

Abstract
Trait-associated genetic variants affect complex phenotypes primarily via regulatory mechanisms on the transcriptome. To investigate the genetics of gene expression, we performed cis- and trans- expression quantitative trait locus (eQTL) analyses, using blood-derived expression from 31,684 individuals through the eQTLGen Consortium. We detected cis-eQTLs for 88% of genes, and these were replicable in numerous tissues. Distal trans-eQTLs (detected for 37% out of 10,317 trait-associated variants tested) showed lower replication rates, partially due to low replication power and confounding by cell-type-composition. However, replication analyses in single-cell RNA-seq data prioritized intracellular trans-eQTLs. Trans-eQTLs exerted their effects via several mechanisms, primarily through regulation by transcription factors. Expression of 13% of the genes correlated with polygenic scores (PGS) for 1,263 phenotypes, pinpointing potential drivers for those traits. In summary, this work represents a large eQTL resource and its results serve as a starting point for in-depth interpretation of complex phenotypes.
Introduction
Expression quantitative trait loci (eQTLs) have become a common tool to interpret the regulatory mechanisms of variants identified by genome-wide association studies (GWAS). In particular, cis-eQTLs, where gene expression levels are affected by a gene-proximal (<1 megabases; Mb) single nucleotide polymorphism (SNP), have been widely used for this purpose. However, cis-eQTLs generally explain only a modest proportion of disease heritability1, suggesting additional routes to disease.
Trans-eQTLs, where the SNP is located distal to the gene (>5 Mb) or on another chromosome, usually have smaller effect sizes than cis-eQTLs and thus require larger sample sizes for detection. However, trans-eQTLs could be relevant for complex traits because, compared to stronger cis-eQTL effects, each individual trans-effect is less likely to be dampened by compensatory post-transcriptional buffering or removed from the population by negative selection2,3. Indeed, genes regulated by weak eQTL effects are estimated to have more impact on the phenotype as compared to those regulated by strong eQTL effects4. Individual trans-eQTL SNPs can affect many genes and have a widespread impact on regulatory networks. Consequently, weak trans-eQTLs have the potential to identify trait-relevant genes1,5–10, and have already been used to prioritize genes that are likely to contribute to disease5.
While trans-eQTLs are useful for the identification of the distal effects of a single variant, a different approach is required to determine the combined consequences of all variants associated with a polygenic trait. Polygenic scores (PGSs) summarize genome-wide combined risk for a complex disease into a single metric that may be used to stratify individuals into groups11,12. The recently proposed omnigenic model13,14 postulates that the heritability of most complex traits is dominated by numerous weak trans-effects and hypothesizes that those effects converge on a smaller set of trait-relevant ‘core’ genes. This suggests that associations between PGSs and gene expression (expression quantitative trait scores, eQTS) could help to prioritize putative trait-relevant genes (Supplementary Note, Liu et al.14). While it remains unclear what fraction of the genome affects complex traits, we here systematically investigated trans-eQTLs and eQTS to determine how genetic effects regulate genes and pathways, and whether these effects could be informative about the biology of the respective traits.
To maximize the statistical power to detect eQTLs and eQTSs, we performed a large-scale meta-analysis in up to 31,684 blood samples from 37 eQTLGen Consortium cohorts. This allowed us to identify cis-eQTLs for 16,987 genes, trans-eQTLs for 6,298 genes and eQTS effects for 2,568 genes (false discovery rate (FDR) <0.05, determined by permutations; Methods, Figure 1). We replicated these eQTLs across gene expression platforms, in other tissues and in single cell (sc)RNA-seq data. While the overall concordance was good, formal replication remained limited, possibly due to the effects of genetics on blood cell composition, the limited sample size of the available replication datasets and the cell-type-specific nature of distal effects. To demonstrate the utility of our resource, we combined the associations with additional data layers to gain biological insights into the mechanisms of blood eQTLs and complex traits.
Figure 1. Overview of the study.
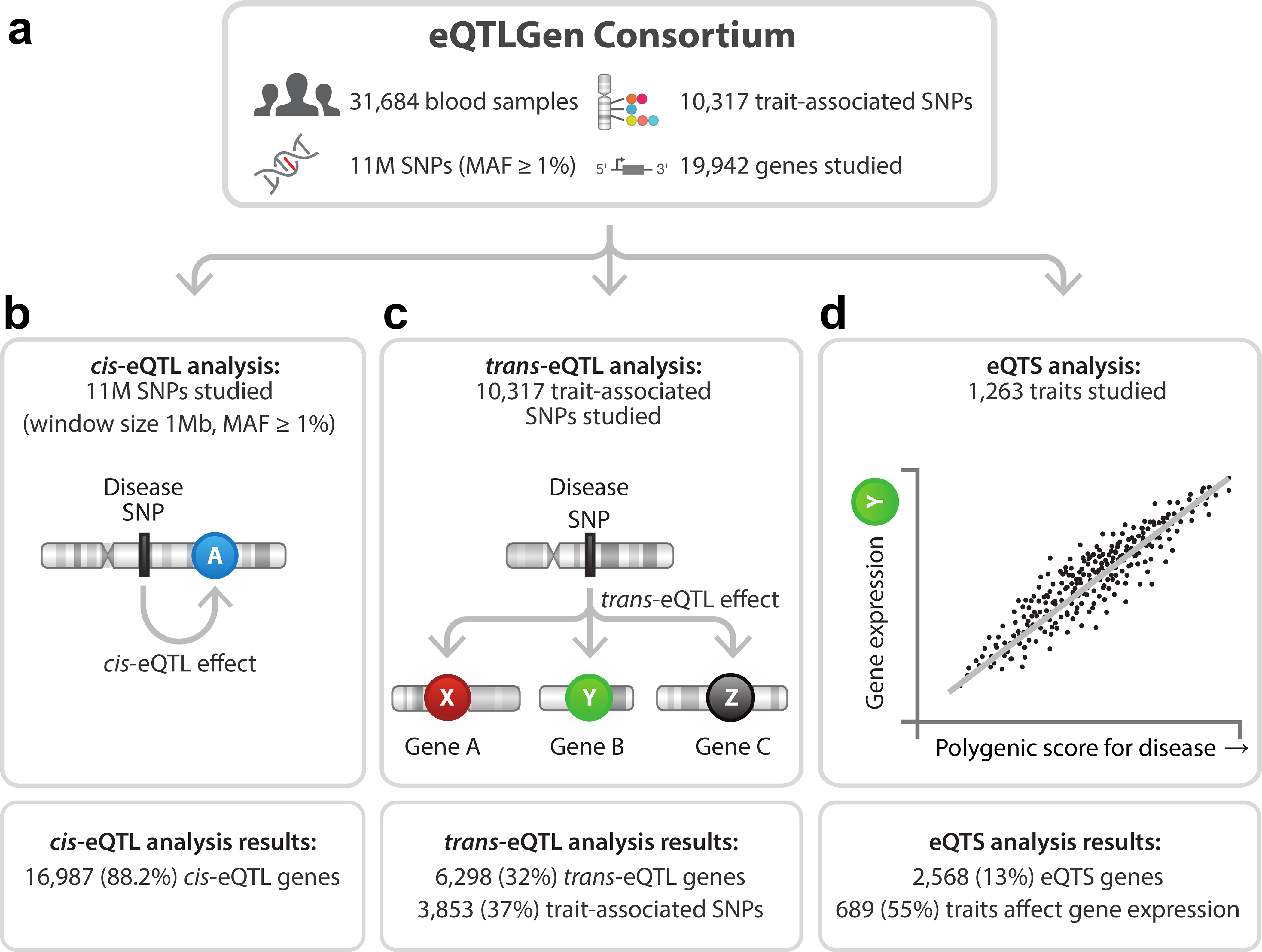
Overview of discovery analyses and their results.
