

Abstract
In eukaryotes, mRNA is modified by the addition of the 7‐methylguanosine (m7G) 5′ cap to protect mRNA from premature degradation, thereby enhancing translation and enabling differentiation between self (endogenous) and non‐self RNAs (e. g., viral ones). Viruses often develop their own mRNA capping pathways to augment the expression of their proteins and escape host innate immune response. Insights into this capping system may provide new ideas for therapeutic interventions and facilitate drug discovery, e. g., against viruses that cause pandemic outbreaks, such as beta‐coronaviruses SARS‐CoV (2002), MARS‐CoV (2012), and the most recent SARS‐CoV‐2. Thus, proper methods for the screening of large compound libraries are required to identify lead structures that could serve as a basis for rational antiviral drug design. This review summarizes the methods that allow the monitoring of the activity and inhibition of enzymes involved in mRNA capping.
Keywords: antiviral agents, high-throughput screening, methyltransferases, mRNA, viral capping
To escape recognition by the host innate immune system, viruses often incorporate 5′ caps into their mRNAs. The differences in mRNA capping make this process an interesting therapeutic target. Here, we review methods to explore the enzymes involved in cap biosynthesis. Such methods allow inhibitor selection, facilitating antiviral drug discovery, which is especially important considering the ongoing COVID‐19 pandemic and possible future outbreaks.

1. Introduction
Eukaryotic mRNA is a complex molecule that consists of various nucleotide regions. The central part of the mRNA sequence is a protein‐coding fragment that is located between the 5′ and 3′ untranslated regions (UTR). The 3′ mRNA end is protected by the poly(A) tail that consists of multiple adenosine monophosphates that stabilizes the transcript and participate in translation. [1] A unique nucleotide structure called a cap is located on the 5′ mRNA end. It has a 7‐methylguanosine constituent linked by a 5′5′‐triphosphate bridge with the first nucleotide of the nascent transcript (Figure 1). Cap biosynthesis occurs co‐transcriptionally and involves three sequential enzymatic reactions mediated by RNA triphosphatase (TPase), guanylyltransferase (GTase), and N7‐guanine methyltransferase (N7‐MTase), ultimately forming the cap 0 structure, which is the dominant form in simple eukaryotic organisms, such as yeasts and plants. [2] Higher organisms, including humans, possess additional methylation of the ribose 2′‐O position of the first transcribed nucleotide by RNA 2′‐O‐methyltransferase (2′‐O‐MTase). Some mRNAs also carry 2′‐O methylation on the second transcribed nucleotide to form the cap 2 structure. The functions of these 2′‐O‐methylations remain unclear; however, they are speculated to protect mRNA from translational shutdown triggered by the innate immune response via the type I interferon signaling pathway. Owing to the binding specificities of human IFIT (Interferon Induced proteins with Tetratricopeptide repeats) proteins, the innate immune system can effectively distinguish between target triphosphate RNAs and cap 0‐carrying mRNAs (from exogenous sources) but not between endogenous cap 1 and cap 2 mRNAs. [3] However, some viruses hijack the capping system of the host or use their own RNA capping machinery to escape host immune response and augment the expression of viral proteins. Although they have different levels of methylation, the biological functions of caps 0, 1, and 2 are essentially a consequence of N7‐methylation of 5′ guanosine and the presence of a negatively charged oligophosphate chain. [4]
Figure 1.
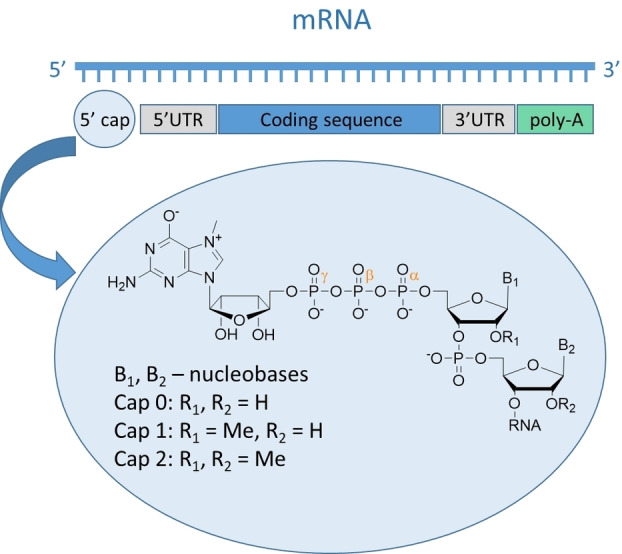
Structure of 5′ mRNA end (so‐called cap).
Here, we review the approaches used to study enzymes engaged in mRNA capping process allowing for identification of their inhibitors for the development of possible antiviral therapies. We focused both on standard methods and assays developed to specifically study selected enzyme, including SARS‐CoV‐2 proteins. This review is divided into sections allowing to understand the process of 5’ mRNA end capping and its therapeutic potential. First, we focused on the mechanism of canonical capping, characteristic for higher eukaryotes, including involved enzymes. Then, we moved to the possible variations of capping mechanism and discussing the role of capping enzymes in the suppression of viral replication. The main part of the review is dedicated to the methods presented in the scientific literature employed for the studies on specific enzymes. To depict the utility of these methods we presented selected inhibitor structures identified with their application.
The review is concluded by providing a personal perspective on the new challenges and potential development on the methods to study the process of mRNA capping.
2. Capping Machinery
In the following section we discuss how canonical capping is proceeding and what are the alternatives for this mechanism, that are utilized e. g., by viruses. The last section is focused on examples of capping enzymes inhibition being a target in antiviral therapies development.
2.1. Canonical Capping
Cap biosynthesis co‐transcriptionally occurs as a step in eukaryotic mRNA maturation. The first modification of RNA is the process of capping, which is transcribed by polymerase II in the nucleus, just after adding the first 25–30 nucleotides to the nascent transcript. [5] Canonical capping mechanism requires the activity of three enzymes (Figure 2A). First, RNA 5′‐triphosphatase (TPase) hydrolyzes 5’‐phosphate of RNA 5′‐triphosphate to release RNA 5′‐diphosphate. The mechanism of TPase action is metal‐dependent in lower eukaryotes, such as fungi, protozoa, [6] and viruses, [7] whereas metazoa, nematodes, and plants do not require metal ions (reviewed in Shuman 2002). [8]
Figure 2.

A) Canonical and B–E) non‐canonical mechanisms of mRNA 5′ cap biosynthesis. The pi and ppi symbols correspond to the phosphate and pyrophosphate, respectively.
The latter possesses a phosphate‐binding loop with the HCXXXXXR(S/T) motif (‘P‐loop’), which is a characteristic of the cysteine phosphatase superfamily. [9] The cysteine residue of the motif is responsible for nucleophilic attack on the ppp‐RNA γ‐phosphate to create a covalent cysteinyl‐S‐phosphate intermediate with simultaneous release of RNA 5′‐diphosphate. [10] The covalent product is subsequently hydrolyzed, and inorganic phosphate leaves the TPase active site.
The next step in cap biosynthesis is the transfer of the GMP molecule to the 5′ RNA end to form the Gppp‐RNA, which is catalyzed by the RNA guanylyltransferase (GTase) utilizing the GTP molecule. [11] The mechanism of catalysis involves the formation of a lysyl‐Nζ‐linked covalent intermediate with a GMP moiety. The lysine backbone in the active site of GTase (K294 in human GTase) carries out nucleophilic attack at the GTP α‐phosphate, which results in the breaking of the α‐β pyrophosphate bond to create a covalent intermediate. The structure of various GTases is flexible on the surface, but the GTP‐binding site, including linking lysine, its electropositivity, and surrounding residues are conserved. The motif KxDG(I/L) is present in almost all GTases. [11]
In the third step, guanosine is specifically methylated at the N7 position by the mRNA cap guanine‐N7‐methyltransferase (N7‐MTase) enzyme. N7‐MTase catalyzes the transfer of a methyl group from S‐adenosyl‐ʟ‐methionine (SAM) to Gppp‐RNA. A by‐product of this reaction is S‐adenosyl‐ʟ‐homocysteine (SAH), which acts as a feedback inhibitor of many methyltransferases. [12] The mechanism of N7‐MTase action relies on optimizing both the proximity and orientation of the substrates and the favorable electrostatic environment inside the binding pocket. [13] The pocket itself consists of two binding sites which is designed for Gppp‐RNA and SAM. Fabrega et al. showed that within the Ecm1 N7‐MTase (Encephalitozoon cuniculi mRNA cap (guanine N‐7) methyltransferase), SAM molecules are stabilized by van der Waals contacts with Tyr124 and Ile95, bidentate hydrogen bonds between 2′‐O and 3′‐O oxygens, and the carboxylate of Asp94. Moreover, additional interactions were observed with Asp122, Ser142, Gln140, Gly72, Lys54, and Asp78. [13] All these amino acids are conserved among different eukaryotic species. Nucleotide substrate is specifically recognized through hydrogen bonding interactions between guanine N1, N3, and N6 atoms and conserved amino‐acid side chains. The crystal structure of the Ecm1‐cap complex shows that guanosine nucleobase is stabilized by van der Waals interactions with hydrophobic amino‐acid side chains, rather than by π‐π stacking, which is a characteristic of cap‐recognizing proteins (i. e., eIF4E [14] ).
The successive reactions catalyzed by TPase, GTase, and N7‐MTase lead to the formation of mRNAs with cap 0 structure at its 5′ end. mRNAs of higher organisms are further methylated at the 2′‐O position of the first transcribed nucleotide by RNA 2′‐O‐methyltransferase (2′‐O‐MTase), which also utilizes the SAM molecule as a methyl group donor. Belanger et al. showed that human and Trypanosoma brucei 2′‐O‐MTases can methylate both mRNA ends with cap 0 and Gppp‐RNA. [15] Viral enzymes possessing 2′‐O‐MTase activity interact with nucleotide substrates in different ways, although the binding pocket is located in the same region of the protein structure. [16] In vaccinia virus VP39 protein, N7‐methylguanosine is oriented with its Hoogsteen bonding site towards the binding pocket floor, thereby requiring the presence of the m7G methyl group.[ 4 , 17 ] Chemically, ribose 2′‐O methylation differs from the N7‐methylation of guanosine as the former requires the deprotonation of the 2′ hydroxyl group to form a nucleophilic oxyanion capable attacking the SAM methyl center or hydrogen bond formation between the 2′‐OH proton and lysine side chain to freeze rotation. [18] Further 2′‐O‐methylation of the second transcribed nucleotide (cap 2) occurs in half of human mRNAs. The cap 2 is hypothesized to participate in the translation enhancement. [19] Some eukaryotes, such as Kinetoplastida, possess even more 2′‐O‐methylations to form caps 3 and 4. [20]
The organization of capping machinery differs among eukaryotic organisms. In metazoa, TPase and GTase are expressed as bifunctional capping enzymes (CE) with an N‐terminal TPase domain and a C‐terminal GTase domain. [21] Human mRNA cap guanine‐N7 methyltransferase (RNMT) is a nuclear protein consisting of a catalytic domain and an N‐terminal domain, which is not required for catalytic activity, but is responsible for binding an RNMT‐activating miniprotein (RAM). [22] The RAM subunit stabilizes the catalytically optimal structure of RNMT. [23] The process of mRNA capping is present in eukaryotes and viruses. The latter either exploits the host capping machinery or encode their own proteins, thus adapting various mRNA capping strategies.
2.2. Viral Capping Machinery
To mask their mRNAs from the host innate immune system and enhance protein synthesis, eukaryotic viruses incorporate a cap structure at the 5′ end. [24] For this purpose, adenoviruses and herpesviruses utilize host‐capping proteins.[ 8 , 25 ] Using capping mechanisms similar to that of canonical capping, other viruses have evolved proteins designed for 5′‐cap biosynthesis. However, many mechanistically distinct pathways have been developed. For instance, the alphaviral protein nsP1 (e. g., chikungunya virus[ 26 , 27 ] or Sindbis virus[ 28 , 29 ]) with N7‐MTase activity directly transfers methyl groups on GTP molecules to release m7GTP. [27] Subsequently, nsP1 GTase incorporates m7GMP molecules from m7GTP into pp‐RNA (generated after nsP2 (TPase)‐catalyzed hydrolysis of ppp‐RNA; Figure 2B). The protein nsP1 forms a covalent complex with the m7GMP molecule instead of GMP. [30] Alphaviral cap biosynthesis does not proceed further than the cap 0 structure.
