

SUMMARY
RNA polymerase II (RNA Pol II) transcription reconstituted from purified factors suggests pre-initiation complexes (PICs) can assemble by sequential incorporation of factors at the TATA box. However, these basal transcription reactions are generally independent of activators and co-activators. To study PIC assembly under more realistic conditions, we used single-molecule microscopy to visualize factor dynamics during activator-dependent reactions in nuclear extracts. Surprisingly, RNA Pol II, TFIIF, and TFIIE can preassemble on enhancer-bound activators before loading into PICs, and multiple RNA Pol II complexes can bind simultaneously to create a localized cluster. Unlike TFIIF and TFIIE, TFIIH binding is singular and dependent on the basal promoter. Activator-tethered factors exhibit dwell times on the order of seconds. In contrast, PICs can persist on the order of minutes in the absence of nucleotide triphosphates, although TFIIE remains unexpectedly dynamic even after TFIIH incorporation. Our kinetic measurements lead to a new branched model for activator-dependent PIC assembly.
In brief
Single-molecule microscopy experiments by Baek et al. show that RNA polymerase II and basal transcription factors TFIIF and TFIIE preassemble on UAS/enhancer-bound activators, poised for loading into initiation complexes, with TFIIH at the core promoter. Transcription activators kinetically enhance factor recruitment, creating a localized cluster of polymerases at the UAS/enhancer.
Graphical abstract
INTRODUCTION
Eukaryotic RNA polymerase II (RNA Pol II) transcribes messenger RNAs (mRNAs) and some non-coding RNA species. RNA Pol II initiates at specialized genomic regions called promoters, yet it does not recognize specific DNA sequences themselves (Nikolov and Burley, 1997). Promoter recognition is conferred by the general transcription factors (GTFs) TFIID, TFIIA, TFIIB, TFIIF, TFIIE, and TFIIH, which assemble RNA Pol II into a preinitiation complex (PIC) (reviewed in Hahn, 2004; Orphanides et al., 1996; Roeder, 1996; Schier and Taatjes, 2020; Thomas and Chiang, 2006). Gene expression is often regulated at the level of PIC assembly, yet details of this process remain unclear.
A spectrum of models has been proposed. The prevailing PIC formation model proposes sequential assembly of factors on the core promoter, beginning with DNA recognition by TFIID, or even just its TATA-binding protein (TBP) subunit. TFIIA and TFIIB join next via direct TBP contacts (Buratowski et al., 1989). RNA Pol II and TFIIF are recruited next, followed by TFIIE and finally TFIIH (He et al., 2013; Luse, 2014). This model of ordered binding is based primarily on electrophoretic mobility shift and footprinting assays (Buratowski et al., 1989; Inostroza et al., 1991; Peterson et al., 1991), which monitor complexes from the perspective of the DNA template. In these early in vitro experiments, PICs were assembled from chromatographically separated and purified GTFs (reviewed in Thomas and Chiang, 2006), usually in the absence of transcription activators, Mediator, or the TBP-associated factors (TAFs). While the factor contacts predicted by the stepwise model have been confirmed by structural studies (He et al., 2013, 2016; Plaschka et al., 2016; Schilbach et al., 2017), it remains unclear whether the partial complexes observed in vitro represent physiologically relevant intermediates.
In alternate models, some GTFs form subcomplexes before joining the PIC. For example, TFIIF was first purified as the RNA Pol II-associated proteins RAP30 and RAP74 (Flores et al., 1988, 1990; Sopta et al., 1985). Similarly, direct physical and functional interactions between TFIIE and TFIIH can be observed in vitro (Ohkuma et al., 1995). At the most extreme end of the model spectrum, co-immunoprecipitation studies in both yeast and mammals lead to proposals that an RNA Pol II “holoenzyme” containing multiple GTFs, Mediator, and often other co-activators, arrives at the promoter as a pre-assembled complex (Kim et al., 1994; Koleske and Young, 1994; Maldonado et al., 1996; Ossipow et al., 1995; Ranish et al., 1999).
The previous biochemical and structural experiments underlying PIC models are limited in that they can only isolate and characterize stable complexes. GTF dynamics are rarely directly measured, and when they are, the results are typically population averaged. In contrast, single-molecule experiments can detect short-lived intermediates and can reveal alternative assembly pathways that would otherwise be convoluted in ensemble assays. Recent single-molecule papers have shown that TFIIB binds the TBP-promoter complex only transiently until RNA Pol II joins the complex (Zhang et al., 2016) and have addressed how TFIIH pulls downstream DNA into the RNA Pol II active site to generate an open complex (Fazal et al., 2015; Tomko et al., 2017).
While important mechanistic insights have emerged, one caveat of the single-molecule experiments published to date is that they used purified GTFs, in which transcription is independent of the activators and co-activators required for gene expression in vivo. In vivo single-molecule imaging has therefore emerged as an essential complementary approach for studying factor dynamics, but comes with its own limitations (Zhang and Tjian, 2018). To bridge this gap, we developed a system for fluorescently labeling and visualizing single transcription factor molecules within yeast nuclear extract (Rosen et al., 2020). Containing the full repertoire of nuclear factors, these extracts approximate physiological conditions better than reactions reconstituted with a limited set of purified factors. We and others previously showed that nuclear extracts faithfully recapitulate activator-dependent PIC assembly, initiation, and elongation on bead-immobilized templates (Joo et al., 2017, 2019; Kang et al., 1995; Rani et al., 2004; Ranish et al., 1999; Sikorski et al., 2012; Yudkovsky et al., 2000). Here, we study late-stage PIC assembly using colocalization single-molecule spectroscopy (CoSMoS), a multi-wavelength single-molecule fluorescence method that measures protein binding at individual DNA molecules tethered on microscope slides (Friedman et al., 2006; Hoskins et al., 2011). The relative dynamics of RNA Pol II and the GTFs TFIIF, TFIIE, and TFIIH on DNA were characterized.
Our results argue against a simple sequential assembly of PICs on the TATA box. We show that one or more RNA Pol II molecules transiently associate with the upstream activating sequence (UAS) tethered by activator, with subsequent transfer to the core promoter/TATA box. TFIIF primarily arrives with RNA Pol II at the UAS, although a substantial fraction of TFIIF associates after RNA Pol II. TFIIE is recruited after RNA Pol II and TFIIF, but its initial binding is also independent of the core promoter DNA. In marked contrast to TFIIF and TFIIE, association of TFIIH with DNA is completely dependent on the core promoter. Therefore, activator-stimulated transcription produces a branched pathway, in which multiple partial complexes containing RNA Pol II, TFIIF, and even TFIIE can pre-assemble on the UAS, locally concentrated to facilitate PIC formation at the core promoter.
RESULTS
Fluorescence imaging of individual PICs in nuclear extract
For imaging single-molecule kinetics of RNA Pol II and GTFs, we constructed a yeast strain in which Rpb1, the largest subunit of RNA Pol II, is genetically fused at the C terminus to the SNAPf tag (Keppler et al., 2003). Rpb1-SNAPf was labeled with SNAP-Surface 549 (green-excited dye DY549) during nuclear extract preparation (Figure S1A), as previously described (Rosen et al., 2020). For labeling individual GTFs, the factor of interest was genetically fused to Escherichia coli dihydrofolate reductase (DHFR) (Calloway et al., 2007). The GTFDHFR was labeled by adding Cy5-TMP, comprising the DHFR inhibitor trimethoprim (TMP) linked to the red-excited dye Cy5, to reactions immediately before imaging (Hoskins et al., 2011). The SNAPf/DHFR double-fusion strains (Table S1) had normal growth and in vitro transcription activity relative to untagged parent strains (Figure S1C). The fusions did not appreciably perturb protein expression levels (Figure S1D).
The transcription template, hereafter referred to as UAS+ promoter, has a five-Gal4-binding site UAS linked to the CYC1 core promoter (Joo et al., 2017, 2019; Figure 1A, top). The downstream end of the 299-bp fragment was labeled with the blue-excited dye Alexa Fluor 488 (AF488), while the other end was biotinylated for tethering to the microscope slide surface (Figure 1B). DNAs were visualized by micromirror multi-wavelength total internal reflection fluorescence (TIRF) microscopy (Friedman et al., 2006). Hundreds of DNA molecules can be simultaneously monitored in a given field of view (Figure 1C). After marking positions of the UAS+promoter template molecules, a second DNA carrying only the UAS (Figure 1A, bottom) was similarly tethered to the same slide surface and imaged (Friedman et al., 2013) to serve as an internal negative control for core promoter-specific binding. To account for non-specific background binding to the slide, at least twice as many locations lacking DNA fluorescence (“off DNA”) as DNA locations were randomly selected as controls.
Figure 1. Single-molecule imaging of RNA Pol II and GTF binding to the DNA template.
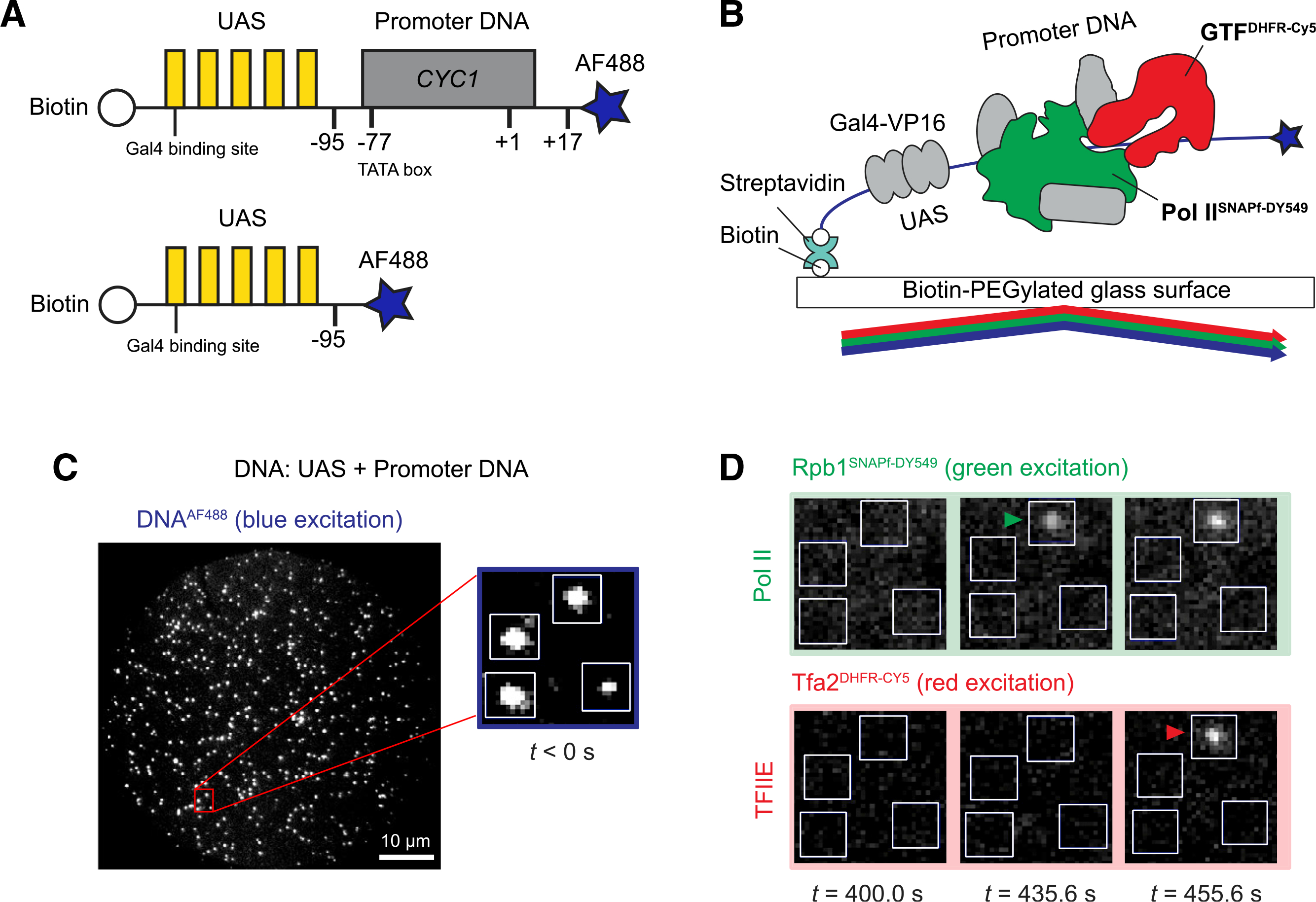
(A) Schematic of DNA templates used. UAS+promoter (top) has five Gal4-binding sites (yellow) upstream of the CYC1 core promoter sequence (gray) carrying a TATA box (−77) and transcription start site (+1). The UAS DNA (bottom) has only the Gal4-binding sites. Both are biotinylated upstream and labeled with Alexa Fluor 488 downstream (blue star) relative to the promoter direction.
(B) Schematic of single-molecule imaging. DNAAF488 molecules were immobilized onto the passivated microscope slide surface via a biotin-streptavidin-biotin linkage and their positions mapped with blue laser excitation. Gal4-VP16 (340 nM) and yeast nuclear extract containing fluorescently labeled RNA Pol IISNAPf-DY549 and GTFDHFR-Cy5 were added and reactions were imaged with alternating excitation with green and red lasers.
(C) Representative image in the blue-excited channel showing hundreds of DNAAF488 molecules that can be simultaneously monitored in a single field of view (65 × 65 μm). The red box marks a magnified area, with 4 DNA molecules (white spots) corresponding to the boxes shown in (D).
(D) Images of the red- and green-excited fluorescence channels at three time points. The indicated time is measured from the initiation of red/green imaging (t = 0 s), which is typically within 2–3 min of extract addition. RNA Pol II and TFIIE arrivals are marked by green and red arrowheads, respectively.
See also Figure S1.
After imaging the positions of DNA molecules, Gal4-VP16 activator (Sadowski et al., 1988) and nuclear extract were introduced into the flow chamber. Unless otherwise noted, no nucleotide triphosphates (NTPs) were added to the extract. There is no RNA synthesis under these conditions (Joo et al., 2017), allowing us to isolate and measure the properties of PICs. For these experiments, ATP was further depleted with hexokinase and glucose to prevent the TFIIH translocase and kinase activities from destabilizing PICs. The arrival and departure of individual Rpb1SNAPf-DY549 and GTFDHFR-Cy5 molecules were monitored by alternating laser excitation at 633 nm (red) and 532 nm (green). Successive images in each channel were captured every 1.4 s (0.5 s/image for each channel, plus switching times) over a time course of 800–1,200 s (i.e., Figure 1D). Colocalization with UAS+promoter or UAS DNAs was determined using fluorescence intensity and proximity thresholds, as previously described (Friedman and Gelles, 2015).
