

Abstract
Lysine acetylation and deacetylation are critical for regulation of many cellular proteins. Despite the importance of this cycle, it is unclear how lysine deacetylase (KDAC) family members discriminate between acetylated proteins to react with a discrete set of substrates. Potential short-range interactions between KDAC8 and a known biologically relevant peptide substrate were identified using molecular dynamics (MD) simulations. Activity assays with a panel of peptides derived from this substrate supported a putative ionic interaction between arginine at the −1 substrate position and KDAC8 D101. Additional assays and MD simulations confirmed this novel interaction, which promotes deacetylation of substrates. Verification that a negatively charged residue at the 101 position is necessary for the ionic interaction and observed reactivity with the substrates was performed using KDAC8 derivatives. Notably, this interaction is specific to KDAC8, as KDAC1 and KDAC6 do not form this interaction and each KDAC has a different specificity profile with the peptide substrates, even though all KDACs could potentially form ionic interactions. When reacted with a panel of putative human KDAC substrates, KDAC8 preferentially deacetylated substrates containing an arginine at the −1 position. KDAC8 D101-R(−1) is a specific enzyme-substrate interaction that begins to explain how KDACs discriminate between potential substrates, and how different KDAC family members can react with different subsets of acetylated proteins in cells. This multi-pronged approach will be extended to identify other critical interactions for KDAC8 substrate binding and determine critical interactions for other KDACs.
Graphical Abstract
Introduction
Lysine acetylation is one of the most prominent types of post-translational modification (PTM). First identified on histone proteins, acetylation has now been found on thousands of proteins, including many that are found only in the cytosol, indicating that this mode of regulation is utilized in many cellular process and reaches far beyond the canonical histone modifications known to regulate gene expression.1-6 Despite the revelation that acetylation is widely present on many proteins that are involved in many cellular processes, the consequences of this PTM on specific non-histone proteins are not well understood.
Like many other PTMs, such as phosphorylation, acetylation is a reversible modification. Toggling between the acetylated and deacetylated state alters the physical properties of the residue, most notably changing the charge state from the positively charged lysine side chain to an uncharged acetyllysine. Lysine deacetylases (KDACs), sometimes referred to as histone deacetylases (HDACs), are a family of enzymes that are responsible for removing acetyl groups, restoring lysine residues to their original state. Of the 18 human enzymes that are capable of catalyzing the hydrolysis of the ϵ-N-acetyllysine, 11 belong to either class I or class II, which are collectively referred to as KDACs (EC 3.5.1.98). These enzymes are metal-dependent, share a well-conserved catalytic domain, and deacetylate substrate proteins through a conserved mechanism.7-9 KDAC8 (UniProtKB: Q9BY41) is a class I deacetylase, a subfamily that also includes KDACs 1, 2, and 3. While deacetylation of modified proteins undoubtedly affects protein function and can easily be envisioned as a regulatory mechanism, specific examples of identified target proteins for specific KDACs are lacking. In fact, out of the several thousand known acetylated human proteins, only a handful of KDAC/substrate pairs have been confidently determined.10
Understanding how particular acetylated substrates interact with KDAC8 is an important step toward understanding KDAC specificity, predicting and identifying substrates of particular KDACs, and ultimately understanding how cells use deacetylation as a mechanism to regulate cellular processes. Structural studies of the KDAC catalytic domain have elucidated several residues in the active site that are important for catalysis.7,11,12 In addition, residues in the L2 loop have been implicated in KDAC8 function through analysis of structural data and computer simulations that indicate that these residues form critical hydrogen bonds with substrates and inhibitors.12-14 However, the identified interactions of these residues, such as hydrogen bonding from an aspartic acid (D101) in the L2 loop to the substrate backbone and hydrogen bonding from a tyrosine (Y306) in the binding pocket to the acetyllysine side chain, involve features of the substrate that would be common to all substrates. Furthermore, the particular residues involved are often entirely conserved within class I KDACs, and often also within some or all members of the class II KDACs. Therefore, the previously reported interactions are unlikely to contribute to substrate discrimination that could lead to the selectivity of different KDACs. The lack of unique identified interactions within KDAC binding sites also limits the ability to develop inhibitors that target only a single KDAC instead of the entire family or class. Although progress has been made in designing specific inhibitors, there is need to identify such interactions for improved inhibitor selectivity.15
For many reasons, including that the catalytic domains of KDACs are generally well conserved and that it is not understood to what extent KDAC family members may have overlapping substrates, performing experiments in cells or organisms has been challenging. It has been established, using the histone proteins as model substrates, that KDACs rely on both short-range and long-range contacts for substrate specificity.16 Several attempts at large scale studies to investigate KDAC8 substrate specificity using unnatural peptide substrates have been conducted.17-19 Overall, these studies revealed patterns for substrate preference for various KDACs, but the conclusions from these studies have been inconsistent with results from studies when more biologically-relevant putative substrates are used.20 Additionally, these large scale studies have not led to significant insights into the molecular determinants of specificity. To better understand mechanisms of KDAC8 substrate specificity, we have used a combination of in vitro deacetylation assays with peptide substrates derived from a known KDAC8 substrate and molecular dynamics (MD) simulations to uncover an important and specific ionic interaction between KDAC8 and putative substrates that enhances deacetylation by KDAC8.
Materials and Methods
KDAC expression and purification.
Recombinant KDAC1 (UniProtKB: Q13547) was purchased from ActivMotif and dialyzed into storage buffer (30 mM 3-(N-morpholino)propanesulfonic acid [MOPS] pH 8.0, 150 mM KCl, 1.0 mM tris(2-carboxyethyl)phosphine [TCEP], 25% glycerol). All other KDACs were recombinantly expressed fused to a tobacco etch virus (TEV) protease cleavage site and a C-terminal His6 tag in either E. coli or insect cells. The constructs used to express KDAC6 (UniProtKB: Q9UBN7) and KDAC8 have been previously described.20 To create KDAC8 derivatives, D101A, D101E, D101N, and D101R mutations were introduced using PCR-based site-directed mutagenesis into either pFastbac1 (D101E) or pJE (D101A, D101N, and D101R) plasmids containing human KDAC8. E. coli expression was performed as previously described to obtain KDAC8 and KDAC8 derivatives.21 For insect cell expression of KDAC6 and KDAC8 D101E, pFastbac1-based plasmids were transformed into DH10Bac cells (Life Technologies) to create bacmids that were isolated using a ZR Bac DNA miniprep kit (Zymo Research). 1 μg of each bacmid was transfected into 106 ExpiSf9 cells (Life Technologies) in a 6-well plate in 3 mL ExpiSf CD media (Life Technologies) using 20 μL Expifectamine transfection reagent (Life Technologies). Transfected cells were incubated at 28 °C for 5 days. Cells and media from the transfected well were transferred to 50-150 mL of ExpiSf9 cells growing in suspension at 28 °C with shaking at a density of 106 cells mL−1. Cells were incubated for 2 days, then harvested by centrifugation at 700 xg for 5 min and stored at −20 °C until purification. ExpiSf9 cells were grown and maintained according to the manufacturer’s instructions.
Purification of KDACs was performed as previously described.21 Briefly, cells were lysed and KDACs were purified based on the presence of the C-terminal His6 tag using TALON metal affinity resin (Takara). KDAC8 and KDAC8 variants, but not KDAC6, were subjected to a second round of metal affinity purification to remove the protease and other impurities, following cleavage of the tag using TEV protease. The resulting protein was quantified using A280. Purified proteins were subjected to polyacrylamide gel electrophoresis (PAGE) to verify identity and purity.
Activity assays.
