

Abstract
Pathological cardiac hypertrophy begins as an adaptive response to increased workload; however, sustained hemodynamic stress will lead it to maladaptation and eventually cardiac failure. Mitochondria, being the powerhouse of the cells, can regulate cardiac hypertrophy in both adaptive and maladaptive phases; they are dynamic organelles that can adjust their number, size, and shape through a process called mitochondrial dynamics. Recently, several studies indicate that promoting mitochondrial fusion along with preventing mitochondrial fission could improve cardiac function during cardiac hypertrophy and avert its progression toward heart failure. However, some studies also indicate that either hyperfusion or hypo-fission could induce apoptosis and cardiac dysfunction. In this review, we summarize the recent knowledge regarding the effects of mitochondrial dynamics on the development and progression of cardiac hypertrophy with particular emphasis on the regulatory role of mitochondrial dynamics proteins through the genetic, epigenetic, and post-translational mechanisms, followed by discussing the novel therapeutic strategies targeting mitochondrial dynamic pathways.
Keywords: mitochondrial dynamics, cardiac hypertrophy, therapeutic potential, recent advances, molecular mechanism
Graphical abstract
This review summarizes the current knowledge regarding the role of mitochondrial dynamics (MD) in cardiac hypertrophy with particular emphasis on the regulatory role of mitochondrial dynamic proteins through the genetic, epigenetic, and post-translational mechanisms, followed by discussing the novel therapeutic strategies targeting MD pathways.
Introduction
Myocardial hypertrophy is characterized by the thickening of the heart muscle as a response to an increase cardiac load by physiological or pathological stimuli.1 Physiological cardiac hypertrophy is an adaptive response to high energy demand during normal growth or pregnancy, and in athletes.2,3 However, pathological cardiac hypertrophy occurs in the presence of chronic stress conditions induced by extrinsic factors such as prolonged and abnormal hemodynamic stress, chronic hypertension, and valvular diseases, or by intrinsic factors including myocardial infarct, ischemia-induced cardiac remodeling, storage diseases, and genetic cardiomyopathy resulting from a mutation in genes encoding sarcomere proteins.2,4,5 In compensated cardiac hypertrophy, cardiomyocytes increase in length and width proportionally with not much change in wall thickness, maintaining their contractile function to meet the high energy requirements of the body; on the contrary, in decompensated hypertrophy, a greater increase in the cardiomyocytes’ length compared to that of the width causes impairment of their contractile function.6 When untreated, pathological cardiac hypertrophy is often followed by cardiac fibrosis and failure, predisposing to myocardial ischemia, arrhythmias, cerebrovascular attacks, and even sudden death.5,7,8
In recent years, heart failure (HF) has been the leading cause of cardiovascular mortality and morbidity9 to the point that is now considered a global pandemic affecting approximately 26 million patients worldwide.10 In the United States, it is expected that the burden of heart failure will continue to increase and that the prevalence of heart failure will rise to approximately more than 46% by 2030. Sustained cardiac hypertrophy is a major contributor to heart failure3,9 and is considered a predictor of cardiovascular morbidity and mortality.11 Furthermore, left ventricular hypertrophy (LVH) alone is regarded as an independent risk factor for heart failure, coronary artery disease, sudden cardiac death, and stroke.12 Mitochondrial dynamics (MD) controls the number, shape, size, and mitochondrial quality,13,14 thereby affecting mitochondrial subcellular transport, mitochondrial function, and the overall cellular energy homeostasis, ultimately determining a cell’s fate (autophagy and apoptosis). Disrupted MD has been found to be associated with the development or progression of several cardiovascular abnormalities, including cardiac hypertrophy,15 ischemia/reperfusion (I/R),16 and heart failure.17,18
Several nuclear and mitochondrial proteins are involved in mitochondrial fusion and fission events,19 and modulating the expression of these proteins can shift the equilibrium either toward fusion or fission. Increasing numbers of studies have shown that enhancing mitochondrial fusion and decreasing fission could prevent or reverse cardiac hypertrophy and improve cardiac function.20, 21, 22 Although cumulative evidence supports targeting MD as a potential therapeutic strategy, its application as an effective therapeutic measure remains far from reaching clinical applications (Boxes 1 and 2). To fill this gap, a thorough understanding of molecular mechanisms of MD and their regulatory approaches are essential and urgent. In this review, we summarize the recent knowledge regarding the effects of MD on the development and progression of cardiac hypertrophy with a particular emphasis on the regulatory role of mitochondrial dynamic proteins through genetic, epigenetic, and post-translational mechanisms, followed by discussing the novel therapeutic strategies targeting mitochondrial dynamic pathways.
Box 1. Facts.
Mitochondrial dynamics is directly linked to mitochondrial function and cellular homeostasis. When impaired, mitochondrial dynamics can influence a wide range of cellular processes, including mitochondrial biogenesis, energy metabolism, mtDNA maintenance, mitochondrial quality control, ROS production, Ca2+ signaling, cell cycle and stem cell regulation, mitophagy, autophagy, and apoptosis.
Increased mitochondrial fragmentation, decreased mitochondrial density, and increased apoptosis are detected in multiple forms of cardiomyopathy and heart failure.
Upregulation of mitochondrial fission factors (Drp1 and Fis1) and downregulation of mitochondrial fusion proteins (Mfn1/2 and Opa1) underline the development of cardiac hypertrophy and progression toward heart failure.
Drugs reducing mitochondrial fission show a significant diminution in cardiac hypertrophy and improvement in cardiac function.
Box 2. Open questions.
How are mitochondrial dynamics and apoptosis regulated in the course of cardiac hypertrophy?
What are the characteristic changes in mitochondrial morphology that determine cardiac hypertrophy?
What are the potential side effects of long-term manipulation of mitochondrial dynamics in patients with cardiac hypertrophy?
How can mitochondrial morphology be effectively detected in patients with cardiac hypertrophy?
Overview of MD
Mitochondria were first discovered in 1989 and the name was given in Greek based on its appearance under light microscopy as mitos (thread) and khon (grain). The morphological dynamism of fusion and fission was acknowledged soon after the discovery.23 Mitochondria are essential cellular organelles, and almost all mammalian cells require mitochondria for their energy production, electron transport chain, cellular signaling, calcium homeostasis, precise functioning, survival, and programmed cell death. MD is crucial for mitochondrial quality controls because (1) MD is directly implicated in the maintenance of mitochondrial integrity24 and turnover,25 (2) MD is involved in the electrical and biochemical connectivity within the cells,26 (3) MD protects and redistributes mitochondrial DNA (mtDNA) during oxidative stress responses and mitotic division,27 and (4) MD also orchestrates the apoptosis pathways.28,29
In a healthy living cell, fluorescence-labeled mitochondria under time-lapse microscopy can be observed as dynamic organelles constantly undergoing cycles of fusion and fission.30 During mitochondrial fission, one mitochondrion is divided into two daughter mitochondria, and the mitochondrial contents are reallocated to generate heterogeneity as well as to eradicate damaged mitochondria. Conversely, during the fusion event, two individual mitochondria merge into one single mitochondrion, and the inner and outer mitochondrial membranes of two mitochondria fuse and allow the mixing and redistribution of the matrix contents as well as mtDNA and prepare the host cells for differentiation and development.13,30 Under normal physiology, the frequencies of fusion and fission events are balanced and mitochondrial shape and number are maintained at equilibrium for the optimal functioning of the cells.30 However, when this balance is disrupted by either endogenous (increase oxidative stress, disturbance in mitochondrial membrane potential, and decrease ATP production) or exogenous stimuli (oncogenic substance, irradiation, environmental toxins), the equilibrium of MD shifts toward mitochondrial fission.31
MD in the heart
With the recent advances in mitochondrial imaging technology, scientists have observed that mitochondrial morphology is highly dynamic and the morphological dynamics show cell types as well as tissue specificities, which are thought to be determined by specific cellular functions or demands.32 For example, in neurons and pancreatic cells, for neurotransmission and endocrine function, mitochondria are constantly making oscillatory movements and fast long-distance movements such as mitochondrial branching, filament extension, retraction, and intracellular translocation.26,33 However, in adult cardiomyocytes and skeletal muscles, to provide bioenergy for muscle contraction, the mitochondrial localization within the cell is relatively stable and well organized.34,35 In addition, mitochondria distribution within a cell varies depending on the cell type; for instance, in neuronal cells, mitochondria are recruited to the pre- and post-synaptic nerve endings, which are areas of high energy demand.36 MD can also alter the cell cycle state. During the S phase of DNA synthesis, mitochondria undergo a high degree of fusion,37 whereas during the M phase of mitosis, mitochondria demonstrate heavy fragmentation.38 Mammalian heart tissue is composed of cardiac myocytes (CMs), cardiac fibroblasts (CFs), vascular smooth muscle cells (VSMCs), and endothelial cells.39 Studies indicate that mitochondrial distribution, function, and morphology in these cells are dissimilar.17,40, 41, 42
Cardiac myocytes are the major functioning units and constitute 30%–40% of the total cell population and account for 70%–80% of the heart volume in the mammalian heart.39 In adult cardiac myocytes, mitochondria are localized in interfibrillar, subsarcolemmal, and perinuclear regions. Mitochondria in the interfibrillar region are uniform in size and shape in order to align with myofibrils for Ca2+ release,40 while in the subsarcolemmal region, due to their implication in the metabolite and electrolyte transport and their involvement in nuclear transcription in the perinuclear region, they are less organized and show greater variation in size and shape.41,42 Disbalance in MD in cardiac myocyte can lead to impairment in mitochondrial respiration, biogenesis, and mitophagy, and it can ultimately lead to cardiomyopathy.43
Cardiac fibroblasts are the most abundant cell type, accounting for 60%–70% of the total cell population in the mammalian heart.39 Cardiac fibroblasts can be differentiated into myofibroblasts and are essential for the maintenance of the extracellular matrix. They are reactive to mechanical and hormonal stress, and changes in MD can affect their function.44 Studies demonstrate that inhibition of mitochondrial fusion in cardiac fibroblasts can impair their oxidative phosphorylation capacity, leading to reduced glucose oxidation and oxygen consumption and subsequently decreased mitochondrial biogenesis.45,46
VSMCs are responsible for the proliferation and production of matrix components of the blood vessel wall to maintain the vascular tone, thereby influencing blood pressure and blood flow to the heart. These differentiated VSMCs have the contractile phenotype, characterized by slight proliferation and minimal secretion of extracellular matrix and specific contractile proteins such as muscle myosin heavy chain, smooth muscle α-actin, and calponin.47 However, upon exposure to endogenous and exogenous stressors, VSMCs are damaged and the differentiated VSMCs are transformed into a synthetic phenotype in which the cells proliferate and migrate toward the injury site.48 The proliferation of VSMCs is induced by several stress-related growth factors such as platelet-derived growth factor (PDGF), insulin-like growth factor 1 (IGF-1), angiotensin II (Ang II), endothelin 1 (ET-1), and transforming growth factor β (TGF-β).49, 50, 51 Studies have shown that the growth factor-induced VSMC proliferation is linked to mitochondrial fragmentation.50,52,53 Notably, the elevation of these growth factors and increased mitochondrial fission are detected in several cardiovascular diseases such as pulmonary hypertension,53 atherosclerosis,54 and cardiac hypertrophy.55
Molecular mechanisms of MD
Mitochondrial fusion machinery
Mitochondrial fusion and fission are controlled by a set of fusion and fission proteins. The mitochondrial fusion protein was first identified in 1997 in Drosophila melanogaster, where fusion factor fuzzy onions (Fzo), an outer mitochondrial membrane (OMM) GTPase, is required for the fusion of mitochondria in spermatogenesis.56 To date, the molecular mechanism of the mitochondrial fusion and fission process is well studied in yeast.24,57,58 Fusion and fission proteins in yeast are also found to be conserved in many other species, and functional homologs are identified in worms,59,60 flies,61 and mammals.62,63 We summarize the different fusion and fission proteins and their reported functions to date in Table 1.
Table 1.
Functions and mechanisms of mitochondrial fusion and fission modulator proteins
| Yeasts | Flies | Rats/mice | Worms | Humans | Location | Mechanism | Ref. |
|---|---|---|---|---|---|---|---|
| Fusion | |||||||
| fzo1p | Fzo | Mfn1/2 Fzo1b/1a | fzo-1 | MFN1/2 | OMM | is involved in tethering of OMM | 24,64,65 |
| mgm1p | Dmel/ Opa1 | Opa1 | eat-3 | OPA1 | IMM | is required for the oligomerization of ATP synthase uniting the IMM | 24,64,66 |
| ugo1p | – | – | – | – | OMM | interacts with Fzo1p and Mgm1p | 68 |
| msto1 | Msto1 | Msto1 | msto-1 | MSTO1 | cytosol, OMM | interacts with mitochondrial fusion proteins as a soluble factor at the cytoplasm-mitochondrial outer membrane interface | 95,96 |
| gdap1 | Gdap1 | Gdap1 | gdap-1 | GDAP1 | OMM, cytosol | is essential for maintenance of the mitochondrial network in a Drp1-dependent manner | 104,105 |
| – | – | Romo1 | – | ROMO1 | IMM | is essential for OPA1 oligomerization | 97 |
| Pld | – | mitoPLD/ Pld6 | – | mitoPLD | MM | converts cardiolipin into PA, which interacts with Opa1 and Mfn | 71, 72, 73 |
| – | PAPLA1 | PA-PLA1 | – | PA-PLA1 | MM | hydrolyzes PA, which can interact with Mfn1 and counteract with mitoPLD and Drp1 | 71,73, 74, 75 |
| CL | – | Crl | – | CL | MM | interacts with Opa1 to induce mitochondrial fusion; can also interact with Drp1 to promote fission | 71,75,76 |
| Fission | |||||||
| Dmn1p | Drp1 | Dnm1l/ Drp1 | drp-1 | DMN1L/ DRP1 | cytosol, OMM | is the major component of the fission complex | 83, 84, 85, 86,89 |
| Fis1p | Fis1 | Fis1 | fis-1/2 | hFIS1 | OMM | binds with Mdv1p to form the Fis-Mdv1p complex | 57,78,79,83,84 |
| Mdv1p | – | – | – | – | OMM, cytosol | possesses an N-terminal Fis1-binding site and a Dmn1 interaction domain | 57,79,264 |
| Caf4p | – | – | – | – | OMM, cytosol | can directly bind to Fis1 and Caf4 to from a fission complex with Dnm1 and Fis1 | 57,79 |
| Num1p | – | – | num-1 | – | MECA | interacts with Drp1 and Mdm36p | 80 |
| Mdm36p | – | – | – | – | MM | regulates Num1p and Dnm1p interaction | 81 |
| Mdm33p | – | – | – | – | IMM | contributes as a component of the mitochondrial fission complex | 82 |
| – | – | Mff | mff-1/2 | MFF | OMM, peroxisome (in CE) | serves as a Drp1 adaptor | 85,86,88 |
| dnm2 | Dyn2 | Dnm2 | Dnm-2 | DNM2 | centrosome, microtubules, MM | catalyzes membrane fission during clathrin-mediated endocytosis (CME) | 90, 91, 92 |
| MiD49 | – | Mief2 | – | MIEF2 | OMM | serves as a Drp1 receptor to promote the association of Drp1 to Fis1 and Mff | 84,88,89 |
| MiD51 | – | Mief1 | – | MIEF1 | OMM | functions similar to MiD49 | 84,88,89 |
| – | March5 | March5/Marchf5/Mitol | – | MARCH5/MITOL | OMM, ER | ubiquitinates Fis1, Dmn1L, and Mfn1/2 | 101,102,265,266 |
| – | mtp-18_1 | Mtfp1/ Mtp18 | mtp-18 | MTFP1/MTP18 | IMM | regulates mitochondrial fission by interacting with Drp1 | 99,148,165,267 |
| Inf2 | Inf-2 | Inf2 | Inf-2 | INF2 | cytoplasm | mediates actin polymerization at ER-mitochondria intersections | 103 |
| – | dSLC25A46 | Slc25a46 | slc25a46 | SLC25A46 | OMM | is involved in cristae maintenance and promotes fission | 106 |
OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane; MM, mitochondrial membrane.
The structural features of mitochondrial fusion proteins are illustrated in Figure 1A. In yeast, mitochondrial fusion is medicated by two core GTPases: Fzo1, which is localized in the OMM, and mgm1, localized in the inner mitochondrial membrane (IMM).64 Fzo1p is responsible for OMM fusion, and mitofusins (MFN1 and MFN2) are its mammalian homologs.24 Mitofusins are double-membrane spanning large GTPases, having cytosolic and mitochondrial domains. They are involved in tethering of the OMM in both cis (also known as homo-oligomeric, i.e., mitofusins of the same mitochondria) and trans (hetero-oligomeric, i.e., mitofusins from different mitochondria) manners.65 The mitochondrial genome maintenance protein 1 (Mgm1p) is a dynamin-related protein and is crucial for IMM fusion. As the name suggests, mgm1 is also involved in the maintenance of mtDNA and cristae morphology.66 Opa1 (optic atrophy 1) is the mammalian ortholog of Mgm1p.24 In yeast, another fusion protein is Ugo1, which resides in the OMM and interacts with Fzo1p and Mgm1p during mitochondrial fusion.67 No mammalian homolog for Ugo1 has yet been identified.
Figure 1.

Schematic representation of the structures of mitochondrial dynamics (MD) proteins (MDPs) and their roles in the mitochondrial dynamics pathway
(A and C) Structural features of mitochondrial fusion (A) and mitochondrial fission (C) proteins. (B and D) Summary of steps involved in the mitochondrial fusion (B) and fission (D) processes. TM, transmembrane; HR1, heptad repeat 1; HR2, heptad repeat 2; GED, GTPase effector domain; PR, proline rich; HS, hydrophobic segment; BSE, bundle signaling element; CC, coiled-coil; RR, repeat region; PH, pleckstrin homology; NTD, nucleotidyl transferase domain; TPR, tetratricopeptide repeat; WD, tryptophan (W) and aspartic acid (D).
Opa1 is a large GTPase IMM protein composed of four domains, namely the transmembrane (TM) domain, the GTPase domain, the middle domains, and the GTPase effector domain (GED), of which the GTPase domain and GED are responsible for GTP hydrolysis, and the roles of the other domains are yet to be defined.68 In addition to regulating mitochondrial fusion, Opa1 is involved in the regulation of cristate stability and apoptosis. Mechanistically, Opa1 maintains mitochondrial cristae stability and enhances mitochondrial respiration, which decreases mitochondrial dysfunction, cytochrome c release, and reactive oxygen species (ROS) production, thereby preventing apoptosis.69 The two mitofusins, Mfn1 and Mfn2, are OMM large GTPases and are essential for OMM fusion. Although the amino acid sequences of these two mitofusins show 80% similarity with those of humans, their structure and mechanism of action are quite different. Both Mfn1 and Mfn2 have one N-terminal GTPase and one heptad repeat (HR)1, two TM, and one C-terminal HR2 domains; however, Mfn2 has an extra proline-rich (PR) domain.68 The HR1 and HR2 regions are involved in tethering of the OMM,68 and the PR domain is thought to be involved in protein-protein interactions and may play a role in intracellular signaling.70 Mitochondrial phospholipase D (mitoPLD), a member of the C-anchored phospholipid D family, can promote mitochondrial fusion by converting cardiolipin into phosphatidic acid (PA) in the OMM, and the resulting PA can interact with Opa1 and Mfn in mitochondrial fusion machinery.71, 72, 73 PA, a cone-shaped phospholipid, can prevent mitochondrial fission and promote mitochondrial fusion by interacting with Mfn1 and counteracting with mitoPLD and Drp1. PA-preferring phospholipase A1 (PA-PLA1) hydrolyzes PA and regulates mitochondrial fusion.71,73, 74, 75 Cardiolipin (CL) can then bind with Opa1 in IMM to induce mitochondrial fusion. CL is derived from PA, which is directly transferred from the endoplasmic reticulum (ER) to the IMM. A small portion of CL can be located to the OMM with the help of mitoPLD.71,75,76
The mitochondrial fusion event can be summarized as a five-step process (Figure 1B) as follows: (1) OMM tethering of two opposing mitochondria, which is mediated by the interaction between the HR domain of one Mfn with the GTPase domains of another Mfn; (2) conformational changes in Mfns: GTP hydrolysis facilitates conformational change in MFNs by allowing HR1 and HR2 to come together, leading to MFNs dimerization; (3) OMM fusion: GTP hydrolysis results in GTP-dependent oligomerization of MFNs; (4) long (L)-Opa1-CL tethering: this is driven by the interaction between L-Opa1 and CL on either side of the membrane to tether the two IMMs via Opa1-dependent GTP hydrolysis; and (5) IMM fusion: short (S)-Opa1 enhances L-Opa1-CL interaction, and fusion of the two opposing membranes is achieved.68,77
Mitochondrial fission machinery
The structural features of mitochondrial fission proteins are demonstrated in Figure 1C. In yeast, the core mitochondrial fission protein is Dnm1p (dynamin-related protein or dynamin-1-like protein), which is generally located in the cytosol. Mitochondrial fission requires the recruitment of Dnm1p to the OMM.78 Dnm1p recruitment is mediated by three proteins, including one outer membrane fission protein, Fis1p, and two fission adaptor proteins, Mdv1p (mitochondrial division protein 1) or Caf4 (CCR4-associated factor 4). Fis1p is a mitochondrial integral outer membrane protein and can directly bind to Dnm1p and is an essential component of the fission complex. The binding of Fis1p to Dnm1p is facilitated by the presence of one molecular adaptor, either Mdv1p or Caf4p.57 Mdv1p possesses an N-terminal Fis1-binding site and a Dmn1 interaction domain. Caf4p can also directly bind to Fis1, but unlike Mdv1, Caf4p is a bona fide fission protein that Caf4 alone is sufficient to form a fission complex with Dnm1p and Fis1p without Mdv1p.79 Num1p is involved in mitochondrial fission by interacting with Dnm1p and Mdm36p, and it is essential for nuclear migration and the accurate distribution of the mitochondrial network.80 Mdm36p regulates Num1p and Dnm1p interaction to form a Num1p-Mdm36p complex for mitochondrial localization of Dnm1p during mitochondrial fission.81 Mdm33p is involved in IMM mitochondrial fission by contributing as a component of the mitochondrial fission complex.82 Dnm1p was first described in yeast78 and C. elegans60 as a crucial mitochondrial fission protein, and it was found to be evolutionally conserved in mammals.83 However, no homologs for Mdv1p and Caf4p have been identified in mammals so far.
