

Abstract
Background:
Renal ischemia-reperfusion injury (IRI) is a major factor contributing to acute kidney injury and it is associated with a high morbidity and mortality if untreated. Renal IRI depletes cellular and tissue adenosine triphosphate (ATP), which compromises mitochondrial function, further exacerbating renal tubular injury. Currently, no treatment for IRI is available. This study investigates the protective role of treprostinil in improving mitochondria biogenesis and recovery during rat renal IRI.
Methods:
Male Sprague Dawley rats were randomly assigned to groups: control, sham, IRI-placebo or IRI-treprostinil and subjected to 45 minutes of bilateral renal ischemia followed by 1–72 hours reperfusion. Placebo or treprostinil (100 ng/kg/min) was administered subcutaneously via an osmotic minipump.
Results:
Treprostinil significantly reduced peak elevated serum creatinine (SCr) levels and accelerated normalization relative to IRI-placebo (p<0.0001). Treatment with treprostinil also inhibited IRI-mediated renal apoptosis, mitochondrial oxidative injury (p<0.05), and the release of cytochrome c (p<0.01) vs. IRI-placebo. In addition, treprostinil preserved renal mitochondrial DNA copy number (p<0.0001) and renal ATP levels (p<0.05) to nearly those of sham-operated animals. Non-targeted semiquantitative proteomics showed reduced levels of ATP synthase subunits in the IRI-placebo group which were restored to sham levels by treprostinil treatment (p<0.05). Furthermore, treprostinil reduced renal IRI-induced upregulated Drp1 and pErk protein levels, and restored Sirt3 and Pgc-1α levels to baseline (p<0.05).
Conclusions:
Treprostinil reduces mitochondrial-mediated renal apoptosis, inhibits mitochondria fission, and promotes mitochondria fusion, thereby accelerating mitochondrial recovery and protecting renal proximal tubules from renal IRI. These results support the clinical investigation of treprostinil as a viable therapy to reduce renal IRI.
Keywords: acute kidney injury, prostacyclin, mitochondria, apoptosis
Graphical Abstract
1. Introduction
Renal ischemia-reperfusion injury (IRI) is a multifactorial process and a major cause of acute kidney injury (AKI). During ischemic conditions, the lack of oxygen inhibits Na+-K+-ATPase activity which increases intracellular Na+ and attracts intracellular water, resulting in cellular edema and apoptosis [1, 2]. Excessive apoptosis contributes to the loss of renal proximal tubular cells and exacerbates IRI-induced AKI [3]. One of the early and major targets of renal IRI is the depletion of adenosine triphosphate (ATP), which disrupts mitochondria structure and function. As the master energy producer enriched in renal proximal tubular cells [4, 5], mitochondria play a central role in kidney function [6].
Mitochondrial homeostasis is maintained by the balance of fission and fusion, which is disrupted during renal IRI. Renal IRI activates fission by the translocation of dynamin related protein 1 (Drp1) to accumulate on the outer membrane of mitochondria causing mitochondrial fragmentation [7]. Normally, mitochondrial fusion compensates for the loss of mitochondria to maintain the number of functional healthy mitochondria [8], however, a lack of mitochondrial fusion during renal IRI exaggerates fission-mediated mitochondria fragmentation [9, 10]. Consequently, cytochrome c is released from the mitochondrial membrane and into the cytosol [3, 11], triggering mitochondria-mediated apoptosis. Maintaining mitochondrial homeostasis is essential to reduce apoptosis and improve the kidneys recovery from renal IRI.
Mitochondrial biogenesis, the generation of new functional mitochondria, require sufficient ATP to meet the energy demands of the kidney, especially renal proximal tubular cells [12]. However, the low turnover of mitochondria during renal IRI depletes ATP, ultimately leading to irreversible kidney damage. Mitochondrial biogenesis is primarily regulated by peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc-1α), and targeting Pgc-1α accelerates mitochondria recovery, thereby expediting proximal tubule repair after renal IRI [13, 14]. In fact, promoting mitochondrial biogenesis by induction of Pgc-1α may be a pharmacological approach to treat renal IRI-induced AKI [15]. Previously, we demonstrated that treprostinil (Remodulin®), an FDA-approved prostacyclin (PGI2) analog, attenuates rat renal IRI [16]. This study investigates the renoprotective role of treprostinil in reducing mitochondria-mediated injury through the Drp1/Sirt3/Pgc-1α pathway during rat renal IRI.
2. Materials and Methods
2.1. IRI Animal model
Male Sprague Dawley rats weighing 200–250 gm (Charles River Laboratories, Wilmington, MA), approximately 7–8 weeks old were housed in a laminar-flow, specific pathogen-free atmosphere in the Central Research Facilities of Rhode Island Hospital (RIH, Providence, RI) with a standard diet and water supplied ad libitum.
Animals were randomly divided into four groups: control, sham, IRI-placebo and IRI-treprostinil. Briefly, animals were anesthetized with isoflurane and subjected to bilateral renal ischemia for 45 minutes, afterward clamps were removed to allow reperfusion for 1–72 hours, as previously described [16]. Control animals were not subjected to any surgical manipulation and serve as baseline, whereas sham-operated animals had incisions only, to account for any influence from basic surgical handling or anesthesia. All surgical procedures were performed by the same surgeon who was blinded to treatment. All procedures involving animals were performed with approval from RIH Institutional Animal Care and Use Committee in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
2.2. Experimental Design
Treprostinil (Remodulin®) and placebo (sodium chloride, metacresol, sodium citrate, water for injection) are manufactured by United Therapeutics, Corp. (Durham, NC, USA). Treprostinil or placebo (100 ng/kg/min) was administrated subcutaneously via osmotic minipumps (Alzet Inc., Cupertino, CA), which were implanted approximately 18–24 hours before renal IRI to ensure steady-state concentrations at the time of IRI. Post-reperfusion, animals were kept under a heating lamp for 2 hours and were given regular food and water ad libitum. The general condition of the rats was checked three times daily. Animals were euthanized at 1, 3, 6, 24, 48, and 72 hours post-IRI by inhalation of carbon dioxide followed by cervical dislocation if needed. Blood samples were collected via tail vein and terminal blood samples were collected via cardiac puncture, centrifuged at 3,000 g for 10 minutes, and the isolated serum was stored at −20 °C. Kidney tissue was immediately snap-frozen in liquid nitrogen then stored at −80 °C for experiment analysis.
2.3. Serum Creatinine Measurements
Serum creatinine (SCr) concentrations were measured using QuantiChrom Creatinine Assay Kit (BioAssay System, Hayward, CA), n=4–10 animals/group.