Peptide substrates containing acetylated lysine residues were custom synthesized, N-terminally acetylated and C-terminally amidated (Genscript). In vitro activity assays were performed by incubating 50 nM KDAC6 or 200 nM other KDACs with 100 μM peptide substrate at 25 °C for 15-60 min in 30 mM potassium phosphate pH 7.6, 5% glycerol (low ionic strength) unless otherwise noted as being under standard high ionic strength conditions (the same buffer with 100 mM KCl). For most substrates, specific activity was determined using fluorescamine as previously described.22 All reported endpoint activity values are the average of n≥3. Except where noted, endpoint activity was normalized by defining the activity of KDAC8 with FRKacRW to be 1 and scaling all other values by the same proportion. For some KDAC8/substrate pairs, kinetic parameters were determined using variable substrate concentrations and sampling the reactions over time.22 Statistical significance was determined using a t-test with a significance threshold of p=0.05, using the Bonferroni correction for multiple testing (0.05 divided by the number of comparisons made to the reference data set in a given group). All t-tests were two-tailed, and assumed unpaired samples with unequal variance unless otherwise noted. All kinetic parameters were calculated using data from 3 timepoints over a 60 min timecourse for each of 5 substrate concentrations. Statistically significant differences in fit values were identified by comparing the 95% confidence intervals of the fits and determining whether the fits overlapped in the regions corresponding to KM or Vmax, which approximately corresponds to when the two values being compared differ by at least 1.4 times the sum of the uncertainties.
For the reactions using FKKacRW and a subset of the FRKacRW reactions, deacetylation was measured using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry instead of fluorescamine. Completed reactions and inhibited reaction controls were diluted 1:50 in TA85 (85% acetonitrile, 0.1% TFA). 1 μl was spotted onto an Anchorchip 384 target plate (Bruker Daltonics). After drying, 0.5 μL matrix solution (saturated α-cyano-4-hydroxycinnamic acid [HCCA] in TA85) was spotted on top of each sample. MALDI-TOF mass spectrometry was performed on each sample using positive reflector mode on an Autoflex speed MALDI-TOF/TOF (Bruker Daltonics). Data from 500 laser shots of each sample were pooled. Peaks were identified and analyzed using Flex Analysis software (Bruker Daltonics). The peaks representing the m/z for the substrate and product were analyzed to determine the fraction of the total peak area for the peak corresponding to the product of each reaction. The corresponding value from the inhibited reaction control was subtracted from the value calculated for the reactions. To create a standard curve, substrate and product (non-acetylated) peptides were mixed such that the product was represented in known ratio compared to the substrate. Standard samples were prepared for mass spectrometry in the same manner as reactions described above. For each concentration of substrate, the percent area for the product peak was divided by the sum of areas of the substrate and product peaks. The percent product in the standard samples was plotted against the average percent area of the product peak (n≥3 for each concentration). A linear fit was performed (QtiPlot) and the slope of the standard curve for each substrate/product pair was used to convert percent product area for the reactions to percent substrate conversion. This value was used to calculate specific activity for each reaction. Statistical analysis was performed as described for fluorescence assays.
Molecular dynamics (MD).
Structures of human KDAC8 and variants with bound substrate were prepared by creating sequence alignments of residues 10-377 (C-terminus) with the desired substrate sequence against inactive KDAC8 variants containing a bound substrate (PDB: 2v5w and 3ewf) and an apo wild-type KDAC8 structure (PDB: 3ew8).12,13 All chains in the crystal structures were included in the alignment. Substrate sequences were aligned as a separate chain such that the acetyllysine residue matched the acetyllysine residue in the active site of the crystal structures. One extra N-terminal amino acid was included on the substrate. The active site water and zinc were retained as rigid bodies, as was the 7-amino-4-methylcoumarin group when present. Models were created with MODELLER, version 9.21 or 9.24, with the CHARMM22 force field, with modified force field parameters to incorporate an adapted version of the acetylated lysine (ALY) residue from CHARMM36.23-25 The resulting structure was modified by removing all atoms from the extra N-terminal substrate residue except the backbone Cα and carbonyl, which were converted to the N-terminal acetyl group, and the 2nd C-terminal oxygen atom was changed to a nitrogen atom to form the C-terminal amide group. When the position of the catalytic water was significantly shifted by MODELLER, it was manually returned to the position of the crystal structures relative to the zinc atom. Structures for human KDAC1 residues 8-376 (PDB: 4bkxB) and human KDAC6 residues 480-835 (PDB: 5eduA) were prepared by first performing a structure-based sequence alignment against KDAC8 (PDB: 2v5wA) using TM-align.26-28 Then sequence alignments were built using the same chains, with incorporation of an additional structure for each KDAC1 (PDB: 5icnB) and KDAC6 (PDB: 5eduB).29 Substrate sequence alignments and MODELLER treatment were the same as done for KDAC8.
Molecular dynamics was performed using GROMACS version 2019.1.30,31 The AMBER03 force field was utilized.32 The force field was modified to include a previously parameterized ALY residue.33 AMBER03 parameters for 7-amino-4-methylcoumarin were calculated using version Jan-2019 of the R.E.D. server with Gaussian03.34 Proteins were placed in a cubic box of length 8.6 nm (KDAC8 and KDAC1) or 9.3 nm (KDAC6) and solvated using the tip3p method. Charges were neutralized with potassium or chloride, and additional potassium and chloride ions were added to a concentration of 150 mM (high ionic strength) or 50 mM (low ionic strength). Simulations utilized low ionic strength unless noted as high ionic strength. The system was energy minimized by steepest descent. All subsequent equilibration and simulation runs utilized 2 fs steps and a Verlet cut-off scheme for interactions, otherwise utilizing default GROMACS settings except as detailed in the following text.35 Frames were recorded every 10 ps during equilibration and every 2 ps during simulations. The system was first equilibrated under constant volume and temperature (NVT) conditions using a Berendsen thermostat for 100 ps at 300 K, with hydrogen bond constraints (LINCS), all non-solvent atoms restrained, and the catalytic water and zinc temperature-regulated as part of the protein and substrate system rather than solvent.36,37 A second equilibration was performed at constant pressure and temperature (NPT) using the V-rescale thermostat and Berendsen pressure couple at 300 K for 100 ps, with hydrogen bond constraints and all non-solvent atoms restrained.38 A final constant pressure and temperature equilibration was performed using the V-rescale thermostat and Parrinello-Rahman pressure couple at 300 K for 100 ps, with hydrogen bond constraints but all atom restraints removed.39 All equilibrations were validated using standard GROMACS tools to ensure stable temperature, pressure, and/or density at each stage after 20 ps. After each equilibration, the distances from the acetyllysine carbonyl to the zinc atom and from the catalytic water to the zinc atom were verified as being less than 0.35 nm and less than 0.25 nm, respectively, to ensure that the substrate was remaining in a fully bound configuration. Failure to pass any equilibration test resulted in a restart of the process. Finally, simulations were run under constant volume and temperature conditions using the V-rescale thermostat at 300 K for 5 ns, with hydrogen bond constraints. The process from solvation through simulation was performed 5 times as independent replicates.
Simulations were analyzed using standard GROMACS tools. Maintenance of overall protein structure was verified by monitoring the root mean square displacement of the protein backbone and the radius of gyration. The minimum distance of every protein and substrate atom to the nearest mirror image atom was verified as being greater than 1.0 nm to confirm the absence of periodic image artifacts. Potential interactions were identified by enzyme residues that were within 0.4 nm of a substrate residue during at least 1% of single simulation, and then further refined for specific contact types. Ionic interactions were identified if the distance from any hydrogen atom of the cationic group to either oxygen of the anionic group was 0.25 nm or less. Hydrogen bonds were identified between two hydrogen bonding groups when the distance between the relevant heavy atoms was 0.35 nm or less and the bond angle incorporating the hydrogen atom was appropriate, with a focus on side chain-side chain (h:h) and enzyme side chain to substrate backbone (h:bb), after atoms previously determined to be ion pairs were discarded. Aromatic ring-cation interactions (π:+) were identified when the centers of mass of the aromatic ring and the cation heavy atom were within 0.4 nm and the ring was at a relative angle of approximately 60 ° or less based on a maximum difference of 0.15 nm from the cation heavy atom to each carbon atoms in the ring. The average percent of time that each interaction was observed was averaged across the five replicate simulations.
Protein structure analysis.
