

Abstract
Uniparental embryos derived from only the mother (gynogenetic [GG]) or the father (androgenetic [AG]) are unique models for studying genomic imprinting and parental contributions to embryonic development. Human parthenogenetic embryos can be obtained following artificial activation of unfertilized oocytes, but the production of AG embryos by injection of two sperm into one denucleated oocyte leads to an extra centriole, resulting in multipolar spindles, abnormal cell division, and developmental defects. Here, we improved androgenote production by transferring the male pronucleus from one zygote into another haploid androgenote to prevent extra centrioles and successfully generated human diploid AG embryos capable of developing into blastocysts with an identifiable inner cell mass (ICM) and trophectoderm (TE). The GG embryos were also generated. The zygotic genome was successfully activated in both the AG and GG embryos. DNA methylome analysis showed that the GG blastocysts partially retain the oocyte transcription-dependent methylation pattern, whereas the AG blastocyst methylome showed more extensive demethylation. The methylation states of most known imprinted differentially methylated regions (DMRs) were recapitulated in the AG and GG blastocysts. Novel candidate imprinted DMRs were also identified. The production of uniparental human embryos followed by transcriptome and methylome analysis is valuable for identifying parental contributions and epigenome memory transitions during early human development.
Uniparental embryos are derived from only the mother (parthenogenetic/gynogenetic [GG]) or the father (androgenetic [AG]). Interfering with the meiotic reduction in oocytes leads to the development of a diploid or haploid embryo with maternal chromosomes only, thus producing parthenogenetic embryos. In contrast, reconstructing zygotes with two paternal pronuclei results in the generation of AG embryos. These uniparental embryos were originally produced to demonstrate the complementary parental contribution to early development (McGrath and Solter 1984; Surani et al. 1984). Mouse uniparental embryos have become a valuable tool for analysis of genomic imprinting, gene function, and epigenetic dynamics (Dean et al. 2001a). However, species-specific epigenetic regulation does exist (Haaf 2006). Major epigenetic reprogramming during preimplantation development differs across mammalian species (Beaujean et al. 2004; Fulka et al. 2004; Shi et al. 2004; Loi et al. 2008). Given the importance of epigenetic modifications and imprinting on human embryonic/placental development and the reported epigenetic errors incurred by assisted reproductive technologies (Maher et al. 2003; Niemitz and Feinberg 2004), it is essential to generate uniparental human diploid embryos to investigate gene imprinting and the contribution of paternal and maternal genomes in human diseases.
Parthenogenetic embryos can be obtained by artificial activation of unfertilized oocytes in diverse mammalian species, whereas the production of AG embryos is a complex and intricate procedure with a low success rate for blastocyst formation. Compared with that of haploid AG embryos, the generation of diploid AG embryos is much more difficult in domestic animals (Matsukawa et al. 2007). In contrast to mice and other rodents, in which the sperm does not contribute a centriole to the oocyte at fertilization, in humans, sheep, and cows, centrioles are paternally inherited and organize the sperm aster after fertilization in zygotes (Sathananthan et al. 1996). Diploid mouse AG blastocysts produced by injecting two sperm into one enucleated mouse oocyte did not compromise the blastocyst formation rate (Latham 2005). However, injection of two sperm into one enucleated domestic animal oocyte resulted in a very poor blastocyst formation rate. Efforts have been made to generate human AG embryos, but introducing an extra centrosome into an enucleated oocyte with fertilization by two sperm showed that direct cleavage was lower in the AG embryos at the first division, and the produced AG morula displayed major differentiation defects with no identifiable inner cell mass (ICM) and limited trophectoderm (TE) development (Leng et al. 2019). Given the poor development of diploid in vitro fertilization (IVF) AG embryos, Matsukawa et al. (2007) diploidized unipronucleated oocytes by pronuclear transfer and found that the blastocyst formation rates of reconstructed AG embryos increased to 18.8% from 2%, and a similar trend was observed in sheep, in which the blastocyst formation rate of diploid AG embryos increased from 11.5% to 31.3%. Mouse AG haploid embryonic stem (ES) cell lines could be established by transferring sperm into an enucleated oocyte to produce live transgenic mice (Li et al. 2012). In humans, parthenogenetic embryos have been generated to derive stem cells (Mai et al. 2007; Revazova et al. 2007), but only haploid cells with limited developmental potential have been generated (Kuznyetsov et al. 2007; Ding et al. 2015; Zhong et al. 2016). For human AG ES cells, haploid embryos were produced to generate AG ES cell lines for investigation of human imprinted genes (Sagi et al. 2019). Hence, an efficient method to generate human diploid AG embryos is essential for analysis of epigenetic reprogramming.
Uniparental embryos were originally produced to demonstrate the complementary parental contribution to early embryonic development and genomic imprinting (McGrath and Solter 1984; Surani et al. 1984; Dean et al. 2001b). Gene imprinting affects fetal development and placental biology before birth and causes postnatal diseases ranging from obesity to psychiatric disorders (Lambertini et al. 2012; Peters 2014). Although this non-Mendelian imprinting phenomenon exists in diverse species ranging from plants to animals (Scott and Spielman 2004), there are major species differences in imprinted genes, with approximately 125 known imprinted genes being identified in mice and approximately 100 in humans (Wilkins et al. 2016). Gene imprinting is controlled by parent-of-origin-dependent, allele-specific DNA methylation (Stelzer et al. 2016). Genome-wide demethylation waves during preimplantation development differ across mammalian species (Beaujean et al. 2004; Fulka et al. 2004; Shi et al. 2004; Loi et al. 2008). The maternal genome is passively demethylated in a replication-dependent manner, whereas some oocyte-specific methylated regions survive and maintain maternal allele-specific methylation at the blastocyst stage during mouse preimplantation development (Smallwood et al. 2011; Kobayashi et al. 2012). Imprinted differentially methylated regions (DMRs) and some transposable elements are specifically protected from demethylation (Messerschmidt et al. 2014). The paternal genome is actively demethylated, and some sperm-specific methylated regions are still maintained at the blastocyst stage (Zhu et al. 2018). Nevertheless, it is difficult to distinguish which genes are expressed exclusively from each chromosome and the differential methylation between parental alleles in human embryos. However, this difficulty could be overcome by the generation of uniparental GG and AG embryos containing only maternal and paternal chromosomes, respectively. Here, we developed a pronuclear transfer strategy to produce human diploid GG and AG blastocysts in vitro. We examined genome-wide copy number variation, DNA methylome, and transcriptome differences using AG, GG, and biparental (Bi-P) blastocysts to investigate potential epigenetic memory transmission from parents to embryos.
