

Abstract
DYRK1A (dual-specificity tyrosine phosphorylation-regulated kinase 1a) is highly expressed in glioma, an aggressive brain tumor, and has been proposed as a therapeutic target for cancer. In the current study, we have used an optimized and validated time-resolved fluorescence energy transfer (TR-FRET)-based DYRK1A assay for high-throughput screening (HTS) in 384-well format. A small-scale screen of the FDA-approved Prestwick drug collection identified the β-carboline, harmine, and four related analogs as DYRK1A inhibitors. Hits were confirmed by dose response and in an orthogonal DYRK1A assay. Harmine’s potential therapeutic use has been hampered by its potent “off-target” activity for monoamine oxidase A (MAO-A) which impacts multiple nervous system targets. Selectivity profiling of harmine and a broader collection of analogs allowed us to map some divergent SAR (structure-activity relationships) for the DYRK1A and MAO-A activities. The panel of harmine analogs had varying activities in vitro in glioblastoma (GBM) cell lines when tested for anti-proliferative effects using a high content imaging assay. In particular, of the identified analogs, harmol was found to have the best selectivity for DYRK1A over MAO-A and, when tested in a glioma tumor xenograft model, harmol demonstrated a better therapeutic window compared to harmine.
Keywords: DYRK1A, harmine, harmol, high throughput screening, monoamine oxidase A, hedgehog, glioma
Graphical Abstract
1. Introduction
Dual-specificity tyrosine phosphorylation-regulated kinase 1a (DYRK1A), a DYRK family member, is part of the CMGC kinome group (Aranda et al., 2011; Hanks and Hunter, 1995). DYRK1A has dual kinase activity; (i) for self-activation, it autophosphorylates on a tyrosine residue within its activation loop (Himpel et al., 2001; Yoshida, 2008), and (ii) it phosphorylates multiple substrates on serine/threonine residues (Dierssen and de Lagrán, 2006), including tau (Coutadeur et al., 2015; Frost et al., 2011; Woods et al., 2001), amyloid precursor protein (Ryoo et al., 2008), DREAM (Litovchick et al., 2011), and GLI1 (Ehe et al., 2017; Javelaud et al., 2012; Mao et al., 2002). DYRK1A is also found in the adult and fetal brain, where it is required for cell proliferation and differentiation and is thus critical for neurodevelopment (Dierssen and de Lagrán, 2006). Knockout DYRK1A mice are embryonic lethal, and under- or over-expression of DYRK1A results in defects in normal brain development (Dierssen and de Lagrán, 2006). With its location on chromosome 21, DYRK1A has been extensively studied as a proposed drug target for Down syndrome (DS), and neurodegenerative diseases including Alzheimer’s and Parkinson’s diseases (Wegiel et al., 2011).
DYRK1A has also been proposed as a target for anti-cancer therapy (Abbassi et al., 2015; Fernandez-Martinez et al., 2015; Ionescu et al., 2012; Radhakrishnan et al., 2016). High levels of DYRK1A have been observed in glioma, an aggressive brain tumor that is resistant to chemo- and radio-therapy, and in a subset of glioblastoma (GBM) cell lines (Pozo et al., 2013). Downregulation of DYRK1A reduces EGFR-dependent glioblastoma growth and decreases viability, increases apoptosis and reduces tumor growth in vivo (Pozo et al., 2013), with tumors generated from shDYRK1A U87 cells being much smaller than control tumors (Pozo et al., 2013). Roles for DYRK1A in tumor progression and tumor suppression have been reported depending on tumor type and cancer model (reviewed in (Boni et al., 2020; Fernandez-Martinez et al., 2015)).
Potent and selective small molecule DYRK1A inhibitors include the β-carboline alkaloid harmine with reported IC50 values of ~100 nM (Bain et al., 2007). Harmine has been utilized as a specific DYRK1A tool compound to probe function in vitro (Göckler et al., 2009) and in vivo (Noll et al., 2009; Pozo et al., 2013). Harmine inhibits DYRK1A-mediated tau phosphorylation in Alzheimer’s disease models (Frost et al., 2011), promotes beta cell mitogenesis/proliferation (Dirice et al., 2016b; Kumar et al., 2018; Shen et al., 2013; Wang et al., 2015), exhibits potent anticancer activities through both autophagy and apoptosis on human gastric cancer cells (Li et al., 2017), and exerts anti-inflammatory effects, likely by inhibition of the NF-κB signaling pathway (Liu et al., 2017a). In addition, a synthetic lethal screen of 41 natural compounds indicates harmine and other compounds that target DYRK1A may also have a therapeutic use in suppressing the cardiotoxicity associated with the widely used chemotherapeutic, doxorubicin (Atteya et al., 2017). However, harmine’s potential therapeutic use has been hampered by its potent activity for monoamine oxidase A (MAO-A) (Boni et al., 2020; Coates and Cox, 1972; Pozo et al., 2013). Harmine has activity on multiple nervous system targets, including 5HT, NMDA receptors, and dopaminergic pathways and, not surprisingly, harmine causes tremors in mice, limiting its in vivo dosing (Airaksinen and Kari, 1981). Indeed, harmine is used in mice as a tremor mouse model (Ahmed and Taylor, 1959; Handforth, 2012).
In the current study, we have used an optimized and validated time-resolved fluorescence energy transfer (TR-FRET)-based DYRK1A assay for a small-scale inhibitor screen of an approved drug and drug-like collection that identified harmine and four of its analogs as DYRK1A inhibitors. Profiling of harmine and a broader collection of β-carbolines allowed us to develop divergent SAR (structure-activity relationships) for the DYRK1A and MAO-A activities. These data along with kinase selectivity profiling and in-cell target engagement assays allowed us to prioritize analogs for in vivo studies. The analog harmol was found to have potent DYRK1A inhibitory activity comparable to harmine while showing greatly reduced MAO-A inhibitory activity. Such a lead compound could provide better safety and therapeutic properties compared to harmine and provide insights into SAR among harmine-related β-carbolines leading to improved DYRK1A inhibition and minimal MAO-A inhibition.
2. Materials and methods
2.1. Materials, compounds and cell lines
Unless otherwise stated all reagents were purchased from Thermo Fisher Scientific (Waltham, MA) or Sigma Aldrich (St. Louis, MO) at the highest level of purity possible. DYRK1A-GST protein (catalog #PR7189B), Kinase Tracer 236 and Europium-anti GST antibody were from Life Technologies (Thermo Fisher Scientific). 384-well low volume black round bottom polystyrene non-binding surface microplates were from Corning, NY (catalog # 4514). The Prestwick Chemical Library was obtained from Prestwick Chemical (Washington, DC). Re-purchased compounds used for confirmation studies were harmine (Thermo Fisher Scientific; Waltham, MA, USA, 302972500), harmaline (VWR; Radnor, PA, USA, H1237), harmalol (VWR, 151226), harmol dihydrate (VWR, J63827), harmane (Sigma-Aldrich; St. Louis, MO, USA, 103276), tetrahydroharmine (Cayman Chemical; Ann Arbor, MI, USA, 14449), and norharmane (Toronto Research Chemicals; North York, ON, Canada, N700000). All compounds were dissolved as 10 mM or in some cases 33 mM (to limit DMSO to < 0.3% for cell studies) stocks in DMSO and stored as aliquots at −20°C. U-87 MG ((U87) catalog #HTB-14), H4 (#HTB-148) and C3H10T1/2 (#CCL-226) cells were obtained directly from ATCC. Both U87 and H4 were derived from male patients (ATCC). U87 has recently been confirmed to be a glioblastoma of CNS origin (Allen et al., 2016). H4 are classified by ATCC as neuroglioma but more recently have been reclassified as a brain-derived astrocytoma (cellosaurus, Expasy.org).
2.2. High-throughput DYRK1A assay of the Prestwick FDA-approved drug collection
To screen for small molecule inhibitors of DYRK1A activity, a time-resolved fluorescence energy transfer (TR-FRET) based-assay for DYRK1A was utilized (LanthaScreen®, Thermo Fisher Scientific). Although the assay employed for this study is commercially available, modifications were made to reduce volumes and modify to a homogenous assay format as detailed in the accompanying MethodsX article (Tarpley et al., 2021a). Briefly, DYRK1A-GST, anti-GST Ab and ATP site-directed tracer 236 were pre-combined and immediately dispensed into 384-well low volume black round bottom polystyrene plates to give a final volume of 7.5 μL per well. For compound addition, 50 nL of each drug from the 1124 Prestwick chemical library (10 mM stocks in DMSO) was then added to the assay wells using a Biomek NX workstation (Beckman-Coulter) equipped with a Pintool array (VP Scientific; San Diego CA) to give final compound concentrations of 6.7 μM. For all plates, the outer pairs of lanes in the plates contained minimum signal (6.7 μM harmine) and maximum signal (DMSO alone) controls to establish an assay window. Plates were covered and then incubated in the dark at room temperature for 1 h before being read with a PHERAstar plate reader (BMG Labtech; Cary, NC, USA) utilizing an HTRF module to detect absorbance at 665 nm (tracer) and 620 nm (Eu-antibody). Primary screen data were recorded, the data files exported and then analyzed using ScreenAble (ScreenAble Inc., Chapel Hill, NC). Z’ scores as an estimate of assay quality were determined by the method of Zhang et. al. (Zhang et al., 1999).
Compounds showing greater than 50% inhibition were repurchased and confirmed by 10-point 2-fold dose response curves using the automated DYRK1A TR-FRET assay. Dose response data were loaded in ScreenAble and IC50 values determined.
2.3. DYRK1A orthogonal33P-ATP assay
DYRK1A hits were confirmed in a radiolabel-based 33P-ATP assay for DYRK1A activity (HotSpot™, Reaction Biology Corp.; Malvern, PA, USA). This biochemical assay measures transfer of 33P-labelled phosphate from ATP to a kinase substrate, which was casein for DYRK1A. Details of the assay are given in (Anastassiadis et al., 2011). Compounds were shipped to the vendor. Briefly, compounds were incubated in dose response (10 point, 3-fold dilution; 10 μM to 0.5 nM for harmine and harmol and 100 μM to 5 nM for other analogs) with full length DYRK1A, casein, ATP (10 μM), and radioisotope-labeled ATP (33P-ɣ-ATP) in reaction buffer for 2 h. Reaction mixtures were spotted on to filter paper and unreacted phosphate removed by washing. DYRK1A kinase activity (% remaining) was calculated relative to DMSO control and after background subtraction. IC50 values and curve fits were carried out using Prism 7 (GraphPad Inc.).
2.4. Monoamine oxidase-A assay
Monoamine oxidase-A (MAO-A) inhibition was determined using a luciferase-based enzyme activity assay (Promega MAO-Glo™) according to the manufacturer’s instructions. In brief, assay buffer and luminogenic MAO substrate were added to all wells of a 384-well plate. Test compounds and controls were then added to the appropriate wells using a pintool array as above, and recombinant MAO-A enzyme (Sigma-Aldrich) added by NanoScreen to all wells except for the negative control wells, which received reaction buffer without enzyme. The plates were then incubated in the dark at room temperature for 1 h, detection reagent added to all wells, and luminescence determined by plate reader (PHERAstar). Percent activity was calculated by normalizing test compound values to DMSO controls.
2.5. Kinase selectivity panel
To assess selectivity, confirmed DYRK1A inhibitors were profiled for selectivity at 1 μM using the radiolabel assay format described above (HotSpot™, Reaction Biology Corp.). The panel consisted of 10 kinases previously identified as kinases frequently inhibited by current DYRK1A inhibitors (Becker and Sippl, 2011). Assay details and substrates are given in (Anastassiadis et al., 2011).
