

ABSTRACT
Hydroxyurea (HU) is classified as a ribonucleotide reductase (RNR) inhibitor and has been widely used to stall DNA replication by depleting deoxyribonucleoside triphosphate (dNTP) pools. Recent evidence in Escherichia coli shows that HU readily forms breakdown products that damage DNA directly, indicating that toxicity is a result of secondary effects. Because HU is so widely used in the laboratory and as a clinical therapeutic, it is important to understand its biological effects. To determine how Bacillus subtilis responds to HU-induced stress, we performed saturating transposon insertion mutagenesis followed by deep sequencing (Tn-seq), transcriptome sequencing (RNA-seq) analysis, and measurement of replication fork progression. Our data show that B. subtilis cells elongate, and replication fork progression is slowed, following HU challenge. The transcriptomic data show that B. subtilis cells initially mount a metabolic response likely caused by dNTP pool depletion before inducing the DNA damage response (SOS) after prolonged exposure. To compensate for reduced nucleotide pools, B. subtilis upregulates the purine and pyrimidine biosynthetic machinery and downregulates the enzymes producing ribose 5-phosphate. We show that overexpression of the RNR genes nrdEF suppresses the growth interference caused by HU, suggesting that RNR is an important target of HU in B. subtilis. Although genes involved in nucleotide and carbon metabolism showed considerable differential expression, we also find that genes of unknown function (y-genes) represent the largest class of differentially expressed genes. Deletion of individual y-genes caused moderate growth interference in the presence of HU, suggesting that cells have several ways of coping with HU-induced metabolic stress.
IMPORTANCE Hydroxyurea (HU) has been widely used as a clinical therapeutic and an inhibitor of DNA replication. Some evidence suggests that HU inhibits ribonucleotide reductase, depleting dNTP pools, while other evidence shows that toxic HU breakdown products are responsible for growth inhibition and genotoxic stress. Here, we use multiple, complementary approaches to characterize the response of Bacillus subtilis to HU. B. subtilis responds by upregulating the expression of purine and pyrimidine biosynthesis. We show that HU challenge reduced DNA replication and that overexpression of the ribonucleotide reductase operon suppressed growth interference by HU. Our results demonstrate that HU targets RNR and several other metabolic enzymes contributing to toxicity in bacteria.
KEYWORDS: hydroxyurea, Tn-seq, Bacillus subtilis, ribonucleotide reductase, genome instability
INTRODUCTION
Understanding how organisms replicate and repair their DNA has been of fundamental importance across biology. Hydroxyurea (HU) is an antineoplastic drug proposed to either specifically inhibit ribonucleotide reductase (RNR) or cause toxicity through non-RNR targets (1). Ribonucleotide reductases convert ribonucleoside diphosphates (rNDPs) into deoxyribonucleoside diphosphates (dNDPs) using a tyrosyl free radical, usually in an iron-sulfur center of the enzyme (for a review, see reference 2). HU has been shown to be a potent inhibitor of class I RNRs from prokaryotes and eukaryotes in vitro (3, 4), resulting in a reduction in deoxyribonucleoside triphosphate (dNTP) pools that are necessary for DNA replication and repair (4). Therefore, based on these studies and others, it has been assumed that RNR is a specific target of HU in bacteria. Due to HU reduction of the RNR center and its subsequent inactivation, it is possible that HU targets other iron-sulfur cluster enzymes more generally, not RNR specifically. Iron-sulfur-containing proteins occur ubiquitously throughout life and are involved in many cellular processes, including gene regulation, central metabolism, DNA repair, DNA replication, and cellular respiration (for a review, see reference 5). It is also well established that iron-sulfur-containing enzymes are sensitive to oxidative stress, leading to physiological effects (for reviews, see references 6 and 7). For example, two major enzymes involved in eukaryotic DNA replication, eukaryotic primase and Pol3, contain iron-sulfur clusters for replication, indicating that more than one replicative target of HU is possible in eukaryotes (for reviews, see references 2 and 8).
Hydroxyurea is also an important clinical therapeutic and has been used to treat sickle cell anemia and chronic myeloproliferative disorders (9, 10). Recent work has shown that the drug concentrations used in patients result in copy number variation (CNV) in human cell culture (11, 12). Additionally, the cellular effect of HU has been studied in Escherichia coli and Bacillus subtilis, since RNRs are essential for growth in both organisms (13, 14). HU has the potential to be both cytotoxic and cytostatic in bacteria (15). Several studies suggest that the depletion of dNTPs through HU challenge causes growth interference (14, 16), although the targets of HU in vivo and the mechanism of toxicity remain unclear (15, 17).
Prolonged exposure of E. coli cells to HU has been shown to induce cell death and lysis through activation of toxin/antitoxin modules (14). A systems level analysis of the physiological responses of E. coli to HU showed induction of the SOS response to DNA damage, iron uptake, and the generation of hydroxyl radicals (14). Thus, high concentrations of HU are cytotoxic in E. coli, resulting in DNA breaks and the production of reactive oxygen species (14). Based on these data, a model was proposed suggesting that HU has several indirect physiological effects that result from inhibition of RNR and the depletion of dNTP pools, including the generation of reactive oxygen species that contribute to HU-induced toxicity (14).
More recently, other studies have shown that cyanide, peroxide, and nitric oxide accumulate when HU is added to a growth medium (15). This work showed that the cytotoxic activity of HU is caused primarily by HU breakdown products that damage DNA directly, inhibiting DNA replication and causing growth interference in repair-compromised E. coli strains (15). In addition, this work showed that aged HU has a higher concentration of breakdown products and that it is the reactive breakdown products that are responsible for the toxicity of HU in E. coli (15). In further support of these findings, recent work by Nazaretyan et al. (17) showed that RNRs involved in oxidative stress and anaerobic growth do not contribute to E. coli growth recovery in the presence of HU, demonstrating that recovery from HU-inhibited DNA synthesis is not dependent on alternative RNRs (17). Nazaretyan et al. also found that cell recovery is independent of RecA but becomes dependent on RecA only when toxic by-products of HU are abundant after HU has been aged (17). When toxic by-products are not abundant, inhibition of DNA replication is only transient, and recovery is independent of alternative RNRs or translesion DNA polymerases, further supporting a model where RNR is not the main target of HU in E. coli (17). The latter study proposes that HU targets enzymes containing an Fe-S center, resulting in transient inhibition of DNA replication, and that cell recovery is mediated by the restoration of Fe-S enzymes (17). Overall, these data clearly show that HU breakdown products are responsible for DNA damage in E. coli and that the inhibition of DNA replication is independent of HU effects on RNRs (15, 17).
Due to differences in DNA replication and DNA repair, as well as differences in metabolic pathways, we asked if B. subtilis responds similarly to E. coli following HU-induced stress. To understand the response of B. subtilis to HU challenge, we utilized several complementary approaches to identify genes critical for managing HU toxicity. We found that B. subtilis mounts multiple physiological changes to cope with HU. During the initial response to freshly prepared HU, the number of RecA-green fluorescent protein (GFP) foci increases significantly, indicating that the cell is experiencing replication fork stress. We found that cells challenged with fresh HU elongate without inducing SOS until 2 h of treatment. Transcriptome sequencing (RNA-seq) analysis revealed significant differential expression in metabolic genes corresponding to the biosynthesis of dNTPs. We performed Tn-seq (transposon insertion mutagenesis followed by deep sequencing) analysis to identify genes that are important for mitigating HU growth interference, and in contrast to the findings of studies with drugs directly damaging DNA (18), the top hits were not involved in DNA replication or repair, indicating that toxic by-products that damage DNA are not a major factor for HU toxicity in B. subtilis. Instead, we found that many genes lacking demonstrated biological functions (y-genes) are important for surviving HU challenge. Additionally, individual knockouts in DNA repair genes sensitized B. subtilis to HU; however, aged HU showed a lower efficacy of growth interference. Further, we show that overexpression of the nrdEF operon restored growth on HU to wild-type (WT) cells, indicating that in B. subtilis, RNR is an important target of HU. With these results, we conclude that HU targets a number of metabolic enzymes in B. subtilis, including RNR.
