

Abstract
Chromosomal translocations constitute driver mutations in solid tumors and leukemias. The mechanisms of how related or even identical gene fusions drive the pathogenesis of various tumor types remain elusive. One remarkable example is the presence of EWSR1 fusions with CREB1 and ATF1, members of the CREB family of transcription factors, in a variety of sarcomas, carcinomas and mesotheliomas. To address this, we have developed in vitro models of oncogenic fusions, in particular, EWSR1-CREB1 and EWSR1-ATF1, in human embryonic stem (hES) cells, which are capable of multipotent differentiation, using CRISPR-Cas9 technology and HDR together with conditional fusion gene expression that allows investigation into the early steps of cellular transformation. We show that expression of EWSR1-CREB1/ATF1 fusion in hES cells recapitulates the core gene signatures, respectively, of angiomatoid fibrous histiocytoma (AFH) and gastrointestinal clear cell sarcoma (GI-CCS), although both fusions lead to cell lethality. Conversely, expression of the fusions in hES cells differentiated to mesenchymal progenitors is compatible with prolonged viability while maintaining the core gene signatures. Moreover, in the context of a mesenchymal lineage, the proliferation of cells expressing the EWSR1-CREB1 fusion is further extended by deletion of the tumor suppressor TP53. We expect the generation of isogenic lines carrying oncogenic fusions in various cell lineages to expand our general understanding of how those single genetic events drive tumorigenesis while providing valuable resources for drug discovery.
Keywords: EWSR1, CREB1, ATF1, CRISPR-Cas9, sarcoma, angiomatoid fibrous histiocytoma, clear cell sarcoma, hES cells, hES-MP cells
INTRODUCTION
Gene fusions involving EWSR1 with members of the cAMP response element binding protein (CREB) family, in particular ATF1 and CREB1, are the main oncogenic drivers of several tumors, including angiomatoid fibrous histiocytoma (AFH), soft tissue and gastrointestinal clear cell sarcoma (CCS), young adult mesothelioma and hyalinizing clear cell carcinoma [1–6]. Although CREB1 and ATF1 are found as gene partners within the same tumor type, there is a propensity for the EWSR1-CREB1 fusion to occur in AFH and EWSR1-ATF1 in soft tissue CCS. An exception is the gastrointestinal CCS (GI-CCS) with the 2 fusions occurring with similar prevalence.
AFH is a mesenchymal neoplasm of borderline malignant potential, typically occurring in the superficial soft tissues of children and adolescents [1, 7]. Soft tissue CCS is a high-grade sarcoma prevalently found in the deep soft tissues of young adults and shows melanocytic differentiation, being positive for MITF and MelanA markers, as well as SOX10 [2, 8]. By contrast, GI-CCS is an aggressive malignant neoplasm that occurs in the gastric or small bowel wall of young adults lacking melanocytic differentiation [3].
Analysis of the etiology of these EWSR1-CREB family translocation-associated tumors and their therapeutic vulnerabilities has been hindered by the paucity of disease models, in particular human cell lines expressing the fusions [9]. The development of genome editing techniques has facilitated the generation of chromosomal translocations in human cell lines [10]. Double strand breaks (DSBs) are introduced into the two genes involved in the fusion, and non-homologous end joinin of the two DSBs results in a chromosomal translocation. However, the process is inefficient, since cells generally repair DSBs without translocation and the recovery of clones harboring the translocation is difficult if the fusion does not induce cellular transformation or impairs cell proliferation.
In this study we developed an approach combining CRISPR-Cas9 technology and homology-directed repair (HDR), which allows the selection of translocation-positive clones and conditional expression of the fusion. Using this strategy we isolated clones harboring the EWSR1-WT1 translocation, the genetic hallmark of desmoplastic small round cell tumor (DSRCT), in human embryonic stem-derived mesenchymal progenitor (hES-MP) cells [11] even if the limited number of passages of the non-immortalized hES-MP cells hampered further investigation on the role of the fusion in sarcomagenesis.
To surmount this limitation, we propose a strategy to generate chromosomal translocations in human embryonic stem (hES) cells that grow almost indefinitely and can differentiate into a variety of cell types. We show that hES cells harboring a chromosomal translocation can be differentiated into hES-MP cells allowing the characterization of the fusion and its role in sarcomagenesis upon conditional expression. Our in vitro models of EWSR1-CREB1 and EWSR1-ATF1 fusions recapitulate the transcriptional profiles found in human tumors with these gene fusions. As the spectrum of tumors driven by these fusions include mesenchymal (AFH and GI-CCS), neuroectodermal (CCS), and epithelial (carcinoma, mesothelioma) lineages, we consider hES cells an optimal system for modeling translocation.
RESULTS
Human AFH and CCS driven by EWSR1-CREB fusions have distinct gene expression profile.
Most cases of AFH (90%) harbor a t(2;22)(q34;q12) translocation (Fig. 1A), with a resulting EWSR1-CREB1 transcript that fuses EWSR1 exons 1–7 with CREB1 exons 7–8 [1, 7]. We have previously defined the gene expression profile of human AFH showing that the top upregulated genes are serum and glucocorticoid-regulated kinase 1 (SGK1), matrix-remodeling-associated protein 5 (MXRA5), and cathepsin B precursor (CTSB) [1]. This expression signature, designated as the ‘AFH core gene signature’, was re-analyzed and confirmed as part of this study compared to a large number of sarcomas and normal tissues available on the same Affymetrix U133A array platform (Fig. 1B).
Figure 1. Gene expression signatures of human AFH and CCS.

(A) AFH is associated with the t(2;22)(q34;q12) translocation, resulting in an EWSR1-CREB1 fusion.
(B)EWSR1-CREB1 fusion-positive AFH tumors have high transcript levels of SGK1, MXRA5, and CTSB (3 cases), as compared to CCS tumors (4 cases), 44 other sarcomas (5 angiosarcoma AS, 3 fibrosarcoma FS, 5 gastrointestinal stromal tumors GIST, 11 paraganglioma CTR, 3 leiomyosarcoma LMS, 3 myxoid liposarcoma MLS, 3 undifferentiated pleomorphic sarcoma MFH, 3 synovial sarcoma SS, 4 solitary fibrous tumor SFT, 4 small blue round cell tumor SBRCT) and 6 normal tissues (testis, brain, adrenal, kidney, small intestine and fetal stomach), (“Others”).
(C) CCS is associated with t(12;22)(q13;q12) translocation, resulting in an EWSR1-ATF1 gene fusion. (D) EWSR1-ATF1 fusion-positive CCS tumors (4 cases) have high transcript levels of several genes, in particular, PMEL and SLC7A5, as compared to AFH tumors and other sarcomas and normal tissues (same as in panel B).
(E) Two additional EWSR1-CREB1 fusion-positive AFH (AFH31.2 and AFH61) samples show high mRNA levels of SGK1, MXRA5, SLC7A5 and DUSP4 and to a lesser extent, SOX10, as compared with normal tissue. CTSB is also high in these two tumors, but it is not present in normal tissue. RT-PCR and Sanger sequencing showing the EWSR1(ex7)-CREB1(ex7) fusion in AFH31.2 is also included.