Circular dichroism spectra of KDAC8 derivatives were collected using 500 nM enzyme and four accumulated scans, as previously described.21 Images of proteins were prepared using PyMOL (Schrödinger), using individual MD frames or the crystal structures and sequence alignments from TM-align output.
Results
An ionic interaction between KDAC8 and substrates promotes activity.
Using a fluorescence-based in vitro assay, we have previously identified several peptide substrates of KDAC8 derived from known acetylated human proteins.22 To investigate how substrates interact with KDAC8, we used the most reactive peptide substrate identified in that study, FRKacRW (a portion of ADAP1 [UniProtKB O75689] with N-terminal acetylation and C-terminal amidation) as a starting point. Using MD simulations, we modeled the interaction between KDAC8 and the peptide substrate under standard reaction conditions (high ionic strength). The simulation was run for 5 ns after the substrate was positioned such that the acetyllysine was in the correct configuration for catalysis, with five independent replicate simulations. While this timescale is insufficient to model catalysis or the binding process, it does allow identification of the close-range interactions between the substrate and enzyme that are potentially relevant for binding in a catalytically relevant configuration. The result of this simulation revealed possible interactions between particular KDAC8 residues and the substrate (Figures 1A and 1B). In this work, we focused on the interactions of the substrate residues adjacent to the acetyllysine, both of which are arginine for this particular substrate: R(−1) and R(+1). As we were interested in features leading to substrate selectivity, we focused on side chain interactions rather than backbone interactions likely to be common to all substrates. However, we did monitor previously reported interactions between D101 and the substrate backbone of the acetyllysine and the +1 residue.13,14 Similarly, we ruled out interactions with the acetyllysine, as that residue is common to all substrates, except for the hydrogen bonding interaction between the side chain of Y306 and the acetyllysine side chain. The Y306 interaction was monitored as a potential indicator of catalytic behavior. The percent of time that each interaction pair spent in position for the interaction to occur was represented as a greyscale heat map (Figure 1C). First, D101 appeared to be hydrogen bonding to the acetyllysine (enzyme side chain to substrate backbone, h:bb), as was Y306 (side chain to side chain, h:h). In addition,Y100 formed an aromatic ring-cation interaction with both arginine residues flanking the acetyllysine (pi-cation, π:+). D101 also interacted with both arginine residues, albeit in different ways: namely ionic interaction with R(−1) and hydrogen bond with R(+1) (h:bb). To determine which of these interactions correlated with deacetylation and contribute to substrate selectivity, we used a combination of in vitro assays and MD analysis of derivatives of the substrate and enzyme to probe the contributions of these interactions.
Figure 1. A novel ionic interaction exists between KDAC8 and FRKacRW.

(A) Snapshot of MD simulation between KDAC8 and FRKacRW demonstrating the interactions of Y100-R(−1) π:+, and previously reported interactions D101-Kac h:bb, D101-R(+1) h:bb, and Y306-Kac h:h (dashed yellow lines). KDAC8 (grey spheres) and side chains of important KDAC8 residues for substrate interaction are shown along with the FRKacRW peptide (sticks colored by atom: carbon green [substrate] or periwinkle [enzyme], oxygen red, nitrogen blue, polar hydrogen white). (B) Snapshot of MD simulation between KDAC8 and FRKacRW demonstrating the interactions of D101-R(−1) ionic, Y100-R(+1) π:+, and Y306-Kac h:h (dashed yellow lines). Coloring is the same as panel A. (C) Results of MD analysis identifying interactions between KDAC8 residues and FRKacRW substrate residues in high ionic strength assay buffer. Shading corresponds to the percent of time a particular interaction was observed during MD simulations. (D) Average normalized activity of KDAC8 with previously identified peptide substrates in either high ionic strength (red) or low ionic strength (blue) buffer. All activity was normalized to FRKacRW in low ionic strength buffer. Error bars represent standard deviations (n≥4), and p-values are shown for statistically significant differences for the comparisons between the activity in the two ionic strengths for each substrate. (E) Results of MD analysis identifying interactions between KDAC8 residues and FRKacRW substrate residues in low ionic strength assay buffer. Shading corresponds to the percent of time a particular interaction was observed during MD simulations.
The ionic interaction of R(−1) and the π-cation interactions identified in our MD analysis have not been previously reported. We reasoned that ionic interactions may be affected by the ionic strength of the buffer, as high concentrations of solvent ions would provide alternative interaction partners and weaken the substrate-enzyme interactions. To determine whether lowering the ionic strength of the buffer would affect KDAC8 activity, we measured the specific activity of KDAC8 with this peptide, as well as other previously reported peptide substrates that do not contain positively charged residues, in a low ionic strength phosphate buffer. These values were compared with our previously reported values for these reactions at higher ionic strength (Figure 1D; absolute specific activity values for all normalized endpoint reactions are reported in Table S1).22 Interestingly, we observed that in the lower ionic strength buffer, KDAC8 showed an approximately 2.5-fold increase in specific activity in an endpoint assay with FRKacRW; however, the specific activity with the other peptides was not affected by the buffer change. Determining the kinetic parameters of KDAC8 with the positively charged FRKacRW peptide substrate revealed that lowering the ionic strength of the buffer resulted in an approximately two-fold decrease in KM and an approximately 2.5-fold increase in catalytic efficiency (Table 1). Thus, we could attribute the increased endpoint activity in the lower ionic strength buffer to an increase in affinity. MD analysis comparing potential interactions in each buffer supported the hypothesis that the increase in affinity was driven by ionic interactions, as all the interactions between D101 and the substrate are increased in the low ionic strength buffer, whereas interactions with Y100 and Y306 were unaffected or decreased (compare Figure 1C with Figure 1E). To maximize our sensitivity to ionic contributions to binding, we conducted all subsequent experiments in the low ionic strength buffer.
Table 1.
Steady-state kinetic parameters of KDAC8 with FRKacRW in buffers of varying ionic strength
| Reaction buffer | Specific activity (s−1) |
KM (μM) |
kcat (s−1) |
kcat/KM (M−1 s−1) |
|---|---|---|---|---|
| High ionic strength* | 0.014 ± 0.003 | 950 ± 110 | 0.162 ± 0.007 | 170 ± 13 |
| Low ionic strength | 0.035 ± 0.007† | 500 ± 50† | 0.233 ± 0.006† | 470 ± 40† |
Previously reported.22
Statistically significant difference from the high ionic strength buffer.
Arginine in the +1 position does not contribute to activity.
To determine whether arginine in both positions are important for KDAC8 activity, we tested the activity of KDAC8 with derivative peptides in which one of the charged residues (R(−1) or R(+1)), was replaced with alanine (A) (Figure 2A). Notably, replacing R(−1) with alanine resulted in a dramatic reduction in deacetylation, while replacing R(+1) resulted in no significant difference in specific activity. In fact, a peptide containing R(−1), but where all other residues have been replaced with alanine (ARKacAA), is sufficient to illicit deacetylation by KDAC8, albeit at reduced levels compared to the biologically relevant peptide FRKacRW. However, a similar peptide where R(+1) has been retained instead (AAKacRA) is not deacetylated by KDAC8 (Figure 2A). These results indicate that R(−1), and not R(+1), is a major driver of KDAC8 activity. MD simulations revealed decreases in several interactions when R(−1) was substituted with alanine, in addition to the loss of the ionic interaction directly associated with the lack of an arginine in the −1 position (Figure 2B). This was not the case when R(+1) was substituted with alanine, as this substitution resulted in only minor changes to the overall interactions with KDAC8. Interestingly, MD analysis suggested that replacing all substrate residues with alanine, except R(+1), does force an ionic interaction between this residue and D101; however, that interaction does not lead to KDAC8 activity, likely because the substrate is no longer in a favorable position for catalysis.
Figure 2. The −1 substrate position participates in an ionic interaction with KDAC8.