Results
Meta-analyses on local and distal gene expression
We performed cis-eQTL, trans-eQTL and eQTS meta-analyses using eQTLGen Consortium data from 31,684 individuals (Figure 1a, Supplementary Table 1, Supplementary Note). Because the consortium contained both array- and sequencing-based expression datasets, we integrated different platforms using co-regulation patterns between genes (Methods). Inter-platform cis-eQTL, trans-eQTL and eQTS replication analyses indicated good concordance between platforms (on average 93.2%, 99.2% and 99.4% for significant cis-, trans- and eQTS effects), enabling integrated gene-level meta-analyses. These analyses also demonstrated that effects identified by our approach replicate between different blood datasets (Methods, Supplementary Note, Supplementary Figure 1a–c). We adopted a permutation-based strategy5,15,16 to account for multiple testing in the discovery meta-analyses (Methods, Supplementary Note), which was more conservative than a Benjamini-Hochberg FDR17 and less stringent than the Bonferroni method (Supplementary Figure 2).
In all the analyses, we accounted for unknown technical confounders (such as batch effects) and biological confounders (such as inter-individual differences in cell-type-composition) by correcting the expression data for up to 25 expression principal components (PCs) that were not associated with genetics (Methods). This correction adjusted for the majority of cell-type-composition effects in a subset of samples (BIOS cohort, N up to 3,831; Supplementary Note, Supplementary Figure 3). Nevertheless, we acknowledge that our dataset may include residual cell-type-composition effects.
Local genetic effects on blood gene expression
We identified cis-eQTLs (SNP gene distance <1 Mb, FDR<0.05; Methods) for 16,987 genes (88.2% of the 19,250 autosomal genes expressed in blood and tested in the cis-eQTL analysis; Figure 1b).
After we observed that cis-eQTLs replicated between whole blood datasets (Supplementary Figure 1a), we investigated the replicability of cis-eQTLs in other tissues. In 47 postmortem tissues (Genotype Tissue Expression (GTEx) data)18, we observed an average replication rate of 14.8% (discovery analysis without GTEx, Benjamini-Hochberg FDR<0.05 in GTEx; median across tissues 15.0%, range 3.6–29.6% when excluding whole blood) and, on average, a 94.9% concordance in allelic directions (median 95.2%, range 86.7–99.2%, when excluding whole blood) among the cis-eQTLs for which the lead SNP effect replicated in GTEx (Supplementary Note, Extended Data Figure 1 and Supplementary Data 1).
Genes highly expressed in blood but without detectable cis-eQTL were more likely to be intolerant to loss-of-function mutations in their coding region19 (two-sided Wilcoxon rank sum test, P=6.5×10−6; Figure 2a, Supplementary Table 2), suggesting that eQTLs on such genes are selectively constrained, as has been recently proposed20.
Figure 2. Results of the cis- and trans-eQTL analysis.
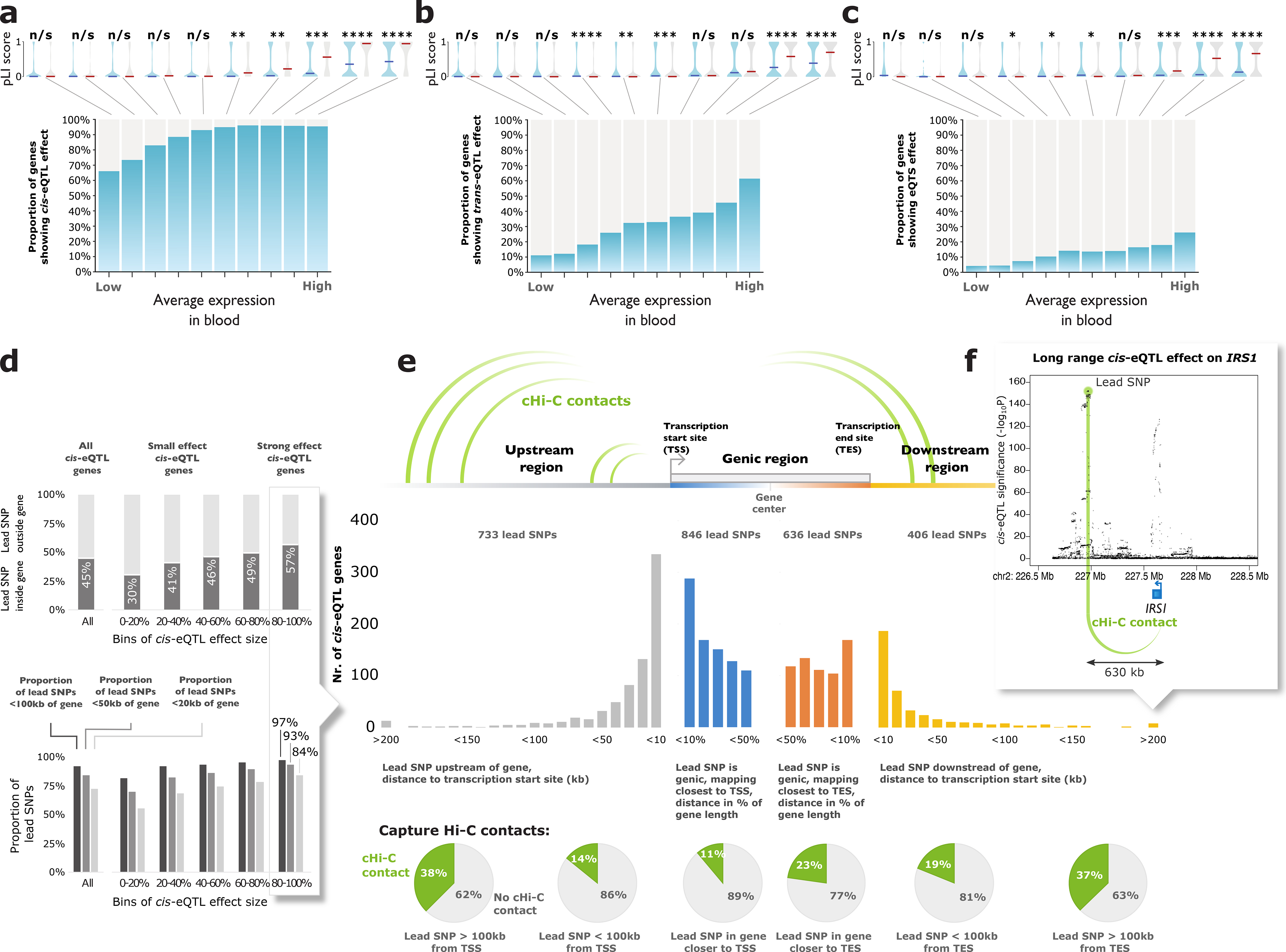
All genes tested in (a) cis-eQTL analysis, (b) trans-eQTL analysis, and (c) eQTS analysis were divided into 10 bins based on their average expression levels in blood (BIOS Cohort). Highly expressed genes without any eQTL effect (grey bars) were less tolerant to loss-of-function variants (two-sided Wilcoxon rank sum test on pLI scores). Indicated are median pLIs per bin. n/s (not significant) P>0.05; * P<0.05; ** P<0.01; *** P<0.001; **** P<1×10−4. (d) Genes with strong effect sizes are more likely to have a lead SNP located within (top panel) or close to the gene (bottom panel) (e) Lead cis-eQTL SNPs overlap capture Hi-C contacts with transcription start sites (TSS). (f) Example of IRS1 locus.
Ninety-two percent of lead cis-eQTL SNPs were located within 100kb of the gene (Figure 2d) and stronger cis-eQTLs typically had a smaller distance between SNP and gene (within 20kb for 84.1% of the top 20% strongest eQTLs).