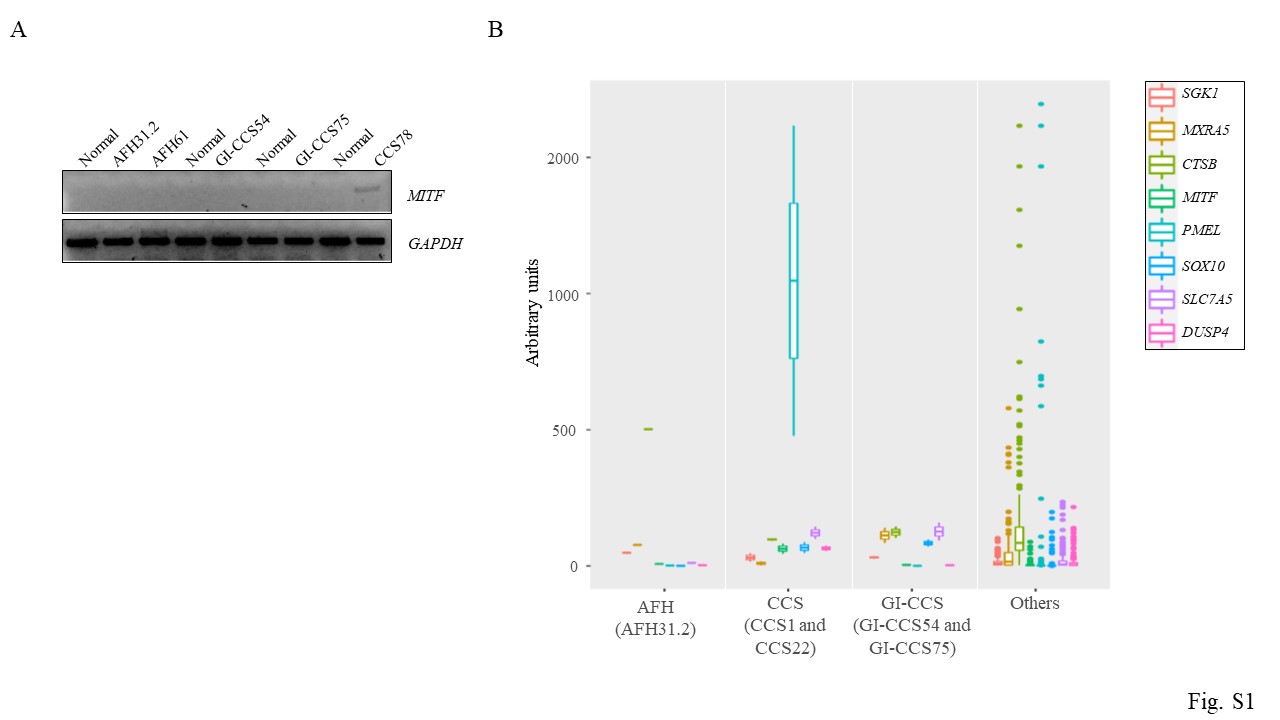
(F) Two additional EWSR1-ATF1 fusion-positive CCS (1T and 22T) cases revealed similarly high levels of expression in the genes shown in (D). RT-PCR and Sanger sequencing showing the in frame EWSR1(ex8)-ATF1(ex4) fusion and MITF expression in these two tumors, but not in normal tissue.
Numerical values indicate fold increase compared to normal tissue. Error bars in this figure represent standard deviation from the mean of at least three technical replicates. Statistical significance is calculated by comparison of tumor samples with normal tissue. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 (paired t-test).
The genetic hallmark of soft tissue CCS is a t(12;22)(q13;q12) (Fig. 1C), resulting in an EWSR1-ATF1 fusion, although rare examples of cases with EWSR1-CREB1 fusions have also been documented [8, 12]. Tumors with EWSR1-ATF1 translocation express multiple isoforms. The most common transcripts include EWSR1 exons 1–8 or 1–7 fused to ATF1 exons 4–7 or 5–7, respectively [2, 8]. Other less common variants have been identified, including an out-of-frame fusion, presumably resulting from alternative splicing (EWSR1 exons 1–7, ATF1 exons 4–7), of questionable oncogenic potential [8]. The soft tissue CCS core gene signature includes several melanocytic markers (MITF and PMEL), the transcription factor SOX10, the amino-acid transporter SLC7A5, and the phosphatase DUSP4 [1] (Fig. 1D).
The above core gene signatures were then tested in two additional AFH tumors expressing the EWSR1-CREB1 fusion (AFH31.2 and AFH61, Fig. 1E), and two additional soft tissue CCS tumors expressing the in-frame EWSR1(ex8)-ATF1(ex4) fusion (1T and 22T; Fig. 1F). By quantitative RT-PCR (qRT-PCR), SGK1 and MXRA5 were confirmed to be upregulated in the AFH tumors but not the CCS tumors (Fig. 1E, F). In contrast, PMEL and SOX10 were upregulated in the CCS tumors compared to the AFH tumors while MITF was only expressed in CCS but not AFH (Fig. 1F and S1A). SLC7A5 and DUSP4 genes were substantially upregulated in the CCS tumors, but also in one of the AFH tumors (AFH61) (Fig. 1E, 1F).
GI-CCS harbors an EWSR1-CREB1 fusion in more than half of the cases, with the remaining showing EWSR1-ATF1 fusion [3, 13]. The data on GI-CCS transcriptional profiling remains limited, with one case harboring an EWSR1-CREB1 fusion previously analyzed by our group with SGK1, MXRA5, SOX10, and SLC7A5 mRNA upregulation [1]. The same tumor (GI-CCS54) was re-analyzed and confirmed to have overexpression of MXRA5, SOX10, and SLC7A5 but not SGK1 by qRT-PCR (Fig. 2A), suggesting a hybrid gene signature, including some genes upregulated in soft tissue CCS (SOX10, SLC7A5), as well as some in AFH (SGK1, MXRA5) [1]. Importantly, GI-CCS lacks the melanocytic signature seen in soft tissue CCS (PMEL, MITF) (Fig. 2A and S1A). In contrast, SOX10 is commonly upregulated in both soft tissue CCS and GI-CCS, being strongly positive by immunohistochemistry in most cases [13, 14], suggesting that high level SOX10 expression can be used to distinguish between AFH and CCS histotypes. Interestingly, qRT-PCR performed in one GI-CCS case expressing the EWSR1(ex8)-ATF1(ex4) fusion (GI-CCS75), also showed upregulation of SOX10, SLC7A5 and MXRA5 and, to a lesser extent, SGK1 and DUSP4 (Fig. 2B and S1A), demonstrating a hybrid signature.
Figure 2. Gene expression signature of GI-CCS tumors expressing EWSR1-CREB1 and EWSR1-ATF1 fusions.

(A) An EWSR1-CREB1 fusion-positive GI-CCS (GI-CCS54) demonstrates upregulation of MXRA5, SOX10 and SLC7A5, and to lesser extent DUSP4, but not SGK1. RT-PCR and Sanger sequencing show the in-frame EWSR1(ex7)-CREB1(ex7) fusion.
(B) An EWSR1-ATF1 fusion-positive GI-CCS (GI-CCS75) also demonstrates high levels of the genes shown in (A), as well as SGK1. RT-PCR and Sanger sequencing show the in-frame EWSR1(ex8)-ATF1(ex4) fusion. Numerical values indicate fold increase compared to normal tissue. Statistical analysis same as in Fig. 1. Fold increase values for PMEL are ≤1 (0.074 and 0.17 respectively).
Whole transcriptome paired-end RNA sequencing was also performed in the subset of cases tested above, including one AFH, 2 CCS and 2 GI-CCS samples. The study group was compared to a large cohort of 186 various tumors types available on the same platform and the core gene signature for each tumor type was confirmed (Fig. S1B).
Engineered hES cells expressing the EWSR1-CREB1 and EWSR1-ATF1 fusions recapitulates the core transcriptional signature of human AFH and GI-CCS tumors.
The approach to generate chromosomal translocations is based on incorporation of a donor template with an hygromycin selection marker (hyg–) and homology arms to guide translocation by HDR after a Cas9-induced DSB (Fig. 3A). Upon correct integration of the donor, a splice acceptor sequence (SA) allows expression of the marker (hyg+) from the promoter upstream of the integration site preventing the immediate expression of the fusion transcript, which can be conditionally activated by Cre recombinase.
Figure 3. Generation and expression of EWSR1-CREB1 and EWSR1-ATF1 fusions in hES cells.

(A) Chromosomal translocation strategy for conditional fusion gene expression. A donor template containing a promoter-less selectable marker hygromycin (hyg–) and two homology arms (shaded regions) is inserted by HDR at the gRNA-mediated DSBs (scissors) at the chromosomal locations of the 5’ and 3’ fusion partners. An in-frame splice acceptor (SA) sequence upstream of hyg– allows expression of the gene upon correct integration of the donor (hyg+), allowing selection in hygromycin. Transcription of the fusion gene is dependent on removal of the selectable marker by Cre recombinase.
(B) Fusion genes are formed upon Cre expression. Left, Schematic representation of the three translocations prior to Cre expression, with the size of the PCR products across the breakpoint junctions before and after (in parenthesis) removal of hyg+. Right, a PCR size shift confirms the formation of the direct fusion for each of the translocations.
(C) The reciprocal translocations confirmed by PCR analysis in the clones harboring the EWSR1(ex7)-CREB1(ex7) and EWSR1(ex7)-ATF1(ex5) translocations.
(D) Dual color FISH analysis showing the EWSR1 and CREB1 loci and the resulting derivative chromosomes, Der 22 (top) and Der 2 (bottom), using the indicated “split” probes (i.e., two for each gene). Note that in the top image, the EWSR1 and CREB1 probes do not match the chromosome coloring scheme). Unrearranged chromosomes 2 and 22 are also indicated.