Results
Generation of human diploid AG and GG blastocysts
Given that bovine diploid AG embryos show better development than their haploid counterparts (Lagutina et al. 2004; Matsukawa et al. 2007), we hypothesized that diploid human AG embryos could develop into blastocysts. To avoid introducing two centrioles into one enucleated oocyte, we performed intracytoplasmic sperm injection (ICSI) for two metaphase II (MII) oocytes. To obtain a haploid AG embryo, we generated Bi-P zygotes, and the female pronucleus was removed before fusion with the male pronucleus from another zygote with the same method (Fig. 1A). At the beginning of pronucleus formation, the female pronucleus is beneath the second polar body, and the male pronucleus is located around the central cytoplasm (Supplemental Fig. S1A). We took advantage of the size differences (the male pronucleus is larger than the female pronucleus) and unique locations (the female pronucleus is closer to the second polar body than the male pronucleus) of the newly formed pronuclei at 6–9 h after sperm injection into mature oocytes for pronucleus manipulation and generated haploid AG zygotes with the female pronuclei being removed (Supplemental Fig. S1B; Supplemental Movie S1). This process was followed by extracting the male pronucleus from a donor zygote and fusing it with the recipient haploid androgenote using the Sendai virus, leading to the formation of a diploid AG embryo (Supplemental Movie S2). The newly produced AG embryos developed at a similar rate as the Bi-P embryos, as shown in Figure 1B. We did not observe a significant difference in the embryo cleavage rate among the artificial AG and GG embryos using pronuclear transfer and Bi-P transfer with the IVF method. Additionally, after culture for 5–6 d, we observed the formation of blastocysts with an identifiable TE and ICM of the uniparental embryos (Fig. 1B), including the AG embryos for the first time. We obtained 18 blastocysts derived from 36 fused AG diploid embryos (50% success). As a control, 12 normally fertilized Bi-P embryos were cultured, with seven blastocysts obtained (58.3% success) (Fig. 1C; Supplemental Table S1). Using the same method, we generated diploid GG embryos as well (Fig. 1B). Thus, we developed a high-efficiency technology to produce human diploid uniparental blastocysts, and the human diploid AG embryos unexpectedly developed to the blastocyst stage with a morphologically intact TE and ICM.
Figure 1.
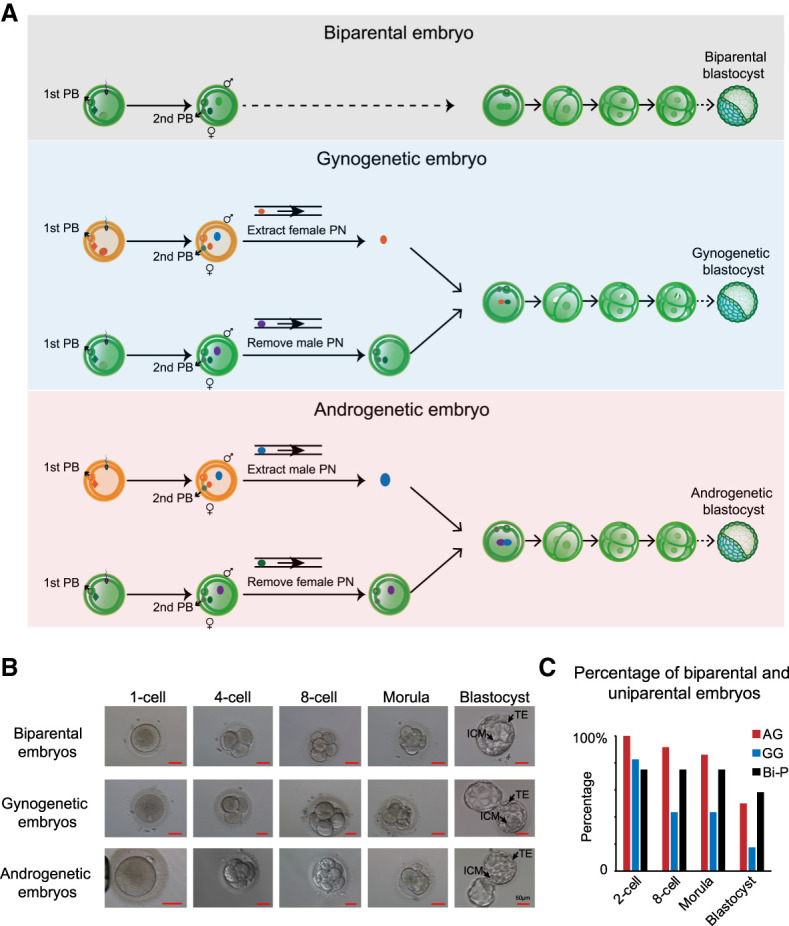
Generation of human diploid AG and GG embryos. (A) Schematic illustration of human diploid Bi-P, GG, and AG embryo generation. Briefly, following ICSI, two oocytes were fertilized. Four to six hours later, female pronuclei were identified based on their proximity to the second (2nd) polar bodies and smaller size compared with male pronuclei. The female pronucleus from individual zygotes was removed to generate haploid androgenotes. The male pronucleus from the donor androgenote was then extracted and mixed with Sendai virus and transferred into the recipient androgenote, leading to the formation of a diploid androgenote for subsequent development into a blastocyst. GG embryos were produced with the same method. (B) The development of Bi-P and uniparental embryos to blastocysts after pronuclei transfer. The uniparental embryos developed blastocysts with identifiable TE and ICM, the same as the Bi-P embryos in vitro. (C) The development ratios of Bi-P, GG, and AG embryos.
Analysis of the parental origin and genome copy number variation of human uniparental blastocysts
We confirmed the origins of AG blastocysts and evaluated their genome copy number variation. We used a SNP array to screen the aneuploidy of AG blastocysts and to reveal their genetic relationship with donor sperm and oocytes (Supplemental Fig. S1C). Among the 15 AG blastocysts analyzed, 13 were found to derive only from the sperm donor (with a probability > 96.6%) without a genetic relationship with oocyte donors, indicating that our AG production strategy was reliable and highly efficient (Supplemental Fig. S1D). Additionally, eight XY, five XX, and no YY blastocysts were produced (Supplemental Fig. S1E), suggesting that YY karyotype embryos were arrested during early embryonic development. We then examined the whole-genome copy number variation of the 13 AG blastocysts using biopsied TE cells, and our data showed that nine embryos were euploid (69%), whereas four were aneuploid (31%) (Supplemental Fig. S1C). Among 13 embryos, two AG blastocysts showed whole-genome loss of heterogeneity (Supplemental Fig. S1E), indicating that their genetic material was inherited exclusively from one male donor.
Parental gene expression of human uniparental embryos
To explore the gene expression of human uniparental blastocysts, we performed RNA sequencing (RNA-seq) for human diploid AG, GG, and Bi-P blastocysts using Smart-seq2. We then compared these data with those of normal human oocytes (GV, MI, and MII), eight-cell embryos, control Bi-P blastocysts, and primed hES cells with genome-wide PCA clustering analysis (Wu et al. 2018) to investigate the differences among the AG, GG, and Bi-P blastocysts. The gene expression profiles of the uniparental blastocysts exhibited a “blastocyst type” pattern similar to that of the Bi-P blastocysts (Fig. 2A; Supplemental Fig. S2A). Oocyte-specific transcripts were degraded in both the AG and GG blastocysts (Fig. 2B; Supplemental Fig. S2B). Zygotic genome activation (ZGA)–related genes were expressed in the AG and GG embryos at comparable levels to those in the Bi-P embryos (Fig. 2B; Supplemental Fig. S2B), indicating the ZGA genes of the uniparental diploid embryos were expressed during development. We found comparable expression of key regulators for early embryonic development and lineage specification such as CDX2, GATA6, POU5F1, and NANOG between the human uniparental and Bi-P blastocysts (Fig. 2C). Furthermore, both ICM- and TE-specific genes were partially expressed in both the AG and GG blastocysts (Supplemental Fig. S2C), confirming the development of both the ICM and the TE. We then evaluated the differentially expressed genes (DEGs) between the uniparental and Bi-P embryos. Here, the Bi-P embryos underwent a normal IVF procedure, which was different from that of the AG and GG groups. There were 64 genes up-regulated in the AG blastocysts compared with Bi-P and 269 genes up-regulated in the GG blastocysts compared with Bi-P, with 23 genes overlapped between the two groups. Genes with up-regulated expression in the AG blastocysts function in RNA modification, whereas those in the GG embryos are related to the cell cycle and cell division. On the other hand, 65 down-regulated genes in AG blastocysts are enriched in transcription, the BMP signaling pathway, and translational elongation, whereas 248 down-regulated genes in GG preferentially function in translational elongation and cell differentiation, (Fig. 2D; Supplemental Table S2). We then compared these DEGs (AG vs. GG) with those from previously published data (paternally expressed genes and maternally expressed genes identified at the cleavage stage from the study of Leng et al. [2019] and paternally expressed genes identified at the morula stage from the study of Zhang et al. [2019]). However, few overlaps were observed (Fig. 2E; Supplemental Table S3), which likely reflects differences in the RNA-seq methods and developmental stages and inherent variations in human embryonic samples.
