

Abstract
Metastatic disease in pheochromocytomas and paragangliomas (PCC/PGL) is not well-understood. The Cancer Genome Atlas discovered recurrent MAML3 fusion genes in a subset of tumors that lacked known germline or somatic driver mutations and were associated with aggressive disease. Here, we aimed to investigate the role of MAML3 in tumorigenesis. Human PCC/PGLs were used for immunohistochemistry and genetic analysis. Three neuroendocrine tumor cell lines, SK-N-SH, QGP-1 and BON-1, were transiently transfected with MAML3 (FL) or exon 1 deleted MAML3 (dEx1; mimicking the fusion), and biologic effects of overexpression were examined in vitro. We found 7% (4/55) of human PCC/PGL have UBTF~MAML3 fusions and all were sporadic cases with metastatic disease. Fusion-positive tumors had intense MAML3 nuclear staining and increased β-catenin by immunohistochemistry and showed increased WNT4 expression. In vitro, overexpression of FL and dEx1 MAML3 increased invasion in SK-N-SH, QGP-1, and BON-1 (all p<0.05) and increased soft agar colony formation in QGP-1 and BON-1 (all p<0.05). Co-transfection with FL or dEx1 MAML3 and β-catenin increased TCF/LEF promoter activation by luciferase activity and co-immunoprecipitation confirmed interaction between MAML3 and β-catenin. These data suggest MAML3 is involved in WNT signaling pathway activation. In summary, UBTF~MAML3 fusions are present in a subset of PCC/PGL and associated with metastatic disease without other known drivers. MAML3 overexpression led to increased tumorigenicity in neuroendocrine tumor cells and the mechanism of action may involve WNT signaling pathways.
Implications:
MAML3 increases tumorigenicity and invasion in neuroendocrine tumor cells and may be a prognostic marker for aggressive disease.
Keywords: neuroendocrine tumors, MAML3, pheochromocytoma, paraganglioma
Introduction
Pheochromocytoma and paraganglioma (PCC/PGL) are unique neuroendocrine tumors derived from the adrenal medulla or paraganglia, respectively. PCC/PGL cause problems due to mass effect and/or secretion of metanephrines and catecholamines, causing cardiovascular morbidity and mortality. Metastatic disease occurs in 15–25% of cases and can present even 20–40 years after the primary tumor diagnosis [1]. While surgery may be curative for primary PCC/PGL, patients with metastatic disease have limited treatment options, none of which are curative. Currently, there are only a few clinical predictors for developing metastatic PCC/PGL including large tumor size (greater than 4–5cm), extra-adrenal location and germline SDHB pathogenic variants [2]. However, metastatic disease can occur from adrenal PCC and sporadic cases as well. Molecular markers associated with metastatic disease include somatic ATRX (Alpha Thalassemia X-linked Intellectual Disability gene) mutations, a pseudohypoxia mRNA expression subtype, TERT (Telomerase Reverse Transcriptase gene) alterations and MAML3 (Mastermind Like Transcriptional Coactivator 3 gene) fusion genes [3–5].
The Cancer Genome Atlas (TCGA) was the first to identify recurrent somatic fusion genes involving MAML3 in 5% of PCC/PGL, establishing a novel marker correlating with metastatic disease [4]. All of the identified MAML3 fusion genes shared the same breakpoint within intron 1 causing deletion of exon 1 regardless of the 5′ fusion partner (either Upstream Binding Transcription Factor – UBTF or Transcription Factor 4 – TCF4) leading to a marked upregulation of an in-frame fusion protein. MAML3 is a member of the Mastermind Like Transcriptional Coactivator (MAML) family which is well conserved [6]. Canonically, MAML proteins bind the NOTCH intracellular domain (NICD) to regulate the transcription of NOTCH pathway genes [7]. However, the NICD binding motif (located in exon 1 of MAML3) is absent from all MAML3 fusion genes in PCC/PGL, suggesting a non-canonical role for MAML3 in the fusion.
Tumors with the MAML3 fusion in TCGA cohort were associated with a WNT-altered mRNA expression pattern [4]. Tumors with the MAML3 fusion had increased mRNA expression of WNT4 as well as classical WNT pathway genes and had increased protein expression of β-catenin by RPPA. PCC/PGL with the MAML3 fusion gene and tumors in the WNT-altered expression subtype were associated with metastatic and aggressive disease [4]. The tumors with MAML3 fusions lacked any other driving germline or somatic alterations, and these tumors also were hypomethylated, differentiating them from other aggressive PCC/PGL which are hypermethylated [4].
We hypothesized that MAML3 is a novel driver in human PCC/PGL and functions through non-canonical pathways. In this study, we confirm the presence of MAML3 fusions in a new cohort of PCC/PGL and determine that overexpression of MAML3 is tumorigenic in neuroendocrine tumor cells.
Methods
Samples
With IRB approval in accordance with ethical guidelines and patient written informed consent (COMIRB #15–0516; UPENN IRB #812495), fresh-frozen and formalin-fixed paraffin-embedded (FFPE) human PCC/PGL were collected from patients undergoing resection of the primary PCC/PGL or metastasis for clinical care. Clinical data was abstracted from the medical record including location of tumor, the presence of metastatic disease, and germline clinical genetic testing results.