(A) Peptides derived from FRKacRW, where residues were substituted with alanine, were reacted with KDAC8. Average specific activity was normalized such that the activity of KDAC8 with FRKacRW was represented as 1. Error bars represent standard deviations (n≥4), and p-values are shown for statistically significant differences between activity with FRKacRW and activity with each other substrate. (B) Results of MD analysis identifying interactions between specific residues of KDAC8 and the derivative peptide substrates reacted in panel A. x(+1) refers to the substrate residue in the +1 position for each peptide. Shading corresponds to the percent of time a particular interaction was observed during MD simulations.
Specific interactions between the enzyme and substrate could affect KDAC8 activity by contributing to binding to the enzyme, rate of catalysis, or both. To distinguish between these possibilities for the ionic interaction we have described, we conducted timecourse experiments at several substrate concentrations with derivative substrates to determine the kinetic parameters for each (Table 2). The arginine to alanine substitution at the −1 position increased the KM approximately 6-fold, indicating a much lower binding affinity when the positively charged residue is not present in that position. In contrast, the kcat decreased less than two-fold, indicating only a small impact on the catalytic process. The approximately 10-fold decrease in KDAC8 catalytic efficiency between these two substrates (FRKacRW vs. FAKacRW) was consistent with the change in specific activity with these substrates calculated from endpoint experiments (Figure 2A). These data indicated that R(−1) is primarily important for substrate binding to the enzyme. Interestingly, substituting R(+1) for alanine slightly increased the kcat without significantly changing KM (Table 2). These measurements confirmed that although there is an arginine residue on either side of the acetyllysine, they are not performing functionally similar roles with respect to interaction with KDAC8 (Table 2 and Figure 2). The MD simulations also indicate that the presence of the Y306 hydrogen bond does not correlate with the activity trend, although a significant presence of this interaction does serve to demonstrate that the peptides remained in catalytically relevant conformations during the simulation.
Table 2.
Steady-state kinetic parameters for derivative substrates compared to FRKacRW
| Substrate |
KM (μM) |
kcat (s−1) |
kcat/KM (M−1 s−1) |
|---|---|---|---|
| FRKacRW | 500 ± 50 | 0.233 ± 0.006 | 470 ± 40 |
| FAKacRW | 3100 ± 1400† | 0.15 ± 0.04† | 48 ± 25† |
| FRKacAW | 680 ± 160 | 0.43 ± 0.04† | 630 ± 160 |
Statistically significant difference from the value for FRKacRW.
KDAC8 is more active with peptides containing an arginine residue in the −1 position.
To determine whether the activity changes were being driven by the R(−1) ionic interaction with D101 or the previously reported D101 hydrogen bonds to the backbone of the acetyllysine and the +1 substrate residue, we compared KDAC8 deacetylation of several derivative peptides where each contained a different residue at the −1 position. To determine the effect of a positively charged substrate that contained lysine instead of arginine, we needed to develop a different method of determining KDAC8 activity, because the fluorescamine assay that we were primarily using to assess KDAC8 activity is insufficiently sensitive when a free amine such as unacetylated lysine is present in the substrate.22 Using MALDI-TOF mass spectrometry, we were able to determine the relative amounts of product and substrate after incubating with the deacetylase. To account for potential differences in sensitivity of detection of the two molecules and to convert the percent substrate conversion to specific activity, we first produced a standard curve by measuring the relative peak area for the product and substrate mixed at known ratios. The standard curves for FRKacRW and FKKacRW (Figures S1A and S1B) had r2 values equal to 0.97 and 0.95 respectively, indicating a reasonable linear relationship between the percent substrate in the mixture and the percent of total signal area. Using the FRKacRW substrate, we were able to validate this technique by comparing the specific activity of single reactions measured using both the fluorescamine assay and the mass spectrometry assay. Because the absolute specific activity calculated using mass spectrometry was somewhat lower, although not statistically different, than the value when using the fluorescamine method (Figure S1C), we report activity for lysine-containing peptides normalized to the activity for FRKacRW obtained from mass spectrometry rather than the fluorescamine activity value used for other normalizations.
The known biologically-relevant peptide was the best KDAC8 substrate. A peptide where R(−1) was replaced with a lysine (K), which retains the positive charge, also retained the ability to react with KDAC8, and was a much better substrate than the equivalent peptide with an alanine substitution (p=0.004 for K to A) (Figure 3A). Consistent with our hypothesis that the −1 substrate residue at this position is interacting with D101 in KDAC8, substituting the arginine residue in the substrate with negatively-charged glutamic acid (E) resulted in no significant deacetylation by KDAC8. Substitution with glutamine (Q), which contains hydrogen bonding groups but lacks charge, resulted in a peptide that can be deacetylated by KDAC8 better than the alanine-containing peptide (p=0.01 for Q to A), but with lower activity than either of the peptides containing positively charged residues (p=0.004 for K to Q). This trend is entirely consistent with the hypothesis that an ionic interaction between the substrate and the D101 residue of KDAC8 promotes deacetylation by KDAC8. MD analysis simulating each of these substrates with KDAC8 was performed. Despite the K(−1)-containing peptide retaining approximately half of its KDAC8 activity, it only barely formed an ionic interaction with D101 in our simulation (Figure 3B). However, the K to R substitution was also accompanied by large changes in interactions at the +2 position (data not shown), indicative of a major shift in substrate position and suggestive that the role of arginine at −1 is more specific than simply providing a positive charge. In contrast, the D101 hydrogen bonding interactions occurred more often with K at −1 than with R, and exhibited no overall correlation with activity of substrates. The Y306 and R(+1) interaction also continued to exhibit no correlation with activity. In contrast, the trend of both Y100 and D101 interactions with R(−1) are both similar to the trend of substrate activity (Figures 2 and 3).
Figure 3. A positive charge at the −1 substrate position is important for KDAC8 activity.

(A) Peptides derived from FRKacRW, where R(−1) was substituted, were reacted with KDAC8. Average specific activity was normalized such that the activity of KDAC8 with FRKacRW was represented as 1. Error bars represent standard deviations (n≥4), and p-values are shown for statistically significant differences between activity with FRKacRW and activity with each other substrate. (B) Results of MD analysis identifying interactions between specific residues of KDAC8 and derivative peptide substrates reacted in panel A. x(−1) refers to the substrate residue in the −1 position for each peptide. Shading corresponds to the percent of time a particular interaction was observed during MD simulations.
D101 forms an ionic interaction with substrates to promote deacetylation.
To determine whether the contribution to catalytic activity by the R(−1) is driven by Y100 or D101, we mutated KDAC8 at the 101 position from aspartic acid (D) to glutamic acid (E), alanine (A), asparagine (N), or arginine (R). We first compared the activity of these derivatives to the activity of wild-type KDAC8 with the FRKacRW peptide (Figure 4A). Only the variant that retained the negative charge at this position (D101E) was active with the substrate, and the activity was not statistically different from that of the wild-type KDAC8. To rule out the possibility of a folding issue, secondary structures of the inactive mutants were compared by circular dichroism. All variants produced spectra that were indistinguishable from wild-type KDAC8 (Figure S2). As expected, replacing the D101 with a positively charged residue (R) or an alanine (A) did not result in activity. Most interestingly, we also did not observe activity for the D101N variant, as asparagine is not charged but otherwise shares properties with aspartic acid, including the ability to form a hydrogen bond. MD simulations revealed that, indeed, a glutamic acid at 101 could interact with the substrate in a similar manner to aspartic acid. In contrast, asparagine lost almost all interactions with the substrate, even through hydrogen bonding (Figure 4B). However, the Y100 interaction with R(−1) was largely retained with all KDAC8 variants, strongly suggesting that the D101 interaction with R(−1) is the essential interaction for explaining the activity trend.
Figure 4. KDAC8 requires a negatively charged 101 residue to deacetylate and interact with FRKacRW.

(A) KDAC8 variants containing substitutions at the 101 position were reacted with FRKacRW. Average specific activity was normalized such that the activity of wild-type KDAC8 with FRKacRW was represented as 1. Error bars represent standard deviations (n≥4), and p-values are shown for statistically significant differences between activity of WT enzyme and variants. (B) Results of MD analysis identifying interactions between specific residues of KDAC8 and FRKacRW reacted in panel A. 101x refers to the residue at position 101 in each KDAC variant. Shading corresponds to the percent of time a particular interaction was observed during MD simulations.