2.6. Cell proliferation assay
Cell seeding densities for PrestoBlue assays in 384-well format were determined to be optimal at 1000 cells per well for U87 and 800 cells per well for H4 (data not shown). Cells were added to plates (Corning 3712) using a multidrop bulk dispenser (Thermo Fisher). After allowing cells to attach overnight at 37° C and 5% CO2, compounds were added to cells by pintool as described above in duplicate, 20-point, 2-fold concentration response (0.0001 μM to 25 μM). Assay plates were incubated for 72 h at which time PrestoBlue reagent, pre-diluted in PBS, was added for a 90 min incubation. Fluorescence was determined by PHERAstar plate reader using λex 560 nm / λem 590 nm. Raw RFU values were converted to relative cell proliferation percentage based on DMSO-treated controls and plotted in Prism GraphPad 7.0.
2.7. High content screening assay for cytotoxicity
High content imaging to assess cell proliferation by nuclear count and cytotoxicity by plasma membrane integrity was carried out essentially as previously described (Abraham et al., 2008; Oladapo et al., 2017; Sauer et al., 2017). This is achieved using two dyes, Hoechst 33342, which is a cell permeable dye that can bind nucleic acid in live cells, and YOYO-1 which is cell impermeable and can only bind nucleic acid when the plasma membrane is compromised. Cell plating densities were optimized for assay window and ideal imaging properties (data not shown). Cells were plated (Corning 3712) using the multidrop at 1000 cells per well for U87, and 1200 cells per well for H4, in a volume of 50 μL per well. The Biomek NX was used for all steps unless stated otherwise. Compounds were added to cells by pintool as described above in duplicate 20-point dose range. After 3 days, media was removed and Hoechst 33342 and YOYO-1 added to the cells to give final dye concentrations of 10 μg/mL and 100 nM, respectively. After 45 min, cells were washed and fixed with formalin as previously described. Plates were imaged using a CellInsight NXT equipped with a 10X, 0.4 NA objective, acquiring data in HCS Scan software (Thermo Fisher Scientific) on nuclear count and nuclear characteristics (Hoechst, λex 386 nm) as well as reduced viability (YOYO-1, λex 485 nm). Data were then accessed using HCS View software (Thermo Fisher Scientific) and analyzed in Excel as a percent effect of DMSO control and plotted in Prism GraphPad.
2.8. In-cell target engagement assay
Direct in-cell target engagement by compounds was assessed using the InCELL Hunter™ cellular assay platform service (DiscoverX, Freemont, CA). Compounds were shipped to the vendor and CLK1 cells used as a surrogate for DYRK1A in their PathHunter® assay. Briefly, compounds were added to cells and allowed to incubate for an assay-dependent period of time in duplicate at concentrations of 0.5 μM, 5 μM, and 50 μM. Data was reported as individual and average RLU values and percent efficiency based on a positive control inhibitor (K252a) for CLK1. Percent activity was reported as background corrected for mean RLU of test sample control.
2.9. C3H10T1/2 cell-based assay
The mouse embryonic fibroblast line C3H10T1/2 (Reznikoff et al., 1973), which has been shown to be hedgehog (Hh) pathway responsive (Nakamura et al., 1997), was used as another measure of in-cell activity. The assay was performed essentially as we have previously described (House et al., 2015; Williams et al., 1999). C3H10T1/2 were maintained in DMEM medium (HyClone SH30081.02) containing 10% FBS and plated in 96-well plates at 5000 cells/well. To assess pathway inhibition, Sonic Hh (Shh) protein was added at its EC50 value (2 μg/ml) and 24 h later compounds were added in concentration response. Cells were incubated for a further 5 days and assayed for alkaline phosphatase (AP) using pNPP as the substrate and plates read at 405 nm. Inhibition constant (IC50) values were determined by non-linear regression in GraphPad 7.0.
2.10. In vivo tumor xenograft
Animal experiments were conducted in accordance with accepted standards of humane animal care and approved by the Animal Care and Use Committee at the University of North Carolina at Chapel Hill. At 6-weeks of age, nude NU/J mice were implanted in the flank with 1×107 U87 cells suspended in Matrigel® and PBS. Mice were randomized into groups for treatments (n = 8 per group). When tumors reached ~100 mm3 in size (~six weeks), animals were treated i.p. with either vehicle control, harmine 15 mg/kg or harmol (15, 30, 45 and 60 mg/kg) q.d.. Tumor response was assessed by caliper measurements every 3–4 days. Animal weights were recorded every 3–4 days. Tumor-bearing mice were euthanized at 4 weeks for morbidity, tumor ulceration or if the tumor was >2 cm in diameter. Changes in tumor growth were determined as the mean relative treatment group tumor volume (% change) ± standard error. Differences in tumor volumes were assessed using Student t-test in GraphPad Prism 7.0. Comparisons in tumor growth were made using linear regression in GraphPad Prism 7.0. Survival data were analyzed by Kaplan–Meier method and the Log-rank (Mantel–Cox) test. Tumor inhibition rates were calculated as described previously (Cao et al., 2004; Cao et al., 2013; Chen et al., 2005; Li et al., 2015) using the formula (C-T/C)*100 where T is the average tumor volume for a treatment group and C is the average treatment volume for the vehicle group.
2.11. Statistical analysis
Inhibition constant (IC50) values were determined by non-linear regression in GraphPad 7.0 (GraphPad Software, Inc). Correlation plots were generated using linear regression in Graphpad Prism 7.0.
3. Results
3.1. DYRK1A kinase inhibition assay and pilot screen
To identify novel DYRK1A inhibitors, we optimized and automated a TR-FRET-based Lanthascreen® kinase binding assay for DYRK1A inhibition and validated it using our previously described (Williams and Scott, 2009). This TR-FRET based assay format (Lebakken et al., 2009) can identify competitive inhibitors of a kinase by displacement of a fluorescently-labeled ATP site-directed tracer, which fluoresces only when proximal to a fluorescently-labeled antibody that recognizes a tag also present on the kinase (see graphical abstract). Assay optimization involved configuring the assay for 384-well format, reducing addition steps and reducing volumes, and is described in detail in the accompanying MethodsX article (Tarpley et al., 2021a). As a pilot test screen under HTS conditions, we first screened the Prestwick collection (at 6.7 μM, single point), which is a commercial compound library composed of 1124 compounds that have established biological activities, 95% of which are approved drugs. All 4 (384-well) plates of the Prestwick collection passed assay acceptance criteria with an average Z’ score of 0.77. Mean coefficient of variance for intra-plate negative controls and positive controls were 5.9% and 8.3%, respectively. This pilot screen identified 37 compounds with ≥ 50% inhibition of DYRK1A (Fig. 1). Twenty-two of these 37 compounds were confirmed as DYRK1A inhibitors by a single-point duplicate re-screen for a final hit-rate of 2.0% and all 22 are listed with associated %-inhibition values in the accompanying Data in Brief article (Tarpley et al., 2021b).
Fig. 1.

DYRK1A kinase pilot screening of the Prestwick chemical compound set. An automated TR-FRET assay for DYRK1A using an optimized LanthaScreen® Eu Kinase Binding Assay for low volume in 384-well format was used to screen the Prestwick collection (at 6.7 μM). The scatterplot shows percent inhibition for the Prestwick set compounds (black circles), negative inhibition controls (DMSO; red inverted triangles), and positive inhibition controls (6.7 μM harmine; blue triangles). Compounds showing ≥ 50% inhibition (dotted red line) were selected for confirmation. β-carbolines are highlighted with red circles.
Among the compounds identified from the pilot HTS as inhibiting DYRK1A were harmine and four associated β-carboline alkaloids (a sub-family of the indole-alkaloids): harmol, harmaline, harmane, and harmalol. These constituted the entirety of the β-carbolines present in the Prestwick library (Fig. 1, annotated with red circles). DYRK1A inhibition was comparable for all the β-carbolines identified with values of 81%, 79%, 78%, 74%, and 71% for harmaline, harmalol, harmol, harmine, and harmane respectively. As an established DYRK1A inhibitor, harmine and other β-carbolines also represented in the Prestwick library, were chosen as the focus of this work. It was hypothesized that a group of compounds based on this β-carboline structure would serve as a starting point for insight into SAR between DYRK1A inhibition and MAO-A inhibition.
3.2. Confirmation of β-carboline family member hits from the Prestwick chemical library by dose response in DYRK1A inhibition assay
To confirm the inhibitory activity, harmine, the four other β-carbolines from the Prestwick screen, and two additional structurally-related compounds (norharmane and tetrahydroharmine) (structures shown in accompanying Data in Brief article (Tarpley et al., 2021b)) were purchased as new powder stocks, and 10-point dose response titrations performed to determine IC50 values in the DYRK1A TR-FRET assay (Fig. 2A and Table 1). All IC50 values were within 3-fold of previously reported values (Table 1). Similar to previously reported data, we determined that harmine was the most potent of these compounds, with an average IC50 of 70 nM, and that tetrahydroharmine was the least potent (IC50 = 9.4 μM). IC50 values in the range of 27–31 nM have been reported for harmine using TR-FRET-based DYRK1A assays (Bálint et al., 2017; Kumar et al., 2020; Kumar et al., 2018). In agreement with published data (Bain et al., 2007), we found a DYRK1A inhibition potency ranking of harmine > harmol >> harmalol >> harmaline ~ harmane > norharmane > tetrahydroharmine.
Fig. 2.

DYRK1A and MAO-A dose response curves for selected harmine-related hits. Dose response curves for β-carbolines in (A) DYRK1A TR-FRET assay and (B) DYRK1A 33P-ATP assay. (C) Correlation of IC50 values between DYRK1A TR-FRET and 33P-ATP assays, linear regression fit. (D) Dose response curves for β-carbolines in MAO-A assay. Color key for harmine-like compounds in all 3 assays shown to the right of panel (B). For each concentration (10 point) run in duplicate, percent inhibition values were calculated and data normalized to controls. Dose response curves were generated using non-linear regression and IC50 values determined in GraphPad Prism 7.
Table 1.
DYRK1A IC50 values (μM) for the identified β-carbolines.
| Compound | TR-FRET | 33P-ATP a | Published values b |
|---|---|---|---|
| Harmine | 0.07 | 0.009 | 0.022 – 0.107 |
| Harmaline | 2.35 | 0.79 | 1.236 – 4.59 |
| Harmalol | 0.63 | 0.18 | Undetermined |
| Harmol | 0.10 | 0.016 | 0.044–0.298 |
| Harmane | 2.68 | 1.00 | 1.82 |
| Tetrahydroharmine | 9.41 | 6.42 | 2.987 |
| Norharmane | 5.46 | 3.19 | 8.746 |
| Staurosporine | 0.002 |
33P-ATP data generated by Reaction Biology Inc.
(Adayev et al., 2011; Bain et al., 2007; Bálint et al., 2017; Bruel et al., 2014; Foucourt et al., 2014; Göckler et al., 2009; Grabher et al., 2012; Kim et al., 2016; Kumar et al., 2018; Liu et al., 2017b) Details in Supplementary Table 1; additional values from National Center for Biotechnology Information; PubChem Database.