Activator-dependent RNA Pol II recruitment does not require the core promoter
Representative time records of Rpb1SNAPf-DY549 fluorescence colocalized with individual UAS+promoter DNA molecules are shown in Figure 2A. As previously observed in the presence of NTPs (Rosen et al., 2020), binding events of various durations were recorded. Individual DNAs often exhibited serial RNA Pol II binding and dissociation events (Figure 2A, top). Interestingly, simultaneous binding of multiple RNA Pol II molecules also occurred (~45% of total RNA Pol II binding events), seen as multiple fluorescence intensity steps (Figure 2A, center and bottom, brackets). To provide an overview of the reaction, binding records for 100 randomly chosen DNA molecules were converted to binary format, indicating when at least one RNA Pol II was present (color) or absent (white) on the DNA. The resulting horizontal time ribbons were then stacked to form a rastergram and sorted by the order of first RNA Pol II binding from bottom to top (Figure 2B, top). Most DNA templates bound RNA Pol II at least once during the 20 min of imaging.
Figure 2. RNA Pol II association kinetics are independent of the core promoter.
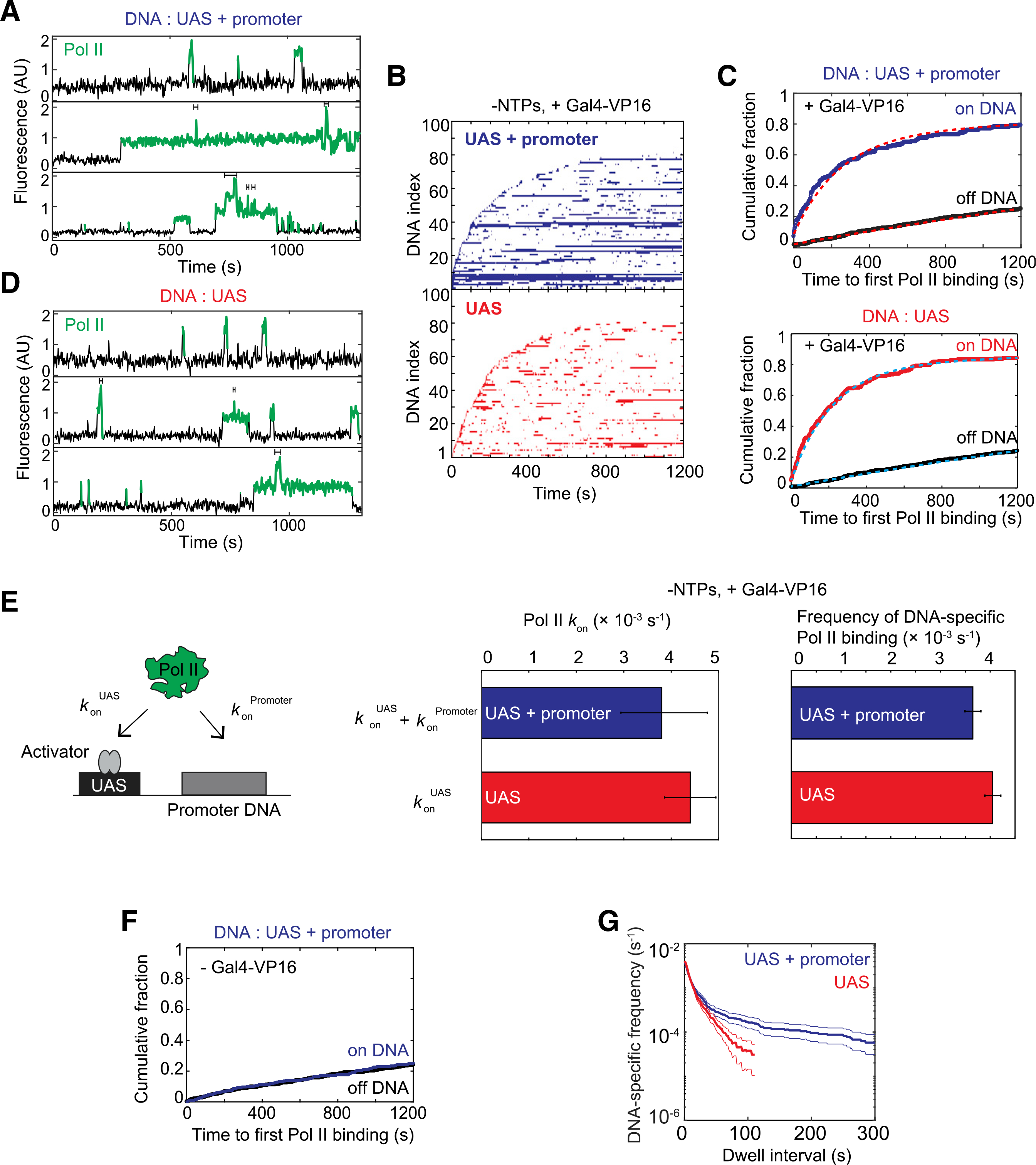
(A) Time records of RNA Pol II fluorescence at three different UAS+promoter locations. Green-colored intervals show times when RNA Pol II colocalizes with the DNA template. Brackets above traces mark when multiple RNA Pol II molecules are simultaneously bound to the same DNA.
(B) Rastergrams show RNA Pol II binding to 100 randomly selected DNA locations, ordered by time of first RNA Pol II detection. Each horizontal line represents a single DNA location, and a colored band indicates ≥1 colocalized RNA Pol II molecules. The top panel data (blue) show UAS+promoter, while the bottom panel data (red) show UAS only. Note that both templates were simultaneously imaged on the same slide surface to ensure identical conditions. This reaction contained Gal4-VP16 activator, but lacked NTPs.
(C) Cumulative fraction of DNA molecules bound at least once by RNA Pol II as a function of time (top: UAS+promoter, blue; bottom: UAS, red). Binding to off DNA sites (black) is shown as a control for background. Data were fit to a single exponential specific binding model (dashed lines). Fit parameters and numbers of observations for all fits are given in (E) (center) and Table S4.
(D) Time records of RNA Pol II fluorescence at three different UAS locations, plotted as in (A).
(E) Schematic at left shows two pathways for initial RNA Pol II association with the DNA template: (1) RNA Pol II recruitment to UAS-bound activator (rate constant konUAS) and (2) direct RNA Pol II recruitment to core promoter (konPromoter). The center panel shows apparent first-order rate constants (±SE) of RNA Pol II initial association with the UAS+promoter (blue) or UAS (red) calculated from curve fitting in (C). The right panel shows DNA-specific binding frequencies (±SE) of total (initial + subsequent) RNA Pol II binding to RNA Pol II-unoccupied DNA.
(F) Cumulative fraction of RNA Pol II-bound DNA versus time for UAS+promoter (blue) or off DNA sites (black), in the absence of Gal4-VP16.
(G) Cumulative distribution of RNA Pol II dwell intervals on UAS+promoter (blue) or UAS (red) with 90% confidence intervals (thin lines). Each continuous time interval with ≥1 labeled RNA Pol II molecules present was scored as a single dwell. Frequency values on the vertical axis include subtraction of off DNA background. Values of ≤0 after background subtraction are not plotted, which is why the UAS curve only extends to ~100 s.
See also Figure S2.
The cumulative fraction of UAS+promoter DNA templates bound at least once by RNA Pol II was plotted as a function of time to first RNA Pol II binding (Figure 2C, top). Whereas 82% ± 3% (SE) of UAS+promoter DNA molecule sites colocalized with RNA Pol II at least once during the 1,200 s imaging interval (blue), only 25% ± 2%(SE) of off DNA locations did (black), demonstrating that RNA Pol II binding was predominantly DNA specific. The data fit well to a single-exponential specific binding model (Friedman and Gelles, 2015; Figure 2C, top, red dashed lines; Table S4), with initial specific binding of RNA Pol II to DNA behaving as a single rate-limiting process with an apparent first-order association rate constant (3.8 ± 1.0) × 10−3 s−1 (Figure 2E, left bar graph, blue), similar to that measured in the presence of NTPs (Rosen et al., 2020). DNA-specific RNA Pol II binding was completely activator dependent, as control experiments lacking Gal4-VP16 showed no DNA binding above the non-specific background (Figure 2F).
Presumably only one PIC can occupy the core promoter at a time, raising the question of how multiple polymerases can bind simultaneously (e.g., Figure 2A, center and bottom, brackets). In our earlier study, we showed that overall RNA Pol II binding to DNA behaved as a single rate-limiting step, yet there was a time lag before binding of the subset of RNA Pol II molecules that proceeded on to elongation. Kinetic modeling suggested the existence of an additional slow step needed to allow RNA Pol II binding to progress to PIC and elongation complexes (Rosen et al., 2020). We postulated that RNA Pol II may initially bind Gal4-VP16 at the UAS, with the arrival of TBP or other GTFs at the core promoter being rate limiting for subsequent RNA Pol II incorporation into the PIC. The initial interaction with the activator at the UAS could also explain simultaneous occupancy by multiple RNA Pol II molecules. In these experiments, the concentration of Gal4-VP16 (340 nM) was in large excess over DNA molecules (<10 pM) and well above the ~1 nM dissociation constant (Taylor et al., 1991). Therefore, each of the five Gal4 binding sites is predicted to be frequently occupied by Gal4-VP16 molecules, and multiple activators present at the UAS could tether multiple RNA Pol II molecules.
To test this idea, we analyzed RNA Pol II binding to a DNA template containing only the UAS in the presence of Gal4-VP16 (Figure 1A, bottom). Supporting the hypothesis, UAS-associated Gal4-VP16 alone was sufficient for both initial Pol II recruitment (Figures 2B and 2C, bottom) and simultaneous binding of multiple molecules (Figure 2D, brackets). Importantly, the yeast Gcn4 activation domain (Gal4-Gcn4) was also sufficient for RNA Pol II recruitment to the UAS (Figure S2A), suggesting that RNA Pol II recruitment to the UAS may be a general mechanism by which activators enhance PIC assembly. RNA Pol II binding to the UAS is activator dependent, as no binding above background was seen in the absence of the activator (Figure S2B). We conclude that one or more RNA Pol II molecules, presumably complexed with Mediator, can bind UAS-bound activators independently of the core promoter.
Is this RNA Pol II binding at the UAS on the pathway to PIC formation or a separate, dead-end pathway? To address this question, the apparent rate constants of initial RNA Pol II binding to UAS (konUAS) and UAS+promoter (konUAS + konPromoter) were compared (Figure 2E, schematic). A direct promoter binding path should increase RNA Pol II binding rate on UAS+promoter relative to UAS by konPromoter. As seen for UAS+promoter, the initial RNA Pol II binding to UAS fit a single exponential model (Figure 2C, bottom, cyan dashed lines; Table S4). The essentially identical rates of first binding at the two DNAs (Figure 2E, left bar graph) suggest that any RNA Pol II going directly to the core promoter cannot be more than a very small fraction of total binding. When we substituted the CYC1 core promoter with the HIS4 core promoter, RNA Pol II was still initially recruited to the UAS and not the core promoter (Figures S2C–S2E). The association rate constant of initial RNA Pol II binding was identical between the CYC1 and HIS4 core promoters (Figure S2E, right; Table S4), suggesting that the initial RNA Pol II recruitment in activator-dependent transcription is independent of the core promoter. This conclusion is further supported by calculating the DNA-specific frequencies for total (rather than only initial) RNA Pol II binding events, where UAS and UAS+promoter were again essentially the same (Figure 2E, right bar graph). These results suggest that, in the context of nuclear extract, RNA Pol II molecules predominantly bind first to UAS-bound activators on the pathway to PIC formation.
One significant difference was seen between UAS and UAS+promoter binding. A cumulative distribution of RNA Pol II dwell times on DNA was plotted after subtracting the off DNA background values (Figure 2G). The curve slopes show different RNA Pol II dwell duration patterns on the two fragments. The nearly straight line seen on the UAS template signifies characteristically short-lived RNA Pol II dwell times. In contrast, RNA Pol II on UAS+promoter showed at least two different characteristic dwell times: one similar to that on UAS alone and a longer-lived component (Figure 2G). This difference is also apparent in rastergrams of the two DNAs, in which many more long duration events can be seen on UAS+promoter (Figures 2B and S2A).
Several observations suggest the long-lived RNA Pol II dwell component results from PIC formation. In addition to dependence on the core promoter, typical RNA Pol II dwell times were strongly decreased by the inclusion of NTPs (Figures S2F and S2G), which trigger PIC dissociation due to initiation and elongation. Furthermore, the GTFs TFIIE and TFIIF show similarly increased stable binding on UAS+promoter relative to UAS (see below). The long-lived RNA Pol II component made up only 10%–20% of total events. This suggests that the formation of complete PICs is relatively inefficient, but we note that this number is similar to estimates of template usage for in vitro transcription in yeast nuclear extracts (Verdier et al., 1990; Yudkovsky et al., 2000).
Although RNA Pol II can directly bind a TBP-TFIIB-TATA complex in the absence of activators when using purified factors (Buratowski et al., 1991; He et al., 2013; Parvin and Sharp, 1993), this does not seem to be the major pathway in the more physiological context of nuclear extract. Our results suggest that initial RNA Pol II association with the template is primarily via transcription activators bound to the UAS/enhancer. We propose that this associated RNA Pol II species can transfer to a PIC at the core promoter when TBP/TFIID, TFIIB, and any other required factors are present. We note that a reservoir of multiple RNA Pol II complexes tethered at the UAS/enhancer could facilitate transcription bursts of closely spaced initiation events.
TFIIF can bind RNA Pol II before or after association with the template
TFIIF was first purified by its high affinity for Pol II (Sopta et al., 1985), and extensive contacts exist between TFIIF and RNA Pol II in the PIC (He et al., 2013; Plaschka et al., 2016). However, RNA Pol II can bind a TBP-TFIIB-TATA complex in vitro in the absence of TFIIF (Buratowski et al., 1991; He et al., 2013; Parvin and Sharp, 1993), and roughly half of the RNA Pol II molecules in yeast nuclear extract are not associated with TFIIF (Rani et al., 2004). Therefore, it remains unclear whether RNA Pol II incorporation into the PIC requires TFIIF pre-association, as is often assumed (Sopta et al., 1985). To determine the timing of RNA Pol II and TFIIF recruitment, we used CoSMoS to examine their relative binding order on DNA templates.