To further probe the role of the 101 residue, we focused on the behavior of the D101E variant to determine whether it was reacting equivalently to wild-type KDAC8. First, we compared the wild-type and D101E activity with the previously tested substrates containing alanine substituted for the arginine residues (Figure 5A). Remarkably, substituting glutamic acid for aspartic acid in the 101 position does not significantly affect the activity with any of these substrates, compared to wild-type KDAC8. Next, we tested activity of the D101E mutant with the set of peptide substrates containing substitutions at the −1 position, which we previously characterized with wild-type KDAC8. As before, the D101E mutant behaved similarly to wild-type KDAC8 with all derivative substrates (Figure 5B). MD simulations demonstrated that the D101E variant had a similar pattern of interactions as the wild-type KDAC8, with the notable exception that D101E more often formed an ionic interaction with K(−1) than wild-type KDAC8 did, to a frequency approaching that of R(−1) with wild-type KDAC8 (Figure 5C). In contrast, the enhanced hydrogen bonding of the 101 position to the acetyllysine seen with the K(−1) peptide and wild-type KDAC8 was not retained with the D101E variant. Glutamic acid hydrogen bonded with the substrate backbone less often than aspartic acid did with all substrates, irrespective of the observed activity.
Figure 5. KDAC8 D101E is equivalent to wild-type KDAC8.

(A) Wild-type KDAC8 (blue) or KDAC8 D101E (red) with FRKacRW and derivative peptides containing arginine to alanine substitutions. Average specific activity was normalized such that the activity of wild-type KDAC8 with FRKacRW was represented as 1. Error bars represent standard deviations (n≥4). No statistically significant differences exist between WT and KDAC8 D101E. (B) Wild-type KDAC8 (blue) or KDAC8 D101E (red) with FRKacRW and derivative peptides containing substitutions at the −1 position. Data are represented as in panel A. Blue bars in panels A and B are the same data presented in Figures 2 and 3, reproduced here to allow comparison. No statistically significant differences exist between WT and KDAC8 D101E. (C) Results of MD analysis comparing interactions between residues of wild-type KDAC8 or KDAC8 D101E and a subset of peptide substrates reacted in panels A and B. Shading corresponds to the percent of time a particular interaction was observed during MD simulations.
While our data are all consistent with the conclusion that an ionic interaction between D101 and a positively charged residue at the −1 position in the substrate promotes deacetylation, previous studies have only observed hydrogen bonding between D101 and the substrate.12-14 However, these studies utilized either unnatural substrates containing 7-amino-4-methylcoumarin (amc) in the +1 position and which did not contain arginine in the −1 position, or inhibitors. To ensure that we could reproduce the reported correlation of D101 hydrogen bonding to the substrate backbone and activity (or binding), we performed MD simulations with substrates containing either amc or tryptophan (W) at the +1 position. As we have previously reported activity data with these substrates, the MD was performed under high ionic strength conditions to match the reported data.20 We observed that for two different substrate sequences, including the substrate present in the crystal structures, the presence of amc in the +1 position results in an increase in hydrogen bonding with the substrate backbone compared to substrates with a tryptophan in that position (Figure 6). Therefore, we are able to reproduce the prior observations using our simulation conditions, which increases our confidence that observations deviating from prior reports are reliable.
Figure 6. Conjugation of amc affects peptide interaction with KDAC8.
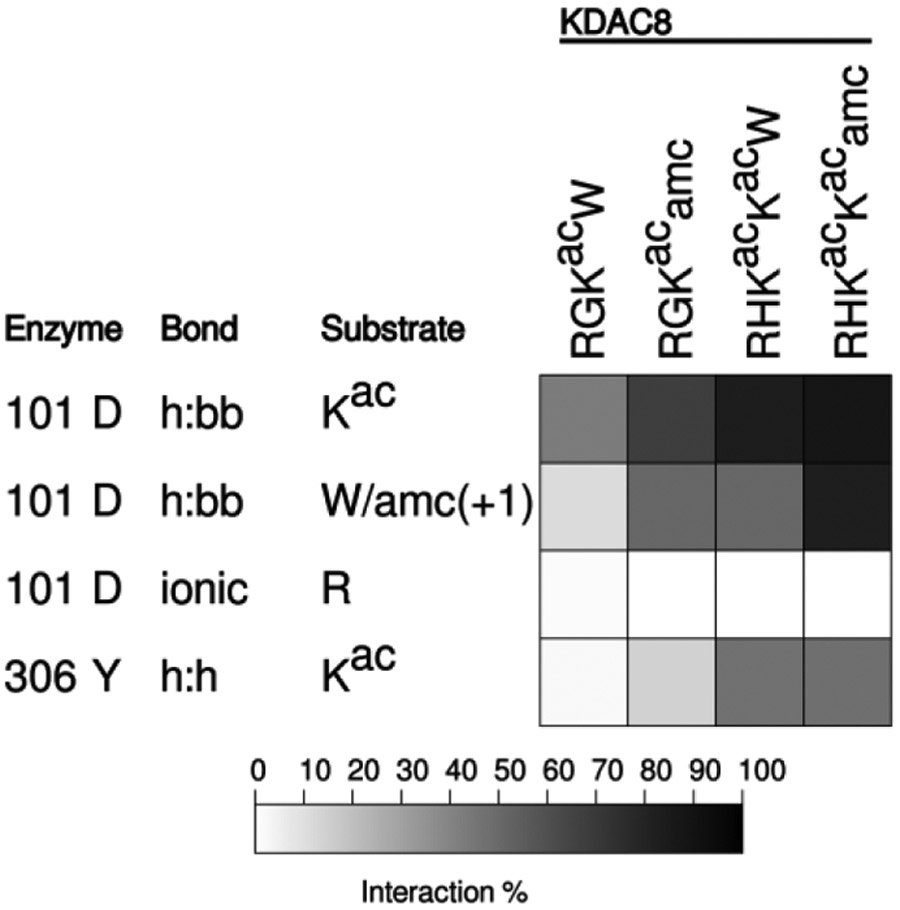
Results of MD analysis comparing interactions between residues of KDAC8 and previously reported peptide substrates with and without amc. Shading corresponds to the percent of time a particular interaction was observed during MD simulations. R indicates the N-terminal arginine of each substrate, in the −2 or −3 position.
The D101 ionic interaction contributes to KDAC8 specificity.