2.4. Histopathology
Kidneys were harvested, bisected through the hilum in the coronal plane, fixed in 10% formalin and paraffin-embedded, n=4–5 animals/group. Whole mount coronal sections (2 μm) were stained with Periodic Acid–Schiff staining reaction (PAS) and evaluated using light microscopy by a renal pathologist blinded to all animal groups (×200, scale = 100 μm). Sections were assessed with respect to the extent of epithelial cell necrosis, as indicated by specific histopathological features of nuclear pyknosis or fragmentation, detachment from basement membranes and accumulation of necrotic debris within tubule lumens, and to the presence of non-lethal tubule injury as indicated by tubular ectasia and loss of apical brush borders in proximal tubule segments. The following semiquantitative grading system was used: 0: <1%; 1: 1–10%; 2:10–25%; 3: 25–50%; 4: >50% of all tubules. Separate injury scores were generated for cortex and outer stripe of outer medulla then combined for a composite score. Images of representative areas for each slide were obtained prior to decoding the animal treatment group from which the respective specimens originated.
2.5. TUNEL Assay
Paraffin-embedded kidney tissue sections (5 μm) were used to perform Click-iT™ Plus TUNEL Assay (Alexa Fluor™ 647 dye, Invitrogen, Carlsbad, CA). The nuclei were counterstained with Hoechst (Invitrogen) according to manufacturer’s instruction. Renal sections were viewed under a Nikon Eclipse Ti2 inverted confocal microscope (Nikon, Tokyo, Japan). Random fields (n=4–5/section) with the same acquisition setting were imaged for each slide (n=3–4 animals/group). Renal tubular DNA fragmentation was semi-quantitated based on the ratio of intensity of TUNEL and Hoechst using Image J (×400, scale bar = 50 μm).
2.6. Tissue ATP concentrations measurements
Snap-frozen kidney tissue (n=3–4 animals/group) was homogenized in ice-cold 2% trichloroacetic acid. The supernatants were collected and neutralized with Tris-acetate (100 mM) and EDTA (2 mM, pH, 7.8). ATP concentrations were determined using the Enliten ATP Assay System Bioluminescence Detection Kit (Promega, Madison, WI).
2.7. SWATH-MS proteomics analysis
ATP synthase subunits were measured using sequential windowed acquisition of all theoretical fragment ion mass spectra (SWATH-MS)-based proteomics [17, 18], n=4–5 animals/group). Protein extraction, pressure cycling technology based protein digestion, and LC-QTOF/MS analysis were performed as described [17]. Briefly, snap-frozen rat kidney tissue was weighed on ice and placed in bead-mill tubes containing fresh urea lysis buffer (8 M urea, 50 mM triethylammonium bicarbonate (Sigma-Aldrich, St. Louis, MO), 10 mM dithiothreitol and protease inhibitor cocktail (Thermo Scientific). Tissues were homogenized using a Bead Ruptor 24 (Omni, Kennesaw, GA) and centrifuged at 12,000 g for 5 min at 4 °C. The samples were diluted to 500 μg/100 μl before digestion. Proteins were digested using pressure cycling technology according to modified methods [17, 18]. Briefly, samples were digested with TPCK-treated trypsin (SCIEX, Framingham, MA) at trypsin/protein ratio of 1:20 under high pressure cycles in a Barocycler (Pressure BioSciences Inc., South Easton, MA). The reaction was stopped by addition of acetonitrile: water (50:50) with 5% formic acid (Fisher Scientific, MA). The supernatant containing peptides was collected for mass spectrometry analysis [17]. Samples were analyzed using a SCIEX 5600 TripleTOF mass spectrometer coupled to Acquity UHPLC HClass system (Waters Corp., Milford, MA) [17]. Subsequently, proteins were identified, and relative quantification was performed through SWATH-MS acquisition, followed by data-dependent analysis using Spectronaut™ (Biognosys AG, Switzerland).
2.8. Oxidative stress
The activity of renal catalase (CAT), superoxide dismutase (SOD), glutathione (GSH), and the protein carbonyl concentration were determined using CAT, SOD, GSH, and Protein Carbonyl Fluorometric assays (Cayman Chemicals). Kidney tissue samples were homogenized (n=4–6/group), and their protein concentrations were calculated using a bovine serum albumin (Pierce) standard prior to analyte determination.
2.9. mtDNA copy number
Total DNA was extracted from snap-frozen kidney tissue (n=4–5 animals/group) using DNeasy Blood & Tissue Kit (Qiagen, MD). DNA concentration was determined using a Nanodrop 2000 (Thermo Scientific, Waltham, MA). Mitochondrial DNA (mtDNA) and nuclear DNA content (nDNA) were determined by mitochondria encoded NADH-ubiquinone oxidoreductase chain 1 (mt-Nd1) and nuclear DNA Gapdh using Taqman® probes (Applied Biosystems, Foster City, CA), respectively. Quantitative real-time PCR was performed using a Vii7 Real-Time PCR System (Life Technologies, Carlsbad, CA). The mtDNA copy number was calculated using the ratio between mitochondrial and nuclear DNA (mt/n) [19, 20]: ΔCt = nDNA Ct − mtDNA Ct
Relative mtDNA content = 2 × 2ΔCt
mtDNA copy number = mtDNA/nDNA
2.10. Cytosolic and mitochondrial fractionation
Snap-frozen kidney tissue was homogenized in 0.25 M sucrose buffer (Millipore Sigma, Burlington, MA), 0.1 mM EDTA (Bio-Rad, Hercules, CA), 10 mM HEPES (Sigma-Aldrich, St. Louis, MO) with protease and phosphatase inhibitor (Thermo Scientific) using a Dounce homogenizer. Supernatant was collected and centrifuged at 13,000 g for 15 mins and collected as the cytosolic fraction. The crude mitochondria pellet was washed twice and suspended in PBS containing protease and phosphatase inhibitor cocktail (Thermo Scientific) and 0.1% Triton X-100, then disrupted twice with a sonicator at 40% of maximum setting for 10 seconds. Following lysate centrifugation, the supernatant was collected as the mitochondria fraction.
2.11. Western blot analysis
Proteins were separated by tris-glycine gel electrophoresis (Thermo Scientific), transferred to a PVDF membrane, and incubated with primary antibodies against Drp-1 (CST-8570, RRID: AB_10950498) and MFF (CST-84580, RRID: AB_2728769), Cytochrome c (sc-13156, RRID: AB_627385), Sirt3 (sc-365175, RRID: AB_10710522), total Erk1/2 (CST-9102, RRID: AB_330744), pErk1/2 (CST-4370, RRID: AB_2315112), Gapdh (sc-32233, RRID: AB_627679), Cox4 (sc-517553, RRID: AB_2797784), followed by IRDye® 680 RD goat anti-mouse (RRID:AB_10956588), or goat anti-rabbit IgG (H+L) (RRID:AB_621841), and IRDye® 800CW goat anti-mouse (RRID:AB_621842) or goat anti-rabbit IgG (H+L) (RRID:AB_621843). Blots were imaged using LI-COR Odyssey® CLx scanner (LI-COR Biosciences, Lincoln, NE). Protein band density was determined using Image J and normalized to Gapdh (cytosol, whole lysate), Cox4 (mitochondria), and expressed as a fold-change vs. sham (n=3–5 animals/group).
