mid‐regional proadrenomedullin to identify disease severity and treatment response to sepsis ‐ a secondary analysis of a large randomised controlled trial. Crit. Care 22, 79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Póvoa, P.et al. Biomarkers kinetics in the assessment of ventilator‐associated pneumonia response to antibiotics ‐ results from the BioVAP study. J. Crit. Care 41, 91–97 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Caironi, P.et al. Pentraxin 3 in patients with severe sepsis or shock: the ALBIOS trial. Eur. J. Clin. Invest. 47, 73–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White, J.O.N.C., Nyle, E.S., Snider, R.H., Becker, K.L. & Habener, J.F. Ubiquitous expression of the calcitonin‐I gene in multiple tissues in response to sepsis. J. Clin. Endocrinol. Metab. 86, 396–404 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Linscheid, P.et al. In vitro and in vivo calcitonin I gene expression in parenchymal cells: a novel product of human adipose tissue. Endocrinology 144, 5578–5584 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Schuetz, P.et al. Procalcitonin to initiate or discontinue antibiotics in acute respiratory tract infections. Cochrane Database Syst. Rev. (2012) 10.1002/14651858.CD007498.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schuetz, P.et al. Effect of procalcitonin‐guided antibiotic treatment on mortality in acute respiratory infections: a patient level meta‐analysis. Lancet Infect. Dis. 18, 95–107 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Baer, G.et al. Procalcitonin guidance to reduce antibiotic treatment of lower respiratory tract infection in children and adolescents (ProPAED): a randomized controlled trial. PLoS One 8, e68419 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stolz, D.et al. Procalcitonin for reduced antibiotic exposure in ventilator‐associated pneumonia: a randomised study. Eur. Respir. J. 34, 1364–1375 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Andriolo, B.N.G., Andriolo, R.B., Salomão, R. & Atallah, Á.N. Effectiveness and safety of procalcitonin evaluation for reducing mortality in adults with sepsis, severe sepsis or septic shock. Cochrane Database Syst. Rev. 1, CD010959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Jong, E.et al. Efficacy and safety of procalcitonin guidance in reducing the duration of antibiotic treatment in critically ill patients: a randomised, controlled, open‐label trial. Lancet Infect. Dis. 16, 819–827 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Shehabi, Y.et al. Procalcitonin algorithm in critically ill adults with undifferentiated infection or suspected sepsis: a randomized controlled trial. Am. J. Respir. Crit. Care Med. 190, 1102–1110 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Hochreiter, M.et al. Procalcitonin to guide duration of antibiotic therapy in intensive care patients: a randomized prospective controlled trial. Crit. Care 13, R83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Póvoa, P., Teixeira‐Pinto, A.M. & Carneiro, A.H. C‐reactive protein, an early marker of community‐acquired sepsis resolution: a multi‐center prospective observational study. Crit. Care 15, R169 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmit, X. & Vincent, J.L. The time course of blood C‐reactive protein concentrations in relation to the response to initial antimicrobial therapy in patients with sepsis. Infection 36, 213–219 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Moreno, M.S., Nietmann, H., Matias, C.M. & Lobo, S.M. C‐reactive protein: a tool in the follow‐up of nosocomial pneumonia. J. Infect. 61, 205–211 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Lisboa, T.et al. C‐reactive protein correlates with bacterial load and appropriate antibiotic therapy in suspected ventilator‐associated pneumonia. Crit. Care Med. 36, 166–171 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Hunter, C.A. & Jones, S.A. IL‐6 as a keystone cytokine in health and disease. Nat. Immunol. 16, 448–457 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Oda, S.et al. Sequential measurement of IL‐6 blood levels in patients with systemic inflammatory response syndrome (SIRS)/sepsis. Cytokine 29, 169–175 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Damas, P.et al. Cytokine serum level during severe sepsis in human IL‐6 as a marker of severity. Ann. Surg. 215, 356–362 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel, R.T., Deen, K.I., Youngs, D., Warwick, J. & Keighley, M.R.B. Interleukin 6 is a prognostic indicator of outcome in severe intra‐abdominal sepsis. Br. J. Surg. 81, 1306–1308 (1994). [DOI] [PubMed] [Google Scholar]