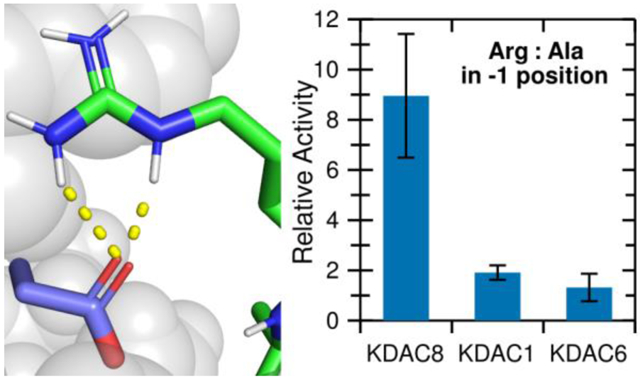
Having established that the ionic interaction between D101 and the substrate R(−1) promotes deacetylation, we hypothesized that this interaction could contribute to KDAC8 substrate specificity. We addressed this hypothesis by comparing KDAC8 to KDAC1 and KDAC6. KDAC1 and KDAC8 are both members of the class I subfamily, while KDAC6 is a class II deacetylase. All three of these enzymes have solved crystal structures.27,28,40 A structure-based sequence alignment demonstrates that the L2 regions have high sequence similarity between KDAC8 and KDAC1 (Figure 7A). One exception is that instead of Y100, KDAC1 contains a glutamic acid at the equivalent position. In contrast, KDAC6 has much lower similarity. Although work published prior to the availability of the KDAC6 crystal structure suggested that the aspartic acid at position 101 in KDAC8 is conserved in all KDACs, the structure alignment indicates that the aspartic acid in the KDAC6 loop is positioned elsewhere, and that a serine residue occupies the physical space analogous to KDAC8 D101 (Figure 7A).14 The overall position of the L2 loop is also much different in KDAC6 than the class I enzymes. To test whether the KDAC8 ionic interaction we have described is conserved among other KDAC family members, we performed assays using KDAC1 and KDAC6 with the peptide substrates containing variations at the −1 position (Figure 7B). Because the absolute activity values varied greatly among enzymes and we were primarily interested in the effects of perturbing the substrate on selectivity, we normalized the activity for each enzyme separately such that the activity of FRKacRW with each enzyme was set to 1. From this limited set of derivative substrates, it is obvious that the specificity profiles for the three KDACs are distinct from one another (compare the patterns of significant differences in Figure 7B). KDAC8 shows a strong preference for a positively charged residue at the −1 position, while the other enzymes do not discriminate in the same manner. MD simulations revealed that ionic interactions were possible between the −1 substrate position and KDAC1, both with D99 (the D101 equivalent position in KDAC1) and with E98 (a second negatively charged residue that occurs in the place of Y100) (Figure 7C). Similarly, KDAC6 can form an ionic interaction between the R(−1) and D567 (analogous sequence position to Y100 although not located in the same physical space), but it is clear that changing the identity of the −1 substrate residue has no effect on activity. While investigating the mechanism behind the specificity preferences of other KDACs is beyond the scope of this work, these data emphasize that the specific interactions described here for KDAC8 are not a general feature of all KDACs, and, therefore, are likely contributing to substrate specificity.
Figure 7. D101 ionic interaction is specific to KDAC8.

(A) Structure (top) and sequence (bottom) alignments of KDAC8 (orange), KDAC1 (blue), and KDAC6 (green) were generated from previously reported crystal structures (PDB: 2v5xA, 4bkxB, and 5eduA).13,27,28 The segment of the structure-guided sequence alignment corresponding to the L2 loop of each KDAC is displayed. The side chain of KDAC8 D101 and the structurally equivalent residues KDAC1 D99 and KDAC6 S568 are shown as stick representations. (B) KDAC8, KDAC1, and KDAC6 were all reacted with FRKacRW (blue), FKKacRW (red), FQKacRW (green), FAKacRW (orange), FEKacRW (yellow), and FRKacAW (purple). Specific activity was normalized so that for each KDAC, activity with FRKacRW was represented as 1. Error bars represent standard deviations (n≥4). p-values are shown for statistically significant differences between activity with FRKacRW and activity with each other substrate within a single enzyme set (top row), and for normalized KDAC8 activity compared to normalized activity of other enzymes for each substrate except FRKacRW (bottom row). KDAC8 data are reproduced from previous figures for comparative purposes. (C) Results of MD analysis comparing interactions between specific residues in the KDACs and the FRKacRW substrate. Shading corresponds to the percent of time a particular interaction was observed during MD simulations.
KDAC8 preferentially deacetylates substrates with arginine in the −1 position.
Together, our data suggested that KDAC8 prefers substrates containing an arginine at the −1 position. To test the general applicability of this observation, we performed activity assays using KDAC8 and a panel of acetyllysine-containing peptides from putative KDAC substrates that were previously identified in the literature and which exhibited a wide range of sequences.16,41-53 These putative substrates were primarily determined by indirect cell-based experiments as described elsewhere.10 While none of these peptides were a better substrate than FRKacRW, and most were not measurably deacetylated by KDAC8 in our assay, KDAC8 was active with all of the peptides containing an arginine at the −1 position (Table 3). With each of these substrates, we observed the D101-R(−1) ionic interaction by MD. Thus, the ionic interaction described here appears to be a clear determinant of KDAC8 specificity for biologically relevant substrates in vitro.
Table 3.
Specific activity of KDAC8 with biologically relevant putative KDAC substrates.
| Substrate | Source protein | UniProtKB | Ref. | Specific activity (s−1)* |
|---|---|---|---|---|
| SVRKacGIM | Cell death activator CIDE-3 | Q96AQ7 | 45 | 0.0036 ± 0.0008 |
| AARKacSAP | Histone H3.1 | P68431 | 47 | 0.0026 ± 0.0002 |
| APRKacQLA | Histone H3.1 | P68431 | 46 | 0.00163 ± 0.00014 |
| GVGKacYIN | RING-type E3 ubiquitin-protein ligase PPIL2 | Q13356 | 44 | 0.0007 ± 0.0004 |
| PEAKacSLL | RAC-alpha serine/threonine-protein kinase | P31749 | 41 | - |
| TGGKacAPR | Histone H3.1 | P68431 | 16,46 | - |
| LSGKacGNP | Catenin beta-1 | P35222 | 42 | - |
| GALKacAPS | Mitotic checkpoint serine/threonine-protein kinase BUB1 beta | O60566 | 43 | - |
| LGGKacQRA | Retinoic acid-induced protein 1 | Q7Z5J4 | 44 | - |
| EIGKacTLA | Zinc finger Ran-binding domain-containing protein 2 | O95218 | 44 | - |
| EVGKacLLN | Centromere protein F | P49454 | 44 | - |
| QTAKacDAG | Heat shock 70 kDa protein 1A/1B | P0DMV8, P0DMV9 | 48 | - |
| GTAKacSVT | Cellular tumor antigen p53 | P04637 | 49-51 | - |
| ITGKacPSG | Serine/threonine-protein kinase TBK1 | Q9UHD2 | 52 | - |
| SHLKacAHL | Transcription factor Sp7 | Q8TDD2 | 53 | - |
- indicates no activity above the limit of reliable detection (0.0004 s−1).
Discussion
While several putative substrates for KDAC8 have been identified both in vitro and in vivo, it remains unclear which of these are bona fide biological substrates, and what features of these potential substrates determine whether and to what extent they can be deacetylated by KDAC8.10 Furthermore, previous attempts to identify important determinants of substrate specificity were hampered by technical limitations, such as including unnatural moieties that affected reactivity and being restricted to evaluating only one side of the acetyllysine.19,20 Through a combination of in vitro peptide assays and molecular modelling, we have uncovered a novel ionic interaction that contributes to deacetylation by KDAC8 (Figure 1). The D101 residue of KDAC8 formed a specific ionic interaction with arginine in the −1 substrate position that resulted in a dramatic increase in KDAC8 activity when a positively-charged residue was present in that position (Figures 2 and 3). Our data indicated that changing the aspartic acid to glutamic acid preserved the ionic interaction with the substrate and resulted in deacetylation; however, mutations to several other amino acids that could not ionically interact did not result in deacetylation (Figures 4 and 5). We have clearly demonstrated the importance of an arginine residue in the −1 position of the substrate for deacetylation by KDAC8, as the enzyme was able to discriminate between residues containing arginine at the −1 position and those that did not in a panel of putative KDAC substrates (Table 3). Importantly, the active putative substrates with R(−1) lacked any other sequence similarity to the original peptide used to establish this interaction. Previously described peptide screens utilizing only a single experimental approach have led to reports that R(−1) is of no significance or enhances activity only when phenylalanine is present at the +1 position.19,54 The contrast with our results and the clear demonstration of the importance of this interaction illustrate the value of our multi-pronged approach, although we acknowledge that validation with full-length protein putative substrates remains as future work.