A yeast strain was created in which Rpb1 is fused to SNAPf, and Tfg1, the large subunit of TFIIF, is fused to DHFR (YSB3551, see Figures S1A, S1C, and S1D; Table S1). The dynamics of Rpb1SNAPf-DY549 and Tfg1DHFR-Cy5 on DNA were monitored in the same reaction by alternating red and green laser excitation, with both UAS and UAS+promoter on the same slide. RNA Pol II and TFIIF colocalizations (i.e., time intervals during which both proteins were present) were frequently observed (Figure 3A). A total of 1,475 colocalizations of RNA Pol II and TFIIF were observed at 415 DNA locations, while only 49 colocalizations were detected at 684 off DNA locations, showing that colocalization of RNA Pol II and TFIIF is DNA specific. TFIIF binding to DNA templates was also highly activator dependent (compare Figures 3F and S3A). Like RNA Pol II, multiple TFIIFs could simultaneously bind a single DNA (~10% of total TFIIF binding events). Of these multiple TFIIF events, at least 80% occurred when multiple RNA Pol II molecules were also present (brackets in Figure 3A and S3C).
Figure 3. Dynamics of RNA Pol II and TFIIF during activator-dependent PIC assembly.
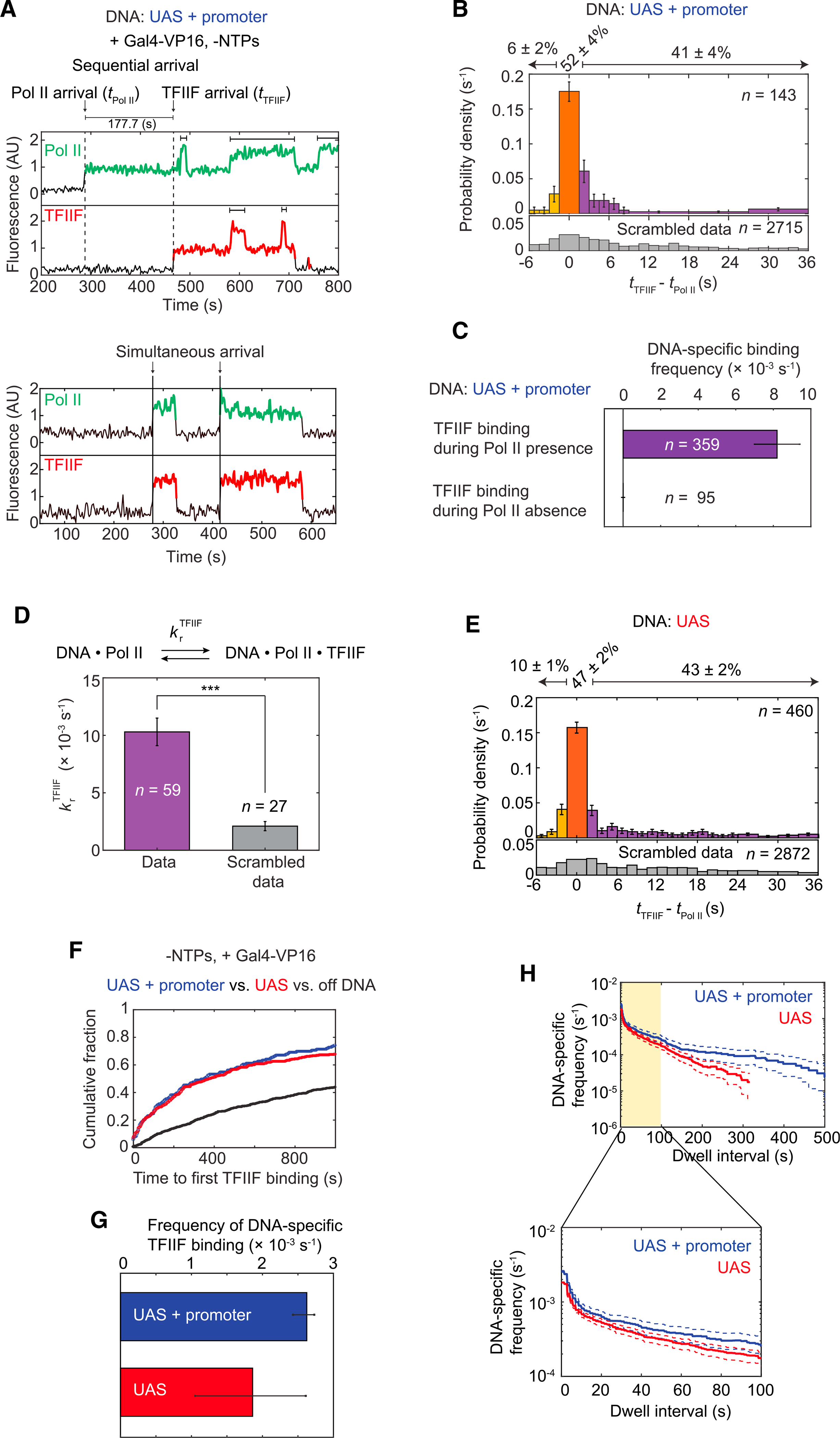
(A) Representative time records of RNA Pol II and TFIIF at two UAS+promoter DNA molecules. Colored intervals are times when RNA Pol II (green) or TFIIF (red) colocalized with DNA. Arrows mark the arrival times of RNA Pol II (tRNA Pol II) or TFIIF (tTFIIF). Dashed lines on the top record show the sequential arrival of RNA Pol II and then TFIIF. Solid lines on the bottom record show two simultaneous arrivals of RNA Pol II and TFIIF. Brackets above traces indicate when multiple RNA Pol II or TFIIF molecules were simultaneously bound to the same DNA.
(B) Histogram shows probability density (±SE) for time differences between TFIIF and RNA Pol II arrival (tTFIIF – tRNA Pol II) at unoccupied UAS+promoter DNA. Note that while 85% of tTFIIF – tRNA Pol II values occur in the range shown, the percentage numbers atop the plot refer to all events. The orange bar represents apparently simultaneous RNA Pol II and TFIIF arrival times—i.e., within ±1 video frame (±1.4 s). The purple bars represent RNA Pol II arrivals before TFIIF, and the yellow bars represent TFIIF arrivals before RNA Pol II. The bottom histogram (gray) shows a control analysis using data scrambled by random pairing of time records. n = total number of the tTFIIF – tRNA Pol II values.
(C) Bars show DNA-specific binding frequencies (±SE) of TFIIF during time intervals when RNA Pol II was present (purple) or absent (yellow) on UAS+promoter templates. n specifies the total number of TFIIF binding events within each category.
(D) Recruitment rate constants (±SE) for TFIIF by RNA Pol II (purple) or in scrambled data (gray). Asterisks indicate p < 0.001, calculated using paired t test.
(E) Histogram shows the probability density (±SE) for tTFIIF – tRNA Pol II values at unoccupied UAS template, plotted as in (B) (81% of the tTFIIF – tRNA Pol II values are shown).
(F) Cumulative fractions over time for UAS+promoter (blue), UAS (red), and off DNA sites (black) bound at least once by TFIIF.
(G) DNA-specific binding frequencies (±SE) of total TFIIF binding events to UAS+promoter (blue) or UAS (red).
(H) Cumulative distribution of TFIIF dwell intervals on UAS+promoter (blue) or UAS (red) with 90% confidence intervals (dashed lines). Frequency values on the vertical axis are after subtraction of off DNA background. Values of ≤0 after background subtraction are not plotted. Bottom: magnified view of the first 100 s (yellow).
All data were taken from an experiment using an Rpb1SNAPf-DY549/Tfg1DHFR-Cy5 yeast nuclear extract from YSB3551 (Table S1).
See also Figure S3.
The binding order of RNA Pol II and TFIIF at individual UAS+promoter DNA molecules was assessed by measuring the intervals between their arrival times (tTFIIF – tRNA Pol II, as diagrammed in Figure 3A, top panel). For this analysis, only de novo colocalization events (i.e., starting with DNA unoccupied by either factor) were included. If the time difference was within the imaging time resolution (±1 video frame, i.e., ±~1.4 s), the corresponding colocalization was scored as an apparent simultaneous arrival (Figure 3B, orange bar). A large fraction of colocalizations (52% ± 4%) showed apparent simultaneous RNA Pol II and TFIIF arrival (i.e., Figure 3A, bottom). In computational simulations where individual TFIIF time records were randomly paired with RNA Pol II time records from different DNA sites (scrambled data), coincidental RNA Pol II and TFIIF binding was ~7% of colocalization events, far below the observed fraction. Therefore, RNA Pol II and TFIIF frequently bind as a preassembled complex or bind in such rapid succession that the separate binding events cannot be resolved in the experiment.
The next largest fraction of colocalizations (41% ± 4%) exhibited the sequential arrival of RNA Pol II and then TFIIF (Figure 3B, purple bars). Most intervals were only a few seconds, but some were on the order of minutes (Figure 3A, top). Although Cy5-TMP dye binds the DHFR tag non-covalently, the fast and tight interaction of TMP ligands with DHFR (Calloway et al., 2007) makes it unlikely that the TFIIF lag is due to reversible labeling. The non-simultaneous arrival events were also not due to non-specific DNA binding, as TFIIF binding to DNA occurred essentially only when RNA Pol II was present (Figure 3C). To confirm that the binding of TFIIF is associated with the prior presence of RNA Pol II in the sequential arrival events, we calculated the TFIIF recruitment rate constant krTFIIF (see STAR Methods). This calculated value of krTFIIF ([10.3 ± 1.2] × 10−3 s−1) was significantly greater than that obtained from a similar analysis of the scrambled data control ([2.1 ± 0.4] × 10−3 s−1) (Figure 3D). Figures 3A–3D demonstrate that TFIIF is primarily recruited to DNA in association with RNA Pol II, but can also arrive after RNA Pol II binding to the template.
Given that RNA Pol II primarily binds first at the UAS, colocalizations of RNA Pol II and TFIIF on UAS and UAS+promoter DNAs were analyzed to determine whether TFIIF binding requires the core promoter. Both simultaneous and sequential arrival of RNA Pol II and TFIIF were observed on UAS (Figures 3E and S3B), and the relative fractions of these two pathways were similar to UAS+promoter (Figures 3B and 3E). There was little difference in the kinetics of initial TFIIF association (Figures 3F and S3D; Table S4). When both initial and subsequent TFIIF binding events were analyzed, association rates were also similar or perhaps slightly increased at UAS+promoter versus UAS alone (Figure 3G, see also the y-intercepts of survival frequency plots of total TFIIF binding in Figure 3H). These results suggest that the majority of RNA Pol II•TFIIF complexes either arrive pre-assembled or form on the UAS. However, some TFIIF molecules may go directly to the core promoter, perhaps joining the PIC after a previous TFIIF molecule has dissociated. Consistent with stable PIC formation on the core promoter, longer dwell intervals in which at least one TFIIF molecule bound were significantly more frequent on UAS+promoter than UAS (Figure 3H).
TFIIE can join RNA Pol II-TFIIF at the UAS or core promoter
Based on gel shift experiments (Inostroza et al., 1991; Peterson et al., 1991), the prevailing stepwise assembly model proposes that TFIIE joins the PIC after both RNA Pol II and TFIIF are bound. However, a reason for TFIIF dependence is not obvious, as TFIIE has far more extensive contact with RNA Pol II than TFIIF (He et al., 2013; Schilbach et al., 2017). We considered whether TFIIF may not be essential for initial TFIIE recruitment, but is needed to stabilize its association enough to survive native gel electrophoresis. To determine the order of factor interactions in our system, TFIIE was imaged in combination with RNA Pol II or TFIIF. To visualize RNA Pol II and TFIIE, nuclear extract was prepared from yeast expressing Rpb1SNAPf and Tfa2, the small subunit of TFIIE, fused to the DHFR tag (YSB3474, see Figures S1A, S1C, and S1D; Table S1). For simultaneous imaging of TFIIF and TFIIE, extract was prepared with Tfg1-SNAPf and Tfa2-DHFR (YSB3553, see Figures S1B–S1D; Table S1). As above, reactions were performed with both the UAS and UAS+promoter templates on the same slide surface, in the absence of NTPs.
Frequent colocalization of TFIIE was seen with both RNA Pol II (Figure 4A) and TFIIF (Figure 4B) at both UAS+promoter (top panels) and UAS (bottom panels). Like RNA Pol II and TFIIF, TFIIE binding was far more frequent at DNA sites compared to off DNA sites (Figure 4D), and was dependent on Gal4-VP16 activator (Figure S4A). Simultaneous binding of multiple TFIIE molecules was sometimes observed during periods when multiple RNA Pol II molecules were bound (Figure S4C). TFIIE bound DNA 24-fold more frequently when RNA Pol II was present than absent, and 26-fold more frequently when TFIIF was present (Figure 4C), supporting the model that both RNA Pol II and TFIIF are needed for TFIIE recruitment. This dependence suggests that TFIIF binding triggers a conformation change in RNA Pol II or TFIIE that allows the two to stably interact, or that TFIIE binding to RNA Pol II without TFIIF is so unstable that dwell times are significantly below the single-frame imaging time (0.5 s). The association rate constants of initial TFIIE binding were the same whether the DNA contained a core promoter (Figures 4D and S4B; Table S4). Therefore, both TFIIE and TFIIF can associate with RNA Pol II at the UAS, independent of a TATA box. As seen for RNA Pol II (Figure 2G), TFIIE dwell times on the UAS were short, while longer dwells, presumably in the PIC, were seen on UAS+promoter (Figure 4E).
Figure 4. Dynamics of TFIIE relative to RNA Pol II and TFIIF during activator-dependent PIC assembly.
