

Summary
Despite the obvious inhibitory outcome of PD-1 signaling, an additional series of functions are activated. We have observed that T cells stimulated through the T cell receptor (TCR) and PD-1 primarily do not proliferate; however, there is a population of cells that proliferates more than through TCR stimulation alone. In this study, we performed flow cytometry and RNA sequencing on individual populations of T cells and discovered that unlike naive T cells, which were inhibited following PD-1 ligation, T cells that proliferated more following PD-1 ligation were associated with effector and central memory phenotypes. We showed that these populations had different gene expression profiles following PD-1 ligation with PD-L1 compared to PD-L2. The presence of transcriptionally and functionally distinct T cell populations responsive to PD-1 ligation provides new insights into the biology of PD-1 and suggest the use of T cell subset-specific approaches to improve the clinical outcome of PD-1 blockade.
Subject areas: Immunology, Cancer, Transcriptomics
Graphical abstract
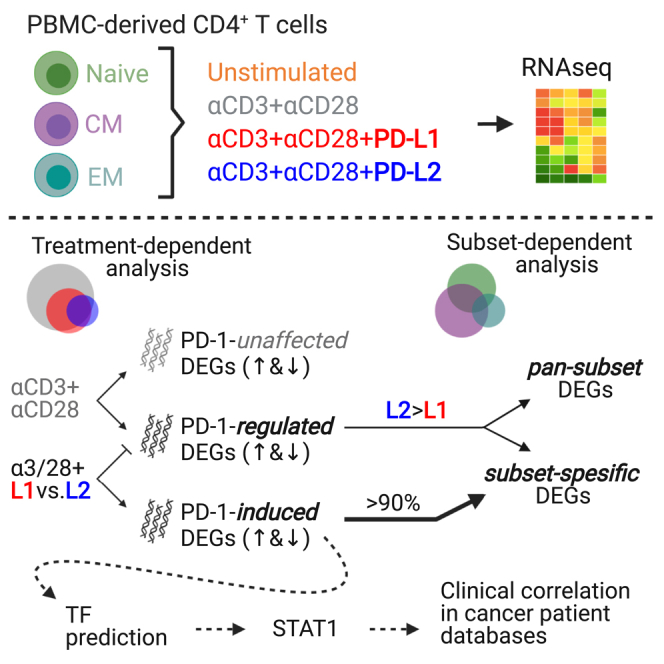
Highlights
-
•
Most of the genes induced or regulated by PD-1 are T cell subsets specific
-
•
PD-1 ligation with PD-L1 or PD-L2 results in diverged transcriptomic signatures
-
•
PD-1-induced STAT1 upregulation correlates with responses to checkpoint blockade
Immunology; Cancer; Transcriptomics
Introduction
Immune checkpoint therapy is a promising technique in cancer immune therapy. Using antibodies, this approach targets immune checkpoints on T cells, such as PD-1, to block the interaction with its ligands (PD-L1 and PD-L2). High expression of PD-1 ligands on tumor cells represents a hijacking of the immune checkpoint system, dampening T cell-mediated tumor clearance. Despite the success of PD-1-targeting checkpoint inhibitors in nearly 30% of patients with a wide array of tumor types, many patients remain unresponsive to this treatment (Li et al., 2019). Within these patients, mounting clinical evidence suggests that a significant portion experience an acceleration of disease following treatment (Tay et al., 2020). This acceleration has been termed hyper progressive disease and is estimated to occur in up to 10% of patients treated with PD-1-targeting therapy. Furthermore, 40% of all patients treated with checkpoint blockade experience immune-related adverse events, likely due to on-target toxicity of the same intervention (Postow et al., 2018).
But progress in the field of immune checkpoints now allows for the identification of numerous factors predictive of responsiveness to PD-1 blockade. While most of these factors, such as mutation burden and PD-L1 expression, pertain to tumor cells (Lagos et al., 2020), differences in PD-1 signaling demonstrate a contribution of the T cell compartment (Thommen et al., 2018). Naturally, PD-1 blocking agents require the expression of PD-1 and the generation of downstream inhibitory signals to function. However, there is an increased understanding that PD-1 is not universally inhibitory for all T cell functions. Like CTLA-4, PD-1 can activate individual T cell functions, or even entire distinct T cell subsets (Shitara and Nishikawa, 2018; Tocheva et al., 2020; Riley, 2009; Hui et al., 2017; Patsoukis et al., 2020). More recently, using phosphoproteomic and genetic approaches, we showed that not all the pathways downstream of PD-1 were inhibitory, and some unique cellular functions were clearly stimulated (Tocheva et al., 2020).
Until now, most studies have focused on CD8 T cell-mediated cytotoxicity as the key to understanding immune checkpoint responsiveness. While cytotoxic CD8 T cells are critically important for an effective anti-tumor immune response, the contribution of other T cell subsets, including effector and cytotoxic CD4 subsets, cannot be overlooked. Mounting evidence indicates that systemic CD4 T cell immunity plays a key role in mediating durable antitumor responses, paving the way for new approaches to target checkpoints on peripheral CD4 T cells (Oh et al., 2020). Despite an increasing number of studies focusing on PD-1 signaling in CD4 T cells, its effects remain unclear (Nagasaki et al., 2020).
To better understand the precise effects of PD-1 signaling on CD4 T cell function, as well as the possible heterogeneity among T cell subsets, we used flow cytometry to measure the expression of CD4 T cell-maturation markers associated with different subsets of proliferating cells in the context of PD-1 ligation. These subsets were sorted and stimulated with PD-L1 or PD-L2, after which RNA sequencing revealed different transcriptional profiles among different populations of cells.