RESULTS
B. subtilis elicits a RecA-dependent, SOS-independent response to HU.
To understand the effect of HU on B. subtilis at the single-cell level, we performed fluorescence microscopy using reporters with translational fusions of GFP to TagC and RecA (designated TagC-GFP and RecA-GFP). TagC is one of the most highly expressed SOS-regulated genes and has been widely used as an SOS reporter (19–21). RecA is loaded onto single-stranded DNA (ssDNA) to facilitate recombinational repair and to trigger the SOS response (for a review, see reference 22). In B. subtilis and many other bacteria, the formation of a RecA/ssDNA nucleoprotein filament stimulates LexA autocleavage, inducing SOS (23–25). Prior work showed that E. coli undergoes the SOS response within 15 min of HU treatment (14). Therefore, to understand how B. subtilis responds to HU, we imaged RecA-GFP and TagC-GFP reporter fusions over time after the addition of HU. We found that B. subtilis cells were able to grow in the presence of 25 mM HU for 1 h without inducing the SOS response (Fig. 1A). In contrast, we show an increase in the proportion of RecA-GFP foci after 1 h of HU treatment (Fig. 1A). The fact that ∼53.2% of cells have RecA-GFP foci indicates problems with DNA replication, although cells have not accumulated enough RecA/ssDNA to elicit SOS. Our prior work has shown that RecA-GFP foci can form readily in B. subtilis without any observation of appreciable SOS induction, in contrast to E. coli, which induces SOS more readily following RecA localization (20). After 2 h of HU challenge of B. subtilis, we observed a 100-fold increase in the percentage of cells that were positive for expression of the SOS reporter (Fig. 1A). Cell elongation has also been used as a proxy for inhibition of DNA replication through induced expression of the SOS-dependent cell division inhibitor YneA (18, 26, 27). We measured cell length and observed only a slight increase in cell length for a small percentage of cells after 1 h of HU challenge (Fig. 1B). The percentage of elongated cells increased at the 2-h incubation time point, supporting the SOS reporter results (Fig. 1B). In conclusion, analysis of both reporters and cell length measurements indicates that replication fork perturbations occur within 1 h of growth in HU, while SOS induction occurs only after prolonged exposure of B. subtilis to HU.
FIG 1.
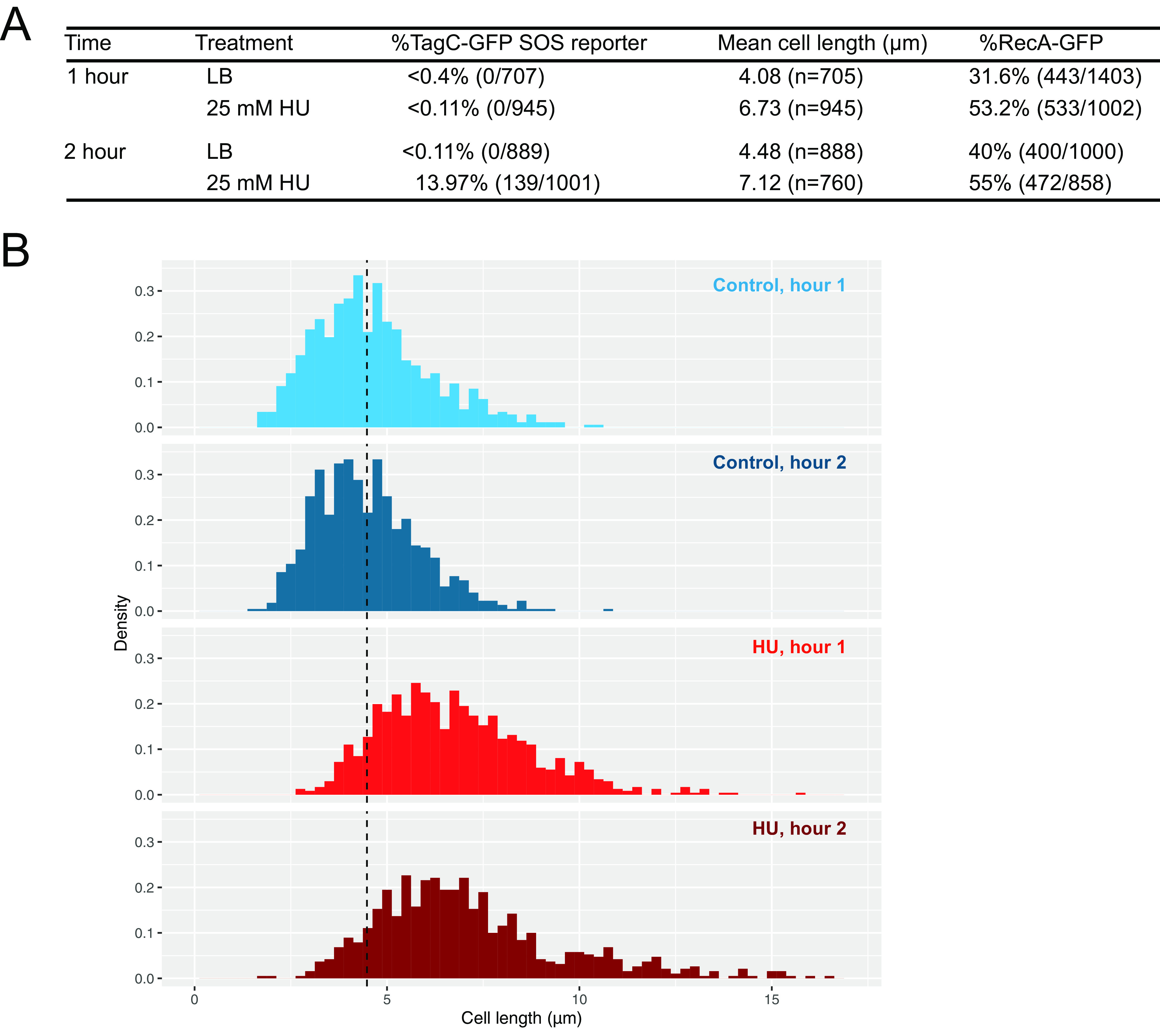
Hydroxyurea at 25 mM causes cell elongation in B. subtilis and SOS induction after 2 h. (A) Significant SOS induction and RecA filamentation after 2 h of HU treatment. The table shows the percentage of SOS-induced (TagC-GFP) cells, mean cell lengths, and the abundance (expressed as a percentage) of RecA-GFP foci. (B) Cell length histograms for control and HU-treated cells after 1 h and 2 h. HU-treated cells are significantly longer than control-treated cells at 1 and 2 h (P, <2.2 × 10−16 by the Wilcoxon rank sum test). The dashed line marks the mean length of control-treated cells at 2 h (4.48 μm).
Metabolic genes are differentially expressed in the B. subtilis response to HU.
After observing the presence of RecA-GFP foci but a lack of SOS induction by HU at the single-cell level within the first hour, we performed RNA sequencing to determine the transcriptional response. We treated cells with a vehicle control or HU for 1 or 2 h, mimicking the conditions of fluorescence microscopy (Fig. 2A). After establishing P value and log2 fold change cutoffs, we visualized genes with increased and decreased expression using a volcano plot (Fig. 2B and Tables 1 and 2; see also Tables S1 and S2 in the supplemental material). We found that 376 of 4,141 genes were significantly upregulated in hour 1 and 351 were downregulated (Table 1, Fig. S1, and Table S1). After 2 h, 441 were upregulated and 359 were downregulated (Fig. 2B). There were 159 genes with increased expression shared between hours 1 and 2, and 132 genes with decreased expression were shared by the two data sets. We next categorized the top 250 differentially expressed genes from hours 1 and 2. Of the top 250, 44% and 38.7% were y-genes, respectively (Fig. 2C and D; Tables 1 and 2), that is, genes in the B. subtilis genome with no demonstrated function. We expected to obtain y-genes given that >40% of the genes in B. subtilis PY79 still lack an experimentally demonstrated function (28). For annotated genes or genes with predicted functions, we showed that genes involved in metabolism were also highly upregulated after 1 and 2 h after HU challenge.
FIG 2.