2.12. Quantitative real-time PCR analysis
RNA was extracted from snap-frozen renal cortex segments (n=3–6 animals/group) using TRIzol™ (Invitrogen,) according to manufacturer’s protocol. The purity and concentration of RNA were measured at 260/280 nm (Nanodrop, Thermo Scientific). Two micrograms of total RNA from each sample were used to generate cDNA using SuperScript™ IV First-Strand Synthesis System kit (Invitrogen). Renal mRNA levels of Nqo1, Gclc, Ppargc1a, Mfn1, Mfn2, Opa1 and Gapdh were analyzed using Taqman® probes (Applied Biosystems, CA). Relative mRNA expression was calculated using the ΔΔCt method and normalized to Gapdh expression. Real-time qPCR was performed using a ViiA 7 Real-Time PCR System (Life Technologies, CA).
2.13. Statistical Analysis.
Data are represented as the mean ± standard deviation (SD), n=3–10 animals/group. Comparisons between the groups were performed using one- or two-way analysis of variance with Turkey’s post-test (GraphPad Prism v7.0, San Diego, CA). Significance was defined as p-value<0.05.
3. Results
3.1. Treprostinil protects the kidneys from renal IRI
Renal IRI results in an increase of serum creatinine (SCr), a surrogate marker of kidney dysfunction. In this study, SCr levels obtained from sham-operated animals (0.6 ± 0.1 mg/dL) were similar to control animals (0.3 ± 0.1 mg/dL), confirming the absence of renal injury in the sham group. In the IRI-placebo group, however, SCr levels were significantly elevated as early as 1-hour post-IRI (1.0 ± 0.2 mg/dL, p<0.0001) and reached peak levels at 24-hour post-reperfusion vs. sham (2.0 ± 0.5 mg/dL, p<0.0001). In contrast, treatment with treprostinil significantly reduced the time to reach peak SCr levels (0.9 ± 0.2 mg/dL) vs. placebo (p<0.0001) to 6-hour post-IRI and returned to baseline levels by 24-hour after IRI (Figure 1A).
Figure 1. Treprostinil attenuates renal IRI.


(A) Serum creatinine (SCr) concentration, measured before ischemia (pre-IRI) and at 1–72 hours post-reperfusion in CTRL, sham, PLB, and TRE-treated animals. (B) Representative histopathological images of PAS-stained paraffin sections of kidneys from rats sacrificed at 6-, 24-, and 48-hour post-reperfusion and from sham-operated animals (×200, scale bar = 100 μm). Black arrows indicate tubular epithelial cell necrosis and detachment from basement membranes in IRI-placebo animals. Semiquantitative analysis of tubular epithelial injury, histological features associated with (C) reversible tubular epithelial cell injury, consisting of tubular ectasia and loss of apical brush borders; (D) irreversible tubular epithelial cell injury, consisting of epithelial cell nuclear pyknosis, detachment of necrotic epithelial cells from basement membranes and filling of tubule lumens by casts comprising necrotic cell debris. Slides were evaluated in a blinded manner using a grading system: 0: <1%; 1: 1–10%; 2: 10–25%; 3: 25–50%; 4: >50% of all tubules. Data are represented as mean ± SD; ***p<0.001 vs. sham, †p<0.05, ††p<0.01, †††p<0.001 vs. placebo (n = 4–10/group). Two-way ANOVA, Tukey’s multiple comparison test. IRI: Ischemia-reperfusion injury; SCr: Serum creatinine; CTRL: control; PLB: IRI-placebo; TRE: IRI-treprostinil; PAS: Periodic acid–Schiff staining reaction.
To evaluate the morphology of renal tubular cells, kidney sections obtained at 6-, 24-, and 48-hour after IRI were stained with PAS to visualize the extracellular matrix structures such as tubule basement membranes and to demonstrate tubular epithelial apical brush borders. Loss of brush borders from proximal tubules of cortex and outer medullary stripe is a sensitive indicator of sublethal epithelial cell injury. Control and sham-operated animals showed normal morphology of the outer stripe of outer medulla at all time points with complete preservation of apical brush borders (purple staining), Figure 1B. In contrast, the IRI-placebo group showed severe ischemic effects characterized by epithelial cell necrosis and detachment from tubular basement membranes and loss of epithelial cell apical brush borders at the 6-hour time point. These features of epithelial injury were most prominent within tubules of the outer stripe of outer medulla and, to a lesser extent, in tubules of deep cortex. Although only occasional necrotic epithelial cells remained evident at 48 hours, tubular ectasia and absence of apical brush borders persisted (Figure 1B – D). The extent of epithelial necrosis, as judged by nuclear pyknosis and detachment of epithelial cells (arrows), was markedly reduced in treprostinil-treated animals relative to placebo following IRI. In addition, only partial attenuation of apical brush borders was observed at 24-hour post-reperfusion, while essentially full reconstitution of normal histology was achieved by 48-hours in the treprostinil-treated animals (Figure 1B – D). Note the preservation of PAS-positive (purple staining) brush borders along apical surfaces of epithelial cells from sham-operated animals and reconstitution of brush borders in treprostinil-treated animals at 48-hour. Brush border preservation indicates the absence of epithelial cell injury. These data suggest that treprostinil protects the kidney against early IRI-induced proximal tubular cell injury, thereby diminishing the severity of AKI.
3.2. Treprostinil inhibits mitochondrial damage and apoptosis
Renal IRI-induced AKI causes mitochondrial damage which contributes to renal tubular epithelial cell apoptosis and kidney damage [6]. The effect of treprostinil on renal tubular apoptosis was examined using a TUNEL assay on kidney sections post-IRI. In IRI-placebo animals, an increase in TUNEL-positive cells, indicating nuclear fragmentation in renal tubular epithelial cells, was observed as early as 6-hour after reperfusion (1.4-fold vs. sham, p<0.05). In contrast, treprostinil significantly reduced peak nuclear fragmentation to that of sham and control levels, reflecting a 42% reduction in IRI-induced apoptosis vs. placebo (p<0.05), Figure 2A, B.
Figure 2. Treprostinil reduces renal tubular apoptosis.