Non‐segmented negative sense (NNS) RNA viruses, such as rhabdoviruses (e. g., rabies virus, [42] vesicular stomatitis virus[ 41 , 53 ]), transfer GDP molecules instead of GMP into the 5′ mRNA end during cap biosynthesis. The RNA‐dependent RNA polymerase L protein responsible for mRNA capping possesses the GDP polyribonucleotidyltransferase (PRNTase) domain, which reacts with the nascent ppp‐RNA, releasing pyrophosphate and covalent intermediate PRNTase‐pN‐RNA (Figure 2C). [54]
Some viruses directly acquire cap structures from host mRNAs via cap snatching. For example, the influenza virus encodes the RNA‐dependent RNA polymerase RdRp, which is composed of three proteins. One of them is the polymerase acidic protein (PA), which releases short‐capped RNA (12–15 nucleotides long) through its endonuclease activity. Afterwards, these short host mRNAs were used as primers by the RdRp enzyme to initiate viral transcription (Figure 2D). [55]
An even more straightforward approach is utilized by the L‐A and L‐BC double‐stranded RNA yeast totiviruses. The viral protein Gag (similar to GTase of canonical capping pathway) removes the m7GMP molecule from the 5′ end of the host mRNA, forming an intermediate covalent product, histydyl‐m7GMP (Gag‐m7GMP). Then, m7GMP was co‐transcriptionally transferred to viral RNA 5′‐diphosphate (Figure 2E). [56]
Coronaviruses (e. g., SARS‐CoV, MERS‐CoV, SARS‐CoV‐2), with one of the most complex and largest genomes, encode unique proteins to cap their mRNAs and presumably utilize a mechanism similar to that of alphaviruses or follow canonical capping pathways. [38] The function of TPase is performed by the nsp13 enzyme, which also acts as an RNA helicase. [57] It is not clear, which enzyme is responsible for GTase activity in RNA capping. However, the recent preprint of Walker et al. [58] indicate that nsp12 RNA polymerase of SARS‐CoV‐2 carries out addition of GTP to the 5′ RNA end. The cap 0 structure is formed upon specific N7‐methylation catalyzed by the nsp14 enzyme, [39] which is highly conserved among coronaviruses. The protein is able to transfer methyl groups on various nucleotide substrates – GTP, dGTP, GpppA, GpppG, and m7GpppG. [59] Moreover, the nsp14 also possesses 3′‐5′ exonuclease (ExoN) activity, thus acting as an RNA proofreading enzyme. [60] The two catalytic domains of the nsp14 – ExoN and N7‐MTase are located at the N‐ and C‐termini, respectively, and function separately; however, amino‐acid sequences 62–527 are required for both activities. The activity of the exonuclease of nsp14 was significantly enhanced upon binding with nsp10, while N7‐MTase activity was not affected. [61] The nascent cap 0 structure can be further methylated by SAM‐dependent nsp16 2′‐O‐methyltransferase, which requires the nsp10 cofactor for its activity. The complex nsp16/nsp10 utilizes the m7GpppA‐RNA substrate to synthesize the cap 1 structure m7GpppAm‐RNA. [62]
2.3. Capping Enzymes As Therapeutic Targets
Due to the reliance of some viruses on their own capping apparatus, the enzymes involved in 5′‐cap biosynthesis can be potential targets for the treatment of many viral infections. Differences in the capping mechanism and participating proteins between viruses and humans can contribute to the development of antiviral drugs. Hence, studies exploring RNA capping processes are crucially important, especially in viruses that cause pandemic outbreaks.
For instance, the non‐structural protein NS5 of flaviviruses, which are widely known for their pathogenic effects (Table 1), has been identified as potential drug target. [63] Compounds from the thioxothiazolidin family could interact with the NS5 GTP‐binding site, thus suppressing the replication of West Nile and yellow fever viruses. [64]
Table 1.
Various families of viruses expressing enzymes involved in RNA capping and the diseases they cause.
|
Virus |
Baltimore classification |
TPase |
GTase |
N7‐MTase |
2′‐O‐MTase |
Disease |
|---|---|---|---|---|---|---|
|
Alphaviruses |
(+)ssRNA |
nsP1 [30] |
nsP1 [32] |
none |
Chikungunya, Sindbis fever |
|
|
Flaviviruses |
(+)ssRNA |
NS3 [33] |
NS5 [34] |
NS5 [35] |
NS5 [36] |
Dengue fever, West Nile fever, Zika fever |
|
Coronaviruses |
(+)ssRNA |
nsp13 [37] |
not identified [38] |
nsp14 [39] |
nsp16/nsp10 [40] |
SARS, MERS, COVID‐19 |
|
Rabdoviruses |
(−)ssRNA |
L‐protein in RdRp [41] |
L‐protein in RdRp (PRNTase) [42] |
L‐protein in RdRp [43] |
L‐protein in RdRp [43] |
Rabies, Vesicular stomatitis |
|
Poxviruses |
dsDNA |
D1/D12 [44] |
D1/D12 [44] |
D1/D12 [45] |
VP39 [46] |
Smallpox, Cowpox |
|
Orbiviruses |
dsRNA |
VP4 [47] |
VP4 [47] |
VP4 [47] |
VP4 [47] |
Bluetongue |
|
Rotaviruses |
dsRNA |
not identified |
VP3 [48] |
VP3 [49] |
VP3 [49] |
gastrointestinal infections |
|
Reoviruses |
dsRNA |
λ1 [50] |
λ2 [51] |
λ2 [52] |
λ2 [52] |
mild respirator and gastrointestinal infections |
Alphaviral nsP1 multifunctional capping protein is an attractive target for antiviral therapies as it utilizes a different host capping mechanism, wherein the m7GMP molecule instead of GMP is transferred to the 5′ mRNA end. Delang et al. showed that some triazolopyrimidinones can suppress replication of the Venezuelan equine encephalitis virus (VEEV) and the Chikungunya virus (CHIKV) in cell culture via inhibiting nsP1 GTase activity. [27]
Another example of a potential antiviral target is the PA domain of the RNA‐dependent RNA polymerase (RdRp) possessing endonuclease activity, which is required to “snatch” the 5′ cap from the host mRNA. There are several examples of small‐molecule inhibitors of RdRp transcription that can also inhibit PA endonucleases.[ 65 , 66 , 67 ]
Non‐structural proteins engaged in the coronavirus capping machinery, namely nsp10, nsp13, nsp14, and nsp16, are specialized to catalyze only one stage of the cap biosynthesis process. Inhibition of any of these activities leads to the suppression of viral replication, making them potential therapeutic targets. [68] The exonuclease activity of nsp14 and 2′‐O‐MTase of nsp16 are positively regulated by the nsp10 subunit. Hence, the nsp10 binding site could also be aimed at suppressing CoV infection development. Wang et al. designed a TP29 peptide based on the sequence of the interaction interface of mouse hepatitis virus (MHV). [69] The peptide showed the ability to inhibit 2′‐O‐MTase activity of MHV in biochemical assays and to enhance IFN response in animal models.
Recently, Dunn et al. reported the association between the activity of human N7‐MTase RNMT‐RAM and carcinogenesis. They observed that a 50 % reduction in RNMT‐RAM cellular activity in breast cancer cell lines reduced cell proliferation and increased apoptosis, whereas the proliferation rate of non‐transformed breast epithelial cells was changed. [70] Hence, the inhibition of RNMT‐RAM activity is also an interesting target for anticancer therapy development.
Considering the significant therapeutic potential of proteins responsible for RNA capping, the discovery of new potent and selective inhibitors of this process is of great interest. Compounds that specifically inhibit crucial viral proteins could potentially be applied in antiviral therapy development. To identify new inhibitors of RNA capping, cost‐effective methods suitable for high‐throughput experiments are required. In the next chapters, currently available methods will be discussed.
3. Triphosphatase Assays
Hydrolysis of RNA triphosphate is the first step in RNA capping in many viruses that utilize various capping mechanisms (Table 1). First, an appropriate enzyme exhibiting TPase activity catalyzes the hydrolysis of pyrophosphate bonds between the β and γ phosphates to release the monophosphate and RNA diphosphate (pp‐RNA). The most common method to study TPase activity is the application of radiolabeled substrates – in this case γ‐32P‐labeled RNA triphosphate.[ 29 , 71 ] The substrate [γ‐32P]ppp‐RNA is then incubated with TPase in the presence of divalent ions (e. g., Mg2+). The reaction products (after different incubation times) are then separated by thin‐layer chromatography (TLC) and quantitatively analyzed using a scintillation counter (Figure 3A). Finally, TPase activity is expressed as the percentage of released phosphate with 32P.
Figure 3.
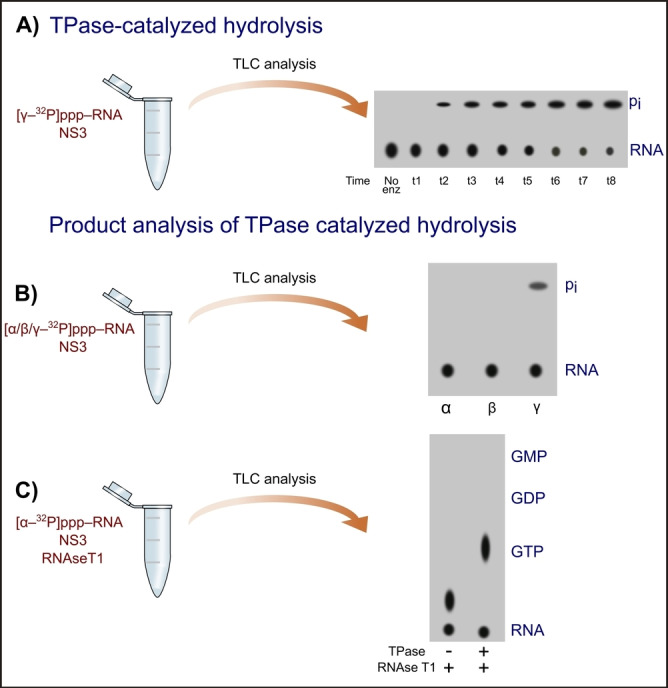
Schematic diagram of radioactive assays for TPase characterization. Illustrations were prepared using Inkscape software based on experimental results from Bartelma et al. 2002. [71] The pi symbol corresponds to the phosphate moiety. A) Monitoring of (NS3) TPase activity using TLC analysis to separate products and autoradiography to visualize products. B) TPase product analysis using α‐, β‐, or γ‐32P‐labelled RNA 5′ triphosphate; C) TPase product analysis using [α‐32P]‐labelled RNA 5′ triphosphate and RNAse T1.
The radioactive approach also allows the identification of the reaction products by comparing TLC analysis of RNA substrate labeled with 32P at different oligophosphate chain positions – α, β, or γ, after incubation with TPase. If the enzyme with the TPase activity can remove 32P from the RNA substrate labeled at the β position, it can cleave the α‐β pyrophosphate bond to release pyrophosphate. The presence of 32P signal from the eliminated phosphate (in the case of the [γ‐32P]ppp‐RNA substrate) indicates the successful cleavage of the β‐γ pyrophosphate bond, as well as monophosphate pi production. An experiment was carried out for flaviviral TPase NS3 (Figure 3B) and resulted in the removal of one phosphate group from the ppp‐RNA substrate. [71] The products of TPase‐catalyzed hydrolysis can also be identified by utilizing the 32P‐labeled RNA in the α position. In this case, the products of [α‐32P]ppp‐RNA incubation with TPase were treated with RNAse T1, which cleaves G located at the 5′ end of RNA. The enzyme hydrolyzes the phosphodiester bond between the 3′‐guanylic acid group and the 5′‐OH of the adjacent nucleotide (‐px‐Gp‐RNA) to produce ‐px‐Gp. [72] The reaction mixture was separated by TLC, with GTP, GDP, and GMP as the control samples (Figure 3C). Results revealed that the product of the two‐step reaction is pp‐Gp, which showed similar migration as GTP, indicating that NS3 catalyzes the cleavage of the β‐γ pyrophosphate bond. [71] Similar radiolabeled assays have been performed for nsP2 of the Sindbis virus, which also exhibited TPase activity as evidenced by the removal of monophosphate from RNA 5′‐triphosphate, [29] and Cet1p from S. cerevisiae. [73]
Radiolabeled RNAs are also utilized in analyses other than the TLC separation of reaction products. Xu et al. proposed the application of charcoal and its unique ability for the selective adsorption of TPase reaction products. [74] In this method, radiolabeled substrate, [γ‐32P]ppp‐RNA, was incubated with proper TPase. After reaction termination, charcoal suspension was added. Then, charcoal adsorbs radiolabeled substrates and RNA products, but not inorganic phosphates, generated upon hydrolysis. Hence, the reaction progress can be monitored by measuring the radioactivity of the supernatant remaining after charcoal centrifugation (Figure 4A). The authors of this work presented the utility of their method on yeast RNA 5′ triphosphatase Cet1p; however, presumably it can also be adapted for the characterization of viral TPases.
Figure 4.
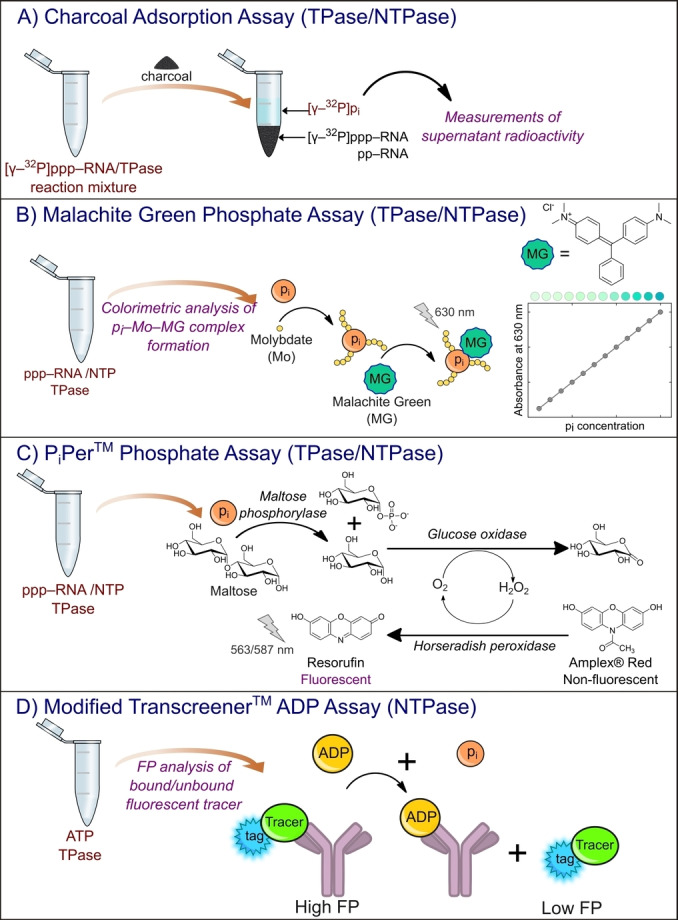
Selected assays used to study for TPase and NTPase activities. A) Charcoal adsorption assay designed for Cet1b triphosphatase and based on the radioactivity of enzymatically radiolabelled RNA interacting with the charcoal surface. [74] The pi symbol corresponds to the phosphate moiety. B) Malachite Green Phosphate assay for the detection of inorganic phosphate released during TPase‐catalyzed hydrolysis of 5′ RNA triphosphate. C) PiPer™ Phosphate assay for the quantification of inorganic phosphate released upon 5′ RNA triphosphate hydrolysis. [85] D) Modified Transcreener™ ADP Assay to determine NTPase activity based on the interactions between released ADP molecules and specific antibodies.