(E) Induction of the EWSR1(ex7)-CREB1(ex7) fusion after Cre expression in hES cells results in upregulation of SGK1 and MXRA5 (ev, empty vector). Histogram represents the mean fold increase compared to empty vector condition of wild type cells as measured by ΔΔCt method and error bars indicate the standard deviation from the mean of 3 independent experiments. Statistical significance is calculated with a paired t-test comparing Cre condition with the corresponding control (ev). *p<0.05; when not indicated, the difference is considered not statistically significant.
(F) Induction of the EWSR1(ex7)-ATF1(ex5) fusion, but not the EWSR1(ex7)-ATF1(ex4), after Cre expression in hES cells results in upregulation of GI-CCS core signature genes, SGK1, MXRA5, SOX10, and DUSP4 (ev, empty vector). Statistical analysis as in panel E, n=3.
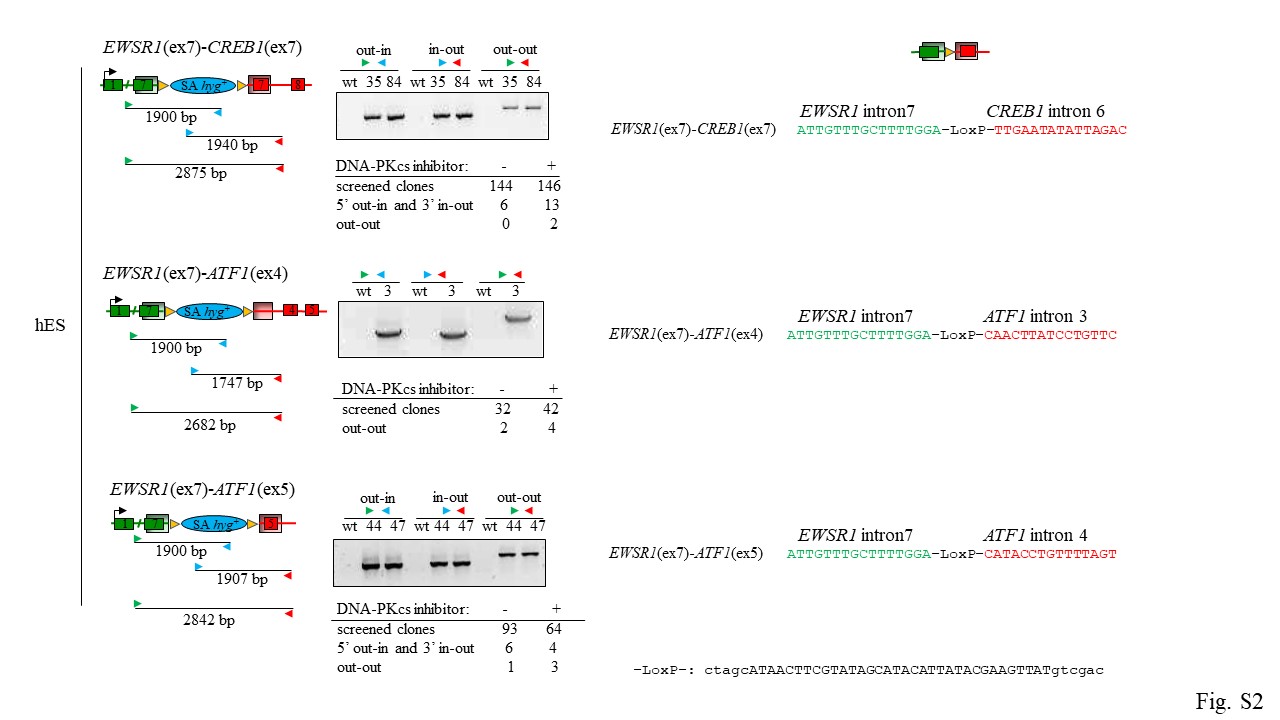
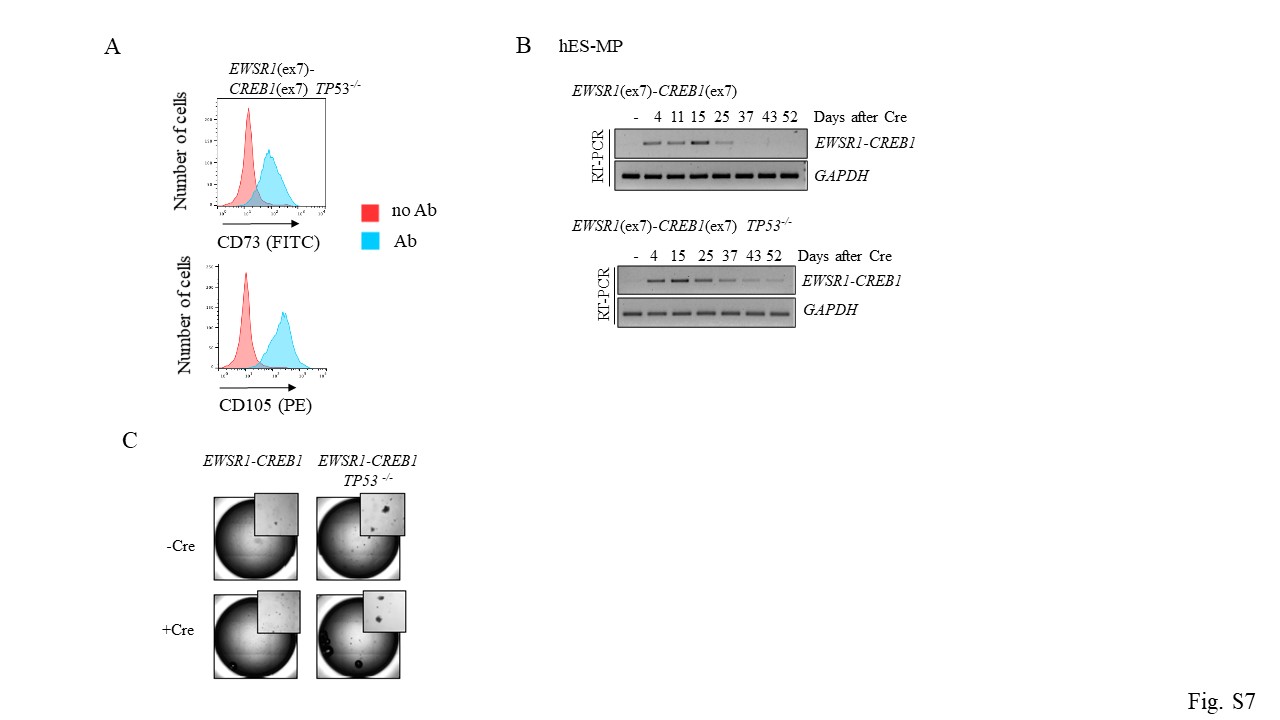
Reflecting the human tumor genotypes, we generated hES cell lines harboring EWSR1 exons 1–7 fusions to CREB1 exons 7–8 (EWSR1(ex7)-CREB1(ex7)) and either ATF1 exons 4–7 or 5–7 (EWSR1(ex7)-ATF1(ex4) and EWSR1(ex7)-ATF1(ex5), respectively) (Fig. 3B). The EWSR1(ex7)-ATF1(ex4) fusion generates the out-of-frame transcript, which would serve as a negative control for our experiments. Hygromycin-resistant colonies were screened for correct integration of the donor at the two translocation loci by out-in, in-out PCRs, and then for the translocation using both primers outside the homology arms (out-out) (Fig. S2). At the time of transfection, cells were treated either with either DNA-PKi (NU7441) to promote HDR [11, 15, 16] or DMSO as control. For the EWSR1(ex7)-CREB1(ex7) fusion, DNA-PKi-treated plates gave 13 out of 146 hyg+ clones positive for both the out-in and in-out PCRs, 2 being also positive for out-out PCR (Fig. S2). Plates treated with DMSO gave 6 out of 144 clones, although none were positive for out-out PCR, possibly due to tandem plasmid integration [11]. We also obtained 4 of 42 hyg+ clones carrying EWSR1(ex7)-ATF1(ex4) and 3 of 64 hyg+ clones carrying EWSR1(ex7)-ATF1(ex5), with somewhat higher efficiency using DNA-PKi (Fig. S2).
Clones harboring translocations were transfected with Cre recombinase and the resulting fusion was verified by the size change of the out-out PCR product (Fig. 3B). The reciprocal translocation (CREB1-EWSR1 or ATF1-EWSR1) was confirmed by PCR and junction sequencing (Fig. 3C and S3A). Finally, these clones were examined at the targeted loci (EWSR1 and CREB1 or ATF1) in the unrearranged chromosomes of each translocation pair. Small indels within the respective introns were identified by Sanger sequencing in all but one of the clones that retained a wild-type allele (Fig. S3B). Protein expression from the unrearranged allele was not impaired as showed by western blot analysis (Fig. S3C). Thus, these chromosomes showed no evidence of additional rearrangements.