Figure 2.

The gene expression of uniparental blastocysts. (A) Clustering RNA-seq of human GV, MI, MII, two-cell, eight-cell, diploid AG/GG/Bi-P, and primed hES cells. (B) Plots showing the oocyte-specific (left) and ZGA-related (right) gene expression levels in Bi-P, AG, and GG blastocysts. The criteria of the “oocyte-specific genes” were oocyte-specific genes identified by selecting those expressed at the oocyte stage (FPKM ≥ 5) but not expressed or expressed at low levels in the embryo stage (FPKM ≤ 1) in both mRNA-seq and total RNA-seq. On the other hand, only the genes that were expressed (FPKM ≥ 5) in the post-ZGA stages but not expressed in the oocyte (FPKM ≤ 1) were identified as ZGA genes. (C) Bar plot showing the expression levels of the marker genes CDX2, GATA6, POU5F1, and NANOG in MII oocytes and Bi-P (two-cell eight-cell and blastocysts), AG, and GG blastocysts. (D) Venn diagrams show the DEGs in the uniparental versus biparental groups. Only the genes expressed at one stage with FPKM ≥ 5 and at least twofold changes between two groups and with a P-value generated by DESeq2 (Love et al. 2014) less than 0.05 were selected as DEGs. (E) Venn diagram showing the comparison of DEGs in our study and previously published studies (Zhang et al. 2019). Only the genes expressed at one stage with FPKM ≥ 5 and at least twofold changes between AG and GG and with a P-value generated by DESeq2 (Love et al. 2014) less than 0.05 were selected as DEGs in our study. (Bi-P) Biparental embryo, (AG) androgenetic, (GG) gynogenetic.
DNA methylation reprogramming of the uniparental and Bi-P blastocysts
Paternal and maternal DNA methylation is globally erased until the blastocyst stage, and remethylation occurs after implantation (Guo et al. 2014). To investigate the DNA methylation reprogramming of uniparental blastocysts, we first confirmed that the expression of both DNA methyltransferases and demethylases (methylation oxidases) was comparable among these embryos (Supplemental Fig. S2D). Then, we conducted postbisulfite methylome profiling for all three types of blastocysts (Farlik et al. 2015). Whole-genome DNA methylomes of the AG, GG, and Bi-P blastocysts were analyzed using Bismark (Krueger and Andrews 2011), and the uniquely mapped reads were analyzed. Global CpG DNA methylation levels of the Bi-P blastocysts were similar to those previously reported (Guo et al. 2014), and global DNA methylation levels for the AG, Bi-P, and GG blastocysts were 22%, 27%, and 41%, respectively (Fig. 3A). Clustering analysis showed that the DNA methylation pattern of AG blastocysts was closer to that of sperm, whereas the GG blastocysts were closer to oocytes, indicating that uniparental embryos partially inherited gametic methylomes (Fig. 3B,C). The genome-wide methylated CpG sites, including those at repeats, showed greater retention in the GG blastocysts than in the AG blastocysts (Supplemental Fig. S3A–C), suggesting that the paternal methylome undergoes stronger demethylation than the maternal genome. GG blastocysts also showed the highest mCH levels (Supplemental Fig. S3D). As reported previously, a transcription-dependent methylation pattern was detected in human oocytes but not in sperm (Smith et al. 2014; Zhou et al. 2019). The GG blastocysts retained this methylation pattern in all replicates (Fig. 3D; Supplemental Fig. S3E). Only ∼29.0% of DNA methylation was erased in the GG blastocysts compared with the oocytes, in contrast to 74.3% in the AG blastocysts compared with the sperm (Supplemental Fig. S3F). As TET proteins were implicated in paternal DNA demethylation, especially in compartment B and the intergenic regions in mouse embryos (Zhang et al. 2016), we speculated that TETs play different roles in the AG and GG human embryos.
Figure 3.

DNA methylome of diploid AG, GG, and Bi-P blastocysts. (A) Global CpG methylation in the AG, Bi-P, GG embryos, and gametes. Note that the AG blastocysts exhibited the lowest level of global DNA methylation, whereas the sperm showed the highest level, as expected. The sperm and oocyte DNA methylation data were used in the Okae et al. (2014) study. (B) Principal component analysis showed a genome-wide DNA methylation relationship among three types of blastocysts and germ cells. (C) Nonsupervised cluster analysis of the DNA methylome from different blastocysts and gametes. (D) The metaplot showing DNA methylation around gene bodies in the AG, Bi-P, GG blastocysts, and gametes. (E) The relationship between TSS (left) and gene body (right) DNA methylation and gene expression of the Bi-P blastocysts and the uniparental blastocysts. (F) Heatmaps showing DEGs in the AG and GG groups and associated gene promoter methylation levels. Only the genes expressed at one stage with FPKM ≥ 5 and at least twofold changes between two groups and with a P-value generated by DESeq2 (Love et al. 2014) less than 0.05 were selected as DEGs. (Bi-P) Biparental embryo, (AG) androgenetic, (GG) gynogenetic.
We further asked how DNA methylation is associated with gene expression. Globally, active and inactive promoters are preferentially hypomethylated and hypermethylated, respectively, in all embryos. Active genes tend to be methylated in gene bodies, although we did not observe a further correlation between gene expression levels and gene body methylation (Fig. 3E), consistent with the notion that global de novo methylation waves have not yet started in blastocysts. Focusing on the DEGs between the AG and GG blastocysts, we found that genes with up-regulated expression in the AG blastocysts tend to have lower promoter DNA methylation than those in the GG blastocysts (Fig. 3F), and these genes are enriched for transcription; the trend for genes with up-regulated expression in the GG blastocysts is less clear (Fig. 3F; Supplemental Table S3). Overall, these data reveal DNA methylation reprogramming in uniparental human blastocysts.
State of imprinted DMRs in uniparental blastocysts
DNA demethylation occurs after fertilization, and global DNA methylation is at the lowest level until the blastocyst stage (Guo et al. 2014; Smith et al. 2014) before implantation. However, germline-imprinted DNA methylation could survive demethylation after fertilization (Guo et al. 2014; Smith et al. 2014). We then sought to determine the states of known imprinted DMRs in the uniparental embryos. As expected, the maternal-specific imprinted regions PEG3 and SNURF remained methylated in the GG blastocysts but were hypomethylated in the AG embryos. The paternally methylated DMRs H19 and IGF2 remained methylated in the AG blastocysts but were hypomethylated in the GG blastocysts (Fig. 4A). We next investigated 67 known imprinted DMRs (Court et al. 2014; Okae et al. 2014) in the AG, GG, and Bi-P methylomes using our data and previously published data (Leng et al. 2019). In most DMRs, DNA methylation in the AG and GG blastocysts was consistent with that in the sperm and oocytes, respectively, and agreed with the published data sets of two- to eight-cell embryos (Fig. 4B; Supplemental Fig. S4; Leng et al. 2019). Previous studies reported 67 human imprinted DMRs: 29 maternal germline DMRs (M-gDMRs), two paternal germline DMRs (P-gDMRs), 15 placenta-specific gDMRs, and 21 secondary DMRs (sDMRs) (Court et al. 2014). Our results confirmed that all M-gDMRs, P-gDMRs, and placental DMRs showed the expected differential methylation in the AG and GG blastocysts (Fig. 4B). Most sDMRs showed hypomethylation in both the AG and GG blastocysts. Two DMRs (ZC3H12C/LIN28B DMRs) were defined as sDMRs by Okae et al. (2014) (and were suspected to be placental DMRs) and placenta-specific DMRs by Court et al. (2014). Both of these DMRs were methylated in the GG but not AG blastocysts, indicating that they are likely placental gDMRs (Fig. 4B; Supplemental Table S4). Finally, allelic gene expression also showed a weak negative correlation with DNA methylation (Fig. 4B; Supplemental Table S4). Our results indicated that our uniparental blastocysts are an efficient tool to identify putative imprinted DMRs.