Results
Proliferating T cells following PD-1 ligation express diverse subset markers
To assess T cell proliferation in the context of PD-1 signaling, we stimulated primary human CD8 T cells, isolated from healthy volunteers, using wells coated with anti-CD3 antibody, with and without the presence of recombinant PD-L1 or PD-L2 (Figure 1A). As expected, most of the cells proliferated after stimulation with anti-CD3 antibody (Figure 1Bi). Interestingly, the addition of PD-L1 or PD-L2 resulted not just in higher number of inhibited cells (Figure 1Bii), but also in more cells that proliferated even more than after treatment with anti-CD3 alone (Figure 1Bi). To uncover the landscape of these unexpected hyper proliferative cells, we measured the expression of CD45RA and CCR7, cell maturation markers, associating these cells with specific T cell subsets (Figure 1C). As shown (Figure 1D), the cells that proliferated more were central memory (CM) and effector memory (EM) T cells and not the naive T cells. The ability of PD-1 to inhibit proliferation in the naive cells or to accelerate proliferation in the EM cells was more striking in the context of PD-L2 over PD-L1 (Figures 1Ei and 1Eii). While stimulation with anti-CD3 antibodies alone resulted in transition of naive cells into CM phenotype, the addition of PD-L1 or PD-L2, surprisingly, resulted in further maturation and transition of the CM cells toward EM phenotype (Figures 1Ei and 1Eii).
Figure 1.

Proliferating T cells following PD-1 ligation express diverse subset markers
(A) A description of the experimental system used for T cell stimulation.
(Bi) Primary human CD8 T cells were stimulated as indicated, with plate bound anti-CD3 and PD-L1 or PD-L2, and proliferation was measured by flow cytometry using celltrace far red proliferation dye dilution. Arrow indicates the cells that proliferated further.
(Bii) Quantification of the previous experiment, showing the percentage of proliferating cells. n = 4, one-way ANOVA, ∗∗∗p < 0.001, ns; not significant.
(C) The flow cytometry markers that were used to define the different cell subsets.
(D) CD8 T cells were stimulated as indicated and flow cytometry was used on day 4 to assess proliferation dye intensity and expression of subset specific markers. Arrows indicate the cell the proliferated further.
(Ei) Dot plot flow analysis of the previous experiment showing the percentages of each subset of T cells in the setting of the different stimulation conditions.
(Eii) Quantification of the percentage of the proliferating cells of same experiments, n = 4, two-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ns; not significant.
(F) A description of the experimental system used for T cell stimulation.
(G and H) Primary human CD3 T cells were stimulated as indicated and flow cytometry was used to asses proliferation and subset distribution.
(Ii) Dot plot flow analysis of CD4 T cells showing the percentages of each subset of T cells in the setting of the different stimulation conditions.
(Iii) Quantification of the percentage of the proliferating cells of same experiments, n = 3, two-way ANOVA, ns; not significant.
(Ji) Dot plot flow analysis of CD8 T cells showing the percentages of each subset of T cells in the setting of the different stimulation conditions.
(Jii) Quantification of the percentage of the proliferating cells of same experiments, n = 3, ns; not significant.
To validate these finding in a more physiological system, we moved from plate bound stimulation into the primary T cell – Raji B cell conjugate system (Figure 1F) where irradiated Raji B cells that stably express either PD-L1 or PD-L2 are loaded with staphylococcus enterotoxin E (SEE), cocultured with labeled primary T cells for four days prior to flow cytometry quantification of proliferation and T cell subset distribution (Figure 1C). As shown in this assay, both CD4 T cells (Figure 1G) and CD8 T cells (Figure 1H) proliferated more in the context of PD-L1 and PD-L2. Quantification of these experiments failed to show statistically significant values, likely due to donor variability, although there was a trend toward higher number of CM cells after treatment with Raji B cells expressing PD-L1 or PD-L2 than after treatment with Raji B cells that express control plasmid (Figures 1Ii, 1Iii, 1Ji, and 1Jii).
Altogether, these findings suggest possible uncoupling of the ability of PD-1 to affect proliferation and maturation. We observed differences in the potency of PD-1 to modulate the function of different T cells subsets and to generate a more comprehensive view of the differential effect of PD-1 signaling on specific T cell subsets, we transitioned to a transcriptomic approach.
PD-1-stimulated T cell subsets exhibit distinct transcriptional profiles
To better uncover the differential signaling of PD-1 in the different subsets, we sorted CD4 T cells into naive, CM, and EM cells, stimulated the cells with immobilized anti-CD3+28 antibodies, PD-L1, or PD-L2, and submitted the 35 samples for RNA sequencing (Figure 2A). The expression levels of PD-1 protein were comparable in all subsets (Figure S1). Principle component analysis confirmed clustering of the expressed genes, not just of the different subsets but also of the diverse treatment conditions among the triplicates (Figure 2B). The distribution of the genes into the different subsets was further validated by the expression levels of naive (naivety markers) and effector (antigen experience markers) genes among the three donors (Figure 2C). Common to all the subsets, the total number of genes that were differentially expressed after stimulation with anti-CD3+28 antibodies were significantly higher compared to stimulation also with PD-L1 or PD-L2 (Figure 2D). Likewise, the expression levels of more genes were altered in the setting of PD-L1 compared to PD-L2 (Figure 2D). These data suggest that even though PD-L1 and PD-L2 bind to the same receptor on the T cells, the extent of their transcriptome profiles is divergent.
Figure 2.

PD-1 stimulated T cell subsets exhibit distinct transcriptional profiles
(A) Experimental design, highlighting the sorted subsets and the stimulation conditions that preceded the RNA sequencing.
(B) Principal components analysis showing the 35 experimental conditions.
(C) Heatmap showing the expression levels of genes that define naivety and antigen experience in unstimulated cells.
(D) Venn diagrams highlighting the number of the genes that were regulated in each of the experimental conditions. See also Figure S1.
PD-1 regulates most of the genes affected by the antigen receptor
There are two groups of genes that are regulated by PD-1 signaling. The first group includes the genes that were initially differently expressed by TCR complex stimulation and subsequently were either upregulated or downregulated by signaling downstream of the PD-1 receptor. This group of genes signifies the effect of PD-1 signaling on fine-tuning TCR responses and is labeled as PD-1-regulated DEGs (Figure 3A). The second group of genes comprises genes that their expressions were not changed by TCR signaling alone but were either upregulated or downregulated secondary to costimulation with PD-L1 or PD-L2. This group of unique genes is termed as PD-1-induced DEGs (Figure 3A). Another group of genes that were not changed at all by PD-1 signaling are named PD-1-unaffected DEGs (Figure 3A).
Figure 3.

PD-1 regulates most of the genes affected by the antigen receptor
(A) Venn diagram classifying the groups of genes between stimulation with anti-CD3 and anti-CD28 antibodies to stimulation with anti-CD3, anti-CD28 antibodies, and PD-Ls.