Drp1/Dnm1L is a large GTPase and the mammalian homolog of Dnm1p. It is structured with the amino-terminal GTPase domain, the middle domain, the variable B-insert domain, and the carboxyl-terminal GTPase effector domain. Drp1 is a cytosolic protein; however, during mitochondrial fission, it is recruited in the OMM and peroxisomal membrane and forms a ring-like structure around the mitochondrial membrane, leading to the constriction of the mitochondrial membrane. The recruitment of Drp1 to the OMM is mediated by multiple signaling pathways, including mitochondrial remodeling, mitophagy, mitosis, and apoptosis. Similar to other dynamin proteins, Drp1 contains BSEs (bundle signaling elements), which are responsible for connecting the GTPase domain with the stalk domain (middle domain), allowing oligomerization in a GTPase-dependent manner.83 Mitochondrial constriction is mediated by the middle domain-forming Drp1-oligomeric helices marking at the two different points of the membrane. Drp1 requires Fis1 for its mitochondrial localization to undergo mitochondrial fission. Drp1 cannot directly bind to phospholipid membrane due to lack of a membrane-binding PH (pleckstrin homology) domain at the C-terminal. Similar to yeast Dnm1p, Drp1 also requires adaptor proteins for its recruitment on OMM. In mammals, four integral proteins, including mitochondrial fission 1 (Fis1) protein,84 tailed-anchor protein mitochondrial fission factor (Mff),85,86 MD proteins (MDPs) of 49 and 51 kDa (MiD49 and MiD51),84,87 function as receptors for Drp1 recruitment on OMM.
Fis1 (mitochondrial fission 1 protein) possesses an N-terminal NTD (maintained in mitochondrial localization), followed by a C-terminal TM domain (distributed to the cytosol).78 Dnm2/Dyn2 (dynamin-2) has an N-terminal GTPase, followed by the middle domain, PH domain, GED, and C-terminal PR domain. With reagard to Mff (mitochondrial fission factor), its N-terminal has two RR domains, followed by CC and C-terminal TM domains where it is attached to mitochondria. The N-terminal residues containing R1 and R2 motifs are responsible for Drp1 recruitment and Drp1-Mff interaction.85,88 Mff serves as a Drp1 adaptor, promoting the recruitment and association of Drp1 to the mitochondrial surface.85,86,88 MiD49/MIEF2 has an N-terminal TM domain that is attached to the OMM, followed by the NTD domain. It serves as a Drp1 receptor in the OMM to regulate mitochondrial organization. It is required for mitochondrial fission and promotes the recruitment and association of Drp1 to the mitochondrial surface independently of Fis1 and Mff.84,88,89 With regard to MiD5/MIEF1, its structure and functions are similar to those of MiD49.84,87,88 In addition to Drp1, Dyn2 is another member of the conventional dynamin family that regulates mitochondrial fission. Dyn2 and Drp1 have structural differences and thus exhibit different functional behavior in the mitochondrial fission machinery. In particular, Dyn2 has a PH domain important for membrane insertion and a PRD responsible for mitochondrial localization, while Drp1 lacks both domains. Hence, Drp1 requires adaptor proteins for mitochondrial fission, whereas Dyn2 can undergo mitochondrial fission without adaptor proteins.90,91 Dyn2 is shown to complete the Drp1-mediated mitochondrial fission initiated at membrane tubule constriction. It can catalyze membrane fission during clathrin-mediated endocytosis (CME), which is critical for cell signaling and survival.90, 91, 92 Intriguingly, Dyn2 was found to be transiently localized in the mitochondrial constriction sites, while Drp1 was more abundant. In addition, after mitochondrial fission, Drp1 is segregated to both daughter mitochondria, whereas Dyn2 tends to appear in only one of the daughter organelles.90,92
Mitochondrial fission is a multi-step process that can also be summarized into five stages (Figure 1D). (1) OMM pre-constriction: in replication of mtDNA before mitochondrial division, the mtDNA in the mitochondrial matrix replicates and marks the site for ER recruitment. (2) IMM pre-constriction: the mitochondria-ER contact site can induce IMM constriction in a Ca2+-dependent manner prior to the Drp1 oligomerization. (3) Oligomerization of Drp1 occurs at the site of pre-initiated constriction mediated by mitochondria-ER contact. Drp1 recruitment at this site is mediated by adaptor proteins Fis1, Mff, MiD49, and MiD51, resulting in oligomerization. (4) GTPase hydrolysis induces the conformational change and induces the OMM constriction. (5) Dnm2 mediated disassembly: Dnm2 is recruited to the Drp1-oligomerized constriction site and terminates the scission process, resulting in two daughter mitochondria. However, the mechanism by which the fission process is disassembled is not yet fully understood.
Fusion and fission coordinators in MD
MD is regulated by organellar interactions that allow the assembly of fusion and fission proteins. Microdomains on the OMM are formed by the interaction of mitochondrial dynamic proteins with the ER.93 In these microdomains, activated Drp1 is brought into proximity and binds non-GTPase partners, such as Fis1, Mff1, MiD49, and MiD51, creating the multimeric fission assembly that divides the organelle.84 Drp1 is a cytosolic protein, and during cellular stress only 3% of Drp1 is translocated to the OMM and participates in mitochondrial fission,94 demonstrating that elevation of Drp1 expression alone is not a sufficient indicator of mitochondrial fission; instead, the translocation and assembly of Drp1 with its adaptor proteins are more important contributors to the fission process.22 Msto1 (misato homolog 1) is a cytoplasmic protein that interacts with mitochondrial fusion proteins as a soluble factor at the cytoplasm-OMM interface and it is essential for OMM fusion.95,96 In HeLa cells, silencing Msto1 without changes in Mfn1, Mfn2, Opa1, and Drp1 expression induces mitochondrial fission and impaired mtDNA distribution.95 This finding is supported by the evidence that patients with Msto1 mutation show a significant reduction in mitochondrial fusion, which is associated with conditions such as myopathy, ataxia, and neurodevelopmental impairments.95,96 In mammals, Romo1 (ROS modulator 1) is essential for Opa1 oligomerization in the redox-depending mitochondrial fusion pathway and normal cristae conformation. Impaired Romo1 expression prevents Opa1-mediated fusion and induces mitochondrial fission under oxidative stress conditions.97 CL can regulate both mitochondrial fusion and fission processes; it not only interacts with Opa1 in the IMM to induce mitochondrial fusion, but it can also interact with Drp1 in the OMM to promote mitochondrial fission.71,75
Mtp18 (mitochondrial 18-kDa protein) or Mtfp1 (mitochondrial fission process protein 1) is an IMM protein that is involved in the regulation of mitochondrial fission and apoptosis in cardiomyocytes, cancer, and neuronal cells.98, 99, 100 We have reported that it can induce Drp1-mediated mitochondrial fission in gastric cancer cells upon doxorubicin (DOX) exposure, promoting the anticancer effect of DOX.99 Mitol (a mitochondrial E3 ubiquitin-protein ligase) and March5 (a mitochondrial E3 ubiquitin-protein ligase) ubiquitinate Fis1, Dnm1L, and Mfn1/2 and play a crucial role in the control of mitochondrial fission.101,102 Mitol is also involved in the regulation of mitochondrial quality control and cellular senescence by blocking Drp1-mediated mitochondrial fission and promoting Mfn1-mediated mitochondrial fusion.102 INF2 (inverted formin 2) is an ER-associated fission protein, mediating actin polymerization at ER-mitochondria intersections and facilitating Drp1 recruitment to mitochondria, which is a critical step in mitochondrial fission.103 Gdap1 is located in the OMM and it is essential for the maintenance of the mitochondrial network by involving in mitochondrial fragmentation in a Drp1-dependent manner. The mitochondrial fission activity of Gdap1 is interfered by mutation in Drp1 (K38A) or counteracted by the mitochondrial fusion proteins such as Mfn1 and Mfn2.104,105 SLC25A46 (solute carrier family 25, member 46) can regulate MD and cristae maintenance by promoting mitochondrial fission and preventing the formation of mitochondrial hyperfilamentation.106
MD and cellular processes
The concept that alterations in mitochondrial morphology may influence mitochondrial function was first described in 1988 even before the discovery of the first mitochondrial-shaping proteins. Under normal physiological conditions, the frequencies of fusion and fission events are balanced, and mitochondrial shape and number are maintained at equilibrium for the optimal functioning of the cells.30 However, when this balance is dramatically changed by either endogenous (increase in oxidative stress, disturbance in mitochondrial membrane potential, and decrease in ATP production) or exogenous (oncogenic substance, irradiation, environmental toxins) stimuli, the equilibrium of MD shifts toward mitochondrial fission.31 Fusion enables the exchange of mitochondrial substances such as proteins and mtDNAs, while fission allows the removal of damaged mitochondria via mitophagy and mitochondrial biogenesis. MD is directly linked to mitochondrial function, and dysfunctional MD can influence a wide range of cellular processes, including mitochondrial biogenesis,107 energy metabolism,42 mtDNA maintenance,108 mitochondrial quality control,109 ROS production,110 Ca2+ signaling,111 cell cycle and stem cell regulation,112 mitophagy,19 autophagy,113 and apoptosis.114
MD and its quality control
Both fusion and fission are essential for the maintenance of the normal functioning of mitochondria. Mitochondrial fusion can reduce cellular oxidative damage, reducing protein oxidation, and mtDNA mutations via a process called complementation. However, when the oxidative damage is excessive, mitochondrial fission is activated and the impaired daughter mitochondria with oxidative damage are removed through mitophagy.115 Pink1 and Parkin are essential for the recruitment of damaged mitochondria in mitophagy. Mutation in Parkin interferes with the degradation of damaged mitochondria and leads to Parkinson disease.116 Disturbance in MD impairs the elimination of damaged mitochondria, leading to progressive injury of the myocardium and heart failure.43 A recent study indicates that parkin-mediated ubiquitination can mark the mitochondria for mitophagy elimination and could be a novel therapeutic strategy to target mitochondrial quality control in cardiovascular diseases, including heart failure and diabetic cardiomyopathy.109
MD and its ATP production
In mammals, mitochondria are the major ATP production machinery in all cell types; therefore, dysfunction in MD can directly influence mitochondrial ATP production. For instance, when Drp1 is depleted in HeLa cells transfected with Drp1 small interfering RNA (siRNA), a reduction of approximately half (44%–54%) of ATP levels in Drp1-depleted cells is observed compared to untreated control cells. Preventing mitochondrial fission by depleting Drp1 expression with short hairpin RNA (shRNA) in HeLa cells led to mitochondrial dysfunction evidenced by increased ROS production, loss of mtDNA, impaired ATP generation, and thus prevented the cells from undergoing mitotic proliferation and autophagy.117 Both clinical studies and animal models showed that progression to heart failure is linked to dysfunction in ATP production and energy depletion. Neubauer118 observed that heart failure patients have reduced concentrations of phosphocreatine, which leads to dysfunction in metabolic homeostasis and ATP production, thereby resulting in energy depletion and disease progression. In addition, growing studies demonstrate that alterations in MD can impact glucose homeostasis and cardiomyocyte metabolism. Oma1 is an enzyme involved in the Opa1 proteolytic process and is crucial for the normal functioning of Opa1119,120 and mitochondrial health.121 Homogeneous Oma1 knockout (Oma1−/−) mice show impaired Opa1 function and reduced mitochondrial fission, resulting in increased insulin resistance and defective mitochondrial glucose homeostasis, as well as altered thermogenesis.122 The phenotype of Oma1−/− mice is similar to those seen in high-fat feeding mice. Recently, Parra et al.123 have indicated that insulin treatment in cardiomyocytes upregulated Opa1 levels and induced mitochondrial fusion, thereby enhancing oxygen consumption and ATP production, demonstrating the role of MD in glucose metabolism and maintaining mitochondrial heath.
MD and mtDNA
The mitochondrial genome of each mitochondrion is organized into one or more nucleoids after a fusion event; one study reported that these nucleoids are motile and have potential interactions.124 Staining of mtDNA nucleoids with anti-DNA antibodies indicated that mtDNA nucleoids are distributed throughout the entire nuclear network in a healthy cell.117 Increasing studies indicate that MD can regulate the distribution and stability of mtDNA nucleoids. For instance, loss of function of mitochondrial fission protein Drp1 can result in severe mtDNA nucleoid clustering and loss of mtDNA.117,125 One recent study reported that increased mitochondrial fission by Drp1 overexpression triggers the release of mtDNA into the cytosol, resulting in cytosolic mtDNA stress and impaired mitochondrial respiration activity in hepatocellular carcinoma (HCC) cells.126 In contrast, disruption of mitochondrial fusion leads to mtDNA instability. For example, conditional deletion of mitofusin (Mfn1 and Mfn2) in skeletal muscle results in rapid accumulation of point mutations and deletions in the mtDNA genome.127 Additionally, lack of Mfn2 in mammalian cells can result in loss of mtDNA nucleoids.128 However, the question as to how mtDNA responds to MD remains unclear. Especially, whether individual mtDNA recombines or to what extent it can interact with each other as well as the impact of these interactions remain elusive, and hence, more research work is required to address these issues.
MD and metabolism
As we mention above, MD can regulate mitochondrial functions. The electrical interconnection created by the long mitochondrial filaments in fibroblasts and mitochondrial clusters in cardiomyocytes allows the movement of energy throughout the myocardium. Interruption in MD from either over-fusion or over-fission can result in impaired mitochondrial function. For example, decreased expression of the fusion mediator Mfn2 is associated with a decrease in substrate oxidation, mitochondrial respiration, and membrane potential, resulting in impairment of mitochondrial energy metabolism followed by increased anaerobic glycolysis with decreased glycogen synthesis.129 Similar phenotypes have also been reported for models with Opa1 gene silencing.45 Also, rat and human models of heart failure showed that Opa1 expression is decreased with increased mitochondrial fragmentation.130 Interestingly, Chen et al.45 indicated that increased expression of Opa1 was not able to improve mitochondrial energy metabolism. Likewise, decreased expression of mitochondrial fission factor Drp1 also negatively impacts mitochondrial metabolism. However, very few studies have investigated the intrinsic role of the changes in MD and their potential relationship with metabolic changes in the hearts of patients with cardiac hypertrophy and heart failure.
MD and mitophagy
Mitophagy is a balancing process between biogenesis and degradation. During this process the damaged or senescent mitochondria are selectively removed to maintain mitochondrial quality control and mitochondrial function. The process of mitophagy consists of three stages: (1) recognition of damaged mitochondria, (2) formation of mitochondrial phagocytic vesicles, and (3) mitolysosome formation by the fusion of mitochondrial phagocytic vesicles and lysosomes. Mitophagy is regulated by the Parkin-dependent pathway and non-canonical parkin-independent pathways, which are mediated by other ubiquitin ligase-mediated, receptor-mediated, or lipid-mediated mitophagy.19 Parkin is a cytosolic protein, and in intact mitochondria it is imported to the IMM and constantly degraded by matrix processing peptidase and presenilin-associated rhomboid-like (PARL) proteins. However, in damaged mitochondria, it is translocated to the OMM by Pink-1-mediated recruitment. The recruited parkin on the surface of damaged mitochondria is able to ubiquitylate MD proteins such as Mfn1 and Mfn2, voltage-dependent anion channel (VDAC), and mitochondrial Rho GTPase 1 (MIRO1) to promote the removal of damaged mitochondrial by lysosomes,19 yet the role of parkin in normal cardiac functioning remains controversial. A study by Billia et al.131 supported that the lack of Pink1 causes increased oxidative stress and can speed the development of left ventricular failure in pathological cardiac hypertrophy, while Kubli et al.132 observed that Parkin-deficient mice show altered mitochondrial morphology and increased susceptibility to ischemia but can maintain myocardial function. The counterregulatory mechanism reserving myocardial function is explained by Dorn and colleagues133 that in Parkin knockout murine hearts, the compensatory upregulation in multiple Parkin-related E3 ubiquitin ligases of RING families can normalize the impairment in mitochondrial morphology and cardiac function by suppressing the mitochondrial fusion, which is mediated in dilated cardiomyopathy.
Increasing studies indicated that changes in MD can affect mitophagy. Both mitochondrial fusion proteins such as Opa1, Mfn1, and Mfn2 and fission factors such as Drp1, Mff, and MIEFs can regulate mitophagy. In Mfn2-deficient mouse cardiac myocytes, an impairment in Parkin-mediated recruitment of damaged mitochondria can induce progressive cardiomyopathy.134 In normal mouse hearts, the expression levels of both parkin mRNA and protein are minimal; however, in cardiac-specific Drp1 knockout mice, Parkin expression is upregulated and parkin-dependent mitophagy is overactivated, leading to severe cardiomyopathy.135 Additionally, Drp1 deficiency can induce mitochondrial elongation and prevent mitochondrial autophagy, leading to cardiac dysfunction and an increase in susceptibility to cardiac injury.136 Furthermore, mitophagy is involved in the exacerbation of compensatory cardiac hypertrophy to heart failure. Drp1 receptors such as Fis1, Mff, and MIEFs also play an important role in mitophagy. The MOM-anchored protein called FUNDC1 can regulate mitophagy through mitochondrial fission pathways by interacting with Drp1 or Opa1. The increased association between FUNDC1 and Drp1 leads to a competitive reduction in its interaction with Opa1, resulting in increased mitochondrial fission and mitophagy.137 The depletion of Fis1 inhibits mitophagy and prevents myeloid differentiation, leading to cell cycle arrest and loss of self-regeneration in human leukemia stem cells.138 Interestingly, Fis1 can also regulate mitophagy in Parkin-independent mechanism by controlling the mitochondrial recruitment of STX17, which can regulate the localization and activation of Drp1.139 In the parkin-independent pathway, mitophagy is regulated by multiple other E3 ligases such as Mitol, Mul1, Arih1, or by mitophagy-specific receptors such as Bcl2, Phb2, Bnip2, Fudnc1, and Nix, which can recognize damaged mitochondria and recruit autophagosomes via interacting with LC3.19 Collectively, it is clear that crosstalk between autophagy factors and MD proteins are implicated in mitophagy, yet their exact role in the pathogenesis and treatment of cardiac hypertrophy remains poorly understood. Further studies exploring their interaction are required to understand their potential therapeutic role in the treatment and prevention of cardiac hypertrophy and heart failure.