(A) Renal apoptosis in situ, measured by TUNEL staining (red fluorescence) of outer medullary region in rat kidney sections at 6-hour post-reperfusion (×400, scale bar = 50 μm). Nuclei were counterstained with Hoechst (blue fluorescence). Random fields (n=4–5/section) with the same acquisition setting were examined for each slide. (B) Apoptosis was determined by the intensity of TUNEL/Hoechst, normalized to the area and quantified with the same threshold using image J software. (C) Renal cytosol cytochrome c protein levels at 1-hour post-reperfusion by western blot, normalized to Gapdh, and quantification was performed using image J software. Data are represented as mean ± SD; *p<0.05, **p<0.01 vs. sham, †p<0.05, ††p<0.01 vs. placebo (n = 3–5/group). One-way ANOVA, Tukey’s multiple comparison test. TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling; cyto: cytosol; mito: mitochondria.
Renal IRI-induced rupture of the mitochondrial membrane promotes the mitochondrial release of cytochrome c into the cytosol, thereby triggering caspase activation and cell death [11]. In this study, cytosolic cytochrome c protein concentration significantly increased in the IRI-placebo group by 3.0-fold vs. sham (p<0.01) at 1-hour post-reperfusion, which treprostinil reduced to that of sham, Figure 2C. We previously demonstrated the inhibitory effects of treprostinil on renal IRI-mediated activation of caspase-3, −8, and −9 [16]. Together, these data demonstrate that treprostinil inhibits the release of mitochondrial cytochrome c early post-reperfusion, thereby limiting its function as a downstream mediator involved in renal apoptosis.
3.3. Treprostinil restores renal ATP and ATP synthase levels
To determine the protective role of treprostinil on renal ischemia-induced ATP depletion, we measured renal ATP concentrations at 1–48 hours post-reperfusion. As early as 1-hour post-reperfusion, renal ATP levels were reduced in both the IRI-placebo and IRI-treprostinil group by 35% (29 ± 4.6 nmol/mg) and 33% (30 ± 9 nmol/mg), respectively, relative to sham (45 ± 10.6 nmol/mg). Reductions in renal ATP levels in the IRI-placebo group continued to decrease to less than 50% of sham at 24-hour post-reperfusion (22 ± 3.5 nmol/mg vs. 41 ± 4.5 nmol/mg, p<0.05), where they remained at 48-hour post-reperfusion (p<0.01). A striking contrast was found in treprostinil-treated animals, where renal ATP levels recovered to 81% of sham by 24-hour post-reperfusion (33 ± 9 nmol/mg) and these levels were sustained through 48-hour post-reperfusion (37 ± 2.7 nmol/mg), Figure 3A. These data indicate that treprostinil restores renal ATP levels after IRI-induced depletion, a key factor in mitigating renal IRI.
Figure 3. Treprostinil restores renal ATP and ATP synthase levels.

(A) Renal ATP levels at 1, 6, 24, and 48-hour after reperfusion measured by bioluminescent assay; (B) ATP synthase subunits at 48-hour post-reperfusion measured by SWATH-MS based proteomics, normalized to Gapdh, and expressed as a fold-change over control. Data are represented as mean ± SD; *p<0.05, **p<0.01, ***p<0.001 vs. sham, †p<0.05, ††p<0.01, †††p<0.001 vs. placebo (n = 3–5/group). Two-way ANOVA, Tukey’s multiple comparison test. ATP: adenosine triphosphate; SWATH-MS: Sequential windowed acquisition of all theoretical fragment ion mass spectra; Atp5f1: ATP synthase F(0) complex subunit B1; Atp5f1c: ATP synthase subunit gamma; Atp5f1d: ATP synthase subunit delta; Atp5f1e: ATP synthase subunit epsilon; Atp5i: ATP synthase subunit e; Atp5j: ATP synthase-coupling factor 6; Atp5fj2: ATP synthase subunit f; Atp5l: ATP synthase subunit g; Atp5o: ATP synthase subunit o; mt-Atp8: ATP synthase protein 8.
To further evaluate the role of treprostinil in preserving mitochondrial ATP after renal IRI, ATP sub-proteins were identified and quantified using SWATH-MS-based proteomics [17]. We found that the ATP synthase subunits were significantly reduced in IRI-placebo after 48-hour reperfusion. Not only did treprostinil treatment attenuate this reduction for ATP 5f1, 5f1c, 5f1d, 5f1e, 5i, 5j, 5j2, 5l, 5o, and Mt-Atp8, but levels were restored to sham levels (Figure 3B). These findings demonstrate that treprostinil upregulates ATP synthase, which is responsible for mitochondrial ATP synthesis and maintaining ATP production, thereby accelerating mitochondrial recovery after renal IRI [21].
3.4. Treprostinil reduces renal oxidative damage
Next, we examined the role of treprostinil in regulating renal oxidative stress during IRI and measured the activity of CAT, SOD, the content of GSH and protein carbonyl. Renal CAT activity was significantly reduced at 24-hour and 48-hour post-reperfusion in the IRI-placebo group vs. sham (972 ± 268 vs. 1399 ± 460 U/mg protein, p<0.05; 596 ± 204 vs. 1316 ± 185 U/mg protein, p<0.0001, respectively). In contrast, treprostinil restored renal CAT activity to sham levels by 48-hour post-reperfusion (1038 ± 426 U/mg protein), Figure 4A. Similarly, SOD activity in the IRI-placebo group was decreased at 48-hour post-reperfusion vs. sham (9.1 ± 1.2 vs. 14.7 ± 0.5 U/mg protein, p<0.0001) and treatment with treprostinil restored SOD to sham levels (12.1 ± 2.7 U/mg protein), Figure 4B. Interestingly, renal GSH levels were significantly reduced at 1 hour after reperfusion in the IRI-placebo group vs. sham (1.7 ± 0.7 vs. 4.5 ± 0.6 μM/mg protein, p<0.0001), whereas treprostinil restored GSH levels to sham levels (3.4 ± 0.3 μM/mg protein), Figure 4C. Lastly, renal protein carbonyl levels were significantly increased in IRI-placebo kidney homogenates after 24-hour reperfusion compared to control (3.5 ± 0.8 vs. 2.4 ± 0.3 nmol/mg protein, p<0.01), which treatment with treprostinil reduced (2.4 ± 0.2 vs. 2.4 ± 0.3 nmol/mg protein, p<0.05 vs. placebo), Figure 4D.
Figure 4. Treprostinil reduces mitochondrial oxidative injury and preserves renal mtDNA copy number.

(A) CAT activity, (B) SOD activity, (C) GSH content, and (D) protein carbonyl content in the kidney compared between control, sham, IRI-placebo and IRI-treprostinil groups at 1-, 6-, 24- and 48-hours post-reperfusion. Renal mRNA expression levels of (E) Nqo1, (F) Gclc. Data are represented as mean ± SD; *p<0.05, **p<0.01, ***p<0.001 vs. sham, †p<0.05, ††p<0.01, †††p<0.001 vs. placebo (n = 4–6/group). Two-way ANOVA, Tukey’s multiple comparison test. CAT: catalase; SOD: superoxide dismutase; GSH: glutathione; Nqo1: NAD(P)H dehydrogenase (quinone 1); Gclc: Glutamate-cysteine ligase catalytic subunit.