- 31.Chenevier‐gobeaux, C., Borderie, D., Weiss, N., Mallet‐coste, T. & Claessens, Y. Presepsin (sCD14‐ST), an innate immune response marker in sepsis. Clin. Chim. Acta 450, 97–103 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Kim, M. & Choi, J. An update on sepsis biomarkers. Infect. Chemother. 52, 1–18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masson, S.et al. Circulating presepsin (soluble CD14 subtype) as a marker of host response in patients with severe sepsis or septic shock: data from the multicenter, randomized ALBIOS trial. Intens. Care Med. 41(1), 12–20 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Yu, H.et al. Evaluating the value of dynamic procalcitonin and presepsin measurements for patients with severe sepsis. Am. J. Emerg. Med. 35, 835–841 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Rawson, T.M.et al. Exploring the use of C‐reactive protein to estimate the pharmacodynamics of vancomycin. Ther. Drug Monit. 40, 315–321 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramos‐Martín, V.et al. Population pharmacokinetics and pharmacodynamics of teicoplanin in neonates: making better use of C‐reactive protein to deliver individualized therapy. J. Antimicrob. Chemother. 71, 3168–3178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spyropoulos, V.et al. Initial immune response in Escherichia coli, Staphylococcus aureus, and Candida albicans Bacteremia. Inflammation 43, 179–190 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Redl, H., Schießer, A., Tögel, E., Assicot, M. & Bohuon, C. Possible role of TNF on procalcitonin release in a baboon model of sepsis. Shock 16, 25–27 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Whang, K.T.et al. Procalcitonin and proinflammatory cytokine interactions in sepsis. Shock 12, 268–273 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Held, F., Hoppe, E., Cvijovic, M., Jirstrand, M. & Gabrielsson, J. Challenge model of TNFα turnover at varying LPS and drug provocations. J. Pharmacokinet. Pharmacodyn. 46, 223–240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thorsted, A.et al. A non‐linear mixed effect model for innate immune response: In vivo kinetics of endotoxin and its induction of the cytokines tumor necrosis factor alpha and interleukin‐6. PLoS One 14, 1–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiers, D.et al. Characterization of a model of systemic inflammation in humans in vivo elicited by continuous infusion of endotoxin. Sci. Rep. 7, 40149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dandona, P.et al. Procalcitonin increase after endotoxin injection in normal subjects. J. Clin. Endocrinol. Metab. 79, 1605–1608 (1994). [DOI] [PubMed] [Google Scholar]
- 44.Weiskopf, D., Weinberger, B. & Grubeck‐Loebenstein, B. The aging of the immune system. Transpl. Int. 22, 1041–1050 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Martín, S., Pérez, A. & Aldecoa, C. Sepsis and immunosenescence in the elderly patient: a review. Front. Med. 4, 20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franceschi, C.et al. Inflamm‐aging: an evolutionary perspective on immunosenescence. Ann. NY Acad. Sci. 908, 244–254 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Delanghe, J.R. & Speeckaert, M.M. Translational research and biomarkers in neonatal sepsis. Clin. Chim. Acta. 451, 46–64 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Sharma, A.A., Jen, R., Butler, A. & Lavoie, P.M. The developing human preterm neonatal immune system: a case for more research in this area. Clin. Immunol. 145, 61–68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charlton, M. & Thompson, J.P. Pharmacokinetics in sepsis. BJA Education 19, 7–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Radke, C.et al. Development of a physiologically based pharmacokinetic modelling approach to predict the pharmacokinetics of vancomycin in critically ill septic patients. Clin. Pharmacokinet. 56, 759–779 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Stuveling, E.M.et al. C‐reactive protein is associated with renal function abnormalities in a non‐diabetic population. Kidney Int. 63, 654–661 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Panichi, V.et al. C‐reactive protein and interleukin‐6 levels are related to renal function in predialytic chronic renal failure. Nephron 91, 594–600 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Carcillo, J.A.et al. Cytochrome P450 mediated‐drug metabolism is reduced in children with sepsis‐induced multiple organ failure. Intensive Care Med. 29, 980–984 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Morgan, E.T.et al. Regulation of drug‐metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab. Dispos. 