The translocations were also tested by fluorescence in situ hybridization (FISH), using probes flanking the EWSR1, CREB1 and ATF1 genes. For the EWSR1-CREB1 fusion, FISH showed the presence of Der22 containing the EWSR1-CREB1 fusion, the reciprocal translocation (Der2) and, intact chromosomes 22 and 2 (Fig. 3D and S3D). The EWSR1(ex7)-ATF1(ex5) translocation and intact chromosomes 22 and 12 were also verified by FISH (Fig. S3D).
To test the induction of the transcriptional core signatures we induced the EWSR1-CREB1 and the two EWSR1-ATF1 fusions by Cre expression. Cells expressing Cre were enriched by cell sorting 2 days after nucleofection with the Cre-linked mCherry marker plasmid and collected at day 4 for RNA extraction. The fusion transcripts (Fig. 3E, 3F) and the chimeric protein (Fig. S3E) were identified in cells with the translocations, but not in parental or control cells. By qRT-PCR, SGK1 and MXRA5 was upregulated in cells expressing EWSR1-CREB1, while PMEL, SOX10, SLC7A5 and DUSP4 mRNAs were not significantly increased (Fig. 3E).
In the case of EWSR1-ATF1 translocations, cells expressing the out-of-frame fusion EWSR1(ex7)-ATF1(ex4) showed no transcriptional changes. In contrast, cells expressing the EWSR1(ex7)-ATF1(ex5) fusion showed upregulation of SGK1, MXRA5, SOX10, and DUSP4, but not the melanocytic gene PMEL or SLC7A5 (Fig. 3F), thus for the most part reiterating the profile of human GI-CCS. Interestingly, cells with the EWSR1(ex7)-ATF1(ex4) fusion revealed also the EWSR1(ex7)-ATF1(ex5) transcript (Fig. S3F), indicating the possibility of alternative splicing, thus recapitulating the multiple transcripts reported in human CCS tumor analysis [17, 18].
Reduced viability of hES cells upon expression of EWSR1(ex7)-CREB1(ex7) or EWSR1(ex7)-ATF1(ex5), which is not rescued by TP53 mutation.
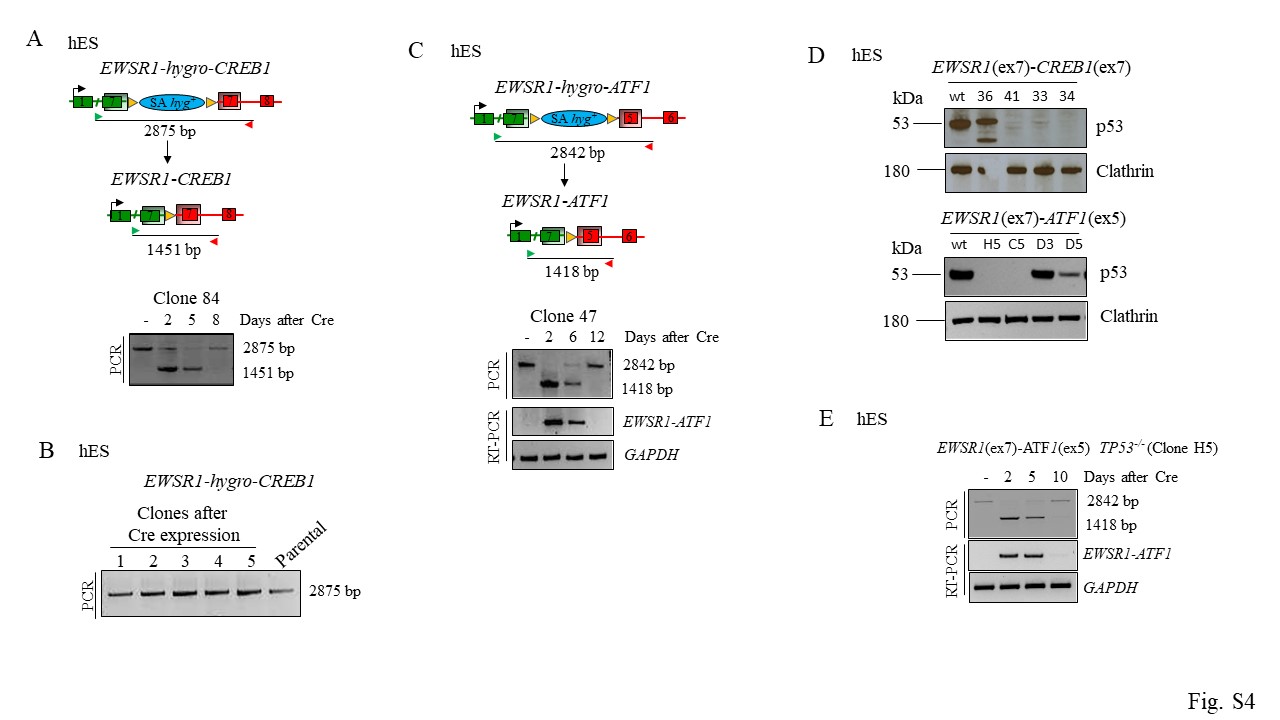
To study the effects of the gene fusions on viability, we performed a time course monitoring the presence of the fusions after Cre expression. For the EWSR1-CREB1 translocation, the fusion product (with deletion of hyg+) was observed at the genomic level 4 and 7 days after Cre expression (Fig. 4A, S4A), but by day 11 it returned to pre-Cre size. Concurrently, the EWSR1-CREB1 transcript was observed at days 4 and 7 after Cre expression but was absent at day 11. These results indicate that the small population of cells that had not undergone Cre-mediated recombination had a significant growth advantage. Concurrent with the decrease in fusion transcript levels, AFH core signature genes were induced by day 4 and 7 but lost at day 11 (Fig. 4B). Thus, expression of the fusion impaired their proliferation and/or survival, enabling the overgrowth of a small population of cells not expressing the fusion. We also attempted single-cell plating to screen for fusion-expressing clones, however, none of the 5 clones recovered had undergone Cre-mediated recombination (Fig. S4B), providing evidence of EWSR1-CREB1 fusion-associated lethality in the hES cell background.
Figure 4. Impaired proliferation of hES cells upon EWSR1(ex7)-CREB1(ex7) or EWSR1(ex7)-ATF1(ex5) fusion expression.

(A) Time course of the EWSR1(ex7)-CREB1(ex7) fusion after Cre expression. The fusion was analyzed using both PCR of genomic DNA and RT-PCR. The lower band in the PCR is the direct fusion, while the upper band indicates that the fusion partners are separated by the hyg+ gene. See Fig. 3B.
(B) Time course for expression of AFH signature genes SGK1 and MXRA5 using qRT-PCR upon EWSR1(ex7)-CREB1(ex7) fusion in sorted cells. Histogram represents the fold increase compared to wild type cells (-Cre condition) of 1 representative experiment (n=3).
(C) Same as panel A for the EWSR1(ex7)-ATF1(ex4) fusion.
(D) Same as panel A for the EWSR1(ex7)-ATF1(ex5) fusion.
(E) Time course for expression analysis using qRT-PCR upon EWSR1(ex7)-ATF1(ex5) fusion in pooled cells. Histogram represents the mean of fold increase compared to wild type cells (-Cre conditions) of 2 independent experiments.
(F) Same as panel A for the EWSR1(ex7)-CREB1(ex7) fusion with, in addition, TP53−/− cells.
(G) Time course for expression of AFH signature genes using qRT-PCR upon EWSR1(ex7)-CREB1(ex7) fusion induction in wild-type and TP53−/− background. Histogram represents the fold increase to wild type of 1 representative experiment (n=3).
(H) Quantification of Annexin V staining for measurement of apoptosis. Annexin V intensity is represented as the ratio of the percentages of Cre and empty vector-transfected cells. Error bars represent standard deviation from the mean of n≥3 independent experiments. Unpaired t-test, *p<0.05, **p<0.01, ***p<0.001, ns=not significant.
The out-of-frame EWSR1(ex7)-ATF1(ex4) fusion was stable for 14 days after Cre expression (Fig. 4C), indicating that translocation itself is not sufficient to affect cell viability. For the in-frame EWSR1(ex7)-ATF1(ex5) translocation the recovery of mCherry-sorted cells after day 4 proved to be difficult, suggesting that the fusion was quite toxic. A time course on a pool of unsorted Cre-transfected cells showed that the fusion was detected at both genomic DNA and mRNA levels at day 2 and 6, but was absent by day 12 (Fig. 4D, Fig. S4C). The induction and decline in fusion transcript expression were accompanied by the rise and fall of the GI-CCS core gene signature (Fig. 4E). Thus, expression of the EWSR1(ex7)-ATF1(ex5) fusion in the hES cell background substantially impairs cell proliferation and/or survival.