MD and apoptosis
Increasing studies have indicated that mitochondrial fission is implicated in several diseases such as vascular and cardiovascular diseases (CVDs),140,141 neurodegenerative diseases,142,143 cancers,142 and aging144,145 by regulating apoptotic pathways. Apoptosis is a form of programmed cell death or cellular suicide that plays a pivotal role in normal cellular development as well as pathological processes. Bcl-2 protein family members, which include pro-apoptotic (such as Bax, Bak, Bid, Bad, and Bik) and anti-apoptotic (such as Bcl-2, Bcl-xL, and Bcl-W) factors, are the key regulators of apoptosis.19,146 Apoptosis is activated through intrinsic pathways (which are activated by oxidative stress, cytochrome c, endoplasmic reticular stress, and increased intracellular calcium iron) and extrinsic pathways (which are mediated through death ligand and receptors by activation of cytokines, cytotoxic drugs, and hormones), and mitochondria are involved in the intrinsic pathways. MD can regulate the apoptotic pathway as a way to remodel the mitochondrial network during cellular stress induced by hypoxia, various forms of cellular pathologies, and even some drug treatments. Studies have shown that mitochondrial fission is linked to apoptosis, while mitochondrial fusion averts it. For instance, the enhanced expression of mitochondrial fission proteins such as Drp1, Fis1, Mff, or MIEF or the depletion of the mitochondrial fusion proteins such as Mfn1, Mfn2, or Opa1 can induce apoptosis.28,114,130 Conversely, the knockdown of mitochondrial fission proteins such as Drp1 or Fis1 and the induced expression of mitochondrial fusion factors such as Mfn1, Mfn2, or Opa1 induce the cell’s resistance to different apoptotic stimuli.114,130,147 In particular, Fis1 is a prerequisite for Bax translocation from the cytosol to mitochondria, and Drp1 is essential for Bax oligomerization and activation.84 Opa1 causes the release of cytochrome c from mitochondrial intermembrane space to the cytosol by regulating cristae remodeling.69 Recently, we found that overexpression of mitochondrial protein 18 (Mtp18), an IMM protein, can induce mitochondrial fission and apoptosis by enhancing dynamin-related protein 1 (Drp1) accumulation in cardiomyocytes. However, when Drp1 was minimally expressed, apoptosis did not occur, indicating that overexpression of Mtp18 alone was not sufficient to execute apoptosis, and that Mtp18 and Drp1 are interdependent in the apoptotic cascade.148 Mitochondrial fission can increase cellular sensitivity to apoptotic stimuli and trigger the release of mitochondrial death factor Smac/Diablo to the cytosol in a MTGM (MIM anchored fission protein)-dependent manner. The Drp1 adaptor proteins MIEF1 and MIEF2 are reported to be the positive regulators of autophagy and apoptosis, and the depletion of MIEF1 can increase cellular resistance to apoptotic stimuli.19 Mitol is found to be a negative regulator of apoptosis. Mitol depletion did not show any significant changes in MD and apoptosis under normal cellular conditions. However, upon exposure to oxidative and cell death stimuli, Mitol triggered the ubiquitin- and proteasome-dependent degradation of MiD49, thereby preventing mitochondrial fission and protecting the cell against stress-induced apoptosis.149 We found that oxidative stress stimuli including hydrogen peroxide and DOX upregulate Mitol expression and that Mitol overexpression can prevent mitochondrial Drp1 accumulation in the cardiomyocytes and subsequently provide protection against mitochondrial fission.150 During apoptotic stimulation, Drp1 is recruited to the OMM and interacts with Bax and Mfn2. However, studies also indicated that mitochondrial hyperfusion can also induce apoptosis. Interestingly, mitochondrial hyperfusion induced by depletion of Drp1/Fis could not prevent cell death upon cellular stress stimuli. In Drp1-deficient cells, mitochondrial hyperfusion still can induce Bax/Bak-dependent apoptosis, and apoptotic mitochondrial fission was observed during apoptosis.19 Collectively, these data suggest that a non-canonical pathway Drp1-independent fission may play an important role in apoptotic mitochondrial fission. Additionally, mitochondrial fission alone may not be sufficient to induce apoptosis, and complex mitochondrial morphogenesis machinery could be involved in apoptosis regulation, which requires further in-depth explanation.
MD in cardiac hypertrophy
Cardiac hypertrophy is characterized by the enlargement of cardiomyocytes; however, the detailed characteristics of cardiac hypertrophy are distinct depending on the pathophysiology. Physiological hypertrophy is characterized by an individual cardiomyocyte depicting a proportional increase in both cardiomyocyte length and width. It often has preserved cardiac contractile function or slightly enhanced contractile function, in some cases without interstitial fibrosis or cell death except for post-natal hypertrophy. Conversely, pathological cardiac hypertrophy during the compensatory phase is often presented as concentric growth of the ventricle. Later, when it progresses to the decompensatory phase, it shows chamber dilatation with wall thickening, where individual cardiomyocytes show a disproportionate increase in length compared to width, leading to contractile dysfunction and heart failure. In addition, the hemodynamic imbalance in pathological cardiac hypertrophy activates the fetal gene program (FGP), resulting in the elevated expression of BNP (brain natriuretic peptide), myosin heavy chain, cardiac muscle β-isoform (MYHCb, MYH7), and skeletal muscle α-actin in pathological cardiac hypertrophy (Figure 2).
Figure 2.

Dysregulation in mitochondrial dynamics during cardiac hypertrophy
(A–D) Key features and mitochondrial dynamic alteration observed in normal (A), compensated (B), and decompensated (C) cardiac hypertrophy, and heart failure (D). (A) Under healthy conditions, the mitochondrial fusion and fission in the heart are in equilibrium. (B and C) During compensated cardiac hypertrophy, an increase in mitochondrial fusion is noted (B), while in decompensated cardiac hypertrophy, an increase in mitochondrial fission is noted (C). (D) Cardiac hypertrophy progressing to heart failure, and extensive mitochondrial fission was induced, leading to cardiac dysfunction and eventually heart failure. (E) Overview of the molecular mechanisms regulating mitochondrial dynamics in cardiac hypertrophy.
Due to high energy demand, the heart is rich in mitochondria. Mitochondria in the adult heart have a heavy load of oxidative phosphorylation and oxidative damage, and, thus, maintaining a healthy mitochondrial network is essential for optimal functioning of cardiomyocytes. Unlike other cell types, mitochondria in fully developed cardiomyocytes show a discrete ovoid shape (not as a connected network as in other cell types), and mitochondrial morphology in adult cardiomyocytes is thought to be less dynamic, yet the balance between fusion and fission is essential for the normal functioning of cardiomyocytes. This notion is supported by the evidence that cardiac-specific knockdown of genes encoding mitochondrial fusion or fission proteins in mammals results in cardiac dysfunction and a threat to life.134,151,152 In contrast, significant fusion and fission events are detected in disease conditions and upon exposure to cellular stress in both beating (animal models) and non-beating cardiac myocytes such as HL-1 and H9c2 cells. Nonetheless, several studies indicate that exposure to extreme stress stimuli can induce abnormal fusion/fission, and several cardiovascular diseases can affect mitochondrial morphology.17,42,141
The presence of mitochondrial morphologic deformity in compensatory cardiac hypertrophy is still controversial. In the spontaneously hypertensive rat (SHR), cardiomyocytes at the phase of compensatory cardiac hypertrophy showed a decrease in mitochondrial fission protein Drp1 and an increase in fusion protein Opa1, indicating that MD is shifting the equilibrium toward fusion to fulfill the cellular burden of high energy demand.153 Mild mitochondrial stress induces a pro-survival response of mitochondrial hyperfusion through upregulating the expression fusion proteins such as the stomatin-like protein 2 (SLP-2), Mfn1, and Opa1, and the process is termed stress-induced mitochondrial hyperfusion (SIMH).154 Alternatively, studies also reported that during compensatory cardiac hypertrophy, no significant changes were observed in mtDNA, mitochondrial contents, and MD.21,155 Hence, the questions as to whether changes in MD only occur in decompensated cardiac hypertrophy and what is the exact molecular mechanism that differentiates the mitochondrial phenotype between compensatory and decompensatory cardiac hypertrophy still require extensive study.
Mitochondrial fusion and cardiac hypertrophy
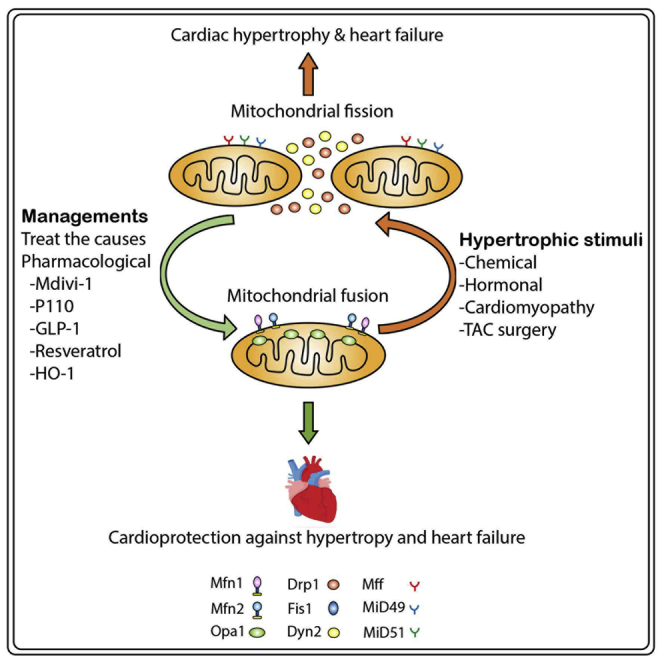
It is well established that mitochondrial fission is involved in the pathogenesis of cardiac hypertrophy and that mitochondrial fusion can prevent cardiac hypertrophy.151,156, 157, 158 Decreased mitochondrial density, upregulation of fission proteins, and downregulation of fusion proteins are the molecular features of a failing heart in mammals.159 For example, mitofusin, also known as hyperplasia suppressor protein, is an essential fusion protein of the OMM component. Mfn1 alone can induce mitochondrial fusion without Mfn2, and cardiac deletion of Mfn2 has shown no significant defect in the normal heart. Embryonic double knockout of Mfn1/Mfn2 was found to be lethal.151 Conditional combined ablation of Mfn1/Mfn2 in adult hearts showed excessive mitochondrial fragmentation and rapid development of lethal dilated cardiomyopathy.151 The Mfn2 expression is downregulated in chemical-induced LVH in neonatal rat cardiomyocytes induced by phenylephrine (PE), and in an in vivo LVH model of spontaneously hypertensive rats, β2-adrenergic transgenic mice, and transverse aortic constriction (TAC).156 Mechanistically, Mfn2 can prevent VSMC proliferation by suppressing extracellular signal-regulated kinase (Erk)1/2 expression, which is implicated in the development of cardiac hypertrophy in both rat and mouse models.156 Likewise, cardiomyocytes incubated with one of the hormonal hypertrophic inducers, Ang II, showed the inhibition of Mfn2 expression and Akt expression that are involved in phosphorylation for Parkin translation and induced mitophagy to maintain mitochondrial quality.157,158 In contrast, overexpression of Mfn2 reversed both chemical- and hormonal-induced LVH in both cellular and animal models.157,158 Cardiac-specific knockout of Mfn2 showed loss of tethering to the ER, which further impaired Ca2+ signaling and enhanced ROS production in cardiomyocytes.134,152
Opa1 is an essential mitochondrial fusion protein and is important for the normal functioning of mitochondria. Life imaging analysis in adult Drosophila indicates that cardiac-specific knockdown of Opa1 induces abnormal mitochondrial morphology and cardiac dilation with significant impairment in cardiomyocyte contractility.160 Downregulation of Opa1 is observed in both human and murine heart failure, and mitochondrial morphological studies in these hearts using electron and confocal microscopy showed small and fragmented mitochondria. MD in the adult heart is maintained by balanced fusion and fission. Notably, both overexpression and knockdown of Opa1 resulted in abnormal MD and induced apoptosis.130 Heterogeneous Opa1 knockout (Opa1+/−) mice showed clusters of the fused mitochondrial network and altered cristae without significant alteration in mitochondrial energy production capacity. However, 6 weeks after the total/TAC procedure, Opa+/− mice experienced a 2-fold higher risk of developing hypertrophy with a significant decrease in ejection fraction compared to wild-type mice.152 In addition, the left ventricle became more susceptible to hypertrophic damage upon TAC when Opa1 was minimally expressed.152 Another study conducted in Opa+/− mice indicated that cardiac function was initially normal; however, the aged Opa+/− mice (12 months old) showed reduced mtDNA copy number and decreased contraction, fractional shortening, and cardiac output. Additionally, the aged OPA1+/− mice showed excessive mitochondrial fission and impaired cardiac function relative to wild-type littermates,161 suggesting that Opa1 dysfunction is associated with late-onset cardiac hypertrophy. These findings indicate the beneficial effects of promoting mitochondrial fusion during cardiac hypertrophy.
Mitochondrial fission and cardiac hypertrophy
Mitochondrial fission is involved in the development of progressive LVH and the development of cardiac failure. Studies indicate a significant correlation between the proliferative phenotype of VSMCs53,162 and cardiomyocytes15,130 with mitochondrial fission. Chang et al.163 has shown that cardiac hypertrophy in cellular (phenylephrine treatment) and animal TAC models induces upregulation of Drp1 expression, which increases mitochondrial fragmentation and activates mitophagy. Drp1 translocates from cytosol to the OMM, where Drp1 docks on its adaptor proteins such as Fis1. In TAC-treated mouse hearts and phenylephrine-treated rat neonatal cardiomyocytes,163 phosphorylation of DRP1 and translocation was detected using phosphoproteomics quantification in myocardial samples at different time points.163 Pennanen et al.15 showed that hypertrophic agonist (norepinephrine [NE]) in neonatal rat cardiomyocytes increases mitochondrial fission with a concomitant decrease in mitochondrial function. Mechanistically, NE acts on α1-adrenergic receptors to increase cytoplasmic Ca2+, which activates calcineurin and promotes Drp1 translation to OMM, facilitating mitochondrial fission.15 Additionally, knockdown of Drp1 using dominant-negative Drp1 (K38A) could prevent mitochondrial fission and cardiomyocyte hypertrophy upon hormonal induction (norepinephrine) of cardiac hypertrophy. Likewise, knockdown of Mfn2 adenoviral-expressing antisense Mfn2 increases mitochondrial fission and activates hypertrophic responses even without NE stimulation.15 Hypertensive cardiac hypertrophy in high salt-fed rats showed increased Drp1 expression and excessive mitochondria fission.164 Pulmonary arterial hypertension (PAH) is one of the most common causes of right ventricular hypertrophy and failure. Pulmonary artery smooth muscle cells obtained from autopsied patients with idiopathic PAH showed increased expression of Drp1 and Fis1 and decreased expression of Mfn2 with an increase in mitochondrial fragmentation compared to those of control patients.53 We have recently reported that Mtp18 or Mtfp1 can bind to Drp1 and facilitate mitochondrial fission in cardiomyocytes98,148,165 under oxidative stress responses. Furthermore, Mtp18 induces the release of cytochrome c, activating the caspase cascade and leading to apoptosis.98,99 We also showed that Mitol could prevent Drp1 accumulation in mitochondria and attenuate mitochondrial fission and apoptosis upon hydrogen peroxide exposure, suggesting that Mitol can play a protective role against oxidative stress-induced cardiac mitochondrial fission and apoptosis.150 Collectively, these data indicate the therapeutic potential of inhibiting mitochondrial fission during cardiac hypertrophy.
MD and heart failure
Pathological cardiac hypertrophy is often followed by heart failure. Impaired mitochondrial function, which is evidenced by decreased mitochondrial oxidative phosphorylation and increased production of ROS, induces the development of heart failure during cardiac hypertrophy.159,166 A recent review on MD in cardiac physiology and pathophysiology emphasized that a significant enhancement in mitochondrial fission and inhibition in mitochondrial fusion were observed in heart failure.167 Increased numbers of fragmented mitochondria are detected in multiple forms of cardiomyopathy42,168,169 and heart failure.42 For example, Chen et al.130 indicated that the expression of the crucial mitochondrial fusion protein Opa1 was decreased in both human and rat models of heart failure. Consistent with the features of reduced fusion, the mitochondria morphologies from the failed heart were small and fragmented. Interestingly, the protein expression levels of Opa1 were significantly reduced in the failed heart, with no change in the expression levels of other MD proteins such as Mfn1, Mfn2, Drp1, or hFis1. Further in-depth analysis showed no significant difference in the Opa1 mRNA expression between normal and failed hearts, suggesting that the reduction in Opa1 protein level is regulated through posttranscriptional modification.130 Notably, they observed a variable expression pattern of MD proteins depending on cardiac pathology. In human ischemia cardiomyopathy, Mfn1, Mfn2, and Drp1 are upregulated but Opa1 is downregulated, with no change in hFis1. In dilated cardiomyopathy, three similar proteins are upregulated with no change in Opa1 and hFis1 levels.130 Alteration in the expression profile of MD proteins is observed when there is progression toward cardiac failure. These findings indicate that a thorough understanding in the regulation of mitochondrial shaping proteins in cardiac hypertrophy and heart failure is crucial for the targeting of MD as an effective therapeutic approach.
Molecular regulation of MD
Cardiomyocytes have relatively sluggish mitochondrial morphological dynamism, which allows the manipulation of MD to achieve a favorable outcome in several cardiovascular pathologies.169 In recent years, extensive studies have been aimed at deciphering the role of MD in a variety of CVDs. Nevertheless, the molecular regulation of mitochondrial fission in the heart remains poorly understood. As we mentioned above, mitochondrial fusion and fission proteins are the key regulators of MD. Regulation of MD proteins can occur at multiple levels including transcriptional, epigenetics, posttranscriptional (which is via non-coding RNAs), and post-translational modification (PTM) (Figure 3).
Figure 3.

Schematic representation of molecular mechanisms regulating mitochondrial dynamics
The left column indicates the mode of molecular dynamics regulation at the genomic level, which includes transcriptional regulation, genetic engineering, mutation, and alternative splicing. The second column demonstrates the epigenetic mechanisms of molecular dynamics regulation. The third column depicts the mechanism of molecular dynamics regulation at the posttranslational level. AD, autosomal dominant; P, phosphorylation; Ub, ubiquitin; SUMO, small ubiquitin-related modifier; OGT, O-linked N-acetylglucosamine (O-GlcNAc) transferase; SNO, S-nitrosylation in serine residue; Ac, acetylation; S1 and S2, two proteolytic cleavage sites to generate a mix of long (L)- and short (S)-OPA1 isoforms. Opa1 possesses two proteolytic cleavage sites (S1 and S2), where their cleavage is mediated by two membrane-bound metalloproteases, OMA1 and YME1L, respectively.
Genetic modifications
-
(1)
Transcriptional regulation. Transcriptional regulation occurs during the process in which genetic information in DNA is converted into RNA. Transcriptional regulation is one of the processes of cellular response to various intra- and extra-cellular signals. During cardiac hypertrophy, studies have indicated that increased hemodynamics or pressure overload in cardiomyocytes modulates the expression of MD proteins. Depending on the number of copies of transcribed RNA, or the temporal control of when the gene is transcribed, the expression level of MD proteins can be varied. The transcription activity is controlled by transcriptional factors that decide whether to activate or repress the transcription. For example, ERRα can bind to the promoter region of mitofusin and transactivate its expression,170 while nuclear factor κB (NF-κB) binds to the promoter region of Opa1 and enhances Opa1 expression.171 Peroxisome proliferator-activated receptor (PPAR)γ coactivator 1 (PGC1-α and PGC1-β) isoforms can coactivate ERRα to enhance the transcriptional activity of mitofusin expression.170 In the adult rodent PAH model, a reduced PGC1-α level downregulates mitofusin-2 expression, leading to reduced mitochondrial fusion and induced hyperproliferation of vascular endothelial smooth muscle, as shown by Marsboom et al.53
-
(2)
Genetic engineering. Genetic engineering, also known as genetic modification or manipulation, is the direct makeup of a MD protein gene using a set of recombination technologies to either increase or knock out the gene expression. Increasing studies indicate that manipulation of MD protein expression during cardiac hypertrophy can induce either a destructive or protective phenotype depending on the target gene function.42,130,152,172,173 As we mentioned above, in general, increased mitochondrial fission or reduced mitochondrial fusion is involved in the pathogenesis of cardiac hypertrophy and heart failure, while reduced mitochondrial fission or increased mitochondrial fusion reduces the progression of cardiac hypertrophy and subsequent heart failure. Indeed, several exciting studies have suggested that inducing mitochondrial fusion along with preventing mitochondrial fission during cardiac hypertrophy do improve cardiac dysfunction and provide a favorable outcome,42,130,152,172,173 suggesting that targeting MD could be a novel therapeutic strategy to combat cardiac hypertrophy and to prevent its progression to heart failure.