Lead cis-eQTL SNPs which located >100kb from the transcription start site (TSS) or transcription end site (TES) of the gene were likely to overlap with capture Hi-C contacts (2.0-fold enrichment compared to when the location of the Hi-C target was flipped relative to the TSS; P<3.3×10−12; two-tailed two-sample test of equal proportions; Methods, Figure 2e, Supplementary Note). This suggests that some long-range cis-eQTLs are caused by physical interactions between the genomic regions of the SNP and the gene. For example, a capture Hi-C contact for IRS1 overlapped the lead eQTL SNP, mapping 630kb downstream from IRS1 (Figure 2f). Similarly, we observed an enriched overlap with Hi-C contacts for short-range cis-eQTL effects (<100kb, 1.3-fold; P<9.1×10−16; two-tailed two-sample test of equal proportions; Figure 2e, Supplementary Note).
When comparing our results to the 5,440 protein-coding cis-eQTL genes previously identified in 5,311 samples5, the lead SNPs typically mapped closer to the cis-eQTL gene (Supplementary Figure 4). In GWAS studies, larger sample sizes and denser imputation panels increase the resolution of signals in associated loci, especially for weaker effects. Additionally, GWAS simulations have indicated that lead GWAS signals map near the causal variant (within 33.5kb in 80% of cases)21. Since the majority of the lead cis-eQTL variants from our study map within 100kb of the TSS and TES, we consider it likely that causal variants for gene expression are also within these regions.
One third of trait-associated variants have distal effects
We focused on 10,317 trait-associated SNPs (GWAS P≤5×10−8; Methods, Supplementary Table 3) and identified 59,786 trans-eQTLs (SNP-gene distance >5 Mb; P<8.3×10−6, corresponding to an FDR<0.05; Supplementary Data 2, Extended Data Figure 2), representing 3,853 SNPs (37% of tested GWAS SNPs) and 6,298 genes (32% of tested genes; Figure 1c). The largest previous trans-eQTL meta-analysis in blood5 (N=5,311) identified trans-eQTLs for 8% of the tested SNPs, indicating that a larger sample size is beneficial for the identification of distal effects. Similar to cis-eQTLs, highly expressed genes without detectable trans-eQTL effects were more likely to be intolerant to loss-of-function variants (two-sided Wilcoxon rank sum test, P=6.4×10−7; Figure 2b), suggesting constrained expression of these genes.
While blood-cell-composition SNPs22 comprised 21% of the tested SNPs, they represented the majority (64%) of trans-eQTL SNPs. Many of the identified trans-eQTL SNPs may regulate the abundance of a specific blood cell type and could result in trans-eQTL effects on genes specifically expressed in that cell type. Although we corrected the individual expression datasets for cell-type composition effects using PCs (Methods, Supplementary Note), the fact that numerous trans-eQTLs emanate from known blood-cell-composition SNPs suggested a residual effect of cell composition.
To prioritize trans-eQTLs caused by intracellular mechanisms (i.e. gene regulation within cells), we applied several analytic strategies (Supplementary Note). Replication in purified cell types and cell lines identified 4,018 (6.7%) trans-eQTLs that replicated in at least one dataset or were supported by DNA methylation QTL data (Benjamini-Hochberg FDR<0.05; 93.3% average allelic concordance; Supplementary Note, Supplementary Figure 5, Supplementary Data 2). The replication rate in GTEx23 tissues was very low (discovery without GTEx, 0.07% of trans-eQTLs replicated in any non-blood tissue, 0.09% in blood, Benjamini-Hochberg FDR<0.05) but the allelic concordance of significant effects was, on average, 66% in non-blood tissues and 100% in blood (Supplementary Data 3). Despite these low replication rates, trans-eQTLs showed an inflation of replication signal in the majority of tissues (Supplementary Figure 6a), most notably in whole blood, esophagus muscularis, liver, heart atrial appendage and non-sun-exposed skin.