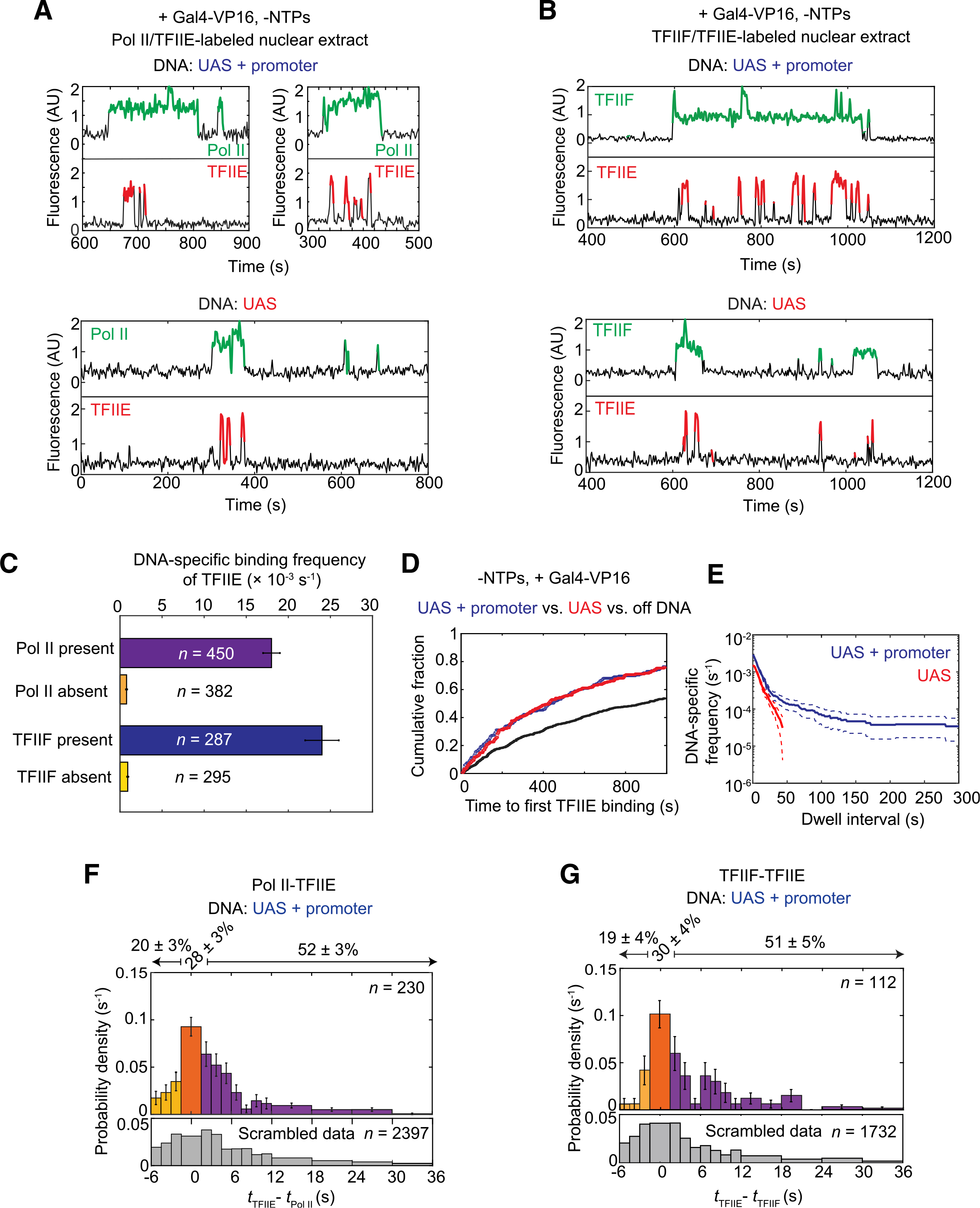
(A) Representative time records at two UAS+promoter molecules (top) and one UAS molecule (bottom) for RNA Pol II (green) and TFIIE (red) colocalization with DNA.
(B) Time records at one UAS+promoter molecule (top) and one UAS molecule (bottom) for TFIIF (green) and TFIIE (red) colocalization with DNA.
(C) DNA-specific binding frequencies (±SE) of TFIIE during time intervals when RNA Pol II was present (purple) or absent (orange), and when TFIIF was present (blue) or absent (yellow) at UAS+promoter. n = number of TFIIE binding events used in each calculation.
(D) Cumulative distributions over time for the fractions of the UAS+promoter (blue), UAS (red), and off DNA sites (black) bound at least once by TFIIE.
(E) Cumulative distribution of TFIIE dwell intervals on UAS+promoter (blue) or UAS (red), with 90% confidence intervals (dashed lines). Frequency values are after subtraction of off DNA background; values of ≤0 are not plotted.
(F) Histogram of probability density (±SE) for time differences between TFIIE and RNA Pol II arrival times (tTFIIE – tRNA Pol II) at unoccupied UAS+promoter DNA, plotted as in Figure 3B (83% of the tTFIIE – tRNA Pol II values are within shown range).
(G) Histogram of probability density (±SE) for time differences between TFIIE and TFIIF arrival times (tTFIIE – tTFIIF) at occupied UAS (80% of the tTFIIE – tTFIIF values shown).
Simultaneous RNA Pol II and TFIIE fluorescence imaging was done using Rpb1SNAPf-DY549/Tfa2DHFR-Cy5 nuclear extract from YSB3474, while TFIIF and TFIIE co-imaging used Tfg1SNAPf-DY549/Tfa2DHFR-Cy5 nuclear extract from YSB3553 (Table S1). Data in (D) and (E) are from YSB3474.
See also Figures S4 and S5.
TFIIE binding showed one striking difference from TFIIF (Figure 3A) and TFIIH (see below), in that multiple cycles of TFIIE association and dissociation were often seen during each TFIIF or RNA Pol II binding event (Figures 4A and 4B). This repetitive binding also manifests in the total TFIIE binding frequency (Figure S5A), in which UAS+promoter had a higher frequency ([2.87 ± 0.15] × 10−3 s−1) than UAS ([1.49 ± 0.11] × 10−3 s−1). Repetitive TFIIE binding is more likely during long-duration RNA Pol II or TFIIF binding, and is therefore preferentially seen on UAS+promoter DNA. These results suggest a model in which TFIIE first joins the RNA Pol II•TFIIF complex primarily at the UAS, but also frequently exchanges on and off RNA Pol II•TFIIF complexes that have transferred to the core promoter.
To determine whether TFIIE pre-binds RNA Pol II or TFIIF before arrival at the template, time differences between RNA Pol II and TFIIE arrival (tTFIIE – tRNA Pol II) (Figure 4F), and between TFIIF and TFIIE (tTFIIE – tTFIIF) (Figure 4G) were calculated. The small fraction of TFIIE arrivals before RNA Pol II or TFIIF (yellow bars in Figures 4F and 4G) may be coincidental independent binding, as frequencies were similar to those calculated using scrambled control data (Figure S5B, yellow bars). In roughly half of the co-binding events on DNA, TFIIE arrived after RNA Pol II or TFIIF (purple bars in Figures 4F and 4G). An additional ~30% of co-binding events were scored as apparently simultaneous within the imaging time resolution (orange bars in Figures 4F and 4G). However, when the sequential binding data (Figure 4F, purple bars) were fit to a first-order sequential binding model curve for RNA Pol II and TFIIE (Figure S5C) and extrapolated back to time 0, ~60% of the apparently simultaneous binding could be accounted for by sequential binding of TFIIE in the time interval 0 ≤ tTFIIE – tRNA Pol II < 1.4 s. We therefore conclude that TFIIE is unlikely to appreciably preassemble with RNA Pol II or TFIIF in yeast nuclear extract.
If TFIIE is unlikely to pre-bind RNA Pol II•TFIIF complexes off the DNA, then what triggers their association on the template? Binding occurs on both UAS and UAS+promoter, with no change in the binding order or kinetics (Figure S5D). Therefore, TFIIE binding is not dependent on RNA Pol II•TFIIF interactions with factors at the TATA box. TFIIE binding is present for ~19% of the time that RNA Pol II or TFIIF is bound to the UAS template. If these factor interactions on DNA, presumably occurring while tethered to Gal4-VP16, were similar in solution, then we would predict an equal fraction of simultaneous arrivals rather than the calculated fraction of RNA Pol II and TFIIE simultaneous arrivals (9% ± 5%) (Figure S5C). This difference suggests that activator interaction with RNA Pol II•TFIIF may somehow promote TFIIE binding. We speculate this could involve activator-induced conformation changes transmitted via Mediator, which links activation domains and RNA Pol II•TFIIF.
TFIIH requires the core promoter DNA for PIC incorporation
To examine whether initial TFIIH association with DNA is also independent of core promoter as seen for RNA Pol II, TFIIF, and TFIIE, we monitored RNA Pol II and TFIIH using fluorescently labeled nuclear extract containing Rpb1SNAPf-DY549 and the tagged TFIIH subunit Tfb1DHFR-Cy5 (Figure S1; Table S1). Like the other GTFs tested, DNA-specific TFIIH binding was activator dependent (compare Figures 5A and S6A). However, in marked contrast to RNA Pol II, TFIIF, and TFIIE, the binding of TFIIH above background was seen only on the UAS+promoter template, not on UAS only (Figure 5A). TFIIH dwell times in the absence of NTPs tend to be long, similar to the other GTFs at the core promoter (Figure 5B). Strikingly, only one molecule of TFIIH associates at a time (Figure S6D). Whereas multiple RNA Pol II, TFIIF, and TFIIE molecules can associate with templates via the five activator-bound Gal4 sites and the core promoter, TFIIH apparently only binds the single PIC expected on the core promoter.
Figure 5. Dynamics of TFIIH relative to RNA Pol II during activator-dependent PIC assembly.
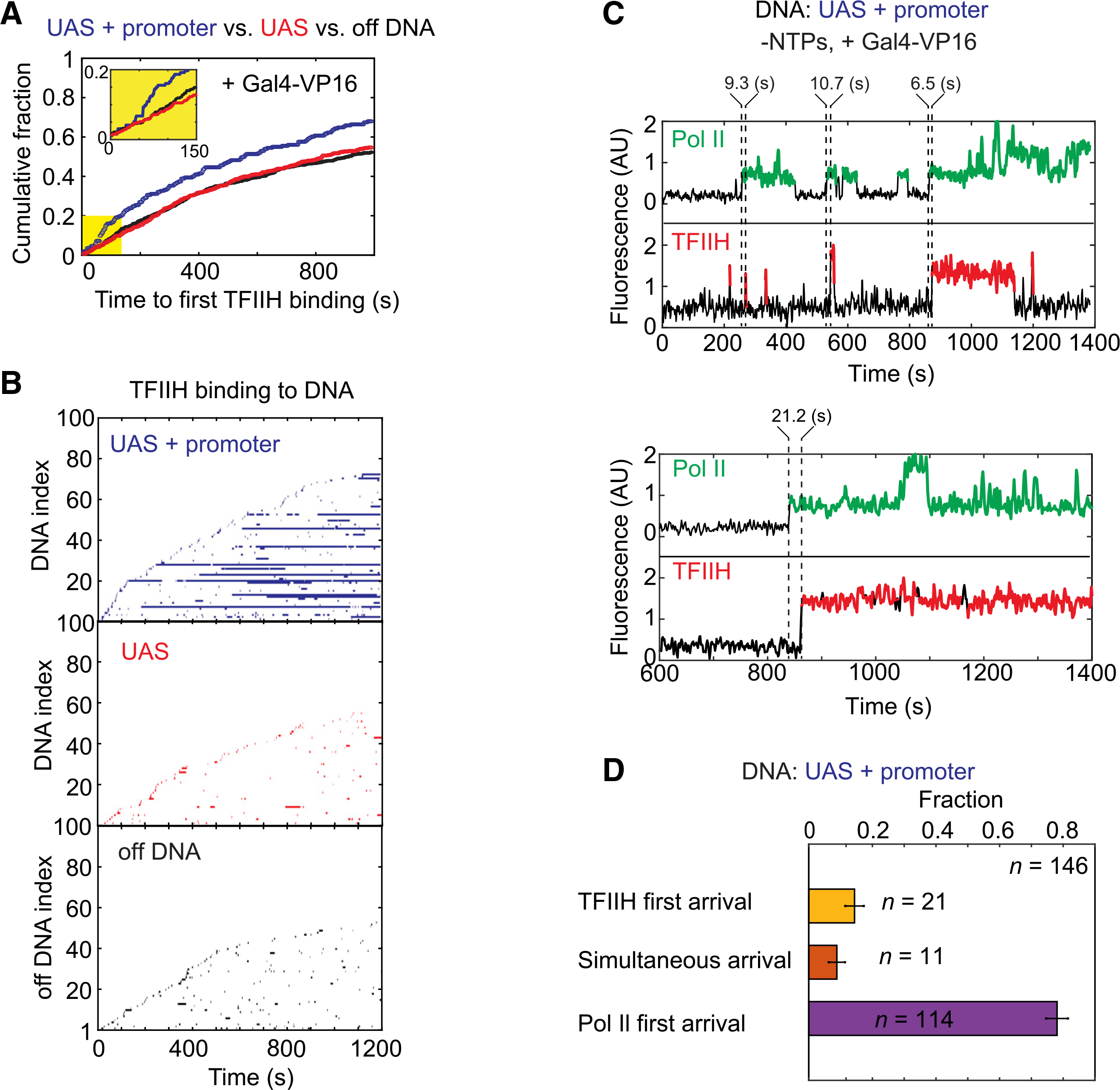
(A) Cumulative fraction versus time for UAS+promoter (blue), UAS (red), and off DNA (black) locations bound at least once by TFIIH. Inset: magnified view of the first 150 s (yellow) showing lag in TFIIH binding.
(B) Rastergrams of TFIIH dwells at 100 randomly selected on or off DNA locations, plotted as in Figure 2B. UAS+promoter (top, blue), UAS only (center, red), off DNA (bottom, black).
(C) Representative time records of RNA Pol II (green) and TFIIH (red) colocalizations at two UAS+promoter molecules. Dashed lines indicate the arrival times of RNA Pol II or TFIIH, with time differences shown above.
(D) Fractions (±SE) of the formation pathways for RNA Pol II•TFIIH complexes on UAS+promoter DNAs. See Figure S6B for time difference histogram. Data were taken from an experiment using NTP-depleted Rpb1SNAPf-DY549/Tfb1DHFR-Cy5 nuclear extract from YSB3473 (Table S1). See also Figure S6.
Importantly, the cumulative fraction plot of times to first TFIIH binding at each DNA template revealed a non-exponential distribution, with a pronounced lag of ~50 s after the start of imaging (Figure 5A, inset). The simplest explanation for the TFIIH lag is that slower binding of one or more other factors must occur first. This slow step is likely to be required for transfer of RNA Pol II and associated factors from the UAS to the core promoter. Notably, Spt5 association shows a similar lag under transcription conditions (Rosen et al., 2020). TFIIH almost exclusively arrived after RNA Pol II (78 ± 3 (SE)% of colocalizations), confirming sequential binding (Figures 5C, 5D, and S6B). Also consistent with a late step in PIC assembly, the delay time between TFIIH and RNA Pol II arrival was significantly longer than that between RNA Pol II and TFIIE (Figure S6C). We thus conclude that TFIIH binding occurs after the transfer of RNA Pol II (likely with TFIIF and TFIIE) from the UAS to the core promoter.