Our results are consistent with previous studies that showed the importance of D101 for deacetylation; however, our work provides a novel role for D101 in substrate binding. The previously solved crystal structures bound to the RHKacKacamc substrate indicated that D101 formed hydrogen bonds with the substrate backbone.12,13 The role of this hydrogen bond was further supported by MD simulations utilizing an inhibitor.14 Our MD analysis performed with amc-containing substrates (including the substrate that was co-crystallized with KDAC8) demonstrated that the presence of the amc moiety greatly enhanced the hydrogen bonding to the backbone, which may account for this observation and its presence in the crystal structures (Figure 6). The difference in behavior for amc-containing peptides is consistent with previous observations that conjugation of amc to peptide substrates greatly affects behavior with several KDACs, including KDAC8, in ways that do not translate to biologically relevant substrates.20 Furthermore, the substrate used for the crystal structures did not contain a charged residue in the −1 position, so the ionic interaction that we observed could not have formed with that particular substrate.12,13 Thus, the hydrogen bonding identified in the previously-published work can be attributed to a combination of the presence of the amc moiety and the lack of arginine at the −1 position. Perhaps unsurprisingly, D101 can only transiently assume a configuration that allows simultaneous hydrogen bonding (to the acetyllysine backbone only) and the ionic interaction. Therefore, to some extent the two interactions are mutually exclusive. Despite this observation, peptides that lack the arginine at the −1 position were observed to have less D101 hydrogen bonding (Figures 2 and 3), suggesting that the ionic interaction might help position the substrate optimally for hydrogen bonding. Of course, it is a certainty that residues other than R(−1) are also important for optimally positioning the substrate, as illustrated by our results with FKKacRW and the observed effect of the amc moiety; such additional contributors to substrate specificity are the subject of ongoing work but beyond the scope of this paper.
Consistent with our observations, previously reported crystal structures of the D101E, D101A, and D101N variants indicated there were no major structural rearrangements caused by the mutations.12 The lack of activity in both the previous work and our work reported here with the D101A and D101N variants, as well as the inability to restore activity by swapping the positive and negative residues between substrate and enzyme as with D101R and E(−1), suggests that some specific interaction, or set of interactions, of the 101 residue is essential. In this study, all active enzyme-peptide combinations had significant frequency of hydrogen bonding from D101 to the acetyllysine, as well as a more variable amount to the +1 residue. Although there is a clear tendency toward greater D101 hydrogen bonding with active substrates than inactive peptide, the hydrogen bonding appears to function more as a switch than a rheostat: a minimum amount of hydrogen bonding to the substrate may be necessary for effective binding and therefore catalysis, but once above some critical threshold, additional frequency of hydrogen bonding does not appear to trend with greater catalytic activity. In contrast, the frequency of ionic interaction between D101 and R(−1) does trend with observed activity, although it is only one of presumably several factors important for determining selectivity. In support of the hypothesis that the ionic interactions of D101 are a major contributor to the selectivity, we observed reducing binding (increased KM) when R(−1) was replaced. In contrast, the D101E mutation had only minor effects. This last observation is in direct contrast to published data on the effect of D101E, in which a 10-fold decrease in activity was observed with one of the amc-containing substrates that lacked arginine at the −1 position.12 Altogether, we propose that the ionic interaction contributes to selectivity and binding, whereas the hydrogen bonding is more reflective of the amount of time the substrate spends in a catalytically relevant configuration as determined by other interactions.
Our combination of experimental and computational approaches described here has revealed an interaction that was not uncovered by previous attempts to observe substrate preferences for KDACs by random substrate screens or by characterization of substrate and/or inhibitor binding in crystals, providing the first explanation of how KDAC8 discriminates between substrates. Our conclusions also provide a data-supported rationale for how these preferences translate to unique specificity between members of the KDAC family. An interaction such as D101-R(−1), which appears to rely on dynamic behavior of the enzyme and is not obvious from crystal structures, could serve as the basis for identifying interactions to lead to more highly selective KDAC inhibitors.15 Our approach can also be extended to probing the preliminary observations of specificity in KDAC1 or KDAC6 (Figure 7), or other KDACs. For example, it appears that KDAC1 has a greater tolerance for residues in the −1 position than does KDAC8, but some discrete preferences are present. The weaker preference of KDAC1 for a positive charge in the −1 substrate position presumably relates to other residues near the active site that are not conserved with KDAC8, and is an area of ongoing research. In contrast, the identity of the −1 residue appears to be of limited importance for KDAC6, at least within the context of the rest of the peptide sequence utilized here. Similarly, comparisons between selectivity of the KDACs may provide insight into the evolutionary pressure leading to natural selection of these enzymes. In particular, we note that although KDAC8 was active with a peptide with lysine in the −1 position, the activity was significantly lower than with arginine. In contrast, both KDAC1 and KDAC6 have equivalent activity with either residue (Figure 7). This observation suggests a hypothesis that the presence of tyrosine at position 100, which is in contrast to the aspartic acid or glutamic acid found in most other KDACs, may have allowed KDAC8 to distinguish between arginine and lysine at the −1 position, whereas other KDACs may be less able to do so. Similarly, the conservation of aspartic acid at the 101 position in all class I KDACs (but not all other KDACs), even to the exclusion of glutamic acid, may relate to selectivity rather than overall catalytic ability. These hypotheses will be the focus of future studies. We also expect to extend this approach to examine longer-range contributions of the type that have been reported to influence activity.16 Understanding how KDACs selectively interact with their substrates will ultimately lead to a more complete understanding of KDAC-substrate pairs and, thus, of both the fundamental biochemistry of KDACS and of how perturbations of various KDACs can lead to or treat diseases.
Supplementary Material
Acknowledgments
We thank Dr. Elena Skripnikova for cloning of pFastBacI-KDAC8-D101E.
Funding
This work was supported by National Science Foundation MCB 1817358 and CHE 1625993; the National Institutes of Health R15GM129682, 5G1200MD7595, TL4GM118968, and UL1GM118967; the U. S. Army Research Laboratory and the U. S. Army Research Office W911NF1310129; the U. S. Department of Education P217A170260; and the Louisiana Cancer Research Consortium.
Abbreviations
- amc
7-amino-4-methylcoumarin
- Kac
acetyllysine
- KDAC
lysine deacetylase
- MALDI
matrix-assisted laser desorption ionization
- MD
molecular dynamics
- PTM
post-translational modification
- TEV
tobacco etch virus
- TOF
time-of-flight
Footnotes
Supporting Information
Specific activity values (Table S1), MALDI-TOF assay controls (Figure S1), and circular dichroism spectra (Figure S2). The Supporting Information is available free of charge on the ACS Publications website at
The authors declare that they have no conflicts of interest with the contents of this article.