Compared to bulk datasets, scRNA-seq eQTL datasets are less impacted by cell-composition and are therefore ideal for trans-eQTL replication. We performed replication analyses in B-cells, CD4+ T-cells, CD8+ T-cells, classical monocytes, non-classical monocytes, dendritic cells, natural killer (NK) cells and plasma cells from up to 1,139 individuals (OneK1K, N=982 and 1M-scBloodNL, N=157; Supplementary Note). Depending on the cell type, we could reliably test between 1,917 and 27,582 discovery trans-eQTLs (Figure 3a) and replicated 35 trans-eQTLs at FDR<0.05 (Supplementary Table 4), with two effects replicating in more than one cell type. For those trans-eQTLs, the allelic concordance between the discovery and the replication analysis was 97%. For 7 out of the 8 cell types, we observed inflation of replication signal (Supplementary Table 5, Supplementary Figure 6a) and greater than expected allelic concordance with the discovery analysis (Figure 3a; Supplementary Table 5; two-sided binomial test P<0.05). Similarly, trans-eQTL effect sizes correlated significantly with replication effects in the scRNA-seq data (rb metric24; Methods, Figure 3a; two-sided P<0.05) for 4 cell types (classical monocytes (P=3.36×10−8, rb=0.514, S.E.=0.093), NK cells (P=3.24×10−4, rb=0.185, S.E.=0.051), CD8+ lymphocytes (P=3.41×10−3, rb=0.454, S.E.=0.155) and B cells (P=5.98×10−3, rb=0.049, S.E.=0.018)). More abundant cell types showed higher correlations with whole blood (Figure 3a, Pearson R2=0.53, two-sided P=0.04) and we observed similar correlations for replication datasets from several purified cell types (Supplementary Figure 7). When confining the analysis to 729 trans-eQTLs with an absolute average Z>1.96 over cell types (corresponding to a nominal P<0.05, Supplementary Table 4), we observed a relatively high effect direction concordance of 84% (Figure 3a, Supplementary Table 5, two-sided binomial test; P=1.25×10−84).
Figure 3. Trans-eQTL replication in scRNA-seq and mechanisms leading to trans-eQTLs.
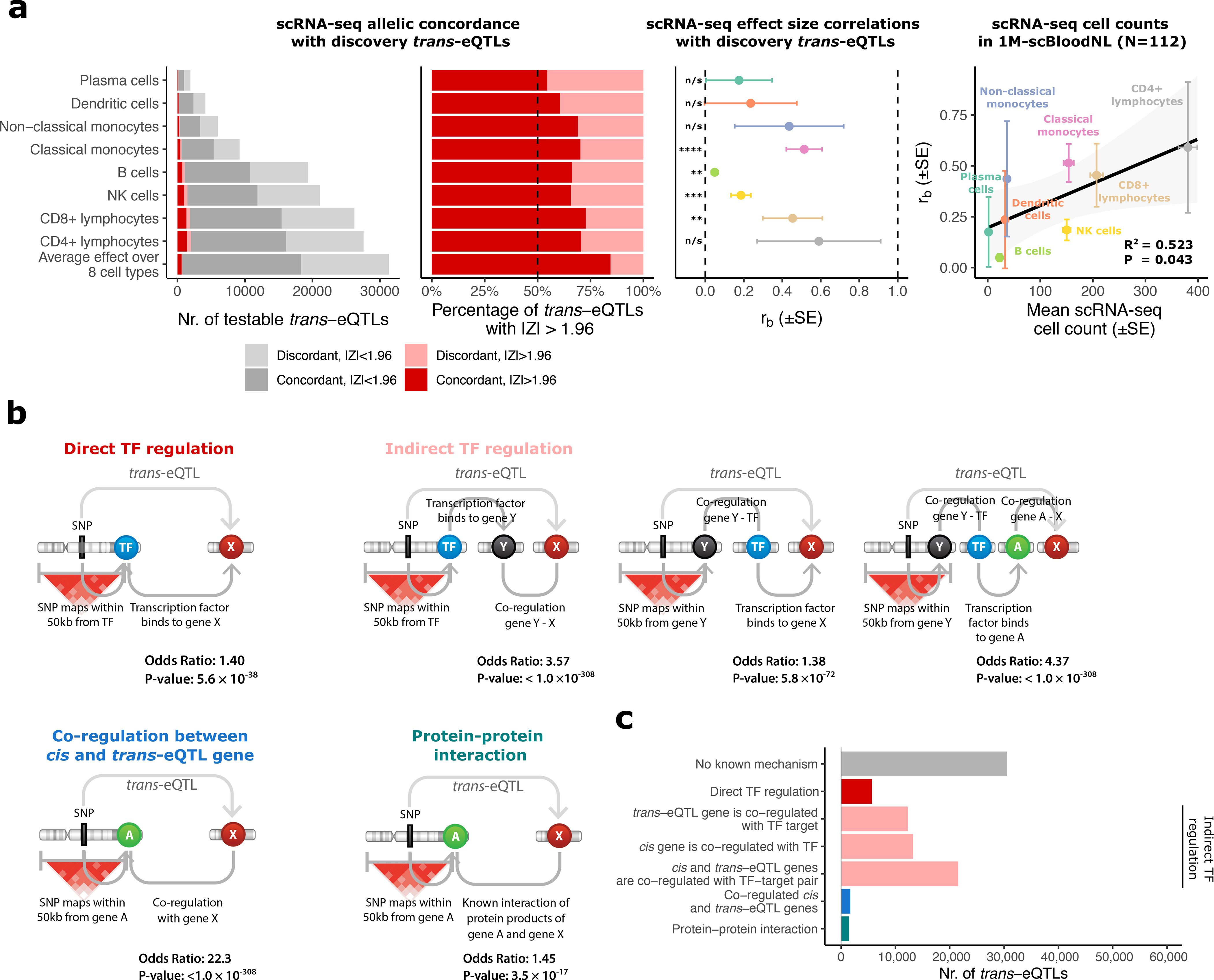
(a) Replication analyses in scRNA-seq of 8 cell types in up to 1,139 individuals. Left panels: allelic concordances relative to trans-eQTL effect direction in the discovery trans-eQTL analysis. Middle panel: correlation estimates (rb) of trans-eQTL effects between the discovery analysis in blood and scRNA-seq blood cell types and corresponding two-sided P-values (Methods). n/s P>0.05; * P<0.05; ** P<0.01; *** P<0.001; **** P<1×10−4. Error bars indicate the standard error (SE) for rb. Right panel: correlation between cell type counts (mean over the subset of samples from 1M-scBloodNL cohort; N=112) and rb estimates. Shown are the squared Pearson correlation coefficient and the two-sided P-value from the Pearson correlation test. Error bars indicate SE for rb and standard error of the mean (SEM) for the cell counts. (b) Enrichment analyses for TF binding, gene co-regulation and protein–protein interactions (PPIs). Cis-acting genes were determined by cis-eQTLs or assigned by the Pascal method (Methods, Supplementary Note). Shown are odds ratio and two-sided P-value from Fisher’s exact test. (c) All 59,786 trans-eQTLs stratified by putative mechanism of action. Hi-C enrichment results are not shown as we only observed enrichment when using a lenient threshold for Hi-C contacts (>0 value for contact). Full results are shown in Supplementary Figure 9.