RT-PCR and quantitative PCR
RNA was extracted from fresh-frozen human PCC/PGL using standard protocols with Trizol (Invitrogen 15596026), then reverse transcribed with iScript cDNA Synthesis Kit (BioRad 1708891). Primers were designed to amplify the most common recurrent MAML3 fusions (UBTF~MAML3 isoform 1 and isoform 2), identified in the TCGA paper [4] (Forward 5′-CCAAGTACAAGGCCCGAGA-3′; Reverse 5′-GAATTCGTTTGCTAGTCGGAGA-3′). TBP, control gene, primers were designed to amplify a 302 bp product (Forward 5′- ACTCTCACAACTGCACCCTT-3′; Reverse 5′- TCTTCACTCTTGGCTCCTGT-3′). WNT4 gene primers were designed to amplify a 142 bp product (Forward 5′- GCCATTGAGGAGTGCCAGTA-3′; Reverse 5′- CCACACCTGCCGAAGAGATG-3′). cDNA was amplified using Power SYBR Green PCR Mix (Applied Biosystems 4367659) at 60 ˚C annealing temperature for all primer sets for 35 cycles on a QuantStudio 3 qPCR machine. The fusion PCR products were visualized on a 1% agarose gel.
Immunohistochemistry (IHC)
IHC on FFPE human PCC/PGL five micrometer sections was performed using standard protocols. In brief, deparaffinization, antigen retrieval, permeabilization and blocking were done according to recommended conditions for the rabbit anti-MAML3 antibody (Bethyl Laboratories A300–684). Sections were incubated with 1:250 primary antibody overnight at 4° C. Slides for β-Catenin were incubated with rabbit anti-β-Catenin D10A8 at 1:100 (Cell Signaling Technologies, 8480). Sections were incubated with 1:200 secondary anti-rabbit antibody (Vector Laboratories Cat# BA-1000) for 1 hour at room temperature. Detection was performed using the VECTASTAIN ABC HRP kit (Vector Laboratories PK-4001) and DAB chromogen (Agilent K346711–2). Slides were then dehydrated and cover-slipped. Images were taken on a Nikon Eclipse microscope equipped with a DS-Fi1-U2 Color camera.
Cell Culture
Since there are no human pheochromocytoma cell lines publicly available, three closely related neuroendocrine tumor (NET) cell lines were utilized, SK-N-SH (neuroblastoma; ATCC), QGP-1 and BON-1 (both pancreatic NET; generous gift from Dr. Christopher Heaphy).
SK-N-SH cells were grown in EMEM (Corning MT10010CV), QGP-1 cells were grown in RPMI (Gibco 11875093) and BON-1 cells were grown in DMEM/F12 (Corning MT10092CV), all supplemented with 10% FBS (Sigma F0926) and Gemini Antibiotic:Antimycotic (F77R001). All experiments were performed using cells within five passages or less between replicates. All cell lines were authenticated with STR profiling (Promega) at least every 10 passages and mycoplasma negative on testing.
Plasmids and Transient Transfections
Cells were transfected using the Lipofectamine 3000 (Invitrogen L3000) reagent according to manufacturer’s protocols. MAML3 plasmids were cloned using the pCMV6-AC vector from Origene (PS100020) using the full-length cDNA MAML3 sequence (FL) and the MAML3 cDNA sequence lacking the first exon (dEx1). A C-terminal V5 tag was added to both constructs. Transfection with the pCMV6-AC vector without either MAML3 sequence was used as the control. TOPFlash and FOPFlash plasmids were purchased through Addgene (#12465, #12457 deposited by Randall Moon) and β-catenin pcDNA3 was purchased through Addgene (#16828 deposited by Eric Fearon) [8, 9]. The pRL-TK Renilla was obtained from Promega (E2241). Mutant constructs of the MAML3 protein were designed by dividing the protein (NP_061187.2) into putative functional domains: notch-binding domain (amino acids 1 to 155), nuclear localization sequence (NLS) domain (N; amino acids 156 to 195), sumoylation domain (S; amino acids 196 to 364), poly-Q track domain (Q; amino acids 365 to 772) and the C-terminal domain (C; amino acids 773 to 1133). Using these domains, constructs were created as follows: FLΔC772 contains amino acids (aa) 1–772, dEx1ΔC772 aa 156–772, FLΔC364 aa 1–364, dEx1ΔC364 aa 156–364, FLΔQ aa 1–364 and 772–1133, dEx1ΔQ aa 156–364 and 772–1133, FLΔS aa 1–195 and 365–1133, dEx1ΔS aa 156–195 and 365–1133, FLΔNΔS aa 1–155 and 365–1133, dEx1ΔNΔS aa 365–1133.
Immunofluorescence
Transfected cells were transferred to 8-chamber slides and allowed to settle for 48 hours. Cells were fixed with methanol overnight at 4°C, then washed in PBS and blocked with 1% BSA in PBS for 1 hour at room temperature. Cells were incubated overnight at 4°C with rabbit anti-MAML3 diluted 1:500 in 0.1% BSA PBS. Secondary antibody goat anti-rabbit AlexaFluor 546 conjugate (Invitrogen, A-11035) was added at 1:500 for one hour at room temperature. For colocalization of β-catenin experiments, slides were additionally stained with a 1:50 dilution of Alexa Fluor 488 mouse anti-β-Catenin Clone 14/Beta-Catenin (BD Biosciences, 563505) and Texas Red-X Phalloidin (Invitrogen, T7471) per manufacturer’s protocol. Slides were mounted with Prolong Gold Antifade containing DAPI (Invitrogen P36931). Images were taken on a Nikon Eclipse microscope equipped with a 100x/1.30 oil Nikon Plan Fluor Objective and 1.5x zoom or a Leica SP8 confocal microscope.