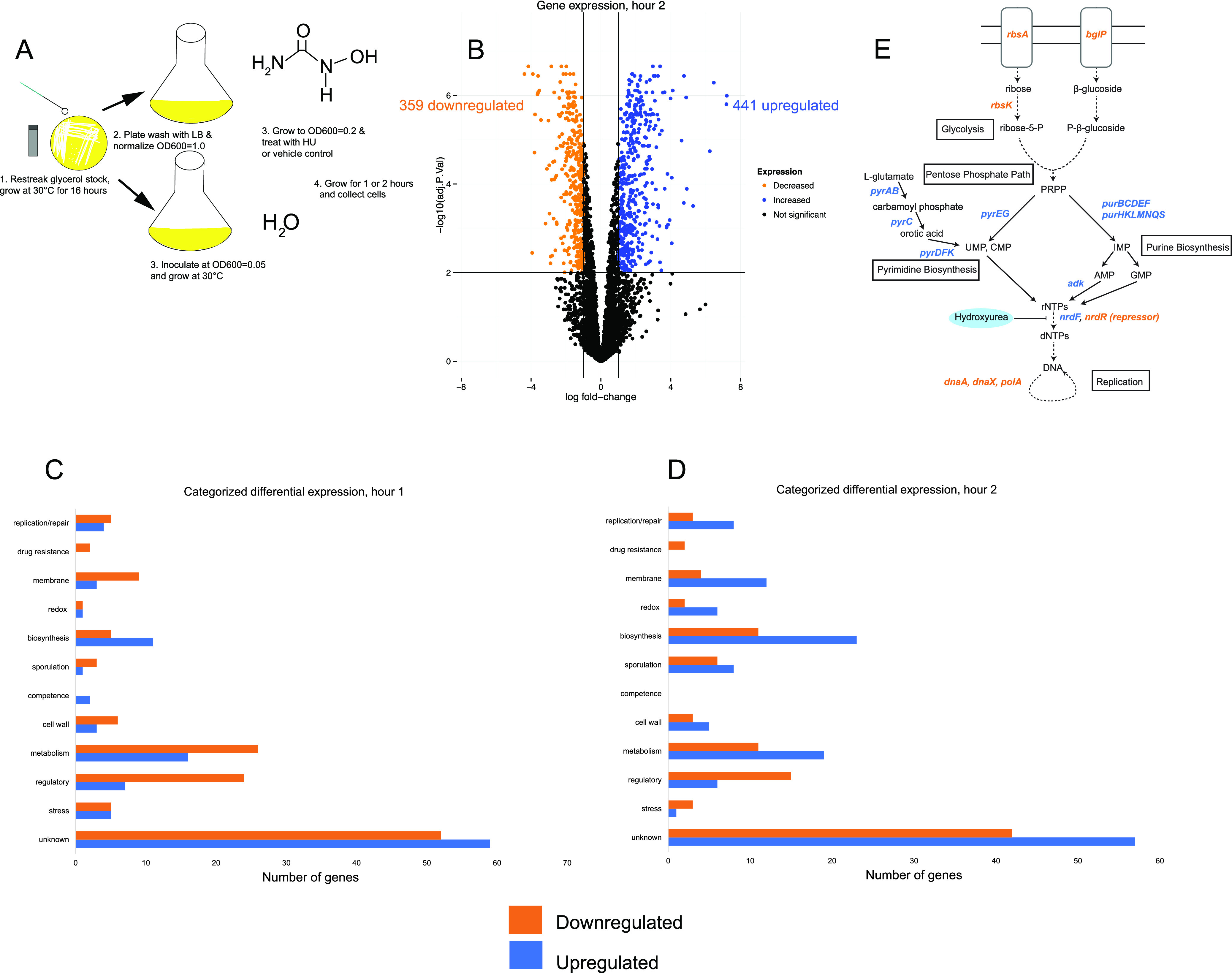
B. subtilis mounts a metabolic response to mitigate the effects of HU before inducing SOS. (A) Experimental scheme of cell growth for RNA-seq. (B) Volcano plot showing differentially expressed genes after 2 h of HU challenge. (C and D) The top 250 differentially expressed genes after 1 h (C) or 2 h (D) of HU treatment, categorized according to Gene Ontology terms. (E) Pathway schematic showing upregulated (blue) and downregulated (orange) genes.
TABLE 1.
Differential gene expression, hour 1 following HU challenge
| Gene | Log2 fold change | Adjusted P value | Function |
|---|---|---|---|
| bglP | −6.14 | 1.99E–07 | PTS system β-glucoside-specific EIIBCA component |
| bglH | −5.47 | 1.50E–07 | Aryl-phospho-β-d-glucosidase BglH |
| rbsB | −5.16 | 5.82E–07 | d-Ribose-binding protein |
| rbsA | −4.72 | 4.54E–06 | Ribose import ATP-binding protein RbsA |
| licA | −4.68 | 1.12E–03 | Lichenan-specific phosphotransferase enzyme IIA component |
| licC | −4.55 | 1.09E–03 | Lichenan permease IIC component |
| rbsR | −4.38 | 4.66E–05 | Ribose operon repressor |
| licH | −4.34 | 1.65E–05 | Probable 6-phospho-β-glucosidase |
| rbsK | −4.12 | 2.27E–05 | Ribokinase |
| purS | 3.48 | 5.82E–07 | UPF0062 protein YexA |
| pyrK | 3.49 | 6.37E–06 | Dihydroorotate dehydrogenase B (NAD+), electron transfer subunit |
| pyrAA | 3.56 | 1.90E–05 | Carbamoyl-phosphate synthase pyrimidine-specific small chain |
| pyrAB | 3.60 | 1.57E–05 | Carbamoyl-phosphate synthase pyrimidine-specific large chain |
| purL | 3.80 | 1.88E–07 | Phosphoribosylformylglycinamidine synthase 2 |
| purM | 4.02 | 4.57E–07 | Phosphoribosylformylglycinamidine cyclo-ligase |
| sboA | 4.04 | 8.88E–04 | Subtilosin A |
| purF | 4.14 | 8.37E–08 | Amidophosphoribosyltransferase |
| purN | 4.44 | 2.65E–06 | Phosphoribosylglycinamide formyltransferase |
| purD | 4.46 | 5.35E–08 | Phosphoribosylamine-glycine ligase |
| purH | 4.57 | 8.37E–08 | Bifunctional purine biosynthesis protein PurH |
TABLE 2.
Differential gene expression, hour 2 following HU challenge
| Gene | Log2 fold change | Adjusted P value | Protein |
|---|---|---|---|
| manA | −4.38 | 1.23E–09 | Mannose-6-phosphate isomerase ManA |
| yjdF | −4.16 | 3.70E–10 | Uncharacterized protein YjdF |
| gntR | −3.94 | 8.6E–04 | Gluconate operon transcriptional repressor |
| manP | −3.90 | 1.21E–09 | PTS mannose-specific EIIBCA component |
| maeN | −3.80 | 1.26E–06 | Na+-malate symporter |
| licA | −3.77 | 7.88E–08 | Lichenan-specific phosphotransferase enzyme IIA component |
| rbsB | −3.63 | 1.32E–08 | d-Ribose-binding protein |
| licH | −3.60 | 1.17E–08 | Probable 6-phospho-β-glucosidase |
| bglH | −3.60 | 1.87E–09 | Aryl-phospho-β-d-glucosidase BglH |
| bglP | −3.53 | 7.0E–09 | PTS β-glucoside-specific EIIBCA component |
| yneN | 4.62 | 1.05E–05 | Thioredoxin-like protein YneN |
| xpf | 4.74 | 02.67E–04 | Positive control factor |
| citB | 4.76 | 3.63E–07 | Aconitate hydratase |
| cimH | 4.88 | 1.11E–06 | Citrate/malate transporter |
| sboA | 4.91 | 1.2E–04 | Subtilosin A |
| pyrC | 5.28 | 3.13E–04 | Dihydroorotase |
| pyrAA | 6.23 | 1.82E–05 | Carbamoyl-phosphate synthase pyrimidine-specific small chain |
| pyrF | 6.46 | 5.15E–07 | Orotidine 5′-phosphate decarboxylase |
| pyrAB | 7.18 | 1.58E–06 | Carbamoyl-phosphate synthase pyrimidine-specific large chain |
| pyrD | 7.21 | 1.02E–06 | Dihydroorotate dehydrogenase B (NAD+), catalytic subunit |
| pyrK | 7.89 | 4.24E–06 | Dihydroorotate dehydrogenase B (NAD+), electron transfer subunit |
| pyrE | 8.13 | 3.29E–07 | Orotate phosphoribosyltransferase |
When genes with the greatest log2 fold change were separated from the top 250 differentially expressed genes, it became evident that most genes differentially expressed were involved in glucose metabolism (Fig. 2C and D). We initially hypothesized that we would observe a change in expression of DNA repair and replication genes. Instead, only 3.6% of the top 250 differentially expressed genes after 1 h of treatment, and 4.3% in hour 2, were involved in DNA repair (Fig. 2D). Interestingly, in both the 1- and 2-h data sets, we found upregulated genes for purine (pur) and pyrimidine (pyr) biosynthesis, as well as antimicrobial peptide production (sboA) (Tables 1 and 2). In hour 2, genes involved in the tricarboxylic acid (TCA) cycle (citB and cimH) were also upregulated, and the TCA cycle uses several iron-dependent enzymes (29) (Table 2). We consistently observed that genes involved in ribose 5-phosphate biosynthesis (rbs) and carbon uptake (man, bgl, and mae) were downregulated. Taking these findings together, it seems that upon HU exposure, B. subtilis senses a loss in dNTPs and attempts to synthesize more dNTPs by upregulation of the nucleotide biosynthesis pathways (model schematic in Fig. 2E). We speculate that the TCA cycle genes are upregulated to create more ATP and iron-dependent enzymes in this pathway that may have been damaged by HU. Carbon utilization genes are downregulated, perhaps to offset the accumulation of dNTP precursors and the lack of dNTPs for DNA replication and repair.
y-genes mitigate the growth interference caused by HU.