Additionally, the mRNA expression of the antioxidant genes NAD(P)H dehydrogenase (quinone 1) (Nqo1) and Glutamate-cysteine ligase catalytic subunit (Gclc) in IRI-placebo animals were significantly reduced to 41% (p<0.05) and 14% of sham (p<0.0001) at 6-hour post-reperfusion, and further declined to 13% and 4% of sham by 48-hour post-reperfusion (p<0.0001), respectively. In stark contrast, treatment with treprostinil improved Nqo1 and Gclc mRNA levels to 79% of sham at 6-hour post-reperfusion and 52% of sham at 48-hour post-reperfusion (p<0.05 vs. placebo), respectively (Figure 4E, 4F). Together, these data indicate that treprostinil protects kidney from extended renal oxidative damage after renal IRI.
3.5. Treprostinil preserves mtDNA and mitochondrial biogenesis
The mtDNA copy number reflects the number of mitochondrial genomes per cell and decreases in renal tissue following renal IRI [22, 23]. In our study, the mtDNA copy number relative to nDNA Gapdh was reduced by 33% in the IRI-placebo group at 3-hour post-reperfusion vs. control (p<0.01). Meanwhile, treatment with treprostinil restored mtDNA copy number to baseline as early as 1-hour post-reperfusion vs. placebo (p<0.01) and preserved these levels throughout the study period (Figure 5A). In addition to reducing renal oxidative injury, these findings suggest that treprostinil promotes mitochondrial recovery early after renal IRI, which, in turn, preserves renal ATP levels.
Figure 5. Treprostinil preserves mtDNA and mitochondrial biogenesis.

(A) Renal mtDNA copy number at 1, 3, and 6 hours after reperfusion determined by the ratio of mt-Nd1/Gapdh, measured by real-time qPCR; (B) Renal mRNA expression of Pgc-1α at 1-, 6-, 24-, and 48-hours post-reperfusion was measured by real-time qPCR, normalized to Gapdh and is expressed as a fold-increase over sham kidney. Data are represented as mean ± SD; *p<0.05 **p<0.01, ***p<0.001 vs. sham, †p<0.05, ††p<0.01, †††p<0.001 vs. placebo (n = 4–5/group). Two-way ANOVA, Tukey’s multiple comparison test. mtDNA: mitochondrial DNA; mt-Nd1: mitochondrially encoded NADH dehydrogenase 1; Pgc-1α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha.
To further examine the role of treprostinil on renal mitochondrial biogenesis, we measured the gene expression of Pgc-1α, the master regulator of mitochondrial biogenesis. In IRI-placebo animals, renal Pgc-1α mRNA levels reduced to 37% of sham at 1-hour and further declined to 26% of sham at 48-hour after reperfusion (p<0.01). Alternatively, animals treated with treprostinil exhibited nearly a two-fold improvement in Pgc-1α mRNA expression to 46% of sham at 1-hour post-reperfusion and restored levels to sham (134%) by 48-hour reperfusion (p<0.0001), Figure 5B. Together, these findings indicate that treprostinil reduces mitochondrial injury early after renal IRI.
3.6. Treprostinil improves mitochondrial dynamics after renal IRI
Mitochondrial homeostasis requires a functional balance between mitochondria turnover, e.g. continuous cycles of fission and fusion [24] and mitochondrial biogenesis [25], both of which renal IRI disrupts. In the IRI-placebo group, mitochondrial Drp1 increased by 3.6-fold vs. sham (p<0.05) at 1-hour post-reperfusion, compared to 1.7-fold in the IRI-treprostinil group, representing a 53% reduction vs. placebo (Figure 6A). In parallel, the protein level of mitochondrial fission factor (Mff) significantly increased by 5.0-fold in the IRI-placebo group vs. sham (p<0.001) which was significantly reduced to 2.8-fold by treprostinil treatment (p<0.05), Figure 6B. In addition, pErk1/2 protein level was increased in IRI-placebo by 14.6-fold vs. sham at 3-hour post-reperfusion (p<0.001), but only increased by 5.3-fold in treprostinil-treated animals, representing a 64% reduction vs. placebo (p<0.0001), Figure 6C. Conversely, in IRI-placebo animals, mitochondrial Sirt3 protein level decreased by 77% from control at 1-hour post-reperfusion, while Sirt3 remained at baseline levels in the IRI-treprostinil group (p<0.05), Figure 6D.
Figure 6. Treprostinil improves mitochondrial dynamics after renal IRI.


Protein expression of mitochondrial (A) Drp1 and (B) Mff at 1-hour post-reperfusion; (C) total Erk1/2 and pErk1/2 at 3-hour post-reperfusion; (D) Sirt3 at 1 hour post-reperfusion by western blot; targets were normalized to Cox4 or Gapdh, and quantification was performed using image J software; renal mRNA expression of (E) Mfn1, (F) Mfn2, (G) Opa1 at 1-, 6-, 24-, and 48-hour post-reperfusion was measured by real-time qPCR, normalized to Gapdh and is expressed as a fold-increase over sham or control. Data are represented as mean ± SD; *p<0.05 **p<0.01, ***p<0.001 vs. sham, †p<0.05, ††p<0.01, †††p<0.001 vs. placebo (n = 4–5/group). Two-way ANOVA, Tukey’s multiple comparison test. Drp1: Dynamin related protein 1; Mff: mitochondrial fission factor; Sirt3: NAD-dependent deacetylase sirtuin-3; Mfn1: Mitofusin-1; Mfn2: Mitofusin-2; Opa1: Mitochondrial Dynamin Like GTPase; pErk1/2: Phosphorylated extracellular signal-regulated kinases; Cox4: Cytochrome c oxidase subunit 4.
In line with the mitochondrial protein changes, the mRNA levels of Mitofusin-1 (Mfn1), Mitofusin-2 (Mfn2), and Mitochondrial Dynamin Like GTPase (Opa1) were significantly reduced to 57% (p<0.05), 16% (p<0.0001), and 71% (p<0.05) of sham levels in IRI-placebo at 1-hour post-reperfusion, which further declined to 36% (p<0.01), 14% (p<0.0001), 31% (p<0.0001) of sham at 48-hour post-reperfusion. In contrast, treatment with treprostinil restored Mfn1, Mfn2, and Opa1 mRNA levels to sham levels by 48-hour post-reperfusion, Figure 6E–G. Altogether, these data support the notion of mitochondrial dynamics protection by treprostinil, which is mediated, in part, by the inhibition of Erk1/2 activation resulting in suppression of Drp1-mediated mitochondrial fission and enhancement of Sirt3-mediated mitochondrial fusion.