These results suggest that some of the trans-eQTLs identified in blood are present in individual cell types, although it remains challenging to prioritize individual effects. The trans-eQTL effect sizes were generally small (median r=0.033; Supplementary Figure 8a, e, i; Supplementary Note). Considering the limited sample sizes of available bulk and scRNA-seq trans-eQTL datasets (N=388–1,480; N=2,905 for methylation QTL data, all <10% of discovery N), the statistical power to replicate these effects was low (Supplementary Figure 8g–h), limiting our ability to reliably distinguish cell-type-composition effects from intracellular effects. Therefore, we used all trans-eQTLs in the interpretive analyses (see also Supplementary Note).
To evaluate whether the trans-eQTLs could be explained by direct or indirect TF action25, protein–protein interactions (PPI)26 or co-regulation between cis- and trans-eQTL genes, we conducted enrichment analyses (Supplementary Note, Supplementary Figure 9, Figure 3b). Cis- and trans-eQTL genes emerging from the same SNP were 1.28-fold enriched by TF - TF target pairs, compared to all other gene pairs (P=4.0×10−21; two-sided Fisher’s exact test; Supplementary Figure 8). When we included additional local genes into the trans-eQTL loci with Pascal27, (Supplementary Note, Supplementary Figure 10) we observed a 1.40-fold enrichment for TFs (P=5.6×10−36; two-sided Fisher’s exact test; Figure 3b). There was also enrichment for genes co-regulated with known TFs (1.38-fold, P=5.8×10−72; two-sided Fisher’s exact test; Figure 3b), genes co-regulated with known target genes (3.57-fold, P<1.0×10−308; two-sided Fisher’s exact test; Figure 3b), and genes co-regulated with both (4.37-fold, P<1.0×10−308; two-sided Fisher’s exact test; Figure 3b), suggesting indirect consequences of transcriptional regulation. We observed a strong 22.3-fold enrichment (P<1.0×10−308; two-sided Fisher’s exact test) of co-regulated gene pairs and a 1.45-fold enrichment of protein–protein interaction (PPI)26 pairs (P=3.5×10−17; two-sided Fisher’s exact test), including co-regulated subunits of the same protein complex (e.g., products of CPSF1 and CPSF7) and receptor-ligand pairs (e.g., products of CSF3 and CSF3R). We note that cell-type-composition effects likely contribute to the enrichment of co-regulated gene pairs, as there was a depletion of co-regulation among 729 effects nominally replicating in scRNA-seq data (OR=0.5, P=0.015, two-sided Fisher’s exact test). Additionally, Hi-C chromatin contacts28 were also enriched for local–distal gene pairs (OR=1.47; P=2.4×10−153; two-sided Fisher’s exact test), suggesting that some trans-eQTLs could be driven by physical contact (Supplementary Figure 9). Altogether, 29,207 (49%) of the reported trans-eQTLs could be assigned a putative biological mechanism (Figure 3c, Supplementary Data 4). However, the trans-eQTL analysis was limited to trait-associated variants, so the observed enrichments might not generalize to all trans-eQTLs.
We identified 1,050 (10.2%) hub SNPs that regulated the expression of >10 genes (Supplementary Table 6). Of these, 196 (18.6%) had a global up- or down-regulating effect on downstream genes (two-sided binomial test, Bonferroni-corrected P<0.05, Supplementary Table 6). We identified 507 (48%) hub SNPs showing enrichment for TF- or miRNA-binding sites (one-sided Fisher’s exact test, Benjamini-Hochberg FDR<0.05; Supplementary Table 7). For nine of these (5 independent loci), we observed that the respective TF was encoded by a gene positioned <1 Mb from the hub SNP, suggesting a TF-mediated mechanism. For example, rs1708733529 (dbSNP 137) affects the expression of 88 neuronal genes, possibly through the neuronal repressor encoded by nearby gene REST (Figure 4, Supplementary Note).
Figure 4. REST locus regulates the expression of 88 trans-eQTL genes.
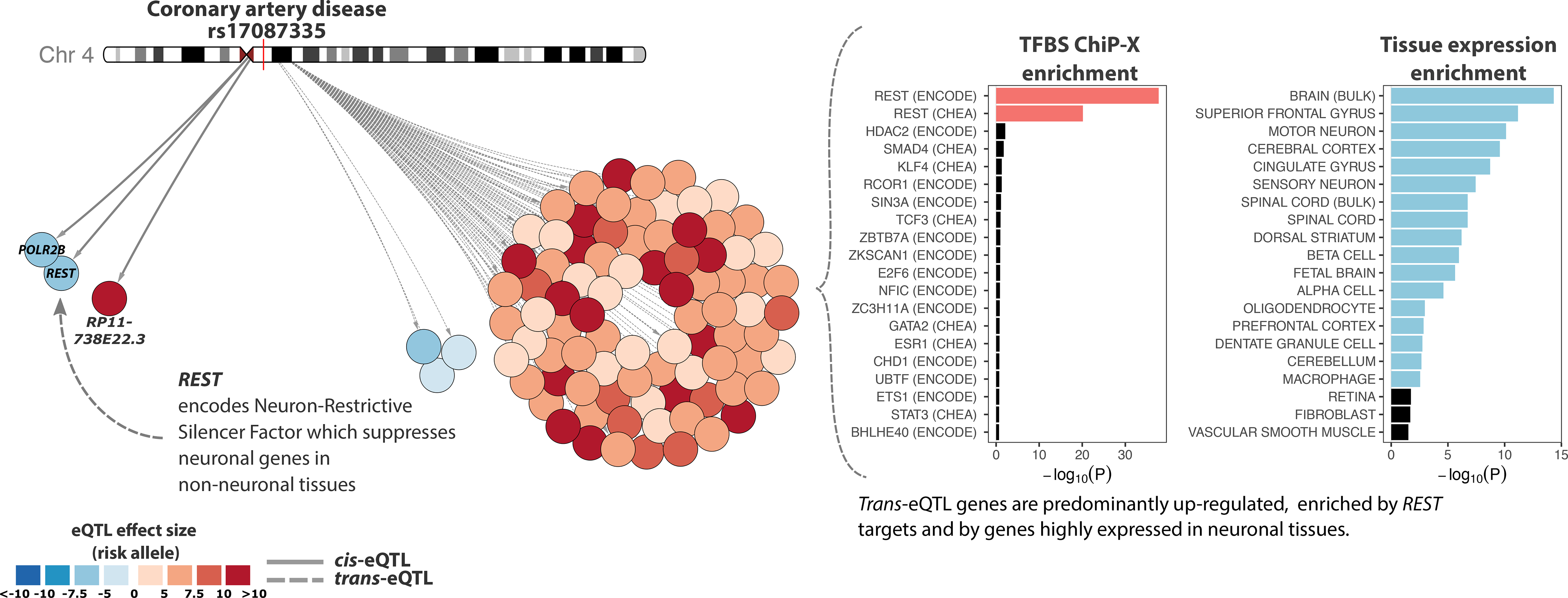
Left, overview of the cis- and trans-eQTL effects for coronary artery disease associated rs17087335. Color of the nodes indicates the trans-eQTL effect direction and size, relative to risk allele. Right, trans-eQTL genes for the REST locus are highly enriched for REST transcription factor targets (TF binding data from ENCODE67,68 and ChEA66) and for the expression of brain-related genes. For each TF and tissue, the length of the bar indicates -log10(P-value) from one-sided Fisher’s exact test (Methods). Twenty most significant effects are visualized.