To further confirm our DYRK1A potency findings for harmine and the selected analogs, compounds were evaluated in an orthogonal radiolabel-based 33P-ATP assay for DYRK1A. This biochemical assay provides a format that directly detects the phosphorylated product and allows validation of inhibitors identified using more indirect assay readouts such as TR-FRET (Ma et al., 2008). Potencies against DYRK1A were determined in dose-response experiments in the radiolabel assay and found to be generally comparable to our TR-FRET data (Fig. 2B and Table 1). Again, harmine was the most potent of the analogs tested with an IC50 value of 9 nM in the 33P-ATP assay (Table 1). Using the same Hotspot assay format, a comparable IC50 value of 22 nM for harmine inhibition of DYRK1A was previously reported (Foucourt et al., 2014). Published values for harmine using a radiolabeled ATP assay format are in the range 22–80 nM (Adayev et al., 2011; Bain et al., 2007; Bruel et al., 2014; Bruel et al., 2012; Foucourt et al., 2014; Göckler et al., 2009; Grabher et al., 2012). Harmine has previously been shown to be an ATP-competitive inhibitor of DYRK1A (Adayev et al., 2011). Details on published assay formats for DYRK1A and IC50 values for harmine and related analogs are provided in Supplementary Table 1. For all the harmine analogs, we observed higher potencies using the 33P-ATP assay compared to the TR-FRET. For harmine and harmol, IC50 values were ~6–7 fold more potent with the 33P-ATP assay versus TR-FRET. For the remaining analogs, IC50 values were within ~1.5–3 fold for the two assay formats.
Although the absolute potencies varied by comparison between the two assays, the data from the orthogonal 33P-ATP assay ranked all 7 alkaloids in the same potency order as determined by TR-FRET for DYRK1A inhibition. A comparison plot of IC50 values from the two DYRK1A enzyme assay formats showed a very high degree of correlation with a calculated linear regression R2 value of 0.9983 (Fig. 2C).
β-carbolines consist of a pyridine ring (ring 1) fused to an indole skeleton, which is itself comprised of a pyrrole ring (ring 2) fused to a benzene ring (ring 3). Structurally harmine and harmol, which showed the most DYRK1A inhibition, have a fully aromatic ring 1 as well as a hydroxyl or methoxyl group at C-7 in ring 3. Less potent DYRK1A inhibition was observed with β-carbolines with a partially or fully saturated ring 1, lacking a methyl side group at position 1, and/or lacking hydroxyl substitution at position 7. Ring position numbering follows IUPAC convention and is as published previously (Ishida et al., 1999).
3.3. Determination of monoamine oxidase A (MAO-A) activity and assessment of relative potencies on DYRK1A versus MAO-A for identified β-carbolines.
We next assessed the selected β-carbolines for their activity against MAO-A. In this assay, originally described in (Valley et al., 2006), a luminogenic substrate is converted to luciferin by MAO-A enzyme. Luciferase in the detection reagent reacts with the luciferin to produce light, with the amount of light being proportional to MAO-A activity. There was potent inhibition of MAO-A by harmine, with an IC50 of 60 nM (Fig. 2D and Table 2), a value comparable to that reported of 107 nM using the same MAO-GLO assay format (Rüben et al., 2015; Wurzlbauer et al., 2020). Harmine is a known inhibitor of MAO-A, and its application as a clinical DYRK1A inhibitor is limited for this reason (Coates and Cox, 1972; Kim et al., 1997). Harmaline, which differs structurally from harmine only by the partial saturation at C-3 and C-4 in ring 1, possessed comparable activity against MAO-A. Harmol (IC50 of 500 nM) and harmane (IC50 of 640 nM) were less potent inhibitors of MAO-A. In addition to the MAO-GLO/bioluminescence-coupled assay format that we and others have used for MAO-A (Reniers et al., 2011; Rüben et al., 2015; Santillo et al., 2014; Valley et al., 2006; Wurzlbauer et al., 2020), MAO-A has also been measured using other enzymatic-based readouts and substrates (Supplementary Table 2). While our IC50 value for harmol is comparable to that of 530 nM obtained using an enzymatic format (Bálint et al., 2017), a relatively wide range of IC50 values (3.5 to 107 nM) has been obtained across various assay formats and conditions for harmine on MAO-A (see Supplementary Table 2). One study undertook a direct comparison of enzymatic and MAO-GLO assays for harmine and three other analogs and determined very close IC50 values, irrespective of assay format (Santillo et al., 2014).
Table 2.
Monoamine oxidase A IC50 values (μM) for the identified β-carbolines.
| Compound | MAO-A Glo | Published a | DYRK1A:MAO-A (IC50 ratio) b |
|---|---|---|---|
| Harmine | 0.06 | 0.003 – 0.107 | 1.2 |
| Harmaline | 0.09 | 0.005 – 0.020 | 26.1 |
| Harmalol | 0.66 | 0.153 | 1.0 |
| Harmol | 0.50 | 0.53 | 0.2 |
| Harmane | 0.64 | 0.029 – 0.337 | 4.2 |
| Tetrahydroharmine | 1.52 | Undetermined | 6.2 |
| Norharmane | 4.29 | 6.47 – 67.1 | 1.3 |
| Staurosporine | No inhibition |
(Bálint et al., 2017; Clarke and Ramsay, 2011; Herraiz and Brandt, 2014; Herraiz and Chaparro, 2005; Ionescu et al., 2012; Kim et al., 1997; Rahman and Rahmatullah, 2010; Reniers et al., 2011; Rommelspacher et al., 1994; Rüben et al., 2015; Santillo et al., 2014; Wurzlbauer et al., 2020). Details in Supplementary Table 2.
Using TR-FRET DYRK1A IC50 values in this work. As we observed a strong correlation between inhibition potencies in both DYRK1A assay formats, we also see a comparable ranking of DYRK1A:MAO-A ratios if we use the DYRK1A values from the 33P-ATP data.
Like harmine, both harmol and harmane have a fully aromatic ring 1 with a methyl group in the C-1 position but differ from harmine and harmaline in that they have either a hydroxyl group substituent at C-7 in ring 3 or no substituent, respectively. Harmalol, which has a partially saturated ring 1 and a hydroxyl group at C-7 in ring 3, showed a relatively intermediate inhibition of MAO-A. The least potent MAO-A inhibitor in the series was norharmane, with an IC50 of 4.3 μM. Tetrahydroharmine, with a fully saturated ring 1 was also found to be a weak MAO-A inhibitor, and these two compounds are either missing a methyl group, as for norharmane, or have an out-of-plane methyl group in that position, which is the case of the chiral center of tetrahydroharmine. Tetrahydroharmine is also completely lacking the planar character of ring 1. The ranking of inhibition potencies of these compounds against MAO-A was different from that observed for DYRK1A, with MAO-A ranking as harmaline ~ harmine >>harmol ~ harmane > harmalol > tetrahydroharmine > norharmane. A comparable ranking of four of these (harmine, harmaline, harmane and harmalol) was also observed by (Santillo et al., 2014).
An IC50 ratio was calculated to assess DYRK1A (TR-FRET data) versus MAO-A inhibition. The two most potent inhibitors of DYRK1A in this set, harmine and harmol, had markedly different activities against MAO-A. While harmine and harmol had nearly equal activity on DYRK1A, harmine had greater than 8-fold more activity against MAO-A than harmol (Table 2). In terms of the DYRK1A:MAO-A activity ratio, norharmane and haramlol were comparable to harmine. Both harmane, tetrahydoharmine and harmaline all had greater potency for MAO-A over DYRK1A, with harmaline ~25 fold more potent on MAO-A. Of the analogs tested, only harmol had a significant potency selectivity for DYRK1A over MAO-A.
3.4. Kinase selectivity of selected β-carbolines
We next determined the selectivity of the β-carbolines against a panel of DYRK1A-related kinases and other kinases known to be inhibited by a range of DYRK1A inhibitor classes (Becker and Sippl, 2011; Rüben et al., 2015). The most potent of the β-carboline DYRK1A inhibitors were also the least specific at the concentration tested (10 μM), e.g., harmine resulted in a 70% or greater reduction in enzyme activity of CK1α1, CLK1, CLK4, DYRK1B, DYRK2, and PIM1 (Fig. 3A and Table 2). CLK1, DYRK1B, and DYRK2 inhibition were essentially 100%, indicating that at the 10 μM test concentration there is minimal harmine selectivity for DYRK1A inhibition over these three other kinases. The selectivity concentrations were similar for harmol, with 7 of the 10 kinases displaying 50% or greater inhibition. There were fewer kinases inhibited by harmalol to the same degree as harmine or harmol. Overall, harmalol was found to be more selective for DYRK1A when considering this panel of kinases compared to harmine. Only CLK1, DYRK1B and DYRK1A were inhibited at nearly 100% when treating with 10 μM harmalol. With regard to the less potent group of β-carboline DYRK1A inhibitors, specificity for DYRK1A/B over other kinases was higher at the concentration tested. None of the β-carbolines demonstrated any apparent selectivity for DYRK1A over DYRK1B. Interestingly, tetrahydroharmine had comparable inhibition of DYRK1A and DYRK1B but minimal effect on DYRK2 indicating some in-family selectivity. In general, these compounds all inhibited the closely homologous DYRK kinases tested, DYRK1A, DYRK1B and DYRK2 (Fig. 3B). The three most potent of which, harmine, harmol and harmalol, all showed comparable inhibition of DYRKs and CLKs. These two kinase families are closely homologous within the CMGC group as highlighted on the kinome tree (Manning et al., 2002) (Fig. 3B).
Fig. 3.

Specificity of the DYRK1A harmine-related alkaloid inhibitors against a kinase panel. (A) To assess selectivity, compounds (at 10 μM) were profiled for selectivity against a panel of 10 kinases (Reaction Biology Corp., n = 2). Reductions in enzyme activity (%) are shown for each compound as a separate bar chart. (B) Human kinome map highlighting CMGC family with eight of the kinase selectivity panel highlighted by black circles and DYRK1A by a red circle. Human Kinome illustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com).
3.5. In vitro cytotoxic effects of β-carbolines in glioma cell lines
To assess the relative effects of selected β-carbolines on blocking cancer cell proliferation, we evaluated their effects on two glioma cell lines, H4 and U87, previously shown to express elevated DYRK1A (Frost et al., 2011; Pozo et al., 2013). Knock down/inhibition studies have demonstrated the dependence of these cells on DYRK1A. DYRK1A knock down and harmine treatment in H4 cells reduces phosphorylation of multiple substrates (Frost et al., 2011). DYRK1A knock down in U87 cells has been shown to reduce tumor growth in vivo (Pozo et al., 2013; Xu et al., 2019), with tumors being less proliferative and having a higher number of apoptotic cells (Pozo et al., 2013).
First, effects on cell viability were assessed using a PrestoBlue assay with compounds being added to cells in dose response for 72 h. Harmine and harmol were the most potent on H4 cells with IC50 values of 4.9 and 10.9 μM respectively (Fig. 4A and Table 3). The other β-carbolines had comparable effects on H4 cell viability, with IC50 values in the 20–50 μM range (Table 3). Tetrahydroharmine was the least effective. For U87 cells, only harmine and harmol generated a full dose response curve giving IC50 values of 45.3 and 19.1 μM, respectively. In this assay, potencies for all the β-carbolines were lower on the U87 compared to H4 cells. A strong correlation (calculated linear regression R2 value of 0.8589) between the potencies in the DYRK1A TR-FRET assay and in the H4 cell proliferation assay was observed for all 7 of these compounds (Fig. 4B).
Fig 4.