DISCUSSION
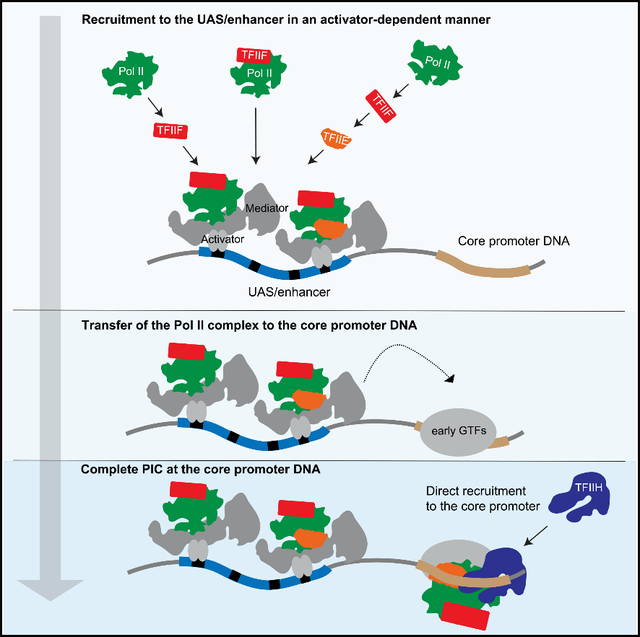
Under physiological conditions, numerous proteins are needed to form RNA Pol II PICs. These include GTFs, transcription activators, and coactivators. However, current PIC assembly models are based primarily on activator-independent studies using only purified GTFs. These experiments led to a simple sequential binding pathway directly on the core promoter. While not excluding this model, our single-molecule experiments reveal that a more complex, activator-dependent pathway predominates in the more physiological context of nuclear extract (Figure 6A). Surprisingly, the initial template association of RNA Pol II, TFIIF, and TFIIE requires only the UAS and activator, independent of the core promoter. However, these initial UAS-bound complexes are relatively short-lived, on the order of a few seconds. The presence of core promoter sequences produces longer duration complexes that exhibit properties expected of PICs, such as sensitivity to NTPs. The similar initial association rates observed on UAS versus UAS+promoter are most simply explained by intermediate complexes assembled on the UAS before transfer to the core promoter (Figure 6A, blue box). Such intermediates also fit a kinetic model derived from our earlier study imaging RNA Pol II and the elongation factor Spt4/5 (Rosen et al., 2020). It is notable that this branched model also fits earlier activator-independent experiments with purified GTFs. The gel-shifted species that led to the classic sequential assembly pathway (Buratowski et al., 1989; Inostroza et al., 1991; Peterson et al., 1991) are encompassed in the core-promoter complexes (Figure 6A, yellow box).
Figure 6. Branched model for activator-dependent PIC assembly pathways.
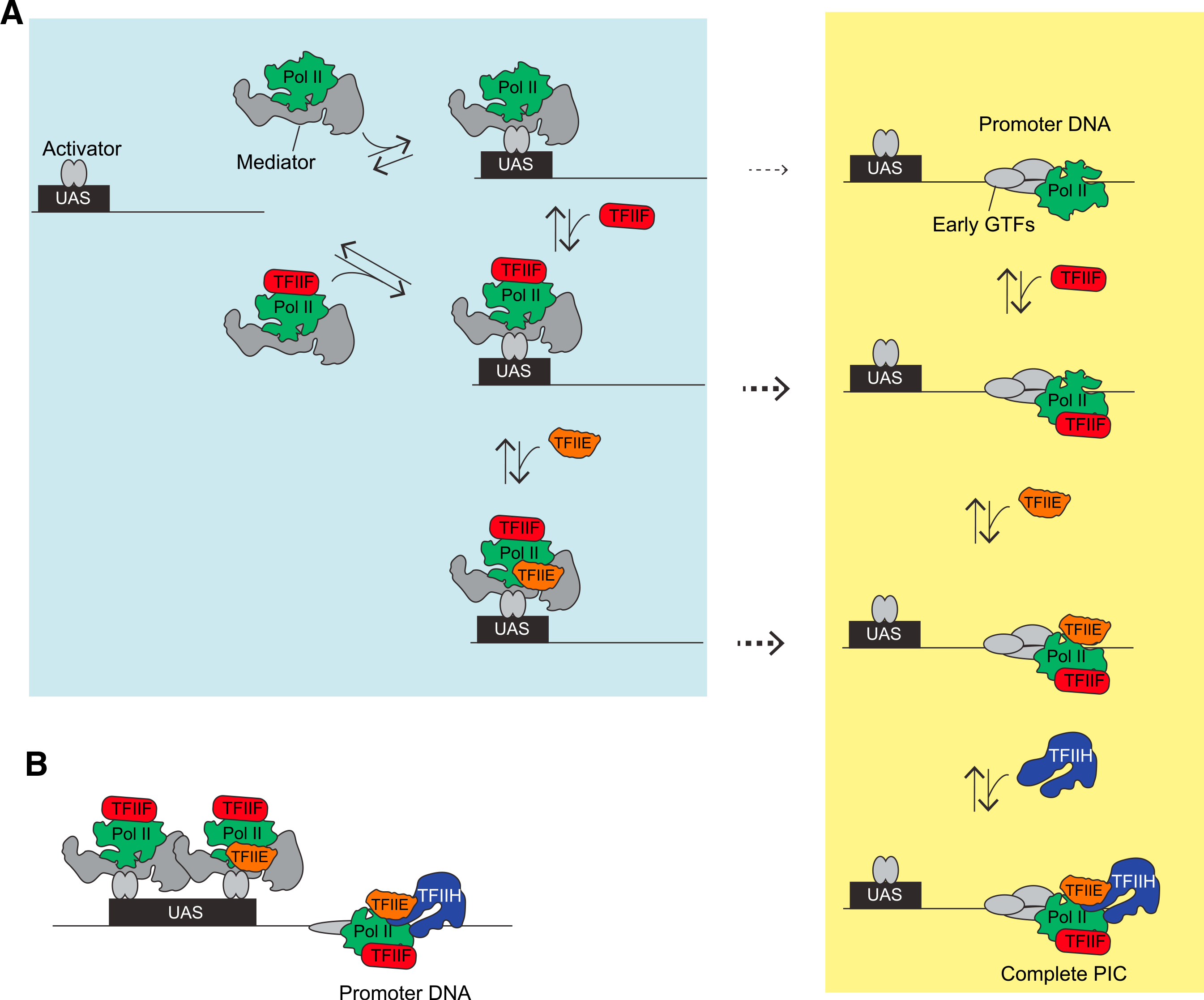
(A) The single-molecule imaging leads to a model in which initial interactions (blue box) of RNA Pol II (green) with the template are primarily via UAS-bound transcription activators (gray ovals), presumably via Mediator (gray), either in association with TFIIF (red) or by itself. TFIIF can also be recruited to RNA Pol II already bound at the UAS. TFIIE (orange) is more dynamic and can join the RNA Pol II•TFIIF complex at the UAS or core promoter. The dashed arrows indicate transfer of RNA Pol II, RNA Pol II•TFIIF, or RNA Pol II•TFIIF•TFIIE from the UAS to the core promoter. TFIIH is recruited directly to the PIC at the core promoter. This model also predicts that in a non-physiological, activator-independent reaction, TFIIF, TFIIE, and TFIIH would incorporate through the intermediates at the core promoter (yellow box), thereby encompassing the previous sequential assembly model.
(B) This activator-dependent PIC assembly model also explains the observation of simultaneous binding of multiple RNA Pol II, TFIIF, or TFIIE molecules on the same DNA, tethered by the multiple activators at the UAS/enhancer plus the single PIC. This phenomenon could provide a mechanism for the in vivo clustering of RNA Pol II observed by microscopy, either in cooperation with or independent of proposed condensate formation.
Could the partial PICs on the UAS correspond to previously proposed RNA Pol II “holoenzyme” complexes? This is unlikely for several reasons. First, a substantial fraction of TFIIF and the majority of TFIIE only bind DNA after RNA Pol II arrives (Figures 3E, 4F, 4G, and S5D), arguing against the pre-assembly of holoenzymes off the DNA, at least in yeast nuclear extract. Second, both yeast and mammalian holoenzyme were reported to contain TFIIH (Kim et al., 1994; Koleske and Young, 1994; Maldonado et al., 1996; Ossipow et al., 1995; Ranish et al., 1999), but this factor does not stably interact with RNA Pol II, TFIIF, and TFIIE at the UAS, and instead requires the core promoter for stable binding (Figures 5A and 5B). The very different dynamics of TFIIF, TFIIE, and TFIIH relative to RNA Pol II and one another are inconsistent with a preassembled holoenzyme.
The association of TFIIF and TFIIE with RNA Pol II before core promoter binding is notable from an evolutionary perspective. Subunits of RNA Pol I and III are generally homologous or identical to those in RNA Pol II. However, several additional RNA Pol I and III subunits have no RNA Pol II counterparts, but instead structurally resemble TFIIF or TFIIE (for a review, see Engel et al., 2018; Khatter et al., 2017; Vannini and Cramer, 2012). Under particular in vitro conditions, TBP and TFIIB are sufficient for RNA Pol II binding to the core promoter and accurate initiation in vitro (Buratowski et al., 1991; Parvin and Sharp, 1993). Although TFIIF and TFIIE are therefore not absolutely required for initial RNA Pol II binding to the TBP-TFIIB-promoter complex, the RNA Pol II (He et al., 2016; Murakami et al., 2015; Plaschka et al., 2016) and RNA Pol III (Han et al., 2018) PIC structures show TFIIF or the homologous RNA Pol III subunit contacting TFIIB or TFIIIB, respectively. This interaction, together with TFIIF and TFIIE encircling and constraining template DNA downstream of the TATA box, may further the position and stabilize the polymerase within the PIC. An interesting evolutionary question is whether the single RNA polymerase in eukaryotic ancestors had integral subunits that branched off in the RNA Pol II system to become TFIIF and TFIIE, or instead began with independent initiation factors that evolved into RNA Pol I and III subunits.
Another unexpected result from our study is that TFIIE shows repetitive cycles of binding to the PIC, perhaps explaining weaker TFIIE binding inferred from crosslinking and cryoelectron microscopy (cryo-EM) studies (Grünberg et al., 2012; He et al., 2013). In the absence of NTPs to trigger promoter melting and initiation, both TFIIF and TFIIH show core promoter-dependent dwells that can last for minutes. In contrast, TFIIE rapidly exchanges on and off, even when RNA Pol II or TFIIF is stably bound (Figures 4A and 4B). TFIIE is reportedly required for TFIIH binding (Compe et al., 2019; Maxon et al., 1994), and PIC structures show direct contacts between the two factors (He et al., 2016; Schilbach et al., 2017). However, long dwell times of TFIIH but not TFIIE are difficult to reconcile with the classic PIC assembly model. It may be that TFIIE promotes initial TFIIH binding, but is not required to retain TFIIH once it makes contact with RNA Pol II and downstream promoter DNA.
Our discovery of partial PIC pre-assembly at the UAS provides important new insight into transcription activation. Activators are often said to “recruit” co-activators and basal machinery (Ptashne and Gann, 1997), but this vague term fits several non-mutually exclusive mechanisms. In the classic cooperative binding model, initial associations of activator and polymerase with DNA are independent, but subsequent contact between the two mutually stabilizes their binding. Transcription activation at many prokaryotic promoters fits this model (van Hijum et al., 2009). In this mechanism, polymerase dissociation rates of one or more intermediate pre-initiation complexes are reduced to increase transcription. In contrast, a kinetic enhancement model postulates that activator increases the initial association rate of RNA polymerase (or other basal factors) with the promoter.
Consistent with kinetic enhancement, Gal4-VP16 or Gal4-Gcn4 activator strongly accelerates the association of RNA Pol II with the template, although initial binding is to the UAS/enhancer rather than the core promoter, as typically pictured in most models. The observations of multiple RNA Pol II, TFIIF, and TFIIE molecules simultaneously bound at the UAS (schematic in Figure 6B, brackets in Figures 2D, S3B, and S4C) definitively demonstrate that activators can increase the local concentration of PIC components. This reservoir of factors can drive PIC formation, and may also contribute to transcription “bursting” if multiple RNA Pol II•TFIIF•TFIIE complexes can rapidly and sequentially transfer to the core promoter during a window of TFIID and TFIIB binding.
The multiple RNA Pol II molecules we observe at a single UAS are reminiscent of polymerase clusters observed in nuclei, which colocalize with Mediator (Cho et al., 2018; Sabari et al., 2018; reviewed in Cramer, 2019). Although these clusters are often interpreted as condensates, our results suggest another plausible mechanism independent of phase separation. A single enhancer comprises multiple activator binding sites, and if each activator (which may itself have multiple activation domains) independently tethers one or more polymerases, a cluster of RNA Pol II would appear in the microscope. Although the cluster would appear long-lived, individual RNA Pol II molecules could exchange if they rapidly associate and dissociate from DNA-bound activator. Our single-molecule studies show that average dwell times of RNA Pol II tethered to the UAS by Gal4-VP16 are on the order of seconds, very similar to timescales seen for fluorescence recovery after photobleaching of RNA Pol II clusters in vivo (Cho et al., 2018).
Our system for visualizing individual transcription factors in nuclear extract has revealed new intermediates not seen with purified factors, indicating that the activator-dependent pathways by which PICs form are more complicated than previously proposed. Future studies with additional labeled factors are likely to provide further new insights into RNA Pol II transcription activation, initiation, and elongation.
Limitations
Like other in vitro transcription systems, the factor concentrations in the extracts used here are lower than those in living cells. Therefore, the parameters measured here are “apparent” association and dissociation rates specific to these conditions. However, the relative ratios of factors and templates appear to be preserved in the reactions, so the relative orders of competing PIC assembly pathways should be similar to those in vivo. Also, the experiments here use naked DNA templates rather than chromatin and therefore measure events that would occur after removal or remodeling of nucleosomes that occlude the promoter in vivo.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen Buratowski (steveb@hms.harvard.edu).
Materials availability
S. cerevisiae strains and plasmids generated in this study (Tables S1 and S2) are available from the lead contact upon request.