-
(3)
Mutation. Mutations in mitochondrial fusion and fission protein-encoding genes have been linked to a variety of diseases. For example, mutations in the Opa1 gene can cause autosomal dominant optic atrophy (ADOA) syndrome,174 as a result of mitochondrial dysfunction and mtDNA loss, leading the cells more susceptible to apoptosis.175 ADOA begins at a young age and slowly progresses to bilateral visual loss, dyschromatopsia, centrocecal scotomas, and temporal optic disc atrophy. However, ADOA shows variable phenotypic expression, ranging from mild visual impairment to blindness.174 Alternatively, mutations in the Mfn2 gene are associated with the Charcot-Marie-Tooth disease type 2A (CMT2A). So far, the phenotypic characteristics of CMT2A remain unclear. CMT2A shows an abnormality in MRI or suggestive CNS and peripheral nervous system (PNS) abnormalities in clinical examination.176 Another example is a novel mutation in Drp1 (C452F) at its M domain, which is linked to the development of autosomal dominant dilated cardiomyopathy in the python mouse, as reported by Ashrafian et al.177 The Drp1’s 452 cysteine (C) residue is responsible for molecular interaction and is highly conserved among yeasts and mammals. The homozygous mutation of cysteine to phenylalanine (F) shows a serious reduction in mitochondrial respiratory enzymes and ATP production, resulting in serious mitochondrial function impairment and embryonic lethality.177
-
(4)
Alternative splicing. Alternative splicing events tend to be cell type- and tissue-specific. The alternative splicing event of transcript encoding mitochondrial fusion and fission proteins can give rise to multiple transcript variants that may exhibit different functions. For example, Opa1 is an essential IMM component to undergo mitochondrial fusion; alternative splicing events in the Opa1 gene can give rise to eight transcripts (mRNA) variants, which exhibit variable expression patterns across different tissues depending on cellular context.178 Additionally, in immune cells, alternative splicing of Drp1 results in the Drp1-x01 isoform, which possesses arginine (Arg) residues in alternative exon, which can specifically target the microtubules, contributing to stabilize the microtubules and prevent apoptosis.179 The Drp1 x01 isoform can be phosphorylated by CKD1, which promotes its release from microtubules and facilitates mitochondrial fission.179 Another example is the Opa1 alternate spliced exon (POA1-exon4b), in which the peptide component is embedded in the IMM and involved in nucleoid attachment to promote mtDNA replication and distribution. Knocking down Opa1-exon4b using siRNA showed inhibition in DNA replication and marked alteration in the distribution of mtDNA and nucleoid throughout the mitochondrial network.180 However, studies reporting the splicing events of MD proteins and their role in different tissue are still scarce. It is worth addressing this issue in future studies.
Epigenetic modifications
Epigenetic modifications regulate the pattern of gene expression by altering DNA accessibility and chromatin structure without altering the DNA sequence. Epigenetic modification is heritable181 and dynamic,182 and it can be either long term (e.g., DNA methylation) or short term (e.g., acetylation and histone modification) in response to nutritional, environmental factors, aging, and diseases.183 Growing evidence has supported that epigenetic modification is linked to the pathogenesis of complex cardiovascular diseases.184 We discuss the role of epigenetics in MD during cardiac hypertrophy and heart failure from three aspects, including methylation and histone modification, non-coding RNAs, and PTM.
-
(1)
Methylation and histone modification. In both nuclear and mtDNA genomes, cytidilic nucleotides can be present in three forms, such as cytosine, 5-methylcytosine (5mC), and 5-hydroxymethylcytosine (5hmC). In mammals, 5mC is highly occupied at the CpG (cytosine juxtaposed to guanine separated by phosphate) contexts; the CG-rich regions, or CpG islands (CGIs), are usually present in the promoter regions. The 5mC modification in promoter regions is often associated with transcriptional repression and silencing of gene expression; however, CpG methylation in the gene body is thought to be involved in gene activation, transcription elongation, and alternative splicing.185 A recently reported post-transcriptional gene regulation is N6-methyladenosine (m6A), which is adenosine methylation at position 6 in mRNA. Several recent studies have observed the changes in global m6A levels in cardiac tissues obtained between normal versus stress or disease states. Genome-wide mapping of DNA methylation and histone H3 lysine 36 trimethylation (H3K36me3) between cardiomyopathic and normal human heart showed a differential pattern in methylation and histone H3K36me3 modifications. The differential methylation profile is enriched in CpG islands of promoter, intragenic, and gene body regions, while the differential H3K36me enrichment was observed in coding regions. Also, distinctive epigenome patterns were observed in the cardiac genome of the end-stage failing heart, suggesting the potential role of using cardiac epigenome to track the disease progression and development of heart failure.186 PPAR is a transcription factor that can directly bind to DNA and has a complex molecular mechanism that interacts with epigenetic modulators in response to developmental, environmental, and pathological changes.187 Cardiac pressure overload induces the recruitment of different epigenetic modulators (either histone PTMs, DNA methylation, or posttranscriptional modifications) to the promoter regions of PPAR target genes and mediates metabolic reprogramming, which is involved in the progression of cardiac hypertrophy and heart failure.188 Taken together, these studies highlight that the epigenetic landscape may be the key system orchestrating the gene expression reprogramming in cardiac hypertrophy.189 However, very few studies have addressed the molecular mechanisms involving changes in methylation and acetylation profiles with the development of cardiac hypertrophy and heart failure. More importantly, studies have rarely addressed the effect that changes in methylation and acetylation play on the modulation of MD. Therefore, epigenetics studies focusing on cardiac MD are needed to understand the molecular mechanism of how the epigenome contributes to disease progression and heart failure. Moreover, improvements in genome-wide technologies, as well as mechanistic studies, are needed for a better understanding of the complex epigenetic signatures of cardiac hypertrophy and heart failure.
-
(2)
Non-coding RNAs. Non-coding RNAs are important epigenetic regulators in the process of replication, transcription, translation, chromatin modification, and post-transcriptional control of gene expression in cardiac hypertrophy.190,191 The expression of non-coding RNAs is cell-, tissue-, and disease-specific and found to be dysregulated in cardiac hypertrophy and heart failure.190, 191, 192, 193, 194 miR-29a-3p,195 miR-129-3p,196 and miR-485-5p197 prevent cardiac hypertrophy by inhibiting mitochondrial fission through repressive expression of mitochondrial fission protiens. In contrast, microRNAs (miRNAs) miR-20b198 and miR-153-3p199 induce cardiac hypertrophy by degrading the mitochondrial fusion proteins. Relative to miRNAs, long non-coding RNAs (lncRNAs) tend to be less conserved among species, and the mechanisms by which lncRNAs regulate gene expression and other cellular processes are more diverse and complex. According to current knowledge, lncRNAs can regulate gene expression by acting as structural scaffolds, or as competing endogenous RNA (ceRNA), and can be involved in RNA processing and chromatin modification. Our knowledge regarding the function and mechanisms of circular RNAs (circRNAs) is still in its infancy. So far, it has been shown that circRNAs have two main mechanisms: (1) circRNAs can function as miRNA sponges, in which binding of circRNA to miRNA impairs the repression of miRNA on its target gene expression; and (2) circRNAs can be involved in alternative splicing and serve as a transcriptional regulator or co-factors of RNA-binding proteins. Increasing evidence highlights the potential of miRNAs, lncRNAs, and circRNAs as novel therapeutic targets, as well as diagnostic and prognostic biomarkers for cardiovascular diseases.200 Although several cardiac hypertrophic related non-coding RNAs have been reported so far, very few have been found to be related to cardiac MD. We summarize all recently identified cardiac MD non-coding RNAs with their phenotype, expression pattern, and mechanisms in Table 2.
Table 2.
The expression patterns, functions, and mechanisms of cardiac mitochondrial dynamics-related non-coding RNAs in cardiac diseases
| Name | Disease model | Levels during disease | Effects on disease | Effects on MF | Mechanism | Ref. |
|---|---|---|---|---|---|---|
| miRNAs | ||||||
| miR-499 | cardiac I/R | down | protect | ↓ | suppresses calcineurin-mediated dephosphorylation of Drp1, thereby decreasing Drp1 accumulation | 268 |
| miR-484 | cardiac A/R and I/R | down | protect | ↓ | attenuates the expression of Fis1, which can induce MF | 269 |
| miR-29a-3p | cardiomyocyte hypertrophy | down | protect | ↓ | targets Drp1 and prevents MF and apoptosis | 195 |
| miR-20b | cardiac hypertrophy | up | induce | ↑ | inhibits Mfn2, which can prevent MF and apoptosis | 198 |
| miR-155-5p | acute MI | up | protect | ↓ | targets the Cab39/AMPK signaling pathway and promotes the expression of Mfn2, p53, and p21 and reduces p-Drp1 | 270 |
| miR-140 | cardiac A/R and I/R | up | induce | ↑ | targets Mfn1, which can induce mitochondrial fusion | 147 |
| miR-324-5p | cardiac A/R and I/R | down | protect | ↓ | suppresses Mtfr1 translation and attenuates MF and apoptosis | 271 |
| miR-761 | cardiac I/R | down | protect | ↓ | represses the expression of Mff, which can induce MF and apoptosis. | 272 |
| miR-30 family (miR-30a/b/c) | H2O2-induced stress | down | protect | ↓ | suppresses the expression of p53, which transcriptionally activates Drp1 | 273 |
| miR-361 | cardiac I/R | up | induce | ↑ | targets prohibitin, which can inhibit MF and apoptosis | 274 |
| miR-532-3p | DOX cardiotoxicity | up | induce | ↑ | represses ARC, which can prevent MF and apoptosis. | 275 |
| miR-129-3p | cardiac hypertrophy | up | protect | ↓ | decreases PKIA activity and prevents MF by targeting Drp. | 196 |
| miR-485-5p | cardiac hypertrophy | down | protect | ↓ | represses mitochondrial-anchored protein ligase (MAPL), which impair its repression on Mfn2 | 197 |
| miR-499-5p | DOX cardiotoxicity | down | protect | ↓ | prevents MF and apoptosis by targeting p21 | 276 |
| miR-153-3p | cardiac hypertrophy | up | induce | ↑ | induces MF by suppressing Mfn-1 expression during cardiac hypertrophy | 199 |
| lncRNAs | ||||||
| CARL | cardiac anoxia | down | protect | ↓ | prevents MF and apoptosis by repressing miR-539-dependent downregulation of prohibitin subunit 2 | 277 |
| Mdrl | cardiac I/R | down | protect | ↓ | targets the miR-361-miR-484 axis; it binds to miR-361 and releases the inhibition of miR-484 expression | 278 |
| circRNAs | ||||||
| MFACR | cardiac I/R | up | protect | ↓ | acts as sponge to miR-652-3p, which suppresses Mtp18 expression, thereby attenuating MF, and cardiomyocyte apoptosis | 98 |
MF, mitochondrial fission; I/R, ischemia/reperfusion; A/R, anoxia/reoxygenation; MI, myocardial infarction; CARL, cardiac apoptosis-related lncRNA; Mdrl, mitochondrial dynamic-related lncRNA; MFACR, mitochondrial fission and apoptosis-related circRNA.
PTMs
In addition to genetic or transcriptional control, the functions of MD proteins are tightly controlled by PTMs. PTM is a process of enzymatic or chemical modifications, such as phosphorylation, SUMOylation, acetylation, glycosylation, and nitrosylation, of a protein that occur after translation and can lead to changes in protein structure, localization, function, or expression level. Similar to epigenetic modification, PTMs are widespread and can be either reversible or irreversible depending on the duration and type of chemical modifications.201 Studies indicate that the structure and function of mitochondrial fusion and fission proteins can be modified post-translationally.22,68,202 Specifically, in most cardiovascular diseases, the predominant PTM mechanisms that regulate MD include phosphorylation, SUMOylation, ubiquitination, nitrosylation, acetylation, and acylation of the key fusion and fission GTPases.
-
(1)
Phosphorylation. Under normal conditions, major percentages of Drp1 are located in the cytosol, and the presence of cellular stress or cell death stimuli can induce Drp1 translocation to the OMM. The localization or activation of cardiac Drp1 is strongly influenced by its phosphorylation status. For example, it has been reported that Drp1 can be phosphorylated at serine 616, 622, 637, or 693 sites and exhibit different effects on MD. Phosphorylation of Drp1 at serine 616 or 622 leads to Drp1 activation and mitochondrial fission;38 however, phosphorylation at serine 637 (S637) leads to Drp1 inhibition and mitochondrial fusion.203,204 Likewise, phosphorylation at S693 can inhibit GTPase activity without affecting the inter-molecular/intra-molecular interaction and prevent the H2O2-induced mitochondrial fission and apoptosis.205 The S637 of Drp1 is phosphorylated by protein kinase A (PKA) and dephosphorylation by the calcium-sensitive phosphatase calcineurin, which is activated by the pathological elevation of intracellular calcium during cardiac stress and I/R. Activation of Drp1 due to dephosphorylation at S637 induces Drp1 translation to the OMM and mitochondrial fission, leading to cell death.206 Phosphorylation at S637 can be mediated by PKA,207 Pim-1,208 CaMKI-α,209 and ROCK1210 depending on the type of stressed stimuli, and phosphorylation at S693 is regulated by GSK3β.205 Recently, Torres et al.211 reported that glucagon-like peptide-1 (GLP-1), a gastrointestinal tract neuroendocrine hormone, is involved in the regulation of cardiac MD via phosphorylation of Drp1. In the VSMC cell line A7r5 derived from embryonic rat aorta, treatment with GLP-1 can prevent cellular migration and proliferation by targeting the PKA-Drp1 axis. GLP-1 phosphorylates Drp1 at S637 and prevents mitochondrial localization of Drp1, thereby preventing PDGF-BB-induced mitochondrial fission and cellular migration even under overexpression of Drp1. Thus, GLP-1 can mimic the Drp1 inhibitory effect of Mdivi-1 and can prevent vascular smooth muscle remodeling.211 Another phosphorylation site of Dr1p is at serine 616, which can be targeted by CDK1/cyclin during mitosis,38 by mitogen-activated protein (MAP) kinase ERK1/2 in response to hyperglycemia,212 and by protein kinase C (PKC)-δ in hypertension encephalopathy.52 Studies indicate that modulation of Drp1 phosphorylation can be a potential therapeutic target that can improve cardiac outcomes. For example, Sharp203 reported that Drp1-mediated fission induces ROS production and impairment of myocardial diastolic dysfunction, and using a Drp1 inhibitor in a Langendorff model showed cardioprotection as evidenced by the preservation of mitochondrial morphology and cytosolic calcium levels. As another example, in adult rats, the overexpression of Pim1208 and administration of Drp inhibitor P110213 showed inhibition of mitochondrial fission and cardioprotection evidenced by a significant reduction in myocardial infarct size and left ventricular remodeling in myocardial infarction (MI).
Studies indicate that phosphorylation in other MD proteins can also influence Drp1 recruitment in the mitochondrial membrane. One example is the phosphorylation of MFF, a mitochondrial outer-membrane receptor that can enhance Drp1 translation to the OMM and induce mitochondrial fission via the AMP-activated protein kinase (AMPK)-mediated fission pathway.214 Another example is that the inner mitochondrial protein Mtfp1, also known as Mtp18, can induce mitochondrial fission by promoting Drp1 phosphorylation. Therefore, knocking down Mtp18 expression plays a cardioprotective role by preventing Drp1 recruitment in the OMM and subsequent mitochondrial fission and apoptosis in DOX-induced cardiotoxicity.165 Lastly, mitochondrial fusion protein Mfn1 can be phosphorylated at T562 of its HR1 domain by ERK. This ERK-mediated phosphorylation of Mfn1 is essential for its oligomerization and for regulation of mitochondrial fusion and apoptosis. Mutation at T562 impairs Mfn1 phosphorylation and prevents the mitochondrial tethering.215
-
(2)
Ubiquitination. Ubiquitination is the addition of ubiquitin, a small regulatory protein ubiquitously present in most eukaryotic tissues, to lysine, serine, and threonine residues on protein substrate and can affect the substrate functions via degradation, localization, or interaction. Ubiquitination is a three-step process including activation (mediated by ubiquitin-activating enzymes, E1s), conjugation (catalyzed by ubiquitin-conjugating enzyme E2s), and ligation (facilitated by ubiquitin ligases E3s) and removed by deubiquitylation enzymes (DUBs). Ubiquitination is one of the commonly found PTMs and is involved in the pathogenesis of cardiac hypertrophy. Recent studies have reported that ubiquitination of key GTPases in MD proteins can alter mitochondrial morphology and influence cellular functions. For example, in mammalian cells, a recently identified mitochondrial ubiquitin ligase, Mitol, is essential for MD by two mechanisms: one is through regulating substrate activation by the K63 ubiquitin chain attachment, and the second is through regulating proteosome-dependent degradation by K48 ubiquitin chain attachment.216 It can reduce Drp1 accumulation in the OMM, thereby preventing mitochondrial fission and apoptosis.150 Knockdown of Mitol can prevent MiD49 ubiquitination and reduces its proteasomal degradation, increasing MiD49 in the OMM and thus inducing mitochondrial fission.149 Reduced expression of Drp1 or Mff can promote Mitol’s activity on MiD49 and prevent mitochondrial fission. Degradation of substrates through Mitol-mediated ubiquitination allows the release of substrates from the OMM. The ubiquitinated degradation of Drp1 and Mff prevents mitochondrial fission.217 Under normal conditions, Drp1 ubiquitination prevents mitochondrial destruction and mediates it to reform into fusion networks after mitotic fission. In contrast, under mitochondrial stress, Mitol ubiquitination to acetylated Mfn1 can promote proteasomal degradation and induce mitochondrial fission.218 In addition to Mitol, other E3 ubiquitin ligases such as Parkin and Huwe1 can ubiquitinate MD proteins to undergo proteasome-mediated degradation. For example, impaired function in Parkin due to mutation or knockdown can prevent Drp1 degradation, and increasing Drp1 levels can induce mitochondrial fission and apoptosis.219 Alternatively, the removal of ubiquitin residues by deubiquitinating enzymes such as Usp30 can prevent its substrate degradation.22 Under stress conditions, ubiquitination of JNK-phosphorylated Mfn2 by Huwe1 leads to degradation of Mfn2 and induces mitochondrial fragmentation and apoptosis.220 However, the effects of ubiquitination of MD proteins on cardiac hypertrophy remain largely unknown.
-
(3)
SUMOylation. Small ubiquitin-related modifier (SUMO) proteins are a member of the large family of ubiquitin-related proteins and covalently bind to lysine in a target protein. SUMOylation is a reversible PTM, which can regulate the function of target proteins by affecting their subcellular localization, activity, or stability. Recent studies indicate that SUMOylaiton is associated with the pathogenesis of several human diseases such as cardiac hypertrophy, neurodegenerative diseases, and cancers.167,221,222 Multiple enzymes can perform SUMOylation, and the most extensively studied ones so far are SUMO-activating enzymes 1 and 2 (SAE1/2, which are E1-activating enzymes), ubiquitin-conjugating enzyme 9 (Ubc9, which is an E2-conjugating enzyme), and E3 ligase. Mitochondrial-anchored protein ligase (MAPL) is the first discovered mitochondrial-anchored SUMO E3. It is an OMM protein that targets Drp1 to stabilize and function as a positive regulator of mitochondrial fission.223 During apoptotic cell death, Bax/Bak mediates the sumoylation of DRP1, leading to stabilization of the association of Drp1 with mitochondria to enhance mitochondrial fission.224 Alternatively, SUMO-2/3-specific protease SENP3 can deSUMOylate Drp1 and induce mitochondrial fission and cell death via Drp1-mediated cytochrome c release during cardiac ischemia.225 These studies collectively suggest that SUMOylation/deSUMOylation of Drp1 plays a significant role in the regulation of cardiac mitochondrial fission and cell death.