Immunoblots
Immunoblots were performed according to standard protocols [10]. The primary antibodies included anti-MAML3 antibody (Bethyl Laboratories A300–684), anti-V5 (Cell Signaling 13202S), anti-β-Catenin D10A8 (Cell Signaling Technologies, 8480), anti-β-Tubulin (Cell Signaling 2146S), anti-PARP (Cell Signaling 9542S) and anti-Caspase 3 (Cell Signaling 9662S). The PARP and Caspase 3 antibodies recognize total and cleaved products of each protein, respectively.
Proliferation and Viability Assays
BrdU proliferation assays were done following standard protocols [11]. Cells were transfected for 24 hours and then counted and seeded at 15,000 cells/well into 8-well chamber slides and allowed to grow for 72 hours before fixation. Given different baseline proliferation rates for each cell line, BrdU was added at various time points before fixation (2 hours prior for SKNSH, 1 hour prior for QGP-1 and 30 mins prior for BON-1). Slides were fixed in 4% paraformaldehyde for 30 minutes, washed in PBS, and incubated in 2N HCl at 37°C for 30 minutes. Primary mouse anti-BrdU 3D4 (BD 555627) was used at 1:500 in blocking buffer at room temperature for 1 hour. Slides were washed in PBS and were incubated with goat anti-mouse secondary (Vector BA-9200) 1:500 dilution for 1 hour. Cells from the same transfection not used in the proliferation assay were plated on a 6cm plate and then protein from cells on the last day of the proliferation assay was assessed by western blot to ensure the presence of the transfected MAML3 protein.
For the viability assay, cells were counted and plated in white 96-well plates at 5,000 cells per well, and transfected with either vector, FL or dEx1 the following day. Twenty-four hours post transfection, plates were evaluated using the Promega CellTiter-Glo 2.0 (G9243) assay according to manufacturer’s protocol on day 1, day 4 and day 5. Plates analyzed on days 4 and 5 had a media change on day 1. All experiments were done with at least N=6 in triplicate and data were analyzed and normalized to empty vector.
Apoptosis assay
Transfected cells were allowed to recover in full serum media for 24 hours and then were switched to serum free media. Cell pellets were collected 48 and 72 hours later, and Western blots were run and probed for cleaved and total PARP and Caspase 3. At least three biological replicates were done per cell line.
Soft Agar Colony Formation
Soft agar colony formation was performed for BON-1 and QGP-1 cells. SK-N-SH cells did not retain transient MAML3 overexpression long enough for this assay. Twenty-four hours post transfection, assays were set up as previously described [12]. QGP-1 cells were seeded at 10,000 cells/well, while BON-1 were seeded at 15,000 cells/well. Each of 4 biological replicates was set up in duplicate and allowed to grow for 7 days before staining with 1 mg/mL nitroblue tetrazolium overnight. Pictures were taken with a digital camera and colonies quantitated in ImageJ. Cells from the same transfection were collected on days 5 and 7 to ensure MAML3 protein expression by Western Blot.
Invasion Assay
Invasion through Matrigel was performed in cells lines according to standard protocols [13]. SK-N-SH cells were seeded at 80,000 cells/chamber and allowed to invade for 48 hours. QGP-1 were seeded at 20,000 cells/chamber and allowed to invade for 54 hours. BON-1 cells were seeded at 10,000 cells/chamber and allowed to invade for 54 hours. After the allotted time, transwell membranes were fixed in 4% paraformaldehyde for 20 minutes, and the cells visualized with DAPI. Pictures were taken of each transwell, and invasion was determined by percent area as calculated in ImageJ. All experiments were done at least N=3 in triplicate. Cells from the same transfection were used to confirm MAML3 expression by immunoblot.
Luciferase Assay
Luciferase assays using a TCF/LEF promoter were performed in all three cell lines. In brief, cells were plated into a 96-well plate at ~50% confluence. The following day, cells were transfected with either empty vector, FL MAML3, or dEx1 MAML3. Twenty-four hours later, wells were co-transfected with equal amounts of both reporter plasmid (either TOPFlash or FOPFlash) and either β-catenin or empty vector. Renilla was included in the second transfection at 1/80th the concentration of the reporter plasmid. Forty-eight hours after that the Promega Dual-Glo kit (E2920) was used according to manufacturer’s protocols in conjunction with a Monolight 3010 luminometer to determine luciferase activity. All experiments were done at least N=3 in triplicate and data were normalized to empty vector.
Co-immunoprecipitation (co-IP) assay
Co-IP was performed in all three cells lines using standard protocols. In brief, two days post-transfection, FL MAML3 and β-catenin co-transfected cells were crosslinked with 1% formaldehyde and quenched with glycine. Cells were harvested on ice in lysis buffer: 5 mM PIPES pH, 8 85 mM KCL, 0.5% NP-40 and Roche protease inhibitors (5892791001) and then incubated with protein-A sepharose resin (Invitrogen 101041). Lysates were immunoprecipitated overnight at 4°C with either rabbit IgG (Cell Signaling 2729) or anti-MAML3 (Bethyl Laboratories A300–684) on protein-A sepharose blocked with 3% BSA. Resins were spun at 500g for 5 minutes, flow through discarded, and then washed 2X with 10 resin-volumes lysis buffer followed by 3X washes with 500 mM LiCl. Proteins were eluted from the column by boiling in Laemmli buffer for 30 minutes. Samples were visualized by immunoblot probed with anti-β-Catenin D10A8 (Cell Signaling Technologies, 8480).