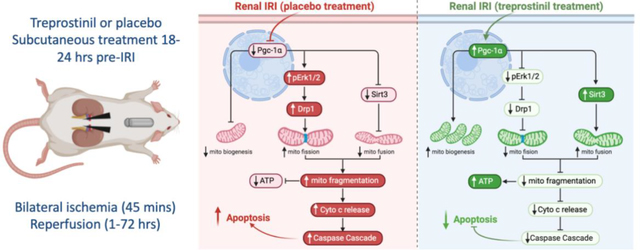
4. Discussion
Renal IRI is a multi-factorial process which limits the clinical efficacy of treatment by a single targeted therapy. Having demonstrated that treprostinil ameliorates renal IRI in vivo [16], the current study investigates the hypothesis that treprostinil reduces mitochondrial-mediated apoptosis to protect the kidneys against renal IRI. Our data identify a novel mechanism by which treprostinil ameliorates renal IRI. Specifically, this study demonstrates that treatment with treprostinil inhibits mitochondrial damage and oxidative injury, which preserves renal proximal tubular cell integrity, restores renal ATP, accelerates mitochondrial biogenesis and recovery by favorably regulating the renal Drp1/Sirt3/Pgc-1α pathway, summarized in Figure 7. Renal proximal tubular cells are particularly vulnerable to mitochondrial-mediated apoptosis post-IRI [26]. Szeto et al. [27] showed the rupture of the mitochondrial membrane and subsequent loss of cristae in the proximal tubules in the outer stripe of the outer medulla after 45-minute bilateral ischemia followed by 24-hour reperfusion. Similarly, in our study, PAS staining reveals that the IRI-placebo group exhibits renal tubular swelling, vacuolization, necrosis, detachment of tubular basement membranes and loss of proximal tubular brush borders in the outer stripe of outer medulla early post-reperfusion, all of which were mitigated by treprostinil. In addition, treprostinil reduced rapid DNA fragmentation, which is exacerbated by renal IRI and associated with apoptosis triggered by renal oxidative stress [28, 29]. These data support the hypothesis that treprostinil mitigates renal IRI-induced mitochondrial damage in vivo.
Figure 7. Summary of treprostinil in reducing mitochondrial injury during rat renal IRI.

IRI: ischemia-reperfusion injury; Pgc-1α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; pErk1/2: Phosphorylated extracellular signal-regulated kinases; Drp1: Dynamin related protein 1; mito: mitochondria; Sirt3: NAD-dependent deacetylase sirtuin-3; ATP: adenosine triphosphate; cyto: cytosol;
Renal ATP concentrations reflect the ability of renal mitochondrial to synthesize and utilize ATP [27]. Depletion of ATP is one of the earliest and most profound changes induced by renal ischemia and a major driving factor involved in mitochondrial injury [30, 31]. During renal IRI, mitochondrial injury reduces ATP synthesis and leads to rapid ATP depletion after ischemia [32] and subsequent Na+/K+ ATPases dysfunction and the accumulation of intracellular sodium and calcium [30, 31, 33]. Prolonged ATP depletion during renal IRI indicates the severe impairment of mitochondria, which further delays kidney function recovery. Prostacyclin has a protective role in reducing oxidative damage by increasing Na+/K+ ATPase activity via EP receptor activation [33, 34]. In this study, treatment with treprostinil restored renal ATP levels to nearly that of control after 24-hour reperfusion, with parallel preservations in renal ATP synthase subunits. Our previous work shows that treprostinil significantly improved hepatic adenine nucleotides levels following rat orthotopic liver transplantation [35], further supporting our current data which demonstrates that treprostinil reverses the loss of renal ATP levels as a necessary mechanism to protect mitochondrial function during renal IRI.
Oxidative injury plays a major role in further aggravating ischemia injury [36] and several studies have shown the importance of preserving antioxidant levels during renal IRI [37–39]. In this study, treprostinil significantly restored the depleted antioxidants, e.g., CAT, SOD and GSH during renal IRI. In parallel, treprostinil markedly reduced elevated renal protein carbonyl levels, suggesting that treprostinil reduces renal oxidative damage.
The normal process of maintaining a balance between mitochondria fission and fusion plays a pivotal role in sustaining the number of functional mitochondria within cells [40]. During renal IRI, however, excessive fission leads to a collapse of the mitochondrial membrane potential, resulting in mitochondrial fragmentation in renal proximal tubular cells [41, 42]. Mitochondrial fragmentation leads to the leakage and damage of mtDNA from the mitochondria and into cytosol, which in turn, exacerbates mitochondrial dysfunction [43]. Moreover, lower mtDNA copies have been significantly associated with the development of cardiovascular disease and all-cause mortality and death due to infections in patients with chronic kidney disease [44]. Here, we found that IRI-placebo animals exhibit a significant decrease in mtDNA early post-renal IRI, indicating the unbalanced mitochondrial dynamics and mitochondrial fragmentation. Notably, treprostinil restored mtDNA content after 1-hour reperfusion, confirming its role in protecting mitochondria in the acute period post-IRI.
Inhibition of Drp1 mitochondrial translocation early during renal IRI abrogates Drp1-mediated mitochondrial fission and subsequent apoptosis to protect proximal tubular cells from renal IRI [10, 41]. Here, we show that renal mitochondrial Drp1 accumulates early post-reperfusion and that treprostinil rescues renal mitochondria from excessive fission by significantly inhibiting renal Drp1 accumulation early post-reperfusion. Acting in concert with Drp1, Sirt3 has been identified as a key regulator of mitochondrial homeostasis due to its antioxidant role and by regulating renal mitochondrial Complex I activity to maintain basal ATP levels [45]. Overexpression of Sirt3 protects against mitochondrial damage during renal IRI by prompting optic atrophy-1 triggered mitochondrial fusion and blocking Drp1 recruitment along the mitochondrial outer membrane [9]. Further supporting our hypothesis of treprostinil-mediated protection against renal IRI-induced mitochondrial injury and apoptosis, our data show that treprostinil restores the renal IRI-induced downregulation of mitochondrial Sirt3, thereby reducing damaged mitochondrial injury and, overall, improving mitochondrial quality.
Lastly, mitochondria biogenesis plays an essential role in maintaining sufficient mitochondria to meet the energy needs of proximal tubules in order to fully function and recover from ischemic injury [46]. Collier et al. [14] demonstrated that early Erk1/2 phosphorylation downregulates the mitochondrial biogenesis regulator Pgc-1α, thereby causing mitochondrial dysfunction. Additionally, Tran et al. [23] found that Pgc-1α accelerates renal recovery by stimulating nicotinamide adenine dinucleotide biosynthesis, which increases the vasodilator prostaglandin E2 after renal IRI. In this study, Erk1/2 phosphorylation is reduced early post-IRI along with upregulated Pgc-1α levels in treprostinil-treated animals, supporting the hypothesis that treprostinil favorably regulates mitochondrial biogenesis. Thus, maintaining mitochondrial homeostasis by treprostinil is essential to reduce apoptosis and improve kidney recovery from renal IRI.