-
(4)
O-GlcNAcylation. O-GlcNAcylation is a form of post-translational modification that has recently emerged as a key regulator of several cardiovascular pathophysiologies.226 The attachment of O-linked N-acetylglucosamine (O-GlcNAc) at serine and threonine residues of nuclear, cytoplasmic, and mitochondrial proteins, is a dynamic process that can be recycled by cells-O-GlcNAc transferase (OGT) for the addition of O-GlcNAC and by O-GlcNAcase (OGA) for its removal.227 The chemical inhibition of OGA can induce O-GlcNAcylation. Several recent studies have indicated that O-GlcNAcylation can regulate multiple cellular processes such as transcription, translation of proteins, signal transduction, calcium handling, bioenergetics, and apoptosis. O-GlcNAcylation in mitochondrial dynamic proteins affects protein function and can influence mitochondrial morphology.226,227 In cardiomyocytes, Drp1 can be O-GlcNAcylated at threonine 585 and 586. Increased O-GlcNAcylation can reduce the phosphorylation of Drp1 at S637, which further activates Drp1 function and promotes Drp1 translocation from the cytoplasm to the OMM, resulting in mitochondrial fission and apoptosis.228,229 In diabetes, continuous hyperglycemia leads to increased O-GlcNAcylation through upregulation of ERK1/2 and cyclin D2 expression230 and is associated with cardiovascular complications and diabetic cardiomyopathy, which can be manifested in cardiac hypertrophy and heart failure. The activation of AMPK reduces O-GlcNAcylation and prevents cardiac hypertrophy.231 Increased O-GlcNAcylation can reduce mitochondrial function observed by decreased mitochondrial membrane potential and complex IV activity.232 Interestingly, an acute increase in O-GlcNAc levels in IR injury was found to protect against cardiac injury.226 In neonatal cardiomyocytes, exposure to high-glucose treatment decreases OGA and increases O-GlcNAcylation in Opa1, followed by downregulation of in OPA1 expression and exhibition of mitochondrial fragmentation. In contrast, overexpression of OGA reduces Opa1 O-GlcNAcylation even in high-glucose exposure and induces mitochondrial fusion. Thus, manipulating OGA and Opa1 expression could improve cardiac dysfunction in diabetes by targeting MD and energy metabolism.232
-
(5)
Nitrosylation. Nitric oxide (NO), also known as an endothelium-derived relaxing factor, plays a multifaceted role in the cardiovascular system by regulating cardiomyocyte contraction, vascular tone, thrombogenicity, endothelial proliferation, and inflammation. NO production is regulated by NO synthase (NOS), which is produced by the myocardium. Myocardium produces three isoforms of NOS, that is, neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS.233 nNOS expression is significantly upregulated and eNOS is downregulated in left ventricular myocytes, whereas the protein level of eNOS is downregulated in Ang II-induced hypertensive rats and hypertrophic myocardium.234 S-nitrosylation of Drp1 and its influence on MD are associated with neurodegenerative diseases such as Alzheimer’s disease and Huntington’s disease (HD). A well-established mediator of Alzheimer’s disease called β-amyloid protein induces the production of NO and causes S-nitrosylation in serine residue of Drp1 (SNO-Drp1). The SNO-Drp1 can activate Drp1 and induce mitochondrial fission, leading to synaptic loss and neuronal damage.235 Likewise, high SNO-Drp1 levels are increased in the brains of transgenic HD mice as well as in HD patients, and SNO-Drp1 induces excessive mitochondrial fission, leading to synaptic damage. SNO-Drp1 can affect Drp1 phosphorylation; an increase in SNO-Drp1 facilitates Drp1 phosphorylation at serine 616, which activates Drp1 function and induces Drp1-mediated mitochondrial fission.236 It is noteworthy that the myocardium can secrete NO, and NO plays an important role in regulating blood vessel tone, thrombogenicity, and cardiac function. NO has been reported as endogenous inhibitors of maladaptive hypertrophy signaling and could protect against cardiac hypertrophy.233 However, the excessive production of NO occurs under cardiac inflammation and cardiac failure.
-
(6)
Acetylation. Histone acetylation and deacetylation comprise a dynamic process in which an acetyl group is added or removed from lysine residues of proteins regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. Histone acetylation loosens the chromatin structure, also known as heterochromatin or open chromatin, and activates transcription, while deacetylation tightens the chromatin, making it less accessible by a transcription factor and repressing gene expression; this state is termed euchromatin or closed chromatin. Histone modification has been shown to play a role in managing stages of organismal development as well as aging by being involved in various cellular processes through regulating the structure of chromosomes and transcription. HATs constitute GNATs (GCN5-related N-acetyltransferases), MYST, and p300/CBP (CREB-binding protein) families.237 So far, there are four classes of HDACs: class I includes HDAC1, 2, 3, and 8); class II constitutes HDAC4, 5, 6, 7, 9, and 10; class III is composed of the sirtuin family (SIRT1–7), the members of which possess mono-ADP-ribosyltransferase or deacylase activity; and class IV HDAC is HDAC11.238 The sirtuin family plays a protective role against several cardiovascular pathologies, including cardiomyopathy, vascular endothelial dysfunction, metabolic syndrome, cardiac ischemia, and even aging. Moreover, using sirtuin-activating medications has been found to improve cardiovascular and metabolic health in human clinical trials.239 Therefore, the health benefits of sirtuins have become one of the most extensively studied research topics, and sirtuins can serve as a potential therapeutic drug for both cardiovascular health and the prevention of aging.
Sirtuin plays a significant role in the regulation of cardiac MD. One study in ischemic mouse livers indicated that SIRT1 exerts its cardioprotective effect by targeting Mfn2, which prevents I/R-induced mitochondrial fission and autophagy.240 Increased mitochondrial fission in diabetic patients has been linked to myocardial contractile dysfunction. A study by Ding et al.,241 in which diabetes was induced in wild-type (WT) and SIRT1-deficient mice with streptozotocin, found that melatonin reduced Drp1 expression through SIRT1/PGC-1α signaling pathway and prevented mitochondrial fission and cardiomyocyte apoptosis, leading to improve cardiac function in WT diabetic mice but not in SIRT1-deficient mice. SIRT3 can deacetylate Opa1 and promote its GTPase activity as evidenced by increased mitochondrial fusion. In pathological stress, Opa1 is overacetylated and mitochondrial fission is activated. In TAC-induced cardiac stress, decreased activity of SIRT3 leads to hyperacetylation of Opa1 and impairs mitochondrial fusion. The SIRT3-deficient cells show acetylation at lysine 926 and 931 residues of Opa1 and extensive mitochondrial fission. However, overexpression of SIRT3 increases Opa1 deacetylation and activates Opa1-dependent mitochondrial fusion, thereby preventing the cardiomyocytes from DOX-mediated cell death.44 These studies collectively suggest that manipulating the acetylation of MD proteins could be a potential therapeutic target to prevent mitochondrial fission-associated cardiac stress conditions.
-
(7)
Proteolytic processing. Opa1 is a crucial IMM component for mitochondrial fusion. Opa1 undergoes proteolytic processing, resulting in short (S-Opa1) and long (L-Opa1) isoforms, both of which are necessary for mitochondrial fusion.120 The interaction between IMS-localized S-OPA1 and IMM-localized L-Opa1 forms an active quaternary structure that allows the IMM to tighten the cristae junction and to interact with OMM fusion protein at ER-mitochondria contact sites to induce mitochondrial fusion.120 The processing of Opa1 is implicated by proteases, including OMA1 metalloprotease,122 YME1L, paraplegin, and the mAAA protease complex ATPase family gene-3, yeast-like-1 (AFG3L1), and presenilin-associated rhomboid-like (PARL) protein.121 Opa1 cleavage can generate at least five isoforms comprising two L-Opa1 and three S-Opa1 forms depending on the cellular context. OPA1 has eight splice variants, with half of them composed of both S1 and S2 proteolytic sites, while the remaining four contain only S1.242 The cleavage of S1 and S2 proteolytic sites are mediated by two membrane-bound metalloproteases, that is, OMA1120 and YME1L,243 respectively; also, healthy human cells contain a mix of L-OPA1 and S-OPA1. Distrubance in mitochondrial function can induce OMA1’s activity, leading to decreased L-OPA1 and increased S-OPA1 forms.242 Notably, the composition of isoforms can influence MD; in particular, an increase in OMA1 cleavage further induces the oxidative phosphorylation via YME1L activation during the stress response.68 Abnormal processing Opa1 mediated by deletion of either OMA1 or YME1L showed dilated cardiomyopathy, ventricular remodeling, and heart failure.244
Therapeutic strategies focusing on MD in cardiac hypertrophy
Treatment of cardiac hypertrophy includes both curative and palliative approaches: curative treatment is that which deals directly with the underlying causes such as surgically repairing aortic valve stenosis, removing endocrine tumors, or treating the renal cause of hypertension, among others. Nonetheless, some underlying causes such as secondary hypertension may not have a curative measure. Hence, palliative treatment that involve treating the symptoms, preventing adverse effects, and lifestyle modifications are commonly prescribed, and thus these measures are crucial. Since decompensatory cardiac hypertrophy often later progresses to heart failure, the pharmacological management of cardiac hypertrophy includes antihypertensives and heart failure medications. The compensatory responses in heart failure are mediated by signaling pathways that initially serve to maintain normal contractility; however, persistent activation of these pathways leads to cardiac dysfunction.245 Due to the significant role of humoral stimuli in disease pathology, angiotensin-converting enzyme (ACE) inhibitors, Ang II receptor blockers (ARBs), G protein-coupled receptor (GPCR) antagonists, calcium channel blockers (CCBs), diuretics, and beta blockers have been the gold standard therapeutic approaches for decades.246,247 However, increasing numbers of studies show that patients receiving neurohormonal blockage later experience ceiling effects with no improvements or, in a few cases, even relapse.248 In addition to conventional cardiac hypertrophy and heart failure treatment, regulating cardiac metabolism using perhexiline, which prevents CPT I and II activity to promote energy generation by shifting the energy substrate to a more efficient fuel carbohydrate, is found to be another promising treatment option. Dietary supplementations such as foods rich in fatty acids (FAs), n-3 long-chain polyunsaturated fatty acids (N3-PUFA), free FAs (FFAs), or l-carnitine have shown to be effective for the management of heart failure.249 Oral administration of chemically synthesized small molecules, including sildenafil and BGF-15, can target heart failure-related intercellular signaling pathways specifically.249 It is also reported that implantation of stem cells into a failing heart can induce cardiac muscle regeneration and improve cardiac function. However, clinical trials using stem cell treatment show a limited effect on cardiac function and no improvement in patient survival.249 Pathological cardiac remodeling is a complex process and is highly interrelated; therefore, targeting a single molecule or process may not be sufficient.250 Hence, a novel therapeutic strategy with fewer side effects and sustainable action that can target multiple pathways is required.
Pharmacological treatments
MD is important for mitochondrial quality control, biogenesis, mitochondrial function, and apoptosis. Homeostasis between fusion and fission enables exchanging contents between the mitochondria and prevents the accumulation of damaged mitochondria. The links between defects in MD and cardiac hypertrophy are particularly intriguing. Increasing numbers of strategies have been developed to modulate the MD, including genetic approaches such as the manipulation of mitochondrial fusion and fission modulators, regulating the expression of various types of non-coding RNAs, and pharmacological treatments that can affect mitochondrial quality control and can modulate post-translational modification. As we have discussed the roles of non-coding RNAs and post-translational modification as a therapeutic potential for regulating MD in cardiac hypertrophy in previous sections, we focus on pharmacological intervention in this section (Figure 4).
Figure 4.

Cardioprotective drugs targeting mitochondrial dynamics pathways
All of these medications have been shown to reduce mitochondrial fission and improve cardiac function in animal models. However, their use in patients requires further extensive studying.
The Drp1 inhibitor Mdivi, also known as mitochondrial division inhibitor 1 (Mdivi-1), inhibits the GTPase function of Drp1 by acting as an allosteric inhibitor of GTPase assembly, decreasing Drp1 translocation to the mitochondria and thereby preventing mitochondrial fission to ameliorate pressure overload-induced cardiac failure. Mdivi can decrease hypertrophic stimuli-mediated ROS production, thereby decreasing intracellular Ca2+ levels and suppressing the activation of Drp1 activators such as calcineurin and Ca2+/calmodulin-dependent kinase II (CaMKII). Treatment with Mdivi-1 can ameliorate LVH and cardiac dysfunction in response to TAC in WT mice by inhibiting mitochondrial fission and autophagy, which is evidenced by decreased Drp1 function and decreased expression of autophagic markers, such as LC3 and p62, and preventing its progression to heart failure.251 Interestingly, studies also reported that Mdivi exhibit a cardioprotective effect in the Drp1-independent pathway by retarding OMM permeability.252,253 Mdivi-1 averts apoptosis by preventing OMM permeability and inhibiting Bid-activated Bax/Bak-dependent cytochrome c release from mitochondria.252 Treatment with Mdivi led to an increase in expression of vascular endothelial growth factor, matrix metalloproteinase-3 (MMP-3), and platelet endothelial cell adhesion molecule or cluster of differentiation 31 (CD31), but with decreased expression of anti-angiogenic factors, MMP-9, and tissue inhibitor of metalloproteinase-3 (TIMP-3).251 Likewise, right ventricular hypertrophy (RVH) mediated by monocrotaline-induced PAH rats showed enhancement in Drp1-Fis interaction and subsequent mitochondrial fission, leading to diastolic dysfunction. P110, a Drp1-Fis interaction peptide inhibitor, can directly bind to Drp1 and prevents Drp1 translocation to mitochondria and mitochondrial fission by interfering with Drp1-oligomer formation. Treatment with P110 can repress PAH-induced mitochondrial fission and RV function impairment.254 A significant cardiac protective effect was also observed in ex vivo I/R models of primary cardiomyocytes treated with ischemia and reoxygenation buffers and a Langendorff coronary perfusion system, and in an in vivo acute MI (AMI) model induced by ligation of the left anterior descending (LAD) artery.213
GLP-1 peptide-mimetic exenatide can phosphorylate Drp1 at the S637 residue and inhibit mitochondrial localization of Drp1, thereby preventing PDGF-BB-induced mitochondrial fission. Torres et al.211 observed that GLP-1 treatment in VSMC cell line A7r5 derived from embryonic rat aorta could induce cardiac regeneration observed by increased cell migration and proliferation. Ding et al.241 showed that melatonin treatment can decrease Drp1 expression through the SIRT1/PGC-1α signaling pathway, thereby preventing mitochondrial fission and reducing oxidative stress and cardiomyocyte apoptosis, leading to improved cardiac function in diabetic mice. However, this cardioprotective effect of melatonin is diminished when SIRT1 is knocked out, indicating that SIRT1 can block Drp1-mediated mitochondrial fission. However, the exact molecular mechanism remains unclear. Melatonin treatment can also protect against post-MI injury in wild-type mice but not in Notch1- or Mfn2-depleted mice, indicating that melatonin can prevent mitochondrial damage in MI by targeting the Notch1/Mfn2 signaling pathway likely due to the inhibition of ROS generation. The cardioprotective effect of melatonin is further confirmed by the usage of a nonselective melatonin receptor antagonist luzindole, which can reverse the melatonin function by preventing Notch1 activation and downregulating Mfn2 expression.255
Resveratrol has been shown to have antiaging and cardiac protective effects in different animal models.256,257 However, little is known about its role in MD. Ren et al.256 showed that resveratrol causes SIRT1 activation, which can prevent mitochondrial fission by downregulating Drp1 expression in d-galactose-induced senescent cardiomyocytes. Additionally, resveratrol can activate Parkin- and PINK1-mediated autophagy, which is evidenced by the increased LC3-II expression and decreased TOM20-labeled mitochondrial contents.256 Cardiac-specific SIRT1 knockout (SIRT1-KO) mice exhibit diabetic cardiomyopathy (DCM) characterized by abnormal glucose metabolism, insulin resistance, cardiac hypertrophy, and dysfunction. The heart tissue extracted from SIRT knockout mice and DCM showed impaired mitochondrial biogenesis and increased apoptosis. Resveratrol can alleviate cardiac dysfunction in the DCM mouse heart by deacetylating PGC-1a, which increases the expression of nuclear respiratory factor 1 (NRF-1), NRF-2, estrogen-related receptor-α (ERR-α), and mitochondrial transcription factor A (TFAM) to improve MD.257 Collectively, these data indicate the potential therapeutic role of resveratrol in cardiac hypertrophy.
HO-1 is a cardioprotective inducible enzyme that degrades pro-oxidant heme into equimolar quantities of carbon monoxide, biliverdin, and iron. Dilated cardiomyopathy induced in wild-type HO-1 knockout and human HO-1 overexpressed mice with IV injection of DOX showed that increased expression of HO-1 can prevent DOX-induced mitochondrial fragmentation by increasing Mfn1/2 expression and decreasing Fis1 expression. Additionally, it promoted mitochondrial biogenesis by increased expression of NRF-1, PGC1α, and TFAM. These findings suggest that HO-1 can protect against oxidative stress-induced cardiac injury and may serve as a novel therapeutic molecule in oxidative stress-induced cardiac injury.258 However, the role of non-GTPase coordinators in cardiac hypertrophy and heart failure remains unclear and, thus, studies addressing this special issue will be worthy of attention to understand the molecular interaction of fusion and fission coordinators in cardiac MD.
Novel therapeutic strategies targeting MD
Several recent studies reported the novel therapeutic strategies for regulating cardiac MD. For example, the cardiac protective approach using anesthetic postconditioning is a novel tactic reported to be effective at preventing subsequent cardiac injury mediated by an I/R event. It is suggested that exposure to a volatile anesthetic such as sevoflurane can help cardiac tissue to be more resilient to I/R-induced injury. In particular, sevoflurane postconditioning (SPostC) can protect primary neonatal rat cardiomyocytes (NCMs) from hypoxia/reoxygenation (H/R) injury by preventing H/R-induced mitochondrial fission. They indicated that SPostC significantly increased Mfn2 and Opa1 expression, but decreased that of Drp1 without affecting Mfn1 and Fis expression.259 Another emerging novel approach shown to have a cardiac-protective effect against cardiac I/R injury via regulating MD is remote ischemic preconditioning (RIPC). Cellier et al.260 conducted a RIPC approach of four cycles of alternative 5-min limb ischemia and 5-min limb reperfusion in male adult rats and immediately after RIPC, and exposed them to 120 min of myocardial ischemia (for both normal and RIPC rats). Their findings indicated that RIPC rats showed an approximately 28% smaller infarct size compared to the control group and had increased expression of Opa1 and less mitochondrial fragmentation. Nonetheless, the mechanism of how RIPC controls MD remains uncertain.
In addition, MD is controlled by mitochondrial inner membrane potential, which is determined by the proton and the electrical gradient across the electron transport chain system. During cardiac pathological conditions, increased oxidative stress and calcium overload induce IMM opening and lead to an imbalance in mitochondrial IMM potential. Disturbance in mitochondrial membrane potential inhibits mitochondrial fusion and induces mitochondrial fission. For example, ionophores can disintegrate the mitochondrial membrane potential, leading to abnormal mitochondrial fission by inhibiting normal mitochondrial fusion.30 Conversely, cyclosporin A (CsA) treatment can inhibit the opening of mPTP during I/R-induced cardiac stress and improve the IMM integrity, thereby maintaining mitochondrial morphology and preventing mitochondrial fission and subsequent apoptosis.261 Intriguingly, treatment with CsA nanoparticles (CsA-NPs), which is CsA incorporated into a poly-lactic/glycolic acid (PLGA) nanoparticle-mediated mitochondrial targeting approach, shows a more promising cardioprotection function. In the murine IR model, intravenous (i.v.) injection of PLGA CsA-NPs at the onset of reperfusion showed reduced infarct size as compared to CsA alone.262 These findings suggest that CsA-NPs could be developed as an effective mPTP opening inhibitor in cardiac injury. Nonetheless, little is known about the therapeutic effect of CsA in cardiac hypertrophy.
Future perspectives and conclusions
MD is a complex process in which the outcome is reflected by the collaborated fusion or fission events that are regulated by an array of fusion and fission proteins. Imbalance increased in either fusion or fission factors can shift the equilibrium toward one end. Dysregulation in mitochondrial fusion and fission proteins can occur at the transcriptional (gene expression), epigenetic, and/or posttranslational levels. Because MD is not a single entity of fusion or fission events, the harmonization between the two is what decides the final cellular function and mitochondrial quality control. The expression pattern of crucial mediators (for example, an increased in fission protein Drp1 and a decrease in fusion protein Mfn2 expression), the fusion and fission coordinator proteins, as well as their functional changes upon posttranslational modification, such as phosphorylation and ubiquitination, are required to be considered as a whole.141
Another challenging question is whether there is a specific characteristic in mitochondrial morphology that could determine whether the changes are due to physiology or pathology. These are very important issues to address prior to the development of drugs targeting MD since they could help prevent unwanted functional deterioration and adverse events during the treatment. Emerging data highlight the potential of targeting MD as a new therapy for combating hypertension and heart failure. Most of these pieces of evidence pay attention to acute inhibition of mitochondrial fission or activation of mitochondrial fusion; however, the main challenge of these approaches is that they will gradually lead to the chronic manipulation of MD. Most importantly, long-term activation or inhibition of either fusion or fission can result in deleterious effects. Additionally, the significance of changes in mitochondrial shaping proteins in patients with cardiac hypertrophy and heart failure as well as whether the inhibition of mitochondrial fission in these settings would be beneficial to mitochondria and cardiac function in the long term remain unclear. For example, Drp1 knockout mice can experience a cardiotoxic effect due to the tonic and complete loss of Drp1, which is essential for normal mitochondrial fission, whereas inhibition of Drp1 with pharmacological inhibitors such as Mdivi-1 or P110 can promote cardioprotective effects because the effect of the pharmacological intervention is transient and provides only partial inhibition of mitochondrial fission.263 However, further work is still needed to better understand the pleiotropic roles that mitochondrial fusion or fission modulators may play in the heart and their long-term effects in the organ systems, providing valuable insights that will guide the clinicians in considering the application of this approach in human disease models.