36, 205–216 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Schmitt, C., Kuhn, B., Zhang, X., Kivitz, A.J. & Grange, S. Disease‐drug‐drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin. Pharmacol. Ther. 89, 735–740 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Vet, N.J.et al. Inflammation and organ failure severely affect midazolam clearance in critically ill children. Am. J. Respir. Crit. Care Med. 194, 58–66 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Tängdén, T.et al. The role of infection models and PK/PD modelling for optimising care of critically ill patients with severe infections. Intensive Care Med. 43, 1021–1032 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Shimoda, M., Kokue, E., Hayama, T. & Vree, T.B. Effect of albumin distribution ‐ a simulation analysis of the effect of altered albumin distribution on the apparent volume of distribution and apparent elimination rate constant of drugs. Pharm. Weekbl. Sci. Ed. 11, 87–91 (1989). [DOI] [PubMed] [Google Scholar]
- 59.Müller, M., Peña, A.D. & Derendorf, H. Issues in pharmacokinetics and pharmacodynamics of anti‐infective agents: distribution in tissue. Antimicrob. Agents Chemother. 48, 1441–1453 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Joukhadar, C.et al. Impaired target site penetration of β‐lactams may account for therapeutic failure in patients with septic shock. Crit. Care Med. 29, 385–391 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Marchand, S., Chauzy, A., Dahyot‐Fizelier, C. & Couet, W. Microdialysis as a way to measure antibiotics concentration in tissues. Pharmacol. Res. 111, 201–207 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Lamer, C.et al. Analysis of vancomycin entry into pulmonary lining fluid by bronchoalveolar lavage in critically ill patients. Antimicrob. Agents Chemother. 37, 281–286 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nau, R., Sörgel, F. & Eiffert, H. Penetration of drugs through the blood‐cerebrospinal fluid/blood‐brain barrier for treatment of central nervous system infections. Clin. Microbiol. Rev. 23, 858–883 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Felton, T.W., Ogungbenro, K., Boselli, E., Hope, W.W. & Rodvold, K.A. Comparison of piperacillin exposure in the lungs of critically ill patients and healthy volunteers. J. Antimicrob. Chemother. 73, 1340–1347 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Arulkumaran, N., Routledge, M., Schlebusch, S., Lipman, J. & Conway Morris, A. Antimicrobial‐associated harm in critical care: a narrative review. Intensive Care Med. 46, 225–235 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tajima, S., Yamamoto, N. & Masuda, S. Clinical prospects of biomarkers for the early detection and/or prediction of organ injury associated with pharmacotherapy. Biochem. Pharmacol. 170, 113664 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Donnell, J.N.O.et al. 24‐Hour pharmacokinetic relationships for vancomycin and novel urinary biomarkers of acute kidney. Injury 61, 1–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vaidya, V.S.et al. Kidney injury molecule‐1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat. Biotechnol. 28, 478–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.González‐Del Castillo, J.et al. Predictive score of haematological toxicity in patients treated with linezolid. Eur. J. Clin. Microbiol. Infect. Dis. 36, 1511–1517 (2017). [DOI] [PubMed] [Google Scholar]
- 70.van Hasselt, J.G.C.et al. Population pharmacokinetic‐pharmacodynamic analysis for eribulin mesilate‐associated neutropenia. Br. J. Clin. Pharmacol. 76, 412–424 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonventre, J.V., Vaidya, V.S., Schmouder, R., Feig, P. & Dieterle, F. Next‐generation biomarkers for detecting kidney toxicity. Nat. Biotechnol. 28, 436–440 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ozer, J.S.et al. A panel of urinary biomarkers to monitor reversibility of renal injury and a serum marker with improved potential to assess renal function. Nat. Biotechnol. 28, 486–494 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Giannini, E.G., Testa, R. & Savarino, V. Liver enzyme alteration: a guide for clinicians. Can. Med. Assoc. J. 172, 367–379 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Campion, S.et al. The current status of biomarkers for predicting toxicity. Expert Opin. Drug Metab. Toxicol. 9, 1391–1408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmidt, E.S. & Schmidt, F.W. Glutamate dehydrogenase: biochemical and clinical aspects of an interesting enzyme. Clin. Chim. Acta 173, 43–55 (1988). [DOI] [PubMed] [Google Scholar]
- 76.O’Brien, P.J., Slaughter, M.R., Polley, S.R. & Kramer, K. Advantages of glutamate dehydrogenase as a blood biomarker of acute hepatic injury in rats. Lab. Anim. 36, 313–321 (2002). [DOI] [PubMed] [Google Scholar]