Figure 4.

DNA methylation of known imprinted regions in the AG, GG, and Bi-P blastocysts. (A) UCSC snapshots showing the previously known DMRs located in PEG3, SNURF, IGF2, and H19. Our results showed that germline DMRs survived demethylation during early embryo development, indicating that our diploid AG and GG blastocysts are suitable models to screen for putative DMRs. The sperm and oocyte DNA methylation data are from Okae et al. (2014). (B) Heatmaps showing the DNA methylation levels of known imprinted DMRs and the related gene expression. DNA methylation in two-, four-, and eight-cell embryos published previously was included as a control (Leng et al. 2019). All replicates for the AG, GG, and Bi-P blastocysts were pooled. These known imprinted DMRs were clarified as previously reported (Okae et al. 2014). (Bi-P) Biparental embryo, (AG) androgenetic, (GG) gynogenetic.
Identification of putative novel DMRs
We then performed a genome-wide search for putative novel DMRs. If the methylation level of a region in the AG blastocysts was higher than that in the Bi-P and GG blastocysts, this region was classified as a paternal DMR. Alternatively, if the methylation level of a region in the GG blastocysts was higher than that in the Bi-P and AG blastocysts, this region was classified as a maternal DMR. We detected 105 putative paternally methylated (AG hypermethylated) DMRs and 506 maternally methylated (GG hypermethylated) DMRs (Fig. 5A; Supplemental Fig. S5A; Supplemental Table S5). Differential methylation of these DMRs was found in the sperm/oocytes and the AG/GG two- to eight-cell embryos (Fig. 5B; Supplemental Fig. S5B). Furthermore, paternally methylated DMRs tended to enrich Alu, whereas maternally methylated DMRs were enriched for ERVK (Supplemental Fig. S5C).
Figure 5.

Putative germline regions with differential DNA methylation. (A) UCSC snapshots showing the novel DMRs. (B) Heatmaps showing the DNA methylation levels for newly identified DMRs and the related gene expression. DNA methylation in two-, four-, and eight-cell embryos published previously was included as a control. The DMRs were firstly identified based on a pairwise comparison between AG and GG blastocysts as previously described (Zhang et al. 2018). Only those DMRs with changes in CG methylation levels between sperm and oocyte greater than 0.1 (e.g., for maternal DMRs, the oocyte showed higher methylation levels than the sperm) were taken as allelic putative DMRs (Methods). One hundred five sperm-specific DMRs and 506 oocyte-specific DMRs were detected. (C) Correlation between mCG difference and nearest gene expression difference between AG and GG blastocysts. (D) GO analysis for the nearest genes of maternal and paternal DMRs.
We then asked whether these novel DMRs participate in gene regulation. The differential methylation of DMRs showed a weak negative correlation with the expression of the nearest genes (R = −0.14) (Fig. 5C). GREAT analysis (McLean et al. 2010) showed that paternally methylated regions are present near genes functioning in potassium ion transmembrane transport and O-glycan processing, whereas maternally hypermethylated regions are near genes involved in neurogenesis and transcription (Fig. 5D). We then compared the uniparental embryos with the Bi-P embryos. Only two DEGs resided near DMRs between the AG and Bi-P groups (one gene with up-regulated expression and one with down-regulated expression). Similarly, only eight DEGs were present near DMRs between the GG and Bi-P groups (six down-regulated and two up-regulated) (Supplemental Fig. S6A). Analysis of the AG and GG groups again revealed little overlap between DEGs and DMRs (Supplemental Fig. S6B). This finding was confirmed when we calculated the distances between DEGs and their nearest DMRs compared with the random control (Supplemental Fig. S6C). These data suggest that most novel DMRs identified here may reflect partial inheritance of gametic methylomes and differential DNA demethylation between the two parental genomes and may not yet regulate gene expression at this early stage. It would be interesting to determine if they are involved in gene regulation at later stages. In sum, we generated a list of potential DMRs that require further validation.
Discussion
To elucidate the unique epigenetic regulation of maternal and paternal genes during early embryonic development (Haaf 2006), we generated human diploid AG, GG, and Bi-P blastocysts and investigated genome-wide methylation reprogramming and gene expression for a better understanding of the differential methylation of genes that could impact fetal and placental development. Our results first showed that human diploid AG embryos can successfully develop to the blastocyst stage and, together with GG embryos, are suitable models to study parental epigenome reprogramming.
Uniparental AG embryos and ES cells have been generated using murine models to assess uniparental-specific imprinting, to perform genetic screening of the haploid genome, and to serve as a vehicle for delivering male genetic material (Mann et al. 1990; Tuorto et al. 2012). Murine AG embryos were generated by denucleation of oocytes before injection of male genetic material because the centriole contribution of zygotes is exclusive to maternal origin (Schatten et al. 1991). In contrast, this approach has been difficult for bovine and ovine species because the sperm from these species, similar to that of humans, contribute functional centrioles that, after recruitment of key maternal components, nucleate tubulin, leading to the organization of the first mitosis (Manandhar et al. 1999). Although human haploid AG embryos have been generated, they were either arrested at the eight-cell stage (Kuznyetsov et al. 2007) or showed low cleavage rates for blastocyst formation (Ding et al. 2015). Because ovine (Matsukawa et al. 2007) and bovine (Lagutina et al. 2004) AG embryos were generated following pronuclear transfer to enhance the success of blastocyst formation, we distinguished male and female pronuclei in one-cell human zygotes based on the unique locations and sizes of male and female pronuclei and generated diploid AG human embryos capable of developing into blastocysts by performing pronuclei transfer. Our results showed that this strategy allowed successful generation of human diploid blastocysts. Proper ZGA is extremely important for early embryonic development, and the ZGA genes of the uniparental embryos we produced expressed during ZGA and maternal-to-zygote transition (MZT). In addition to preventing the introduction of an extra centrosome into the oocyte, the use of male pronuclei for transfer in our study might also enhance sperm chromatin reprogramming by retaining the oocyte genome during the first few hours of fertilization. We also identified several DEGs in uniparental blastocysts, allowing analysis of paternal and maternal contributions to embryonic development. We also observed that the blastocyst formation rate of our diploid AG embryos was lower than that of Bi-P, normally fertilized embryos. One possibility is that one-quarter of diploid AG embryos contained lethal YY Chromosomes and were likely arrested during development (Latham 2005).
Our results demonstrated that human AG and GG blastocysts showed different patterns of methylation. The maternal genome exhibits a hypermethylation pattern. TET might not function optimally in the maternal genome. In addition, EHMT2- and SETDB1-dependent H3K9 methylation plays important roles in protecting CpG methylation in the maternal pronucleus in mice (Ramsahoye et al. 2000; Zeng et al. 2019), and a similar mechanism may help protect the maternal methylome in human embryos.