As a result of the action of many TPases, the monophosphate moiety is released. Hence, the designed methods for quantifying inorganic phosphate (Pi) can be potentially used to monitor the activity of specific TPases. One of the most widely employed assay designs for this purpose is the malachite green (MG) phosphate assay. The assay is a simple colorimetric method using absorbance readouts (630 nm) of the complex, in which phosphate (released during reaction) forms with molybdate and malachite green (Figure 4B). [75] The experiment was carried out in two steps. First, the reaction mixture was incubated with molybdate to form phosphomolybdate (pi‐Mo) complexes with nascent orthophosphates. Then, malachite green was added, and the final turquoise complex was created and quantified. The assay has already been used to study various phosphatases, such as nsP2 from chikungunya virus, [26] NS3 from bovine viral diarrhea virus, [76] cvRtp1 from chlorella virus, [77] NS3 from murine norovirus, [78] nsp13 from SARS‐CoV, [79] SARS‐CoV‐2, [80] and triphosphatase TbCet1 from Trypanosoma brucei parasites.[ 81 , 82 ] However, in some cases, for example, for hydrophobic amines such as papaverine and sildenafil (inhibitors of phosphodiesterase PDE), [83] signal interference appears due to the formation of amine‐phosphomolybdate aggregates that reduce the amount of phosphomolybdate accessible to MG. Feng et al. showed that if the reaction is initially incubated with MG, and then molybdate is added, there are no interferences resulting from the presence of hydrophobic amines. In this scenario, MG competes with the interfering amine and forms a complex with an absorbance peak at 630 nm, before aggregation of phosphomolybdate with that amine occurs. [84]
Many other assays designed for the quantification of inorganic phosphate are offered by manufacturers and could be potentially applied to study RNA triphosphatases. For instance, the Molecular Probes’ PiPer™ Phosphate Assay was designed for monophosphate spectrophotometric/fluorometric detection involving the utilization of maltose phosphorylase, which converts maltose to glucose 1‐phosphate and glucose in the presence of pi. The other enzyme, glucose oxidase, catalyzes the oxidation of glucose to gluconolactone and produces H2O2. Then, the nascent H2O2 was detected by oxidation of 10‐acetyl‐2,7‐dihydroxyphenoxazine (Amplex® Red) to resorufin, with absorption and emission maxima at approximately 563 nm and 587 nm, respectively (Figure 4C). [85] Thus, the increase in absorption or fluorescence is proportional to the amount of pi, created as a result of TPase action.
Unlike their mammalian counterparts, viral RNA triphosphatases often possess nucleoside‐triphosphatase activity (NTPase) within the same active center.[ 7 , 26 , 86 , 87 ] Such an activity can be studied in a manner similar to that previously described for TPase, using radiolabeled substrate [γ‐32P]NTP hydrolysis together with product analysis by TLC.[ 7 , 87 ] Moreover, NTPase activity can be also exploited using another class of assays employing fluorescence polarization measurements. An example is the Transcreener™ assay technology developed by BellBrook Labs (Madison, WI), [88] based on the detection of ADP molecules produced as a result of ATP hydrolysis. The method relies on a decrease in fluorescence polarization of fluorescently labeled tracer bound to anti‐ADP antibody upon competitive binding with nascent ADP (Figure 4D). This methodology has been adapted to study RNA triphosphatase TbCet1 from Trypanosoma brucei parasite, [81] and NS3 from hepatitis C virus. [89]
The active site of TPases can also be studied with binding assays, which allows the determination of physical parameters of RNA‐protein interactions. For this purpose, a decrease in intrinsic protein fluorescence upon binding is often exploited.[ 90 , 91 ] Protein at a constant concentration is titrated with RNA in the absence of metal ions, together with fluorescence intensity readouts at approximately 338 nm (excitation 290 nm). The NS3 TPase of West Nile virus possesses five tryptophanes, and approximately 37 % of its emission is accessible to the quencher RNA substrate. [92] The dissociation constant (K D) of the 30‐nt‐long RNA‐NS3 complex determined with time‐synchronized fluorescence quenching titration (ts‐FQT) experiment was 6 μM. A similar value was obtained with electrophoretic mobility shift assay (EMSA), a method based on different migration of radiolabeled RNA in bound and unbound states in polyacrylamide gel in the presence of an electric field. The authors also used circular dichroism spectroscopy (CD) and thermal unfolding experiments to characterize the stability of West Nile virus NS3 TPase upon RNA substrate binding. The same methodology (fluorescence titration, CD, and equilibrium unfolding experiments) has also been applied to study RNA binding with S. cerevisiae TPase Cet1. [91] Studies on TPase‐ligand binding have also been performed using fluorescence‐based thermal stability shift assays. [93] This method is based on fluorescent dye binding with the hydrophobic site of the protein, which emits fluorescence, while protein unfolding is caused by thermal shift.
4. Guanylyltransferase Assays
The second stage of the 5′ RNA cap biosynthesis is catalyzed by an enzyme with guanylyltransferase activity. Its function is to transfer the guanosine nucleotide moiety to the 5′ diphosphate (canonical capping, Alphaviridae‐like non‐canonical capping) or monophosphate (GDP RNA non‐canonical capping) of TPase‐processed RNA to form GpppX‐RNA or m7GpppX‐RNA (cap 0). RNA guanylyltransferase reaction involves a two‐step mechanism: (i) GTase reacts with GTP or m7GTP to form a covalent intermediate with GMP, m7GMP, or GDP moiety (Figure 2) with simultaneous release of pi or pp, and (ii) transfer of the moiety on 5′ RNA di‐ or monophosphate. Both reaction steps are reversible and require divalent cations. [94]
One of the most widely used approaches to study GTase activity is radioactive labeling of the substrate. GTP radiolabeled with 32P in the α position ([α‐32P]GTP) incubated with proper GTase forms [α‐32P]GpE (E – enzyme) covalent complex, which can be detected by different migration in gel using SDS‐PAGE and autoradiography visualization method (Figure 5A). The percentage of formed [α‐32P]GpE complexes corresponded to the GTase activity. There are many examples of studies on various GTases using radioactive assays. Soulière et al. used [α‐32P]GTP for kinetic and thermodynamic studies on RNA GTase from Paramecium bursaria Chlorella virus 1. [95] To determine the physical parameters of the guanylyltransferase reaction (e. g., Michaelis‐Menten constant, and association and dissociation rate constants), the authors separately studied two steps of the GTase‐catalyzed reaction. The formation of the GpE intermediate was examined as described above using [α‐32P]GTP substrate and SDS‐PAGE electrophoretic analysis. The nascent pyrophosphate is known for its inhibition of GTase activity; hence, pyrophosphatase has to be added to the reaction mixture. [96] For the second reaction step, the radiolabeled [32P] GpE complex was isolated and incubated with 81 nt RNA 5′ diphosphate. The reaction products were then analyzed by electrophoretic separation on polyacrylamide gel and autoradiography visualization (Figure 5A). These radioactive assays for monitoring GpE complex formation and GMP transfer to pp‐RNA have also been performed for NS5 protein with GTase activity from flaviviruses such as West Nile virus, dengue virus, and yellow fever virus in another publication of Bisaillon group (Issur et al.). [34] Another example is the application of [α‐32P]GTP to monitor the LEF‐4 subunit of RNA polymerase with GTase activity from Autographa californica baculovirus [97] and nsP1 GTase of VEEV. [98] If the viral capping machinery follows the GDP RNA capping pathway instead of 32P‐labelled GTP, guanosine diphosphate is used. [α‐32P]GDP and pppAACAG oligo‐RNA have been used for GTase activity studies of the L protein of vesicular stomatitis virus. [41]
Figure 5.
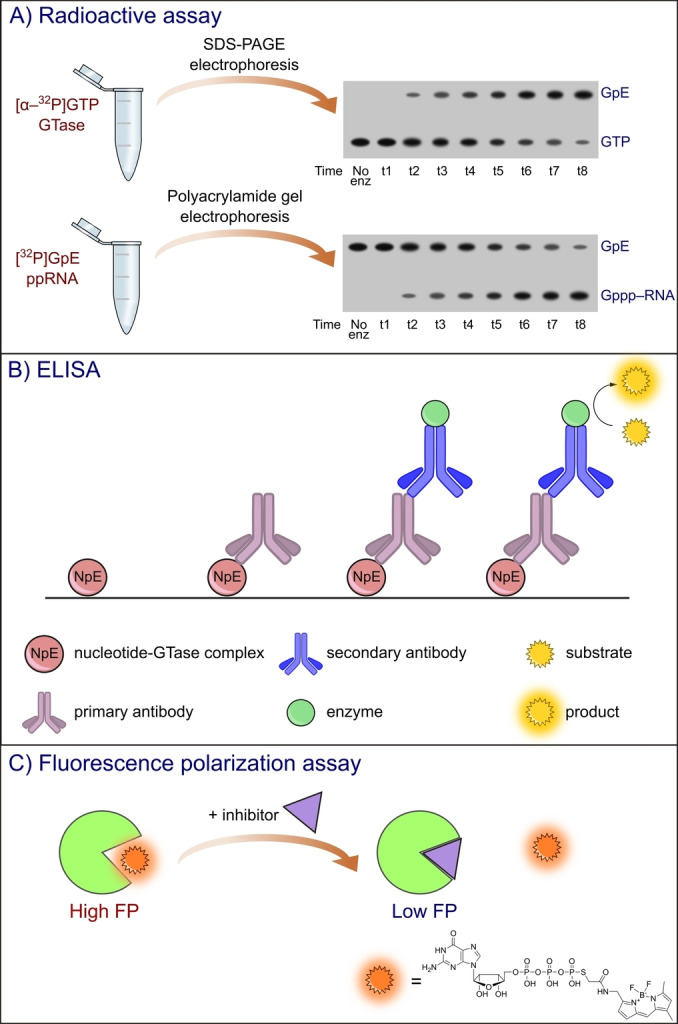
Schematic representation of selected assays for GTase activity. A) Radioactive assay based on two steps of guanylyltransferase reaction: 1) formation of GpE covalent complex and 2) GMP transfer to 5′ RNA diphosphate. Illustration was prepared using Inkscape software based on the experimental results of Soulière et al. 2008. B) ELISA assay for GTase characterization. C) Fluorescence polarization assay based on BODIPY‐labelled GTP.
Although it is still unclear which enzyme is responsible for GTase activity in Coronaviridae, in the recent preprint of Walker et al. 2021 [58] identified nsp12 RNA polymerase of SARS‐CoV‐2 to carry out GTP addition to the 5′ end of viral RNA in vitro. The reaction was catalyzed by the nidovirus RdRP‐associated nucleotidyltransferase (NiRAN) domain of the nsp12 enzyme. The products of nsp12‐catalyzed guanylylation in a mixture of [α‐32P]GTP and diphosphorylated 20 nt RNA were analyzed using denaturing PAGE and autoradiography. Similar findings about the nsp12 NiRAN domain with GTase activity and potential involvement in RNA capping have been presented by Lehmann et al. (2015). [99]
A radioactive assay for GTase activity monitoring can also utilize tritium‐labeled nucleotide substrates. Martin et al. [100] reported the application of [8‐3H]GTP for the characterization of a Vaccinia virus multifunctional capping protein. The authors used RNA isolated from the virus [100] or synthetic 5′ diphosphate poly(A) as a substrate. [101] To determine the amount of incorporated GMP, the reaction mixture was filtered through DEAE‐cellulose filters or precipitated using trichloroacetic acid and collected onto nitrocellulose filters. Unreacted GTP molecules were removed by washing, and the filters were dried and counted in toluene‐based scintillation fluid. [102]
Sample visualization via radiolabeling can sometimes be problematic. This requires special equipment to work with radioactive substances. Geiss et al. [103] proposed a different approach for studies on GTases based on the application of fluorescently labeled GTP (GTP‐ATTO‐680) for efficient visualization of guanylylation reaction products separated by SDS‐PAGE electrophoresis. The GTP molecule was labeled at the C8‐guanosine position; hence, the adduct GpE also possessed fluorescence properties. The authors applied this method for characterization of the NS5 enzyme from dengue virus. This method has also been applied to studies on nsP1 GTase from Chikungunya virus [104] and for inhibitor screening of flaviviral NS5 enzyme. [64]
The products of 5′ RNA guanylylation could also be detected by the application of specific antibodies. Hence, another popular methodology for kinetic studies of the second step of cap biosynthesis is the enzyme‐linked immunosorbent assay (ELISA). [105] Immunoassays rely on antigen detection using antibodies with highly specific antibody‐antigen interactions. In ELISA, antigens can be immobilized directly on a solid surface (direct and indirect ELISA) or bound to a specific capture antibody coated on the surface. Then, the primary antibody specifically binding to the antigen was added, followed by the addition of the secondary antibody conjugated with the enzyme that catalyzes the reaction of colorful product formation (Figure 5B). The advantage of ELISA over radioactive assay is the possibility of performing the experiment in a high‐throughput screening (HTS) format in a multi‐well plate (which also provides a solid surface). ELISA has been previously used for studies on nsP1 of Venezuelan equine encephalitis alphavirus (VEEV).[ 27 , 106 ] VEEV follows the m7GTP RNA capping pathway (characteristic of Alphaviridae) in which nsP1 catalyzes GTP N7‐methylation (N7‐MTase) and then forms a covalent adduct with m7GMP. To monitor only GTase activity, the N7‐MTase inhibitor SAH was added to the reaction mixture, while m7GTP was utilized as a reaction substrate. m7GMP‐nsP1 was then detected on a plate by the addition of anti‐m3G/anti‐m7G antibody and secondary antibody coupled with peroxidase. Another example of viral GTase characterized by ELISA with anti‐m3G/anti‐m7G antibody is the nsP1 enzyme from Chikungunya virus.[ 27 , 107 ]
As mentioned before, the disadvantage of radioactive assays is the requirement for product separation; hence, they are not adaptable to the HTS format (although they provide invaluable insight into the function of proteins involved in cap biosynthesis). However, in order to identify potent inhibitors of the capping process, fast and efficient screening methods are required. Fluorescence techniques are one of the simplest approaches for evaluating protein‐ligand interactions They involve measurements of the fluorescence intensity (FLINT), fluorescence polarization/anisotropy (FP/FA), time‐resolved fluorescence (TRF), and microscale thermophoresis (MST). [108] The development of such fluorescence assays is often based on molecular probes containing a part that specifically interacts with the protein and the other is responsible for the generation of a measurable analytical signal. Geiss et al.[ 103 , 109 ] designed a fluorescence polarization assay that allowed the characterization of the GTP‐binding site of NS5 capping enzymes from dengue virus, West Nile virus, and yellow fever virus, possessing active sites for both GTase and N7‐MTase (Table 1). [110] In this work, GTP fluorescently labeled with BODIPY dye via a terminal thiophosphate group has been exploited as a molecular probe with a K D value of 126±15 nM. The probe in the bound state had high FP values, which decreased as a result of competition with a potential inhibitor (Figure 5C). There are examples of applications of this FP assay for studies on nsP1 GTase from Chikungunya virus in order to find new small‐molecule inhibitors.[ 104 , 111 ]
Intrinsic GTase fluorescence originating from protein tryptophanes and tyrosines provides measurable analytical signals that can be used for determination of ligand binding affinity. Titration of the protein solution with the ligand caused a decrease in the protein fluorescence intensity. Soulière et al. used this approach to determine the dissociation constant of A103R GTase from Paramecium bursaria Chlorella virus 1. [95] The protein requires divalent cations for catalytic activity, but not for nucleotide binding. Hence, in order to study purely binding, divalent ions were not used in the assay.