Given the wide range of gene ontology classes undergoing differential gene expression, we chose to identify gene knockouts in B. subtilis that were important for mitigating the effects of HU. To this end, we performed transposon insertion mutagenesis followed by deep sequencing (Tn-seq) (18). A library of 120,000 unique insertions was created in the B. subtilis PY79 genome, and this mutant library was challenged with HU over three growth periods as described elsewhere (18) (Fig. 3A). Using read counts of HU-treated cells compared to vehicle control as a proxy for fitness, we identified gene mutants with the greatest fitness decrease in HU, suggesting that these genes are important for mitigating HU stress. By principal-component analysis (PCA), we showed that the fitness of cells from growth periods 2 and 3 differed the most from that of the starting culture (Fig. S2). Therefore, we used the top gene hits in growth periods 2 and 3 to guide our subsequent analyses (Table 3; Tables S3 and S4). Gene deletions of top hits from Tn-seq were created. Strains were then spotted onto plates containing HU to test for sensitivity (Fig. 3B). We found that isogenic strains with ΔyaaA and ΔytoI showed growth interference in the presence of HU. The yaaA gene is annotated as contributing to ribosomal assembly or stabilization, and ytoI is a putative transcriptional regulator. If HU is inhibiting cell growth, it is logical that deletion of genes important for translation, and broader transcriptional regulation would sensitize cells.
FIG 3.
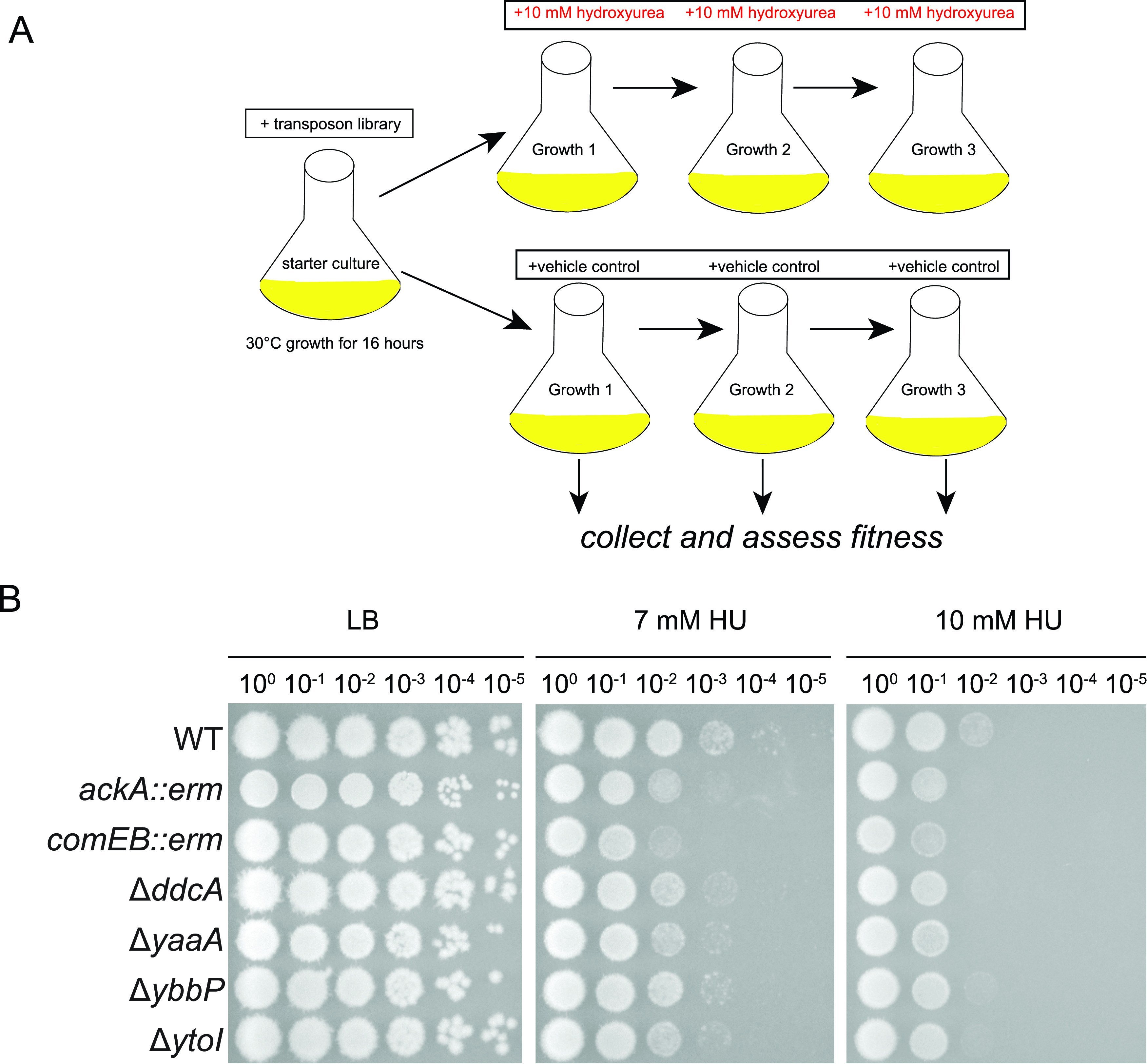
Genes with decreased fitness in Tn-seq with HU treatment. (A) Experimental scheme of cell growth and treatment for Tn-seq. (B) Follow-up analysis of individual gene disruptions or gene deletions in a spot titer assay. Individual gene interruptions (gene::erm) and in-frame deletions (Δgene) were chosen for a subset of the top gene hits from growth periods 2 and 3 in Tn-seq.
TABLE 3.
Summary of top six gene hits from Tn-seq validated using spot titer assays with resulting phenotypes on HU platesa
| Mutant | Function |
|---|---|
| ackA::erm | Acetate kinase |
| comEB::erm | dCMP deaminase |
| ΔddcA | DNA damage checkpoint agonist |
| ΔyaaA | Putative ribosome assembly factor |
| ΔybbP | Diadenylate cyclase |
| ΔytoI | Putative transcriptional regulator |
Gene hits were identified from Tn-seq growth periods 2 and 3. The complete gene validation list is presented in Table S5 in the supplemental material. The complete Tn-seq data sets are presented in Tables S3 and S4 (Excel files).
Other sensitive hits include ddcA (formerly ysoA), which regulates the DNA damage checkpoint protein YneA in B. subtilis (30). We showed previously (30) that deletion of ddcA sensitizes cells to DNA damage, and ddcA insertions showed a fitness decrease in growth period 3 of Tn-seq, as well as increased ddcA expression in hours 1 and 2 of RNA-seq. Deletion of ddcA sensitizes cells to HU, likely because its presence is necessary to prevent YneA from enforcing the DNA damage checkpoint until a threshold of damage is reached (30). We showed that ackA::erm, comEB::erm, and ΔybbP also sensitize cells to HU. ackA encodes acetate kinase to generate ATP, comEB encodes a dCMP deaminase for nucleotide scavenging, and ybbP encodes a diadenylate cyclase. We conclude that several genes of unknown function do indeed sensitize B. subtilis to HU, while other genes have minor effects or polar effects on downstream genes (Fig. 3B and Fig. S3). An important feature of these data is that several single deletions in different pathways have moderate effects on growth in the presence of HU, suggesting that HU inhibits several enzymatic processes in vivo and that Bacillus has a number of pathways that contribute to coping with HU-induced stress.
HU slows the replication of B. subtilis.
Nazaryetan et al. showed in 2018 that HU transiently slowed replication and reduced the amount of total DNA in E. coli (17); however, replication resumed, and the pause in DNA synthesis was transient (17). Based on this finding in E. coli and our observations of cell elongation and increased RecA-GFP foci upon the addition of HU, we chose to further investigate the effect of HU on DNA replication. To do so, we performed whole-genome resequencing as a proxy for DNA synthesis and replication fork fate. We compared vehicle control-treated cells with HU-treated cells to assess chromosomal DNA abundance in duplicate. As expected, control cells exhibited normal replication, with a higher abundance of reads occurring at the origin of replication and tapering off as replisomes moved toward the terminus (Fig. 4A and Fig. S4). Cells treated with HU for 1 h had less DNA than paired vehicle control-treated cells, as shown by the decreased enrichment in DNA copy number across the length of the genome (Fig. 4B and Fig. S4). Cells treated with HU for 2 h showed a much more drastic decline in DNA copy number from the origin to the terminus, indicative of laggard fork movement (31–33). Based on the decrease in chromosome marker frequency as measured by Illumina-based DNA sequencing, we conclude that replication fork progression is slowed, if not stalled, in B. subtilis after treatment with HU.