- 77.Watkins, P.B.The DILI‐sim initiative: insights into hepatotoxicity mechanisms and biomarker interpretation. Clin. Transl. Sci. 12, 122–129 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Hasselt, J.et al. Disease progression‐clinical outcome model for castration‐resistant prostate cancer in patients treated with eribulin. CPT Pharmacometrics Syst. Pharmacol. 4, 386–395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scheer, C.S.et al. Impact of antibiotic administration on blood culture positivity at the beginning of sepsis: a prospective clinical cohort study. Clin. Microbiol. Infect. 25, 326–331 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Palsson, S.et al. The development of a fully‐integrated immune response model (FIRM) simulator of the immune response through integration of multiple subset models. BMC Syst. Biol. 7, 95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Diep, J.K., Russo, T.A. & Rao, G.G. Mechanism‐based disease progression model describing host‐pathogen interactions during the pathogenesis of Acinetobacter baumannii pneumonia. CPT Pharmacometrics Syst. Pharmacol. 7, 507–516 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heilmann, E.et al. Association of kidney function with effectiveness of procalcitonin‐guided antibiotic treatment: a patient‐level meta‐analysis from randomized controlled trials. Clin. Chem. Lab. Med. 59, 441–453 (2021). [DOI] [PubMed] [Google Scholar]
- 83.Assink‐de Jong, E.et al. Stop Antibiotics on guidance of Procalcitonin Study (SAPS): a randomised prospective multicenter investigator‐initiated trial to analyse whether daily measurements of procalcitonin versus a standard‐of‐care approach can safely shorten antibiotic duration. BMC Infect. Dis. 13, 178 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iankova, I.et al. Efficacy and safety of procalcitonin guidance in patients with suspected or confirmed sepsis: a systematic review and meta‐analysis. Crit. Care Med. 46, 691–698 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Nobre, V., Harbarth, S., Graf, J.D., Rohner, P. & Pugin, J. Use of procalcitonin to shorten antibiotic treatment duration in septic patients: a randomized trial. Am. J. Respir. Crit. Care Med. 177, 498–505 (2008). [DOI] [PubMed] [Google Scholar]
- 86.Bouadma, L.et al. Use of procalcitonin to reduce patients’ exposure to antibiotics in intensive care units (PRORATA trial): a multicentre randomised controlled trial. Lancet 375, 463–474 (2010). [DOI] [PubMed] [Google Scholar]
- 87.Meisner, M., Schmidt, J., Hüttner, H. & Tschaikowsky, K. The natural elimination rate of procalcitonin in patients with normal and impaired renal function. Intensive Care Med. 26, S212–S216 (2000). [DOI] [PubMed] [Google Scholar]
- 88.Monnet, E.et al. Evidence of NI‐0101 pharmacological activity, an anti‐TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin. Pharmacol. Ther. 101, 200–208 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Vigushin, D.M., Pepys, M.B. & Hawkins, P.N. Metabolic and scintigraphic studies of radioiodinated human C‐reactive protein in health and disease. J. Clin. Invest. 91, 1351–1357 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nakamura, M.et al. Early elevation of plasma soluble CD14 subtype, a novel biomarker for sepsis, in a rabbit cecal ligation and puncture model. Crit. Care 12, P194 (2008). [Google Scholar]
- 91.Knapp, S.et al. Cutting edge: expression patterns of surface and soluble triggering receptor expressed on myeloid cells‐1 in human endotoxemia. J. Immunol. 173, 7131–7134 (2004). [DOI] [PubMed] [Google Scholar]
- 92.De Kruif, M.D.et al. The influence of corticosteroids on the release of novel biomarkers in human endotoxemia. Intensive Care Med. 34, 518–522 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hinrichs, S.et al. Precursor proadrenomedullin influences cardiomyocyte survival and local inflammation related to myocardial infarction. Proc. Natl. Acad. Sci. USA. 115, E8727–E8736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shi, Q., Hong, H., Senior, J. & Tong, W. Biomarkers for drug‐induced liver injury. Expert Rev. Gastroenterol. Hepatol. 4, 225–234 (2010). [DOI] [PubMed] [Google Scholar]
- 95.Murayama, H., Ikemoto, M., Fukuda, Y., Tsunekawa, S. & Nagata, A. Advantage of serum type‐I arginase and ornithine carbamoyltransferase in the evaluation of acute and chronic liver damage induced by thioacetamide in rats. Clin. Chim. Acta. 375, 63–68 (2007). [DOI] [PubMed] [Google Scholar]
- 96.Dear, J.W.et al. Risk stratification after paracetamol overdose using mechanistic biomarkers: results from two prospective cohort studies. Lancet Gastroenterol. Hepatol. 3, 104–113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Fig S1
Fig S2
Supinfo S1‐S2