In addition, recent studies mainly focused on the quantitative alterations of mitochondrial morphological dynamics, while temporal consideration has been less appreciated. It is suggested that molecular deletion of a fission and fusion mediator in a healthy versus diseased organ may show different effects, and that both transient and partial inhibition tend to be more beneficial to prevent permanent cardiotoxic effects.263 Hence, the pharmacological intervention that often generates transient and partial inhibition of activated fission pathway is a more promising approach. A better understanding of the molecular mechanisms that may regulate cardiac hypertrophy is essential for its clinical implications. Thus, further studies focusing on temporal changes of MD upon genetic manipulation or pharmacological management are required for the development of a more efficient therapeutic strategy.
MD is essential for normal cardiac function and plays a crucial role in cardiac homeostasis as well as in cardiovascular diseases. Disruption in MD is linked to various types of cardiac hypertrophy, and thus treatments targeting MD have shown promising results in animal models. However, due to the lack of efficient tools to study MD in humans, very little is known about the feasibility and usefulness of these strategies in patients with cardiac hypertrophy and heart failure. For the clinical translation of this approach, it is essential to develop tools to better evaluate MD in patients and couple them with biopsy analyses. Notably, changes in MD can also be reflected in a decrease in cardiac ATP production and mitophagy or an increase in cardiac apoptosis. Thus, the development of indirect biomarkers that can detect the levels of key mitochondrial modulators, or measuring cardiac ATP production, will help to evaluate the state of MD in patients with CVDs. Therefore, it is critical to develop non-invasive tools that could assess MD, allowing the clinical translation of MD modulation as a convincing approach.
Acknowledgments
This project was supported by the National Natural Science Foundation of China Research Fund for International Young Scientists (82050410450 and 81850410551) and the Natural Science Foundation of Shandong Province (ZR2019BH014).
Author contributions
L.H.H.A. generated the idea and wrote the manuscript. J.C.C.J. revised the manuscript. Y.W. and P.L. helped to revise the manuscript. All authors have read and approved the final submission.
Declaration of interests
The authors declare no competing interests.
References
- 1.Shimizu I., Minamino T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016;97:245–262. doi: 10.1016/j.yjmcc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura M., Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018;15:387–407. doi: 10.1038/s41569-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 3.Lips D.J., deWindt L.J., van Kraaij D.J., Doevendans P.A. Molecular determinants of myocardial hypertrophy and failure: Alternative pathways for beneficial and maladaptive hypertrophy. Eur. Heart J. 2003;24:883–896. doi: 10.1016/s0195-668x(02)00829-1. [DOI] [PubMed] [Google Scholar]
- 4.Li L., Xu J., He L., Peng L., Zhong Q., Chen L., Jiang Z. The role of autophagy in cardiac hypertrophy. Acta Biochim. Biophys. Sin. (Shanghai) 2016;48:491–500. doi: 10.1093/abbs/gmw025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marian A.J., Braunwald E. Hypertrophic cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 2017;121:749–770. doi: 10.1161/CIRCRESAHA.117.311059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunter J.J., Chien K.R. Signaling pathways for cardiac hypertrophy and failure. N. Engl. J. Med. 1999;341:1276–1283. doi: 10.1056/NEJM199910213411706. [DOI] [PubMed] [Google Scholar]
- 7.Shenasa M., Shenasa H. Hypertension, left ventricular hypertrophy, and sudden cardiac death. Int. J. Cardiol. 2017;237:60–63. doi: 10.1016/j.ijcard.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Geske J.B., Ommen S.R., Gersh B.J. Hypertrophic cardiomyopathy: Clinical update. JACC Heart Fail. 2018;6:364–375. doi: 10.1016/j.jchf.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 9.Kass D.A., Bronzwaer J.G., Paulus W.J. What mechanisms underlie diastolic dysfunction in heart failure? Circ. Res. 2004;94:1533–1542. doi: 10.1161/01.RES.0000129254.25507.d6. [DOI] [PubMed] [Google Scholar]
- 10.Savarese G., Lund L.H. Global public health burden of heart failure. Card. Fail. Rev. 2017;3:7–11. doi: 10.15420/cfr.2016:25:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kavey R.E. Left ventricular hypertrophy in hypertensive children and adolescents: Predictors and prevalence. Curr. Hypertens. Rep. 2013;15:453–457. doi: 10.1007/s11906-013-0370-3. [DOI] [PubMed] [Google Scholar]
- 12.Gradman A.H., Alfayoumi F. From left ventricular hypertrophy to congestive heart failure: Management of hypertensive heart disease. Prog. Cardiovasc. Dis. 2006;48:326–341. doi: 10.1016/j.pcad.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Patrushev M.V., Mazunin I.O., Vinogradova E.N., Kamenski P.A. Mitochondrial fission and fusion. Biochemistry (Mosc.) 2015;80:1457–1464. doi: 10.1134/S0006297915110061. [DOI] [PubMed] [Google Scholar]
- 14.Youle R.J., Karbowski M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 15.Pennanen C., Parra V., López-Crisosto C., Morales P.E., Del Campo A., Gutierrez T., Rivera-Mejías P., Kuzmicic J., Chiong M., Zorzano A. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell Sci. 2014;127:2659–2671. doi: 10.1242/jcs.139394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lesnefsky E.J., Chen Q., Tandler B., Hoppel C.L. Mitochondrial dysfunction and myocardial ischemia-reperfusion: Implications for novel therapies. Annu. Rev. Pharmacol. Toxicol. 2017;57:535–565. doi: 10.1146/annurev-pharmtox-010715-103335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vásquez-Trincado C., García-Carvajal I., Pennanen C., Parra V., Hill J.A., Rothermel B.A., Lavandero S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016;594:509–525. doi: 10.1113/JP271301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marín-García J., Akhmedov A.T. Mitochondrial dynamics and cell death in heart failure. Heart Fail. Rev. 2016;21:123–136. doi: 10.1007/s10741-016-9530-2. [DOI] [PubMed] [Google Scholar]
- 19.Yu R., Lendahl U., Nistér M., Zhao J. Regulation of mammalian mitochondrial dynamics: Opportunities and challenges. Front. Endocrinol. (Lausanne) 2020;11:374. doi: 10.3389/fendo.2020.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin Y., Li A., Liu B., Jiang W., Gao M., Tian X., Gong G. Mitochondrial fusion mediated by fusion promotion and fission inhibition directs adult mouse heart function toward a different direction. FASEB J. 2020;34:663–675. doi: 10.1096/fj.201901671R. [DOI] [PubMed] [Google Scholar]
- 21.Ong S.B., Kalkhoran S.B., Cabrera-Fuentes H.A., Hausenloy D.J. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur. J. Pharmacol. 2015;763(Pt A):104–114. doi: 10.1016/j.ejphar.2015.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nan J., Zhu W., Rahman M.S., Liu M., Li D., Su S., Zhang N., Hu X., Yu H., Gupta M.P., Wang J. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim. Biophys. Acta Mol. Cell Res. 2017;1864:1260–1273. doi: 10.1016/j.bbamcr.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Liesa M., Palacín M., Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 24.Zhao J., Lendahl U., Nistér M. Regulation of mitochondrial dynamics: Convergences and divergences between yeast and vertebrates. Cell. Mol. Life Sci. 2013;70:951–976. doi: 10.1007/s00018-012-1066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirihai O.S., Song M., Dorn G.W., 2nd How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 2015;116:1835–1849. doi: 10.1161/CIRCRESAHA.116.306374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hollenbeck P.J., Saxton W.M. The axonal transport of mitochondria. J. Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishra P., Chan D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014;15:634–646. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landes T., Martinou J.C. Mitochondrial outer membrane permeabilization during apoptosis: the role of mitochondrial fission. Biochim. Biophys. Acta. 2011;1813:540–545. doi: 10.1016/j.bbamcr.2011.01.021. [DOI] [PubMed] [Google Scholar]
- 29.Ong S.-B., Gustafsson Å.B. New roles for mitochondria in cell death in the reperfused myocardium. Cardiovasc. Res. 2012;94:190–196. doi: 10.1093/cvr/cvr312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Detmer S.A., Chan D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 31.Ferree A., Shirihai O. Mitochondrial dynamics: The intersection of form and function. Adv. Exp. Med. Biol. 2012;748:13–40. doi: 10.1007/978-1-4614-3573-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuznetsov A.V., Hermann M., Saks V., Hengster P., Margreiter R. The cell-type specificity of mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009;41:1928–1939. doi: 10.1016/j.biocel.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 33.Boldogh I.R., Pon L.A. Mitochondria on the move. Trends Cell Biol. 2007;17:502–510. doi: 10.1016/j.tcb.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 34.Anmann T., Guzun R., Beraud N., Pelloux S., Kuznetsov A.V., Kogerman L., Kaambre T., Sikk P., Paju K., Peet N. Different kinetics of the regulation of respiration in permeabilized cardiomyocytes and in HL-1 cardiac cells. Importance of cell structure/organization for respiration regulation. Biochim. Biophys. Acta. 2006;1757:1597–1606. doi: 10.1016/j.bbabio.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Anesti V., Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta. 2006;1757:692–699. doi: 10.1016/j.bbabio.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Chang D.T.W., Honick A.S., Reynolds I.J. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 2006;26:7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitra K., Wunder C., Roysam B., Lin G., Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taguchi N., Ishihara N., Jofuku A., Oka T., Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 39.Zhou P., Pu W.T. Recounting cardiac cellular composition. Circ. Res. 2016;118:368–370. doi: 10.1161/CIRCRESAHA.116.308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iglewski M., Hill J.A., Lavandero S., Rothermel B.A. Mitochondrial fission and autophagy in the normal and diseased heart. Curr. Hypertens. Rep. 2010;12:418–425. doi: 10.1007/s11906-010-0147-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimada T., Horita K., Murakami M., Ogura R. Morphological studies of different mitochondrial populations in monkey myocardial cells. Cell Tissue Res. 1984;238:577–582. doi: 10.1007/BF00219874. [DOI] [PubMed] [Google Scholar]
- 42.Ong S.B., Hausenloy D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010;88:16–29. doi: 10.1093/cvr/cvq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu Z., Wei Y., Song Q., Du B., Wang H., Chu Y., Hu Y. The role of myocardial mitochondrial quality control in heart failure. Front. Pharmacol. 2019;10:1404. doi: 10.3389/fphar.2019.01404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samant S.A., Zhang H.J., Hong Z., Pillai V.B., Sundaresan N.R., Wolfgeher D., Archer S.L., Chan D.C., Gupta M.P. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell. Biol. 2014;34:807–819. doi: 10.1128/MCB.01483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H., Chomyn A., Chan D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 46.Chen L., Knowlton A.A. Mitochondrial dynamics in heart failure. Congest. Heart Fail. 2011;17:257–261. doi: 10.1111/j.1751-7133.2011.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Owens G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 48.Johnson J.L., van Eys G.J., Angelini G.D., George S.J. Injury induces dedifferentiation of smooth muscle cells and increased matrix-degrading metalloproteinase activity in human saphenous vein. Arterioscler. Thromb. Vasc. Biol. 2001;21:1146–1151. doi: 10.1161/hq0701.092106. [DOI] [PubMed] [Google Scholar]
- 49.Björkerud S. Effects of transforming growth factor-beta 1 on human arterial smooth muscle cells in vitro. Arterioscler. Thromb. 1991;11:892–902. [PubMed] [Google Scholar]
- 50.Watanabe T., Saotome M., Nobuhara M., Sakamoto A., Urushida T., Katoh H., Satoh H., Funaki M., Hayashi H. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp. Cell Res. 2014;323:314–325. doi: 10.1016/j.yexcr.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 51.Pukac L., Huangpu J., Karnovsky M.J. Platelet-derived growth factor-BB, insulin-like growth factor-I, and phorbol ester activate different signaling pathways for stimulation of vascular smooth muscle cell migration. Exp. Cell Res. 1998;242:548–560. doi: 10.1006/excr.1998.4138. [DOI] [PubMed] [Google Scholar]
- 52.Salabei J.K., Hill B.G. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. 2013;1:542–551. doi: 10.1016/j.redox.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marsboom G., Toth P.T., Ryan J.J., Hong Z., Wu X., Fang Y.-H., Thenappan T., Piao L., Zhang H.J., Pogoriler J. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012;110:1484–1497. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lacolley P., Regnault V., Nicoletti A., Li Z., Michel J.B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012;95:194–204. doi: 10.1093/cvr/cvs135. [DOI] [PubMed] [Google Scholar]
- 55.Qi J., Wang F., Yang P., Wang X., Xu R., Chen J., Yuan Y., Lu Z., Duan J. Mitochondrial fission is required for angiotensin II-induced cardiomyocyte apoptosis mediated by a Sirt1-p53 signaling pathway. Front. Pharmacol. 2018;9:176. doi: 10.3389/fphar.2018.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hales K.G., Fuller M.T. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–129. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- 57.Shaw J.M., Nunnari J. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 2002;12:178–184. doi: 10.1016/s0962-8924(01)02246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jakobs S., Martini N., Schauss A.C., Egner A., Westermann B., Hell S.W. Spatial and temporal dynamics of budding yeast mitochondria lacking the division component Fis1p. J. Cell Sci. 2003;116:2005–2014. doi: 10.1242/jcs.00423. [DOI] [PubMed] [Google Scholar]
- 59.Kanazawa T., Zappaterra M.D., Hasegawa A., Wright A.P., Newman-Smith E.D., Buttle K.F., McDonald K., Mannella C.A., van der Bliek A.M. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008;4:e1000022. doi: 10.1371/journal.pgen.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Labrousse A.M., Zappaterra M.D., Rube D.A., van der Bliek A.M. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell. 1999;4:815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- 61.Rana A., Oliveira M.P., Khamoui A.V., Aparicio R., Rera M., Rossiter H.B., Walker D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017;8:448. doi: 10.1038/s41467-017-00525-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S.O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 63.Smirnova E., Griparic L., Shurland D.L., van der Bliek A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hermann G.J., Thatcher J.W., Mills J.P., Hales K.G., Fuller M.T., Nunnari J., Shaw J.M. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J. Cell Biol. 1998;143:359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Legros F., Lombès A., Frachon P., Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell. 2002;13:4343–4354. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meeusen S., DeVay R., Block J., Cassidy-Stone A., Wayson S., McCaffery J.M., Nunnari J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–395. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 67.Sesaki H., Jensen R.E. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J. Cell Biol. 2001;152:1123–1134. doi: 10.1083/jcb.152.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tilokani L., Nagashima S., Paupe V., Prudent J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018;62:341–360. doi: 10.1042/EBC20170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Varanita T., Soriano M.E., Romanello V., Zaglia T., Quintana-Cabrera R., Semenzato M., Menabò R., Costa V., Civiletto G., Pesce P. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015;21:834–844. doi: 10.1016/j.cmet.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Freund C., Schmalz H.G., Sticht J., Kühne R. Proline-rich sequence recognition domains (PRD): Ligands, function and inhibition. Handb. Exp. Pharmacol. 2008;186:407–429. doi: 10.1007/978-3-540-72843-6_17. [DOI] [PubMed] [Google Scholar]
- 71.Kameoka S., Adachi Y., Okamoto K., Iijima M., Sesaki H. Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. 2018;28:67–76. doi: 10.1016/j.tcb.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Q., Tamura Y., Roy M., Adachi Y., Iijima M., Sesaki H. Biosynthesis and roles of phospholipids in mitochondrial fusion, division and mitophagy. Cell. Mol. Life Sci. 2014;71:3767–3778. doi: 10.1007/s00018-014-1648-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baba T., Kashiwagi Y., Arimitsu N., Kogure T., Edo A., Maruyama T., Nakao K., Nakanishi H., Kinoshita M., Frohman M.A. Phosphatidic acid (PA)-preferring phospholipase A1 regulates mitochondrial dynamics. J. Biol. Chem. 2014;289:11497–11511. doi: 10.1074/jbc.M113.531921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adachi Y., Itoh K., Yamada T., Cerveny K.L., Suzuki T.L., Macdonald P., Frohman M.A., Ramachandran R., Iijima M., Sesaki H. Coincident phosphatidic acid interaction restrains Drp1 in mitochondrial division. Mol. Cell. 2016;63:1034–1043. doi: 10.1016/j.molcel.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frohman M.A. Role of mitochondrial lipids in guiding fission and fusion. J. Mol. Med. (Berl.) 2015;93:263–269. doi: 10.1007/s00109-014-1237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Joshi A.S., Thompson M.N., Fei N., Hüttemann M., Greenberg M.L. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J. Biol. Chem. 2012;287:17589–17597. doi: 10.1074/jbc.M111.330167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Formosa L.E., Ryan M.T. Mitochondrial fusion: Reaching the end of mitofusin’s tether. J. Cell Biol. 2016;215:597–598. doi: 10.1083/jcb.201611048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bleazard W., McCaffery J.M., King E.J., Bale S., Mozdy A., Tieu Q., Nunnari J., Shaw J.M. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo Q., Koirala S., Perkins E.M., McCaffery J.M., Shaw J.M. The mitochondrial fission adaptors Caf4 and Mdv1 are not functionally equivalent. PLoS ONE. 2012;7:e53523. doi: 10.1371/journal.pone.0053523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cerveny K.L., Studer S.L., Jensen R.E., Sesaki H. Yeast mitochondrial division and distribution require the cortical Num1 protein. Dev. Cell. 2007;12:363–375. doi: 10.1016/j.devcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 81.Hammermeister M., Schödel K., Westermann B. Mdm36 is a mitochondrial fission-promoting protein in Saccharomyces cerevisiae. Mol. Biol. Cell. 2010;21:2443–2452. doi: 10.1091/mbc.E10-02-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Messerschmitt M., Jakobs S., Vogel F., Fritz S., Dimmer K.S., Neupert W., Westermann B. The inner membrane protein Mdm33 controls mitochondrial morphology in yeast. J. Cell Biol. 2003;160:553–564. doi: 10.1083/jcb.200211113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fröhlich C., Grabiger S., Schwefel D., Faelber K., Rosenbaum E., Mears J., Rocks O., Daumke O. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013;32:1280–1292. doi: 10.1038/emboj.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Losón O.C., Song Z., Chen H., Chan D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell. 2013;24:659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gandre-Babbe S., van der Bliek A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Otera H., Wang C., Cleland M.M., Setoguchi K., Yokota S., Youle R.J., Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Palmer C.S., Osellame L.D., Laine D., Koutsopoulos O.S., Frazier A.E., Ryan M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu R., Chan D.C. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol. Biol. Cell. 2015;26:4466–4477. doi: 10.1091/mbc.E15-08-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Samangouei P., Crespo-Avilan G.E., Cabrera-Fuentes H., Hernández-Reséndiz S., Ismail N.I., Katwadi K.B., Boisvert W.A., Hausenloy D.J. MiD49 and MiD51: New mediators of mitochondrial fission and novel targets for cardioprotection. Cond. Med. 2018;1:239–246. [PMC free article] [PubMed] [Google Scholar]