(Bi) Venn diagrams displaying the number of PD-1-regulated DEGs across the three subsets of T cells.
(Bii) Quantification of the previous experiment highlighting the proportion of antigen receptor affected genes that were regulated by either PD-L1 or PD-L2 in each of the T cell subsets.
(Ci) Venn diagrams showing the common and the diverse genes between the different subsets in cell stimulated with anti-CD3 and anti-CD28 antibodies.
(Cii) Comparison between the proportion of PD-1-regulated DEGs out of the genes that were shared between the subsets vs. the genes that were specific to only one of the subsets.
Remarkably, more than half of the genes that were initially regulated by the TCR were subsequently modified by treating the cells with PD-L1 or PD-L2 and this was more evident in the naive and CM cells and to a lesser extent also in the EM cells (Figures 3Bi and 3Bii). Including all the subsets, 1,129 TCR genes were downregulated by PD-L1 and 1,574 TCR genes were downregulated by PD-L2. Similarly, 712 genes were upregulated by PD-L1 and 1,010 genes were upregulated by PD-L2 (Figures 3Bi and 3Bii). Similar number of shared genes were either upregulated or downregulated (499 vs. 520) downstream of TCR signaling in all the subsets together (Pan-subset DEGs; Figure 3Ci). These genes were better represented in the naive and CM cells (Figure 3Cii). Complementary to that, TCR signaling resulted is higher number of subset-specific differently expressed genes in the EM cells (Subset-specific DEGs; Figures 3Ci and 3Cii).
Thus, PD-1 signaling had a remarkable effect on modifying TCR regulated transcriptome; the number of the genes that were differentially regulated among the specific subsets was higher than the number of genes that were shared, and these numbers were higher for PD-L2 over PD-L1 (Figure 3Cii).
The effects of PD-1-regulated genes in the different T cell subsets are associated with specific cellular function
To better understand not just the extent of the number of the genes, but the functional meaning of the PD-1-regulated DEGs in the different T cell subsets, we performed GO enrichment analysis (Zhao et al., 2020) (Figure 4A). In terms of metabolism, it was mainly in the naive and CM cells where the PD-1-regulated genes were associated with amino acid metabolic process, ATP generation and proteoglycan biosynthetic process (Figure 4B). PD-1 signaling also regulated genes associated with cell cycle regulation, such as DNA replication and cell cycle phase transition in the naive and CM cells, but less in the EM cells (Figure 4C). Similar results in the naive and CM cells were observed in T cell activation and differentiation genes such as those related to TCR activation and CD4 T cell differentiation (Figure 4D) as well as within genes linked to the production of cytokines such as IL-4, 7, 10, and 12 (Figure 4E). Interestingly, it was the EM cells that dominated the genes associated with IL-8 and MCP-1 (Figure 4E). Within the same EM cells, PD-1 signaling also regulated genes controlling effector functions, such as cell adhesion and migration (Figure 4F), but not apoptosis (Figure 4G). Mixed responses among the subsets were observed in genes related to signaling (Figure 4H) and inflammatory responses (Figure 4I) in the context of PD-1.
Figure 4.

The effects of PD-1-regulated genes in the different T cell subsets are associated with specific cellular functions
(A) Venn diagram focusing on the PD-1 regulated gene group.
(B–I) Heatmaps showing the relative enrichment of genes that constituted specific functional GO annotated groups.
Altogether, the abilities of PD-1 signaling to modify TCR responses are diverse in the different T cell subsets and are likely context dependent.
PD-1-induced genes are not shared between the different T cell subsets
The genes that were exclusively induced by PD-1 signaling and that were not shared with the TCR signaling (Figure 5A) offer the opportunity to identify PD-1 pathway that might have translational implications. We discovered 517 genes that were induced upon PD-L1 ligation in all the subsets, compared to 350 genes that were induced by PD-L2 in the same cells (Figure 5B). In contrast to the PD-1-regulated DEGs (Figure 3Bi), very little overlap was observed among the PD-1-induced DEGs within the different T cell subsets (Figure 5B). While in the case of the PD-1-regulated genes, PD-L2 governed PD-L1 in terms of the number of affected genes (Figure 3Bii), in the case of the PD-1-induced genes, it was actually PD-L1 that changed the expression levels of more genes, at least in the CM and EM cells (Figure 5C). In the naive cells, PD-1-induced DEGs were enriched with genes associated with T cell migration and chemotaxis (Figure 5D) and in the CM cell there was enrichment of genes connected with T helper responses and differentiation (Figure 5D). Interestingly, the PD-1-induced genes in the EM cells were related to IL-4 responses, calcium signaling, and JAK-STAT pathways (Figure 5D).
Figure 5.

PD-1-induced genes are not shared between the different T cell subsets
(A) Venn diagram focusing on the PD-1 induced genes group.
(B) Venn diagrams showing the number of the genes that were induced by either PD-L1 or PD-L2 across the three subsets of T cells.
(C) Venn diagram comparing the number of the genes that were induced between PD-L1 and PD-L2 in each of the T cell subsets.
(D) Heatmap showing the relative enrichment of the PD-1 induced genes and their allocation into selected GO functional groups.
PD-1 signaling changes the expression levels of genes associated with functional T cell subsets
To assess the effects of PD-1-ligation on different functional T cell subsets, we analyzed the expression levels of master transcription factors (TFs) of Th1, Th2, Th17, Th22, Tfh (follicular helper), and Treg (regulatory) lineages (Figure S2). In the naive cells, PD-1 ligation resulted in higher levels of BCL6, a master TF of Tfh cells (Figure 6A). Moreover, the levels of other genes associated with Tfh, such as MAF, CXCL13, CD84, and IL-4 were also elevated (Figure 6A). PD-1 failed to considerably increase the expression of any lineage specific TF in the CM cells (Figure 6B). However, in EM cells, PD-1 signaling induced higher levels of AHR and FOXP3, master TFs of Th22, and Treg cells, respectively. Other genes commonly associated with Th22 cells, such as BATF, IL22, IL13, and CCL7 were also upregulated in EM cells specifically downstream of PD-1 (Figure 6C). Conversely, the expression of several Treg-associated genes did not show a similar trend of upregulation following PD-1 ligation (Figures 6C and S2). Altogether, it is suggested that PD-1 signaling is unique not just in antigen experienced subsets but also in functional subsets such as Tfh and Th22.