5. Methyltransferase Assays
The structure of cap 0 (m7GpppX‐RNA) is eventually formed as a result of specific N7‐methylation of guanosine in GpppX‐RNA or GTP (Alphavirus non‐canonical capping) substrate. In order to avoid the innate immune system, many viruses proceed to the nascent transcript even further by 2′‐O methylation of the first transcribed nucleoside to obtain cap 1 structure (m7GpppXm‐RNA). Both methylation reactions require a methyl group donor, which is S‐adenosyl‐ʟ‐methionine (SAM) and a proper nucleotide substrate. Hence, MTase assays can exploit molecular probes based on the structure of either SAM co‐substrates or nucleotide substrates. SAM‐based assays should be applicable for both N7‐MTase and 2′‐O‐MTase, as the transfer of the methyl group in both cases involves nucleophilic substitution of the N7‐ or 2′‐O‐positions, respectively.
Similar to TPase and GTase studies, the primary method for monitoring N7‐MTase or 2′‐O‐MTase activity is substrate radiolabeling, either of SAM methyl group with tritium ([methyl‐3H]‐SAM) or nucleotide/RNA with phosphorus 32. While the second approach can be used only for MTases accepting 5′‐capped RNA fragments/nucleotides, the first one is a versatile tool for characterization of all SAM‐utilizing methyltransferase families. The reaction was carried out between [methyl‐3H]‐SAM and short GpppX‐RNAs. Quantitative analysis of the transferred [3H]CH3 groups was then carried out using TLC product separation followed by radioactivity counting. By using radiolabeled RNAs/nucleotides, it was possible to observe changes that appeared within the nucleotide substrate structure. Radiolabeled RNA was prepared by capping the RNA template with [32P]‐GTP. After purification, RNA was digested with nuclease P1 and alkaline phosphatase, and the products were separated on polyethyleneimine cellulose TLC plates. An example of a radioactive assay application is the characterization of the NS5 enzyme of dengue virus, possessing both N7‐MTase and 2′‐O‐MTase activity. The scheme of the TLC plate after separation of the reaction mixture of [32P]GpppA and NS5 is presented in Figure 6A. In the first step, the m7GpppA (cap 0) signal appeared as a result of substrate N7‐methylation. Then, m7GpppA is further methylated at the adenosine 2′‐O‐position to form the main product m7GpppAm (cap 1), which leads to the disappearance of the m7GpppA signal.[ 35 , 117 ]
Figure 6.

Assays used to determine MTase activity. A) Schematic representation of radioactive assays used to monitor N7‐MTase and 2′‐O‐MTase activities. Illustration was prepared using Inkscape software based on the experimental results of Chung et al. 2010. [112] B) Fluorescence polarization immunoassay (FPIA) based on the competitive binding of nascent SAH molecule and fluorescently labelled SAH probe to specific anti‐SAH antibodies. [113] C) Fluorescence polarization binding assay based on FAM‐labeled short capped RNA for ligand interaction with nsp16 2′‐O‐MTase from SARS‐CoV‐2 in complex with the nsp10 subunit. [114] D) SAM‐fluoro: SAM methyltransferase assay (G‐Biosciences®) for the quantification of SAH molecules released following methylation. [107] E) Time‐resolved Förster resonance energy transfer (TR‐FRET) assay based on competitive binding of nascent SAH molecule and fluorescently labelled SAH‐d2 probe to the specific anti‐SAH antibody, fluorescently labelled with L4‐Tb forming FRET pair with d2 dye. [115] F) Yeast‐based assay designed for N7‐MTase activity monitoring in‐vivo. [116]
Radioactivity measurements are also the basis of a filter binding assay, in which methylation reaction products are separated with DEAE‐cellulose filters. After separation, the filter was dried, and the radioactivity was measured by liquid scintillation counting. This approach has been used for studies on nsP1 N7‐MTase of Venezuelan equine encephalitis virus, [106] nsP1 N7‐MTase of Chikungunya virus, [27] nsp16/nsp10 2′‐O‐MTase of Middle East Respiratory Syndrome (MERS) Coronavirus, [118] dengue virus NS5 (N7‐MTase and 2′‐O‐MTase), [119] and human FTSJ3 rRNA 2′‐O MTase recruited by human immunodeficiency virus (HIV). [120] Product separation can also be performed by electrophoresis in a gel mobility shift assay.
Another example of the application of radioactivity counting is the scintillation proximity assay (SPA), which is widely used for sensitive biochemical assays and adaptable to HTS format. The method uses coated microbeads with scintillation liquid that emits light as a result of energy conversion from radioactive decay of particles in close proximity to the bead (i. e., scintillation liquid). To selectively catch RNA, beads were coated with streptavidin, which binds tightly to biotin‐labeled RNAs. The emitted light is proportional to the amount of reaction product (N7 and/or 2′‐O‐methylated mRNA). Streptavidin‐coated beads have been used to monitor the N7 and 2′‐O‐methylation activity of the dengue virus NS5.[ 112 , 121 , 122 ]
An interesting technique for RNA methylation studies is the thermal shift assay (TSA), which is based on measurements of changes in thermal denaturation (protein melting temperature), resulting from ligand binding. Thermal shift measurements are often coupled with differential scanning fluorimetry (DSF) and the application of fluorescent dyes. High‐throughput screening TSA with SYPRO Orange dye was designed and optimized by Lo et al. [123] The dye binds non‐specifically to the hydrophobic regions of proteins that are exposed through protein unfolding, which results in an increase in fluorescence intensity (Ex/Em: 490/530 nm). The assay has been applied in a fragment‐based approach to identify small molecule inhibitors of the dengue virus N7‐MTase domain. [124] The tighter the ligand binds to the protein active site, the higher the N7‐MTase protein melting temperature value.
In the previous chapter, the application of ELISA to detect the m7GMP‐nsP1 adduct was presented. Alphaviral protein nsP1 catalyzes both GTase and N7‐MTase; hence, this method can also be used for N7‐MTase activity monitoring. [106] Besides ELISA, there are other examples of modified immunoassays designed for the detection of methylation reaction products without the need to amplify analytical signals by using secondary antibodies. Graves et al. [113] modified the fluorescence polarization immunoassay (FPIA), first described by Dandliker et al. [125] which enables studies on all SAM‐utilizing MTases. The method is based on fluorescence polarization changes of SAH (product of SAM demethylation) labeled with fluorescein (SAH‐FAM) upon competitive binding with anti‐SAH antibody. Before the reaction, fluorescent SAH is bound to the antibody; hence, the fluorescence polarization is high. When new non‐fluorescent SAH molecules are produced upon the reaction of methylation, they displace SAH‐FAM, leading to a decrease in its fluorescence polarization (Figure 6B).
Changes in fluorescence polarization are often used to study the formation of protein‐ligand complexes. This approach has been recently applied to develop an FP method suitable for HTS binding experiments of compounds to 2′‐O MTase nsp16/nsp10 from SARS‐CoV‐2 (causing COVID‐19 disease). [114] For this purpose, an FAM‐labeled RNA probe was designed with a fluorescent tag located at its 3′ end (5 m7GpppACCCCC‐FAM 3). The scheme illustrating the FP assay is shown in Figure 6C. The main limitation of such a binding assay is the use of only one MTase substrate, which prevents identification of inhibitors interacting with proteins in the SAM binding pocket.
Quantification of SAH molecules released upon methylation reactions can also be performed with assays using a chain of consecutive enzymatic reactions, enabling continuous MTase activity monitoring. Nascent SAH molecules are degraded by SAH (AdoHcy) nucleosidase to release S‐ribosylhomocysteine and adenine. Both reaction products can be further quantified using different enzyme mixtures, together with colorimetric detection. One of the first reports of an enzyme‐coupled assay for SAH quantification was reported by Hendricks et al. for studies on salicylic acid carboxyl methyltransferase (SAMT). [126] The authors employed enzyme S‐ribosylcysteinase to obtain homocysteine, which can be quantified using Ellman's reagent and absorption measurements. This approach was adopted with modifications for the characterization of various MTases. Wang et al. [127] used SAH hydrolase to produce thiol homocysteine that cleaves cystamine in a specially designed fluorescent probe (fluoresceine‐cystamine‐methyl red), leading to an increase in FAM fluorescence. Adenine molecule, the second product of SAH degradation by SAH nucleotidase, can also be used to detect the amount of SAH produced upon methylation. Adenine deaminase and xanthine oxidase proceed further, resulting in H2O2 release that horseradish peroxidase uses for oxidation of non‐fluorescent Amplex® Red to fluorescent Resorufin (Figure 6D).[ 107 , 128 ]
Another possibility of fluorescence detection of SAH was demonstrated by Aouadi et al. [115] In this work, the time‐resolved Förster resonance energy transfer (TR‐FRET) methodology has been applied to studies on nsp14 N7‐MTase from SARS‐CoV and MERS‐CoV. To generate a FRET signal, two labels forming a FRET pair must be used. For this purpose anti‐SAH antibody labeled with lumi4 terbium cryptate (L4‐Tb; emission maximum at 620 nm) was designed together with d2‐labelled SAH (emission maximum at 665 nm). In such a system, FRET occurs between tags and can be observed as a fluorescence emission at 665 nm. Upon N7‐methylation, SAH molecules are released and compete with SAH‐d2 for binding with antibody, which leads to a decrease in the FRET signal at 665 nm and an increase in emission at 620 nm resulting from SAH‐d2 dissociation. If the N7‐MTase inhibitor was present in the reaction mixture, a strong FRET signal would be registered (Figure 6E).
Many other detection techniques have been reported for monitoring the SAM‐utilizing methyltransferase activity. In addition to the assays discussed above, it is worth mentioning approaches such as LC/MS [129] or ITC. [130] The recent COVID‐19 pandemic in 2019 entailed the development of new methods investigating the activity and inhibition of crucial SARS‐CoV‐2 proteins. For example, RapidFire MS technology exploit nsp14 N7‐methyltransferase by quantification of nascent SAH products. [131]
All MTase assays described so far were performed in vitro using recombinant proteins. A more advanced approach was reported by Sun et al. [116] who proposed the application of yeast cells to study SARS‐CoV, MERS‐CoV, transmissible gastroenteritis virus (TGEV), murine hepatitis virus (MEV), and infectious bronchitis virus (IBV) N7‐MTase nsp14. Saccharomyces cerevisiae yeast cell strain YBS40 had a deleted chromosomal locus Abd1 encoding yeast N7‐MTase and plasmid p360 containing the Abd1 gene and URA3 selection marker. Another plasmid carrying the coronavirus nsp14 gene and TRP1 marker has been used for cell transformation. The 5‐FOA selection [132] was performed to counter‐select for URA3 carrying plasmid, allowing the growth of yeast cells that have foreign genes that can functionally complement the yeast Abd1 gene (encoded in TRP1 plasmid). mRNA transcription levels of N7‐MTases and spectrophotometric measurements of cell density confirmed that viral N7‐MTases were expressed correctly. The authors then applied the assay for yeast growth suppression and inhibition studies in HTS format (Figure 6F). In the presence of the inhibitor, cell growth is limited, which is visible in low cell density determined by registering absorption at 595 nm.
Recently, we developed a direct fluorescence intensity (FLINT) assay for N7‐MTase activity monitoring and screening of compound libraries. [133] This method is based on the difference in interactions between the N7‐methylation reaction substrate (guanosine nucleotide) and product (7‐methylguanosine nucleotide) with a proper fluorescent tag. For the tested N7‐MTases – Ecm1 from Encephalitozoon cuniculi parasite, human RNMT‐RAM, and VCE of vaccinia virus, we observed that the minimal substrate length for efficient catalysis was dinucleotide (GpppA). Among the different tested fluorescent tags (pyrene analogues, fluorescein, cyanines, BODIPY), only the pyrene‐labeled GpppA probe changed its fluorescent properties upon N7‐methylation. This results from stronger quenching of pyrene emission by the reaction product – m7GpppA analogue, leading to a measurable decrease in fluorescence intensity (Figure 7).