Effect of harmine and harmol on glioma cancer cell proliferation. (A) H4 and U87 cells were incubated with harmine and harmol (0.0001 – 25 μM) for 72 h, and proliferation assessed by Presto Blue. For each concentration, percent inhibition values were calculated and data normalized to vehicle. Dose response curves were generated using non-linear regression and IC50 values determined in GraphPad Prism 7. (B) Correlation of IC50 values between DYRK1A TR-FRET and H4 proliferation assays, linear regression fit. Dose response curves for the other analogs are shown in the accompanying Data in Brief article (Tarpley et al. 2021b).
Table 3.
Cell viability IC50 values (μM) for harmine analogs.
| Compound | H4 (cell proliferation) a |
H4 (nuclear count) b |
U87 (cell proliferation) a |
U87 (nuclear count) b |
|---|---|---|---|---|
| Harmine | 4.9 | 14.91 | 45.3 | 17.6 |
| Harmaline | 19.6 | 14.87 | 14% c | ~66% c |
| Harmalol | 23.7 | 25.2 | 37% c | <50% c |
| Harmol | 10.9 | 20.0 | 19.1 | 20.8 |
| Harmane | 33.5 | 17.1 | 30% c | ~50% c |
| Tetrahydroharmine | 38%c | 26.9 | No inhibition c | ~50% c |
| Norharmane | 49 | 29.1 | No inhibition c | No inhibition c |
PrestoBlue cell viability assay.
Hoechst 33342 high content imaging assay.
% inhibition at highest dose tested.
We next assessed the effects of the β-carbolines using a multi-parametric high content assay that provides a direct measure of cell proliferation (Abraham et al., 2008). H4 and U87 cells were incubated with compounds in dose response for 72 h, Hoechst 33342 and YOYO-1 dyes added, and cells imaged. A nuclear mask was established (Hoechst 33342) to determine nuclear characteristics (nuclear count and nuclear area) that provided measures of cell proliferation and cell health. Nuclear staining by the membrane impermeable dye YOYO-1 (area inside nuclear mask) provided a measure of plasma membrane integrity. Changes in nuclear morphology such as increased or decreased nuclear area are indicative of changes in cell health such as altered transcriptional activity, mitotic arrest, and cytotoxicity (Abraham et al., 2008). In this assay, all the evaluated β-carbolines had comparable effects on H4 nuclear count per unit area (a measure of cell number) with IC50 values in the 15–30 μM range (Table 3). On U87 cells, harmine and harmol had IC50 values of 17.6 and 20.8 μM respectively for nuclear count, similar potencies to those observed with H4 cells (dose response curves Fig. 5A-B and representative images in Fig. 5C). Differences in the response of β-carbolines in H4 and U87 in the PrestoBlue assay compared to the high content assay (nuclear count) may reflect differences in the metabolic activity of the U87 and H4 cells (Quent et al., 2010). In U87 cells, the other β-carbolines had weaker potencies with 50% inhibition only being observed at the highest dose tested (66 μM). The cytotoxic potency of harmine on U87 cells is comparable to that previously demonstrated (Ishida et al., 1999). In our high content assay, harmine and harmol had significant effects on nuclear area and plasma membrane integrity at doses greater than 10 μM indicating cytotoxicity with effects more pronounced in H4 cells.
Fig 5.

Effect of harmine and harmol on glioma cancer cell cytotoxicity. For high content imaging, H4 and U87 cells were incubated with harmine (A) or harmol (B) at varying concentrations (0.1 – 66 μM) for 72 h and cells then stained with Hoechst and YOYO-1 dyes. Nuclear count, nuclear area and YOYO-1 staining were measured, data normalized to DMSO vehicle control and dose response curves generated by non-linear regression using GraphPad Prism 7. (C) Representative high content imaging fields (10x; overlay of Hoechst=blue, YOYO-1=green) for vehicle (DMSO), harmine (16 μM) and harmol (16 μM) on H4 and U87 cells. Dose response curves and images for the other harmine and harmol doses tested and for the other analogs are shown in the accompanying Data in Brief article (Tarpley et al. 2021b).
3.6. In-cell target engagement and hedgehog cell-based assays
To assess compound entry into a cell and demonstrate intracellular target engagement of harmine and harmol, the InCELL Hunter™ cellular assay system was used. The principle of the assay is that engagement of a compound with a protein target in the cell increases the stability of the protein, thus reducing its degradation. The InCELL Hunter assay is based on enzyme fragment complementation which uses two fragments of β-galactosidase (β-gal) consisting of a small enzyme donor (ED) fragment and a large enzyme acceptor (EA) fragment. When separate, these two pieces of β-gal are inactive. When brought together, they complement and form active β-gal, which can be detected using a chemiluminescence (CL) substrate. Cells are engineered to express a relevant target protein fused with the small ED fragment of β-gal. For the assay, these cells are incubated with compound and then with the larger EA fragment of β-gal added with the detection reagents. Compound binding to the target in the cell promotes complementation and stabilization of the two fragments of β-gal, resulting in an increase in β-gal activity (described in (Schürmann et al., 2016)) and a CL signal is detected. At the time of this study, DYRK target engagement assays were not available in any format. Hence, as we had shown above that both harmine and harmol have comparable inhibitory activity on DYRK1A and CLK1, we chose to use the available CLK1 InCELL Hunter™ assay to provide a surrogate measure of DYRK1A cellular target engagement. In this assay, increasing concentrations of harmine and harmol caused an increase in β-gal activity indicating target engagement, with harmol having a higher efficiency compared to harmine (Fig. 6A).
Figure 6.

Assessment of in-cell and in vivo antitumor activity of harmine and harmol. (A) Cellular target engagement assay for harmine and harmol in PathHunter® cell assay for CLK1 (DiscoverX). Harmine and harmol were incubated at 3 concentrations with the cells and effects on β-gal activity determined. Inset; K252a as a positive control inhibitor gave an EC50 value of 8.7 nM. (B) Inhibitory activities of harmine and harmol in C3H10T1/2 Hh cell assay. C3H10T1/2 were induced with ShhN protein and harmine and harmol added at the indicated concentrations. After 5 days, the effects on alkaline phosphatase (AP) activity were measured at 405 nm using pNPP. (C-F) Vehicle, harmine (15 mg/kg daily) and harmol (15 and 60 mg/kg daily) were dosed intraperitoneally in a U87 xenograft nude mouse model and assessed for in vivo effects on body weight (C), survival (D), tumor growth (E) and tumor inhibition (F).
The activity of the harmine analogs was also assessed in a cellular assay for Hh signaling, the C3H10T1/2 assay. C3H10T1/2 is a mesenchymal stem cell line that is responsive to Hh as assessed by induction of alkaline phosphatase (AP) activity, a marker for differentiation into an osteoblast lineage (Nakamura et al., 1997). We have previously shown that this cell-based assay provides a simple system for measuring inhibition of Hh pathway activity (House et al., 2015; Williams et al., 1999). Although DYRK1A is reported as a regulator of Hh signaling, its exact role appears to be cell context-dependent (reviewed in (Boni et al., 2020; Singh and Lauth, 2017)). Both harmine and harmol inhibited Hh-stimulated AP activity in this cell assay with IC50 values of 14.1 and 5.2 μM, respectively (Fig. 6B). The activity of the other harmine analogs in this Hh cell-based assay are shown in Supplemental Figure 1. No effect on cell viability from the harmine analogs was observed in this assay for concentrations <50 μM (data not shown).
3.7. Glioma tumor xenograft models
Next, we chose to assess the anti-tumor effects of a subset of these β-carbolines in an in vivo tumor model. Based on its inhibition potency for DYRK1A, anti-proliferative activity in the glioma cell lines, along with its selectivity for DYRK1A over MAO-A compared to the other analogs we proposed to test harmol in vivo over a range of doses. Harmine was also included in the study at its reported upper limit of 15 mg/kg in vivo for not causing tremors (Pozo et al., 2013). Prior to the in vivo studies, the masses and structures of harmine and harmol were confirmed by mass spectrometry and NMR, respectively (Supplemental Fig. 2). U87 cells were chosen for the in vivo analysis as they are a more established in vivo glioma tumor model (Du et al., 2013; Li et al., 2019; Pozo et al., 2013; Swenson et al., 2018; Tseng et al., 2008; Xu et al., 2019). Further, U87s have been termed highly aggressive and invasive, and H4 as less aggressive and invasive (Louca et al., 2019), with U87 cells reported as tumorigenic in vivo while H4 are not (ATCC).
To assess the effects of harmol on tumor growth, a xenograft glioma tumor model was carried out as follows. Nude NU/J mice were implanted in the flank with U87 human glioblastoma cells, and after reaching ~100 mm3 size, dosed daily i.p. with harmol or harmine. Tumor volumes, survival and mouse weights were followed every 3–4 days for 4 weeks. Treatment up to 21 days (q.d.) with low (15 mg/kg) and high (60 mg/kg) harmol or harmine (15 mg/kg) did not lead to significant differences in body weight (Fig 6C). Increased toxicity was observed after 4 weeks. Compared to vehicle, for the 60 mg/kg harmol treatment, while not statistically significant as assessed by Kaplan-Meier analysis, there was a trend towards improved survival (Fig. 6D). A modest decrease in tumor growth, while not reaching significance, was observed with the high dose harmol at day 21 (Fig. 6E). In this U87 xenograft model, when analyzed for tumor inhibition rates, harmine demonstrated modest anti-tumor activity of 16.5% while harmol exhibited more potent tumor inhibition activity with increasing doses; 28.2% at 15 mg/kg and 38.1% at 60 mg/kg (Fig. 6F).
4. Discussion
There is strong evidence for DYRK1A as a potential target for pharmacological intervention in a number of therapeutic areas including Down syndrome (Becker et al., 2014), neurodegenerative diseases (Wegiel et al., 2011), diabetes (Dirice et al., 2016a; Rachdi et al., 2014) and cancer (Ionescu et al., 2012). Our current study describes a high-throughput screen of 1124 approved drugs and drug-like compounds using an optimized and validated TR-FRET-based DYRK1A assay (see accompanying MethodsX article (Tarpley et al., 2021a)). Validation of the screen was demonstrated by the identification of harmine, a known DYRK1A inhibitor (Bain et al., 2007; Göckler et al., 2009), and four of its analogs from this pilot screen of the Prestwick collection. A total of 22 confirmed hits were identified for a relatively high hit rate of 2%, potentially due to the Prestwick library containing only biologically active, FDA-approved drugs and drug-like compounds. Harmine and its analogs were the focus of this study as multiple β-carboline analogs were identified providing preliminary SAR direct from a primary screen. The other DYRK1A inhibitors identified in this screen (see accompanying Data in Brief article (Tarpley et al., 2021b)), will be evaluated as part of a more extensive DYRK1A screen in future studies. A strong correlation (calculated linear regression R2 value of 0.8589) between the potencies in the DYRK1A enzyme assay and in the glioma (H4) proliferation assay was observed for all 7 of the harmine-related compounds. Harmol, which demonstrated the best selectivity for DYRK1A over MAO-A inhibition, was tested in vivo in a glioma xenograft model and demonstrated better safety characteristics than harmine as well as modest efficacy.