Transcriptome analysis
SK-N-SH, BON-1 and QGP-1 cells were transfected with vector control, FL MAML3 or dEx1 MAML3. Three biological replicates were plated in triplicate. On day three post transfection, RNA was harvested using Trizol (Invitrogen) and submitted to the Genomics and Microarray Core at the University of Colorado for library prep using an Illumina TruSeq mRNA library kit (Illumina, San Diego, CA, USA). Sequencing was performed on a NanoSeq 6000 (Illumina).
During analysis, adapter sequences were removed with the help of Trimmomatic trimmer [14], and the “Tuxedo” pipeline was used for transcript expression analysis, as previously described [15]. RNAseq reads were aligned to the hg19 reference genome with HISAT2 [15]; stringtie was used for transcriptome assembly and transcript abundance estimation [15]. Differential expression analysis was done using DeSeq2 package in R [16]. Gene set enrichment analysis (GSEA) was performed using the R package fgsea (version 1.16.0) [17] and msigdbr (version 7.2.1) (https://CRAN.R-project.org/package=msigdbr) for the H collection of hallmark gene sets and C6 collection of oncogenic signature gene sets (http://www.gsea-msigdb.org/gsea/msigdb/collection_details.jsp) [18]. The transcriptome data have been deposited in Gene Expression Omnibus (GEO accession GSE174701).
Statistics
All assays were performed in triplicate with at least three biological replicates unless otherwise stated. When comparing differences between two means, Student’s T-test was used. ANOVA with Bonferroni correction were used to detect differences between multiple group means. P-values less than 0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism software.
Results
MAML3 fusion confirmed in a cohort of human PCC/PGL tumors
To determine the presence of the UBTF-MAML3 fusion in a novel cohort of PCC/PGL tumor samples, 55 fresh-frozen tumors were screened by RT-PCR for the two isoforms detected in TCGA [4]. Similar to the TCGA cohort, 7% of tumors (n=4 of 55) had UBTF-MAML3 fusions (Fig. 1A). Of the four fusion-positive tumors, one had an isoform with a breakpoint after exon 17 of UBTF, whereas the remaining three had a breakpoint after exon 19. The four fusion-positive tumors were from patients with sporadic PCC/PGL (i.e. no known germline susceptibility gene pathogenic variant) and all four had metastatic disease. The fusion-positive PCC/PGL samples had intense nuclear staining with MAML3 IHC compared to fusion-negative tumors (Fig. 1B). Similar to the TCGA data showing that MAML3-fusion-positive PCC clustered in the Wnt-altered expression subtype with WNT4 mRNA being consistently highly expressed [4], our cohort demonstrated high WNT4 mRNA expression in the MAML3 fusion-positive tumors compared with fusion-negative tumors (Fig. 1C).
Figure 1. Validating the UBTF-MAML3 fusion in a new PCC/PGL cohort.

A, RT-PCR identification of UBTF-MAML3 fusion-positive tumors (PCPG-021, PCPG-057, PP-103, and PCPG-130) and fusion negative tumors (PP-115, PP-126). Two fusion products were identified representing the two major UBTF-MAML3 fusions reported in the TCGA dataset. TBP is a positive control for the RT-PCR reaction. B, MAML3 immunohistochemistry showed prominent nuclear staining in the fusion positive tumors and weak staining in the fusion negative tumors (scale bar = 100 μm). C, RT-qPCR analysis of WNT4 showed transcriptional upregulation in fusion-positive tumors. Error bars represent standard deviation.
In vitro overexpressed MAML3 is localized to the nucleus
Having confirmed the presence of recurrent MAML3 fusions in a separate cohort of PCC/PGL, we asked if overexpression of MAML3 was tumorigenic in vitro. Over-expression plasmids expressing FL MAML3 and dEx1 MAML3 (version lacking exon 1 and hence lacking the NICD binding motif to mimic the fusion gene) were created (Fig. 2A). Since there are no human PCC/PGL cell lines publicly available, three other neuroendocrine tumor (NET) cell lines with low to no endogenous expression of MAML3 were utilized for the functional experiments. SK-N-SH is a human neuroblastoma cell line; neuroblastoma is a tumor derived from the same embryologic origin as PCC/PGL and shares a similar mRNA profile [4, 19]. BON-1 and QGP-1 are pancreatic neuroendocrine tumor cell lines. Following transient transfection, both FL and dEx1 MAML3 were localized to the nucleus as expected in all cell lines (Fig. 2B and Supplementary Fig. S1). Interestingly, the immunofluorescence experiments detected a punctate nuclear pattern with FL MAML3 compared to a more diffuse nuclear pattern associated with dEx1 MAML3 overexpression in all cell lines.
Figure 2. MAML3 constructs and in vitro cellular localization.