Figure 7.
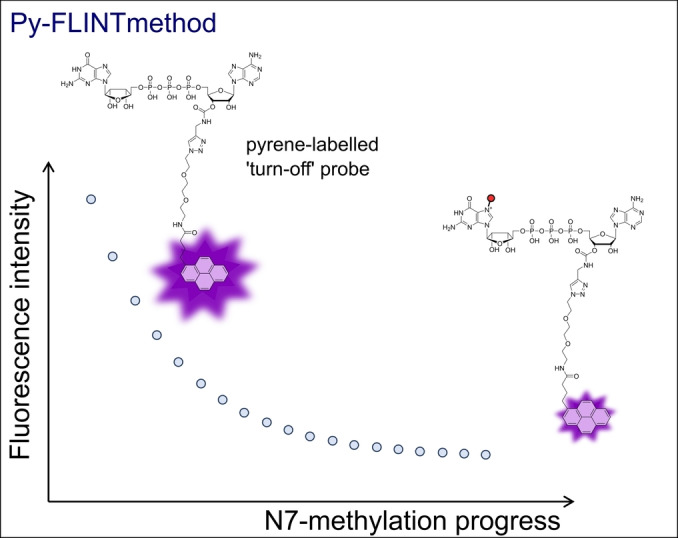
Schematic showing the principles of Py‐FLINT assay for the monitoring of N7‐MTase activity. [133]
The pyrene‐based FLINT (Py‐FLINT) assay satisfied the requirements of the HTS method (z factor=0.74, Ecm1; z factor 0.67, RNMT‐RAM) and was used for screening and inhibition experiments of small in‐house set of modified nucleotides and commercially available library of pharmacologically active compounds LOPAC®1280. The assay was further adjusted to investigate the activity and inhibition of nsp14 N7‐MTase from SARS‐CoV‐2. [134] Additionally, we developed two fluorescence polarization binding assays based on either FAM‐labeled GpppA or SAH analogues. Inhibitor characterization using these FP assays allowed the discrimination of the area of nsp14 binding site (nucleotide or SAM binding site).
6. Cap Snatching Endonuclease Assays
Instead of employing complex TPase‐GTase‐MTase capping machinery, some viruses possess proteins with either endonuclease or GTase activity to acquire the 5′ cap of the host mRNA for the synthesis of nascent viral mRNA in a process called “cap snatching” (Figure 2D–E). For instance, the influenza virus (Orthomyxoviridae) encodes a polymerase with three subunits, namely PB1, PB2, and PA, where PB1 acts as polymerase and PB2 – cap‐binding protein. The PA subunit possesses endonuclease activity and catalyzes the cleavage of cell mRNA after 10–13 nucleotides in the presence of divalent cations. [135] The PA endonuclease of influenza virus also preferentially cleaves mRNAs with cap 1 over cap 0 but could not cleave non‐methylated ones. [136] The snatched 5′ mRNA end serves as a primer for RNA‐dependent RNA polymerase (RdRp), thus initiating the synthesis of viral mRNA.
Radioactive labelling, together with urea PAGE analysis, can be applied for studies analyzing many proteins involved in cap metabolism, including the endonuclease responsible for cap‐snatching reaction catalysis. Shibagaki et al. [137] proposed a combination of radioactivity readouts with product separation utilizing specific and strong biotin‐streptavidin interactions to study cap snatching endonuclease of influenza virus. The proposed pull‐down assay could potentially be automated using magnetic beads or streptavidin‐coated multi‐well plates. The authors synthesized 3′‐biotin‐labeled 32 nt RNA with a cap 1 structure at the 5′ end containing [32P] radiolabel. After the reaction, the samples were incubated with streptavidin‐coated paramagnetic beads (Dynabeads® M‐280 Streptavidin) and filtered. The ratio of cleaved RNA to the total capped RNA was determined by the quantification of Dynabead‐bound and unbound fractions using Cerenkov radiation measurements.
One approach to study PA endonuclease activity is the use of fluorescently labeled RNA substrate, together with electrophoresis. Omoto et al. used a 42 nt ssRNA substrate labeled with FAM at the 5′ end to study the inhibition of PA derived from influenza A and B viruses. [138] The reaction products were separated by denaturing urea polyacrylamide gel electrophoresis and visualized by FAM excitation (520 nm).
While useful for visualizing the cleavage patterns of PA, product separation by gel electrophoresis is labor‐consuming and does not allow real‐time monitoring of the reaction. Hence, an additional dye is sometimes used to form a FRET pair with the first dye. For instance, Kowalinski et al. [66] designed a PA endonuclease assay using a 20 nt RNA substrate dual‐labeled with 5′‐FAM and 3′‐BHQ1 (Black Hole Quencher 1). FAM fluorescence was quenched by BHQ1 and released upon RNA degradation by PA endonuclease.
Yuan et al. [67] employed a similar approach to study influenza A PA endonuclease. However, instead of the 3′ quencher, they used the TAMRA dye to generate a FRET signal with 5′‐FAM before enzymatic degradation. The same fluorescent labels incorporated into 33 nt RNA have been exploited by Noble et al. to characterize endonucleases from influenza virus. [139] Recently, Wang et al. [140] used a FRET assay for the real‐time monitoring of the endonucleases of severe fever with thrombocytopenia syndrome virus (SFTSV) and Heartland virus (HRTV) using 21 nt ss‐RNA dual‐labeled with 5′‐FAM and 3′‐lowa Black® FQ (dark quencher).
Exploiting the structures of known PA inhibitors allows the design of a competition fluorescence polarization assay with HTS potential. Baughman et al. proposed a fluorescent probe based on the structure of an L‐742,001 inhibitor (4‐substitued 2,4‐dioxobutanoic acid), wherein piperazine nitrogen is substituted by fluorescein. [141]
The previously described thermal shift assay is also applicable for PA endonuclease characterization as reported by Omoto et al., [138] Wang et al., [140] and Fernandez‐Garcia et al. [142] They characterized the enzymes from influenza A and B viruses, SFTSV and HRTV, and several Bunyavirales, respectively.
7. Capping Inhibition
Many enzymes engaged in viral mRNA capping have already been identified as therapeutic targets. A better understanding of 5′ cap biosynthesis of viral mRNAs can provide new ideas for antiviral therapies and facilitate drug design and development. Thus, biophysical methods are required to investigate capping enzyme activity and its inhibition. Inhibition experiments involving assays described in the previous chapters have led to the discovery of new classes of viral capping inhibitors. In this section, we present the selected capping inhibitors identified using a specific assay.
Substrate structures are often elucidated to develop compounds that can efficiently compete for protein‐binding sites. The radioactive assay used for NS3 DENV characterization revealed its TPase activity and its inhibition by ATP and its analogue adenosine‐5′‐O‐(3‐thiotriphosphate) (Figure 8A). [71] Application of TPase/NTPase assays based on quantification of released inorganic phosphate, such as the charcoal adsorption assay and malachite green assay, revealed the inhibitory properties of even smaller substrate analogues, such as pyrophosphate (Figure 8B) and tripolyphosphate (Figure 8C). Gong et al. applied hydrolysis of radiolabeled [γ‐32P]ATP together with TLC separation for inhibition studies of cvRtp1 TPase from Chlorella virus and found that tripolyphosphate (IC50=0.6 μM) is a better inhibitor of cvRtp1 than pyrophosphate IC50 2.4 μM). [77]
Figure 8.

Structures of RNA‐capping inhibitors identified by assays dedicated for monitoring the activity of enzymes involved in cap biosynthesis: A) ATPγS; [71] B) Pyrophosphate; [77] C) Tripolyphosphate; [77] D) Irigenol;[ 81 , 82 ] E) 2′,2′‐Bisepigallocatechin monogallate;[ 81 , 82 ] F) Suramin;[ 74 , 133 ] G) Ellagic acid;[ 81 , 82 , 115 ] H) Chembridge3 5660163; [103] I) Maybridge5 GK 02514; [103] J) Chembridge3 7871678; [103] K) Benzbromarone; [104] L) Pyrantel pamoate; [104] M) Pyrimethamine; [106] N) (3‐Fluorobenzyl)‐N6‐SAH (X=F), (3‐chlorobenzyl)‐N6‐SAH (X=Cl) and (3‐methylbenzyl)‐N6‐SAH (X=CH3); [122] O) NF 023; [133] P) Aurintricarboxylic acid; [133] Q) Reactive Blue 2; [133] R) Myricetin;[ 115 , 133 , 134 ] S) Quercetin;[ 115 , 133 , 134 ] T) SCH 202676 HBr; [134] U) Thimerosal; [134] V) Nitazoxanide; [131] W) Adenosine dinucleotide/SAM analogue (bisubstrate inhibitor); [145] X) 2‐Hydroxy‐4‐oxo‐4‐phenyl‐2‐butenoic acid; [137] Y) Baloxavir acid (BXA); [138] Z) P‐30; [67] and AA) PA‐48. [67]
Assays suitable for HTS experiments are useful for the rapid screening of large‐compound libraries to identify lead structures that serve as basis of rational drug development. Fluorescence‐based methods are often applied due to their high sensitivity and selectivity. A 1536‐well fluorescence polarization assay was used to screen 3000 compounds against TbCet1 TPase/NTPase from Trypanosoma brucei using Transcreener™ technology. [81] The screening revealed 23 inhibitors with IC50<10 μM, including flavonoids irigenol (Figure 8D; Table 2), 2′,2′‐bisepigallocatechin monogallate (Figure 8E), and ellagic acid (Figure 8G).
Table 2.
Inhibitors of various enzymes involved in RNA capping and the various assays used for their screening.
|
Inhibitor[a] |
Method (activity) |
Enzyme (organism) |
IC50 [μM] |
|---|---|---|---|
|
ATPγS |
Radioactive assay (TPase) |
NS3 (DENV) |
∼500 [71] |
|
Pyrophosphate |
Radioactive assay (NTPase) |
cvRtp1 (Chlorella virus) |
2.4 [77] |
|
Tripolyphosphate |
Radioactive assay (NTPase) |
cvRtp1 (Chlorella virus) |
0.6 [77] |
|
Irigenol |
Transcreener™ ADP assay (NTPase) |
TbCet1 (Trypanosoma brucei) |
0.065 [81] |
|
2′,2′‐Bisepigallocatechin monogallate |
Transcreener™ ADP assay (NTPase) |
TbCet1 (Trypanosoma brucei) |
0.010 [81] |
|
Suramin |
Charcoal adsorption assay (TPase) Py‐FLINT (N7‐MTase) Py‐FLINT (N7‐MTase) Py‐FLINT (N7‐MTase) |
Cet1p (Saccharomyces cerevisae) VCE (Vaccinia virus) Ecm1 (E. cuniculi) RNMT‐RAM (Homo sapiens) |
1.9±0.3 [74] 0.083±0.015 [133] 0.046±0.006 [133] 0.70±0.08 [133] |
|
Ellagic acid |
Transcreener™ ADP assay (NTPase) HRTF (N7‐MTase) |
TbCet1 (Trypanosoma brucei) nsp14 (SARS‐CoV) |
|
|
Chembridge3 5660163 |
FP (GTase) |
NS5 (DENV) |
7.1±1.4 [103] |
|
Maybridge5 GK 02514 |
FP (GTase) |
NS5 (DENV) |
9.8±1.0 [103] |
|
Chembridge3 7871678 |
FP (GTase) |
NS5 (DENV) |
7.4±0.8 [103] |
|
Benzbromarone |
FP (GTase) |
NS5 (DENV) nsP1 (CHIKV) |
|
|
Pyrantel pamoate |
FP (GTase) |
NS5 (DENV) nsP1 (CHIKV) |
|
|
Pyrimethamine |
Western blot assay (GTase) Filter binding assay (N7‐MTase) |
nsP1 (VEEV) |
|
|
(3‐Fluorobenzyl)‐N6‐SAH |
SPA (N7‐MTase) SPA (2′‐O‐MTase) |
NS5 (DENV) |
|
|
(3‐Chlorobenzyl)‐N6‐SAH |
SPA (N7‐MTase) SPA (2′‐O‐MTase) |
NS5 (DENV) |
|
|
(3‐Methylbenzyl)‐N6‐SAH |
SPA (N7‐MTase) SPA (2′‐O‐MTase) |
NS5 (DENV) |
|
|
NF 023 |
Py‐FLINT (N7‐MTase) |
Ecm1 (E. cuniculi) |
0.015±0.005 [133] |
|
Aurintricarboxylic acid |
Py‐FLINT (N7‐MTase) |
Ecm1 (E. cuniculi) |
0.031±0.005 [133] |
|
Reactive Blue 2 |
Py‐FLINT (N7‐MTase) |
nsp14 (SARS‐CoV‐2) Ecm1 (E. cuniculi) |
|
|
Myricetin |
Py‐FLINT (N7‐MTase) HRTF (N7‐MTase) Py‐FLINT (N7‐MTase) |
nsp14 (SARS‐CoV‐2) nsp14 (SARS‐CoV) Ecm1 (E. cuniculi) |
|
|
Quercetin |
Py‐FLINT (N7‐MTase) HRTF (N7‐MTase) Py‐FLINT (N7‐MTase) |
nsp14 (SARS‐CoV‐2) nsp14 (SARS‐CoV) Ecm1 (E. cuniculi) |
|
|
SCH 202676 HBr |
Py‐FLINT (N7‐MTase) |
nsp14 (SARS‐CoV‐2) |
1.50±0.20 [134] |
|
Thimerosal |
Py‐FLINT (N7‐MTase) |
nsp14 (SARS‐CoV‐2) |
1.05±0.08 [134] |
|
Nitazoxanide |
RapidFire MS (N7‐MTase) |
nsp14 (SARS‐CoV‐2) |
9.7 [131] |
|
Adenosine dinucleotide SAM analogue |
Filter binding assay (N7‐MTase) |
nsp14 (SARS‐CoV) |
0.6±0.1 [145] |
|
2‐Hydroxy‐4‐oxo‐4‐phenyl‐2‐butenoic acid |
Radioactive assay (endonuclease) |
PA endonuclease (influenza virus) |
65.0 [137] |
|
Diketo acid analogue |
TSA (endonuclease) |
PA endonuclease (Andes virus) |
3.4±0.5 [142] |
|
Baloxavir acid (BXA) |
TSA and Urea PAGE with fluorescence visualization (endonuclease) |
PA endonuclease (influenza virus) |
n.d. [138] |
|
P‐30 |
FRET assay (endonuclease) |
PA endonuclease (influenza virus) |
1.5±0.2 [67] |
|
ANA‐0 |
FRET assay (endonuclease) |
PA endonuclease (influenza virus) |
0.8±0.1 [67] |
Geiss et al. [103] employed another fluorescence polarization‐based assay for a pilot screen of 43 323 commercially available compounds and applied BODIPY‐labeled GTP. They identified 11 strong inhibitors (>30 % inhibition) of the flaviviral NS5 enzyme, including the compounds Chembridge3 5660163 (Figure 8H), Maybridge7 GK 02514 (Figure 8I), and Chembridge 7871678 (Figure 8J). Using the same method, Feibelman et al. reported benzobromarone (Figure 8K), pyrantel pamoate (Figure 8L), garcinolic acid, and lobaric acid as inhibitors of CHIKV nsP1 and DENV NS5 GTase activity. [104] They screened 2320 compounds from the Spectrum Collection library using MicroSource Discovery Systems.