- 90.Lee J.E., Westrate L.M., Wu H., Page C., Voeltz G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. 2016;540:139–143. doi: 10.1038/nature20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Doyon J.B., Zeitler B., Cheng J., Cheng A.T., Cherone J.M., Santiago Y., Lee A.H., Vo T.D., Doyon Y., Miller J.C. Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nat. Cell Biol. 2011;13:331–337. doi: 10.1038/ncb2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Joshi A.U., Mochly-Rosen D. Mortal engines: Mitochondrial bioenergetics and dysfunction in neurodegenerative diseases. Pharmacol. Res. 2018;138:2–15. doi: 10.1016/j.phrs.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lackner L.L., Ping H., Graef M., Murley A., Nunnari J. Endoplasmic reticulum-associated mitochondria-cortex tether functions in the distribution and inheritance of mitochondria. Proc. Natl. Acad. Sci. USA. 2013;110:E458–E467. doi: 10.1073/pnas.1215232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ji W.K., Chakrabarti R., Fan X., Schoenfeld L., Strack S., Higgs H.N. Receptor-mediated Drp1 oligomerization on endoplasmic reticulum. J. Cell Biol. 2017;216:4123–4139. doi: 10.1083/jcb.201610057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gal A., Balicza P., Weaver D., Naghdi S., Joseph S.K., Várnai P., Gyuris T., Horváth A., Nagy L., Seifert E.L. MSTO1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol. Med. 2017;9:967–984. doi: 10.15252/emmm.201607058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nasca A., Scotton C., Zaharieva I., Neri M., Selvatici R., Magnusson O.T., Gal A., Weaver D., Rossi R., Armaroli A. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum. Mutat. 2017;38:970–977. doi: 10.1002/humu.23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Norton M., Ng A.C., Baird S., Dumoulin A., Shutt T., Mah N., Andrade-Navarro M.A., McBride H.M., Screaton R.A. ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci. Signal. 2014;7:ra10. doi: 10.1126/scisignal.2004374. [DOI] [PubMed] [Google Scholar]
- 98.Wang K., Gan T.Y., Li N., Liu C.Y., Zhou L.Y., Gao J.N., Chen C., Yan K.W., Ponnusamy M., Zhang Y.H., Li P.F. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 2017;24:1111–1120. doi: 10.1038/cdd.2017.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aung L.H.H., Li R., Prabhakar B.S., Maker A.V., Li P. Mitochondrial protein 18 (MTP18) plays a pro-apoptotic role in chemotherapy-induced gastric cancer cell apoptosis. Oncotarget. 2017;8:56582–56597. doi: 10.18632/oncotarget.17508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kreymerman A., Buickians D.N., Nahmou M.M., Tran T., Galvao J., Wang Y., Sun N., Bazik L., Huynh S.K., Cho I.J. MTP18 is a novel regulator of mitochondrial fission in CNS neuron development, axonal growth, and injury responses. Sci. Rep. 2019;9:10669. doi: 10.1038/s41598-019-46956-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nakamura N., Kimura Y., Tokuda M., Honda S., Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–1022. doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Park Y.Y., Lee S., Karbowski M., Neutzner A., Youle R.J., Cho H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J. Cell Sci. 2010;123:619–626. doi: 10.1242/jcs.061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Korobova F., Ramabhadran V., Higgs H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–467. doi: 10.1126/science.1228360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Niemann A., Ruegg M., La Padula V., Schenone A., Suter U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J. Cell Biol. 2005;170:1067–1078. doi: 10.1083/jcb.200507087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pedrola L., Espert A., Wu X., Claramunt R., Shy M.E., Palau F. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum. Mol. Genet. 2005;14:1087–1094. doi: 10.1093/hmg/ddi121. [DOI] [PubMed] [Google Scholar]
- 106.Suda K., Ueoka I., Azuma Y., Muraoka Y., Yoshida H., Yamaguchi M. Novel Drosophila model for mitochondrial diseases by targeting of a solute carrier protein SLC25A46. Brain Res. 2018;1689:30–44. doi: 10.1016/j.brainres.2018.03.028. [DOI] [PubMed] [Google Scholar]
- 107.Yu S.B., Pekkurnaz G. Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J. Mol. Biol. 2018;430:3922–3941. doi: 10.1016/j.jmb.2018.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Silva Ramos E., Motori E., Brüser C., Kühl I., Yeroslaviz A., Ruzzenente B., Kauppila J.H.K., Busch J.D., Hultenby K., Habermann B.H. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 2019;15:e1008085. doi: 10.1371/journal.pgen.1008085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oh C.M., Ryu D., Cho S., Jang Y. Mitochondrial quality control in the heart: New drug targets for cardiovascular disease. Korean Circ. J. 2020;50:395–405. doi: 10.4070/kcj.2019.0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ježek J., Cooper K.F., Strich R. Reactive oxygen species and mitochondrial Dynamics: The yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants. 2018;7:13. doi: 10.3390/antiox7010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Szabadkai G., Simoni A.M., Bianchi K., De Stefani D., Leo S., Wieckowski M.R., Rizzuto R. Mitochondrial dynamics and Ca2+ signaling. Biochim. Biophys. Acta. 2006;1763:442–449. doi: 10.1016/j.bbamcr.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 112.Seo B.J., Yoon S.H., Do J.T. Mitochondrial dynamics in stem cells and differentiation. Int. J. Mol. Sci. 2018;19:3893. doi: 10.3390/ijms19123893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Twig G., Elorza A., Molina A.J.A., Mohamed H., Wikstrom J.D., Walzer G., Stiles L., Haigh S.E., Katz S., Las G. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Suen D.F., Norris K.L., Youle R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Twig G., Shirihai O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011;14:1939–1951. doi: 10.1089/ars.2010.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Geisler S., Holmström K.M., Treis A., Skujat D., Weber S.S., Fiesel F.C., Kahle P.J., Springer W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6:871–878. doi: 10.4161/auto.6.7.13286. [DOI] [PubMed] [Google Scholar]
- 117.Parone P.A., Da Cruz S., Tondera D., Mattenberger Y., James D.I., Maechler P., Barja F., Martinou J.-C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE. 2008;3:e3257. doi: 10.1371/journal.pone.0003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Neubauer S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 119.Griparic L., Kanazawa T., van der Bliek A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ehses S., Raschke I., Mancuso G., Bernacchia A., Geimer S., Tondera D., Martinou J.C., Westermann B., Rugarli E.I., Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McBride H., Soubannier V. Mitochondrial function: OMA1 and OPA1, the grandmasters of mitochondrial health. Curr. Biol. 2010;20:R274–R276. doi: 10.1016/j.cub.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 122.Quirós P.M., Ramsay A.J., Sala D., Fernández-Vizarra E., Rodríguez F., Peinado J.R., Fernández-García M.S., Vega J.A., Enríquez J.A., Zorzano A., López-Otín C. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012;31:2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Parra V., Verdejo H.E., Iglewski M., Del Campo A., Troncoso R., Jones D., Zhu Y., Kuzmicic J., Pennanen C., Lopez-Crisosto C. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 signaling pathway. Diabetes. 2014;63:75–88. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Legros F., Malka F., Frachon P., Lombès A., Rojo M. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 2004;117:2653–2662. doi: 10.1242/jcs.01134. [DOI] [PubMed] [Google Scholar]
- 125.Ota A., Ishihara T., Ishihara N. Mitochondrial nucleoid morphology and respiratory function are altered in Drp1-deficient HeLa cells. J. Biochem. 2020;167:287–294. doi: 10.1093/jb/mvz112. [DOI] [PubMed] [Google Scholar]
- 126.Bao D., Zhao J., Zhou X., Yang Q., Chen Y., Zhu J., Yuan P., Yang J., Qin T., Wan S., Xing J. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene. 2019;38:5007–5020. doi: 10.1038/s41388-019-0772-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen H., Vermulst M., Wang Y.E., Chomyn A., Prolla T.A., McCaffery J.M., Chan D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen H., McCaffery J.M., Chan D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 129.Pich S., Bach D., Briones P., Liesa M., Camps M., Testar X., Palacín M., Zorzano A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005;14:1405–1415. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 130.Chen L., Gong Q., Stice J.P., Knowlton A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009;84:91–99. doi: 10.1093/cvr/cvp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Billia F., Hauck L., Konecny F., Rao V., Shen J., Mak T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA. 2011;108:9572–9577. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kubli D.A., Zhang X., Lee Y., Hanna R.A., Quinsay M.N., Nguyen C.K., Jimenez R., Petrosyan S., Murphy A.N., Gustafsson A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bhandari P., Song M., Chen Y., Burelle Y., Dorn G.W., 2nd Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ. Res. 2014;114:257–265. doi: 10.1161/CIRCRESAHA.114.302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen Y., Dorn G.W., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Song M., Gong G., Burelle Y., Gustafsson Å.B., Kitsis R.N., Matkovich S.J., Dorn G.W., 2nd Interdependence of Parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ. Res. 2015;117:346–351. doi: 10.1161/CIRCRESAHA.117.306859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ikeda Y., Shirakabe A., Maejima Y., Zhai P., Sciarretta S., Toli J., Nomura M., Mihara K., Egashira K., Ohishi M. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015;116:264–278. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 137.Chen M., Chen Z., Wang Y., Tan Z., Zhu C., Li Y., Han Z., Chen L., Gao R., Liu L., Chen Q. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12:689–702. doi: 10.1080/15548627.2016.1151580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pei S., Minhajuddin M., Adane B., Khan N., Stevens B.M., Mack S.C., Lai S., Rich J.N., Inguva A., Shannon K.M. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell. 2018;23:86–100.e6. doi: 10.1016/j.stem.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xian H., Yang Q., Xiao L., Shen H.-M., Liou Y.-C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 2019;10:2059. doi: 10.1038/s41467-019-10096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ong S.B., Hall A.R., Hausenloy D.J. Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox Signal. 2013;19:400–414. doi: 10.1089/ars.2012.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sharp W.W., Archer S.L. Mitochondrial dynamics in cardiovascular disease: Fission and fusion foretell form and function. J. Mol. Med. (Berl.) 2015;93:225–228. doi: 10.1007/s00109-015-1258-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Serasinghe M.N., Chipuk J.E. Mitochondrial fission in human diseases. Handb. Exp. Pharmacol. 2017;240:159–188. doi: 10.1007/164_2016_38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Santos D., Esteves A.R., Silva D.F., Januário C., Cardoso S.M. The impact of mitochondrial fusion and fission modulation in sporadic Parkinson’s disease. Mol. Neurobiol. 2015;52:573–586. doi: 10.1007/s12035-014-8893-4. [DOI] [PubMed] [Google Scholar]
- 144.Liu Y.J., McIntyre R.L., Janssens G.E., Houtkooper R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020;186:111212. doi: 10.1016/j.mad.2020.111212. [DOI] [PubMed] [Google Scholar]
- 145.Biala A.K., Dhingra R., Kirshenbaum L.A. Mitochondrial dynamics: Orchestrating the journey to advanced age. J. Mol. Cell. Cardiol. 2015;83:37–43. doi: 10.1016/j.yjmcc.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 146.Reed J.C. Bcl-2 family proteins. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 147.Li J., Li Y., Jiao J., Wang J., Li Y., Qin D., Li P. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol. Cell. Biol. 2014;34:1788–1799. doi: 10.1128/MCB.00774-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Aung L.H.H., Li Y.Z., Yu H., Chen X., Yu Z., Gao J., Li P. Mitochondrial protein 18 is a positive apoptotic regulator in cardiomyocytes under oxidative stress. Clin. Sci. (Lond.) 2019;133:1067–1084. doi: 10.1042/CS20190125. [DOI] [PubMed] [Google Scholar]
- 149.Xu S., Cherok E., Das S., Li S., Roelofs B.A., Ge S.X., Polster B.M., Boyman L., Lederer W.J., Wang C., Karbowski M. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol. Biol. Cell. 2016;27:349–359. doi: 10.1091/mbc.E15-09-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang J., Aung L.H., Prabhakar B.S., Li P. The mitochondrial ubiquitin ligase plays an anti-apoptotic role in cardiomyocytes by regulating mitochondrial fission. J. Cell. Mol. Med. 2016;20:2278–2288. doi: 10.1111/jcmm.12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen Y., Liu Y., Dorn G.W., 2nd Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Piquereau J., Caffin F., Novotova M., Prola A., Garnier A., Mateo P., Fortin D., Huynh H., Nicolas V., Alavi M.V. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc. Res. 2012;94:408–417. doi: 10.1093/cvr/cvs117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tang Y., Mi C., Liu J., Gao F., Long J. Compromised mitochondrial remodeling in compensatory hypertrophied myocardium of spontaneously hypertensive rat. Cardiovasc. Pathol. 2014;23:101–106. doi: 10.1016/j.carpath.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 154.Das R., Chakrabarti O. Mitochondrial hyperfusion: A friend or a foe. Biochem. Soc. Trans. 2020;48:631–644. doi: 10.1042/BST20190987. [DOI] [PubMed] [Google Scholar]
- 155.Dorn G.W., 2nd Mitochondrial dynamism and heart disease: Changing shape and shaping change. EMBO Mol. Med. 2015;7:865–877. doi: 10.15252/emmm.201404575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fang L., Moore X.L., Gao X.M., Dart A.M., Lim Y.L., Du X.J. Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life Sci. 2007;80:2154–2160. doi: 10.1016/j.lfs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 157.Yu H., Guo Y., Mi L., Wang X., Li L., Gao W. Mitofusin 2 inhibits angiotensin II-induced myocardial hypertrophy. J. Cardiovasc. Pharmacol. Ther. 2011;16:205–211. doi: 10.1177/1074248410385683. [DOI] [PubMed] [Google Scholar]
- 158.Xiong W., Ma Z., An D., Liu Z., Cai W., Bai Y., Zhan Q., Lai W., Zeng Q., Ren H., Xu D. Mitofusin 2 participates in mitophagy and mitochondrial fusion against angiotensin II-induced cardiomyocyte injury. Front. Physiol. 2019;10:411. doi: 10.3389/fphys.2019.00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhou B., Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Invest. 2018;128:3716–3726. doi: 10.1172/JCI120849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dorn G.W., 2nd, Clark C.F., Eschenbacher W.H., Kang M.-Y., Engelhard J.T., Warner S.J., Matkovich S.J., Jowdy C.C. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ. Res. 2011;108:12–17. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chen L., Liu T., Tran A., Lu X., Tomilov A.A., Davies V., Cortopassi G., Chiamvimonvat N., Bers D.M., Votruba M., Knowlton A.A. OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 2012;1:e003012. doi: 10.1161/JAHA.112.003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chalmers S., Saunter C., Wilson C., Coats P., Girkin J.M., McCarron J.G. Mitochondrial motility and vascular smooth muscle proliferation. Arterioscler. Thromb. Vasc. Biol. 2012;32:3000–3011. doi: 10.1161/ATVBAHA.112.255174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chang Y.W., Chang Y.T., Wang Q., Lin J.J., Chen Y.J., Chen C.C. Quantitative phosphoproteomic study of pressure-overloaded mouse heart reveals dynamin-related protein 1 as a modulator of cardiac hypertrophy. Mol. Cell. Proteomics. 2013;12:3094–3107. doi: 10.1074/mcp.M113.027649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hasan P., Saotome M., Ikoma T., Iguchi K., Kawasaki H., Iwashita T., Hayashi H., Maekawa Y. Mitochondrial fission protein, dynamin-related protein 1, contributes to the promotion of hypertensive cardiac hypertrophy and fibrosis in Dahl-salt sensitive rats. J. Mol. Cell. Cardiol. 2018;121:103–106. doi: 10.1016/j.yjmcc.2018.07.004. [DOI] [PubMed] [Google Scholar]
- 165.Aung L.H.H., Li R., Prabhakar B.S., Li P. Knockdown of Mtfp1 can minimize doxorubicin cardiotoxicity by inhibiting Dnm1l-mediated mitochondrial fission. J. Cell. Mol. Med. 2017;21:3394–3404. doi: 10.1111/jcmm.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Osterholt M., Nguyen T.D., Schwarzer M., Doenst T. Alterations in mitochondrial function in cardiac hypertrophy and heart failure. Heart Fail. Rev. 2013;18:645–656. doi: 10.1007/s10741-012-9346-7. [DOI] [PubMed] [Google Scholar]
- 167.Adaniya S.M., O-Uchi J., Cypress M.W., Kusakari Y., Jhun B.S. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am. J. Physiol. Cell Physiol. 2019;316:C583–C604. doi: 10.1152/ajpcell.00523.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schaper J., Froede R., Hein S., Buck A., Hashizume H., Speiser B., Friedl A., Bleese N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83:504–514. doi: 10.1161/01.cir.83.2.504. [DOI] [PubMed] [Google Scholar]
- 169.Dorn G., II Mitochondrial fission/fusion and cardiomyopathy. Curr. Opin. Genet. Dev. 2016;38:38–44. doi: 10.1016/j.gde.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Martin O.J., Lai L., Soundarapandian M.M., Leone T.C., Zorzano A., Keller M.P., Attie A.D., Muoio D.M., Kelly D.P. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 2014;114:626–636. doi: 10.1161/CIRCRESAHA.114.302562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Müller-Rischart A.K., Pilsl A., Beaudette P., Patra M., Hadian K., Funke M., Peis R., Deinlein A., Schweimer C., Kuhn P.H. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol. Cell. 2013;49:908–921. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- 172.Liu C.F., Tang W.H.W. Epigenetics in cardiac hypertrophy and heart failure. JACC Basic Transl. Sci. 2019;4:976–993. doi: 10.1016/j.jacbts.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Papanicolaou K.N., Khairallah R.J., Ngoh G.A., Chikando A., Luptak I., O’Shea K.M., Riley D.D., Lugus J.J., Colucci W.S., Lederer W.J. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ranieri M., Del Bo R., Bordoni A., Ronchi D., Colombo I., Riboldi G., Cosi A., Servida M., Magri F., Moggio M. Optic atrophy plus phenotype due to mutations in the OPA1 gene: Two more Italian families. J. Neurol. Sci. 2012;315:146–149. doi: 10.1016/j.jns.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Olichon A., Guillou E., Delettre C., Landes T., Arnauné-Pelloquin L., Emorine L.J., Mils V., Daloyau M., Hamel C., Amati-Bonneau P. Mitochondrial dynamics and disease, OPA1. Biochim. Biophys. Acta. 2006;1763:500–509. doi: 10.1016/j.bbamcr.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 176.Feely S.M.E., Laura M., Siskind C.E., Sottile S., Davis M., Gibbons V.S., Reilly M.M., Shy M.E. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology. 2011;76:1690–1696. doi: 10.1212/WNL.0b013e31821a441e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ashrafian H., Docherty L., Leo V., Towlson C., Neilan M., Steeples V., Lygate C.A., Hough T., Townsend S., Williams D. A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. 2010;6:e1001000. doi: 10.1371/journal.pgen.1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Delettre C., Griffoin J.M., Kaplan J., Dollfus H., Lorenz B., Faivre L., Lenaers G., Belenguer P., Hamel C.P. Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 2001;109:584–591. doi: 10.1007/s00439-001-0633-y. [DOI] [PubMed] [Google Scholar]
- 179.Strack S., Wilson T.J., Cribbs J.T. Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J. Cell Biol. 2013;201:1037–1051. doi: 10.1083/jcb.201210045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Elachouri G., Vidoni S., Zanna C., Pattyn A., Boukhaddaoui H., Gaget K., Yu-Wai-Man P., Gasparre G., Sarzi E., Delettre C. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 2011;21:12–20. doi: 10.1101/gr.108696.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Holliday R. DNA methylation and epigenetic inheritance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990;326:329–338. doi: 10.1098/rstb.1990.0015. [DOI] [PubMed] [Google Scholar]
- 182.Rando O.J., Verstrepen K.J. Timescales of genetic and epigenetic inheritance. Cell. 2007;128:655–668. doi: 10.1016/j.cell.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 183.Harvey A.J. Mitochondria in early development: Linking the microenvironment, metabolism and the epigenome. Reproduction. 2019;157:R159–R179. doi: 10.1530/REP-18-0431. [DOI] [PubMed] [Google Scholar]