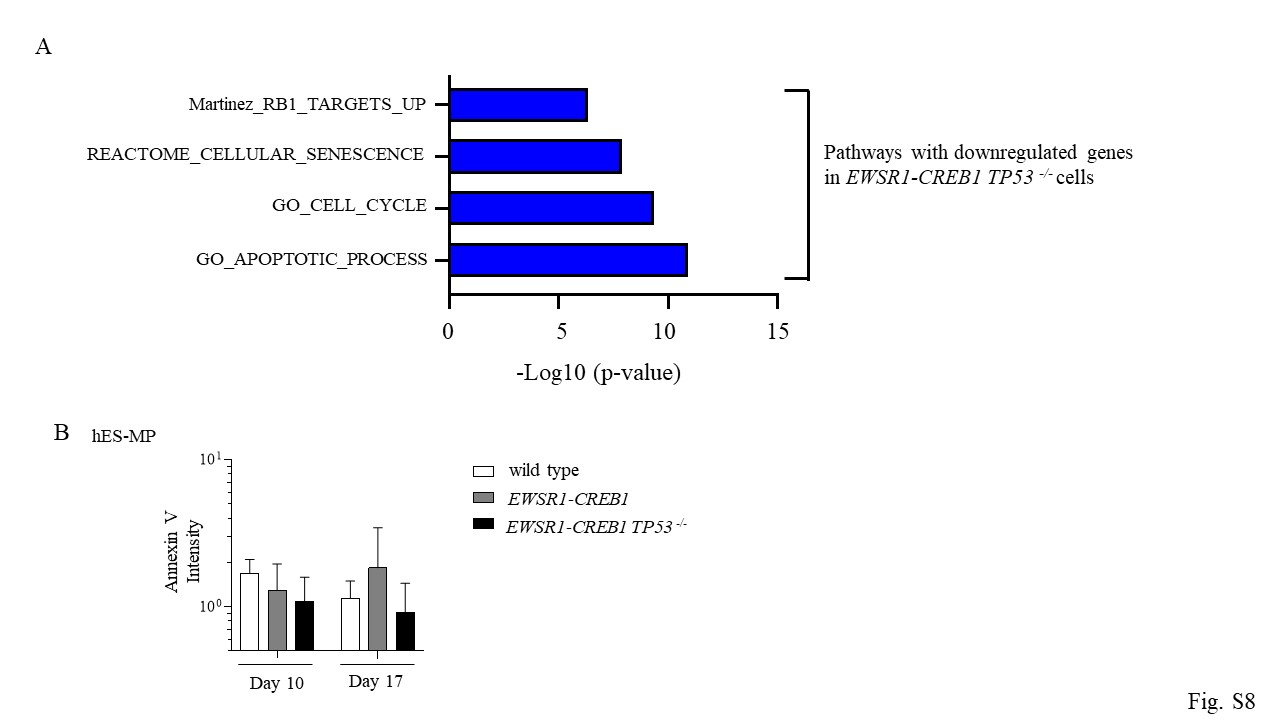
Mutations in the tumor suppressor TP53 are frequently detected in soft tissue sarcoma [19]. To test whether loss of TP53 would rescue cell proliferation/viability we generated null alleles of TP53 in cells harboring the EWSR1(ex7)-CREB1(ex7) and EWSR1(ex7)-ATF1(ex5) translocations (Fig. S4D). Deletion of TP53did not alter the time course of expression of the fusions or the AFH gene signature, and importantly, it did not rescue the viability of hES cells (Fig. 4F, G, Fig S4E). Thus, we measured the AnnexinV 4 days after Cre-mediated fusion induction and the number of AnnexinV positive cells was increased even in TP53−/− cells (Fig. 4H), suggesting a TP53-independent induction of apoptosis mediated by the oncogenic fusions in hES cells.
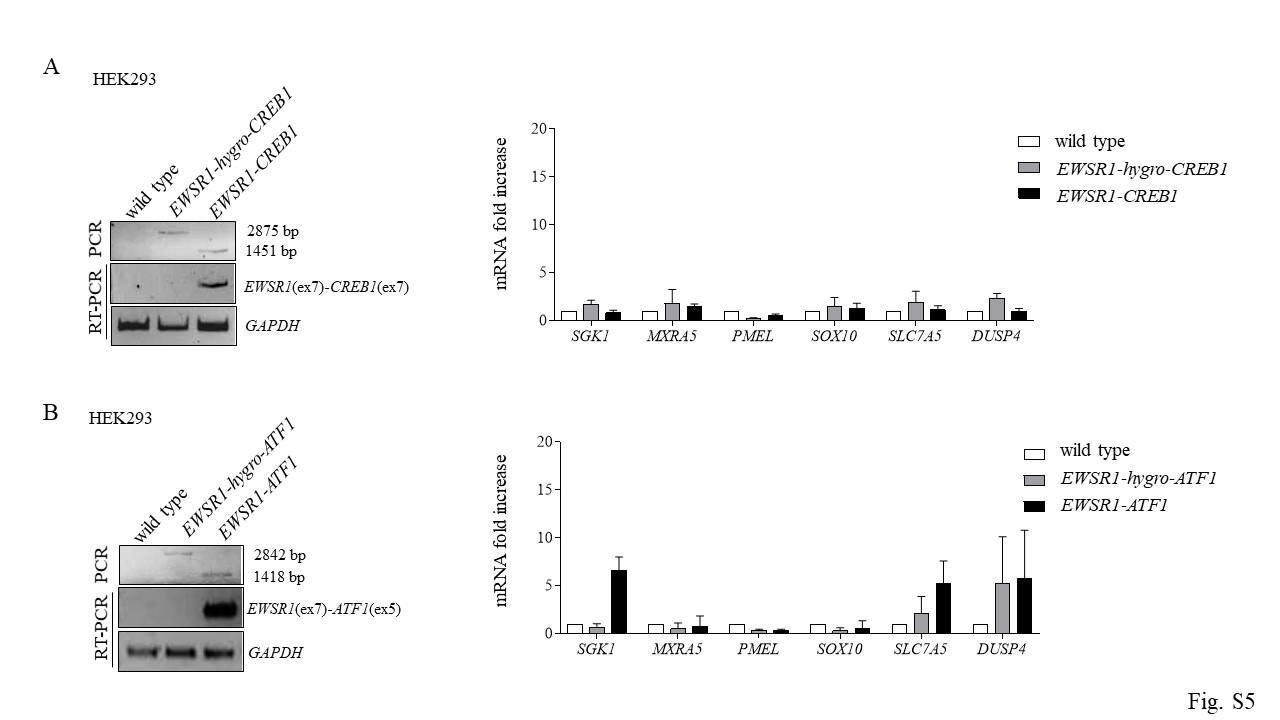
Given the inability to propagate hES cells expressing the fusions, we asked whether these translocations could be propagated in the immortalized human embryonic kidney 293 (HEK293) that are often used for molecular studies including generation of translocations[10]. We were able to generate both translocations in HEK293 cells (Fig. S5A, B) and to recover colonies uniformly expressing both fusions with no impact on cell viability; however, these cells did not recapitulate the expression signature observed in AFH, CCS and GI-CCS tumor samples (Fig. S5A, B). Thus, HEK293 cells are not an appropriate model for these translocations.
Prolonged viability of hES-derived mesenchymal cells expressing the EWSR1-CREB1 or EWSR1-ATF1 fusions while maintaining the core gene signature.
Although the cell of origin of most translocation-associated sarcomas remains elusive, a potential derivation from mesenchymal stem cell progenitors has been suggested [20]. However, the use of non-immortalized mesenchymal cells to model and characterize cancer relevant chromosomal translocations has been hindered by their limited ability to be passaged [11]. Therefore, we differentiated hES cells harboring the EWSR1-CREB1/ATF1 translocations (prior to Cre expression) to hES-derived mesenchymal progenitor (hES-MP) cells [21]. Differentiated cells were confirmed to be mesenchymal stem cells by staining for two surface markers, CD73 and CD105 (Fig. 5A). These cells were infected with a lentivirus expressing the Cre recombinase and collected at different time points to monitor fusion expression. hES-MP cells expressing the EWSR1-CREB1 fusion remained viable longer (~1 month) compared to the hES cells, while also inducing expression of SGK1 and MXRA5 (Fig. 5B). Interestingly, the expression levels of SLC7A and DUSP4 that were upregulated by qRT-PCR in the AFH tumors (Fig. 1E), but not in hES cells (Fig. 3E), were increased in this cellular context. A similar observation was also seen for hES-MP cells expressing the EWSR1-ATF1 fusion and the GI-CCS gene core signature (Fig. 5C). Thus, hES-MP cells represent a suitable system to study the EWSR1-CREB1 and EWSR1-ATF1 translocations and their role in cellular transformation.