Figure 6.

PD-1 signaling changes the expression levels of genes associated with functional T cell subsets
(A) Expression levels of genes associated with functional subset of T cells are shown in naive T cells.
(B) Expression levels of genes associated with functional subset of T cells are shown in CM cells.
(C) Expression levels of genes associated with functional subset of T cells are shown in EM cells. See also Figure S2.
PD-1 signaling-induced STAT1 and STAT2 upregulation correlates with clinical response to checkpoint blockade
To better understand the implication of the PD-1-induced genes among the different T cell subsets, we performed integrative enrichment analysis using ChEA3 (https://maayanlab.cloud/chea3), a ChIP-seq enrichment analysis tool made of published gene set libraries (Gheorghe et al., 2019; Keenan et al., 2019) (Figures 7A and 7B). These analyses predict that different subset of T cells up regulated similar, but no identical, set of TF secondary to PD-1 signaling. Intriguingly, the TF were not identical when the same PD-1 was ligated with either PD-L1 or PD-L2. From this predicative analysis we also learned that STAT1 and STAT2 were more prevalent across all three subsets of T cells, compared to the other TF. Furthermore, we validated the expression level of STAT1 and STAT2 using our own RNA sequencing data across all the subsets (Figure 7C). Not less interesting, and unlike the other TF, STAT1, and STAT2 levels increased not just upon TCR ligation, but also significantly further upon PD-L1 and PD-L2 treatment (Figure 7C).
Figure 7.

PD-1 signaling-induced STAT1 and STAT2 upregulation correlates with clinical response to checkpoint blockade
(A and B) Upregulated PD-1 induced genes were analyzed using ReMap project data (A) and Mean Rank score (B) to predict specific transcription factors across the different subset and the experiential conditions that might regulated key genes in the relevant groups.
(C) Heatmap showing the relative expression of key transcription factors through the different experimental conditions.
(D) Curated single cell data was used to calculate the expression of STAT1 and STAT2 among melanoma patients the either responded, or not to PD-1 blockade.
(E) Kaplan Myer plots showing 5-year survival of patient with lung adenocarcinoma in relation to STAT1 expression in the tumor tissue, and in different immune compartments. See also Figure S3.
We hypothesized that since STAT1 and STAT2 expression in T cells were upregulated upon intact PD-1 signaling, this would inversely correlate with clinical outcome and response to immunotherapy. This premise is supported by the notion that interference to PD-1 signaling is associated with improved T cell antitumor response, whether achieved by low expression of PD-1, altered downstream signaling, or ligand binding blockade with checkpoint inhibitors. To test this, we analyzed a publicly available single-cell RNA sequencing data set (GSE120575) of patients with melanoma that were treated with pembrolizumab, a PD-1-targeting antibody (Sade-Feldman et al., 2019). The CD4 T cells were identified and further segregated into those found in the patients that responded, or not, to anti-PD-1 antibodies (Figure 7Di). As shown, lower expression of both STAT1 and STAT2 were associated with better response to the checkpoint blockade (Figure 7Dii). Through a meta-analysis of samples included in The Cancer Genome Atlas, Gene Expression Omnibus, and European Genome–phenome Archive (EGA), we found that 5-year overall survival of patients with lung adenocarcinoma inversely correlates with STAT1 expression (Figure 7E). This was more significant when we restricted the analysis to tumor samples where the CD4 T cells were enriched (p value of 0.024). That is, some patients with lower expression of STAT1 in T cells demonstrated increased overall survival and this inverse correlation is consistent with an inhibitory role in T cells.
Discussion
We have observed distinct instances in which PD-1 ligation leads to inconsistency in inhibiting cell proliferation. We propose that these differences can be attributed to heterogeneity within the stimulated population, wherein a subpopulation of cells may be inhibited, while another is not. Whole population-based methods of detection will not appreciate these differences. By sorting naive, CM, and EM CD4 T cell subsets first, followed by PD-1 ligation and transcriptome analysis, we are able to associate PD-1 genetic signatures with specific T cell subsets. Our ability to identify T cell subsets that respond differentially to PD-1 ligation is a promising translational path for future studies.
Previously, we have shown that the stimulatory coreceptor CD28 can inhibit T cell adhesion (Strazza et al., 2015), while the coinhibitory receptor CTLA-4 positively stimulates selected T cell functions (Kloog and Mor, 2014). Interestingly, CD28 and CTLA-4 bind to the same ligands, CD80 and CD86, resulting in an opposite outcome; one which is likely to be context-dependent (Greenwald et al., 2005). This dichotomous signaling could be explained as a cell-intrinsic process, or as is the case for PD-1, by subset-specific seclusion of the same receptors. For example, CTLA-4 signaling inhibits many pathways in effector T cells, but the same CTLA-4 can clearly activate other pathways in regulatory T cells (Oyewole-Said et al., 2020). More recently, we have reported that PD-1 ligation has led to an increased phosphorylation at several tyrosine sites, some of which were clearly associated with an activation of cellular functions (Tocheva et al., 2020; Pedoeem et al., 2014). In this context, the observation of a population of T cells with enhanced proliferation following PD-1 ligation is reasonably predictable. In a more recent study, we found inconsistencies in follicular helper T cell proliferation and cytokine secretion, in the context of PD-1 ligation (Strazza et al., 2021). With our currently ongoing research, we have demonstrated PD-1’s ability to differently affect cell proliferation, maturation, and diverse transcript signatures generated by the same PD-1 in different subsets of cells. This has important implications, due to the essential role proliferation plays in T cell function and immune responses to tumors. Understanding the differences between the proliferating and nonproliferating populations holds promise for identifying unique PD-1 signaling mediators that may enhance current therapeutic targeting strategies.