- 184.Handy D.E., Castro R., Loscalzo J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation. 2011;123:2145–2156. doi: 10.1161/CIRCULATIONAHA.110.956839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jones P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 186.Movassagh M., Choy M.K., Knowles D.A., Cordeddu L., Haider S., Down T., Siggens L., Vujic A., Simeoni I., Penkett C. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124:2411–2422. doi: 10.1161/CIRCULATIONAHA.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kanherkar R.R., Bhatia-Dey N., Csoka A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014;2:49. doi: 10.3389/fcell.2014.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Warren J.S., Oka S.I., Zablocki D., Sadoshima J. Metabolic reprogramming via PPARα signaling in cardiac hypertrophy and failure: From metabolomics to epigenetics. Am. J. Physiol. Heart Circ. Physiol. 2017;313:H584–H596. doi: 10.1152/ajpheart.00103.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Papait R., Cattaneo P., Kunderfranco P., Greco C., Carullo P., Guffanti A., Viganò V., Stirparo G.G., Latronico M.V.G., Hasenfuss G. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 2013;110:20164–20169. doi: 10.1073/pnas.1315155110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Dong Y., Xu S., Liu J., Ponnusamy M., Zhao Y., Zhang Y., Wang Q., Li P., Wang K. Non-coding RNA-linked epigenetic regulation in cardiac hypertrophy. Int. J. Biol. Sci. 2018;14:1133–1141. doi: 10.7150/ijbs.26215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wang L., Wang J., Li G., Xiao J. Non-coding RNAs in physiological cardiac hypertrophy. Adv. Exp. Med. Biol. 2020;1229:149–161. doi: 10.1007/978-981-15-1671-9_8. [DOI] [PubMed] [Google Scholar]
- 192.Li Y., Liang Y., Zhu Y., Zhang Y., Bei Y. Noncoding RNAs in cardiac hypertrophy. J. Cardiovasc. Transl. Res. 2018;11:439–449. doi: 10.1007/s12265-018-9797-x. [DOI] [PubMed] [Google Scholar]
- 193.Ottaviani L., da Costa Martins P.A. Non-coding RNAs in cardiac hypertrophy. J. Physiol. 2017;595:4037–4050. doi: 10.1113/JP273129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dangwal S., Schimmel K., Foinquinos A., Xiao K., Thum T. Noncoding RNAs in heart failure. Handb. Exp. Pharmacol. 2017;243:423–445. doi: 10.1007/164_2016_99. [DOI] [PubMed] [Google Scholar]
- 195.Xie Y., Hu J., Zhang X., Li C., Zuo Y., Xie S., Zhang Z., Zhu S. Neuropeptide Y induces cardiomyocyte hypertrophy via attenuating miR-29a-3p in neonatal rat cardiomyocytes. Protein Pept. Lett. 2020;27:878–887. doi: 10.2174/0929866527666200416144459. [DOI] [PubMed] [Google Scholar]
- 196.Sotomayor-Flores C., Rivera-Mejías P., Vásquez-Trincado C., López-Crisosto C., Morales P.E., Pennanen C., Polakovicova I., Aliaga-Tobar V., García L., Roa J.C. Angiotensin-(1–9) prevents cardiomyocyte hypertrophy by controlling mitochondrial dynamics via miR-129-3p/PKIA pathway. Cell Death Differ. 2020;27:2586–2604. doi: 10.1038/s41418-020-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhao Y., Ponnusamy M., Liu C., Tian J., Dong Y., Gao J., Wang C., Zhang Y., Zhang L., Wang K., Li P. miR-485-5p modulates mitochondrial fission through targeting mitochondrial anchored protein ligase in cardiac hypertrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2017;1863:2871–2881. doi: 10.1016/j.bbadis.2017.07.034. [DOI] [PubMed] [Google Scholar]
- 198.Qiu Y., Cheng R., Liang C., Yao Y., Zhang W., Zhang J., Zhang M., Li B., Xu C., Zhang R. MicroRNA-20b promotes cardiac hypertrophy by the inhibition of mitofusin 2-mediated inter-organelle Ca2+ cross-talk. Mol. Ther. Nucleic Acids. 2020;19:1343–1356. doi: 10.1016/j.omtn.2020.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wang T., Zhai M., Xu S., Ponnusamy M., Huang Y., Liu C.Y., Wang M., Shan C., Shan P.P., Gao X.Q. NFATc3-dependent expression of miR-153-3p promotes mitochondrial fragmentation in cardiac hypertrophy by impairing mitofusin-1 expression. Theranostics. 2020;10:553–566. doi: 10.7150/thno.37181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bronze-da-Rocha E. MicroRNAs expression profiles in cardiovascular diseases. BioMed Res. Int. 2014;2014:985408. doi: 10.1155/2014/985408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Prabakaran S., Lippens G., Steen H., Gunawardena J. Post-translational modification: Nature’s escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012;4:565–583. doi: 10.1002/wsbm.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ikeda Y., Shirakabe A., Brady C., Zablocki D., Ohishi M., Sadoshima J. Molecular mechanisms mediating mitochondrial dynamics and mitophagy and their functional roles in the cardiovascular system. J. Mol. Cell. Cardiol. 2015;78:116–122. doi: 10.1016/j.yjmcc.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sharp W.W. Dynamin-related protein 1 as a therapeutic target in cardiac arrest. J. Mol. Med. (Berl.) 2015;93:243–252. doi: 10.1007/s00109-015-1257-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cereghetti G.M., Stangherlin A., Martins de Brito O., Chang C.R., Blackstone C., Bernardi P., Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chou C.H., Lin C.C., Yang M.C., Wei C.C., Liao H.D., Lin R.C., Tu W.Y., Kao T.C., Hsu C.M., Cheng J.T. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE. 2012;7:e49112. doi: 10.1371/journal.pone.0049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sharp W.W., Fang Y.H., Han M., Zhang H.J., Hong Z., Banathy A., Morrow E., Ryan J.J., Archer S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014;28:316–326. doi: 10.1096/fj.12-226225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cribbs J.T., Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Din S., Mason M., Völkers M., Johnson B., Cottage C.T., Wang Z., Joyo A.Y., Quijada P., Erhardt P., Magnuson N.S. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc. Natl. Acad. Sci. USA. 2013;110:5969–5974. doi: 10.1073/pnas.1213294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Han X.J., Lu Y.F., Li S.A., Kaitsuka T., Sato Y., Tomizawa K., Nairn A.C., Takei K., Matsui H., Matsushita M. CaM kinase Iα-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wang W., Wang Y., Long J., Wang J., Haudek S.B., Overbeek P., Chang B.H., Schumacker P.T., Danesh F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186–200. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Torres G., Morales P.E., García-Miguel M., Norambuena-Soto I., Cartes-Saavedra B., Vidal-Peña G., Moncada-Ruff D., Sanhueza-Olivares F., San Martín A., Chiong M. Glucagon-like peptide-1 inhibits vascular smooth muscle cell dedifferentiation through mitochondrial dynamics regulation. Biochem. Pharmacol. 2016;104:52–61. doi: 10.1016/j.bcp.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yu T., Jhun B.S., Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid. Redox Signal. 2011;14:425–437. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Disatnik M.H., Ferreira J.C., Campos J.C., Gomes K.S., Dourado P.M., Qi X., Mochly-Rosen D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013;2:e000461. doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Toyama E.Q., Herzig S., Courchet J., Lewis T.L., Jr., Losón O.C., Hellberg K., Young N.P., Chen H., Polleux F., Chan D.C., Shaw R.J. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Pyakurel A., Savoia C., Hess D., Scorrano L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell. 2015;58:244–254. doi: 10.1016/j.molcel.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Shiiba I., Takeda K., Nagashima S., Yanagi S. Overview of mitochondrial E3 ubiquitin ligase MITOL/MARCH5 from molecular mechanisms to diseases. Int. J. Mol. Sci. 2020;21:3781. doi: 10.3390/ijms21113781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cherok E., Xu S., Li S., Das S., Meltzer W.A., Zalzman M., Wang C., Karbowski M. Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5-dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol. Biol. Cell. 2017;28:396–410. doi: 10.1091/mbc.E16-04-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Park Y.Y., Nguyen O.T., Kang H., Cho H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 2014;5:e1172. doi: 10.1038/cddis.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wang H., Song P., Du L., Tian W., Yue W., Liu M., Li D., Wang B., Zhu Y., Cao C. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 2011;286:11649–11658. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Leboucher G.P., Tsai Y.C., Yang M., Shaw K.C., Zhou M., Veenstra T.D., Glickman M.H., Weissman A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell. 2012;47:547–557. doi: 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sarge K.D., Park-Sarge O.-K. SUMO and its role in human diseases. Int. Rev. Cell Mol. Biol. 2011;288:167–183. doi: 10.1016/B978-0-12-386041-5.00004-2. [DOI] [PubMed] [Google Scholar]
- 222.Yan K., Wang K., Li P. The role of post-translational modifications in cardiac hypertrophy. J. Cell. Mol. Med. 2019;23:3795–3807. doi: 10.1111/jcmm.14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Braschi E., Zunino R., McBride H.M. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wasiak S., Zunino R., McBride H.M. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Guo C., Hildick K.L., Luo J., Dearden L., Wilkinson K.A., Henley J.M. SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 2013;32:1514–1528. doi: 10.1038/emboj.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wright J.N., Collins H.E., Wende A.R., Chatham J.C. O-GlcNAcylation and cardiovascular disease. Biochem. Soc. Trans. 2017;45:545–553. doi: 10.1042/BST20160164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Nie H., Yi W. O-GlcNAcylation, a sweet link to the pathology of diseases. J. Zhejiang Univ. Sci. B. 2019;20:437–448. doi: 10.1631/jzus.B1900150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gawlowski T., Suarez J., Scott B., Torres-Gonzalez M., Wang H., Schwappacher R., Han X., Yates J.R., 3rd, Hoshijima M., Dillmann W. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J. Biol. Chem. 2012;287:30024–30034. doi: 10.1074/jbc.M112.390682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Jin J.Y., Wei X.X., Zhi X.L., Wang X.H., Meng D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin. 2020;42:655–664. doi: 10.1038/s41401-020-00518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ding F., Yu L., Wang M., Xu S., Xia Q., Fu G. O-GlcNAcylation involvement in high glucose-induced cardiac hypertrophy via ERK1/2 and cyclin D2. Amino Acids. 2013;45:339–349. doi: 10.1007/s00726-013-1504-2. [DOI] [PubMed] [Google Scholar]
- 231.Gélinas R., Mailleux F., Dontaine J., Bultot L., Demeulder B., Ginion A., Daskalopoulos E.P., Esfahani H., Dubois-Deruy E., Lauzier B. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 2018;9:374. doi: 10.1038/s41467-017-02795-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Makino A., Suarez J., Gawlowski T., Han W., Wang H., Scott B.T., Dillmann W.H. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;300:R1296–R1302. doi: 10.1152/ajpregu.00437.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Massion P.B., Feron O., Dessy C., Balligand J.-L. Nitric oxide and cardiac function: Ten years after, and continuing. Circ. Res. 2003;93:388–398. doi: 10.1161/01.RES.0000088351.58510.21. [DOI] [PubMed] [Google Scholar]
- 234.Jeong E.M., Jin C.Z., Jang J.H., Zhao Z.H., Jin C.L., Lee J.H., Lee K.B., Kim S.J., Kim I.G., Zhang Y.H. S-nitrosylation of transglutaminase 2 impairs fatty acid-stimulated contraction in hypertensive cardiomyocytes. Exp. Mol. Med. 2018;50:1–11. doi: 10.1038/s12276-017-0021-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Cho D.H., Nakamura T., Fang J., Cieplak P., Godzik A., Gu Z., Lipton S.A. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lee D.S., Kim J.E. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. 2018;9:869. doi: 10.1038/s41419-018-0910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ali I., Conrad R.J., Verdin E., Ott M. Lysine acetylation goes global: From epigenetics to metabolism and therapeutics. Chem. Rev. 2018;118:1216–1252. doi: 10.1021/acs.chemrev.7b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Xu S., Kamato D., Little P.J., Nakagawa S., Pelisek J., Jin Z.G. Targeting epigenetics and non-coding RNAs in atherosclerosis: From mechanisms to therapeutics. Pharmacol. Ther. 2019;196:15–43. doi: 10.1016/j.pharmthera.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kane A.E., Sinclair D.A. Sirtuins and NAD+ in the development and treatment of metabolic and cardiovascular diseases. Circ. Res. 2018;123:868–885. doi: 10.1161/CIRCRESAHA.118.312498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Biel T.G., Lee S., Flores-Toro J.A., Dean J.W., Go K.L., Lee M.H., Law B.K., Law M.E., Dunn W.A., Jr., Zendejas I. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner. Cell Death Differ. 2016;23:279–290. doi: 10.1038/cdd.2015.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ding M., Feng N., Tang D., Feng J., Li Z., Jia M., Liu Z., Gu X., Wang Y., Fu F., Pei J. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J. Pineal Res. 2018;65:e12491. doi: 10.1111/jpi.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.MacVicar T., Langer T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016;129:2297–2306. doi: 10.1242/jcs.159186. [DOI] [PubMed] [Google Scholar]
- 243.Song Z., Chen H., Fiket M., Alexander C., Chan D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wai T., García-Prieto J., Baker M.J., Merkwirth C., Benit P., Rustin P., Rupérez F.J., Barbas C., Ibañez B., Langer T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. 2015;350:aad0116. doi: 10.1126/science.aad0116. [DOI] [PubMed] [Google Scholar]
- 245.Harvey P.A., Leinwand L.A. The cell biology of disease: Cellular mechanisms of cardiomyopathy. J. Cell Biol. 2011;194:355–365. doi: 10.1083/jcb.201101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Anderson T.S., Odden M., Penko J., Kazi D.S., Bellows B.K., Bibbins-Domingo K. Generalizability of clinical trials supporting the 2017 American College of Cardiology/American Heart Association blood pressure guideline. JAMA Intern. Med. 2020;180:795–797. doi: 10.1001/jamainternmed.2020.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Willenbrock R., Philipp S., Mitrovic V., Dietz R. Neurohumoral blockade in CHF management. J. Renin Angiotensin Aldosterone Syst. 2000;1(Suppl 1):24–30. doi: 10.3317/JRAAS.2000.030. [DOI] [PubMed] [Google Scholar]
- 248.Mehra M.R., Uber P.A., Francis G.S. Heart failure therapy at a crossroad: Are there limits to the neurohormonal model? J. Am. Coll. Cardiol. 2003;41:1606–1610. doi: 10.1016/s0735-1097(03)00245-6. [DOI] [PubMed] [Google Scholar]
- 249.Tham Y.K., Bernardo B.C., Ooi J.Y., Weeks K.L., McMullen J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015;89:1401–1438. doi: 10.1007/s00204-015-1477-x. [DOI] [PubMed] [Google Scholar]
- 250.Burchfield J.S., Xie M., Hill J.A. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Givvimani S., Munjal C., Tyagi N., Sen U., Metreveli N., Tyagi S.C. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS ONE. 2012;7:e32388. doi: 10.1371/journal.pone.0032388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Cassidy-Stone A., Chipuk J.E., Ingerman E., Song C., Yoo C., Kuwana T., Kurth M.J., Shaw J.T., Hinshaw J.E., Green D.R., Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ong S.B., Subrayan S., Lim S.Y., Yellon D.M., Davidson S.M., Hausenloy D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 254.Tian L., Neuber-Hess M., Mewburn J., Dasgupta A., Dunham-Snary K., Wu D., Chen K.H., Hong Z., Sharp W.W., Kutty S., Archer S.L. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J. Mol. Med. (Berl.) 2017;95:381–393. doi: 10.1007/s00109-017-1522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Pei H., Du J., Song X., He L., Zhang Y., Li X., Qiu C., Zhang Y., Hou J., Feng J. Melatonin prevents adverse myocardial infarction remodeling via Notch1/Mfn2 pathway. Free Radic. Biol. Med. 2016;97:408–417. doi: 10.1016/j.freeradbiomed.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 256.Ren X., Chen L., Xie J., Zhang Z., Dong G., Liang J., Liu L., Zhou H., Luo P. Resveratrol Ameliorates Mitochondrial Elongation via Drp1/Parkin/PINK1 Signaling in Senescent-Like Cardiomyocytes. Oxid. Med. Cell. Longev. 2017;2017:4175353. doi: 10.1155/2017/4175353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ma S., Feng J., Zhang R., Chen J., Han D., Li X., Yang B., Li X., Fan M., Li C. SIRT1 activation by resveratrol alleviates cardiac dysfunction via mitochondrial regulation in diabetic cardiomyopathy mice. Oxid. Med. Cell. Longev. 2017;2017:4602715. doi: 10.1155/2017/4602715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Hull T.D., Boddu R., Guo L., Tisher C.C., Traylor A.M., Patel B., Joseph R., Prabhu S.D., Suliman H.B., Piantadosi C.A. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight. 2016;1:e85817. doi: 10.1172/jci.insight.85817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Yu J., Wu J., Xie P., Maimaitili Y., Wang J., Xia Z., Gao F., Zhang X., Zheng H. Sevoflurane postconditioning attenuates cardiomyocyte hypoxia/reoxygenation injury via restoring mitochondrial morphology. PeerJ. 2016;4:e2659. doi: 10.7717/peerj.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Cellier L., Tamareille S., Kalakech H., Guillou S., Lenaers G., Prunier F., Mirebeau-Prunier D. Remote ischemic conditioning influences mitochondrial dynamics. Shock. 2016;45:192–197. doi: 10.1097/SHK.0000000000000500. [DOI] [PubMed] [Google Scholar]
- 261.Leshnower B.G., Kanemoto S., Matsubara M., Sakamoto H., Hinmon R., Gorman J.H., 3rd, Gorman R.C. Cyclosporine preserves mitochondrial morphology after myocardial ischemia/reperfusion independent of calcineurin inhibition. Ann. Thorac. Surg. 2008;86:1286–1292. doi: 10.1016/j.athoracsur.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ikeda G., Matoba T., Nakano Y., Nagaoka K., Ishikita A., Nakano K., Funamoto D., Sunagawa K., Egashira K. Nanoparticle-mediated targeting of cyclosporine A enhances cardioprotection against ischemia-reperfusion injury through inhibition of mitochondrial permeability transition pore opening. Sci. Rep. 2016;6:20467. doi: 10.1038/srep20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Knowlton A.A., Liu T.T. Mitochondrial dynamics and heart failure. Compr. Physiol. 2015;6:507–526. doi: 10.1002/cphy.c150022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Tieu Q., Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J. Cell Biol. 2000;151:353–366. doi: 10.1083/jcb.151.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Nagashima S., Tokuyama T., Yonashiro R., Inatome R., Yanagi S. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J. Biochem. 2014;155:273–279. doi: 10.1093/jb/mvu016. [DOI] [PubMed] [Google Scholar]
- 266.Karbowski M., Neutzner A., Youle R.J. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Tondera D., Czauderna F., Paulick K., Schwarzer R., Kaufmann J., Santel A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J. Cell Sci. 2005;118:3049–3059. doi: 10.1242/jcs.02415. [DOI] [PubMed] [Google Scholar]
- 268.Wang J.X., Jiao J.Q., Li Q., Long B., Wang K., Liu J.P., Li Y.R., Li P.F. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011;17:71–78. doi: 10.1038/nm.2282. [DOI] [PubMed] [Google Scholar]
- 269.Wang K., Long B., Jiao J.-Q., Wang J.-X., Liu J.-P., Li Q., Li P.-F. miR-484 regulates mitochondrial network through targeting Fis1. Nat. Commun. 2012;3:781. doi: 10.1038/ncomms1770. [DOI] [PubMed] [Google Scholar]
- 270.Hong Y., He H., Jiang G., Zhang H., Tao W., Ding Y., Yuan D., Liu J., Fan H., Lin F. miR-155-5p inhibition rejuvenates aged mesenchymal stem cells and enhances cardioprotection following infarction. Aging Cell. 2020;19:e13128. doi: 10.1111/acel.13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wang K., Zhang D.L., Long B., An T., Zhang J., Zhou L.Y., Liu C.Y., Li P.F. NFAT4-dependent miR-324-5p regulates mitochondrial morphology and cardiomyocyte cell death by targeting Mtfr1. Cell Death Dis. 2015;6:e2007. doi: 10.1038/cddis.2015.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Long B., Wang K., Li N., Murtaza I., Xiao J.Y., Fan Y.Y., Liu C.Y., Li W.H., Cheng Z., Li P. miR-761 regulates the mitochondrial network by targeting mitochondrial fission factor. Free Radic. Biol. Med. 2013;65:371–379. doi: 10.1016/j.freeradbiomed.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Li J., Donath S., Li Y., Qin D., Prabhakar B.S., Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010;6:e1000795. doi: 10.1371/journal.pgen.1000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Wang K., Liu C.Y., Zhang X.J., Feng C., Zhou L.Y., Zhao Y., Li P.F. miR-361-regulated prohibitin inhibits mitochondrial fission and apoptosis and protects heart from ischemia injury. Cell Death Differ. 2015;22:1058–1068. doi: 10.1038/cdd.2014.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Wang J.X., Zhang X.J., Feng C., Sun T., Wang K., Wang Y., Zhou L.Y., Li P.F. MicroRNA-532-3p regulates mitochondrial fission through targeting apoptosis repressor with caspase recruitment domain in doxorubicin cardiotoxicity. Cell Death Dis. 2015;6:e1677. doi: 10.1038/cddis.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wan Q., Xu T., Ding W., Zhang X., Ji X., Yu T., Yu W., Lin Z., Wang J. miR-499-5p attenuates mitochondrial fission and cell apoptosis via p21 in doxorubicin cardiotoxicity. Front. Genet. 2019;9:734. doi: 10.3389/fgene.2018.00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Wang K., Long B., Zhou L.-Y., Liu F., Zhou Q.-Y., Liu C.-Y., Fan Y.-Y., Li P.-F. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat. Commun. 2014;5:3596. doi: 10.1038/ncomms4596. [DOI] [PubMed] [Google Scholar]
- 278.Wang K., Sun T., Li N., Wang Y., Wang J.-X., Zhou L.-Y., Long B., Liu C.-Y., Liu F., Li P.-F. MDRL lncRNA regulates the processing of miR-484 primary transcript by targeting miR-361. PLoS Genet. 2014;10:e1004467. doi: 10.1371/journal.pgen.1004467. [DOI] [PMC free article] [PubMed] [Google Scholar]