We also identified 47 GWAS traits for which at least four independent variants affected the same gene in trans (Supplementary Table 8–10), 3.4-times higher than expected by chance (P=0.001; two-tailed two-sample test of equal proportions). For systemic lupus erythematosus (SLE)30, the expression of IFIT1, IFI44L, HERC5, IFI6, IFI44, RSAD2, MX1, ISG15, ANKRD55, OAS3, OAS2, OASL and EPSTI1 was affected by at least three SLE-associated genetic variants (FDR<0.05). These genes are nearly all known interferon genes in the SLE interferon signature31–33 (Supplementary Table 11), reflecting the involvement of interferon signaling in SLE pathophysiology (Figure 5). While the trans-eQTL analysis identified only one novel interferon signature gene, it helped to pinpoint SLE GWAS loci that collectively affect interferon signature genes.
Figure 5. SNPs associated with systemic lupus erythematosus (SLE) converge on a shared cluster of interferon-response genes.
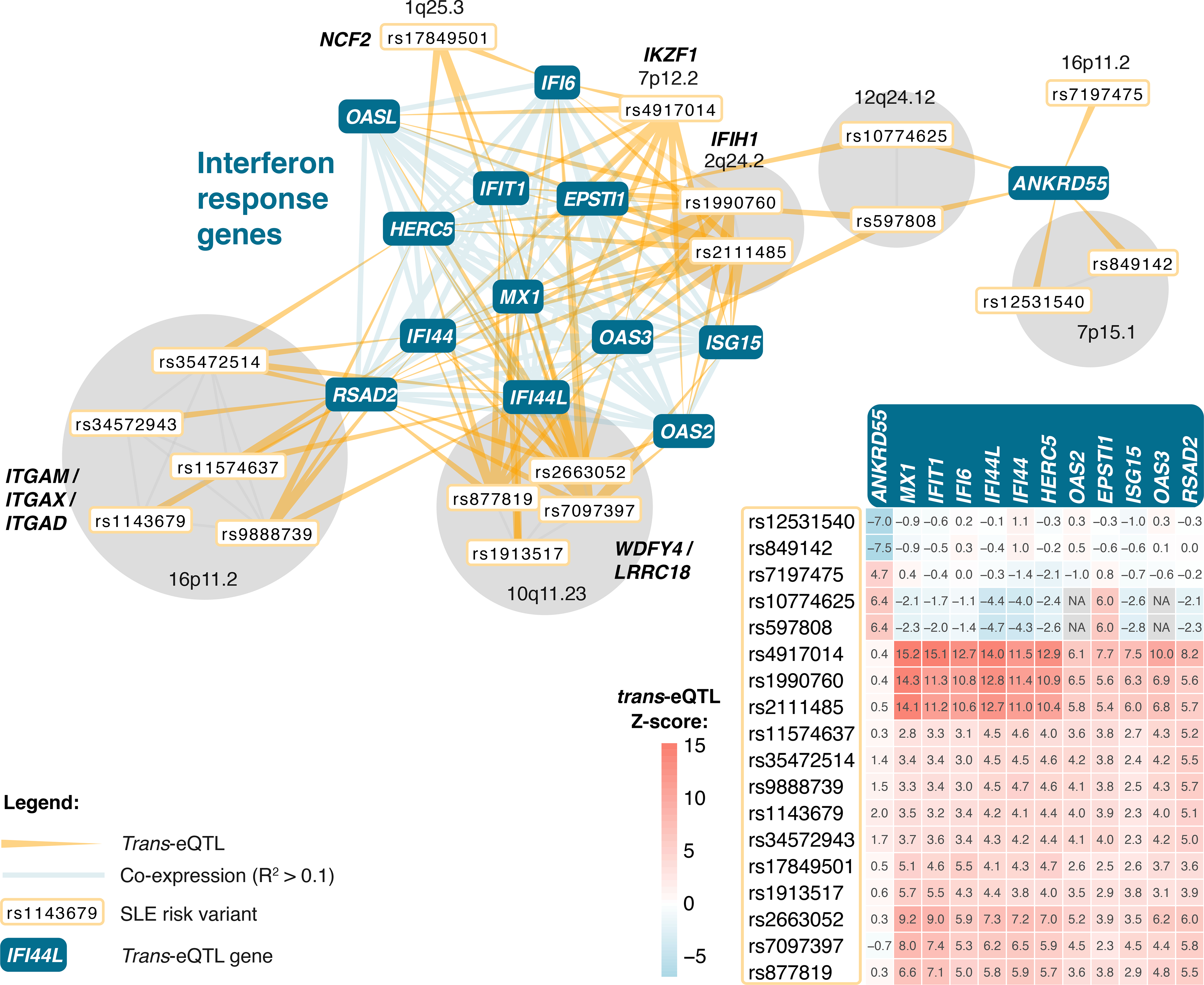
The genes shown are those affected by at least three independent GWAS SNPs. SNPs in the HLA region are not visualized and SNPs in partial linkage disequilibrium are grouped together. The heatmap indicates the direction and strength of individual trans-eQTL effects (Z-scores), relative to the SLE risk allele.