The contribution of specific subsets of T cells in mounting an effective antitumor immune response is now better appreciated across the field (Mami-Chouaib et al., 2018; Tanaka and Sakaguchi, 2017). It is widely accepted that CD8 T cells play a central role in mediating antitumor immunity, by recognizing tumor-associated antigens presented on MHC-I through their expressed T cell receptors. However, it is also becoming increasingly clear that CD8 T cell-binding of neoantigens is insufficient for mounting a powerful antitumor response, and immunotherapy-induced antitumor responses require the added presence of CD4 T cells (Kennedy and Celis, 2008). Multiple studies have indicated that CD4 T cells mediate antitumor effects through mechanisms that vary according to tumor microenvironments (Ngiow and Young, 2020). In some settings, CD4 T cells became cytotoxic in the presence of IL-2, or in the absence of regulatory T cells (Zuazo et al., 2020). More recent studies have indicated that cytotoxic CD4 T cells reach up to 25% of all tumor-infiltrating lymphocytes, supporting an essential role which contributes toward better survival (Maibach et al., 2020). In accordance, our CD4 T cell transcriptomic data provide a better understanding of these cells in the context of PD-1 signaling and suggests that specific subsets of CD4 T cells may be promising targets for cancer immunotherapies.
Understanding the populations of T cells that respond differently to PD-1 ligation has implications beyond proliferation. Even the characterization of PD-1 expression as a marker of T cell exhaustion is complex, since PD-1 blockade could restore function in some “exhausted” T cells. It has so far been convenient to classify PD-1 as a T cell function inhibitor. However, the true signaling events underlying the functional consequences of PD-1 ligation are proving to be far more nuanced (Kamada et al., 2019; Xiong et al., 2018).
Naive T cells differentiate in the thymus, and subsequently undergo positive and negative selection. These cells are immature and, unlike effector cells, have not encountered their cognate antigen within the periphery. They are commonly found within the lymph nodes and in the peripheral circulation. Accordingly, and as demonstrated in our research, it was predicted that the pathways altered by PD-1 signaling were associated with T cell differentiation and activation. Peripheral EM cells express CD45RO, but do not express CCR7 and other lymph-node-homing receptors. Since their primary role is to migrate and expand at the sites of inflammation, it was fitting that PD-1 affects genes related to adhesion and migration. Likewise, CM T cells express CD45RO and CCR7 and are commonly found in the lymph nodes, where they communicate with other immune cells. It is not unexpected that PD-1 signaling in these cells regulates genes related to interferon and other cytokine signaling.
Our use of PD-1-ligands, rather than antibodies, to engage the PD-1 pathway is one of our work’s strengths. Through this approach, we were not only able to uncover a more physiological transcriptome but also to disclose differences between the two known ligands of PD-1, PD-L1, and PD-L2. While the former is ubiquitously expressed in inflamed tissues, the latter is restricted to antigen-presenting cells. PD-L2 binds to PD-1 with a 3-fold stronger affinity, as compared to PD-L1, but we have recently shown that this advantage is not translated to stronger effector functions (Philips et al., 2020). In the current work, we identify for the first time the different genes regulated by either PD-L1 or PD-L2. Even though both bind to the same receptor, their gene signatures are quite diverse. In all subsets, stimulation via PD-L2 led to more powerful inhibition of the TCR-regulated genes, an observation that, again, cannot be explained by differences in the affinity of these ligands (Philips et al., 2020). We were surprised to learn that very few PD-1-induced genes were shared between PD-L1 and PD-L2, suggesting the contribution of other factors to the downstream signaling of PD-1, such as PD-L1-CD80 (Sugiura et al., 2019) and PD-L2-RGMb interactions (Nie et al., 2018).
STAT1 is a protein involved in cellular responses to interferon signaling (Najjar and Fagard, 2010). Its role in cancer biology has been the subject of investigations for many years, and according to most published data, STAT1 supports tumor-suppressing pathways (Zhang and Liu, 2017). However, other studies indicate that STAT1 could enhance the growth of some tumors. Thus, the roles of STAT1 in tumor biology are complexed and require better understanding (Zhang and Liu, 2017). While most published data focus on STAT1 expression and functions in tumor cells, we were determined to uncover its role in T cells. Having different roles in tumor and immune cells could explain the inconsistences regarding its antitumor properties. The fact that STAT1 expression level in tumor tissue correlates with infiltrating CD4 T cells proportion (Figure S3) supports a possible contribution of STAT1 not only to the function of the tumor cells, but also to the T cell compartment. Under these conditions and in association with PD-1 signaling, lower levels of STAT1 were clearly associated with better survival and an improved response to checkpoint blockade. We would hypothesize that the inhibition of STAT1 with small molecules might sensitize anti-PD-1-resistant tumors, or even enhance the therapeutic effect of PD-1 blockade (Qu et al., 2017).