Many examples of SAM and SAH analogues that can inhibit various MTases have been previously identified. One of the most extensively studied is sinefungin, a natural purine nucleoside. [143] However, such inhibitors could be non‐specific and act on many MTases utilizing the SAM molecule as a donor of the methyl group, which subsequently leads to increased cytotoxicity. [144] To increase inhibitor selectivity, some approaches utilize the specific properties of the binding pocket. For example, Lim et al. [122] designed a series of SAH analogues that have been modified in the N6 adenosine position with substituents that can extend into an uncovered flavivirus‐conserved cavity, which is located next to the cofactor binding site of the NS5 enzyme. Moreover, a scintillation‐proximity assay [121] was applied to identify halogen‐modified benzyl substituents at the meta position, including (3‐fluorobenzyl)‐N6‐SAH, (3‐chlorobenzyl)‐N6‐SAH, and (3‐methylbenzyl)‐N6‐SAH (Figure 8N). These novel potent N7‐ and 2′‐O‐MTase inhibitors showed improved selectivity toward human RNMT N7‐MTase.
Recently, we developed a fluorescent assay based on pyrene‐labeled Gp3A (nucleotide substrate analogue) for the real‐time monitoring of the N7‐MTase reaction. [133] The method was applied to screen commercially available LOPAC1280 library, and we identified four compounds with inhibitory potency towards Ecm1 N7‐MTase (IC50<50 nM), including NF 023 (Figure 8O), aurintricarboxylic acid (Figure 8P), Reactive Blue 2 (Figure 8Q), and suramin (Figure 8F). Suramin could also inhibit the VCE capping enzyme of vaccinia virus. The second set of inhibitors involved flavonoids, such as galloflavin, myricetin (Figure 8R), and quercetin (Figure 8S). The last two have also been found to inhibit SARS‐CoV nsp14 N7‐MTase in a study utilizing the HRTF assay applied to screen the Prestwick Chemical Library® with 2000 compounds. [115] In this study, ellagic acid (Figure 8G) was identified as an even more potent inhibitor of SARS‐CoV nsp14 N7‐MTase than sinefungin. Due to their relatively low molecular weight, all flavonoid‐like compounds are potential lead structures for future drug development.
We further modified our Py‐FLINT method to determine the N7‐MTase activity of nsp14 from SARS‐CoV‐2. [134] Five commercially available libraries were screened (7039 compounds in total) to identify 83 nsp14 inhibitors (IC50<50 μM) acting with either nucleotide‐site targeting or bisubstrate mode of action. We identified three compounds that can inhibit viral replication in the Huh 7 cell model, including pyridostatin, Reactive Blue 2, and Evans Blue. Pyridostatin exhibited the best inhibition property (EC50=3.58±0.16 μM, similar to that of the parent nucleoside of remdesivir GS‐441524) and a selectivity index of 16.6, although whether these properties were due to its interaction with N7‐MTase remains unclear. Some of the other most potent inhibitors include thiadiazole compound SCH‐202676 (Figure 8T), polyphenols, tannic acid, theaflavin, catechin analogues, and the FDA‐approved drugs thimerosal (Figure 8U), gastrodenol, and ebselen.
Although many computational approaches have been employed to screen compound libraries against SARS‐CoV‐2 N7‐MTase nsp14, only a few inhibitor examples have been identified with experimental assays. For example, using another HTS assay based on RapidFire Mass Spectrometry, Pearson et al. [131] 2021 screened the FDA‐approved drug library with 1771 compounds and identified that nitaxozanide (Figure 8V) exhibits inhibitory properties against the SARS‐CoV‐2 nsp14 enzyme.
An alternative approach for SARS‐CoV MTase inhibitor design has been presented by Ahmed‐Belkacem et al. [145] They proposed a bisubstrate inhibitor structure (adenosine dinucleotides) that mimics the transition state of the methyl transfer reaction, resulting in cap 1 formation (Figure 8W). Developed inhibitors have been aimed at 2′‐O‐MTases; however, their activity against SARS‐CoV enzymes is negligible. Some structures showed significant inhibition of N7‐MTases, including the nsp14 of SARS‐CoV.
Multiply assays have been conducted to identify inhibitors of capping machinery employing the ‘cap‐snatching’ mechanism, in particular the PA endonuclease of influenza virus. Discovered inhibitors can then be used for other assays. For example, 2‐hydroxy‐4‐oxo‐4‐phenyl‐2‐butenoic acid (Figure 8X), [146] a known inhibitor of the cap‐snatching reaction, was used to validate the pull‐down assay that combines radioactive labeling with specific biotin‐streptavidin interactions. [137] Fernandez‐Garcia et al. [142] exploited further structures of diketo acids to develop potent PA endonuclease inhibitors of selected Bunyavirales, such as La Crosse virus, Rift Valley Fever virus, and Andes virus. Baloxavir acid (Figure 8Y) and its prodrug, baloxavir marboxil containing additional phenolic hydroxyl groups, have been synthesized as alternatives for influenza treatment.[ 138 , 147 ] Using Thermal Stability assay and fluorescently labelled RNA, BXA (baloxavir acid) selectively inhibited the activity of cap‐dependent influenza A virus endonuclease.
The fluorescence FRET assay has been used to screen a chemical library consisting of 950 compounds, and new classes of influenza PA inhibitors have been revealed. Among all tested compounds, the compound P‐30 (Figure 8Z) exhibited the highest selectivity index (defined as the ratio of 50 % cellular cytotoxicity concentration CC50 to IC50). [67] Based on the structural properties of the identified inhibitors, another set of compounds was designed, and compound ANA‐0 (Figure 8AA) was selected as a potent inhibitor of influenza virus PA endonuclease. Both ANA‐0 and P‐30 inhibited the growth of various subtypes of influenza A virus (H1 N1 and H5 N1) in cell cultures in a dose‐dependent manner. Furthermore, ANA‐0 inhibited viral growth in mouse lung tissues, which showed a significant reduction in viral loads.
8. Summary and Outlook
Cap biosynthesis is an essential step in mRNA maturation. Owing to its implications to the development and treatment of many viral infections, it has become a research hotspot. Further elucidation of the differences in the capping machinery between eukaryotes and viruses can contribute to the development of more effective antiviral therapies. Hence, new potent small‐molecule inhibitors of viral capping are desired for the further development of antiviral therapies. Here, we described selected methods for inhibitor evaluation (IC50 determination), established for enzymes that catalyze one or more steps of the cap biosynthesis pathway, mostly from pathogenic viruses.
Radioactive methods (i. e. radiolabeled RNAs with TLC/SDS‐PAGE separation, filter binding and SPA assays) provide invaluable insights into the function of capping enzymes. Here, the RNA substrate is labeled with groups identical to those of natural substrates. However, radioactive assays are time‐consuming and labor‐intensive and requires the separation of reaction products, making them unsuitable for high‐throughput screening experiments.
Fluorescence techniques (i. e. fluorescence intensity, fluorescence polarization) are highly sensitive and straightforward. Because fluorescence detection is not time‐consuming and labor‐intensive, it is suitable for HTS experiments. Hence, many capping assays employ fluorescence detection with a properly designed probe. These methods require dye applications that do not disrupt protein‐probe interactions and maintain their emission properties during enzymatic reactions.
Cell‐based assays provide a suitable environment for studying viral growth inhibition. However, such assays are often laborious; therefore, large compound library screening using methods that utilize homogenous proteins is more preferable.
We speculate that due to the resemblance of capping enzymes, the assays could potentially be adapted to explore not only the target enzyme itself but also analogous enzymes from other organisms.
The development of new methods that allow for studies on processes crucial for virus replication, such as RNA capping, allows the identification of potential drugs for antiviral therapies. Testing of large compound number to verify if they induce appropriate effect constitute one of the first steps in drug development. Hence, methods suitable for HTS experiments are particularly desired for this purpose. Inhibitor selection, together with structure‐activity analysis, could facilitate rational drug design and discovery, which are particularly crucial considering the ongoing COVID‐19 pandemic and potential future outbreaks. Finally, assays for studies on viral proteins allows also for a better understanding of their life cycle, resulting in discovery of new therapeutic targets.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Renata Kasprzyk studied biophysics and chemistry at the University of Warsaw (Poland) where she received her PhD in 2021 in the group of Prof. Jemielity. She works on the synthesis of fluorescently labelled nucleotides and development of new fluorescent assays for high‐throughput screening applications. Her main research interest is exploration of modified nucleotides in studies on therapeutically relevant molecular targets.

Biographical Information
Prof. Jacek Jemielity is the head of the Laboratory of Bioorganic Chemistry at Centre of New Technologies, University of Warsaw. His main research interests are the synthesis, properties and applications of biologically relevant analogues of nucleotides. He is also co‐inventor of several patented and licensed technologies improving therapeutic properties of mRNA.

Acknowledgements
This work was financially supported by the National Science Centre (grant number UMO‐2020/01/0/ST4/00124) to J.J. and (grant numbers UMO‐2019/32/T/ST4/00091 and UMO‐2016/23/N/ST4/03169) to R.K., and Foundation for Polish Science (grant number TEAM/2016‐2/13) to J.J.