Despite those results, most blood trans-eQTLs remain unexplained. To aid interpretation, we provide the results of per-phenotype gene ontology (GO) term enrichment analysis (Supplementary Note, Supplementary Table 12). In the Supplementary Note we also highlight additional examples of trans-eQTL variants associated with age of menarche34 (ZNF131 locus), lipid levels35 (FADS1/2 locus), IBD36 and SLE37 (IFIH1 locus), asthma38 (GSDMB locus), and height39 (CLOCK locus), and explore their potential biological mechanisms to show how trans-eQTL results can be used to generate hypotheses for further research (Supplementary Figure 11a–e).
eQTSs identify potential driver genes for polygenic traits
To ascertain the coordinated effects of trait-associated variants on gene expression, we used GWAS summary statistics to calculate PGSs for 1,263 traits in 28,158 samples (Methods, Supplementary Table 13). We reasoned that when the PGS for a trait correlates with the expression of a gene, the trans-eQTL effects of the individual risk variants (Figure 6a) converge on that gene, and it can be prioritized as a putative driver of the disease (Figure 6b).
Figure 6. eQTS analyses.
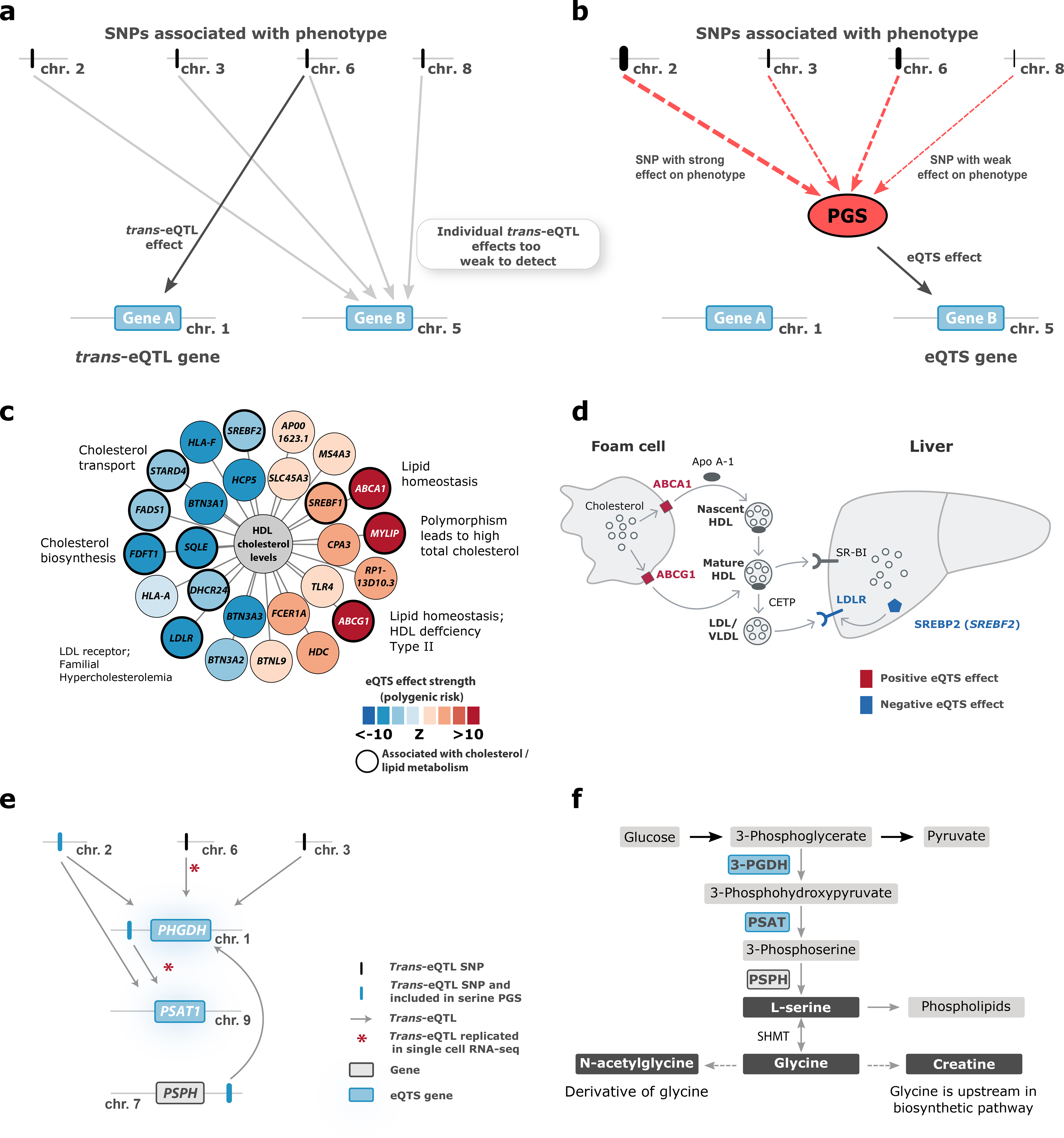
(a) In trans-eQTL analysis, individual SNPs are associated with gene expression. (b) In eQTS analysis, the effect sizes and directions of individual trait-associated SNPs are combined into a polygenic score (PGS) that is associated with gene expression. Here, we outline the case where eQTS analysis identifies a gene not detectable in the trans-eQTL analysis. Other scenarios we observed include: Gene A also being identified by eQTS analysis, Gene B being identified by both methods, or the combined effect of PGS yielding no significant eQTS. (c) The PGS for high density lipoprotein (HDL) associates to lipid metabolism genes. (d) The role of ABCA1, ABCG1, LDLR and SREBF2 in cholesterol transport. (e) Both trans-eQTLs and the serine PGS associate with the known serine biosynthesis genes PHGDH and PSAT1. (f) Serine biosynthesis pathway.
We identified 18,210 eQTSs (FDR<0.05) representing 689 unique traits (55% of tested traits) and 2,568 genes (13% of tested genes; Supplementary Data 5, Figure 1d). Of these genes, 719 (28%) were not identified in the trans-eQTL analysis, emphasizing the value of analyzing eQTS in addition to trans-eQTLs (Figure 6a–b). Median eQTS effect sizes were smaller than for cis-eQTLs and similar to trans-eQTLs (median r=0.037; Supplementary Figure 8a, e, i).
Ten eQTSs replicated in LCLs (Benjamini-Hochberg FDR<0.05), and 9 also had the same effect direction as in the discovery dataset (Supplementary Figure 12a, Supplementary Data 5), while 78 replicated in the induced pluripotent stem cells (iPSCs; Benjamini-Hochberg FDR<0.05) with 71 (91%) showing the same direction (Supplementary Figure 12b, Supplementary Data 5). We also identified 19 replicating eQTSs (Benjamini-Hochberg FDR<0.05, same effect direction) in the European subset of GTEx samples (Supplementary Note, Supplementary Data 6) and observed an inflation of replication signal in some tissues, primarily in blood (Supplementary Figure 6b).