Figure 5. Viability of hES-derived mesenchymal cells expressing EWSR1-CREB1 and EWSR1-ATF1 while maintaining the AFH and CCS core gene signatures.

(A) hES-MP cells express mesenchymal surface markers CD73 and CD105. EWSR1(ex7)-CREB1(ex7) and EWSR1(ex7)-ATF1(ex5) refer to cells carrying the translocation prior to Cre expression.
(B) Time course in days of EWSR1-CREB1 fusion expression in hES-MP cells after Cre expression. The AFH core gene signature is detected in hES-MP cells expressing the EWSR1(ex7)-CREB1(ex7) fusion over time. Histogram represents the fold increase compared to wild type cells (-Cre condition) and the error bars the standard deviation from the mean of 4 independent experiments. Statistical significance is calculated with a paired t-test comparing +Cre with the corresponding -Cre condition. *p<0.05, **p<0.01; when not indicated the difference is considered not statistically significant.
(C) Same as panel B for the EWSR1(ex7)-ATF1(ex5) fusion. qRT-PCR in hES-MP cells expressing the EWSR1(ex7)-ATF1(ex5) at different time points. Statistical analysis as in panel B, n=5. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
EWSR1-WT1 fusion expression is associated with a distinct transcriptional signature from the EWSR1-CREB fusion in isogenic lines.
DSRCT is an aggressive sarcoma of presumed mesenchymal origin characterized by a t(11;22)(p13;q12) translocation, with breakpoints within the EWSR1 and WT1 genes (Fig. S6A) [22].
To compare with the CREB family fusions, we generated the EWSR1 exons 1–7-WT1 exons 8–10 (EWSR1(ex7)-WT1(ex8)) fusion in hES cells (Fig. S6B). The fusion transcript and the PCR product from the fusion were detected 2 days after Cre expression, but both were reduced over time and undetectable by day 11 (Fig. S6C).
We then induced differentiation of hES cells harboring the EWSR1(ex7)-WT1(ex8) translocation to hES-MP cells and expressed Cre recombinase. As observed with the EWSR1-CREB1 and EWSR1-ATF1 fusions, expression of EWSR1-WT1 was tolerated by hES-MP cells for ~ 1 month (Fig. S6D). Importantly, in cells expressing EWSR1-WT1 none of the genes of the AFH, CCS and GI-CCS core signatures were upregulated, while PDGFA, a known DSRCT target gene [23], was upregulated (Fig. S6E). Thus, these cells appear to represent a valid model to study DSRCT and provide an otherwise isogenic system to compare different EWSR1 fusions found in distinct tumor types.
EWSR1-CREB1 fusion expression, the AFH core signature, and viability are maintained longer in a TP53−/− background.
Although the EWSR1-CREB1 fusion is better tolerated in hES-MP cells than in hES cells, fusion expression is nevertheless reduced after several weeks. Therefore, we investigated the effect of TP53 deletion in this cellular context by differentiating TP53−/− hES cells harboring the EWSR1-CREB1 translocation to mesenchymal precursors (Fig. S7A). After Cre recombinase expression, we found that fusion transcripts (Fig. 6A, S7B) and the AFH core gene signature (Fig. 6B) were maintained longer (~7 weeks) with TP53 mutation. Thus, in hES-MP cells TP53 deletion partially rescues the lethality caused by expression of the EWSR1-CREB1 oncogenic fusion, unlike in hES cells. We then tested if the prolonged viability resulted in anchorage-independent cell growth in a soft agar assay. Cells were plated in soft agar 3 days after infection with Cre recombinase and colony formation was monitored. Expression of the fusion was not enough to induce cellular transformation, however, colonies formed in the TP53−/− background regardless of the expression of the fusion, making it difficult to determine a specific role for fusion in this context (Fig. S7C).
Figure 6. Prolonged viability of hES-derived mesenchymal cells expressing EWSR1-CREB1 in TP53−/− background.

(A) Time course in days for EWSR1-CREB1 fusion expression and (B) AFH core gene signature in wild type and TP53−/− genetic background. Histogram represents the fold increase to wild type (-Cre condition) of 1 representative experiment (n=2).
(C)CDKN1A expression is measured 15 days after the induction of the EWSR1-CREB1 fusion in wild type and TP53−/− cells. Histogram represents the fold increase of the +Cre condition compared to the corresponding -Cre condition and the error bars the standard deviation from the mean of 3 independent experiments. Statistical significance is calculated with a paired t-test comparing the +Cre with the corresponding -Cre condition. ***p<0.001, ns=not significant. (D) EWSR1(ex7)-CREB1(ex7) fusion expression impairs the cell proliferation/viability of hES cell after 7 days, regardless of TP53 deletion. On the contrary, in hES-MP the viability is prolonged and is further expanded by deletion of TP53 (see discussion for details).
To dissect the mechanism leading to extended viability in TP53−/− genetic background, gene set enrichment analysis (GSEA) was performed on whole transcriptome RNAseq comparing wild type and TP53−/− cells expressing the EWSR1-CREB1 fusion. TP53−/− cells show downregulation of genes involved in cell cycle, apoptosis, and cellular senescence (Fig. S8A). Among these, CDKN1A, the gene coding for the TP53 target p21, is of interest, because its role in cell cycle progression [24], apoptosis [25] and it is frequently downregulated in many types of cancers [26]. RT-PCR on hES-MP cells expressing the EWSR1-CREB1 fusion confirmed the upregulation of CDKN1A, which is suppressed by TP53 depletion (Fig. 6C). The level of Annexin V measured 10 and 17 days after infection was not increased in fusion expressing cells (Fig. S8B) indicating that the induction of p21 is not associated with apoptosis and suggesting that p21 may be acting instead to control cell cycle progression.
DISCUSSION
The histogenesis of translocation-associated sarcomas remains elusive, in most cases lacking a defined cell of origin [27]. Furthermore, it has become apparent that certain gene fusions are promiscuous, driving tumor pathogenesis in various cell lineages, akin to the fusions of the EWSR1-CREB family, which can give rise to sarcomas, carcinomas, and mesotheliomas. In this study we model EWSR1-CREB translocations in hES cells, an advantageous cell type for modelling oncogenic translocations, given their immortal growth, clonability, and ability to give rise to a diverse spectrum of cell types, and thus tumor types.
To reproduce the human disease, we generated EWSR1 (exons 1–7)-CREB1 (exons 7–8) and EWSR1 (exons 1–7)-ATF1 (exons 5–7) fusions commonly detected in AFH and CCS. Results showed that hES cell lines conditionally expressing the EWSR1-CREB1 and EWSR1-ATF1 fusions recapitulate the core transcriptional signature, respectively, of AFH and GI-CCS. However, prolonged expression of these oncogenic fusions (<12 days) impairs cell proliferation, in agreement with the embryonic lethality induced in mice expressing the EWSR1-ATF1 fusion [28]. By contrast, HEK293 cells expressing the fusions continue to proliferate but do not recapitulate the gene signatures, likely due to the altered transcriptional output of this cell line and/or mutations in tumor suppressors oroncogenes that abrogate cell death.
While fusion induction in hES cells is sufficient to drive upregulation of genes found in AFH, CCS and GI-CCS, that by itself does not translate into cellular transformation, likely requiring additional secondary genetic events and/or epigenetic modifications, to initiate the transformation process. Based on next generation sequencing a subset of human AFH and CCS tumors is accompanied by additional mutations in tumor suppressor genes (e.g., TP53, CDKN2A/B) (MSK-IMPACT, Antonescu personal communication). Typically, TP53 plays important roles in response to genotoxic stress and oncogene activation through cell cycle arrest, apoptosis, and senescence. However, deletion of TP53 did not rescue cell proliferation or suppress apoptosis upon expression of either the EWSR1-CREB1 or EWSR1-ATF1 fusion in hES cells. A possible explanation lies in the additional role that TP53 plays in hES cells [29][30, 31] in suppressing the expression of pluripotency factors in cells under oncogenic stress and eliminating these cells by the self-renewing pool [32].