The “imprintome” refers to the differentially methylated regulatory elements that control the parent-of-origin, monoallelic expression of genes (Skaar et al. 2012). Researchers have attempted to define imprinted genes in diverse species, but the identification of the entire repertoire of imprinted genes is difficult because the monoallelic expression of an imprinted gene may occur only in one of several isoforms in a tissue- or stage-specific manner. The present study based on methylome analyses of uniparental human blastocysts represents one approach to provide a genome-wide analysis of potentially imprinted DMRs and genes to fully understand imprinting-related diseases and disorders such as autism, cancer, diabetes, obesity, and schizophrenia. Analysis of known imprinted DMRs confirmed our uniparental embryos as ideal models to identify novel imprinted DMRs, although follow-up studies are needed.
Because epigenetic programming involves both DNA methylation and histone methylation without changing DNA sequences, future studies on chromatin accessibility and histone modifications in human AG and GG embryos are of interest. The generation of human AG blastocysts and future derivation of AG ES cells may pave the way for human imprinting and regenerative medicine–related applications. In summary, we successfully obtained human uniparental blastocysts, especially AG blastocysts with an identifiable TE and ICM, for the first time. These data shed light on parental-specific epigenetic memory reprogramming during early human development.
Methods
Human subject recruitment and controlled ovarian stimulation
This study was approved by the medical ethical committee of the First Affiliated Hospital of Zhengzhou University (approval number 2013-KY-01) in accordance with the measures of the People's Republic of China on the administration of human assisted reproductive technology, the ethical principles of the human assisted reproductive technology, and the Human Sperm Bank and the Helsinki Declaration. Our research followed the guiding principles of the human embryonic stem cell ethics issued by the MOST and MOH of China and was regularly reviewed by the medical ethics committee of The First Affiliated Hospital of Zhengzhou University. Informed consent was obtained from IVF patients who had more than 15 oocytes retrieved, and after the selection of at least 12 oocytes for IVF and cryopreservation, they were willing to donate surplus oocytes for research. After oocyte donation, at least 12 oocytes were kept for patients’ IVF procedures. Anonymous donated semen samples from the Henan Sperm Bank Institute, the First Affiliated Hospital of Zhengzhou University, were used to derive AG embryos. The controlled ovarian hyperstimulation protocol consisted of down-regulation of the expression of gonadotropin-releasing hormone (GnRH) agonist followed by follicle-stimulating hormone/human menopausal gonadotropins (FSH/HMG) and GnRH antagonist treatment protocols. Self-administration of injectable FSH Gonal F (Merck-Serono) or Menopur (Ferring) commenced on cycle day 2 or 3 of the cycle and continued for 8–12 d. Final oocyte maturation was induced by injecting 10,000 IU of human chorionic gonadotrophin (hCG) (Pregnyl, Organon) when at least three follicles reached >18 mm in diameter under ultrasound. Transvaginal ultrasound-guided oocyte retrieval was performed 36 h after hCG administration. Retrieved cumulus-oocyte complexes (COCs) were washed free of follicular fluid and blood before being placed in groups of four in 0.5 mL of equilibrated G-Fert (Vitrolife) covered with mineral oil. COCs were then cultured in a humidified atmosphere of 6% CO2, 5% O2, and 89% N2 for 2–4 h at 37°C until the removal of cumulus cells before ICSI.
Donated COCs were exposed to 80 IU/mL hyaluronidase (Vitrolife) for 1 min (Jin et al. 2014), after which surrounding cumulus cells were removed by stepwise mechanical stripping with pulled Pasteur pipettes. The maturational status of stripped oocytes was evaluated, and MII oocytes were subjected to ICSI. The injected oocytes were rinsed and transferred to 20 µL drops of pre-equilibrated G1 medium (Vitrolife) supplemented with 5% (V/V) human serum albumin (HSA; Vitrolife).
Production of AG embryos
Removal of the female pronucleus and generation of haploid AG zygotes
Timing is important for accurate removal of the female pronucleus from a fertilized oocyte. Because it is difficult to differentiate the female pronucleus from the male pronucleus in human oocytes once pronuclear fusion occurs, removal of the female pronucleus was performed before pronuclei fusion. Time-lapse recording showed that male and female pronuclei were visible as early as 3 h after sperm injection (Payne et al. 1997), and female pronucleus formation occurred at 4.5–5 h after sperm injection. The female pronucleus was distinguished from the male pronucleus based on their size differences and distances from the second polar body. The newly formed female pronucleus was adjacent to the second polar body, and the male pronucleus was close to the center of the cytoplasm. In addition, the newly formed male pronucleus was larger than the female pronucleus. For embryos with visible female and male pronuclei, removal of the female pronucleus was immediately performed. For those without a visible female and/or male pronucleus appearance, a second evaluation was followed 0.5 h later. Locating the second polar body following gentle shaking of the embryo culture assisted the identification of the female pronucleus. At 4 h after sperm injection, the pronucleus was observed in 13% of oocytes, whereas 48% and 84% of fertilized oocytes showed pronuclear formation at 4.5 and 6 h after injection, respectively. Both pronuclei then increased in size and moved toward the center of the oocyte cytoplasm, where pronuclear fusion occurred ∼7 h after injection.
Removal of the female pronucleus was performed in G-MOPS (Vitrolife) containing 5 mg/mL cytochalasin B (CB; Sigma-Aldrich) and 0.2 µg/mL nocodazole (Sigma-Aldrich) (Supplemental Movie S1). The oocyte was first fixed on a holding pipette so that the second polar body and newly formed female pronucleus were situated at the 3-o’-clock position, after which a 10-µm hole was made in the zona pellucida by laser drilling (XYClone laser system, Hamilton-Thorne Biosciences). The female pronucleus was then aspirated from the oocyte using a blunt 20-µm biopsy pipette (Vitrolife). The haploid androgenote containing only the male pronucleus was then cultured in LifeGlobal medium supplemented with 5% HSA before generating diploid AG embryos.
Production of diploid androgenotes using pronuclear transfer
Because injecting two sperm into the same human oocytes results in access to the centrosome and subsequent abnormal embryonic development (Schatten and Sun 2009), diploid AG embryos were constructed using pronuclear transfer, in which the donor male pronucleus was first removed from a haploid androgenote, transferred into the perivitelline space of a recipient haploid androgenote, and then fused with the recipient haploid androgenote (Supplemental Movie S1).
Following removal of the female pronucleus, one haploid androgenote was cultured until 12 h after sperm injection, at which time the male pronucleus from another haploid androgenote was transferred to generate a diploid AG embryo. To obtain the second male pronucleus, we first incubated the donor haploid androgenote in MOPS containing CB (5 mg/mL) and nocodazole (2.5 µg/mL) for 5 min at 37°C. A biopsy pipette with an inner diameter of 25 µm (Vitrolife) was then inserted through the hole made by laser drilling. The male pronucleus containing minimal cytoplasm was aspirated into the pipette as a membrane-bound donor karyoplast. Donor pronucleus karyoplasts were transferred within the biopsy pipette to a 1 µL drop of HVJ-E containing Sendai virus (GenomONE-CFEX HVJ envelope cell fusion kit, Cosmo Bio). The pipette was then moved to a drop containing a recipient haploid androgenote before insertion into the zygote through the same hole made when the female pronucleus was removed. Fusion of the pronucleus karyoplast with the recipient zygote was confirmed visually and usually occurred within 10–30 min. The reconstructed diploid androgenote was transferred to global medium with 5% HSA and cultured individually until day 7 of embryonic development. Embryo quality on day 4 and blastocyst development from day 5 through day 7 were recorded.