FIG 4.

DNA replication is stalled by HU. Whole-genome resequencing log2 fold abundance was plotted against the genomic position for mock-treated (A) and HU-treated (B) cells after 1 and 2 h. The data shown are from two replicates. Both independent replicates are shown in Fig. S4. Blue indicates the 1-h time point, and red indicates the 2-h time point.
B. subtilis is less sensitive to aged HU than to fresh HU.
Prior studies have clearly shown that aged HU is more toxic to E. coli than freshly prepared HU, due to the accumulation of breakdown products that damage DNA directly and impair cellular respiration (15, 17). Prior work has also demonstrated that the combination of peroxide and cyanide or nitric oxide accumulates in the micromolar range (15). In all the experiments presented above, HU was freshly prepared immediately before use to reduce the likelihood that breakdown products would occur in our experiments. Therefore, we tested the effect of fresh HU compared to that of aged HU on strains with DNA repair gene deletions in order to determine if toxic by-products from aged HU contribute to interference with the growth of B. subtilis (15, 17). We found that ΔrecA cells were sensitive to freshly prepared HU. These results support an important contribution of RecA to the initial HU response in B. subtilis (Fig. 5A). Additionally, we tested a deletion in the gene for DNA polymerase I (ΔpolA), a disruption of a Holliday junction endonuclease (recU::erm), a deletion of the gene for RNase HIII (ΔrnhC), and a deletion of the gene for nucleotide excision repair (ΔuvrA) (Fig. 5A). We used ΔuvrA as a test to determine if HU breakdown products that damage DNA are repaired through nucleotide excision repair. We found that deletions or disruptions of all genome maintenance genes except for uvrA conferred sensitivity greater than that of the WT to freshly prepared HU. We used the same protocol described previously to create HU breakdown products through a 48-h heat-aging process (15, 17). Interestingly, WT, ΔpolA, ΔrnhC, and recU::erm strains showed less growth interference in the presence of aged HU than in the presence of fresh HU (Fig. 5B). Therefore, in our experiments using B. subtilis, we find that aged HU is less potent than fresh HU, suggesting that breakdown products are not a major factor contributing to HU toxicity in B. subtilis. Further, we show that genes deficient in DNA repair exhibited increased growth interference in the presence of HU. Together, these data suggest that HU challenge compromises genome integrity, supporting our results above for the SOS and RecA-GFP reporters. However, HU breakdown products do not seem to make a major contribution to toxicity in B. subtilis (see Discussion).
FIG 5.

DNA repair genes are sensitive to HU, and aged HU desensitizes cells, likely due to increased detoxification. DNA repair gene deletions (ΔrecA, ΔpolA, ΔuvrA, ΔrnhC, and recU::erm) were tested for sensitivity on fresh HU (A) and 48-h heat-aged HU (B).
Overexpression of RNR mitigates growth interference caused by HU.
B. subtilis contains a class 1b ribonucleotide reductase (RNR) expressed from the nrdI-nrdE-nrdF-ymaB operon (nrdIEF-ymaB). The NrdI, NrdE, NrdF, and YmaB proteins have been purified and characterized biochemically (34, 35). NrdE and NrdF represent the α2 and β2 subunits responsible for nucleotide reduction and the dimanganese-tyrosyl radical cofactor, respectively (34, 36). NrdI is a flavodoxin that helps maintain the NrdF dimanganese-tyrosyl radical, while purified YmaB does not affect the catalytic activity of NrdEF, and the function of this protein is unknown (35). Prior work with E. coli showed that deletion of two alternative RNRs did not increase HU toxicity (35) and that overexpression of RNR in E. coli reduced HU toxicity (37). Therefore, we asked if overexpression of the RNR operon, nrdIEF-ymaB (34), from an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter (amyE::Pspac-nrdIEF-ymaB), in WT and DNA repair-deficient backgrounds (recU::erm, ΔpolA, ΔrnhC, and ΔuvrA) influenced growth in B. subtilis. We found that overexpression of RNR rescued the sensitivity of WT and ΔpolA cells while providing feeble rescue to ΔrnhC and recU::erm cells. This result, in conjunction with the changes in gene expression showing an increase in nucleotide biosynthesis and the reporter results demonstrating an increase in SOS induction, suggests that in B. subtilis, RNR is indeed an in vivo target of HU (Fig. 6).
FIG 6.

DNA repair gene deletion sensitivity is suppressed by ribonucleotide reductase overexpression. The nrdIEF-ymaB genes were overexpressed using an IPTG-inducible promoter at amyE and were inserted into WT and DNA repair deletion backgrounds. (A) LB control plates; (B) HU-treated cells without IPTG (left) or with IPTG and nrdIEF-ymaB overexpression (right).
DISCUSSION
Hydroxyurea has been studied in many organisms as a bactericidal drug and a clinical therapeutic (2, 11, 14, 17, 38–40). The in vivo target(s) and cellular processes used to cope with HU are unclear. Prior work has shown that HU inhibits ribonucleotide reductase (2, 41), impairs iron-dependent enzymes (39, 42), and directly damages DNA through HU breakdown products (15, 17). Here, we used a combination of single-cell reporters, RNA-seq, Tn-seq, and genetic approaches to understand the HU stress response in B. subtilis. In the first 2 h of HU treatment, B. subtilis experiences replication fork stress as RecA-GFP coalesces into foci to stabilize forks. B. subtilis induces the expression of genes involved in glucose metabolism and purine and pyrimidine biosynthesis. The observation that RecA-GFP foci persist suggests that the effect of HU on DNA replication is not transient but prolonged. In addition, the RNR operon (nrdIEF-ymaB) is differentially expressed during HU treatment. B. subtilis does not elicit the SOS response as readily as E. coli, a finding consistent with our previous results (20). The initial response of regulating genes involved in nucleotide biosynthesis and RNR appears to be futile and results in induction of the SOS response at 2 h after HU exposure. Therefore, our work indicates that the early response to HU in B. subtilis is a change in metabolism that is driven by a need for nucleotide biosynthesis.
Previous studies using E. coli unequivocally showed that DNA repair deletion strains are not sensitive to HU unless the HU is aged and breakdown products that damage DNA directly accumulate (15, 17). Using B. subtilis, we found that DNA repair-compromised strains are sensitive to fresh HU and that heat-aged HU is much less potent, yielding differences from E. coli (15, 17). Furthermore, deletion of the nrdEF or nrdG gene, encoding alternative RNRs that are HU insensitive in E. coli, did not delay recovery in HU-treated cells (17). We show that overexpression of RNR in B. subtilis does recover HU sensitivity for the WT and the polA deletion strain. One possibility is that the differences observed between E. coli and B. subtilis can be explained by the response pathways used by B. subtilis during HU challenge. Upon close examination of the differentially expressed genes, we found that catalases and peroxidases (katA, katE, katX, ahpF, ahpC, and ohrB) are upregulated between hours 1 and 2. Additionally, iron storage genes are upregulated (mrgA and dps). It is possible that the enzymes required to detoxify HU by-products are highly upregulated and more abundant in HU-stressed B. subtilis cells, while iron storage genes are also upregulated to limit Fenton chemistry caused when HU destabilizes Fe-S cluster enzymes. We also suggest that B. subtilis is more adept at clearing toxic by-products using transporters or efflux pumps. The RNA-seq data show as many as 14 transporters upregulated 2 h after HU challenge. The upregulated transporters are predicted to be involved in drug resistance and nutrient metabolism. Taking the findings together, peroxidases, catalases, iron storage proteins, and drug efflux pumps could enable B. subtilis to mitigate the effects of aged HU breakdown products, allowing intact HU to inhibit in vivo targets. In contrast to findings for E. coli, we did not find upregulation of toxin-antitoxin modules in HU-treated B. subtilis cells (14). It is possible that B. subtilis relies more heavily on detoxification of HU rather than inducing toxin-antitoxin systems, further underscoring the different mechanisms each organism employs during HU stress. Based on the differential gene expression data, we find that the machinery to produce ribose 5-phosphate is downregulated while purine and pyrimidine biosynthetic genes are upregulated. This could be a response to depleted dNTPs while conversion from rNDPs to dNDPs is inhibited by HU.