Limitations of the study
As with any study, this work is not without limitations, namely the low number of donors for RNA sequencing. We were able to confirm the expression of STAT1 through available clinical data sets, but a larger study that will prospectively document the level of key transcription factors in peripheral CD4 T cells is needed. This study focused on CD4 T cells, and an expansion into the CD8 population would enhance it. It must be noted that single-cell RNA sequencing would lend further power to this study, and is a promising direction for future studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ultra-LEAF™ Purified anti-human CD3 (clone UCHT1) | BioLegend | Cat#300465 |
| Ultra-LEAF™ Purified anti-human CD28 (clone CD28.2) | BioLegend | Cat#302934 |
| Ultra-LEAF™ Purified Mouse IgG1, κ Isotype Ctrl (clone MG1-45) | BioLegend | Cat#401407 |
| APC/Cyanine7 anti-human CD3 (clone HIT3A) | BioLegend | Cat#300318 |
| Alexa Fluor® 700 anti-human CD4 (clone RPA-T4) | BioLegend | Cat#300526 |
| PE/Cyanine7 anti-human CD8a (clone RPA-T8) | BioLegend | Cat#301012 |
| Brilliant Violet 605™ anti-human CD45RA (clone HI100) | BioLegend | Cat#304134 |
| Alexa Fluor® 488 anti-human CD197 (CCR7) (clone G043H7) | BioLegend | Cat#353206 |
| PE/Dazzle™ 594 anti-human CD197 (CCR7) (clone G043H7) | BioLegend | Cat#353236 |
| PerCP/Cyanine5.5 anti-human CD19 (clone HIB19) | BioLegend | Cat#302230 |
| Biological Samples | ||
| Whole blood of healthy donors | New York Blood Center | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human B7-H1 (PD-L1, CD274)-Fc Chimera (carrier-free) | BioLegend | Cat#762506 |
| Recombinant Human PD-L2 (B7-DC, CD273)-Fc Chimera (carrier-free) | BioLegend | Cat#772508 |
| Staphylococcus Enterotoxin E (SEE) | Toxin Technology | Cat#ET404 |
| Lymphoprep density gradient | STEMCELL Technologies | Cat#07851 |
| RosetteSep™ Human T Cell Enrichment Cocktail | STEMCELL Technologies | Cat#15061 |
| CFSE Cell Division Tracker Kit | BioLegend | Cat#423801 |
| CellTrace™ Far Red Cell Proliferation Kit | Thermo Fisher Scientific | Cat#C34572 |
| Zombie UV™ Fixable Viability Kit | BioLegend | Cat#423108 |
| CD8 MicroBeads, human | Miltenyi Biotec | Cat#130-045-201 |
| Naive CD4+ T Cell Isolation Kit II, human | Miltenyi Biotec | Cat#130-094-131 |
| Memory CD4+ T Cell Isolation Kit, human | Miltenyi Biotec | Cat#130-091-893 |
| Deposited Data | ||
| bulk RNAseq data | This manuscript | GEO: GSE174860 |
| Single-cell RNAseq data | https://doi.org/10.1016/j.cell.2018.10.038 | GEO: GSE120575 |
| Experimental Models: Cell Lines | ||
| Raji (ATCC® CCL-86™) | ATCC | ATCC® CCL-86™ |
| Recombinant DNA | ||
| pHR-PD-L1-mCherry | Hui et al.Science. 2017 Mar 31; 355(6332):1428-1433 | N/A |
| pHR-PD-L2-mCherry | Philips et al.J Biol Chem. 2020 Apr 3; 295(14): 4372–4380 | N/A |
| pHR-control | This manuscript | N/A |
| Software and Algorithms | ||
| bulk RNAseq data analysis | https://app.basepairtech.com/ | N/A |
| Gene ontology enrichment analysis | http://www.pantherdb.org | N/A |
| Transcription factor enrichment analysis | https://maayanlab.cloud/chea3 | N/A |
| Single cell RNAseq analysis | https://bioturing.com | N/A |
| Survival analysis | kmplot.com | N/A |
| Gene expression associations in TCGA | http://timer.cistrome.org | N/A |
| GraphPad Prism 9 | https://www.graphpad.com | N/A |
| FlowJo 10.1r7 | https://www.flowjo.com | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to Lead Contact, Adam Mor (am5121@cumc.columbia.edu).
Materials availability
The pHR-control vector generated in this study will be made available upon request.
Experimental model and subject details
Human subjects
Whole blood of healthy donors was obtained from the New York Blood Center. The study was approved by the Institutional Review Board at Columbia University Medical Center and all donors provided informed consent (AAAB3287). Due to procedural reasons related to the operation of the blood center, the age and gender identity of the donors are not reported.
Cell culture
Raji cells were grown in RPMI 1640 supplemented with 10% heat inactivated fetal bovine serum and 1% penicillin – streptomycin. Human primary T cells were grown in enriched media (RPMI 1640, 10% heat inactivated fetal bovine serum, 1% penicillin - streptomycin, 1X non-essential amino acids, and 1mM sodium pyruvate). All cells were kept at 37°C with 5% CO2.
Method details
General reagents
RPMI 1640 medium, Dulbecco’s PBS, and FBS were purchased from Life Technologies. Staphylococcus Enterotoxin E (SEE) was acquired from Toxin Technology.
Isolation of human CD8 T cells and plate bound anti-CD3 proliferation assay
48 well plates (Corning) were coated with either 1.5 μg/mL anti-CD3 (Biolegend) and 5 μg/mL Isotype control antibody, 1.5 μg/mL anti-CD3 and 5 μg/mL PD-L1 (Biolegend), or 1.5 μg/mL anti-CD3 and 5 μg/mL PD-L2 (Biolegend) for 16 hours at 4°C. Mononuclear cells were isolated from whole blood of healthy donors by Lymphoprep density gradient (Stemcell). CD8 T cells were then purified by CD8 Microbeads (Miltenyi). Prior to stimulation, 5 x 105 cells per condition were stained with 2 μM CellTrace Far Red proliferation dye (Thermo) for 20 minutes at 37°C protected from light. Excess proliferation dye was neutralized with warm media, the stained cells were washed in PBS, resuspended in enriched media, and then added to the appropriate stimulation wells at 0.5 mL per well. Cells were incubated for 4 days at 37°C with 5% CO2. On day 4 the cells were analyzed for proliferation dye intensity and surface protein expression by flow cytometry as described below.
Isolation of human CD3 T cells and co-culture with Raji B cells
CD3 T cells were isolated from whole blood of healthy donors by RosetteSep human T cell enrichment cocktail (Stemcell) and Lymphoprep density gradient (Stemcell). Isolated cells were allowed to rest in enriched media for 24 hours and then were stained with 1 μM of the proliferation dye carboxyfluorescein succinimidyl ester (CFSE; Biolegend) for 20 minutes at 37°C protected from light. Excess CFSE was neutralized with warm media, the stained cells were washed in PBS, and resuspended in enriched media at 1 x 106 cells/mL. Raji cells, stably expressing either pHR-control vector, pHR-PD-L1-mCherry vector, or pHR-PD-L2-mCherry vector, were irradiated (50 Gy) and pre-incubated for 30 minutes with 100pg/ml SEE. CD3 T cells and Raji cells were mixed in a 1:1 ratio (1 x 105 cells each) in 96 U-bottom shaped wells, in a final volume of 0.2 mL of enriched media. Each experimental condition was plated in several wells which were pooled together after 4 days of co-culture and analyzed for proliferation dye intensity and surface protein expression by flow cytometry as described below. pHR-control vector was generated by site-directed mutagenesis of pHR-PD-L2-mCherry vector.