5. Conclusion
In conclusion, this study identifies a novel mechanism of treprostinil, an FDA approved prostacyclin analog, in reducing mitochondrial injury during renal IRI. Specifically, our data show that treprostinil maintains mitochondrial homeostasis via inhibiting Drp-1-mediated mitochondrial fission and Erk1/2 phosphorylation, and upregulating mitochondrial Sirt3 and Pgc-1α to promote mitochondrial fusion and biogenesis, thereby counteracting mitochondrial damage early post-renal IRI. These findings support the potential of treprostinil to serve as a therapeutic agent to treat clinical renal IRI.
Supplementary Material
Highlights.
What is already known about this subject: Renal IRI significantly contributes to mortality in AKI, currently there is no treatment.
What this study adds: Treprostinil inhibits renal IRI-induced mitochondrial injury and apoptosis in vivo.
Clinical significance: Treprostinil is a novel and viable treatment to ameliorate renal IRI-induced AKI.
Acknowledgement
We thank Dr. Benjamin J. Barlock for technical support
FUNDING
The material presented herein is supported by Institutional Development Award (IDeA) #U54GM115677 from the National Institute of General Medical Sciences of the National Institutes of Health, which funds Brown University Advance-CTR (U54GM115677), and University of Rhode Island Core Lab (P20GM103430). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- IRI
Ischemia-reperfusion injury
- AKI
Acute kidney injury
- PGI2
Prostacyclin
- SCr
Serum creatinine
- mtDNA
Mitochondrial DNA
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- PAS
Periodic acid–Schiff staining reaction
- CAT
Catalase
- SOD
Superoxide dismutase
- GSH
Glutathione
- Nqo1
NAD(P)H dehydrogenase (quinone 1)
- Gclc
Glutamate-cysteine ligase catalytic subunit
- Drp1
Dynamin related protein 1
- Mff
Mitochondrial fission factor
- Mfn1
Mitofusin-1
- Mfn2
Mitofusin-2
- Opa1
Mitochondrial Dynamin Like GTPase
- Sirt3
NAD-dependent deacetylase sirtuin-3
- pErk1/2
Phosphorylated extracellular signal-regulated kinases
- Pgc-1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- Cox IV
Cytochrome c oxidase subunit IV
- SWATH-MS
Sequential windowed acquisition of all theoretical fragment ion mass spectra
Footnotes
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Molinas SM, Trumper L, Serra E, Elias MM, Evolution of renal function and Na+, K +-ATPase expression during ischaemia-reperfusion injury in rat kidney, Mol Cell Biochem 287(1–2) (2006) 33–42. [DOI] [PubMed] [Google Scholar]
- [2].Coux G, Trumper L, Elias MM, Renal function and cortical (Na(+)+K(+))-ATPase activity, abundance and distribution after ischaemia-reperfusion in rats, Biochim Biophys Acta 1586(1) (2002) 71–80. [DOI] [PubMed] [Google Scholar]
- [3].Kaushal GP, Basnakian AG, Shah SV, Apoptotic pathways in ischemic acute renal failure, Kidney Int 66(2) (2004) 500–6. [DOI] [PubMed] [Google Scholar]
- [4].Weinberg JM, Venkatachalam MA, Roeser NF, Saikumar P, Dong Z, Senter RA, Nissim I, Anaerobic and aerobic pathways for salvage of proximal tubules from hypoxia-induced mitochondrial injury, Am J Physiol Renal Physiol 279(5) (2000) F927–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen Y, Fry BC, Layton AT, Modeling glucose metabolism and lactate production in the kidney, Math Biosci 289 (2017) 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Emma F, Montini G, Parikh SM, Salviati L, Mitochondrial dysfunction in inherited renal disease and acute kidney injury, Nat Rev Nephrol 12(5) (2016) 267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li N, Wang H, Jiang C, Zhang M, Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway, Exp Cell Res 369(1) (2018) 27–33. [DOI] [PubMed] [Google Scholar]
- [8].Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L, OPA1 requires mitofusin 1 to promote mitochondrial fusion, Proc Natl Acad Sci U S A 101(45) (2004) 15927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang Q, Xu J, Li X, Liu Z, Han Y, Xu X, Li X, Tang Y, Liu Y, Yu T, Li X, Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway, J Cell Physiol 234(12) (2019) 23495–23506. [DOI] [PubMed] [Google Scholar]
- [10].Perry HM, Huang L, Wilson RJ, Bajwa A, Sesaki H, Yan Z, Rosin DL, Kashatus DF, Okusa MD, Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI, J Am Soc Nephrol 29(1) (2018) 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ow YP, Green DR, Hao Z, Mak TW, Cytochrome c: functions beyond respiration, Nat Rev Mol Cell Biol 9(7) (2008) 532–42. [DOI] [PubMed] [Google Scholar]
- [12].Szeto HH, Liu S, Soong Y, Birk AV, Improving mitochondrial bioenergetics under ischemic conditions increases warm ischemia tolerance in the kidney, Am J Physiol Renal Physiol 308(1) (2015) F11–21. [DOI] [PubMed] [Google Scholar]
- [13].Funk JA, Schnellmann RG, Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury, Toxicol Appl Pharmacol 273(2) (2013) 345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Collier JB, Whitaker RM, Eblen ST, Schnellmann RG, Rapid Renal Regulation of Peroxisome Proliferator-activated Receptor gamma Coactivator-1alpha by Extracellular Signal-Regulated Kinase 1/2 in Physiological and Pathological Conditions, J Biol Chem 291(52) (2016) 26850–26859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ishimoto Y, Inagi R, Mitochondria: a therapeutic target in acute kidney injury, Nephrol Dial Transplant 31(7) (2016) 1062–9. [DOI] [PubMed] [Google Scholar]
- [16].Ding M TE, Birkenbach M, Gohh R, Akhlaghi F, and Ghonem NS, Treprostinil, a prostacyclin analog, ameliorates renal ischemia-reperfusion injury: Preclinical studies in a rat model of acute kidney injury, Nephrol Dial Transpl (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Barlock BJ, Use of Hyphenated Mass Spectrometry to Uncover True NAFLD Effect on Human Drug Disposition Proteins, Open Access Dissertations, 2019. [Google Scholar]
- [18].Jamwal R, Barlock BJ, Adusumalli S, Ogasawara K, Simons BL, Akhlaghi F, Multiplex and Label-Free Relative Quantification Approach for Studying Protein Abundance of Drug Metabolizing Enzymes in Human Liver Microsomes Using SWATH-MS, J Proteome Res 16(11) (2017) 4134–4143. [DOI] [PubMed] [Google Scholar]