Castro-Cerritos et al. performed liquid chromatography-tandem mass spectrometry (LC–MS-MS) on WT cells and cells with a deletion of nrdR (encoding the ribonucleotide reductase regulator) that were starved of histidine, methionine, and leucine for 5 days (43). They found differentially abundant proteins involved in nucleotide metabolism, mRNA degradation, DNA repair, and the stringent response (43). Interestingly, when we compared our lists of differentially expressed RNA-seq hits and Tn-seq hits, we found that nearly 60% of the data reported by Castro-Cerritos et al. overlapped with our data sets, with most overlap occurring in nucleotide metabolism and the stringent response (43). Therefore, we suggest that starving ΔnrdR cells have a stress response similar to the response to HU treatment in B. subtilis, further supporting RNR as a target of HU. Based on our findings, we suggest a mechanism for the B. subtilis response to HU (Fig. 7). Upon initial HU exposure, RNR in B. subtilis is inhibited, and the cells sense the lack of dNTPs. To restore the rNTP-dNTP balance, cells divert carbon metabolism into the creation of rNTPs. During this time, replication is slowed and RecA accumulates in the cells at laggard replication forks. Although the cell attempts to increase rNTP synthesis, dNTP conversion is inefficient due to the inhibition of RNR. Two hours after HU exposure, the futile metabolic response results in SOS induction as replication fork movement becomes increasingly impaired. Around 2 h of HU exposure, other Fe-S cluster enzymes are inhibited, causing greater metabolic stress on B. subtilis. For B. subtilis, RNR seems to be a major target of HU, since overexpression of nrdEF-ymaB rescues the growth interference for WT and polA deletion cells. Evidence from E. coli and a Sulfolobus sp. suggests that other iron-sulfur cluster enzymes are targeted (17, 39). Based on our transcriptomic and Tn-seq results, we suggest that HU has targets other than RNR leading to the changes in gene expression that we observe and to effects on carbon metabolism.
FIG 7.

Mechanism of HU action in B. subtilis cells. (A) HU inhibits RNR, preventing the conversion of rNDPs into dNDPs. (B) As a result, DNA replication is stalled, while purine and pyrimidine biosynthetic genes are upregulated to compensate for the lack of dNTPs (C). (D) B. subtilis cells upregulate efflux pumps and detoxification enzymes when challenged with HU. (E) RecA (green circles) binds ssDNA and eventually elicits the SOS response (F) when the threshold of ssDNA is reached. Red ovals represent catalase and peroxidases; yellow and orange membrane-bound proteins represent efflux pumps.
It is interesting that in our study, aged HU has a restorative effect on growth, whereas in E. coli, aged HU was clearly more toxic (15, 17). As discussed above, one possibility is that B. subtilis is better suited to detoxify HU by-products with the help of peroxidases, catalases, and drug resistance efflux pumps. Another possibility is that the commercial source of HU is critical, and some sources may accelerate the accumulation of HU by-products. Data from studies in E. coli (15, 17), in addition to our finding that growth of the recU-deficient strain is only partially rescued by nrdIEF-ymaB overexpression, support the model that other enzymes are targeted and/or that by-products from HU can damage DNA directly, requiring homologous recombination for repair. Based on our data, this effect is not increased by aging HU but is instead mitigated through the aging process. We conclude that RNR in B. subtilis is a target of HU. Additionally, HU likely has many other targets that contribute to the toxicity of this therapeutic. We speculate that the inhibition of RNR and other iron-sulfur cluster enzymes, and some direct damage caused by HU by-products or reactive oxygen species from the Fenton reaction, contribute to the cytotoxicity of HU.
MATERIALS AND METHODS
General bacteriology.
Bacterial strains are listed in Table S6 in the supplemental material. Erythromycin disruptions of gene candidates in B. subtilis 168 were acquired from the Bacillus Genetic Stock Center (http://www.bgsc.org). Chromosomal DNA was prepared (see “Genomic DNA extraction” below) and was used to transform PY79 (44) by standard procedures (45). Clean deletions were made by transforming the recipient strain with pDR244, containing Cre recombinase driven by a temperature-sensitive origin of replication. Once the erythromycin cassette is integrated, incubation at 45°C causes it to be excised from the genome, leaving loxP scars and a markerless deletion.
(i) Tn-seq.
The B. subtilis Tn insertion library was grown at 30°C overnight in LB medium supplemented with 100 ng/ml spectinomycin and was back diluted into triplicate flasks. Three samples were treated with a vehicle control (water), and three samples were treated with 10 mM HU, over three growth periods. Once the cell density reached 1.5, cells were collected for DNA extraction, serially diluted, and plated for viable plate counts.
(ii) Microscopy.
Strains for microscopy were plate washed, the optical density (OD) was normalized to 1.0, and flasks were inoculated to a starting OD at 600 nm (OD600) of 0.05 in fresh LB medium. Strains were then grown at 30°C in a shaking water bath (225 rpm) until they reached an OD600 of 0.2, at which point 25 mM HU was added, and strains were grown for 1 to 2 h. Slides were prepared for imaging as described previously (18). For RNA sequencing, PY79 was grown to an OD600 of 0.2 in LB medium at 30°C, and the medium was supplemented with 25 mM HU for 1 and 2 h before the collection of cell pellets for nucleic acid extraction.
Microscopy and data quantification.
After 1 and 2 h of growth at 30°C in HU, 1.5 μl FM 4-64FX membrane stain was added to 300-μl aliquots of cells. Next, cells were fixed onto S750 agarose pads, and microscopy was performed. A 1-s exposure time was used for red fluorescent protein (RFP), and a 300- to 500-ms exposure time was used for GFP. Cell lengths were measured using ImageJ.
Whole-genome sequencing.
Two LB plates with restreaked PY79 were washed with LB, and the OD600 was normalized to 1.0. Two flasks containing 20 ml LB per 250-ml flask were inoculated at a final density of 0.05 for each respective plate (4 flasks total: 2 for control treatment and 2 for HU treatment). Cells were grown at 30°C in flasks in a water bath with shaking at 200 rpm. Once cultures reached an OD600 of 0.2, either 25 mM HU or water (for the vehicle-treated control) was added. Cells from one control flask and one HU flask were grown for an hour, and cells at an OD600 of 10 were pelleted at 4,000 × g for 10 min. After 2 h, cells from the remaining control flask and HU-treated flask were collected at an OD600 equivalent to 10 and were centrifuged at 4,000 × g for 10 min to pellet cells. The supernatant was removed, and the genomic DNA was subsequently extracted. Genomic DNA was sequenced using an Illumina MiSeq (50 cycles).
For analysis, reads were aligned to the PY79 genome using the Burrows-Wheeler Aligner (BWA), v 1.7.0_51. Reads with mapping quality lower than 20 were removed from the BAM files using SAMtools (v 0.1.18). The resulting alignments were sorted by read name using SortSam, and duplicates of reads were removed using MarkDuplicates. Subsequently, the BAM files were converted to BED format. Consecutive 1-kb window regions were created across the entire PY79 genome using makewindows from BEDTools (v 2.15.0). Alignment counts for each window were calculated from each alignment using annotateBed from BEDTools. Using R, raw alignment counts were divided by the total number of alignments in millions to control for differences in sample coverage and were then log2 transformed. The 1-kb windows were reordered such that the origin of replication was in the center and log2-normalized counts were plotted against their respective genomic positions. Two replicates for each treatment and time point were averaged to create Fig. 4.
Genomic DNA extraction.
Cells were centrifuged at 8,000 × g for 5 min at room temperature to pellet them, the supernatant was removed, and pellets were exposed to 200 μl lysis buffer (50 mM Tris-HCl [pH 8], 10 mM EDTA [pH 8], 1% Triton X-100, 0.5 mg/ml RNase A, 5 mg/ml lysozyme) for 30 min at 37°C. Next, 30 μl of 10% SDS was added and mixed well, followed by 40 μl proteinase K (10 mg/ml in Tris-EDTA with 10% glycerol). Samples were placed at 55°C for 30 min with occasional flicking to ensure even heating. Five hundred microliters of PB buffer (5 M guanidine-HCl, 30% [vol/vol] isopropanol) was added, and the sample was applied to a column and centrifuged for 1 min at 10,000 × g. The flowthrough was discarded, and 500 μl PB was used to wash the column. Seven hundred fifty microliters of PE (10 mM Tris-HCl [pH 7.5], 80% [vol/vol] ethanol) was added for a final wash, followed by centrifugation of the dry column for an additional 60 s at 13,000 × g. One hundred microliters of ultrapure water was used to elute the DNA.
RNA extraction and sequencing.
The protocol for RNA extraction was adapted from the RNAsnap method as described elsewhere (46). The Bacillus cell pellet was resuspended in 150 μl RNA extraction solution by pipetting up and down (the extraction solution comprised 18 mM EDTA, 0.025% SDS, 1% β-mercaptoethanol, and 95% formamide). Cells were transferred to a 0.5-ml screw-cap tube with 250 μl chilled zirconia beads and were lysed by beating on a vortex mixer for 10 min. Cellular debris was pelleted using centrifugation (16,000 × g for 5 min at room temperature). rRNA was depleted using an Illumina Ribo-Zero rRNA depletion kit according to the manufacturer’s specifications by the University of Michigan Sequencing Core. Samples treated with the vehicle control and HU in triplicate were sequenced using an Illumina HiSeq 4000 system (50 cycles).