When hES cells were differentiated to mesenchymal progenitors, we found that expression of the EWSR1-CREB1 and EWSR1-ATF1 fusions was similarly associated with specific gene signatures of AFH and GI-CCS, but also with prolonged cell proliferation, in contrast to the rapid cell death of hES cell. Moreover, deletion of TP53 in hES-MP cells expressing the EWSR1-CREB1 fusion further enhanced proliferation and/or viability, in accordance with the different role of this tumor suppressor in somatic and embryonic stem cells (Fig. 6D). TP53 exerts its tumor suppressor functions in different ways depending on the cell type, the genetic background, and the nature of the stress [33]. We provide evidence that in hES-MP cells expressing the EWSR1-CREB1 fusion, p21, a well-known TP53 target, is upregulated. Its role in controlling cell cycle and replication speed may explain the reduced proliferation in these cells, even if other TP53-dependent or independent functions remain to be uncovered to fully rescue cell proliferation or to induce cellular transformation.
It is notable that the majority of translocation-associated sarcomas lack clear evidence of further genetic alterations, raising the possibility that epigenetic and transcriptional alterations provide an important component leading to cellular transformation. Thus, an integrated analysis of fusion positive isogenic cell lines in various differentiated backgrounds are expected to elucidate steps in cellular transformation and vulnerabilities in sarcomagenesis. Further, the approach holds promise to dissect the promiscuity observed for EWSR1-CREB fusions in the pathogenesis of various tumor types.
As in our work, two mouse studies have pointed to the importance of cellular background for EWSR1-ATF1 transformation, although with seemingly contradictory results. In one model, tumors arise only when the fusion is expressed in a mesenchymal compartment [34]. In the second, however, tumors arise only in neural crest-derived peripheral nerve cells, while expression in other tissues induces senescence [35]. Thus, developing human models in different cellular contexts and differentiation stages becomes critical for resolving the issues.
In summary, our findings highlight the flexibility of the stepwise, combined approaches of CRISPR-Cas9 technology with HDR in the hES cellular context, differentiation to an appropriate cell type, and conditional fusion gene expression from the endogenous locus. More generally, this system has the potential to investigate the cell of origin in human neoplasia lacking a clear line of differentiation from phenotypic or immunohistochemical analyses, including the large group of translocation-associated sarcomas.
MATERIALS AND METHODS
Mammalian cell culture.
All experiments were approved by the Tri-SCI Embryonic Stem Cell Research Oversight Committee (ESCRO). Human embryonic stem cells (WA01, H1) were cultured in E8 medium (Life technologies, #A1517001) on vitronectin-coated plates (Life technologies, #A14700). HEK293 cells were cultured in DME-HG with 10% fetal bovine serum. hES-MP cells were generated and maintained following the manufacturer’s instructions (STEMCELL Technologies, #05240).
Tumor Samples.
Human samples were collected from consenting patients (Institutional Review Board 02–060).
Gene expression and whole transcriptome sequencing.
Affymetrix U133A was performed comparing 3 AFH, 4 CCS cases, to a group of 44 tumors of various histotypes and 6 normal tissues, using log2-fold change threshold of 1, FDR 0.01 [36, 37]. Total RNA was extracted using RNeasy Plus Mini (Qiagen) and prepared for RNA sequencing following Illumina protocol [38]. All reads were aligned with STAR (ver 2.3) and BowTie2 against the human reference genome (hg19). The mRNA expression of the genes in the core signatures of AFH, CCS, and GI-CCS [1] was investigated on the 5 study group samples and 186 of various tumor types. Pathway enrichment analyses were performed using online curated gene set, gene ontology and transcription factors binding site collections on the Gene Set Enrichment Analysis software (http://software.broadinstitute.org/gsea/msigdb/index.jsp). Data are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168562.
Generation of cell lines.
Cells were transfected by Amaxa nucleofector with 2.5 μg of each plasmid (gRNAs and donor plasmid), plated and treated with 0.1μM DNA-PKcs inhibitor (NU7441) or DMSO for 24 h, before selection with 150 μg/ml hygromycin. Hygromycin-resistant colonies were picked in 96-well plate for PCR analysis. TP53 knockout clones were generated by nucleofection of a vector expressing TP53 gRNA [39] and confirmed by western blot.
Generation of plasmids.
Donors were obtained from plasmid previously described [11]. Homology arms (HAs) were amplified from hES cells and cloned at NotI-NheI (EWSR1) or SalI-ApaI (CREB1, ATF1 and WT1) cloning sites. gRNAs were cloned into pSpCas9(BB)-2A-Puro (PX459, Addgene 48139) as previously described [40]. Primers and oligos are listed in Tables S1 and S2.
PCR analysis.
PCRs were performed using Jeffrey’s Buffer PCR buffer with Taq Polymerase [41] under condition previously described [40]. The primers, the annealing temperature (Ta) and extension time for each reaction are listed in Table S3.
RT-PCR.
cDNA was synthesized by SuperScript IV First-Strand Synthesis System (Invitrogen, #18091050). RT-PCR conditions were previously described [11] qRT-PCR was done on QuantStudio 3Real-Time PCR system with Powerup SYBER green master mix (Applied Biosystem, #A25743). Quantification was performed with ΔΔCt method using GAPDH as standard. No-template reactions were performed as negative controls. Primers used for RT-PCR and qRT-PCR are listed in Tables S4 and S5.
Flow cytometry.
Staining for surface markers was done as previously described [11] with antibodies listed in Table S6. Acquisition was done using a Becton Dickinson FACScan and the analysis by FlowJo software.
Cre Expression.
hES cells were transfected with 3 μg of a plasmid expressing Cre and mCherry marker [11] and harvested 48 h after for enrichment by FACS of mCherry-positive cells. hES-MP were infected with a self-deleting lentivirus expressing Cre [42]. Cells were collected for DNA and RNA extraction.
Apoptosis assay.
Cells were collected 4 days after transfection (hES) or 10 and 17 days after infection (hES-MP) with Cre and labeled using FITC AnnexinV kit (Biolegend, 640905) according to manufacturer’s instruction and propidium iodide, followed by flow cytometry and quantification by FlowJo software.
Cell Transformation assay.
Cell Transformation Assay Kit (ab235698) was performed as instructed by manufacturer. Briefly, 4 day after Cre-infection hES-MP cells were resuspended in 75μL of DMEM 10% FBS mixed with 0.3% (wt/vol) agarose and added to 96-well plate pre-coated with 75μL media mixed with 0.6% (wt/vol) agarose (10,000 cells/well).
Western Blot.
Cell lysate was prepared as previously described [11] and separated on 4–12% Bis-Tris Protein gel. After transfer and blocking in PBS 5% (wt/vol) milk, nitrocellulose membrane was probed over-night with primary antibodies (Table S6) and incubated with secondary antibody for 1 h. Proteins were visualized with Plus-ECL Enhanced Chemiluminescence.
Fluorescence in situ hybridizatio.
Custom BAC probes (Table S7) for EWSR1, CREB1 and ATF1 were applied on slides with metaphase spreads, using standard protocol [43]. Metaphases were imaged using the metasystem (Zeiss Imager.2 and ISIS 5.2) (Metasystems, Boston, USA).
Statistical analysis.
Analysis was performed using the Graphpad software. Number of experiments is indicated in figure legends. Numerical data are shown as the mean±s.d and differences between groups were determined using paired or unpaired t-test. p-value <0.05 was considered statistically significant. If not specified, the analysis is not significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments.
We thank Chew-Li Soh and Xian Zhang for technical assistance and reagents; members of the Jasin Laboratory for discussions and suggestions; and Rohit Prakash, Travis White and Pei Xin Lim for critical reading of the manuscript. This work was supported in part by: R35 CA253174 (MJ), P50 CA140146 (CRA), P50 CA217694 (CRA), Cycle for Survival (CRA), Kristin Ann Carr Foundation (CRA), St Baldrick Foundation (CRA). Core facilities at MSK are supported by the Cancer Center Support Grant (NIH P30 CA008748).
Footnotes
Competing interests.
The authors declare no competing interests.