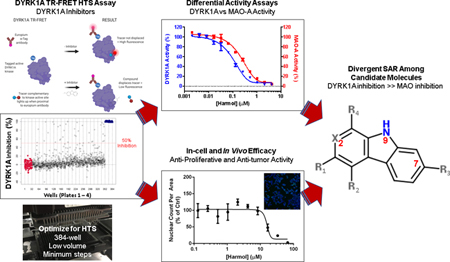
Harmine was initially characterized as a potent and selective inhibitor of DYRK1A (Bain et al., 2007; Göckler et al., 2009). However, harmine’s potential therapeutic use has been hampered by its potent “off-target” activity for MAO-A (Chen et al., 2005; Clarke and Ramsay, 2011; Coates and Cox, 1972; Herraiz and Chaparro, 2005; Kim et al., 1997; Reniers et al., 2011; Rommelspacher et al., 1994; Santillo et al., 2014), impacting multiple nervous system targets. In our study, we were particularly interested in examining whether there was a preference by any of the harmine analogs we identified from our screen for inhibition of DYRK1A over MAO-A, such that one could be dosed at a level sufficient to achieve anti-tumor effects in vivo while minimizing neurotoxicity side effects. Such information could provide insights into medicinal chemistry approaches that would yield a more selective and safer β-carboline-based DYRK1A inhibitor. The seven identified β-carbolines were profiled via dose response in two orthogonal DYRK1A enzyme assays. From these assays, a range of DYRK1A and MAO-A inhibitory activities emerged among the evaluated compounds. The data from our kinase and MAO-A assays were then utilized to illuminate divergent SAR around the three-ringed heterocycle common to all β-carbolines (summarized in Fig. 7).
Fig. 7.

Summary of divergent SAR for DYRK1A and MAO-A inhibition by identified harmine analogs. (A) Schematic of core β-carboline structure highlighting R substituents and positions 2, 7 and 9 (in red). (B) Table of substituents, IC50 values for DYRK1A, MAO-A with compounds ranked by inhibition ratio (DYRK1A IC50: MAO IC50) for the harmine analogs.
In considering the range of activities against DYRK1A and MAO-A and focusing on the structural relationship between the most potent DYRK1A inhibitors in this evaluation (harmine and harmol), there were trends identified in particular structural properties of β-carboline-derived compounds that appear important in their properties for inhibiting both DYRK1A and MAO-A. In the compounds evaluated, there were 3 areas in which the structures varied (Fig. 7); (i) The degree of saturation in ring 1, (ii) the presence or absence of a co-planar methyl group at R4, and (iii) the presence of a hydroxyl group or hydroxymethyl at R3. In our study, harmine and harmol, with comparable activity in the 70–100 nM range, were the most potent of the β-carbolines in the DYRK1A assay. Moving from a methoxy at R3 (harmine) to hydroxyl (harmol) had minimal effect on DYRK1A inhibition but a more pronounced effect on MAO-A inhibition, with harmol being 10-fold weaker than harmine for MAO-A. Harmaline, with the same substituents as harmine at positions R3, R4 and X but with saturation at the 3 and 4 positions of ring 1, has ~30-fold less activity for DYRK1A but comparable activity for MAO-A. Harmalol is similar in structure to harmol except that ring 1 is partially saturated. There are no published IC50 values for harmalol against DYRK1A, and we observed that its activity against DYRK1A is ~6-fold lower than harmol although it is still one of the more active compounds against DYRK1A in this series. Both harmane and norharmane have higher reported IC50 values against DYRK1A (see Table 1) and are structurally unique in this series in that they have no substitution on ring 3.
Harmol, in particular, was the only one of the 7 analogs that we observed to have a strong preference for DYRK1A inhibition over MAO-A, as was also observed by (Bálint et al., 2017). In contrast, harmine and harmaline were the most potent MAO-A inhibitors with similar activity in the 60–90 nM range, with harmaline having a ~30-fold preference for MAO-A over DYRK1A. This is supported by the observation that harmine and harmaline have been shown to be comparable in their tremorgenic effects in a number of animal models (Handforth, 2012). It has been reported that the non-planar conformation of these N-2 hydrogen bond acceptor groups, such as in harmaline, restricts binding to the ATP pocket of DYRK1A (Wang et al., 2015). Harmol, harmalol and harmane have comparable activities for MAO-A in the 500–640 nM range. Changes at the R3 position away from methoxy affect MAO-A activity. Harmalol, with a methyl group in the first ring ortho position but with a more saturated first ring, is less potent against DYRK1A. Norharmane, lacking a methyl group in the first ring and an oxygen-containing group in the third ring, and tetrahydroharmine, with a fully saturated first ring and chiral methyl group, were the weakest DYRK1A inhibitors in this assay. Norharmane (unsubstituted) had comparable weak activity on both DYRK1A and MAO-A. In a three-dimensional model of harmine’s interaction with DYRK1A, it appears as a relatively planar molecule that fits neatly into the ATP-binding site of DYRK1A (Ogawa et al., 2010; Uhl et al., 2018). In contrast, tetrahydroharmine has a fully saturated first ring and is reportedly a relatively weak inhibitor of DYRK1A and a weak or non-inhibitor of MAO-A. With a completely non-planar ring 1 and the chiral carbon and out-of-plane methyl group in ring 1, the interaction between this molecule and either DYRK1A or MAO may be minimized. To better understand the influence of the chiral center at C-1 would require testing of the enantiomer.
Thus, our findings indicate that some loss of planar aromatic character with sp3 carbons at R1 and R2 impact DYRK1A inhibition but do not affect MAO-A. In contrast, changes at R3 affect both DYRK1A and MAO-A inhibition, with the methoxy being critical for MAO-A inhibition and both the hydroxyl and methoxy substantially increasing DYRK1A inhibition. While both of these substituents could be acting as an H-bond acceptor, the addition of the methyl group is clearly providing some additional beneficial interaction with MAO-A. Other groups have reported that the methoxy at position 7 (R3) is vital for neurotoxicity (Bálint et al., 2017) and that adding an alkoxy group at R3 eliminates neurotoxicity (Cao et al., 2013). Others have shown that changes at the R3 position are limited for further optimization of DYRK1A inhibition (Kumar et al., 2020). A number of studies have explored the β-carboline scaffold, and a number of groups have gone on to employ computational and/or synthetic chemistry routes with harmine as a starting point to design novel β-carboline compounds with improved potency and selectivity for DYRK1A over MAO-A (Akabli et al., 2020; Bálint et al., 2017; Drung et al., 2014; Haider et al., 2018; Kumar et al., 2020; Kumar et al., 2018; Rüben et al., 2015; Wurzlbauer et al., 2020).
Harmine exhibits anti-cancer activity in vitro (Cao et al., 2004; Cao et al., 2013; Ishida et al., 1999; Li et al., 2015), including in glioma (Pozo et al., 2013), breast (Ma and Wink, 2010; Zhao and Wink, 2013) and gastric (Li et al., 2017) models, although the proposed mechanisms of action do not always indicate DYRK1A as the primary target (Cao et al., 2005; Ishida et al., 1999; Li et al., 2017; Zhao and Wink, 2013). In our study, two potent DYRK1A inhibitors identified from a pilot DYRK1A enzyme inhibitor screen, harmine and harmol, were also the most potent in the in vitro cytotoxicity assays when utilizing H4 and U87 glioma cell lines. The strong correlation between the IC50 values observed for the harmine analogs for DRYRK1A inhibition as well as the H4 and U87 anti-proliferative effects would suggest that blocking DYRK1A in these cells leads to a decrease in proliferation. In our high-content imaging assay, harmine and harmol had significant effects on increasing nuclear area and disrupting plasma membrane integrity. This assay can be used to provide some mechanistic insight, as compounds that increase nuclear size and disrupt plasma membrane integrity typically include cell cycle inhibitors, agents that induce DNA damage and compounds disrupting mitochondrial function (Abraham et al., 2008; Towne et al., 2012). In cancer models, harmine and related analogs have been shown to be anti-proliferative and to induce apoptosis (Cao et al., 2011; Kumar et al., 2018; Li et al., 2017; Liu et al., 2013; Uhl et al., 2018) with these being caspase-mediated (caspase-3/7 and caspase-9) (Cao et al., 2011; Uhl et al., 2018). Harmine also induces DNA damage and triggers apoptosis by a mitochondria-mediated pathway (Mota et al., 2020). Mitochondrial dysregulation, via increasing reactive oxygen species, has been proposed as a mechanism for harmine’s cytotoxicity (Atteya et al., 2017; Mota et al., 2020; Réus et al., 2012). Harmine and its derivatives are thought to bind to DNA and interfere with topoisomerase activities (Pagano et al., 2017). Other reported mechanisms for harmine’s anti-cancer efficacy include induction of pro-death autophagy (Li et al., 2017) and inhibition of angiogenesis (Dai et al., 2012). Several potential mechanisms for harmine’s inhibition of tumor growth via DYRK1A have been reported. DYRK1A increases cell proliferation and the expression of anti-apoptotic pathways (Zhou et al., 2017). Caspase 9, the apoptotic initiator, is directly phosphorylated by DYRK1A restraining apoptosis. Harmine is able to block this DYRK1A-mediated phosphorylation of caspase 9 (Seifert et al., 2008). Crosstalk between DYRK1A and p53 can also be proposed as another mechanism for harmine’s anti-tumor activity. p53 function is inhibited by high levels of oncoprotein MDM2 (Nag et al., 2013) and so inhibiting this MDM2-p53 interaction can be a potential approach for an anti-cancer therapy. Harmine has been shown to be a novel activator of p53 by inducing phosphorylation of p53, with increasing harmine concentrations dramatically decreasing the amount of MDM2 associated with p53 (Dai et al., 2012). Harmine-mediated disruption of p53-MDM2 led to inhibition of tumor growth in a lung cancer xenograft model (Dai et al., 2012). Further supporting a link between p53 and DYRK1A is the observation that disruption of p53/MDM2 binding directly has been shown to reduce DYRK1A levels and inhibit tumor growth in a U87 xenograft model (Xu et al., 2019). Interestingly, in a recent report, a high content screen of a Prestwick compound library using an osteosarcoma cell line with an androgen receptor (AR) reporter identified harmol and subsequently showed it be a competitive inhibitor of AR with efficacy in prostate cancer cell models (Dellal et al., 2020). Our high content data would support a potential mechanism of harmine and harmol acting to induce DNA and mitochondrial damage leading to cell death. Identifying the exact mechanism will require further study.
Our findings and a number of other studies have explored the β-carboline scaffold (Cao et al., 2004; Cao et al., 2013; Cao et al., 2005; Ishida et al., 1999; Li et al., 2015) to increase anti-tumor activity and/or reduce neurotoxicity indicating the therapeutic potential of these compounds. We further demonstrated that in an in-cell target engagement assay, harmine and harmol were both active with harmol being more effective than harmine. In another cell-based assay, harmine and harmol were also able to inhibit Hh-induced alkaline phosphatase activity in the C3H10T1/2 cell model for Hh signaling. A role for DYRK1A in Hh signaling has been observed (reviewed in (Boni et al., 2020; Heretsch et al., 2010; Ionescu et al., 2012; Javelaud et al., 2012; Singh and Lauth, 2017)) including direct phosphorylation of GLI1 by DYRK1A (Ehe et al., 2017; Mao et al., 2002), and in modulating GL1 nuclear translocation (Mao et al., 2002; Shimokawa et al., 2013; Shimokawa et al., 2008). DYRK1A inhibitors block Hh activity in vitro (Wang et al., 2012) and block DYRK1A mediated phosphorylation of GLI1 (Ehe et al., 2017). DYRK1B has also been reported as a regulator of Hh signaling (Gruber et al., 2016; Lauth et al., 2010; Singh et al., 2017; Singh and Lauth, 2017). More recently, a role for DYRK2 in modulating Hh signaling during embryogenesis has been reported (Yoshida et al., 2020). Determining whether in our Hh model, harmine and its analogs are acting through DYRK1A, or also through related kinases DYRK1B and DYRK2 will require further investigation.