Generation of GG embryos
During the reconstruction of diploid AG embryos, one male pronucleus of the first 2PN zygote was extracted and fused with the second 2PN zygote whose female pronucleus was removed, leaving the female pronucleus of the first zygote and the second zygotes whose male pronucleus was removed and contained only the female pronucleus. For diploid parthenogenetic embryos, the female pronucleus of the first zygote was transferred to a drop of media containing HVJ-E with Sendai virus for ∼10 sec by using a biopsy needle with an inner diameter of 25 µm. The female pronucleus was then transferred into the perivitelline space of the second zygote through the same hole through which a male pronucleus was removed. Fusion of the female pronucleus of the first zygote with the zygote containing only the female pronucleus was confirmed visually and usually occurred within 10–30 min. GG embryos were cultured in G1 medium (Vitrolife) containing 5% HSA between day 1 and day 3 and then in G2 medium (Vitrolife) supplemented with 6% HSA on day 3 and further cultured to the blastocyst stage.
Confirmation of the parent–child relationship between AG embryos and sperm donors
AG blastocysts were genetically identified using a human SNP array (HumanCytoSNP-12 v2.1 BeadChip, Illumina). Two to four TE cells were biopsied from AG blastocysts with the assistance of a laser, after which whole-genome amplification was performed by using the REPLI-g midi kit (Qiagen 150045). The genome amplification product was used to identify SNPs using HumanCytoSNP-12 v2.1 BeadChip. The parent–child relationship between the AG blastocysts and the sperm/oocyte donors was assessed to confirm a lack of relationship between AG blastocysts and oocyte donors, as well as a close relationship with the sperm donor. Experimental data were analyzed using the Edit parental relationship and Create reproducibility combined with heritable report module of Genome Studio to calculate the relationship. A P-C heritability-free (parent–child relationship) above 99% was needed for the verification of genetic parent–child relationships.
Single-cell DNA methylation profile and data analysis
We used the postbisulfite PCR method (Farlik et al. 2015) to establish the single-cell methylome library. Briefly, AG, GG, and Bi-P blastocysts were lysed using lysis buffer supplied by an EZ DNA methylation-direct kit (Zymo Research D5020). Bisulfite treatment was directly performed using a lysis mix with approximately 1:400 lambda DNA as a spike-in control to estimate the bisulfite conversion rate. All protocols were performed according to the manufacturer's manual. The eluted bisulfite DNA was amplified using random primers and ligated adapter by using Ampure XP beads purification. Qubit and Agilent 2100 were used to assess the library quality. The HiSeq 2500 PE 2×125 mode was used for sequencing to obtain 90 Gb data per sample. We used hg19 as a reference genome in our previously published work, and the data we downloaded from previously published studies used hg19. Furthermore, we used the GRCh38 reference using the liftOver tool (UCSC) and then analyzed the known imprinted region. The conclusion was the same as that obtained using the h19 reference. Thus, we used hg19 genome reference as well.
RNA-seq library generation and sequencing
The Smart-seq2 libraries of human AG and GG blastocysts were prepared as previously reported (Picelli et al. 2014). The blastocysts were lysed in lysis buffer containing RNase inhibitor according to the Smart-seq v4 ultra low input RNA kit user manual for sequencing (Clontech 634888). The library was quantified using a Qubit and Agilent 2100 system before being subjected to sequencing. The raw sequencing file was demultiplexed using the CASAVA (Illumina) 1.8.2 script.
Identification of DEGs and stage-specific genes
Only the genes expressed at one stage with FPKM ≥ 5 and at least twofold changes between two groups and with a P-value generated by DESeq2 (Love et al. 2014) of less than 0.05 were selected as DEGs. For the identification of the stage-specific genes in eight cells, ICM, and TE, the Shannon entropy-based method was used as previously described (Wu et al. 2018). Maternally expressed genes (FPKM ≥ 1 in GV or MII oocytes) were first removed. For the remaining genes, only the genes with entropy scores less than two and FPKM greater than 10 at the expressed stage but not at other stages were identified as stage-specific genes.
Low-input DNA methylation data processing
All data sets were mapped to the hg19 reference genome by Bismark v0.16.3 (Krueger and Andrews 2011) with the default parameters (Krueger and Andrews 2011). After mapping, the reads mapped to the mitochondrial genome were removed. Then, the multimapped reads and PCR duplicates were removed with the Bismark command deduplicate_bismark. Bismark_methylation_extractor was used to calculate the single CpG methylation level. The CpG sites that were covered at least three times were kept for downstream analysis. For each single CpG site, the methylation level was calculated as the total methylated counts divided by the total counts within this CpG. The methylation levels for pooled cells were generated from merged BAM files for both MII oocytes and in vitro maturation (IVM) oocytes. For the quantification of CpG methylation for bins, such as 200-bp, 1-kb, or 1-Mb bins, the methylation value was calculated as the total methylated counts divided by the total counts within each bin.
Correlation calculation, hierarchical clustering, and PCA of the DNA methylome
The average methylation value for the 10-kb bin within the whole genome of all gametes and embryos was used for Pearson's correlation analysis. The average methylation value for the 10-kb bin within the whole genome of all gametes and embryos was used for the clustering analysis. This hierarchical clustering was conducted with Cluster 3.0 with –e 2 (Pearson's correlation). The average methylation value for the 10-kb bin of the entire genome for all gametes and embryos was used for the PCA. The princomp command in R (R Core Team 2019) was used for this analysis.
The DNA methylation level distribution at different genomic elements
For individual genes, the gene body was defined as the region from the transcription start site (TSS) to the transcription end site (TES). To calculate the methylation around the active and inactive gene body, we expanded the gene body region (from 10 kb downstream from the TSS to the TSS and TES to 10 kb upstream of the TES). The whole expanded regions were divided into 50 bins, and average methylation levels were calculated for each bin with single CpG methylation files for all tissues. The promoters were identified from 2.5 kb downstream from the TSS to 2.5 kb upstream of the TSS. The promoter or gene body methylation levels were calculated as above. The methylation value was calculated as the total methylated count divided by the total count within each promoter or gene body. To determine the DNA methylation levels in different genomic elements, we segmented the genome into CpG islands (CGIs), 5′ untranslated regions (5′ UTRs), promoters (from 2.5 kb downstream from the TSS to 2.5 kb upstream of the TSS), exons, introns, 3′ untranslated regions (3′ UTRs), and different repeat families, including Alu, ERVK, LINE, and SINE, using annotations combining the RefSeq, UCSC Known Gene, and Repbase databases. The average methylation levels were calculated for each genomic element with single CpG methylation files for all tissues.
Known and putative DMR identification
First, we identified differentially methylated CGs based on a pairwise comparison between the GG, AG, and Bi-P blastocysts as previously described (Zhang et al. 2018). Briefly, a single CG covered by at least five reads was selected for downstream analyses. Two-tailed Fisher's exact test was performed to evaluate the significance of differentially methylated CG sites between two types of blastocysts. Only CG sites with a P-value less than 0.05 and a change in CG methylation levels between two types greater than 0.1 were identified and used for downstream analyses. We then selected differentially methylated bins (2 kb) containing at least three differentially methylated CGs. These bins that were no more than 2 kb away were further merged into DMRs. For exploration of the genomic distribution of these DNA DMRs, the genome was segmented into TSSs, exons, introns, TESs, and intergenic regions using annotations combining the RefSeq, UCSC Known Gene, Ensemble, and GENCODE databases as described above. A set of random regions was generated with a length equal to each hypermethylated region to assess the significance of hypermethylated regions falling into a certain category. The numbers of regions that fell into each category were calculated, and the significance was computed as the log ratio of observed numbers divided by those for random regions (observed/expected).