R. Kasprzyk, J. Jemielity, ChemBioChem 2021, 22, 3236.
References
- 1. Dreyfus M., Régnier P., Cell 2002, 111, 611–613. [DOI] [PubMed] [Google Scholar]
- 2.Y. Furuichi, in eLS, Wiley, Chichester 2014, DOI: 10.1002/9780470015902.a0000891.pub3. [DOI]
- 3. Daffis S., Szretter K. J., Schriewer J., Li J., Youn S., Errett J., Lin T. Y., Schneller S., Zust R., Dong H., Thiel V., Sen G. C., Fensterl V., Klimstra W. B., Pierson T. C., Buller R. M., Gale M., Shi P. Y., Diamond M. S., Nature 2010, 468, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu G., Gershon P. D., Hodel A. E., Quiocho F. A., Proc. Natl. Acad. Sci. USA 1999, 96, 7149–7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Shatkin A. J., Manley J. L., Nat. Struct. Biol. 2000, 7, 838–842; [DOI] [PubMed] [Google Scholar]
- 5b. Moteki S., Price D., Mol. Cell 2002, 10, 599–609. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Gong C., Martins A., Shuman S., J. Biol. Chem. 2003, 278, 50843–50852; [DOI] [PubMed] [Google Scholar]
- 6b. Pei Y., Schwer B., Hausmann S., Shuman S., Nucleic Acids Res. 2001, 29, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ho C. K., Pei Y., Shuman S., J. Biol. Chem. 1998, 273, 34151–34156. [DOI] [PubMed] [Google Scholar]
- 8. Shuman S., Nat. Rev. Mol. Cell Biol. 2002, 3, 619–625. [DOI] [PubMed] [Google Scholar]
- 9. Takagi T., Moore C. R., Diehn F., Buratowski S., Cell 1997, 89, 867–873. [DOI] [PubMed] [Google Scholar]
- 10. Changela A., Ho C. K., Martins A., Shuman S., Mondragón A., EMBO J. 2001, 20, 2575–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chu C., Das K., Tyminski J. R., Bauman J. D., Guan R., Qiu W., Montelione G. T., Arnold E., Shatkin A. J., Proc. Natl. Acad. Sci. USA 2011, 108, 10104–10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richon V. M., Johnston D., Sneeringer C. J., Jin L., Majer C. R., Elliston K., Jerva L. F., Scott M. P., Copeland R. A., Chem. Biol. Drug Des. 2011, 78, 199–210. [DOI] [PubMed] [Google Scholar]
- 13. Fabrega C., Hausmann S., Shen V., Shuman S., Lima C. D., Mol. Cell 2004, 13, 77–89. [DOI] [PubMed] [Google Scholar]
- 14. Niedzwiecka A., Marcotrigiano J., Stepinski J., Jankowska-Anyszka M., Wyslouch-Cieszynska A., Dadlez M., Gingras A. C., Mak P., Darzynkiewicz E., Sonenberg N., Burley S. K., Stolarski R., J. Mol. Biol. 2002, 319, 615–635. [DOI] [PubMed] [Google Scholar]
- 15. Bélanger F., Stepinski J., Darzynkiewicz E., Pelletier J., J. Biol. Chem. 2010, 285, 33037–33044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smietanski M., Werner M., Purta E., Kaminska K. H., Stepinski J., Darzynkiewicz E., Nowotny M., Bujnicki J. M., Nat. Commun. 2014, 5, 3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hodel A. E., Gershon P. D., Quiocho F. A., Mol. Cell 1998, 1, 443–447. [DOI] [PubMed] [Google Scholar]
- 18. Li C., Xia Y., Gao X., Gershon P. D., Biochemistry 2004, 43, 5680–5687. [DOI] [PubMed] [Google Scholar]
- 19. Werner M., Purta E., Kaminska K. H., Cymerman I. A., Campbell D. A., Mittra B., Zamudio J. R., Sturm N. R., Jaworski J., Bujnicki J. M., Nucleic Acids Res. 2011, 39, 4756–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Bangs J. D., Crain P. F., Hashizume T., McCloskey J. A., Boothroyd J. C., J. Biol. Chem. 1992, 267, 9805–9815; [PubMed] [Google Scholar]
- 20b. Zamudio J. R., Mittra B., Foldynová-Trantírková S., Zeiner G. M., Lukes J., Bujnicki J. M., Sturm N. R., Campbell D. A., Mol. Cell. Biol. 2007, 27, 6084–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Hâkansson K., Wigley D. B., Proc. Natl. Acad. Sci. USA 1998, 95, 1505–1510; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Wen Y., Yue Z., Shatkin A. J., Proc. Natl. Acad. Sci. USA 1998, 95, 12226–12231; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Fabrega C., Shen V., Shuman S., Lima C. D., Mol. Cell 2003, 11, 1549–1561. [DOI] [PubMed] [Google Scholar]
- 22. Byszewska M., Śmietański M., Purta E., Bujnicki J. M., RNA Biol. 2014, 11, 1597–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Varshney D., Petit A. P., Bueren-Calabuig J. A., Jansen C., Fletcher D. A., Peggie M., Weidlich S., Scullion P., Pisliakov A. V., Cowling V. H., Nucleic Acids Res. 2016, 44, 10423–10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furuichi Y., Shatkin A. J., Adv. Virus Res. 2000, 55, 135–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harwig A., Landick R., Berkhout B., Viruses 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karpe Y. A., Aher P. P., Lole K. S., PLoS One 2011, 6, e22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delang L., Li C., Tas A., Quérat G., Albulescu I. C., De Burghgraeve T., Guerrero N. A., Gigante A., Piorkowski G., Decroly E., Jochmans D., Canard B., Snijder E. J., Pérez-Pérez M. J., van Hemert M. J., Coutard B., Leyssen P., Neyts J., Sci. Rep. 2016, 6, 31819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tomar S., Narwal M., Harms E., Smith J. L., Kuhn R. J., Protein Expression Purif. 2011, 79, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vasiljeva L., Merits A., Auvinen P., Kääriäinen L., J. Biol. Chem. 2000, 275, 17281–17287. [DOI] [PubMed] [Google Scholar]
- 30. Ahola T., Kääriäinen L., Proc. Natl. Acad. Sci. USA 1995, 92, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rikkonen M., Peränen J., Kääriäinen L., J. Virol. 1994, 68, 5804–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ahola T., Laakkonen P., Vihinen H., Kääriäinen L., J. Virol. 1997, 71, 392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wengler G., Virology 1993, 197, 265–273. [DOI] [PubMed] [Google Scholar]
- 34. Issur M., Geiss B. J., Bougie I., Picard-Jean F., Despins S., Mayette J., Hobdey S. E., Bisaillon M., RNA 2009, 15, 2340–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou Y., Ray D., Zhao Y., Dong H., Ren S., Li Z., Guo Y., Bernard K. A., Shi P. Y., Li H., J. Virol. 2007, 81, 3891–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Dong H., Chang D. C., Hua M. H., Lim S. P., Chionh Y. H., Hia F., Lee Y. H., Kukkaro P., Lok S. M., Dedon P. C., Shi P. Y., PLoS Pathog. 2012, 8, e1002642; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36b. Egloff M. P., Benarroch D., Selisko B., Romette J. L., Canard B., EMBO J. 2002, 21, 2757–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ivanov K. A., Ziebuhr J., J. Virol. 2004, 78, 7833–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen Y., Guo D., Virol. Sin. 2016, 31, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen Y., Cai H., Pan J., Xiang N., Tien P., Ahola T., Guo D., Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.
- 40a. Decroly E., Debarnot C., Ferron F., Bouvet M., Coutard B., Imbert I., Gluais L., Papageorgiou N., Sharff A., Bricogne G., Ortiz-Lombardia M., Lescar J., Canard B., PLoS Pathog. 2011, 7, e1002059; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40b. Lugari A., Betzi S., Decroly E., Bonnaud E., Hermant A., Guillemot J. C., Debarnot C., Borg J. P., Bouvet M., Canard B., Morelli X., Lécine P., J. Biol. Chem. 2010, 285, 33230–33241; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40c. Decroly E., Imbert I., Coutard B., Bouvet M., Selisko B., Alvarez K., Gorbalenya A. E., Snijder E. J., Canard B., J. Virol. 2008, 82, 8071–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ogino T., Banerjee A. K., Mol. Cell 2007, 25, 85–97. [DOI] [PubMed] [Google Scholar]
- 42. Ogino M., Ito N., Sugiyama M., Ogino T., Viruses 2016, 8, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Testa D., Banerjee A. K., J. Virol. 1977, 24, 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kyrieleis O. J., Chang J., de la Peña M., Shuman S., Cusack S., Structure 2014, 22, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.
- 45a. De la Peña M., Kyrieleis O. J., Cusack S., EMBO J. 2007, 26, 4913–4925; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45b. Mao X., Shuman S., J. Biol. Chem. 1994, 269, 24472–24479. [PubMed] [Google Scholar]
- 46. Barbosa E., Moss B., J. Biol. Chem. 1978, 253, 7698–7702. [PubMed] [Google Scholar]
- 47. Sutton G., Grimes J. M., Stuart D. I., Roy P., Nat. Struct. Mol. Biol. 2007, 14, 449–451. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. Pizarro J. L., Sandino A. M., Pizarro J. M., Fernández J., Spencer E., J. Gen. Virol. 1991, 72, 325–332; [DOI] [PubMed] [Google Scholar]
- 48b. Liu M., Mattion N. M., Estes M. K., Virology 1992, 188, 77–84. [DOI] [PubMed] [Google Scholar]
- 49. Chen D., Luongo C. L., Nibert M. L., Patton J. T., Virology 1999, 265, 120–130. [DOI] [PubMed] [Google Scholar]
- 50. Bisaillon M., Lemay G., J. Biol. Chem. 1997, 272, 29954–29957. [DOI] [PubMed] [Google Scholar]
- 51. Mao Z., Jokliki W. K., Virology 1991, 185, 377–386. [DOI] [PubMed] [Google Scholar]
- 52. Bujnicki J. M., Rychlewski L., Genome Biol. 2001, 2, research0038.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ogino T., Yadav S. P., Banerjee A. K., Proc. Natl. Acad. Sci. USA 2010, 107, 3463–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neubauer J., Ogino M., Green T. J., Ogino T., Nucleic Acids Res. 2016, 44, 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Plotch S. J., Bouloy M., Ulmanen I., Krug R. M., Cell 1981, 23, 847–858. [DOI] [PubMed] [Google Scholar]
- 56.
- 56a. Fujimura T., Esteban R., Proc. Natl. Acad. Sci. USA 2011, 108, 17667–17671; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56b. Fujimura T., Esteban R., J. Biol. Chem. 2013, 288, 23716–23724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ivanov K. A., Thiel V., Dobbe J. C., van der Meer Y., Snijder E. J., Ziebuhr J., J. Virol. 2004, 78, 5619–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walker A. P., Fan H., Keown J. R., Grimes J. M., Fodor E., bioRxiv 2021, 10.1101/2021.03.17.435913. [DOI] [Google Scholar]
- 59. Jin X., Chen Y., Sun Y., Zeng C., Wang Y., Tao J., Wu A., Yu X., Zhang Z., Tian J., Guo D., Virus Res. 2013, 176, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eckerle L. D., Becker M. M., Halpin R. A., Li K., Venter E., Lu X., Scherbakova S., Graham R. L., Baric R. S., Stockwell T. B., Spiro D. J., Denison M. R., PLoS Pathog. 2010, 6, e1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.
- 61a. Bouvet M., Debarnot C., Imbert I., Selisko B., Snijder E. J., Canard B., Decroly E., PLoS Pathog. 2010, 6, e1000863; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61b. Bouvet M., Imbert I., Subissi L., Gluais L., Canard B., Decroly E., Proc. Natl. Acad. Sci. USA 2012, 109, 9372–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Debarnot C., Imbert I., Ferron F., Gluais L., Varlet I., Papageorgiou N., Bouvet M., Lescar J., Decroly E., Canard B., Acta Crystallogr. Sect. F 2011, 67, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lim S. P., Noble C. G., Shi P. Y., Antiviral Res. 2015, 119, 57–67. [DOI] [PubMed] [Google Scholar]
- 64. Stahla-Beek H. J., April D. G., Saeedi B. J., Hannah A. M., Keenan S. M., Geiss B. J., J. Virol. 2012, 86, 8730–8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. DuBois R. M., Slavish P. J., Baughman B. M., Yun M. K., Bao J., Webby R. J., Webb T. R., White S. W., PLoS Pathog. 2012, 8, e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kowalinski E., Zubieta C., Wolkerstorfer A., Szolar O. H., Ruigrok R. W., Cusack S., PLoS Pathog. 2012, 8, e1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yuan S., Chu H., Singh K., Zhao H., Zhang K., Kao R. Y., Chow B. K., Zhou J., Zheng B. J., Sci. Rep. 2016, 6, 22880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tong T. R., Infect. Disord.: Drug Targets 2009, 9, 223–245. [DOI] [PubMed] [Google Scholar]
- 69. Wang Y., Sun Y., Wu A., Xu S., Pan R., Zeng C., Jin X., Ge X., Shi Z., Ahola T., Chen Y., Guo D., J. Virol. 2015, 89, 8416–8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dunn S., Lombardi O., Lukoszek R., Cowling V. H., Open Biol. 2019, 9, 190052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bartelma G., Padmanabhan R., Virology 2002, 299, 122–132. [DOI] [PubMed] [Google Scholar]
- 72. Egami F., Takahashi K., Uchida T., Prog. Nucleic Acid Res. Mol. Biol. 1964, 3, 59–101. [DOI] [PubMed] [Google Scholar]
- 73. Igarashi M., Sawa R., Yamasaki M., Hayashi C., Umekita M., Hatano M., Fujiwara T., Mizumoto K., Nomoto A., J. Antibiot. 2017, 70, 582–589. [DOI] [PubMed] [Google Scholar]
- 74. Xu Y., Triantafyllou I., Cable M., Palermo R., Anal. Biochem. 2008, 372, 89–95. [DOI] [PubMed] [Google Scholar]
- 75.
- 75a. Itaya K., Ui M., Clin. Chim. Acta 1966, 14, 361–366; [DOI] [PubMed] [Google Scholar]
- 75b. Penney C. L., Anal. Biochem. 1976, 75, 201–210; [DOI] [PubMed] [Google Scholar]
- 75c. Fisher D. K., Higgins T. J., Pharm. Res. 1994, 11, 759–763. [DOI] [PubMed] [Google Scholar]
- 76. Gu B., Liu C., Lin-Goerke J., Maley D. R., Gutshall L. L., Feltenberger C. A., Del Vecchio A. M., J. Virol. 2000, 74, 1794–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gong C., Shuman S., J. Biol. Chem. 2002, 277, 15317–15324. [DOI] [PubMed] [Google Scholar]
- 78. Han K. R., Lee J. H., Kotiguda G. G., Jung K. H., Chung M. S., Kang S., Hwang S., Kim K. H., J. Gen. Virol. 2018, 99, 1482–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tanner J. A., Watt R. M., Chai Y. B., Lu L. Y., Lin M. C., Peiris J. S., Poon L. L., Kung H. F., Huang J. D., J. Biol. Chem. 2003, 278, 39578–39582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shu T., Huang M., Wu D., Ren Y., Zhang X., Han Y., Mu J., Wang R., Qiu Y., Zhang D. Y., Zhou X., Virol. Sin. 2020, 35, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Antczak C., Shum D., Radu C., Seshan V. E., Djaballah H., Comb. Chem. High Throughput Screening 2009, 12, 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Smith P., Ho C. K., Takagi Y., Djaballah H., Shuman S., mBio 2016, 7, e00058–00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rahimi R., Ghiasi S., Azimi H., Fakhari S., Abdollahi M., Cytokine 2010, 49, 123–129. [DOI] [PubMed] [Google Scholar]
- 84. Feng J., Chen Y., Pu J., Yang X., Zhang C., Zhu S., Zhao Y., Yuan Y., Yuan H., Liao F., Anal. Biochem. 2011, 409, 144–149. [DOI] [PubMed] [Google Scholar]
- 85.