Data and code availability
Original/source data for single molecule experiments are provided as “intervals” files. The data files have been deposited at Zenodo and are publicly available as of the date of publication. A DOI is listed in the Key resources table. “Intervals” files can be read and manipulated by the MATLAB program “Imscroll,” which is available at https://github.com/gelles-brandeis/CoSMoS_Analysis.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal anti-Rpb1 CTD (8WG16) | (Thompson et al., 1990) | N/A |
| Monoclonal anti-HA, peroxidase conjugated (3F10) | Roche | Cat#12013819001; RRID: AB_2314622 |
| Polyclonal anti-Rpb4 (2Y14) | Biolegend | Cat#665106 RRID: AB_2861051 |
| Polyclonal anti-Tfa2 | (Kuldell and Buratowski, 1997) | N/A |
| Polyclonal anti-Tfb1 | (Matsui et al., 1995) | N/A |
| Bacterial and Virus Strains | ||
| BL21(DE3) | (Haraszti and Braun, 2020) | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Zymolyase 100T | Amsbio | Cat#120493–1 |
| Ficoll 400 | Accurate Chemical and Scientific corp. | Cat#AN-228520 |
| Spermidine trihydrochloride | Sigma | Cat#S2501 |
| Spermine tetrahydrochloride | Sigma | Cat#S1141 |
| Aprotinin | Gold Biotechnology | Cat#A-655–100 |
| Leupeptin | Peptides International | Cat#ILP-4041 |
| Peptatin A | Peptides International | Cat#IPA-4397 |
| Antipain | Peptides International | Cat#IAP-4062 |
| SNAP-Surface 549 | New England BioLabs | Cat#S9112S |
| Pierce NHS-activated agarose, Dry | Thermofisher Scientific | Cat#26196 |
| Pierce centrifuge column, 5 mL | Thermofisher Scientific | Cat#89897 |
| Creatine kinase | Sigma | Cat#C3755 |
| Phosphocreatine | Sigma | Cat#P7936 |
| RNasin | Promega | Cat#N2515 |
| Rnase T1 | Life Technologies | Cat#EN0541 |
| Proteinase K | Applied Biosystems | Cat#2544 |
| Protocatechuate dioxygenase | Sigma | Cat#P8279 |
| Protocatechuic acid | Sigma | Cat#03930590 |
| Trolox | Sigma | Cat#238813 |
| Propyl gallate | Sigma | Cat#02370 |
| 4-nitrobenzyl alcohol | Sigma | Cat#N12821 |
| Cy5-TMP | (Hoskins et al., 2011) | N/A |
| Hexokinase | Sigma | Cat#H4502 |
| mPEG-SG2000 | Laysan Bio | Cat#MPEG-SG-2000–1GR |
| Biotin-PEG-SVA5000 | Laysan Bio | Cat# BIO-PEG-SVA-5K/mPEG-SVA-5K |
| TransFlouSpheres Streptavidin-Labeled Microspheres, 0.04 μm | Thermofisher Scientific | Cat#T10711 |
| Critical Commercial Assays | ||
| DNA SizeSelector-I | ALINE Biosciences | Cat#Z-6001 |
| Pierce Coomassie (Bradford) protein assay kit | Thermofisher Scientific | Cat#23200 |
| Deposited Data | ||
| Analyzed data (“Intervals files”) | This paper | http://doi.org/10.5281/zenodo.5043636 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae strains, see Table S1 | This paper | N/A |
| Oligonucleotides | ||
| Primers for strain, plasmid, DNA template generation, see Table S3 | This paper | N/A |
| Recombinant DNA | ||
| Plasmids, see Table S2 | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | http://imagej.nih.gov/ij |
| Glimpse | Gelles lab | https://github.com/gelles-brandeis/Glimpse |
| LabView | National Instruments | https://www.ni.com/ |
| MATLAB | The MathWorks | https://www.mathworks.com/ |
| Imscroll | (Friedman and Gelles, 2015) | https://github.com/gelles-brandeis/CoSMoS_Analysis |
| Other | ||
| Phosphoscanner/fluorescence imager Amersham Typhoon 5 | GE healthcare | Model: 29187191 |
| Homogenizer | Wheaton | Cat#62400–802 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
S. cerevisiae strain information is in Table S1. All S. cerevisiae strains for nuclear extract preparation were grown in YPD (Yeast extract Peptone 3% Dextrose) media at 30°C until OD600 reached 3–4.
METHOD DETAILS
Generation of HA3 - DHFR/SNAPf tagging plasmids
A hygromycin resistant DHFR tag which also encodes for an HA tag was created by an isothermal assembly reaction with the following four DNA fragments: (1) pBlueScript (Stratagene, #212205; see Table S2) plasmid digested with EcoRI and BamHI, (2) a DNA fragment containing a HA tag and GSG linker repeats amplified from pFA6a-HA-KIURA3 with primers 3XHA-Forward and 3XHA-Reverse, (3) a DHFR tag amplified from pAAH2 with primers DHFR-Forward and DHFR-Reverse and (4) a hygromycin resistance cassette amplified from pAAH2 with primers TEFpro-Forward and TEFterm-Reverse. It should be noted that the DHFR tag is followed by a 39 bp synthetic terminator which prevents transcription read-through toward the hygromycin resistance cassette. The resulting plasmid pBS-SKII-3XHA-eDHFR-Hygromycin (YV317) was confirmed by sequencing.
A nourseothricin resistant fast SNAP (SNAPf) tag which also encodes for an HA tag was created by an isothermal assembly reaction with the following four DNA fragments: (1) pBlueScript plasmid digested with EcoRI and BamHI, (2) a DNA fragment containing a HA tag and GSG linker repeats amplified from pFA6a-HA-KIURA3 with primers 3XHA-Forward and 3XHA-Reverse, (3) a SNAPf tag amplified from pAAH13 with primers SC-Forward and SC-Reverse and (4) a nourseothricin resistance cassette amplified from pAAH6 with primers TEFpro-Forward and TEFterm-Reverse. It should be noted that the SNAPf tag is followed by a 39 bp synthetic terminator which prevents transcription read-through toward the nourseothricin resistance cassette. The resulting plasmid pBS-SKII-3XHA-fSNAP-NAT (YV309) was confirmed by sequencing.
Plasmid information is given in Table S2. Primers used to generate the plasmids are listed in Table S3.
Yeast strains, plasmids, and oligonucleotides
S. cerevisiae strains used in this study are listed in Table S1. To create doubly-fused yeast strains (YSB3473, YSB3474, YSB3551 and YSB3553 in Table S1), two DNA cassettes for yeast transformation were prepared by PCR amplification of YV317 (Table S2) and YV309 (Table S2) for DHFR and SNAPf tagging, respectively, with the appropriate primer pairs from Table S3. The amplified DHFR- and SNAPf-containing fragments were sequentially transformed into the protease-deficient strain, YF702 (Table S1). After each round of transformation, positive clones were selected for the marker. Additionally, selected clones were checked for presence of the insert by colony PCR with the appropriate primer pairs from Table S3. In-frame fusion protein expression and stability was confirmed by immunoblotting for the target protein. The fusion strains had similar growth to the YF702 wild-type as determined by a spotting assay (Figure S1C).
Yeast nuclear extract preparation
Yeast nuclear extracts were prepared as previously described (Rosen et al., 2020; Sikorski et al., 2012). Briefly, yeast cells were grown in 4 l of YPD (3% dextrose) medium at 30°C to an OD600 of 3–4 and were harvested. Yeast cell walls were digested with 15 mg of Zymolyase 100T (Amsbio, #120493–1) until ~80%–90% of cells became spheroplasts. The digestion times varied from 30 min to 1.5 hr depending on the strain. The spheroplasts were suspended in 250 mL of YPD+1 M sorbitol and incubated at 30°C for 30 min for recovery. After residual Zymolyase 100T was removed by serial centrifugations, the spheroplasts were resuspended in Buffer A (lysis buffer; 18% (w/v) Ficoll 400, 10 mM Tris-HCl pH 7.5, 20 mM potassium acetate, 5 mM magnesium acetate, 1 mM EDTA (ethylenediamine tetraacetic acid), 0.5 mM spermidine, 0.15 mM spermine, 3 mM DTT, 1 μg/mL each of aprotinin, leupeptin, pepstatin A, and antipain). The resuspended spheroplasts were lysed with a motorized homogenizer (Wheaton, #62400–802) and the supernatant was collected by four sequential centrifugations (twice at 5000 x g for 8 min followed by twice at 5000 x g for 5 min). Crude nuclei from the lysed spheroplasts were pelleted by centrifugation at 25,000 x g for 30 min and were suspended in Buffer B (100 mM Tris-acetate pH 7.9, 50 mM potassium acetate, 10 mM magnesium sulfate, 10% glycerol, 3 mM DTT, 2 mM EDTA, 1 μg/mL each of aprotinin, leupeptin, pepstatin A, and antipain). Nuclear proteins were extracted by the addition of ammonium sulfate solution (pH 7.5) to a final concentration of 0.4 M, followed by rotating for 30 min at 4°C. After centrifugation in a Beckman type 71 rotor at 37,500 rpm (~120,000 x g) for 1.5 hr at 4°C, the soluble fraction was collected. Nuclear proteins were precipitated by the addition of solid ammonium sulfate granules to ~70% saturation (0.35 g to 1 mL of nuclear protein solution). The ammonium sulfate precipitate was recovered by centrifugations at 13,000 rpm (~16,000 x g) for 20 min and then 5 min. The pooled protein pellet was weighed and resuspended in Buffer C’ (20 mM HEPES pH 7.6, 10 mM MgSO4, 1 mM EGTA (ethylene glycol-bis(β-aminoethylether) tetraacetic acid), 10% glycerol, 3 mM DTT, 1 μg/mL each of aprotinin, leupeptin, pepstatin A, and antipain) at a ratio of ~400 mg protein pellet to a 1 mL Buffer C’.
SNAPf fusion proteins in nuclear extracts were labeled by adding SNAP-Surface 549 (New England BioLabs, #S9112S) to a final concentration of 0.4 μM (unless otherwise specified) and incubating at 4°C for 1 hr on a rotator in the dark. The suspension was dialyzed three times (1 hr, 1.5 hr and 2 hr) each time against 500 mL of Buffer C’ supplemented with 75 mM ammonium sulfate (nuclear extract dialysis buffer). Residual unreacted SNAP-Surface 549 was removed from SNAP-labeled extracts as previously described (Haraszti and Braun, 2020; Rosen et al., 2020). Briefly, one fourth of the extract volume of SNAPf-coupled agarose beads were added to the extract and incubated at 4°C for 1 hr on a rotator, after which the beads were removed by centrifugation at 1,000 x g for 2 min at 4°C. Finally, the extract was aliquoted, frozen in liquid nitrogen, and stored at −80°C. Labeling of SNAPf fusion proteins and depletion of residual dye were confirmed by in-gel fluorescence imaging on a Typhoon imager (GE Healthcare). In vitro transcription activity of the final nuclear extract was measured by the in vitro transcription assay described below.
Preparation of SNAPf-coupled agarose beads
His-SNAPf proteins were prepared as previously described (Rosen et al., 2020). Specifically, BL21(DE3) cells containing the His-SNAPf construct were grown at 37°C in Luria-Bertani media to an OD600 of 0.8, followed by the induction of protein expression with 0.4 mM IPTG (Isopropyl pβ-d-1-thiogalactopyranoside) at 18°C overnight, His-SNAPf proteins were purified with Ni2+-NTA-agarose resin. NHS-activated agarose beads (Thermofisher Scientific #26196) were incubated with purified His-SNAPf protein at a ratio of 90 mg resin to 10 mg protein for 2–3 hr at room temperature. The beads were transferred to a 5 mL Pierce centrifuge column (Thermofisher Scientific, #89897). After washing with 1X PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.4) until residual uncoupled His-SNAPf protein was no longer detectable in the effluent using Protein Detection Reagent (Pierce PI23200), resin was quenched with 1 M Tris-HCl, pH 7.5 for 30 min at room temperature. The beads were equilibrated with nuclear extract dialysis buffer supplemented with 3 mM DTT for long-term storage at 4°C. Prior to use, efficiency of the dye depletion by the beads was tested. SNAP-Surface 549 (New England Biolabs, #S9112S) was suspended in yeast nuclear extract containing no SNAPf-fusion proteins to a final concentration of 1 μM. The extent of a decrease in the dye concentration after dye depletion was measured by in-gel fluorescence imaging on a Typhoon imager (GE Healthcare). The residual dye concentration was determined by comparison against a standard curve of known free dye concentrations. The SNAPf-coupled bead treatment typically reduced SNAP-Surface 549 concentration in extract from 1 μM to 50 nM.
In vitro bulk transcription assay
In vitro transcription assay was performed as previously described (Sikorski et al., 2012). Briefly, the plasmid pUC18-G5CYC1 G- (SB649; Table S2) (Joo et al., 2019) was used as the DNA template. The DNA template (100 ng) was incubated with Gal4-VP16 (340 nM), the ATP regeneration system consisting of creatine kinase (0.1–0.2 units, Sigma #C3755) and phosphocreatine (10 mM, Sigma #P7936), and nuclear extract (typically 8 μL to 10 μL in 50 μL reaction). 400 μM each of ATP, CTP, UTP and 100 μM of 3′-O-methyl-GTP (chain terminator) along with 32P-labeled UTP were added to initiate the reaction. RNasin (Promega, #N2515) also was added to prevent transcript degradation. After 45 min reaction at room temperature, transcripts were treated with RNase T1 (Life Technologies, #EN0541) and Proteinase K (Applied Biosystems, #2544), extracted with phenol-chloroform, ethanol precipitated, separated by gel electrophoresis (8 M urea 5% polyacrylamide gel), and analyzed by autoradiography and/or phosphorimager (GE Healthcare).
Preparation of DNA templates for single molecule assays
Upstream biotinylated and downstream AF488 labeled DNA templates (Figure 1A) were prepared by PCR from pUC18-G5CYC1 G- (SB649; Table S2) with Platinum Taq DNA polymerase (Invitrogen) and primers Biotin-universal and AF488-G-less-PL_REV for the UAS and AF488-Mid G-less d2 REV(+17) for the UAS+promoter templates (primer sequence information in Table S3). The DNA templates used to test effects of the core promoters and 4NTPs were prepared by PCR from pUC18-G5CYC1 G- (SB649; Table S2) and pUC18-G5-HIS4 G- (SB1964; Table S2) for the CYC1 and HIS4 core promoter, respectively with Platinum Taq DNA polymerase (Invitrogen) and primers Biotin-universal and AF488-M13 rev2 (primer sequence information in Table S3). The PCR product was purified using DNA SizeSelector-I SPRI magnetic beads (Aline Biosciences). The DNA template sequence information is in Table S5.
Single-molecule microscopy
Single-molecule imaging experiments were performed on a multi-wavelength single-molecule total internal reflection fluorescence (TIRF) inverted microscope with a pair of micromirrors positioned just beneath the objective. A 785 nm IR beam was used to maintain focus throughout imaging (Crawford et al., 2013; Friedman and Gelles, 2015).