References
- (1).Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang X-J, and Zhao Y (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618. [DOI] [PubMed] [Google Scholar]
- (2).Basu A, Rose KL, Zhang J, Beavis RC, Ueberheide B, Garcia BA, Chait B, Zhao Y, Hunt DF, Segal E, Allis CD, and Hake SB (2009) Proteome-wide prediction of acetylation substrates. Proc. Natl. Acad. Sci. U.S.A 106, 13785–13790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, and Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- (4).Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, and Guan K-L (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, and Olsen JV (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schölz C, Weinert BT, Wagner SA, Beli P, Miyake Y, Qi J, Jensen LJ, Streicher W, McCarthy AR, Westwood NJ, Lain S, Cox J, Matthias P, Mann M, Bradner JE, and Choudhary C (2015) Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol 33, 415–423. [DOI] [PubMed] [Google Scholar]
- (7).Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, and Pavletich NP (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193. [DOI] [PubMed] [Google Scholar]
- (8).Chen K, Zhang X, Wu Y-D, and Wiest O (2014) Inhibition and mechanism of HDAC8 revisited. J. Am. Chem. Soc 136, 11636–11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gantt SL, Gattis SG, and Fierke CA (2006) Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry 45, 6170–6178. [DOI] [PubMed] [Google Scholar]
- (10).Toro TB, and Watt TJ (2020) Critical review of non-histone human substrates of metal-dependent lysine deacetylases. FASEB J. 34, 13140–13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkühler C, Gallinari P, and Carfí A (2008) Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J. Biol. Chem 283, 26694–26704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dowling DP, Gantt SL, Gattis SG, Fierke CA, and Christianson DW (2008) Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry 47, 13554–13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, De Francesco R, Steinkuhler C, and Di Marco S (2007) Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 8, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kunze MBA, Wright DW, Werbeck ND, Kirkpatrick J, Coveney PV, and Hansen DF (2013) Loop interactions and dynamics tune the enzymatic activity of the human histone deacetylase 8. J. Am. Chem. Soc 135, 17862–17868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Melesina J, Simoben CV, Praetorius L, Bülbül EF, Robaa D, and Sippl W (2021) Strategies to design selective histone deacetylase inhibitors. ChemMedChem 16, 1336–1359. [DOI] [PubMed] [Google Scholar]
- (16).Castañeda CA, Wolfson NA, Leng KR, Kuo Y-M, Andrews AJ, and Fierke CA (2017) HDAC8 substrate selectivity is determined by long- and short-range interactions leading to enhanced reactivity for full-length histone substrates compared with peptides. J. Biol. Chem 292, 21568–21577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Alam N, Zimmerman L, Wolfson NA, Joseph CG, Fierke CA, and Schueler-Furman O (2016) Structure-Based Identification of HDAC8 Non-histone Substrates. Structure 24, 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gurard-Levin ZA, Kilian KA, Kim J, Bahr K, and Mrksich M (2010) Peptide arrays identify isoform-selective substrates for profiling endogenous lysine deacetylase activity. ACS Chem. Biol 5, 863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Riester D, Hildmann C, Grunewald S, Beckers T, and Schwienhorst A (2007) Factors affecting the substrate specificity of histone deacetylases. Biochem. Biophys. Res. Commun 357, 439–445. [DOI] [PubMed] [Google Scholar]
- (20).Toro TB, Bryant JR, and Watt TJ (2017) Lysine deacetylases exhibit distinct changes in activity profiles due to fluorophore-conjugation of substrates. Biochemistry 56, 4549–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Toro TB, Painter RG, Haynes RA, Glotser EY, Bratton MR, Bryant JR, Nichols KA, Matthew-Onabanjo AN, Matthew AN, Bratcher DR, Perry CD, and Watt TJ (2018) Purification of metal-dependent lysine deacetylases with consistently high activity. Protein Expr. Purif 141, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Toro TB, and Watt TJ (2015) KDAC8 substrate specificity quantified by a biologically-relevant, label-free deacetylation assay. Protein Sci. 24, 2020–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sali A, and Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- (24).MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, and Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (25).Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, and Mackerell AD (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang Y, and Skolnick J (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, Fairall L, and Schwabe JWR (2013) Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 51, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hai Y, and Christianson DW (2016) Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Watson PJ, Millard CJ, Riley AM, Robertson NS, Wright LC, Godage HY, Cowley SM, Jamieson AG, Potter BVL, and Schwabe JWR (2016) Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat. Commun 7, 11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- (31).Bekker H, Berendsen HJC, Dijkstra EJ, Achterop S, van Drunen R, van der Spoel D, Sijbers A, Keegstra H, and Renardus MKR (1993) Gromacs: A parallel computer for molecular dynamics simulations, in Physics computing (Groot de, A. R, and Nardchal J, Eds.), pp 252–256. World Scientific, Singapore. [Google Scholar]
- (32).Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, and Kollman P (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem 24, 1999–2012. [DOI] [PubMed] [Google Scholar]
- (33).Khoury GA, Thompson JP, Smadbeck J, Kieslich CA, and Floudas CA (2013) Forcefield_PTM: Ab initio charge and AMBER forcefield parameters for frequently occurring post-translational modifications. J. Chem. Theory Comput 9, 5653–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Vanquelef E, Simon S, Marquant G, Garcia E, Klimerak G, Delepine JC, Cieplak P, and Dupradeau F-Y (2011) R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 39, W511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Páll S, and Hess B (2013) A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun 184, 2641–2650. [Google Scholar]
- (36).Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, and Haak JR (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys 81, 3684–3690. [Google Scholar]
- (37).Hess B, Bekker H, Berendsen HJC, and Fraaije JGEM (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 18, 1463–1472. [Google Scholar]
- (38).Bussi G, Donadio D, and Parrinello M (2007) Canonical sampling through velocity rescaling. J. Chem. Phys 126, 014101. [DOI] [PubMed] [Google Scholar]
- (39).Parrinello M, and Rahman A (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys 52, 7182–7190. [Google Scholar]
- (40).Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, and Di Marco S (2004) Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U.S.A 101, 15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Iaconelli J, Lalonde J, Watmuff B, Liu B, Mazitschek R, Haggarty SJ, and Karmacharya R (2017) Lysine deacetylation by HDAC6 regulates the kinase activity of AKT in human neural progenitor cells. ACS Chem. Biol 12, 2139–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Li Y, Zhang X, Polakiewicz RD, Yao T-P, and Comb MJ (2008) HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem 283, 12686–12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Watanabe Y, Khodosevich K, and Monyer H (2014) Dendrite development regulated by the schizophrenia-associated gene FEZ1 involves the ubiquitin proteasome system. Cell Rep 7, 552–564. [DOI] [PubMed] [Google Scholar]
- (44).Olson DE, Udeshi ND, Wolfson NA, Pitcairn CA, Sullivan ED, Jaffe JD, Svinkina T, Natoli T, Lu X, Paulk J, McCarren P, Wagner FF, Barker D, Howe E, Lazzaro F, Gale JP, Zhang Y-L, Subramanian A, Fierke CA, Carr SA, and Holson EB (2014) An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem. Biol 9, 2210–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Qian H, Chen Y, Nian Z, Su L, Yu H, Chen F-J, Zhang X, Xu W, Zhou L, Liu J, Yu J, Yu L, Gao Y, Zhang H, Zhang H, Zhao S, Yu L, Xiao R-P, Bao Y, Hou S, Li P, Li J, Deng H, Jia W, and Li P (2017) HDAC6-mediated acetylation of lipid droplet-binding protein CIDEC regulates fat-induced lipid storage. J. Clin. Invest 127, 1353–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yan K, Cao Q, Reilly CM, Young NL, Garcia BA, and Mishra N (2011) Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J. Biol. Chem 286, 28833–28843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ha S-D, Reid C, Meshkibaf S, and Kim SO (2016) Inhibition of interleukin 1β (IL-1β) expression by anthrax lethal toxin (LeTx) is reversed by histone deacetylase 8 (HDAC8) inhibition in murine macrophages. J. Biol. Chem 291, 8745–8755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yang Y, Fiskus W, Yong B, Atadja P, Takahashi Y, Pandita TK, Wang H-G, and Bhalla KN (2013) Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. U.S.A 110, 6841–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sen N, Kumari R, Singh MI, and Das S (2013) HDAC5, a key component in temporal regulation of p53-mediated transactivation in response to genotoxic stress. Mol. Cell 52, 406–420. [DOI] [PubMed] [Google Scholar]
- (50).Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, Zhai Y, Kossenkov AV, Vara-Ailor A, Rauscher FJ, Zou W, Speicher DW, Huntsman DG, Conejo-Garcia JR, Cho KR, Christianson DW, and Zhang R (2017) ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol 19, 962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Park S-Y, Phorl S, Jung S, Sovannarith K, Lee S-I, Noh S, Han M, Naskar R, Kim J-Y, Choi Y-J, and Lee J-Y (2017) HDAC6 deficiency induces apoptosis in mesenchymal stem cells through p53 K120 acetylation. Biochem. Biophys. Res. Commun 494, 51–56. [DOI] [PubMed] [Google Scholar]
- (52).Li X, Zhang Q, Ding Y, Liu Y, Zhao D, Zhao K, Shen Q, Liu X, Zhu X, Li N, Cheng Z, Fan G, Wang Q, and Cao X (2016) Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol 17, 806–815. [DOI] [PubMed] [Google Scholar]
- (53).Lu J, Qu S, Yao B, Xu Y, Jin Y, Shi K, Shui Y, Pan S, Chen L, and Ma C (2016) Osterix acetylation at K307 and K312 enhances its transcriptional activity and is required for osteoblast differentiation. Oncotarget 7, 37471–37486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gurard-Levin ZA, Kim J, and Mrksich M (2009) Combining mass spectrometry and peptide arrays to profile the specificities of histone deacetylases. ChemBioChem 10, 2159–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.