A, Diagram of MAML3 constructs showing predicted sites for posttranslational modifications, motifs, and nuclear localization signals. B, Immunofluorescence of SK-N-SH cells transfected with either FL or dEx1 MAML3 and stained for MAML3 show the nuclear localization. FL MAML3 showed punctate areas in the nucleus, whereas dEx1 MAML3 showed a more diffuse staining pattern. No endogenous MAML3 could be detected in transfected controls. (100x/1.30 oil Nikon Plan Fluor Objective 1.5x Zoom, AlexaFluor546, Scale bar represents 10 μm)
MAML3 overexpression is associated with cell invasion and colony formation
Since MAML3 fusions in human PCC/PGL are associated with metastatic disease, we hypothesized that MAML3 expression would contribute to a more tumorigenic and invasive cellular phenotype. Overexpression of FL and dEx1 MAML3 increased invasion through Matrigel compared to controls (Fig. 3A: SK-N-SH: 53% (p=0.0004) and 25% (p=0.0324) increase with FL and dEx1, respectively. QGP-1: 81% (p< 0.0001) and 50% (p=0.0020) increase; BON-1: 38% (p<0.0001) and 35% (p<0.0001) increase).
Figure 3. FL and dEx1 MAML3 contribute to a more aggressive phenotype in vitro.

A, All three neuroendocrine tumor cell lines had higher invasion through Matrigel when transfected with either FL or dEx1 MAML3.B, Soft agar colony formation was increased in QGP-1 and BON-1 cell lines transfected with MAML3 FL or dEx1 compared to empty vector controls. SK-N-SH did not retain transient transfection long enough for this assay. C, Cell viability increased in all three cell lines with FL or dEx1 MAML3 transfection. Error bars represent SEM across at least three separate experiments done in at least triplicate for invasion and cell viability, and four separate experiments done in duplicate for colony formation. (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001)
We also investigated the effect of MAML3 on soft agar colony formation. MAML3 overexpression led to increased soft agar colony formation in both QGP-1 and BON-1 cells (Fig. 3B; QGP-1: 94% (p< 0.0001) and 68% (p=0.0009) increase with FL and dEx1, respectively; BON-1: 60% (p=0.0313) and 100% (p=0.0017) increase). The transient expression was not maintained in SK-N-SH cells for an extended length of time to allow for performing of the colony formation assay. There was no increase in the rate of proliferation as measured by BrdU IHC in any of the cell lines transfected with FL or dEx1 MAML3 (Supplementary Fig. S2A). It is possible that no differences were seen in this assay due to the nature of the short time points and lower overall transfection rates for transient transfections, thereby washing out small differences. There also was no decrease in apoptosis as measured by cleaved PARP or caspase 3 by Western blot (Supplementary Fig. S2B) in any of the cell lines transfected with FL or dEx1 MAML3. In fact, no cleaved caspase 3 was detected at all. This suggests MAML3 does not alter apoptosis at least through PARP and caspase 3. Despite these data, there was, however, a statistically significant increase in cell viability in all cell lines when either FL or dEx1 MAML3 was overexpressed (all at least p<0.01) (Fig. 3C and Supplementary Fig. S3).
MAML3 interacts with β-catenin in neuroendocrine cells and activates WNT signaling pathways
Next, we sought to elucidate the mechanism of increased tumorigenicity and invasion in cells with overexpressed FL and dEx1 MAML3. Because the MAML3 fusion-positive human PCC/PGLs clustered with the Wnt-altered mRNA expression subtype and had increased β-catenin protein through RPPA in the TCGA cohort, we hypothesized that overexpressed FL and dEx1 MAML3 may interact with β-catenin and activate WNT signaling target genes. We confirmed the TCGA finding that MAML3 fusion-positive human PCC/PGL samples showed increased protein expression of β-catenin compared to fusion-negative tumors, this time by IHC (Fig. 4A). There was increased membranous and some increased cytoplasmic expression of β-catenin in the fusion-positive tumors compared with the fusion-negative tumors. This association is unlikely to be the result of direct transcriptional activation of β-catenin by MAML3 given β-catenin mRNA was not increased in the TCGA study in fusion-positive tumors despite the increase in β-catenin protein [4].
Figure 4. β-catenin is overexpressed in UBTF-MAML3 fusion-positive human tumors and interacts with FL MAML3 in vitro.

A, β-catenin IHC expression in four UBTF-MAML3 fusion positive tumors (PCPG-021, PCPG-057, PP-103, and PCPG-130) and two fusion negative tumors (PP-115 and PP-126). B, TCF/LEF driven luciferase (TOP) activity relative to negative control promoter activation (FOP) was drastically increased in neuroendocrine cell lines co-transfected with β-catenin and MAML3 compared to β-catenin alone. (*** p<0.001,**** p<0.0001) Error bars represent SEM of at least three separate experiments done in triplicate. C, Neuroendocrine tumor cell lines were co-transfected with β-catenin and FL MAML3. Immunoprecipitation of MAML3 demonstrated co-precipitation of β-catenin. Representative blot from at least three separate experiments.
To study the role of β-catenin in MAML3 overexpressing cells, we co-transfected β-catenin with FL or dEx1 MAML3 in vitro. In all three cell lines, co-transfection of FL or dEx1 MAML3 with β-catenin statistically significantly increased TCF/LEF luciferase activity (p<0.001), suggesting activation of the TCF/LEF target gene expression is a major outcome of MAML3 overexpression (Fig. 4B; SK-N-SH: 23- and 14-fold increase with FL and dEx1, respectively. QGP-1: 65- and 45-fold increase; BON-1: 23- and 24-fold increase. All with p<0.001). Further supporting these data, when either FL or dEx1 MAML3 was co-transfected with β-catenin, both proteins co-localized in the nucleus as seen by immunofluorescent confocal microscopy, whereas transfection of β-catenin alone resulted in a diffuse staining pattern across the entirety of the cell (Supplementary Fig. S4). This diffuse pattern seen with β-catenin transfection alone was similar to that seen with endogenous β-catenin, although the expression of the endogenous protein was quite low, requiring an increase in exposure to visualize (Supplementary Fig. S4). Nevertheless, when either FL or dEx1 MAML3 were transfected alone, there was faint nuclear co-localization with endogenous β-catenin (Supplementary Fig. S4). To further support the interaction of the two proteins, when MAML3 and β-catenin were co-transfected in QGP-1 and BON-1 cells, both FL and dEx1 MAML3 were detected in the same protein complex as β-catenin as shown by co-immunoprecipitation (Fig. 4C and Supplementary Fig. S5). In SK-N-SH cells, the interaction was only seen with FL MAML3 (Fig. 4C).