- [19].Quiros PM, Goyal A, Jha P, Auwerx J, Analysis of mtDNA/nDNA Ratio in Mice, Curr Protoc Mouse Biol 7(1) (2017) 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE, Mayer GD, Greenamyre JT, Meyer JN, PCR based determination of mitochondrial DNA copy number in multiple species, Methods Mol Biol 1241 (2015) 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pfanner N, Warscheid B, Wiedemann N, Mitochondrial proteins: from biogenesis to functional networks, Nat Rev Mol Cell Biol 20(5) (2019) 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sun Z, Zhang X, Ito K, Li Y, Montgomery RA, Tachibana S, Williams GM, Amelioration of oxidative mitochondrial DNA damage and deletion after renal ischemic injury by the KATP channel opener diazoxide, Am J Physiol Renal Physiol 294(3) (2008) F491–8. [DOI] [PubMed] [Google Scholar]
- [23].Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, Parikh SM, PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection, Nature 531(7595) (2016) 528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chang CR, Blackstone C, Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1, Ann N Y Acad Sci 1201 (2010) 34–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rasbach KA, Schnellmann RG, PGC-1alpha over-expression promotes recovery from mitochondrial dysfunction and cell injury, Biochem Biophys Res Commun 355(3) (2007) 734–9. [DOI] [PubMed] [Google Scholar]
- [26].Bonventre JV, Yang L, Cellular pathophysiology of ischemic acute kidney injury, J Clin Invest 121(11) (2011) 4210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY, Seshan SV, Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury, J Am Soc Nephrol 22(6) (2011) 1041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Beeri R, Symon Z, Brezis M, Ben-Sasson SA, Baehr PH, Rosen S, Zager RA, Rapid DNA fragmentation from hypoxia along the thick ascending limb of rat kidneys, Kidney Int 47(6) (1995) 1806–10. [DOI] [PubMed] [Google Scholar]
- [29].Iwata M, Myerson D, Torok-Storb B, Zager RA, An evaluation of renal tubular DNA laddering in response to oxygen deprivation and oxidant injury, J Am Soc Nephrol 5(6) (1994) 1307–13. [DOI] [PubMed] [Google Scholar]
- [30].Malis CD, Bonventre JV, Mechanism of calcium potentiation of oxygen free radical injury to renal mitochondria. A model for post-ischemic and toxic mitochondrial damage, J Biol Chem 261(30) (1986) 14201–8. [PubMed] [Google Scholar]
- [31].Wilson DR, Arnold PE, Burke TJ, Schrier RW, Mitochondrial calcium accumulation and respiration in ischemic acute renal failure in the rat, Kidney Int 25(3) (1984) 519–26. [DOI] [PubMed] [Google Scholar]
- [32].Van Waarde A, Avison MJ, Thulin G, Gaudio KM, Shulman RG, Siegel NJ, Role of nucleoside uptake in renal postischemic ATP synthesis, Am J Physiol 262(6 Pt 2) (1992) F1092–9. [DOI] [PubMed] [Google Scholar]
- [33].Shinmura K, Tamaki K, Sato T, Ishida H, Bolli R, Prostacyclin attenuates oxidative damage of myocytes by opening mitochondrial ATP-sensitive K+ channels via the EP3 receptor, Am J Physiol Heart Circ Physiol 288(5) (2005) H2093–101. [DOI] [PubMed] [Google Scholar]
- [34].Koksel O, Ozdulger A, Aytacoglu B, Tamer L, Polat A, Sucu N, Yildirim C, Degirmenci U, Kanik A, The influence of iloprost on acute lung injury induced by hind limb ischemia-reperfusion in rats, Pulm Pharmacol Ther 18(4) (2005) 235–41. [DOI] [PubMed] [Google Scholar]
- [35].Ghonem N, Yoshida J, Stolz DB, Humar A, Starzl TE, Murase N, Venkataramanan R, Treprostinil, a prostacyclin analog, ameliorates ischemia-reperfusion injury in rat orthotopic liver transplantation, Am J Transplant 11(11) (2011) 2508–16. [DOI] [PubMed] [Google Scholar]
- [36].Dobashi K, Ghosh B, Orak JK, Singh I, Singh AK, Kidney ischemia-reperfusion: modulation of antioxidant defenses, Mol Cell Biochem 205(1–2) (2000) 1–11. [DOI] [PubMed] [Google Scholar]
- [37].Dare AJ, Bolton EA, Pettigrew GJ, Bradley JA, Saeb-Parsy K, Murphy MP, Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ, Redox Biol 5 (2015) 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Park EJ, Dusabimana T, Je J, Jeong K, Yun SP, Kim HJ, Kim H, Park SW, Honokiol Protects the Kidney from Renal Ischemia and Reperfusion Injury by Upregulating the Glutathione Biosynthetic Enzymes, Biomedicines 8(9) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Walker LM, York JL, Imam SZ, Ali SF, Muldrew KL, Mayeux PR, Oxidative stress and reactive nitrogen species generation during renal ischemia, Toxicol Sci 63(1) (2001) 143–8. [DOI] [PubMed] [Google Scholar]
- [40].Karbowski M, Youle RJ, Dynamics of mitochondrial morphology in healthy cells and during apoptosis, Cell Death Differ 10(8) (2003) 870–80. [DOI] [PubMed] [Google Scholar]
- [41].Brooks C, Wei Q, Cho SG, Dong Z, Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models, J Clin Invest 119(5) (2009) 1275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tang C, Dong Z, Mitochondria in Kidney Injury: When the Power Plant Fails, J Am Soc Nephrol 27(7) (2016) 1869–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Carelli V, Maresca A, Caporali L, Trifunov S, Zanna C, Rugolo M, Mitochondria: Biogenesis and mitophagy balance in segregation and clonal expansion of mitochondrial DNA mutations, Int J Biochem Cell Biol 63 (2015) 21–4. [DOI] [PubMed] [Google Scholar]
- [44].Fazzini F, Lamina C, Fendt L, Schultheiss UT, Kotsis F, Hicks AA, Meiselbach H, Weissensteiner H, Forer L, Krane V, Eckardt KU, Kottgen A, Kronenberg F, Investigators G, Mitochondrial DNA copy number is associated with mortality and infections in a large cohort of patients with chronic kidney disease, Kidney Int 96(2) (2019) 480–488. [DOI] [PubMed] [Google Scholar]
- [45].Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T, A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis, Proc Natl Acad Sci U S A 105(38) (2008) 14447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Weinberg JM, Mitochondrial biogenesis in kidney disease, J Am Soc Nephrol 22(3) (2011) 431–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.