For putative allelic DMR selection, we further evaluated the DNA methylation levels in both sperm and oocytes within all the DMRs identified above. Only those DMRs with changes in CG methylation levels between sperm and oocytes greater than 0.1 (e.g., for maternal DMRs, the oocyte showed higher methylation levels than the sperm) were taken as allelic putative DMRs. DAVID v6.7 (Dennis et al. 2003) was used for GO analysis for the closest genes for each allelic DMR.
Data access
All raw and processed sequencing data generated in this study have been submitted to the Genome Sequence Archive in National Genomics Data Center (China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences) (Wang et al. 2017; CNCB-NGDC Members and Partners 2021; http://bigd.big.ac.cn/gsa-human) under accession number HRA000323.
Supplementary Material
Acknowledgments
This work was supported by the National Key R&D Program of China (2019YFA0110900 to Y. Sun and J.X., 2019YFA0802200 to J.X.) and the National Natural Science Foundation of China (31870817 to J.X.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.273318.120.
Competing interest statement
The authors declare no competing interests.
References
- Beaujean N, Taylor J, Gardner J, Wilmut I, Meehan R, Young L. 2004. Effect of limited DNA methylation reprogramming in the normal sheep embryo on somatic cell nuclear transfer. Biol Reprod 71: 185–193. 10.1095/biolreprod.103.026559 [DOI] [PubMed] [Google Scholar]
- CNCB-NGDC Members and Partners. 2021. Database resources of the National Genomics Data Center, China National Center for Bioinformation in 2021. Nucleic Acids Res 49: D18–D28. 10.1093/nar/gkaa1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, Simon C, Moore H, Harness JV, et al. 2014. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res 24: 554–569. 10.1101/gr.164913.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean W, Santos F, Stojkovic M, Zakhartchenko V, Walter J, Wolf E, Reik W. 2001a. Conservation of methylation reprogramming in mammalian development: aberrant reprogramming in cloned embryos. Proc Natl Acad Sci 98: 13734–13738. 10.1073/pnas.241522698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean WL, Kelsey G, Reik W. 2001b. Generation of monoparental embryos for investigation into genomic imprinting. Methods Mol Biol 181: 1–19. 10.1385/1-59259-211-2:1 [DOI] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: P3. 10.1186/gb-2003-4-5-p3 [DOI] [PubMed] [Google Scholar]
- Ding C, Huang S, Qi Q, Fu R, Zhu W, Cai B, Hong P, Liu Z, Gu T, Zeng Y, et al. 2015. Derivation of a homozygous human androgenetic embryonic stem cell line. Stem Cells Dev 24: 2307–2316. 10.1089/scd.2015.0031 [DOI] [PubMed] [Google Scholar]
- Farlik M, Sheffield NC, Nuzzo A, Datlinger P, Schönegger A, Klughammer J, Bock C. 2015. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep 10: 1386–1397. 10.1016/j.celrep.2015.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulka H, Mrazek M, Tepla O, Fulka J Jr. 2004. DNA methylation pattern in human zygotes and developing embryos. Reproduction 128: 703–708. 10.1530/rep.1.00217 [DOI] [PubMed] [Google Scholar]
- Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y, Yan J, Ren X, Lin S, Li J, et al. 2014. The DNA methylation landscape of human early embryos. Nature 511: 606–610. 10.1038/nature13544 [DOI] [PubMed] [Google Scholar]
- Haaf T. 2006. Methylation dynamics in the early mammalian embryo: implications of genome reprogramming defects for development. Curr Top Microbiol Immunol 310: 13–22. 10.1007/3-540-31181-5_2 [DOI] [PubMed] [Google Scholar]
- Jin H, Shu Y, Dai S, Peng Z, Shi S, Sun Y. 2014. The value of second polar body detection 4 hours after insemination and early rescue ICSI in preventing complete fertilisation failure in patients with borderline semen. Reprod Fertil Dev 26: 346–350. 10.1071/RD12369 [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. 2012. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet 8: e1002440. 10.1371/journal.pgen.1002440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznyetsov V, Kuznyetsova I, Chmura M, Verlinsky Y. 2007. Duplication of the sperm genome by human androgenetic embryo production: towards testing the paternal genome prior to fertilization. Reprod Biomed Online 14: 504–514. 10.1016/S1472-6483(10)60900-5 [DOI] [PubMed] [Google Scholar]
- Lagutina I, Lazzari G, Duchi R, Galli C. 2004. Developmental potential of bovine androgenetic and parthenogenetic embryos: a comparative study. Biol Reprod 70: 400–405. 10.1095/biolreprod.103.021972 [DOI] [PubMed] [Google Scholar]
- Lambertini L, Marsit CJ, Sharma P, Maccani M, Ma Y, Hu J, Chen J. 2012. Imprinted gene expression in fetal growth and development. Placenta 33: 480–486. 10.1016/j.placenta.2012.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham KE. 2005. X chromosome imprinting and inactivation in preimplantation mammalian embryos. Trends Genet 21: 120–127. 10.1016/j.tig.2004.12.003 [DOI] [PubMed] [Google Scholar]
- Leng L, Sun J, Huang J, Gong F, Yang L, Zhang S, Yuan X, Fang F, Xu X, Luo Y, et al. 2019. Single-cell transcriptome analysis of uniparental embryos reveals parent-of-origin effects on human preimplantation development. Cell Stem Cell 25: 697–712.e6. 10.1016/j.stem.2019.09.004 [DOI] [PubMed] [Google Scholar]
- Li W, Shuai L, Wan H, Dong M, Wang M, Sang L, Feng C, Luo GZ, Li T, Li X, et al. 2012. Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature 490: 407–411. 10.1038/nature11435 [DOI] [PubMed] [Google Scholar]
- Loi P, Beaujean N, Khochbin S, Fulka J Jr, Ptak G. 2008. Asymmetric nuclear reprogramming in somatic cell nuclear transfer? Bioessays 30: 66–74. 10.1002/bies.20684 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher ER, Afnan M, Barratt CL. 2003. Epigenetic risks related to assisted reproductive technologies: epigenetics, imprinting, ART and icebergs? Hum Reprod 18: 2508–2511. 10.1093/humrep/deg486 [DOI] [PubMed] [Google Scholar]
- Mai Q, Yu Y, Li T, Wang L, Chen MJ, Huang SZ, Zhou C, Zhou Q. 2007. Derivation of human embryonic stem cell lines from parthenogenetic blastocysts. Cell Res 17: 1008–1019. 10.1038/cr.2007.102 [DOI] [PubMed] [Google Scholar]
- Manandhar G, Simerly C, Salisbury JL, Schatten G. 1999. Centriole and centrin degeneration during mouse spermiogenesis. Cell Motil Cytoskeleton 43: 137–144. [DOI] [PubMed] [Google Scholar]
- Mann JR, Gadi I, Harbison ML, Abbondanzo SJ, Stewart CL. 1990. Androgenetic mouse embryonic stem cells are pluripotent and cause skeletal defects in chimeras: implications for genetic imprinting. Cell 62: 251–260. 10.1016/0092-8674(90)90363-J [DOI] [PubMed] [Google Scholar]
- Matsukawa K, Turco MY, Scapolo PA, Reynolds L, Ptak G, Loi P. 2007. Development of sheep androgenetic embryos is boosted following transfer of male pronuclei into androgenetic hemizygotes. Cloning Stem Cells 9: 374–381. 10.1089/clo.2006.0016 [DOI] [PubMed] [Google Scholar]