To determine which domains of MAML3 were required for co-activation of TCF/LEF promoters with β-catenin, multiple mutant constructs of FL and dEx1 MAML3 were created. These constructs expressed stable protein products by immunoblot (Supplementary Fig. S6A and S6B). Deletion of either the sumoylated domain (S) with or without the nuclear localization sequence (N), the glutamine tracts (Q) or the C-terminal domain (C) decreased luciferase activity (Fig. 5, Supplementary Fig. S6C), suggesting that MAML3 has multiple protein binding domains necessary for the effects and likely mediates its effects through a large multi-protein complex to drive dysregulated transcription.
Figure 5. TCF/LEF promoter activation by MAML3 deletion constructs in SK-N-SH cells.

Diagrams of each MAML3 construct are shown to the left of their relative promoter activation by luciferase assay. All MAML3 constructs were co-transfected with β-catenin. Cells transfected with β-catenin alone, or with neither MAML3 nor β-catenin are shown as controls, in addition to the negative control FOP vector for every combination. (**** p<0.0001) Error bars represent SEM from at least three different experiments done in triplicate.
MAML3 upregulation is associated with differentially expressed pathways involved in tumorigenesis
To determine potential downstream targets of FL and dEx1 MAML3, transcriptome analysis was performed in all three cell lines with overexpression of FL or dEx1 MAML3 compared with control empty vector. Principal component analysis showed tight clustering by cell line (Fig. 6A), suggesting a high level of reproducibility of the transcriptome profiling. To isolate the effects of the FL or dEx1 MAML3 overexpression and account for cell line specific differences in expression, we employed a multi-factor DESeq2 experimental design using cell line as a covariate [16] (raw data in Supplementary Table S1). Eight differentially expressed genes were shared between both constructs across the three cell lines (Fig. 6B). Seven out of eight genes were downregulated by FL and dEx1 overexpression. Interestingly, the long non-coding RNA, Linc00659, was upregulated in both FL and dEx1 overexpressing cells. GSEA with the MSigDB Hallmark pathways showed several pathways were dysregulated by overexpression of FL and/or dEx1 MAML3 (Fig. 6C). Most notably, both FL and dEx1 MAML3 overexpression led to statistically significant enrichment of pathways involved in proliferation, such as G2M check point, E2F targets, and MYC targets (Fig. 6C). When using the C6 Oncogenic pathway analysis in MSigDB, the WNT pathway was shown to be statistically significantly upregulated in cell overexpressing FL MAML3, and the SHH pathway was upregulated in cell overexpressing either FL or dEx1 MAML3 (Supplementary Fig. S7). These latter data support that from TCGA analysis showing these two pathways are the most highly overexpressed in the MAML3 fusion-positive human PCC/PGL [4]. The Wnt and SHH signaling pathways did not reach statistical significance when using the Hallmark analysis.
Figure 6. Transcriptomics reveal NOTCH independent roles for MAML3 shared across three cell lines.
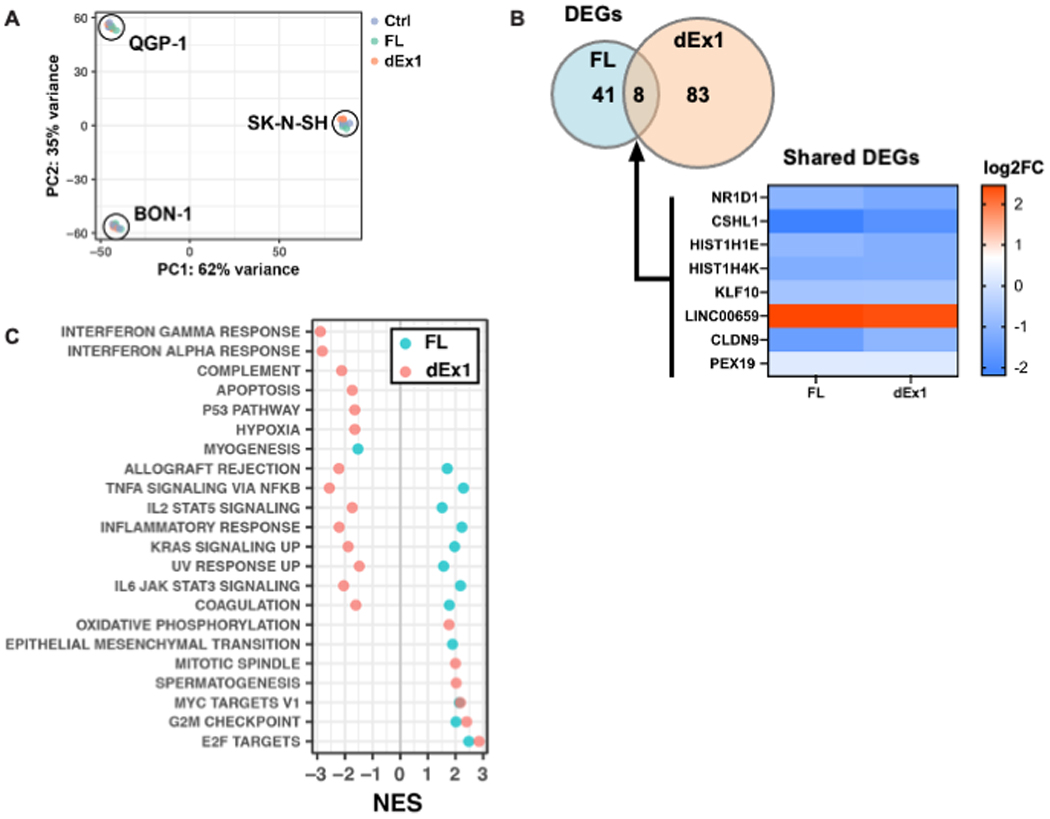
A, mRNA sequencing of all three cell lines transfected with either control, FL MAML3 or dEx1 MAML3 was analyzed using principal component analysis (PCA) showing tight clustering according to cell line (N=3 for each cell line and each construct). We therefore used DESeq2 to account for cell line differences and isolate effects of the MAML3 overexpression for the rest of the analyses. B, Differentially expressed genes (DEGs) shared between cells with FL or dEx1 MAML3 are highly correlated (Pearson r = 0.98). C, GSEA of the MSigDB Hallmark collection shows enriched pathways related to proliferation, overlapping for both FL and dEx1 MAML3 overexpression (NES - Normalized Enrichment Score; all adjusted p-values < 0.05).
Discussion
Our data confirm for the first time, to our knowledge, in a separate cohort of human PCC/PGL, that recurrent UBTF-MAML3 fusion genes occur in approximately 7% of PCC/PGL and are associated with metastatic disease, independent of other known driver mutations or hereditary pathogenic variants. MAML3 protein is highly expressed in human PCC/PGL containing the MAML3 fusion and associated with higher expression of β-catenin protein and WNT4 mRNA. These data suggest that MAML3 plays a role in the activation of the WNT signaling pathway in human PCC/PGL.