For the analysis, sequencing reads were mapped to the B. subtilis PY79 genome using the BWA (47). Differential expression analysis was performed using the Limma package in R with a log2 fold change cutoff at 1.0 and a Benjamini-Hochberg adjusted P value cutoff at 0.05. To visualize all genes from RNA sequencing while highlighting those with the greatest differential expression, volcano plots were created. No P value cutoffs were applied to the initial data set, but upon the generation of the volcano plots, genes with a log2 fold change of >1.0 and an adjusted P value of >–log10(0.01) were marked as “increased” or “decreased” in expression. The top 250 differentially expressed genes were assigned a biological category and were represented using bar graphs.
Tn-seq and analysis.
A library of 120,000 mariner transposon insertions used for transposon insertion mutagenesis followed by deep sequencing (Tn-seq) with HU was created as described elsewhere (18). DNA was extracted as outlined under “Genomic DNA extraction” above. To ligate barcoded adapters, DNA was digested with MmeI and purified with calf intestinal alkaline phosphatase. The adapters were then ligated to the ends, and the samples were again purified. Quantitative PCR was performed with a subset of the samples to determine the starting volume to amplify linearly during sequencing. Using an Illumina HiSeq 2500 high-output sequencer (V4 single end; 50 cycles), a library of DNA sequences was generated. We trimmed adapter ends, aligned reads to the B. subtilis PY79 genome, and performed an EdgeR analysis on genes in R. A negative log fold change is a proxy for the insertion fitness of HU-treated cells compared to that of WT cells.
For the creation of Tn insertion plots, insertion frequency for each gene was calculated by summing the number of unique insertions and normalizing against the length of the gene. Next, the insertion fitness for each growth period and treatment was calculated using the equation described previously (18), where the following variables were input into the equation: the number of cells at the start and end of each growth period (No and Nf) and the frequency of transposons at the start and end of each growth period (Fo and Ff).
Bacillus competency and transformation.
Bacillus was made competent by first inoculating a single colony into 2 ml LM medium (LB with 1 M MgSO4) in a 14-ml test tube and growing for 3 h at 37°C. After ∼3 h, 20 μl of turbid LM medium was inoculated into 500 ml MD medium (1× PC buffer [10× PC buffer is 107 g/liter potassium hydrate phosphate {anhydrous}, 174.2 g/liter potassium dihydrate phosphate {anhydrous}, 10 g/liter trisodium citrate {pentahydrate}, up to 1 liter H2O], 50% [wt/vol] glucose, 10 mg/ml L-tryptophan, 2.2 g/ml ferricammonium citrate, 100 mg/ml potassium aspartate, 1 M MgSO4 , 10 mg/ml phenylalanine) and was grown for an additional 5 h at 37°C. One microliter of genomic DNA (∼200 ng/ml) was added to MD medium and was incubated for 90 min at 37°C. Aliquots (200 μl) of cells were plated on medium with 0.5 μg/ml erythromycin and incubated at 30°C overnight.
Serial dilution and spot titer assays.
A single colony of B. subtilis was inoculated into 1 ml LB medium and was grown with shaking at 37°C for ∼3 h. Once cultures reached a density between 0.5 and 0.7, they were back diluted to 0.5 in 200 μl of sterile saline (0.85% [wt/vol] NaCl). In a 96-well plate, 20 μl of dilute culture was serially diluted into 180 μl sterile saline. Five microliters of the dilution series was plated onto a plate, allowed to dry, and then incubated at 30°C overnight. Spot titer LB plates were made fresh on the same day they were used, with HU and xylose (if applicable) added directly to LB agar. Plates were dried at 37°C for 3 h before use. Hydroxyurea was freshly resuspended in water before being added to molten LB agar. Spot titer assays were performed at least three times.
Data availability.
All RNA-seq, Tn-seq, and whole-genome sequencing data used to measure replication forks are available through SuperSeries accession number GSE169565 in the Gene Expression Omnibus database by NCBI (48). All other strains and resources are available upon request.
ACKNOWLEDGMENTS
We thank Peter Burby for help with setting up the Tn-seq study. We thank Kevin Myers (University of Wisconsin—Madison) and Weisheng Wu (University of Michigan Bioinformatics Core) for help with whole-genome sequencing analysis. We also thank the Bacillus Genetic Stock Center and Daniel Zeigler for strain curation and distribution and the Stubbe Lab at MIT for providing us with the nrd overexpression strain.
Work in the Simmons lab is funded by National Institutes of Health grant R35GM131772. K.J.W. was funded in part by an NIH Cellular Biotechnology Training Program grant (award T32 GM008353) and a predoctoral fellowship from the National Science Foundation (award DEG 1256260).
Footnotes
Supplemental material is available online only.
Contributor Information
Lyle A. Simmons, Email: lasimm@umich.edu.
Tina M. Henkin, Ohio State University
REFERENCES
- 1.Rosenkranz HS, Winshell EB, Mednis A, Carr HS, Ellner CJ. 1967. Studies with hydroxyurea. VII. Hydroxyurea and the synthesis of functional proteins. J Bacteriol 94:1025–1033. 10.1128/JB.94.4.1025-1033.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh A, Xu YJ. 2016. The cell killing mechanisms of hydroxyurea. Genes 7:99. 10.3390/genes7110099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinha NK, Snustad DP. 1972. Mechanism of inhibition of deoxyribonucleic acid synthesis in Escherichia coli by hydroxyurea. J Bacteriol 112:1321–1324. 10.1128/JB.112.3.1321-1334.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimanyi CM, Chen PY, Kang G, Funk MA, Drennan CL. 2016. Molecular basis for allosteric specificity regulation in class Ia ribonucleotide reductase from Escherichia coli. Elife 5:e07141. 10.7554/eLife.07141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khodour Y, Kaguni LS, Stiban J. 2019. Iron-sulfur clusters in nucleic acid metabolism: varying roles of ancient cofactors. Enzymes 45:225–256. 10.1016/bs.enz.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Imlay JA. 2006. Iron-sulphur clusters and the problem with oxygen. Mol Microbiol 59:1073–1082. 10.1111/j.1365-2958.2006.05028.x. [DOI] [PubMed] [Google Scholar]
- 7.Imlay JA. 2014. The mismetallation of enzymes during oxidative stress. J Biol Chem 289:28121–28128. 10.1074/jbc.R114.588814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baranovskiy AG, Siebler HM, Pavlov YI, Tahirov TH. 2018. Iron-sulfur clusters in DNA polymerases and primases of eukaryotes. Methods Enzymol 599:1–20. 10.1016/bs.mie.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGann PT, Ware RE. 2015. Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf 14:1749–1758. 10.1517/14740338.2015.1088827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Randi ML, Ruzzon E, Luzzatto G, Tezza F, Girolami A, Fabris F. 2005. Safety profile of hydroxyurea in the treatment of patients with Philadelphia-negative chronic myeloproliferative disorders. Haematologica 90:261–262. [PubMed] [Google Scholar]
- 11.Arlt MF, Ozdemir AC, Birkeland SR, Wilson TE, Glover TW. 2011. Hydroxyurea induces de novo copy number variants in human cells. Proc Natl Acad Sci U S A 108:17360–17365. 10.1073/pnas.1109272108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arlt MF, Wilson TE, Glover TW. 2012. Replication stress and mechanisms of CNV formation. Curr Opin Genet Dev 22:204–210. 10.1016/j.gde.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shivakumar AG, Dubnau D. 1978. Differential effect of hydroxyurea on the replication of plasmid and chromosomal DNA in Bacillus subtilis. J Bacteriol 136:1205–1207. 10.1128/JB.136.3.1205-1207.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies BW, Kohanski MA, Simmons LA, Winkler JA, Collins JJ, Walker GC. 2009. Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol Cell 36:845–860. 10.1016/j.molcel.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuong KJ, Kuzminov A. 2009. Cyanide, peroxide and nitric oxide formation in solutions of hydroxyurea causes cellular toxicity and may contribute to its therapeutic potency. J Mol Biol 390:845–862. 10.1016/j.jmb.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Godoy VG, Jarosz DF, Walker FL, Simmons LA, Walker GC. 2006. Y-family DNA polymerases respond to DNA damage-independent inhibition of replication fork progression. EMBO J 25:868–879. 