REFERENCES
- 1.Antonescu CR, Dal Cin P, Nafa K, Teot LA, Surti U, Fletcher CD et al. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer.2007; 46: 1051–1060. [DOI] [PubMed] [Google Scholar]
- 2.Antonescu CR, Tschernyavsky SJ, Woodruff JM, Jungbluth AA, Brennan MF, Ladanyi M. Molecular diagnosis of clear cell sarcoma: detection of EWS-ATF1 and MITF-M transcripts and histopathological and ultrastructural analysis of 12 cases. The Journal of molecular diagnostics : JMD.2002; 4: 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antonescu CR, Nafa K, Segal NH, Dal Cin P, Ladanyi M. EWS-CREB1: a recurrent variant fusion in clear cell sarcoma--association with gastrointestinal location and absence of melanocytic differentiation. Clin Cancer Res.2006; 12: 5356–5362. [DOI] [PubMed] [Google Scholar]
- 4.Thway K, Nicholson AG, Lawson K, Gonzalez D, Rice A, Balzer B et al. Primary pulmonary myxoid sarcoma with EWSR1-CREB1 fusion: a new tumor entity. Am J Surg Pathol.2011; 35: 1722–1732. [DOI] [PubMed] [Google Scholar]
- 5.Desmeules P, Joubert P, Zhang L, Al-Ahmadie HA, Fletcher CD, Vakiani E et al. A Subset of Malignant Mesotheliomas in Young Adults Are Associated With Recurrent EWSR1/FUS-ATF1 Fusions. Am J Surg Pathol.2017; 41: 980–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonescu CR, Katabi N, Zhang L, Sung YS, Seethala RR, Jordan RC et al. EWSR1-ATF1 fusion is a novel and consistent finding in hyalinizing clear-cell carcinoma of salivary gland. Genes Chromosomes Cancer.2011; 50: 559–570. [DOI] [PubMed] [Google Scholar]
- 7.Rossi S, Szuhai K, Ijszenga M, Tanke HJ, Zanatta L, Sciot R et al. EWSR1-CREB1 and EWSR1-ATF1 fusion genes in angiomatoid fibrous histiocytoma. Clin Cancer Res.2007; 13: 7322–7328. [DOI] [PubMed] [Google Scholar]
- 8.Wang WL, Mayordomo E, Zhang W, Hernandez VS, Tuvin D, Garcia L et al. Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1 chimeric transcripts in clear cell sarcoma (melanoma of soft parts). Mod Pathol.2009; 22: 1201–1209. [DOI] [PubMed] [Google Scholar]
- 9.Braunreiter CL, Hancock JD, Coffin CM, Boucher KM, Lessnick SL. Expression of EWS-ETS fusions in NIH3T3 cells reveals significant differences to Ewing’s sarcoma. Cell cycle.2006; 5: 2753–2759. [DOI] [PubMed] [Google Scholar]
- 10.Brunet E, Jasin M. Induction of Chromosomal Translocations with CRISPR-Cas9 and Other Nucleases: Understanding the Repair Mechanisms That Give Rise to Translocations. Advances in experimental medicine and biology.2018; 1044: 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanoli F, Tomishima M, Feng W, Lamribet K, Babin L, Brunet E et al. CRISPR-Cas9-guided oncogenic chromosomal translocations with conditional fusion protein expression in human mesenchymal cells. Proc Natl Acad Sci U S A.2017; 114: 3696–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hisaoka M, Ishida T, Kuo TT, Matsuyama A, Imamura T, Nishida K et al. Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol.2008; 32: 452–460. [DOI] [PubMed] [Google Scholar]
- 13.Stockman DL, Miettinen M, Suster S, Spagnolo D, Dominguez-Malagon H, Hornick JL et al. Malignant gastrointestinal neuroectodermal tumor: clinicopathologic, immunohistochemical, ultrastructural, and molecular analysis of 16 cases with a reappraisal of clear cell sarcoma-like tumors of the gastrointestinal tract. Am J Surg Pathol.2012; 36: 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karamchandani JR, Nielsen TO, van de Rijn M, West RB. Sox10 and S100 in the diagnosis of soft-tissue neoplasms. Applied immunohistochemistry & molecular morphology : AIMM / official publication of the Society for Applied Immunohistochemistry.2012; 20: 445–450. [DOI] [PubMed] [Google Scholar]
- 15.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes & development.2001; 15: 3237–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vriend LE, Prakash R, Chen CC, Vanoli F, Cavallo F, Zhang Y et al. Distinct genetic control of homologous recombination repair of Cas9-induced double-strand breaks, nicks and paired nicks. Nucleic acids research.2016; 44: 5204–5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jakubauskas A, Valceckiene V, Andrekute K, Seinin D, Kanopka A, Griskevicius L. Discovery of two novel EWSR1/ATF1 transcripts in four chimerical transcripts-expressing clear cell sarcoma and their quantitative evaluation. Exp Mol Pathol.2011; 90: 194–200. [DOI] [PubMed] [Google Scholar]
- 18.Panagopoulos I, Mertens F, Debiec-Rychter M, Isaksson M, Limon J, Kardas I et al. Molecular genetic characterization of the EWS/ATF1 fusion gene in clear cell sarcoma of tendons and aponeuroses. Int J Cancer.2002; 99: 560–567. [DOI] [PubMed] [Google Scholar]
- 19.Toguchida J, Yamaguchi T, Ritchie B, Beauchamp RL, Dayton SH, Herrera GE et al. Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res.1992; 52: 6194–6199. [PubMed] [Google Scholar]
- 20.Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer cell.2007; 11: 421–429. [DOI] [PubMed] [Google Scholar]
- 21.Liang Q, Monetti C, Shutova MV, Neely EJ, Hacibekiroglu S, Yang H et al. Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature.2018; 563: 701–704. [DOI] [PubMed] [Google Scholar]
- 22.Gerald WL, Rosai J, Ladanyi M. Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc Natl Acad Sci U S A.1995; 92: 1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SB, Kolquist KA, Nichols K, Englert C, Maheswaran S, Ladanyi M et al. The EWS-WT1 translocation product induces PDGFA in desmoplastic small round-cell tumour. Nat Genet.1997; 17: 309–313. [DOI] [PubMed] [Google Scholar]
- 24.Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nature reviews Molecular cell biology.2013; 14: 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther.2002; 1: 639–649. [PubMed] [Google Scholar]
- 26.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer.2009; 9: 400–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mertens F, Antonescu CR, Mitelman F. Gene fusions in soft tissue tumors: Recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer.2016; 55: 291–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haldar M, Hancock JD, Coffin CM, Lessnick SL, Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer cell.2007; 11: 375–388. [DOI] [PubMed] [Google Scholar]
- 29.Qin H, Yu T, Qing T, Liu Y, Zhao Y, Cai J et al. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem.2007; 282: 5842–5852. [DOI] [PubMed] [Google Scholar]
- 30.Filion TM, Qiao M, Ghule PN, Mandeville M, van Wijnen AJ, Stein JL et al. Survival responses of human embryonic stem cells to DNA damage. J Cell Physiol.2009; 220: 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aladjem MI, Spike BT, Rodewald LW, Hope TJ, Klemm M, Jaenisch R et al. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol.1998; 8: 145–155. [DOI] [PubMed] [Google Scholar]
- 32.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nature cell biology.2005; 7: 165–171. [DOI] [PubMed] [Google Scholar]
- 33.Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol.2009; 1: a001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Straessler KM, Jones KB, Hu H, Jin H, van de Rijn M, Capecchi MR. Modeling clear cell sarcomagenesis in the mouse: cell of origin differentiation state impacts tumor characteristics. Cancer cell.2013; 23: 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komura S, Ito K, Ohta S, Ukai T, Kabata M, Itakura F et al. Cell-type dependent enhancer binding of the EWS/ATF1 fusion gene in clear cell sarcomas. Nature communications.2019; 10: 3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hajdu M, Singer S, Maki RG, Schwartz GK, Keohan ML, Antonescu CR. IGF2 over-expression in solitary fibrous tumours is independent of anatomical location and is related to loss of imprinting. J Pathol.2010; 221: 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antonescu CR, Yoshida A, Guo T, Chang NE, Zhang L, Agaram NP et al. KDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitors. Cancer Res.2009; 69: 7175–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antonescu CR, Le Loarer F, Mosquera JM, Sboner A, Zhang L, Chen CL et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer.2013; 52: 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng W, Jasin M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nature communications.2017; 8: 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanoli F, Jasin M. Generation of chromosomal translocations that lead to conditional fusion protein expression using CRISPR-Cas9 and homology-directed repair. Methods.2017; 121–122: 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Vanoli F, LaRocque JR, Krawczyk PM, Jasin M. Biallelic targeting of expressed genes in mouse embryonic stem cells using the Cas9 system. Methods.2014; 69: 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfeifer A, Brandon EP, Kootstra N, Gage FH, Verma IM. Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo. Proc Natl Acad Sci U S A.2001; 98: 11450–11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kao YC, Sung YS, Zhang L, Chen CL, Vaiyapuri S, Rosenblum MK et al. EWSR1 Fusions With CREB Family Transcription Factors Define a Novel Myxoid Mesenchymal Tumor With Predilection for Intracranial Location. Am J Surg Pathol.2017; 41: 482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.