Overexpression of MAML3 is a driver of tumorigenesis in neuroendocrine tumor cells. Both FL MAML3 and dEx1 MAML3 (to mimic the region found in the fusion gene) led to increased cell invasion and colony formation. This may occur through WNT signaling pathways as our data suggest that MAML3 can bind in a complex with β-catenin and activate TCF/LEF promoters (Fig. 7). GSEA data suggested upregulation of pathways involved in proliferation and cell growth, which is consistent with the increase in cell viability seen across the cell lines. Overexpression of FL or dEx1 MAML3 led to upregulation of pathways involving MYC and E2F target genes and genes involved in progression through the G2M checkpoint. In addition, both FL and dEx1 MAML3 overexpression led to significant upregulation of Linc00659, which in other studies has been shown to prevent the degradation of β-catenin, allowing for increased expression of cyclin D1 leading to cell cycle progression [20].
Figure 7. Proposed model of MAML3 function in neuroendocrine tumor cells.

The left frame illustrates the canonical role of MAML3 in NOTCH signaling. The right frame illustrates a proposed model of FL (oval) and dEx1 MAML3 (ridged oval) function when overexpressed in neuroendocrine tumor cells.
The concept that overexpression of MAML3 contributes to metastatic disease has been suggested previously. In gastric cancer cells, silencing MAML3 with siRNA decreased expression of proteins involved in EMT [21], suggesting MAML3 has a role in metastasis. Furthermore, human gastric cancers with high MAML3 expression have a worse survival than those with low MAML3 expression [21]. In patients with well-differentiated small intestine neuroendocrine tumors, high MAML3 protein expression in serum was associated with the presence of metastatic disease to the liver and lymph nodes [22]. Perhaps in neuroendocrine tumor cells, overexpression of MAML3 increases the cell cycle pathways, enhances β-catenin stabilization, allows for binding in a complex with β-catenin, and activates downstream target genes, leading to increased tumorigenesis and metastatic potential. Future studies are needed to confirm this hypothesis.
Our study is limited by the lack of model systems of human PCC/PGL. We therefore used three different neuroendocrine tumor cell lines including a neuroblastoma cell line which is derived from the same embryologic origin. Prior work has shown that neuroblastoma cells treated with retinoic acid and a spontaneous truncation of MAML3 after exon 1 had similar expression pattern to human PCC/PGL with MAML3-fusions [4, 19]. Despite this, it is possible that the 5′ partner of MAML3 plays a role in the function of the fusion gene. UBTF is a transcription factor and may bring other partners to a transcriptional complex or have alternative DNA binding sites than MAML3 overexpressed alone, either FL or dEx1. Nevertheless, we hypothesize that the overexpression of MAML3 itself is the driving event, based on the facts that neuroblastoma cells with spontaneous truncation of MAML3 after exon 1 had similar expression patterns to human PCC/PGL with the MAML3 fusions [4, 19] and that different types of MAML protein fusions with different upstream partners have been identified in other human cancers. For example, biphenotypic sinonasal sarcomas have a recurrent PAX3-MAML3 fusion gene [23]. Interestingly, in this fusion, the MAML3 breakpoint is in intron 1, thereby similarly deleting the NCID binding motif in exon 1 as seen in PCC/PGL. In mucoepidermoid carcinomas, recurrent CRTC1-MAML2 fusions are detected and have a gain-of-function interaction with Myc [24], consistent with MYC overexpression in MAML3 fusion positive PCC/PGL [4]. These data, together with the TCGA data on MAML3 fusions with UBTF or rarely with TCF as the 5′ partner in PCC/PGL, support the hypothesis that high expression of MAML3 is the driving event of the fusion. Ongoing studies in our lab are investigating the function of the composite UBTF-MAML3 fusion gene.