10.1038/sj.emboj.7600986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nazaretyan SA, Savic N, Sadek M, Hackert BJ, Courcelle J, Courcelle CT. 2018. Replication rapidly recovers and continues in the presence of hydroxyurea in Escherichia coli. J Bacteriol 200:e00713-17. 10.1128/JB.00713-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burby PE, Simmons ZW, Schroeder JW, Simmons LA. 2018. Discovery of a dual protease mechanism that promotes DNA damage checkpoint recovery. PLoS Genet 14:e1007512. 10.1371/journal.pgen.1007512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goranov AI, Kuester-Schoeck E, Wang JD, Grossman AD. 2006. Characterization of the global transcriptional responses to different types of DNA damage and disruption of replication in Bacillus subtilis. J Bacteriol 188:5595–5605. 10.1128/JB.00342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simmons LA, Goranov AI, Kobayashi H, Davies BW, Yuan DS, Grossman AD, Walker GC. 2009. Comparison of responses to double-strand breaks between Escherichia coli and Bacillus subtilis reveals different requirements for SOS induction. J Bacteriol 191:1152–1161. 10.1128/JB.01292-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Britton RA, Kuster-Schock E, Auchtung TA, Grossman AD. 2007. SOS induction in a subpopulation of structural maintenance of chromosome (Smc) mutant cells in Bacillus subtilis. J Bacteriol 189:4359–4366. 10.1128/JB.00132-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simmons LA, Foti JJ, Cohen SE, Walker GC. 25July2008, posting date. The SOS regulatory network. EcoSal Plus 2008. 10.1128/ecosalplus.5.4.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LovettCM, Jr, Love PE, Yasbin RE, Roberts JW. 1988. SOS-like induction in Bacillus subtilis: induction of the RecA protein analog and a damage-inducible operon by DNA damage in Rec+ and DNA repair-deficient strains. J Bacteriol 170:1467–1474. 10.1128/jb.170.4.1467-1474.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LovettCM, Jr, O’Gara TM, Woodruff JN. 1994. Analysis of the SOS inducing signal in Bacillus subtilis using Escherichia coli LexA as a probe. J Bacteriol 176:4914–4923. 10.1128/jb.176.16.4914-4923.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LovettCM, Jr, Cho KC, O’Gara TM. 1993. Purification of an SOS repressor from Bacillus subtilis. J Bacteriol 175:6842–6849. 10.1128/jb.175.21.6842-6849.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawai Y, Moriya S, Ogasawara N. 2003. Identification of a protein, YneA, responsible for cell division suppression during the SOS response in Bacillus subtilis. Mol Microbiol 47:1113–1122. 10.1046/j.1365-2958.2003.03360.x. [DOI] [PubMed] [Google Scholar]
- 27.Mo AH, Burkholder WF. 2010. YneA, an SOS-induced inhibitor of cell division in Bacillus subtilis, is regulated posttranslationally and requires the transmembrane region for activity. J Bacteriol 192:3159–3173. 10.1128/JB.00027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroeder JW, Simmons LA. 2013. Complete genome sequence of Bacillus subtilis strain PY79. Genome Announc 1(16): e01085-13. 10.1128/genomeA.01085-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smaldone GT, Revelles O, Gaballa A, Sauer U, Antelmann H, Helmann JD. 2012. A global investigation of the Bacillus subtilis iron-sparing response identifies major changes in metabolism. J Bacteriol 194:2594–2605. 10.1128/JB.05990-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burby PE, Simmons ZW, Simmons LA. 2019. DdcA antagonizes a bacterial DNA damage checkpoint. Mol Microbiol 111:237–253. 10.1111/mmi.14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nye TM, McLean EK, Burrage AM, Dennison DD, Kearns DB, Simmons LA. 2021. RnhP is a plasmid-borne RNase HI that contributes to genome maintenance in the ancestral strain Bacillus subtilis NCIB 3610. Mol Microbiol 115:99–115. 10.1111/mmi.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simmons LA, Breier AM, Cozzarelli NR, Kaguni JM. 2004. Hyperinitiation of DNA replication in Escherichia coli leads to replication fork collapse and inviability. Mol Microbiol 51:349–358. 10.1046/j.1365-2958.2003.03842.x. [DOI] [PubMed] [Google Scholar]
- 33.Walsh BW, Bolz SA, Wessel SR, Schroeder JW, Keck JL, Simmons LA. 2014. RecD2 helicase limits replication fork stress in Bacillus subtilis. J Bacteriol 196:1359–1368. 10.1128/JB.01475-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Stubbe J. 2011. Bacillus subtilis class Ib ribonucleotide reductase is a dimanganese(III)-tyrosyl radical enzyme. Biochemistry 50:5615–5623. 10.1021/bi200348q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartig E, Hartmann A, Schatzle M, Albertini AM, Jahn D. 2006. The Bacillus subtilis nrdEF genes, encoding a class Ib ribonucleotide reductase, are essential for aerobic and anaerobic growth. Appl Environ Microbiol 72:5260–5265. 10.1128/AEM.00599-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cotruvo JA, Stubbe J. 2011. Class I ribonucleotide reductases: metallocofactor assembly and repair in vitro and in vivo. Annu Rev Biochem 80:733–767. 10.1146/annurev-biochem-061408-095817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Babu VMP, Itsko M, Baxter JC, Schaaper RM, Sutton MD. 2017. Insufficient levels of the nrdAB-encoded ribonucleotide reductase underlie the severe growth defect of the Δhda E. coli strain. Mol Microbiol 104:377–399. 10.1111/mmi.13632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbé J, Villaverde A, Guerrero R. 1987. Induction of the SOS response by hydroxyurea in Escherichia coli K12. Mutat Res 192:105–108. 10.1016/0165-7992(87)90105-9. [DOI] [PubMed] [Google Scholar]
- 39.Liew LP, Lim ZY, Cohen M, Kong Z, Marjavaara L, Chabes A, Bell SD. 2016. Hydroxyurea-mediated cytotoxicity without inhibition of ribonucleotide reductase. Cell Rep 17:1657–1670. 10.1016/j.celrep.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Navarra P, Preziosi P. 1999. Hydroxyurea: new insights on an old drug. Crit Rev Oncol Hematol 29:249–255. 10.1016/S1040-8428(98)00032-8. [DOI] [PubMed] [Google Scholar]
- 41.Slater ML. 1973. Effect of reversible inhibition of deoxyribonucleic acid synthesis on the yeast cell cycle. J Bacteriol 113:263–270. 10.1128/JB.113.1.263-270.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang ME, Facca C, Fatmi Z, Baille D, Benakli S, Vernis L. 2016. DNA replication inhibitor hydroxyurea alters Fe-S centers by producing reactive oxygen species in vivo. Sci Rep 6:29361. 10.1038/srep29361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castro-Cerritos KV, Lopez-Torres A, Obregon-Herrera A, Wrobel K, Wrobel K, Pedraza-Reyes M. 2018. LC-MS/MS proteomic analysis of starved Bacillus subtilis cells overexpressing ribonucleotide reductase (nrdEF): implications in stress-associated mutagenesis. Curr Genet 64:215–222. 10.1007/s00294-017-0722-7. [DOI] [PubMed] [Google Scholar]
- 44.Youngman P, Perkins JB, Losick R. 1984. Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid 12:1–9. 10.1016/0147-619x(84)90061-1. [DOI] [PubMed] [Google Scholar]
- 45.Hardwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. John Wiley & Sons, Chichester, United Kingdom. [Google Scholar]
- 46.Stead MB, Agrawal A, Bowden KE, Nasir R, Mohanty BK, Meagher RB, Kushner SR. 2012. RNAsnap: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res 40:e156. 10.1093/nar/gks680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wozniak KJ, Simmons LA. 2021. RNA-seq, Tn-seq, and whole genome sequencing of hydroxyurea treated Bacillus subtilis cells. Gene Expression Omnibus https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169565 (accession no. GSE169565).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S5, captions to Tables S1 to S4, and Tables S5 and S6. Download JB00171-21-s0001.pdf, PDF file, 1.09 MB (1.1MB, pdf)
Table S1. Download JB00171-21-s0002.xlsx, XLSX file, 97 KB (96.6KB, xlsx)
Table S2. Download JB00171-21-s0003.xlsx, XLSX file, 103 KB (103KB, xlsx)
Table S3. Download JB00171-21-s0004.xlsx, XLSX file, 37 KB (36KB, xlsx)
Table S4. Download JB00171-21-s0005.xlsx, XLSX file, 3s6 KB (35.6KB, xlsx)
Data Availability Statement
All RNA-seq, Tn-seq, and whole-genome sequencing data used to measure replication forks are available through SuperSeries accession number GSE169565 in the Gene Expression Omnibus database by NCBI (48). All other strains and resources are available upon request.