- McGrath J, Solter D. 1984. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37: 179–183. 10.1016/0092-8674(84)90313-1 [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messerschmidt DM, Knowles BB, Solter D. 2014. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev 28: 812–828. 10.1101/gad.234294.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemitz EL, Feinberg AP. 2004. Epigenetics and assisted reproductive technology: a call for investigation. Am J Hum Genet 74: 599–609. 10.1086/382897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okae H, Chiba H, Hiura H, Hamada H, Sato A, Utsunomiya T, Kikuchi H, Yoshida H, Tanaka A, Suyama M, et al. 2014. Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet 10: e1004868. 10.1371/journal.pgen.1004868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne D, Flaherty SP, Barry MF, Matthews CD. 1997. Preliminary observations on polar body extrusion and pronuclear formation in human oocytes using time-lapse video cinematography. Hum Reprod 12: 532–541. 10.1093/humrep/12.3.532 [DOI] [PubMed] [Google Scholar]
- Peters J. 2014. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet 15: 517–530. 10.1038/nrg3766 [DOI] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R. 2014. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9: 171–181. 10.1038/nprot.2014.006 [DOI] [PubMed] [Google Scholar]
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. 2000. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci 97: 5237–5242. 10.1073/pnas.97.10.5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2019. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/. [Google Scholar]
- Revazova E, Turovets N, Kochetkova O, Kindarova L, Kuzmichev L, Janus J, Pryzhkova M. 2007. Patient-specific stem cell lines derived from human parthenogenetic blastocysts. Cloning Stem Cells 9: 432–449. 10.1089/clo.2007.0033 [DOI] [PubMed] [Google Scholar]
- Sagi I, De Pinho JC, Zuccaro MV, Atzmon C, Golan-Lev T, Yanuka O, Prosser R, Sadowy A, Perez G, Cabral T, et al. 2019. Distinct imprinting signatures and biased differentiation of human androgenetic and parthenogenetic embryonic stem cells. Cell Stem Cell 25: 419–432.e9. 10.1016/j.stem.2019.06.013 [DOI] [PubMed] [Google Scholar]
- Sathananthan AH, Ratnam SS, Ng SC, Tarin JJ, Gianaroli L, Trounson A. 1996. The sperm centriole: its inheritance, replication and perpetuation in early human embryos. Hum Reprod 11: 345–356. 10.1093/HUMREP/11.2.345 [DOI] [PubMed] [Google Scholar]
- Schatten H, Sun QY. 2009. The role of centrosomes in mammalian fertilization and its significance for ICSI. Mol Hum Reprod 15: 531–538. 10.1093/molehr/gap049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatten G, Simerly C, Schatten H. 1991. Maternal inheritance of centrosomes in mammals? Studies on parthenogenesis and polyspermy in mice. Proc Natl Acad Sci 88: 6785–6789. 10.1073/pnas.88.15.6785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RJ, Spielman M. 2004. Epigenetics: imprinting in plants and mammals—the same but different? Curr Biol 14: R201–R203. 10.1016/j.cub.2004.02.022 [DOI] [PubMed] [Google Scholar]
- Shi W, Dirim F, Wolf E, Zakhartchenko V, Haaf T. 2004. Methylation reprogramming and chromosomal aneuploidy in in vivo fertilized and cloned rabbit preimplantation embryos. Biol Reprod 71: 340–347. 10.1095/biolreprod.103.024554 [DOI] [PubMed] [Google Scholar]
- Skaar DA, Li Y, Bernal AJ, Hoyo C, Murphy SK, Jirtle RL. 2012. The human imprintome: regulatory mechanisms, methods of ascertainment, and roles in disease susceptibility. ILAR J 53: 341–358. 10.1093/ilar.53.3-4.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G. 2011. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet 43: 811–814. 10.1038/ng.864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Humm KC, Karnik R, Mekhoubad S, Regev A, Eggan K, Meissner A. 2014. DNA methylation dynamics of the human preimplantation embryo. Nature 511: 611–615. 10.1038/nature13581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer Y, Wu H, Song Y, Shivalila CS, Markoulaki S, Jaenisch R. 2016. Parent-of-origin DNA methylation dynamics during mouse development. Cell Rep 16: 3167–3180. 10.1016/j.celrep.2016.08.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surani MA, Barton SC, Norris ML. 1984. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308: 548–550. 10.1038/308548a0 [DOI] [PubMed] [Google Scholar]
- Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F. 2012. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol 19: 900–905. 10.1038/nsmb.2357 [DOI] [PubMed] [Google Scholar]
- Wang Y, Song F, Zhu J, Zhang S, Yang Y, Chen T, Tang B, Dong L, Ding N, Zhang Q, et al. 2017. GSA: Genome Sequence Archive. Genom Proteom Bioinform 15: 14–18. 10.1016/j.gpb.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins JF, Úbeda F, Van Cleve J. 2016. The evolving landscape of imprinted genes in humans and mice: conflict among alleles, genes, tissues, and kin. BioEssays 38: 482–489. 10.1002/bies.201500198 [DOI] [PubMed] [Google Scholar]
- Wu J, Xu J, Liu B, Yao G, Wang P, Lin Z, Huang B, Wang X, Li T, Shi S, et al. 2018. Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature 557: 256–260. 10.1038/s41586-018-0080-8 [DOI] [PubMed] [Google Scholar]
- Zeng TB, Han L, Pierce N, Pfeifer GP, Szabó PE. 2019. EHMT2 and SETDB1 protect the maternal pronucleus from 5mC oxidation. Proc Natl Acad Sci 116: 10834–10841. 10.1073/pnas.1819946116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xia W, Wang Q, Towers AJ, Chen J, Gao R, Zhang Y, Yen CA, Lee AY, Li Y, et al. 2016. Isoform switch of TET1 regulates DNA demethylation and mouse development. Mol Cell 64: 1062–1073. 10.1016/j.molcel.2016.10.030 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiang Y, Yin Q, Du Z, Peng X, Wang Q, Fidalgo M, Xia W, Li Y, Zhao ZA, et al. 2018. Dynamic epigenomic landscapes during early lineage specification in mouse embryos. Nat Genet 50: 96–105. 10.1038/s41588-017-0003-x [DOI] [PubMed] [Google Scholar]
- Zhang W, Chen Z, Yin Q, Zhang D, Racowsky C, Zhang Y. 2019. Maternal-biased H3K27me3 correlates with paternal-specific gene expression in the human morula. Genes Dev 33: 382–387. 10.1101/gad.323105.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Zhang M, Yin Q, Zhao H, Wang Y, Huang S, Tao W, Wu K, Chen ZJ, Li J. 2016. Generation of human haploid embryonic stem cells from parthenogenetic embryos obtained by microsurgical removal of male pronucleus. Cell Res 26: 743–746. 10.1038/cr.2016.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Wang R, Yuan P, Ren Y, Mao Y, Li R, Lian Y, Li J, Wen L, Yan L, et al. 2019. Reconstituting the transcriptome and DNA methylome landscapes of human implantation. Nature 572: 660–664. 10.1038/s41586-019-1500-0 [DOI] [PubMed] [Google Scholar]
- Zhu P, Guo H, Ren Y, Hou Y, Dong J, Li R, Lian Y, Fan X, Hu B, Gao Y, et al. 2018. Single-cell DNA methylome sequencing of human preimplantation embryos. Nat Genet 50: 12–19. 10.1038/s41588-017-0007-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.