RNA sequencing and data analysis
Mononuclear cells were isolated from whole blood of healthy donors by Lymphoprep density gradient (Stemcell). CD4 Naive, CM and EM T cell subsets were isolated by the appropriate microbeads kits (Miltenyi), following the manufacturer’s protocols. Isolated cells were allowed to rest in enriched media for 24 hours. The cells were then resuspended at 1 x 106 cells/mL of enriched media containing 1 μg/mL anti-CD28 (Biolegend) and plated on pre-coated 48 well plates as described earlier with either 5 μg/mL anti-CD3 and 5 μg/mL Isotype control antibody, 5 μg/mL anti-CD3 and 5 μg/mL PD-L1 (Biolegend), or 5 μg/mL anti-CD3 and 5 μg/mL PD-L2 (Biolegend) for 18 hours at 0.5 mL media per well.
Total RNA was isolated from the stimulated cells using RNeasy Mini Prep (Qiagen). Low input library preparation and sequencing was performed by Genewiz with mRNA enrichment, mRNA fragmentation, random priming, first and second strand cDNA synthesis, end repair, 5’ phosphorylation, dA-tailing, adaptor ligation, PCR enrichment, and paired-end sequencing using Illumina HiSeq 3000, PE 2x150. The RNA-Seq data was analyzed using Basepair software (https://www.basepairtech.com) with a pipeline that included the following steps: Reads were aligned to the transcriptome derived from UCSC genome assembly (hg19) using STAR with default parameters. Read counts for each transcript was measured using feature Counts. Differentially expressed genes (DEGs) were determined using DESeq2. A cut-off of Log2FC ≥ 2 and adjusted p-value (corrected for multiple hypotheses testing) ≤ 0.05 was used for defining creating lists and heatmaps. Significant DEGs are included in heatmaps used to depict Z-scores for each gene (row). Gene ontology enrichment analysis was performed via Panther web services (http://www.pantherdb.org). Transcription factor enrichment analysis was generated using on-line tool kit (https://maayanlab.cloud/chea3) using the ReMap project data (Figure 7A) and the Mean Rank score (Figure 7B). Single cell RNA sequencing data (GEO: GSE120575) was queried through Bio Turing server (https://bioturing.com).
Flow cytometry
For protein expression analysis following proliferation, CD8 or CD4 T cells were isolated and stimulated as described above. On day 4 of the proliferation assay cells were collected and stained with the following antibodies for surface protein expression: CD3 APC-Cy7 (Biolegend), CD4 AF-700 (Biolegend), CD8 PE-Cy7 (Biolegend), CD45RA BV-605 (Biolegend), CCR7 AF-488 (Biolegend) or CCR7 PE-Dazzle594 (Biolegend), and CD19 PerCP-Cy5.5 (Biolegend). Dead cells were excluded from the analysis by using Zombie-UV (Biolegend). Doublets and double positive CD4/CD8 cells were removed through sequential gating. Flow cytometry acquisition was done using the BD LSRII with BD FACSDiva and Cyteck Aurora. Data was analyzed by FlowJo 10.1r7 and GraphPad Prism 9.
Survival analysis
Kaplan Myer plots showing five-year overall survival of patients were generated with KM-plotter (kmplot.com) using data from The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), and European Genome-phenome Archive (EGA). We compared the survival of patients in the upper vs. lower trident of STAT1 expression, with or without enrichment of different immune cell types.
Quantification and statistical analysis
Graphs depict mean ± SD. Statistical analyses for graphs were performed using ANOVA on GraphPad Prism 9. ∗ p<0.05, ∗∗ p<0.01, ∗∗∗ p<0.001, ns; not significant.
Acknowledgments
We would like to thank Marianne Strazza of Columbia University for productive discussions. This work was supported by grants from the NIH (AI125640, CA231277, AI150597), Cancer Research Institute, and Lisa M. Baker Autoimmunity Innovation Fund. Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, NIH under awards S10RR027050 and S10OD020056.
Author contributions
Conceptualization, S.L., A.S.T, A.M.; Methodology, S.L., K.A., S.M.B., A.M.; Formal Analysis, S.L.; Investigation, S.L., A.S.T.; Writing, A.M., S.L; Funding acquisition, A.M.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103020.
Supplemental information
Data and code availability
-
•
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table. Flow cytometry data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Gheorghe M., Sandve G.K., Khan A., Chèneby J., Ballester B., Mathelier A. A map of direct TF-DNA interactions in the human genome. Nucleic Acids Res. 2019;47:e21. doi: 10.1093/nar/gky1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald R.J., Freeman G.J., Sharpe A.H. The B7 family revisited. Annu. Rev. Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- Hui E., Cheung J., Zhu J., Su X., Taylor M.J., Wallweber H.A., Sasmal D.K., Huang J., Kim J.M., Mellman I., Vale R.D. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada T., Togashi Y., Tay C., Ha D., Sasaki A., Nakamura Y., Sato E., Fukuoka S., Tada Y., Tanaka A. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. 2019;116:9999–10008. doi: 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan A.B., Torre D., Lachmann A., Leong A.K., Wojciechowicz M.L., Utti V., Jagodnik K.M., Kropiwnicki E., Wang Z., Ma'ayan A. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019;47:W212–W224. doi: 10.1093/nar/gkz446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy R., Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol. Rev. 2008;222:129–144. doi: 10.1111/j.1600-065X.2008.00616.x. [DOI] [PubMed] [Google Scholar]
- Kloog Y., Mor A. Cytotoxic-T-lymphocyte antigen 4 receptor signaling for lymphocyte adhesion is mediated by C3G and Rap1. Mol. Cell Biol. 2014;34:978–988. doi: 10.1128/MCB.01024-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos G.G., Izar B., Rizvi N.A. Beyond tumor PD-L1: emerging genomic biomarkers for checkpoint inhibitor immunotherapy. Am. Soc. Clin. Oncol. Educ. Book. 2020;40:1–11. doi: 10.1200/EDBK_289967. [DOI] [PubMed] [Google Scholar]
- Li B., Chan H.L., Chen P. Immune checkpoint inhibitors: basics and challenges. Curr. Med. Chem. 2019;26:3009–3025. doi: 10.2174/0929867324666170804143706. [DOI] [PubMed] [Google Scholar]