As described in Rosen et al. (2020), glass flow chambers on the microscope slide was passivated with a mPEG-SG-2000:biotin-PEG-SVA5000 (Laysan Bio) 200:1 mixture. Streptavidin-coated fluorescent beads (TransFluoSpheres, Thermofisher Scientific T10711) were added to use as fiducial markers for stage drift correction. After the slide surface was coated with 0.01 mg/mL streptavidin (Thermofisher Scientific 21122), 10 pM of the biotinylated/AF488-labeled DNA template was introduced along with an oxygen scavenging system. The oxygen scavenging system (Crawford et al., 2008), used to minimize photobleaching, contains protocatechuate dioxygenase (Sigma P8279) at a final concentration of 0.9 units/ml and protocatechuic acid (Sigma 03930590) at a final concentration of 5 mM. DNA images were acquired with the 488 nm laser at 1.2 mW. In the experiments with two different DNA templates tethered to the same slide surface, they were sequentially introduced and two DNA images were acquired: one with the fluorescent spots of the first DNA template and the other with fluorescent spots of both DNA templates. After acquiring DNA images, PIC assembly reactions were performed in a flow chamber at room temperature in a mixture in transcription buffer (20 mM HEPES (pH 7.6), 100 mM potassium acetate, 5 mM magnesium acetate, 1 mM EDTA) containing yeast nuclear extract, Gal4-VP16 or Gal4-Gcn4, and other reagents with the following final concentrations: extract (6–8 mg protein/mL), Gal4-VP16 or Gal4-Gcn4 (340 nM, (Joo et al., 2019)), oxygen scavenging system, triplet state quenchers (Dave et al., 2009) (1 mM Trolox (Sigma # 238813), 1 mM propyl gallate (Sigma #02370), and 2 mM 4-nitrobenzyl alcohol (Sigma #N12821)), 20 nM Cy5-TMP (Hoskins et al., 2011), 20 μM acetyl-CoA (Sigma #A2056), E. coli genomic DNA (0.02 μg/μL), and an ATP depletion system (20 mM glucose and 2 units hexokinase (Sigma H4502)). In the experiments carried out in the presence of 4 NTPs, 400 μM each ATP, UTP, CTP and GTP were added and the ATP-depletion system was omitted.
Successive images in each channel were captured every 1.4 s (0.5 s/frame for each channel, plus switching times) over a time course of 800–1200 s. Laser powers were 800 μW and 600 μW for the 532 nm and 633 nm laser, respectively. The cited laser powers were measured at an intermediate point in the excitation beam path before the objective lens. Custom software Glimpse implemented with LabView (National Instruments; Austin, TX) operated microscope, laser shutters, filter wheels, the camera and image acquisition (https://github.com/gelles-brandeis/Glimpse).
General image analysis procedure
Image data analysis was performed with custom software implemented with MATLAB (The MathWorks; Natick, MA) as previously described (Friedman and Gelles, 2015) https://github.com/gelles-brandeis/CoSMoS_Analysis. Briefly, DNA locations were identified by automatically selecting fluorescent spots in each DNA image followed by manual removal of overlapping spots. Off DNA locations were also selected in the same image by automatically picking areas that do not contain fluorescent spots. The data were corrected for spatial stage drift that occurred during the experiment. The images from the same field of view created at different channels (red, green and blue channels) were spatially mapped by correcting for spatial displacement between the channels. This correction was done using hundreds of reference fluorescent spot pairs. The reference spots were obtained using surface-immobilized oligonucleotides that were labeled with all three dye molecules (AF488, Cy3, and Cy5). Fluorescence emission from the protein channels (red or green channel) at the pre-defined locations corresponding to template DNA spots was integrated from ~0.16 μm2 areas (3 × 3 pixels) to obtain fluorescence intensity time records at each DNA location (Figures 2A, 2D, 3A, 4A, 4B, 5C, S3B, S3C, S4C, and S6D). Images of protein fluorescence at each DNA location were scored to determine the presence or absence of a fluorescent spot, indicating a “bound” or “unbound” state, respectively. Rastergrams of bound and unbound states were plotted using custom MATLAB scripts (Figures 2B, 5B, and S2A).
QUANTIFICATION AND STATISTICAL ANALYSIS
Rate constants of PIC factor association with DNA
To analyze the DNA-specific apparent first-order rate constants for factor associations with the DNA template (Figures 2C, S2D, S2E, S3D, and S4B), we performed the analysis as described in Friedman and Gelles (2015) and Rosen et al. (2020). Briefly, we assumed that not all DNA molecules were capable of factor binding. We termed the fraction of DNA molecules that are capable of factor binding to be the active fraction (Af). We used the times to first binding at each DNA to minimize the artifacts caused by photobleaching and/or photoblinking. We also accounted for nonspecific binding of factor to the flow chamber surface. The nonspecific association rate constant (kns) was first determined by analyzing off DNA locations and we then measured the DNA-specific apparent first-order association rate constant (kon) by fitting the wait intervals from the DNA locations to a model that included both exponential nonspecific binding (with kns held fixed at the previously determined value) and exponential specific binding. Standard errors of fit parameters were determined by bootstrapping. Fit parameters are reported in Table S4.
DNA-specific frequency of factor association with DNA
Total binding frequencies (right panel in Figures 2E (right), 3G, and S5A)
We measured the total binding frequency by dividing the total number of binding events by the sum of absent times at all locations. To determine the DNA-specific total binding frequency, we subtracted the total binding frequency at off-DNA locations from that at DNA locations. The vertical axis intercept of the cumulative distribution of dwell interval frequencies (Figure 2G, 3H, 4E, and S2F) corresponds to the DNA-specific total binding frequency. Standard error in counting was calculated as the standard deviation of a binomial distribution (if P is close to 0 or 1) where N is the total number of observed binding events, and P is the event probability in time.
Binding frequencies of GTFs during presence or absence of Pol II (Figures 3C and 4C)
To examine whether GTF binding preferentially occurs when Pol II is already bound, the DNA-specific binding frequencies of GTFs during Pol II presence or absence were calculated. The Pol II presence time was defined as the sum of the intervals when Pol II, but not the GTF of interest, was present. The binding frequency was calculated as the number of GTF binding events occurring during Pol II presence divided by the Pol II presence time. The binding frequency during Pol II absence was similarly determined by dividing the number of GTF binding events occurring during Pol II absence by the total Pol II absence time. The Pol II absence time was defined as the sum of the intervals when neither Pol II nor the GTF of interest was present. Apparent simultaneous arrival events were not included in this analysis. To determine the DNA-specific binding frequencies, the binding frequencies at off DNA locations were subtracted from those at DNA locations. Standard errors were calculated as the standard deviation of a binomial distribution. We also applied this analysis to determine the DNA-specific binding frequencies of TFIIE during presence or absence of TFIIF (Figure 4C).
Cumulative distributions of factor dwell times (Figures 2G, 3H, 4E, S2F, and S2G)
To compare the dwell intervals of DNA·factor complexes between the UAS and UAS+promoter DNA templates, we plotted the cumulative DNA-specific binding frequency distributions for the UAS and UAS+promoter DNA. To visually examine whether two cumulative distributions are statistically different, we generated 1000 bootstrap sample sets of the experimental data for the DNA and off DNA locations. We subtracted the off DNA binding frequencies from the DNA binding frequencies and plotted the 90% confidence interval envelope of the bootstrapped samples.
Analyzing orders of factor addition during complex assembly (Figures 3B, 3E, 4F, 4G, 5D, S5D, and S6B)
Cy5- and DY549-labeled factors (i.e., GTFDHFR-Cy5 and Rpb1SNAPf-DY549) were imaged by alternating laser excitation at 633 nm and 532 nm. The time between two consecutive frames in the same channel was ~1.4 s (0.5 s/frame for each channel, plus switching times). To analyze the order of binding of Cy5- and DY549-labeled factors to DNA during their complex formation, time intervals in which both factors were present simultaneously for at least part of their bindings to the same DNA were scored as a colocalization. Only de novo colocalizations were included for this analysis. For the selected events, the delay times between arrival times of Cy5- and DY549-labeled factors (e.g., tTFIIF – tPol II) were determined. Probability density histograms of the delay times were plotted (e.g., Figure 3B, top). Standard errors of the bar heights were calculated from the binomial distribution. The events with a time difference between −1.4 s and +1.4 s (±1 frame) were scored as apparent simultaneous arrivals and were put in the same bin (e.g., Figure 3B, orange bar).
To rule out the possibility that colocalization of two factors on the same DNA molecules occurred by chance, we generated the control scrambled data by performing simulations in which the time record of protein fluorescence from each DNA location was randomly paired with the record of the second protein taken from a different DNA location. We performed 30–50 such simulations, yielding 2000–3000 coincident appearances of two factors. These scrambled control data were then analyzed in the same way as the experimental data.
We classified the complex assembly pathway based on the order of factor binding. Events with a delay time greater than 1.4 s (+1 frame) were scored as a first arrival of the DY549-labeled factor (e.g., Figure 3B, purple bars). Events with a delay time less than −1.4 s (−1 frame) were scored as a first arrival of the Cy5-labeled factor (e.g., Figure 3B, yellow bars).
Estimating the fraction of Pol II and TFIIE sequentially arriving at DNA within the experimental time resolution (Figure S5C)
We fit the part of Figure 4F delay time distribution where Pol II arrival is followed by TFIIE arrival using a maximum likelihood approach that accounts for contributions by both sequential and simultaneous arrival events. Delay times between Pol II and TFIIE arrivals are modeled using a bi-exponential distribution and we account for the fraction of simultaneous arrivals by including a parameter S in the Equation 1 expression for the likelihood L(a, r1, r2, S).
| (Equation 1) |
where
a is the relative amplitude, r1 and r2 are two characteristic rates, and S is the ratio of simultaneous to sequential arrival events.
In this dataset we recorded 119 intervals of duration τj > tmin, and these data arise from arrival of Pol II followed by a TFIIE arrival with Pol II still present. Each factor of (ar1 exp[−r1τj] + (1 − a)r2 exp[−r2τj]) / (1 + S) in Equation 1 is proportional to the probability of observing one such interval of duration τj separating the sequential arrival of Pol II and TFIIE. Alternating image acquisition for the two proteins means that initial detections of Pol II and TFIIE that are separated by only one frame (separation interval τ < tmin) may be due to either a simultaneous or sequential landing by the Pol II and TFIIE. This dataset contained 36 such events, and each A(a, r1, r2, S) factor in Equation 1 is the probability of observing one such event for which the Pol II/TFIIE landings might have been either simultaneous or sequential. The a, r1 and r2 parameters for the bi-exponential distribution describing sequential Pol II/TFIIE landings along with the S parameter are determined by optimizing the value of the likelihood function L(a, r1, r2, S) in Equation 1. The number of sequential arrival events was calculated as 155/(1+S) and the number of simultaneous arrival events was calculated as (155 S) / (1+S), where 155 is the total number of observations in this dataset. The standard errors of the a, r1, r2 and S fit parameters were determined by bootstrapping.
Recruitment rate constant analysis (Figure 3D)
To determine the recruitment rate constant of TFIIF by pre-bound Pol II, the number of TFIIF bindings occurring during Pol II presence (purple bars in Figure 3B) was divided by the total dwell times of Pol II up to the point, if any, that TFIIF bound. Only the first TFIIF binding at each Pol II was included. The recruitment rate constant determined here may underestimate the true recruitment rate constant of TFIIF by pre-bound Pol II since the sequential binding of Pol II followed by TFIIF within the experimental time resolution were not included. Similar analysis was performed for the scrambled control data.
Supplementary Material
Highlights.
Single-molecule microscopy reveals unexpected dynamics of RNA Pol II and GTFs
Multiple RNA Pol IIs cluster on enhancer-bound activators before core promoter binding
RNA Pol II, TFIIF, and TFIIE, but not TFIIH, can pre-assemble at the UAS/enhancer
Activators increase the rates of RNA Pol II and GTF association with DNA
ACKNOWLEDGMENTS
We thank members of the Buratowski and Gelles labs for helpful discussions and Yujin Chun for technical support. This work was supported by NIH grants R01GM046498, to S.B.; R01GM081648, to J.G.; and R01CA246500, to S.B. and J.G. I.B. was partially supported by the Van Maanen Fellowship from Harvard Medical School.
Footnotes
DECLARATION OF INTERESTS
S.B. is a member of the Molecular Cell Advisory Board.
SUPPORTING CITATIONS
The following references appear in the supplemental information: Inada et al. (2002); Johnson et al. (2009); Sung et al. (2008).
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.07.025.