In conclusion, we confirm that UBTF-MAML3 fusions are expressed in 7% of human PCC/PGL and lead to increased expression of MAML3 and β-catenin protein and Wnt4 mRNA. The fusion protein is associated with aggressive metastatic disease. In functional assays, we show evidence that overexpression of MAML3 increases tumorgenicity and invasion. Together these data suggest that strong nuclear MAML3 IHC expression may be a useful prognostic marker in human PCC/PGL. Future prospective studies using large sample sets are needed.
Supplementary Material
Acknowledgements
QGP-1 and BON-1 cells were a generous gift from Dr. Christopher Heaphy. We thank Hannah Haas for technical assistance with the viability assays. Funding support to L. Fishbein from the American Cancer Society MRSG-15-063-01-TBG and the Cancer League of Colorado and to K. Kiseljak-Vassiliades from NIH/NCI K08CA222620.
Funding Support: Funding support to L. Fishbein from the American Cancer Society MRSG-15-063-01-TBG and the Cancer League of Colorado and to K. Kiseljak-Vassiliades from NIH/NCI K08CA222620.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
References
- 1.Fishbein L, Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment. Hematol Oncol Clin North Am, 2016. 30(1): p. 135–50. [DOI] [PubMed] [Google Scholar]
- 2.Ayala-Ramirez M, et al. , Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab, 2011. 96(3): p. 717–25. [DOI] [PubMed] [Google Scholar]
- 3.Fishbein L, et al. , Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun, 2015. 6: p. 6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fishbein L, et al. , Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell, 2017. 31(2): p. 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Job S, et al. , Telomerase Activation and ATRX Mutations Are Independent Risk Factors for Metastatic Pheochromocytoma and Paraganglioma. Clin Cancer Res, 2019. 25(2): p. 760–770. [DOI] [PubMed] [Google Scholar]
- 6.McElhinny AS, Li JL, and Wu L, Mastermind-like transcriptional co-activators: emerging roles in regulating cross talk among multiple signaling pathways. Oncogene, 2008. 27(38): p. 5138–47. [DOI] [PubMed] [Google Scholar]
- 7.Kitagawa M, Notch signalling in the nucleus: roles of Mastermind-like (MAML) transcriptional coactivators. J Biochem, 2016. 159(3): p. 287–94. [DOI] [PubMed] [Google Scholar]
- 8.Kolligs FT, et al. , Neoplastic transformation of RK3E by mutant beta-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol Cell Biol, 1999. 19(8): p. 5696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veeman MT, et al. , Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol, 2003. 13(8): p. 680–5. [DOI] [PubMed] [Google Scholar]
- 10.Kiseljak-Vassiliades K, et al. , Elucidating the Role of the Maternal Embryonic Leucine Zipper Kinase in Adrenocortical Carcinoma. Endocrinology, 2018. 159(7): p. 2532–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fishbein L, et al. , In vitro studies of steroid hormones in neurofibromatosis 1 tumors and Schwann cells. Mol Carcinog, 2007. 46(7): p. 512–523. [DOI] [PubMed] [Google Scholar]
- 12.Borowicz S, et al. , The soft agar colony formation assay. J Vis Exp, 2014(92): p. e51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Justus CR, et al. , In vitro cell migration and invasion assays. J Vis Exp, 2014(88). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolger AM, Lohse M, and Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014. 30(15): p. 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pertea M, et al. , Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc, 2016. 11(9): p. 1650–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Love MI, Huber W, and Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 2014. 15(12): p. 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korotkevich G, Skukhov V, and Sergushichiev A, Fast gene set enrichment analysis, in bioRxiv. 2019. [Google Scholar]
- 18.Liberzon A, et al. , The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst, 2015. 1(6): p. 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heynen GJ, et al. , Mastermind-like 3 controls proliferation and differentiation in neuroblastoma. Mol Cancer Res, 2016. [DOI] [PubMed] [Google Scholar]
- 20.Tsai KW, et al. , Linc00659, a long noncoding RNA, acts as novel oncogene in regulating cancer cell growth in colorectal cancer. Mol Cancer, 2018. 17(1): p. 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, et al. , MiR-2392 suppresses metastasis and epithelial-mesenchymal transition by targeting MAML3 and WHSC1 in gastric cancer. FASEB J, 2017. 31(9): p. 3774–3786. [DOI] [PubMed] [Google Scholar]
- 22.Darmanis S, et al. , Identification of candidate serum proteins for classifying well-differentiated small intestinal neuroendocrine tumors. PLoS One, 2013. 8(11): p. e81712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, et al. , Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat Genet, 2014. 46(7): p. 666–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amelio AL, et al. , CRTC1/MAML2 gain-of-function interactions with MYC create a gene signature predictive of cancers with CREB-MYC involvement. Proc Natl Acad Sci U S A, 2014. 111(32): p. E3260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.