An assessment of the relative effectiveness of harmine and harmol in vivo showed some promising trends that may warrant further analysis of the β-carbolines or their derivatives as leads for anti-cancer therapeutics. Since the standard dose chosen for inducing tremors in rodents with harmine is > 15 mg/kg (Pozo et al., 2013), we surmised that due to the superior selectivity of harmol for DYRK1A over MAO-A inhibition, that harmol dosed as high as 60 mg/kg may not be tremorgenic. In a glioma xenograft model, we observed over 4-weeks of treatment, that overall animal weight was not significantly affected between vehicle and either harmine or harmol treatment. However, there was a significant increase in tumor growth inhibition for harmine and harmol vs vehicle. Additionally, 60 mg/kg harmol was more effective in tumor inhibition than the 15 mg/kg harmol treatment, indicating a potential dose effect. Also encouraging to note is that no tremor activity was anecdotally observed in these mice for the duration of the study. There were several other parameters evaluated in this in vivo study that, although not significant, showed trends in a promising direction. These include apparent improvements in animal survival when treated with harmine or increasing doses of harmol as well as reductions in mean tumor volume. Although not significant, in both cases the 60 mg/kg harmol treatment was more effective compared to 15 mg/kg harmol and harmine.
The additional status of harmine as an MAO-A inhibitor (Kim et al., 1997) might initially appear to limit its in vivo application as a DYRK1A inhibitor. However, it has been speculated that harmine could serve as a promising lead compound for the development of an optimized novel DYRK1A inhibitor devoid of MAO-inhibiting activity (Becker and Sippl, 2011; Göckler et al., 2009). Our in vivo and SAR data and other groups’ characterization of novel harmine analog series with reduced or no MAO-A inhibitory activity (Haider et al., 2018; Rüben et al., 2015) supports this intriguing premise. Our observations confirm that specific activities, such as the anti-tumor activity and acute toxicity as well as neurotoxic effect, of β-carboline derivatives are substituent-dependent (Cao et al., 2004; Cao et al., 2005). An SAR analysis by Chen et al. (Chen et al., 2005) indicated that harmine derivatives with specific substitutions led to high anti-tumor activity and low toxicity while at the same time providing their own possible lead molecules for further development. Li et al. (Li et al., 2015) also developed novel harmine derivatives with promising potential for a targeted cancer therapy. Their derivatives displayed higher anti-tumor efficiency and lower systemic and neural toxicity than unmodified harmine (Li et al., 2015).
In conclusion, our in vitro and in vivo assessments of the relative effectiveness of harmine and harmol have produced some promising data that warrant further analysis of the β-carbolines or other indole alkaloids as leads for anti-cancer therapeutics. Furthermore, our findings have highlighted some interesting structural characteristics of these β-carbolines, that is, changes at positions 3 and 4 impact DYRK1A inhibition but do not affect MAO-A while changes at position 7 do not affect DYRK1A but do impact MAO-A inhibition. These insights may have implications in the further development of these β-carboline compounds as therapeutic molecules, i.e., as DYRK1A inhibitors with an absent MAO-inhibiting activity.
Supplementary Material
Supplementary Fig. 2. Harmine and harmol structural confirmation. Chemical structures of harmine and harmol used for in vivo studies were confirmed by LC/MS using an Agilent 1200 HPLC/Agilent Quadruple LC/MS and by NMR using a 500 MHz Varian instrument. Mass (upper) and NMR (lower) spectra for harmine (A) and harmol (B).
Supplementary Fig. 1. Activities of selected β-carbolines in C3H10T1/2 Hh cell assay. C3H10T1/2 were induced with ShhN protein and β-carbolines added at the indicated concentrations. After 5 days, the effects on alkaline phosphatase (AP) activity were measured at 405 nm using pNPP. As full dose response curves were not achieved for these 5 inhibitors, a 3-parameter curve fit was done. Inset is the maximal inhibition observed (InhibitionMax) at the highest compound concentration tested (20 μM). THH = tetrahydroharmine.
Acknowledgments
This study was supported in part by funding from NIH/NCI awards R15CA208651 and P20CA202924; NIH/NIMHD RCMI U54MD012392 (K.P.W.), and project support from NIH award U54CA137844 (K.P.W., R.U.O. and D.M.D.), and Komen Graduate Training in Disparities Research award GTDR16377604 (K.P.W. and H.O.H.) with additional support from the Golden LEAF Foundation and the BIOIMPACT Initiative of the State of North Carolina. The authors would like to thank Ginger Smith and David Lamson (BRITE) for technical assistance and Dr. C. Ryan Miller from UNC Lineberger for technical advice on in vivo glioma models. Human Kinome illustration reproduced by permission of Cell Signaling Technology, Inc. (www.cellsignal.com). The assay schematic image in the graphical abstract was created with BioRender.com.
References
- Abbassi R, Johns TG, Kassiou M, Munoz L, 2015. DYRK1A in neurodegeneration and cancer: molecular basis and clinical implications. Pharmacology & therapeutics 151, 87–98. [DOI] [PubMed] [Google Scholar]
- Abraham VC, Towne DL, Waring JF, Warrior U, Burns DJ, 2008. Application of a high-content multiparameter cytotoxicity assay to prioritize compounds based on toxicity potential in humans. Journal of Biomolecular Screening 13, 527–537. [DOI] [PubMed] [Google Scholar]
- Adayev T, Wegiel J, Hwang YW, 2011. Harmine is an ATP-competitive inhibitor for dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A). Arch Biochem Biophys 507, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed A, Taylor NR, 1959. The analysis of drug-induced tremor in mice. British journal of pharmacology and chemotherapy 14, 350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airaksinen MM, Kari I, 1981. Beta-carbolines, psychoactive compounds in the mammalian body. Part I: Occurrence, origin and metabolism. Med Biol 59, 21–34. [PubMed] [Google Scholar]
- Akabli T, Toufik H, Lamchouri F, 2020. In silico modeling studies of N9 -substituted harmine derivatives as potential anticancer agents: combination of ligand-based and structure-based approaches. Journal of Biomolecular Structure and Dynamics, 1–14. [DOI] [PubMed] [Google Scholar]
- Allen M, Bjerke M, Edlund H, Nelander S, Westermark B, 2016. Origin of the U87MG glioma cell line: Good news and bad news. Science translational medicine 8, 354re353–354re353. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR, 2011. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nature biotechnology 29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda S, Laguna A, de la Luna S, 2011. DYRK family of protein kinases: evolutionary relationships, biochemical properties, and functional roles. The FASEB Journal 25, 449–462. [DOI] [PubMed] [Google Scholar]
- Atteya R, Ashour ME, Ibrahim EE, Farag MA, El-Khamisy SF, 2017. Chemical screening identifies the β-Carboline alkaloid harmine to be synergistically lethal with doxorubicin. Mechanisms of ageing and development 161, 141–148. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P, 2007. The selectivity of protein kinase inhibitors: a further update. The Biochemical journal 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bálint B, Wéber C, Cruzalegui F, Burbridge M, Kotschy A, 2017. Structure-Based Design and Synthesis of Harmine Derivatives with Different Selectivity Profiles in Kinase versus Monoamine Oxidase Inhibition. ChemMedChem 12, 932–939. [DOI] [PubMed] [Google Scholar]
- Becker W, Sippl W, 2011. Activation, regulation, and inhibition of DYRK1A. FEBS Journal 278, 246–256. [DOI] [PubMed] [Google Scholar]
- Becker W, Soppa U, J Tejedor F, 2014. DYRK1A: a potential drug target for multiple Down syndrome neuropathologies. CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders) 13, 26–33. [DOI] [PubMed] [Google Scholar]
- Boni J, Rubio-Perez C, López-Bigas N, Fillat C, de la Luna S, 2020. The DYRK family of kinases in cancer: Molecular functions and therapeutic opportunities. Cancers 12, 2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel A, Bénéteau R, Chabanne M, Lozach O, Le Guevel R, Ravache M, Bénédetti H, Meijer L, Logé C, Robert J-M, 2014. Synthesis of new pyridazino [4, 5-b] indol-4-ones and pyridazin-3 (2H)-one analogs as DYRK1A inhibitors. Bioorganic & medicinal chemistry letters 24, 5037–5040. [DOI] [PubMed] [Google Scholar]
- Bruel A, Logé C, de Tauzia M-L, Ravache M, Le Guevel R, Guillouzo C, Lohier J-F, Sopkova-de Oliveira Santos J, Lozach O, Meijer L, 2012. Synthesis and biological evaluation of new 5-benzylated 4-oxo-3, 4-dihydro-5H-pyridazino [4, 5-b] indoles as PI3Kα inhibitors. European journal of medicinal chemistry 57, 225–233. [DOI] [PubMed] [Google Scholar]
- Cao M-R, Li Q, Liu Z-L, Liu H-H, Wang W, Liao X-L, Pan Y-L, Jiang J-W, 2011. Harmine induces apoptosis in HepG2 cells via mitochondrial signaling pathway. Hepatobiliary & Pancreatic Diseases International 10, 599–604. [DOI] [PubMed] [Google Scholar]
- Cao R, Chen Q, Hou X, Chen H, Guan H, Ma Y, Peng W, Xu A, 2004. Synthesis, acute toxicities, and antitumor effects of novel 9-substituted beta-carboline derivatives. Bioorganic & medicinal chemistry 12, 4613–4623. [DOI] [PubMed] [Google Scholar]
- Cao R, Fan W, Guo L, Ma Q, Zhang G, Li J, Chen X, Ren Z, Qiu L, 2013. Synthesis and structure–activity relationships of harmine derivatives as potential antitumor agents. European journal of medicinal chemistry 60, 135–143. [DOI] [PubMed] [Google Scholar]
- Cao R, Peng W, Chen H, Hou X, Guan H, Chen Q, Ma Y, Xu A, 2005. Synthesis and in vitro cytotoxic evaluation of 1,3-bisubstituted and 1,3,9-trisubstituted beta-carboline derivatives. European journal of medicinal chemistry 40, 249–257. [DOI] [PubMed] [Google Scholar]
- Chen Q, Chao R, Chen H, Hou X, Yan H, Zhou S, Peng W, Xu A, 2005. Antitumor and neurotoxic effects of novel harmine derivatives and structure-activity relationship analysis. Int J Cancer 114, 675–682. [DOI] [PubMed] [Google Scholar]
- Clarke SED, Ramsay RR, 2011. Dietary inhibitors of monoamine oxidase A. Journal of Neural Transmission 118, 1031–1041. [DOI] [PubMed] [Google Scholar]
- Coates GH, Cox B, 1972. Harmine tremor after brain monoamine oxidase inhibition in the mouse. Eur J Pharmacol 18, 284–286. [DOI] [PubMed] [Google Scholar]
- Coutadeur S, Benyamine H, Delalonde L, de Oliveira C, Leblond B, Foucourt A, Besson T, Casagrande AS, Taverne T, Girard A, 2015. A novel DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: effect on Tau and amyloid pathologies in vitro. Journal of neurochemistry 133, 440–451. [DOI] [PubMed] [Google Scholar]