- 85a. Zhou M., Diwu Z., Panchuk-Voloshina N., Haugland R. P., Anal. Biochem. 1997, 253, 162–168; [DOI] [PubMed] [Google Scholar]
- 85b. Mohanty J. G., Jaffe J. S., Schulman E. S., Raible D. G., J. Immunol. Methods 1997, 202, 133–141. [DOI] [PubMed] [Google Scholar]
- 86. Benarroch D., Selisko B., Locatelli G. A., Maga G., Romette J. L., Canard B., Virology 2004, 328, 208–218. [DOI] [PubMed] [Google Scholar]
- 87. Wang C. C., Huang Z. S., Chiang P. L., Chen C. T., Wu H. N., FEBS Lett. 2009, 583, 691–696. [DOI] [PubMed] [Google Scholar]
- 88. Lowery R. G., Kleman-Leyer K., Expert Opin. Ther. Targets 2006, 10, 179–190. [DOI] [PubMed] [Google Scholar]
- 89. Sweeney N. L., Shadrick W. R., Mukherjee S., Li K., Frankowski K. J., Schoenen F. J., Frick D. N., J. Biol. Chem. 2013, 288, 19949–19957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bougie I., Parent A., Bisaillon M., Biochem. J. 2004, 384, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bisaillon M., Bougie I., J. Biol. Chem. 2003, 278, 33963–33971. [DOI] [PubMed] [Google Scholar]
- 92. Benzaghou I., Bougie I., Picard-Jean F., Bisaillon M., FEBS Lett. 2006, 580, 867–877. [DOI] [PubMed] [Google Scholar]
- 93. Takagi Y., Kuwabara N., Dang T. T., Furukawa K., Ho C. K., J. Biol. Chem. 2020, 295, 9076–9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Venkatesan S., Moss B., Proc. Natl. Acad. Sci. USA 1982, 79, 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Soulière M. F., Perreault J. P., Bisaillon M., Biochemistry 2008, 47, 3863–3874. [DOI] [PubMed] [Google Scholar]
- 96. Ho C. K., Van Etten J. L., Shuman S., J. Virol. 1996, 70, 6658–6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Guarino L. A., Jin J., Dong W., J. Virol. 1998, 72, 10003–10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rabah N., Ortega Granda O., Quérat G., Canard B., Decroly E., Coutard B., Antiviral Res. 2020, 182, 104883. [DOI] [PubMed] [Google Scholar]
- 99. Lehmann K. C., Gulyaeva A., Zevenhoven-Dobbe J. C., Janssen G. M., Ruben M., Overkleeft H. S., van Veelen P. A., Samborskiy D. V., Kravchenko A. A., Leontovich A. M., Sidorov I. A., Snijder E. J., Posthuma C. C., Gorbalenya A. E., Nucleic Acids Res. 2015, 43, 8416–8434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Martin S. A., Paoletti E., Moss B., J. Biol. Chem. 1975, 250, 9322–9329. [PubMed] [Google Scholar]
- 101. Martin S. A., Moss B., J. Biol. Chem. 1975, 250, 9330–9335. [PubMed] [Google Scholar]
- 102. Venkatesan S., Gershowitz A., Moss B., J. Biol. Chem. 1980, 255, 903–908. [PubMed] [Google Scholar]
- 103. Geiss B. J., Stahla-Beek H. J., Hannah A. M., Gari H. H., Henderson B. R., Saeedi B. J., Keenan S. M., J. Biomol. Screening 2011, 16, 852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Feibelman K. M., Fuller B. P., Li L., LaBarbera D. V., Geiss B. J., Antiviral Res. 2018, 154, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Engvall E., Perlmann P., J. Immunol. 1972, 109, 129–135. [PubMed] [Google Scholar]
- 106. Ferreira-Ramos A. S., Li C., Eydoux C., Contreras J. M., Morice C., Quérat G., Gigante A., Pérez Pérez M. J., Jung M. L., Canard B., Guillemot J. C., Decroly E., Coutard B., Antiviral Res. 2019, 163, 59–69. [DOI] [PubMed] [Google Scholar]
- 107. Kaur R., Mudgal R., Narwal M., Tomar S., Virus Res. 2018, 256, 209–218. [DOI] [PubMed] [Google Scholar]
- 108.
- 108a. Rossi A., Taylor C., Nat. Protoc. 2011, 6, 365–387; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108b. Palmier M., Van Doren S., Anal. Biochem. 2007, 371, 43–51; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108c. Breen C., Raverdeau M., Voorheis H., Sci. Rep. 2016, 6, 25769; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108d. Wienken C., Baaske P., Rothbauer U., Braun D., Duhr S., Nat. Commun. 2010, 1, 100. [DOI] [PubMed] [Google Scholar]
- 109. Geiss B. J., Thompson A. A., Andrews A. J., Sons R. L., Gari H. H., Keenan S. M., Peersen O. B., J. Mol. Biol. 2009, 385, 1643–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.
- 110a. Ferrero D. S., Ruiz-Arroyo V. M., Soler N., Usón I., Guarné A., Verdaguer N., PLoS Pathog. 2019, 15, e1007656; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110b. Bollati M., Milani M., Mastrangelo E., Ricagno S., Tedeschi G., Nonnis S., Decroly E., Selisko B., de Lamballerie X., Coutard B., Canard B., Bolognesi M., J. Mol. Biol. 2009, 385, 140–152. [DOI] [PubMed] [Google Scholar]
- 111. Bullard-Feibelman K. M., Fuller B. P., Geiss B. J., PLoS One 2016, 11, e0158923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chung K. Y., Dong H., Chao A. T., Shi P. Y., Lescar J., Lim S. P., Virology 2010, 402, 52–60. [DOI] [PubMed] [Google Scholar]
- 113. Graves T. L., Zhang Y., Scott J. E., Anal. Biochem. 2008, 373, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Perveen S., Khalili Yazdi A., Devkota K., Li F., Ghiabi P., Hajian T., Loppnau P., Bolotokova A., Vedadi M., SLAS Discov. 2021, 26, 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Aouadi W., Eydoux C., Coutard B., Martin B., Debart F., Vasseur J. J., Contreras J. M., Morice C., Quérat G., Jung M. L., Canard B., Guillemot J. C., Decroly E., Antiviral Res. 2017, 144, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sun Y., Wang Z., Tao J., Wang Y., Wu A., Yang Z., Wang K., Shi L., Chen Y., Guo D., Antiviral Res. 2014, 104, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.
- 117a. Lim S. P., Bodenreider C., Shi P. Y., Methods Mol. Biol. 2013, 1030, 249–268; [DOI] [PubMed] [Google Scholar]
- 117b. Dong H., Ren S., Zhang B., Zhou Y., Puig-Basagoiti F., Li H., Shi P. Y., J. Virol. 2008, 82, 4295–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Aouadi W., Blanjoie A., Vasseur J. J., Debart F., Canard B., Decroly E., J. Virol. 2017, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Barral K., Sallamand C., Petzold C., Coutard B., Collet A., Thillier Y., Zimmermann J., Vasseur J. J., Canard B., Rohayem J., Debart F., Decroly E., Antiviral Res. 2013, 99, 292–300. [DOI] [PubMed] [Google Scholar]
- 120. Ringeard M., Marchand V., Decroly E., Motorin Y., Bennasser Y., Nature 2019, 565, 500–504. [DOI] [PubMed] [Google Scholar]
- 121. Lim S. P., Wen D., Yap T. L., Yan C. K., Lescar J., Vasudevan S. G., Antiviral Res. 2008, 80, 360–369. [DOI] [PubMed] [Google Scholar]
- 122. Lim S. P., Sonntag L. S., Noble C., Nilar S. H., Ng R. H., Zou G., Monaghan P., Chung K. Y., Dong H., Liu B., Bodenreider C., Lee G., Ding M., Chan W. L., Wang G., Jian Y. L., Chao A. T., Lescar J., Yin Z., Vedananda T. R., Keller T. H., Shi P. Y., J. Biol. Chem. 2011, 286, 6233–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lo M. C., Aulabaugh A., Jin G., Cowling R., Bard J., Malamas M., Ellestad G., Anal. Biochem. 2004, 332, 153–159. [DOI] [PubMed] [Google Scholar]
- 124. Coutard B., Decroly E., Li C., Sharff A., Lescar J., Bricogne G., Barral K., Antiviral Res. 2014, 106, 61–70. [DOI] [PubMed] [Google Scholar]
- 125. Dandliker W. B., Kelly R. J., Dandliker J., Farquahar J., Levin J., Immunochemistry 1973, 10, 219–227. [DOI] [PubMed] [Google Scholar]
- 126. Hendricks C. L., Ross J. R., Pichersky E., Noel J. P., Zhou Z. S., Anal. Biochem. 2004, 326, 100–105. [DOI] [PubMed] [Google Scholar]
- 127. Wang C., Leffler S., Thompson D. H., Hrycyna C. A., Biochem. Biophys. Res. Commun. 2005, 331, 351–356. [DOI] [PubMed] [Google Scholar]
- 128.
- 128a. Akhtar M. K., Vijay D., Umbreen S., McLean C. J., Cai Y., Campopiano D. J., Loake G. J., Front. Bioeng. Biotechnol. 2018, 6, 146; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128b. Wooderchak W. L., Zhou Z. S., Hevel J., Current Protocols in Toxicology, Wiley, 2008, Chapter 4, Unit4.26. [DOI] [PubMed] [Google Scholar]
- 129. Salyan M. E., Pedicord D. L., Bergeron L., Mintier G. A., Hunihan L., Kuit K., Balanda L. A., Robertson B. J., Feder J. N., Westphal R., Shipkova P. A., Blat Y., Anal. Biochem. 2006, 349, 112–117. [DOI] [PubMed] [Google Scholar]
- 130. Du X., Gao Z. Q., Geng Z., Dong Y. H., Zhang H., J. Virol. 2020, 95, 749–756. [Google Scholar]
- 131. Pearson L. A., Green C. J., Lin, Petit A. P., Gray D. W., Cowling V. H., Fordyce E. A. F., SLAS Discov. 2021, 26, 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Boeke J. D., Trueheart J., Natsoulis G., Fink G. R., Methods Enzymol. 1987, 154, 164–175. [DOI] [PubMed] [Google Scholar]
- 133. Kasprzyk R., Fido M., Mamot A., Wanat P., Smietanski M., Kopcial M., Cowling V. H., Kowalska J., Jemielity J., Chem. Eur. J. 2020, 26, 11266–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kasprzyk R., Spiewla T. J., Smietanski M., Golojuch S., Vangeel L., De Jonghe S., Jochmans D., Neyts J., Kowalska J., Jemielity J., ChemRxiv. 2021, 10.26434/chemrxiv.14484933.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.
- 135a. Dias A., Bouvier D., Crépin T., McCarthy A. A., Hart D. J., Baudin F., Cusack S., Ruigrok R. W., Nature 2009, 458, 914–918; [DOI] [PubMed] [Google Scholar]
- 135b. Reich S., Guilligay D., Pflug A., Malet H., Berger I., Crépin T., Hart D., Lunardi T., Nanao M., Ruigrok R. W., Cusack S., Nature 2014, 516, 361–366. [DOI] [PubMed] [Google Scholar]
- 136. Wakai C., Iwama M., Mizumoto K., Nagata K., J. Virol. 2011, 85, 7504–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Shibagaki Y., Ikuta N., Iguchi S., Takaki K., Watanabe S., Kaihotsu M., Masuda C., Maeyama K., Mizumoto K., Hattori S., J. Virol. Methods 2014, 202, 8–14. [DOI] [PubMed] [Google Scholar]
- 138. Omoto S., Speranzini V., Hashimoto T., Noshi T., Yamaguchi H., Kawai M., Kawaguchi K., Uehara T., Shishido T., Naito A., Cusack S., Sci. Rep. 2018, 8, 9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Noble E., Cox A., Deval J., Kim B., Virology 2012, 433, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wang W., Shin W. J., Zhang B., Choi Y., Yoo J. S., Zimmerman M. I., Frederick T. E., Bowman G. R., Gross M. L., Leung D. W., Jung J. U., Amarasinghe G. K., Cell Rep. 2020, 30, 153–163.e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Baughman B. M., Jake Slavish P., DuBois R. M., Boyd V. A., White S. W., Webb T. R., ACS Chem. Biol. 2012, 7, 526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Fernández-García Y., Horst S. T., Bassetto M., Brancale A., Neyts J., Rogolino D., Sechi M., Carcelli M., Günther S., Rocha-Pereira J., Antiviral Res. 2020, 183, 104947. [DOI] [PubMed] [Google Scholar]
- 143.
- 143a. Hausmann S., Zheng S., Fabrega C., Schneller S. W., Lima C. D., Shuman S., J. Biol. Chem. 2005, 280, 20404–20412; [DOI] [PubMed] [Google Scholar]
- 143b. Hercik K., Brynda J., Nencka R., Boura E., Arch. Virol. 2017, 162, 2091–2096; [DOI] [PubMed] [Google Scholar]
- 143c. Jain R., Butler K. V., Coloma J., Jin J., Aggarwal A. K., Sci. Rep. 2017, 7, 1632; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143d. Pugh C. S., Borchardt R. T., Stone H. O., J. Biol. Chem. 1978, 253, 4075–4077. [PubMed] [Google Scholar]
- 144. Zhang J., Zheng Y. G., ACS Chem. Biol. 2016, 11, 583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Ahmed-Belkacem R., Sutto-Ortiz P., Guiraud M., Canard B., Vasseur J. J., Decroly E., Debart F., Eur. J. Med. Chem. 2020, 201, 112557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Tomassini J., Selnick H., Davies M. E., Armstrong M. E., Baldwin J., Bourgeois M., Hastings J., Hazuda D., Lewis J., McClements W., Antimicrob. Agents Chemother. 1994, 38, 2827–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Noshi T., Kitano M., Taniguchi K., Yamamoto A., Omoto S., Baba K., Hashimoto T., Ishida K., Kushima Y., Hattori K., Kawai M., Yoshida R., Kobayashi M., Yoshinaga T., Sato A., Okamatsu M., Sakoda Y., Kida H., Shishido T., Naito A., Antiviral Res. 2018, 160, 109–117. [DOI] [PubMed] [Google Scholar]