REFERENCES
- Buratowski S, Hahn S, Guarente L, and Sharp PA (1989). Five intermediate complexes in transcription initiation by RNA polymerase II. Cell 56, 549–561. [DOI] [PubMed] [Google Scholar]
- Buratowski S, Sopta M, Greenblatt J, and Sharp PA (1991). RNA polymerase II-associated proteins are required for a DNA conformation change in the transcription initiation complex. Proc. Natl. Acad. Sci. USA 88, 7509–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloway NT, Choob M, Sanz A, Sheetz MP, Miller LW, and Cornish VW (2007). Optimized fluorescent trimethoprim derivatives for in vivo protein labeling. ChemBioChem 8, 767–774. [DOI] [PubMed] [Google Scholar]
- Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, and Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compe E, Genes CM, Braun C, Coin F, and Egly JM (2019). TFIIE orchestrates the recruitment of the TFIIH kinase module at promoter before release during transcription. Nat. Commun 10, 2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P (2019). Organization and regulation of gene transcription. Nature 573, 45–54. [DOI] [PubMed] [Google Scholar]
- Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, and Moore MJ (2008). Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA 14, 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, and Moore MJ (2013). Single-molecule colocalization FRET evidence that spliceosome activation precedes stable approach of 5′ splice site and branch site. Proc. Natl. Acad. Sci. USA 110, 6783–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave R, Terry DS, Munro JB, and Blanchard SC (2009). Mitigating unwanted photophysical processes for improved single-molecule fluorescence imaging. Biophys. J 96, 2371–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel C, Neyer S, and Cramer P (2018). Distinct Mechanisms of Transcription Initiation by RNA Polymerases I and II. Annu. Rev. Biophys 47, 425–446. [DOI] [PubMed] [Google Scholar]
- Fazal FM, Meng CA, Murakami K, Kornberg RD, and Block SM (2015). Real-time observation of the initiation of RNA polymerase II transcription. Nature 525, 274–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores O, Maldonado E, Burton Z, Greenblatt J, and Reinberg D (1988). Factors involved in specific transcription by mammalian RNA polymerase II. RNA polymerase II-associating protein 30 is an essential component of transcription factor IIF. J. Biol. Chem 263, 10812–10816. [PubMed] [Google Scholar]
- Flores O, Ha I, and Reinberg D (1990). Factors involved in specific transcription by mammalian RNA polymerase II. Purification and subunit composition of transcription factor IIF. J. Biol. Chem 265, 5629–5634. [PubMed] [Google Scholar]
- Friedman LJ, and Gelles J (2015). Multi-wavelength single-molecule fluorescence analysis of transcription mechanisms. Methods 86, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LJ, Chung J, and Gelles J (2006). Viewing dynamic assembly of molecular complexes by multi-wavelength single-molecule fluorescence. Biophys. J 91, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LJ, Mumm JP, and Gelles J (2013). RNA polymerase approaches its promoter without long-range sliding along DNA. Proc. Natl. Acad. Sci. USA 110, 9740–9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünberg S, Warfield L, and Hahn S (2012). Architecture of the RNA polymerase II preinitiation complex and mechanism of ATP-dependent promoter opening. Nat. Struct. Mol. Biol 19, 788–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S (2004). Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol 11, 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Yan C, Fishbain S, Ivanov I, and He Y (2018). Structural visualization of RNA polymerase III transcription machineries. Cell Discov 4, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraszti RA, and Braun JE (2020). Preparation of SNAPf-Beads for Colocalization Single-Molecule Spectroscopy (CoSMoS) of RNA-Protein Complexes. Methods Mol. Biol. 2113, 17–22. [DOI] [PubMed] [Google Scholar]
- He Y, Fang J, Taatjes DJ, and Nogales E (2013). Structural visualization of key steps in human transcription initiation. Nature 495, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Yan C, Fang J, Inouye C, Tjian R, Ivanov I, and Nogales E (2016). Near-atomic resolution visualization of human transcription promoter opening. Nature 533, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, and Moore MJ (2011). Ordered and dynamic assembly of single spliceosomes. Science 331, 1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada T, Winstall E, Tarun SZ Jr., Yates JR 3rd, Schieltz D, and Sachs AB (2002). One-step affinity purification of the yeast ribosome and its associated proteins and mRNAs. RNA 8, 948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inostroza J, Flores O, and Reinberg D (1991). Factors involved in specific transcription by mammalian RNA polymerase II. Purification and functional analysis of general transcription factor IIE. J. Biol. Chem 266, 9304–9308. [PubMed] [Google Scholar]
- Johnson A, Li G, Sikorski TW, Buratowski S, Woodcock CL, and Moazed D (2009). Reconstitution of heterochromatin-dependent transcriptional gene silencing. Mol. Cell 35, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo YJ, Ficarro SB, Soares LM, Chun Y, Marto JA, and Buratowski S (2017). Downstream promoter interactions of TFIID TAFs facilitate transcription reinitiation. Genes Dev 31, 2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo YJ, Ficarro SB, Chun Y, Marto JA, and Buratowski S (2019). In vitro analysis of RNA polymerase II elongation complex dynamics. Genes Dev 33, 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JJ, Auble DT, Ranish JA, and Hahn S (1995). Analysis of the yeast transcription factor TFIIA: distinct functional regions and a polymerase II-specific role in basal and activated transcription. Mol. Cell. Biol 15, 1234–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, and Johnsson K (2003). A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol 21, 86–89. [DOI] [PubMed] [Google Scholar]
- Khatter H, Vorländer MK, andMüller CW (2017). RNA polymerase I and III: similar yet unique. Curr. Opin. Struct. Biol 47, 88–94. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Björklund S, Li Y, Sayre MH, and Kornberg RD (1994). A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell 77, 599–608. [DOI] [PubMed] [Google Scholar]
- Koleske AJ, and Young RA (1994). An RNA polymerase II holoenzyme responsive to activators. Nature 368, 466–469. [DOI] [PubMed] [Google Scholar]
- Kuldell NH, and Buratowski S (1997). Genetic analysis of the large subunit of yeast transcription factor IIE reveals two regions with distinct functions. Mol. Cell. Biol 17, 5288–5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luse DS (2014). The RNA polymerase II preinitiation complex. Through what pathway is the complex assembled? Transcription 5, e27050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado E, Shiekhattar R, Sheldon M, Cho H, Drapkin R, Rickert P, Lees E, Anderson CW, Linn S, and Reinberg D (1996). A human RNA polymerase II complex associated with SRB and DNA-repair proteins. Nature 381, 86–89. [DOI] [PubMed] [Google Scholar]
- Matsui P, DePaulo J, and Buratowski S (1995). An interaction between the Tfb1 and Ssl1 subunits of yeast TFIIH correlates with DNA repair activity. Nucleic Acids Res 23, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxon ME, Goodrich JA, and Tjian R (1994). Transcription factor IIE binds preferentially to RNA polymerase IIa and recruits TFIIH: a model for promoter clearance. Genes Dev 8, 515–524. [DOI] [PubMed] [Google Scholar]
- Murakami K, Tsai KL, Kalisman N, Bushnell DA, Asturias FJ, and Kornberg RD (2015). Structure of an RNA polymerase II preinitiation complex. Proc. Natl. Acad. Sci. USA 112, 13543–13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov DB, and Burley SK (1997). RNA polymerase II transcription initiation: a structural view. Proc. Natl. Acad. Sci. USA 94, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuma Y, Hashimoto S, Wang CK, Horikoshi M, and Roeder RG (1995). Analysis of the role of TFIIE in basal transcription and TFIIH-mediated carboxy-terminal domain phosphorylation through structure-function studies of TFIIE-alpha. Mol. Cell. Biol 15, 4856–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, Lagrange T, and Reinberg D (1996). The general transcription factors of RNA polymerase II. Genes Dev 10, 2657–2683. [DOI] [PubMed] [Google Scholar]
- Ossipow V, Tassan JP, Nigg EA, and Schibler U (1995). A mammalian RNA polymerase II holoenzyme containing all components required for promoter-specific transcription initiation. Cell 83, 137–146. [DOI] [PubMed] [Google Scholar]
- Parvin JD, and Sharp PA (1993). DNA topology and a minimal set of basal factors for transcription by RNA polymerase II. Cell 73, 533–540. [DOI] [PubMed] [Google Scholar]
- Peterson MG, Inostroza J, Maxon ME, Flores O, Admon A, Reinberg D, and Tjian R (1991). Structure and functional properties of human general transcription factor IIE. Nature 354, 369–373. [DOI] [PubMed] [Google Scholar]
- Plaschka C, Hantsche M, Dienemann C, Burzinski C, Plitzko J, and Cramer P (2016). Transcription initiation complex structures elucidate DNA opening. Nature 533, 353–358. [DOI] [PubMed] [Google Scholar]
- Ptashne M, and Gann A (1997). Transcriptional activation by recruitment. Nature 386, 569–577. [DOI] [PubMed] [Google Scholar]
- Rani PG, Ranish JA, and Hahn S (2004). RNA polymerase II (Pol II)-TFIIF and Pol II-mediator complexes: the major stable Pol II complexes and their activity in transcription initiation and reinitiation. Mol. Cell. Biol 24, 1709–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranish JA, Yudkovsky N, and Hahn S (1999). Intermediates in formation and activity of the RNA polymerase II preinitiation complex: holoenzyme recruitment and a postrecruitment role for the TATA box and TFIIB. Genes Dev 13, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder RG (1996). The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci 21, 327–335. [PubMed] [Google Scholar]
- Rosen GA, Baek I, Friedman LJ, Joo YJ, Buratowski S, and Gelles J (2020). Dynamics of RNA polymerase II and elongation factor Spt4/5 recruitment during activator-dependent transcription. Proc. Natl. Acad. Sci. USA 117, 32348–32357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I, Ma J, Triezenberg S, and Ptashne M (1988). GAL4-VP16 is an unusually potent transcriptional activator. Nature 335, 563–564. [DOI] [PubMed] [Google Scholar]
- Schier AC, and Taatjes DJ (2020). Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev 34, 465–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilbach S, Hantsche M, Tegunov D, Dienemann C, Wigge C, Urlaub H, and Cramer P (2017). Structures of transcription pre-initiation complex with TFIIH and Mediator. Nature 551, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski TW, Joo YJ, Ficarro SB, Askenazi M, Buratowski S, and Marto JA (2012). Proteomic analysis demonstrates activator- and chromatin-specific recruitment to promoters. J. Biol. Chem 287, 35397–35408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopta M, Carthew RW, and Greenblatt J (1985). Isolation of three proteins that bind to mammalian RNA polymerase II. J. Biol. Chem 260, 10353–10360. [PubMed] [Google Scholar]
- Sung MK, Ha CW, and Huh WK (2008). A vector system for efficient and economical switching of C-terminal epitope tags in Saccharomyces cerevisiae. Yeast 25, 301–311. [DOI] [PubMed] [Google Scholar]
- Taylor IC, Workman JL, Schuetz TJ, and Kingston RE (1991). Facilitated binding of GAL4 and heat shock factor to nucleosomal templates: differential function of DNA-binding domains. Genes Dev 5, 1285–1298. [DOI] [PubMed] [Google Scholar]
- Thomas MC, and Chiang CM (2006). The general transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol 41, 105–178. [DOI] [PubMed] [Google Scholar]
- Thompson NE, Aronson DB, and Burgess RR (1990). Purification of eukaryotic RNA polymerase II by immunoaffinity chromatography. Elution of active enzyme with protein stabilizing agents from a polyol-responsive monoclonal antibody. J. Biol. Chem 265, 7069–7077. [PubMed] [Google Scholar]
- Tomko EJ, Fishburn J, Hahn S, and Galburt EA (2017). TFIIH generates a six-base-pair open complex during RNAP II transcription initiation and start-site scanning. Nat. Struct. Mol. Biol 24, 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hijum SA, Medema MH, and Kuipers OP (2009). Mechanisms and evolution of control logic in prokaryotic transcriptional regulation. Microbiol. Mol. Biol. Rev 73, 481–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A, and Cramer P (2012). Conservation between the RNA polymerase I, II, and III transcription initiation machineries. Mol. Cell 45, 439–446. [DOI] [PubMed] [Google Scholar]
- Verdier JM, Stalder R, Roberge M, Amati B, Sentenac A, and Gasser SM (1990). Preparation and characterization of yeast nuclear extracts for efficient RNA polymerase B (II)-dependent transcription in vitro. Nucleic Acids Res 18, 7033–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkovsky N, Ranish JA, and Hahn S (2000). A transcription reinitiation intermediate that is stabilized by activator. Nature 408, 225–229. [DOI] [PubMed] [Google Scholar]
- Zhang Z, and Tjian R (2018). Measuring dynamics of eukaryotic transcription initiation: challenges, insights and opportunities. Transcription 9, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, English BP, Grimm JB, Kazane SA, Hu W, Tsai A, Inouye C, You C, Piehler J, Schultz PG, et al. (2016). Rapid dynamics of general transcription factor TFIIB binding during preinitiation complex assembly revealed by single-molecule analysis. Genes Dev 30, 2106–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original/source data for single molecule experiments are provided as “intervals” files. The data files have been deposited at Zenodo and are publicly available as of the date of publication. A DOI is listed in the Key resources table. “Intervals” files can be read and manipulated by the MATLAB program “Imscroll,” which is available at https://github.com/gelles-brandeis/CoSMoS_Analysis.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal anti-Rpb1 CTD (8WG16) | (Thompson et al., 1990) | N/A |
| Monoclonal anti-HA, peroxidase conjugated (3F10) | Roche | Cat#12013819001; RRID: AB_2314622 |
| Polyclonal anti-Rpb4 (2Y14) | Biolegend | Cat#665106 RRID: AB_2861051 |
| Polyclonal anti-Tfa2 | (Kuldell and Buratowski, 1997) | N/A |
| Polyclonal anti-Tfb1 | (Matsui et al., 1995) | N/A |
| Bacterial and Virus Strains | ||
| BL21(DE3) | (Haraszti and Braun, 2020) | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Zymolyase 100T | Amsbio | Cat#120493–1 |
| Ficoll 400 | Accurate Chemical and Scientific corp. | Cat#AN-228520 |
| Spermidine trihydrochloride | Sigma | Cat#S2501 |
| Spermine tetrahydrochloride | Sigma | Cat#S1141 |
| Aprotinin | Gold Biotechnology | Cat#A-655–100 |
| Leupeptin | Peptides International | Cat#ILP-4041 |
| Peptatin A | Peptides International | Cat#IPA-4397 |
| Antipain | Peptides International | Cat#IAP-4062 |
| SNAP-Surface 549 | New England BioLabs | Cat#S9112S |
| Pierce NHS-activated agarose, Dry | Thermofisher Scientific | Cat#26196 |
| Pierce centrifuge column, 5 mL | Thermofisher Scientific | Cat#89897 |
| Creatine kinase | Sigma | Cat#C3755 |
| Phosphocreatine | Sigma | Cat#P7936 |
| RNasin | Promega | Cat#N2515 |
| Rnase T1 | Life Technologies | Cat#EN0541 |
| Proteinase K | Applied Biosystems | Cat#2544 |
| Protocatechuate dioxygenase | Sigma | Cat#P8279 |
| Protocatechuic acid | Sigma | Cat#03930590 |
| Trolox | Sigma | Cat#238813 |
| Propyl gallate | Sigma | Cat#02370 |
| 4-nitrobenzyl alcohol | Sigma | Cat#N12821 |
| Cy5-TMP | (Hoskins et al., 2011) | N/A |
| Hexokinase | Sigma | Cat#H4502 |
| mPEG-SG2000 | Laysan Bio | Cat#MPEG-SG-2000–1GR |
| Biotin-PEG-SVA5000 | Laysan Bio | Cat# BIO-PEG-SVA-5K/mPEG-SVA-5K |
| TransFlouSpheres Streptavidin-Labeled Microspheres, 0.04 μm | Thermofisher Scientific | Cat#T10711 |
| Critical Commercial Assays | ||
| DNA SizeSelector-I | ALINE Biosciences | Cat#Z-6001 |
| Pierce Coomassie (Bradford) protein assay kit | Thermofisher Scientific | Cat#23200 |
| Deposited Data | ||
| Analyzed data (“Intervals files”) | This paper | http://doi.org/10.5281/zenodo.5043636 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae strains, see Table S1 | This paper | N/A |
| Oligonucleotides | ||
| Primers for strain, plasmid, DNA template generation, see Table S3 | This paper | N/A |
| Recombinant DNA | ||
| Plasmids, see Table S2 | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | http://imagej.nih.gov/ij |
| Glimpse | Gelles lab | https://github.com/gelles-brandeis/Glimpse |
| LabView | National Instruments | https://www.ni.com/ |
| MATLAB | The MathWorks | https://www.mathworks.com/ |
| Imscroll | (Friedman and Gelles, 2015) | https://github.com/gelles-brandeis/CoSMoS_Analysis |
| Other | ||
| Phosphoscanner/fluorescence imager Amersham Typhoon 5 | GE healthcare | Model: 29187191 |
| Homogenizer | Wheaton | Cat#62400–802 |