- Dai F, Chen Y, Song Y, Huang L, Zhai D, Dong Y, Lai L, Zhang T, Li D, Pang X, 2012. A natural small molecule harmine inhibits angiogenesis and suppresses tumour growth through activation of p53 in endothelial cells. PloS one 7, e52162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellal H, Boulahtouf A, Alaterre E, Cuenant A, Grimaldi M, Bourguet W, Gongora C, Balaguer P, Pourquier P, 2020. High Content Screening Using New U2OS Reporter Cell Models Identifies Harmol Hydrochloride as a Selective and Competitive Antagonist of the Androgen Receptor. Cells 9, 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierssen M, de Lagrán MM, 2006. DYRK1A (dual-specificity tyrosine-phosphorylated and-regulated kinase 1A): a gene with dosage effect during development and neurogenesis. The Scientific World Journal 6, 1911–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, Dančík V, Burns SM, Gilbert TJ, Olson DE, Clemons PA, Kulkarni RN, Wagner BK, 2016a. Inhibition of DYRK1A stimulates human β-cell proliferation. Diabetes 65, 1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drung B, Scholz C, Barbosa VA, Nazari A, Sarragiotto MH, Schmidt B, 2014. Computational & experimental evaluation of the structure/activity relationship of β-carbolines as DYRK1A inhibitors. Bioorganic & medicinal chemistry letters 24, 4854–4860. [DOI] [PubMed] [Google Scholar]
- Du WZ, Feng Y, Wang XF, Piao XY, Cui YQ, Chen LC, Lei XH, Sun X, Liu X, Wang HB, 2013. Curcumin Suppresses Malignant Glioma Cells Growth and Induces Apoptosis by Inhibition of SHH/GLI 1 Signaling Pathway in Vitro and Vivo. CNS neuroscience & therapeutics 19, 926–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehe BK, Lamson DR, Tarpley M, Onyenwoke RU, Graves LM, Williams KP, 2017. Identification of a DYRK1A-mediated phosphorylation site within the nuclear localization sequence of the hedgehog transcription factor GLI1. Biochemical and biophysical research communications 491, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Martinez P, Zahonero C, Sanchez-Gomez P, 2015. DYRK1A: the double-edged kinase as a protagonist in cell growth and tumorigenesis. Molecular & cellular oncology 2, e970048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foucourt A, Hédou D, Dubouilh-Benard C, Girard A, Taverne T, Casagrande A-S, Désiré L, Leblond B, Besson T, 2014. Design and synthesis of thiazolo [5, 4-f] quinazolines as DYRK1A inhibitors, part II. Molecules 19, 15411–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost D, Meechoovet B, Wang T, Gately S, Giorgetti M, Shcherbakova I, Dunckley T, 2011. β-carboline compounds, including harmine, inhibit DYRK1A and tau phosphorylation at multiple Alzheimer’s disease-related sites. PLoS One 6, e19264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göckler N, Jofre G, Papadopoulos C, Soppa U, Tejedor FJ, Becker W, 2009. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS Journal 276, 6324–6337. [DOI] [PubMed] [Google Scholar]
- Grabher P, Durieu E, Kouloura E, Halabalaki M, Skaltsounis LA, Meijer L, Hamburger M, Potterat O, 2012. Library-based discovery of DYRK1A/CLK1 inhibitors from natural product extracts. Planta medica 78, 951–956. [DOI] [PubMed] [Google Scholar]
- Gruber W, Hutzinger M, Elmer DP, Parigger T, Sternberg C, Cegielkowski L, Zaja M, Leban J, Michel S, Hamm S, 2016. DYRK1B as therapeutic target in Hedgehog/GLI-dependent cancer cells with Smoothened inhibitor resistance. Oncotarget 7, 7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider S, Alhusban M, Chaurasiya ND, Tekwani BL, Chittiboyina AG, Khan IA, 2018. Isoform selectivity of harmine-conjugated 1, 2, 3-triazoles against human monoamine oxidase. Future medicinal chemistry 10, 1435–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handforth A, 2012. Harmaline tremor: underlying mechanisms in a potential animal model of essential tremor. Tremor and other hyperkinetic movements 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Hunter T, 1995. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification 1. The FASEB journal 9, 576–596. [PubMed] [Google Scholar]
- Heretsch P, Tzagkaroulaki L, Giannis A, 2010. Modulators of the hedgehog signaling pathway. Bioorganic & medicinal chemistry 18, 6613–6624. [DOI] [PubMed] [Google Scholar]
- Herraiz T, Brandt SD, 2014. 5-(2-Aminopropyl) indole (5-IT): a psychoactive substance used for recreational purposes is an inhibitor of human monoamine oxidase (MAO). Drug testing and analysis 6, 607–613. [DOI] [PubMed] [Google Scholar]
- Herraiz T, Chaparro C, 2005. Human monoamine oxidase is inhibited by tobacco smoke: β-carboline alkaloids act as potent and reversible inhibitors. Biochemical and biophysical research communications 326, 378–386. [DOI] [PubMed] [Google Scholar]
- Himpel S, Panzer P, Eirmbter K, Czajkowska H, Sayed M, Packman LC, Blundell T, Kentrup H, Grotzinger J, Joost HG, Becker W, 2001. Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. The Biochemical journal 359, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House AJ, Daye LR, Tarpley M, Addo K, Lamson DS, Parker MK, Bealer WE, Williams KP, 2015. Design and characterization of a photo-activatable hedgehog probe that mimics the natural lipidated form. Archives of Biochemistry and Biophysics 567, 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu A, Dufrasne F, Gelbcke M, Jabin I, Kiss R, Lamoral-Theys D, 2012. DYRK1A kinase inhibitors with emphasis on cancer. Mini reviews in medicinal chemistry 12, 1315–1329. [DOI] [PubMed] [Google Scholar]
- Ishida J, Wang H-K, Bastow KF, Hu C-Q, Lee K-H, 1999. Antitumor agents 201.< sup> 1 Cytotoxicity of harmine and β-carboline analogs. Bioorganic & Medicinal Chemistry Letters 9, 3319–3324. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Pierrat M-J, Mauviel A, 2012. Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Letters 586, 2016–2025. [DOI] [PubMed] [Google Scholar]
- Kim H, Lee K-S, Kim A-K, Choi M, Choi K, Kang M, Chi S-W, Lee M-S, Lee J-S, Lee S-Y, 2016. A chemical with proven clinical safety rescues Down-syndrome-related phenotypes in through DYRK1A inhibition. Disease models & mechanisms 9, 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Sablin SO, Ramsay RR, 1997. Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch Biochem Biophys 337, 137–142. [DOI] [PubMed] [Google Scholar]
- Kumar K, Wang P, Swartz E, Khamrui S, Secor C, B Lazarus M, Sanchez R, Stewart A, DeVita R, 2020. Structure–Activity Relationships and Biological Evaluation of 7-Substituted Harmine Analogs for Human β-Cell Proliferation. Molecules 25, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K, Wang P, Sanchez R, Swartz EA, Stewart AF, DeVita RJ, 2018. Development of kinase-selective, harmine-based DYRK1A inhibitors that induce pancreatic human β-cell proliferation. Journal of Medicinal Chemistry 61, 7687–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauth M, Bergström Å, Shimokawa T, Tostar U, Jin Q, Fendrich V, Guerra C, Barbacid M, Toftgård R, 2010. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nature structural & molecular biology 17, 718. [DOI] [PubMed] [Google Scholar]
- Lebakken CS, Riddle SM, Singh U, Frazee WJ, Eliason HC, Gao Y, Reichling LJ, Marks BD, Vogel KW, 2009. Development and applications of a broad-coverage, TR-FRET-based kinase binding assay platform. Journal of biomolecular screening 14, 924–935. [DOI] [PubMed] [Google Scholar]
- Li C, Wang Y, Wang C, Yi X, Li M, He X, 2017. Anticancer activities of harmine by inducing a pro-death autophagy and apoptosis in human gastric cancer cells. Phytomedicine 28, 10–18. [DOI] [PubMed] [Google Scholar]
- Li S, Wang A, Gu F, Wang Z, Tian C, Qian Z, Tang L, Gu Y, 2015. Novel harmine derivatives for tumor targeted therapy. Oncotarget 6, 8988–9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tang K, Huang L, Liu C, Du Q, Li T, Yan C, Feng Z, Li X, 2019. Anticancer effects of FL34 through the inhibition of GLI1 in glioblastoma. Journal of cancer research and therapeutics 15, 889. [DOI] [PubMed] [Google Scholar]
- Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA, 2011. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes & development 25, 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Han D, Liu Y, Hou X, Wu J, Li H, Yang J, Shen C, Yang G, Fu C, 2013. Harmine hydrochloride inhibits Akt phosphorylation and depletes the pool of cancer stem-like cells of glioblastoma. Journal of neuro-oncology 112, 39–48. [DOI] [PubMed] [Google Scholar]
- Liu X, Li M, Tan S, Wang C, Fan S, Huang C, 2017a. Harmine is an inflammatory inhibitor through the suppression of NF-κB signaling. Biochemical and biophysical research communications 489, 332–338. [DOI] [PubMed] [Google Scholar]
- Liu Y, Adayev T, Hwang Y-W, 2017b. An ELISA DYRK1A non-radioactive kinase assay suitable for the characterization of inhibitors. F1000Research 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louca M, Stylianou A, Minia A, Pliaka V, Alexopoulos LG, Gkretsi V, Stylianopoulos T, 2019. Ras suppressor-1 (RSU-1) promotes cell invasion in aggressive glioma cells and inhibits it in non-aggressive cells through STAT6 phospho-regulation. Scientific reports 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Deacon S, Horiuchi K, 2008. The challenge of selecting protein kinase assays for lead discovery optimization. Expert opinion on drug discovery 3, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Wink M, 2010. The beta-carboline alkaloid harmine inhibits BCRP and can reverse resistance to the anticancer drugs mitoxantrone and camptothecin in breast cancer cells. Phytotherapy Research 24, 146–149. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S, 2002. The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Mao J, Maye P, Kogerman P, Tejedor FJ, Toftgard R, Xie W, Wu G, Wu D, 2002. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. Journal of Biological Chemistry 277, 35156–35161. [DOI] [PubMed] [Google Scholar]
- Mota NS, Kviecinski MR, Felipe KB, Grinevicius VM, Siminski T, Almeida GM, Zeferino RC, Pich CT, Pedrosa RC, 2020. β-carboline alkaloid harmine induces DNA damage and triggers apoptosis by a mitochondrial pathway: study in silico, in vitro and in vivo. International Journal of Functional Nutrition 1, 1–1. [Google Scholar]
- Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R, 2013. The MDM2-p53 pathway revisited. Journal of biomedical research 27, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Aikawa T, Iwamoto-Enomoto M, Iwamoto M, Higuchi Y, Maurizio P, Kinto N, Yamaguchi A, Noji S, Kurisu K, 1997. Induction of Osteogenic Differentiation by Hedgehog Proteins. Biochemical and Biophysical Research Communications 237, 465–469. [DOI] [PubMed] [Google Scholar]
- Noll C, Planque C, Ripoll C, Guedj F, Diez A, Ducros V, Belin N, Duchon A, Paul J-L, Badel A, 2009. DYRK1A, a novel determinant of the methionine-homocysteine cycle in different mouse models overexpressing this Down-syndrome-associated kinase. PLoS One 4, e7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Nonaka Y, Goto T, Ohnishi E, Hiramatsu T, Kii I, Yoshida M, Ikura T, Onogi H, Shibuya H, 2010. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nature communications 1, 86. [DOI] [PubMed] [Google Scholar]