- Maibach F., Sadozai H., Seyed Jafari S.M., Hunger R.E., Schenk M. Tumor-infiltrating lymphocytes and their prognostic value in cutaneous melanoma. Front. Immunol. 2020;11:2105. doi: 10.3389/fimmu.2020.02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mami-Chouaib F., Blanc C., Corgnac S., Hans S., Malenica I., Granier C., Tihy I., Tartour E. Resident memory T cells, critical components in tumor immunology. J. Immunother. Cancer. 2018;6:87. doi: 10.1186/s40425-018-0399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaki J., Togashi Y., Sugawara T., Itami M., Yamauchi N., Yuda J., Sugano M., Ohara Y., Minami Y., Nakamae H. The critical role of CD4+ T cells in PD-1 blockade against MHC-II-expressing tumors such as classic Hodgkin lymphoma. Blood Adv. 2020;4:4069–4082. doi: 10.1182/bloodadvances.2020002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar I., Fagard R. STAT1 and pathogens, not a friendly relationship. Biochimie. 2010;92:425–444. doi: 10.1016/j.biochi.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngiow S.F., Young A. Re-education of the tumor microenvironment with targeted therapies and immunotherapies. Front. Immunol. 2020;11:1633. doi: 10.3389/fimmu.2020.01633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X., Chen W., Zhu Y., Huang B., Yu W., Wu Z., Guo S., Zhu Y., Luo L., Wang S., Chen L. B7-DC (PD-L2) costimulation of CD4(+) T-helper 1 response via RGMb. Cell. Mol. Immunol. 2018;15:888–897. doi: 10.1038/cmi.2017.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh D.Y., Kwek S.S., Raju S.S., Li T., McCarthy E., Chow E., Aran D., Ilano A., Pai C.S., Rancan C. Intratumoral CD4(+) T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell. 2020;181:1612–1625.e13. doi: 10.1016/j.cell.2020.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyewole-Said D., Konduri V., Vazquez-Perez J., Weldon S.A., Levitt J.M., Decker W.K. Beyond T-cells: functional characterization of CTLA-4 expression in immune and non-immune cell types. Front. Immunol. 2020;11:608024. doi: 10.3389/fimmu.2020.608024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsoukis N., Wang Q., Strauss L., Boussiotis V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020;6:eabd2712. doi: 10.1126/sciadv.abd2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedoeem A., Azoulay-Alfaguter I., Strazza M., Silverman G.J., Mor A. Programmed death-1 pathway in cancer and autoimmunity. Clin. Immunol. 2014;153:145–152. doi: 10.1016/j.clim.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Philips E.A., Garcia-España A., Tocheva A.S., Ahearn I.M., Adam K.R., Pan R., Mor A., Kong X.P. The structural features that distinguish PD-L2 from PD-L1 emerged in placental mammals. J. Biol. Chem. 2020;295:4372–4380. doi: 10.1074/jbc.AC119.011747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow M.A., Sidlow R., Hellmann M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018;378:158–168. doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- Qu S., Guo Y., Huang S.T., Zhu X.D. Inhibition of STAT1 sensitizes radioresistant nasopharyngeal carcinoma cell line CNE-2R to radiotherapy. Oncotarget. 2017;9:8303–8310. doi: 10.18632/oncotarget.19690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009;229:114–125. doi: 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M., Yizhak K., Bjorgaard S.L., Ray J.P., de Boer C.G., Jenkins R.W., Lieb D.J., Chen J.H., Frederick D.T., Barzily-Rokni M. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2019;176:404. doi: 10.1016/j.cell.2018.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitara K., Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Ann. N.Y. Acad. Sci. 2018;1417:104–115. doi: 10.1111/nyas.13625. [DOI] [PubMed] [Google Scholar]
- Strazza M., Azoulay-Alfaguter I., Dun B., Baquero-Buitrago J., Mor A. CD28 inhibits T cell adhesion by recruiting CAPRI to the plasma membrane. J. Immunol. 2015;194:2871–2877. doi: 10.4049/jimmunol.1401492. [DOI] [PubMed] [Google Scholar]
- Strazza M., Bukhari S.M., Tocheva A.S., Mor A. PD-1 induced proliferating T cells exhibit a distinct transcriptional signature. Immunology. 2021 doi: 10.1111/imm.13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura D., Maruhashi T., Okazaki I.M., Shimizu K., Maeda T.K., Takemoto T., Okazaki T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science. 2019;364:558–566. doi: 10.1126/science.aav7062. [DOI] [PubMed] [Google Scholar]
- Tanaka A., Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–118. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay C., Qian Y., Sakaguchi S. Hyper-progressive disease: the potential role and consequences of T-regulatory cells foiling anti-PD-1 cancer immunotherapy. Cancers (Basel) 2020;13:48. doi: 10.3390/cancers13010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen D.S., Koelzer V.H., Herzig P., Roller A., Trefny M., Dimeloe S., Kiialainen A., Hanhart J., Schill C., Hess C. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018;24:994–1004. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocheva A.S., Peled M., Strazza M., Adam K.R., Lerrer S., Nayak S., Azoulay-Alfaguter I., Foster C.J.R., Philips E.A., Neel B.G. Quantitative phosphoproteomic analysis reveals involvement of PD-1 in multiple T cell functions. J. Biol. Chem. 2020;295:18036–18050. doi: 10.1074/jbc.RA120.014745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong D., Wang Y., Singavi A.K., Mackinnon A.C., George B., You M. Immunogenomic landscape contributes to hyperprogressive disease after anti-PD-1 immunotherapy for cancer. iScience. 2018;9:258–277. doi: 10.1016/j.isci.2018.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu Z. STAT1 in cancer: friend or foe? Discov. Med. 2017;24:19–29. [PubMed] [Google Scholar]
- Zhao Y., Wang J., Chen J., Zhang X., Guo M., Yu G. A literature review of gene function prediction by modeling gene ontology. Front. Genet. 2020;11:400. doi: 10.3389/fgene.2020.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuazo M., Arasanz H., Bocanegra A., Fernandez G., Chocarro L., Vera R., Kochan G., Escors D. Systemic CD4 immunity as a key contributor to PD-L1/PD-1 blockade immunotherapy efficacy. Front. Immunol. 2020;11:586907. doi: 10.3389/fimmu.2020.586907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table. Flow cytometry data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.