- Oladapo HO, Tarpley M, Sauer SJ, Addo KA, Ingram SM, Strepay D, Ehe BK, Chdid L, Trinkler M, Roques JR, 2017. Pharmacological targeting of GLI1 inhibits proliferation, tumor emboli formation and in vivo tumor growth of inflammatory breast cancer cells. Cancer letters 411, 136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano B, Caterino M, Filosa R, Giancola C, 2017. Binding of harmine derivatives to DNA: a spectroscopic investigation. Molecules 22, 1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo N, Zahonero C, Fernández P, Liñares JM, Ayuso A, Hagiwara M, Pérez A, Ricoy JR, Hernández-Laín A, Sepúlveda JM, 2013. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. The Journal of Clinical Investigation 123, 2475–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quent VM, Loessner D, Friis T, Reichert JC, Hutmacher DW, 2010. Discrepancies between metabolic activity and DNA content as tool to assess cell proliferation in cancer research. Journal of cellular and molecular medicine 14, 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachdi L, Kariyawasam D, Guez F, Aiello V, Arbones ML, Janel N, Delabar JM, Polak M, Scharfmann R, 2014. Dyrk1a haploinsufficiency induces diabetes in mice through decreased pancreatic beta cell mass. Diabetologia 57, 960–969. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A, Nanjappa V, Raja R, Sathe G, Puttamallesh VN, Jain AP, Pinto SM, Balaji SA, Chavan S, Sahasrabuddhe NA, 2016. A dual specificity kinase, DYRK1A, as a potential therapeutic target for head and neck squamous cell carcinoma. Scientific reports 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman T, Rahmatullah M, 2010. Proposed structural basis of interaction of piperine and related compounds with monoamine oxidases. Bioorganic & medicinal chemistry letters 20, 537–540. [DOI] [PubMed] [Google Scholar]
- Reniers J, Robert S, Frederick R, Masereel B, Vincent S, Wouters J, 2011. Synthesis and evaluation of β-carboline derivatives as potential monoamine oxidase inhibitors. Bioorganic & medicinal chemistry 19, 134–144. [DOI] [PubMed] [Google Scholar]
- Réus GZ, Stringari RB, Gonçalves CL, Scaini G, Carvalho-Silva M, Jeremias GC, Jeremias IC, Ferreira GK, Streck EL, Hallak JE, 2012. Administration of harmine and imipramine alters creatine kinase and mitochondrial respiratory chain activities in the rat brain. Depression research and treatment 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikoff CA, Brankow DW, Heidelberger C, 1973. Establishment and characterization of a cloned line of C3H mouse embryo cells sensitive to postconfluence inhibition of division. Cancer research 33, 3231–3238. [PubMed] [Google Scholar]
- Rommelspacher H, May T, Salewski B, 1994. Harman (1-methyl-β-carboline) is a natural inhibitor of monoamine oxidase type A in rats. European journal of pharmacology 252, 51–59. [DOI] [PubMed] [Google Scholar]
- Rüben K, Wurzlbauer A, Walte A, Sippl W, Bracher F, Becker W, 2015. Selectivity profiling and biological activity of novel β-carbolines as potent and selective DYRK1 kinase inhibitors. PloS one 10, e0132453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo SR, Cho HJ, Lee HW, Jeong HK, Radnaabazar C, Kim YS, Kim MJ, Son MY, Seo H, Chung SH, 2008. Dual-specificity tyrosine (Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer’s disease. Journal of neurochemistry 104, 1333–1344. [DOI] [PubMed] [Google Scholar]
- Santillo MF, Liu Y, Ferguson M, Vohra SN, Wiesenfeld PL, 2014. Inhibition of monoamine oxidase (MAO) by β-carbolines and their interactions in live neuronal (PC12) and liver (HuH-7 and MH1C1) cells. Toxicology in Vitro 28, 403–410. [DOI] [PubMed] [Google Scholar]
- Sauer SJ, Tarpley M, Shah I, Save AV, Lyerly HK, Patierno SR, Williams KP, Devi GR, 2017. Bisphenol A activates EGFR and ERK promoting proliferation, tumor spheroid formation and resistance to EGFR pathway inhibition in estrogen receptor-negative inflammatory breast cancer cells. Carcinogenesis 38, 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürmann M, Janning P, Ziegler S, Waldmann H, 2016. Small-molecule target engagement in cells. Cell chemical biology 23, 435–441. [DOI] [PubMed] [Google Scholar]
- Seifert A, Allan LA, Clarke PR, 2008. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS Journal 275, 6268–6280. [DOI] [PubMed] [Google Scholar]
- Shen W, Tremblay MS, Deshmukh VA, Wang W, Filippi CM, Harb G, Zhang YQ, Kamireddy A, Baaten JE, Jin Q, Wu T, Swoboda JG, Cho CY, Li J, Laffitte BA, McNamara P, Glynne R, Wu X, Herman AE, Schultz PG, 2013. Small-molecule inducer of beta cell proliferation identified by high-throughput screening. Journal of the American Chemical Society 135, 1669–1672. [DOI] [PubMed] [Google Scholar]
- Shimokawa T, Rahman MF-U, Tostar U, Sonkoly E, Ståhle M, Pivarcsi A, Palaniswamy R, Zaphiropoulos PG, 2013. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biology 10, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa T, Tostar U, Lauth M, Palaniswamy R, Kasper M, Toftgård R, Zaphiropoulos PG, 2008. Novel human glioma-associated oncogene 1 (GLI1) splice variants reveal distinct mechanisms in the terminal transduction of the hedgehog signal. Journal of Biological Chemistry 283, 14345–14354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Dhanyamraju PK, Lauth M, 2017. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 8, 833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Lauth M, 2017. Emerging Roles of DYRK Kinases in Embryogenesis and Hedgehog Pathway Control. Journal of developmental biology 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson S, Minea RO, Tuan CD, Thein T-Z, Chen TC, Markland FS, 2018. A novel venom-derived peptide for brachytherapy of glioblastoma: preclinical studies in mice. Molecules 23, 2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpley M, Caligan TB, Onyenwoke R, Williams KP, 2021a. Optimization and validation of a DYRK1A TR-FRET assay for high-throughput screening. MethodsX 8, 101383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpley M, Oladapo HO, Caligan TB, Onyenwoke R, Williams KP, 2021b. Data supporting a pilot high throughput screen of a drug library for identification of DYRK1A inhibitors and high-content imaging analysis of identified harmine analogs. Data in Brief 37, 107189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towne DL, Nicholl EE, Comess KM, Galasinski SC, Hajduk PJ, Abraham VC, 2012. Development of a high-content screening assay panel to accelerate mechanism of action studies for oncology research. Journal of biomolecular screening 17, 1005–1017. [DOI] [PubMed] [Google Scholar]
- Tseng JR, Kang KW, Dandekar M, Yaghoubi S, Lee JH, Christensen JG, Muir S, Vincent PW, Michaud NR, Gambhir SS, 2008. Preclinical efficacy of the c-Met inhibitor CE-355621 in a U87 MG mouse xenograft model evaluated by 18F-FDG small-animal PET. Journal of Nuclear Medicine 49, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl KL, Schultz CR, Geerts D, Bachmann AS, 2018. Harmine, a dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) inhibitor induces caspase-mediated apoptosis in neuroblastoma. Cancer Cell International 18, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valley MP, Zhou W, Hawkins EM, Shultz J, Cali JJ, Worzella T, Bernad L, Good T, Good D, Riss TL, 2006. MAO papers 2021A bioluminescent assay for monoamine oxidase activity. Analytical biochemistry 359, 238–246. [DOI] [PubMed] [Google Scholar]
- Wang D, Wang F, Tan Y, Dong L, Chen L, Zhu W, Wang H, 2012. Discovery of potent small molecule inhibitors of DYRK1A by structure-based virtual screening and bioassay. Bioorganic & medicinal chemistry letters 22, 168–171. [DOI] [PubMed] [Google Scholar]
- Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocana A, Stewart AF, 2015. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nature medicine 21, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Gong CX, Hwang YW, 2011. The role of DYRK1A in neurodegenerative diseases. The FEBS journal 278, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Scott J, 2009. Enzyme assay design for high-throughput screening. Methods in molecular biology (Clifton, NJ) 565, 107–126. [DOI] [PubMed] [Google Scholar]
- Williams KP, Rayhorn P, Chi-Rosso G, Garber EA, Strauch KL, Horan GS, Reilly JO, Baker DP, Taylor FR, Koteliansky V, Pepinsky RB, 1999. Functional antagonists of sonic hedgehog reveal the importance of the N terminus for activity. J Cell Sci 112, 4405–4414. [DOI] [PubMed] [Google Scholar]
- Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Xuemin W, Proud CG, 2001. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bɛ at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochemical Journal 355, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurzlbauer A, Rüben K, Gürdal E, Chaikuad A, Knapp S, Sippl W, Becker W, Bracher F, 2020. How to Separate Kinase Inhibition from Undesired Monoamine Oxidase A Inhibition—The Development of the DYRK1A Inhibitor AnnH75 from the Alkaloid Harmine. Molecules 25, 5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Liu Q, Zhang C, Ren S, Xu L, Zhao Z, Dou H, Li P, Zhang X, Gong Y, 2019. Inhibition of DYRK1A-EGFR axis by p53-MDM2 cascade mediates the induction of cellular senescence. Cell death & disease 10, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, 2008. Role for DYRK family kinases on regulation of apoptosis. Biochem Pharmacol 76, 1389–1394. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Aoki K, Fujiwara K, Nakakura T, Kawamura A, Yamada K, Ono M, Yogosawa S, Yoshida K, 2020. The novel ciliogenesis regulator DYRK2 governs Hedgehog signaling during mouse embryogenesis. Elife 9, e57381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J-H, Chung TD, Oldenburg KR, 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of biomolecular screening 4, 67–73. [DOI] [PubMed] [Google Scholar]
- Zhao L, Wink M, 2013. The β-carboline alkaloid harmine inhibits telomerase activity of MCF-7 cells by down-regulating hTERT mRNA expression accompanied by an accelerated senescent phenotype. PeerJ 1, e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Phoa AF, Abbassi RH, Hoque M, Reekie TA, Font JS, Ryan RM, Stringer BW, Day BW, Johns TG, 2017. Structural optimization and pharmacological evaluation of inhibitors targeting dual-specificity tyrosine phosphorylation-regulated kinases (DYRK) and CDC-like kinases (CLK) in glioblastoma. Journal of Medicinal Chemistry 60, 2052–2070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 2. Harmine and harmol structural confirmation. Chemical structures of harmine and harmol used for in vivo studies were confirmed by LC/MS using an Agilent 1200 HPLC/Agilent Quadruple LC/MS and by NMR using a 500 MHz Varian instrument. Mass (upper) and NMR (lower) spectra for harmine (A) and harmol (B).
Supplementary Fig. 1. Activities of selected β-carbolines in C3H10T1/2 Hh cell assay. C3H10T1/2 were induced with ShhN protein and β-carbolines added at the indicated concentrations. After 5 days, the effects on alkaline phosphatase (AP) activity were measured at 405 nm using pNPP. As full dose response curves were not achieved for these 5 inhibitors, a 3-parameter curve fit was done. Inset is the maximal inhibition observed (InhibitionMax) at the highest compound concentration tested (20 μM). THH = tetrahydroharmine.