Most eQTS associations (72.8%) represented blood-cell traits (Extended Data Figure 3, Supplementary Data 5). For instance, the PGS for mean corpuscular volume40 correlated positively with the expression of genes specifically expressed in erythrocytes, e.g. genes encoding hemoglobin subunits (HBG1 and HBG2, both FDR<0.05). eQTS genes were most enriched by GO terms involved in cellular secretion, blood cell traits and intercellular signaling (Supplementary Table 14).
Because there was no strong replication signal in non-blood tissues and the majority of eQTSs were observed for blood-related traits, we speculate that these effects are highly tissue- or cell-type-specific and that eQTS analysis would yield the most informative results if conducted in the trait-relevant tissue. However, power analyses suggest that the limited replication in other tissues could also be the result of a lack in statistical power explained by the small effect sizes of eQTSs (Supplementary Figure 8i) and moderately sized replication datasets.
Still, in our blood data, we also identified eQTSs for non-blood PGSs, including metabolite- and lipid-levels, anthropometric traits and diseases such as asthma, celiac disease and coronary artery disease (Supplementary Note; Supplementary Figure 13a–c; Supplementary Data 5). For example, 11 out of the 26 eQTS genes that were associated with the PGS for high-density lipoprotein levels (HDL41,42; all FDR<0.05; Figure 6c) have previously been linked to lipid or cholesterol metabolism (Supplementary Table 15). ABCA1 and ABCG1, which were positively correlated with the PGS for high HDL, mediate the efflux of cholesterol from macrophage foam cells and participate in HDL formation. In macrophages, downregulation of ABCA1 and ABCG1 reduces reverse cholesterol transport into the liver by HDL43 (Figure 6d). The PGS for high HDL was also negatively correlated with the expression of the low-density lipoprotein receptor LDLR (strongest eQTS P=3.35×10−20), mutations in which are known to cause familial hypercholesterolemia44. Similarly, SREBF2, the gene encoding the TF SREBP-2, which increases the expression of LDLR, was downregulated (strongest eQTS P=3.08×10−7). The negative correlation between SREBF2 expression and measured HDL levels has been described before15, indicating that the eQTS reflects an association with an actual phenotype. Zhernakova et al.15 proposed a model where down-regulation of SREBF2 results in lower expression of its target gene, FADS2. However, we did not observe an HDL eQTS effect on FADS2 (all eQTS P>0.07), possibly because the indirect effect was too small to detect. We hypothesize that higher blood HDL levels can result in stronger reverse cholesterol transport via HDL (Figure 6d), which may result in downregulation of LDLR45.
eQTS can also identify pathways known to be associated with monogenic diseases. For example, the PGSs for serine, glycine, the glycine derivative N-acetylglycine and creatine56,57 were negatively associated with the expression of PHGDH, PSAT1 and AARS (P<5.3×10−7). The PGSs for these traits are driven by SNPs near CPS1 (2q34), PHGDH (1p12) and PSPH (7p11.2) (Supplementary Table 16–18) that influence expression of PHGDH and PSAT1 in trans. We nominally replicated these trans-eQTLs in scRNA-seq data (absolute average Z>1.96 across tested cell types, part of the 729 trans-eQTLs replicating in the scRNA-seq data; Supplementary Table 4, Figure 6e), suggesting that this eQTS is driven by multiple genetic loci, but independent of cell-type-composition. PHGDH and PSAT1 encode enzymes that regulate the synthesis of serine and, in turn, glycine46, while N-acetylglycine and creatine form downstream of glycine47 (Figure 6f). Mutations in PSAT1 and PHGDH can result in monogenic conditions with defective serine biosynthesis, which are characterized by low concentrations of serine and glycine in blood and severe neuronal manifestations48–50. Unexpectedly, the PGS for higher levels of these amino acids was associated with lower expression of PHGDH, PSAT1 and AARS, implying the presence of a negative feedback loop that controls serine synthesis.
Discussion
We performed cis-eQTL, trans-eQTL and eQTS analyses in 31,684 blood samples - a six-fold increase in sample size over earlier studies5,9. Of the genes expressed in blood, 88.2% showed a cis-eQTL effect, 32% showed a trans-eQTL effect and 13% showed an eQTS effect.
Most studies prioritizing genes for complex traits have considered only cis-eQTL effects and our blood cis-eQTLs can be used for that purpose. However, cis-eQTL effects have been estimated to contribute to a limited fraction of the heritability of gene expression, while the combination of many weak trans-eQTL effects is estimated to explain the majority51, emphasizing the importance of distal effects. At the same time, the interpretation of trans-eQTLs in blood remains challenging: limited replication and the influence of blood-cell-composition suggest that the effects are highly cell-type-specific. Nevertheless, the replication analyses we carried out in PBMC scRNAs-seq data prioritized 729 trans-eQTLs, and half of the identified trans-eQTLs were assigned to a putative biological mechanism of action, with transcriptional regulation through TF activity being the most prevalent.
To identify genes that are coordinately affected by multiple independent trait-associated SNPs, we performed eQTS analysis. We identified eQTS associations for 2,568 genes and have outlined several examples where the associated genes point to interpretable biology. One possible interpretation of these eQTS associations is in the context of the recently proposed omnigenic model13,14. As explained by Liu et al.14, many weak distal effects could converge on the trait-relevant ‘core’ genes, and eQTS analysis might help to prioritize such genes (Supplementary Note). However, an important limitation is that eQTS analysis can also identify genes which are merely co-regulated with the trait-relevant ones. Therefore, it remains challenging to systematically evaluate which fraction of the detected eQTS genes is causal. While our analysis does not formally prove or disprove the validity of the model by Liu et al.14, and the true implications of this model remain to be investigated, our results can serve as a starting point to follow up on the eQTS genes and to ascertain their role in complex traits. Our eQTS analysis provides a comprehensive resource in blood that can be used to interpret the effects of PGS on a molecular level.
There are some important limitations that require consideration when using our resource for hypothesis generation. First, we limited our cis-eQTL analysis to variants within 1 Mb of the gene center, and limited our trans-eQTL analysis on variants >5 Mb from genes on the same chromosome. We acknowledge the possibility that these thresholds may have excluded distal cis-eQTLs (e.g. those caused by distal enhancers or chromatin loops), and trans-eQTLs on nearby genes. We chose these thresholds to ensure that the trans-eQTLs we observed were not driven by long-range cis-eQTLs. While our approach might have excluded long-range cis-eQTLs, we observed that for 95.6% of genes, the lead cis-eQTL SNP maps within 100kb of the gene, suggesting that long-range cis-eQTLs reflect only a small proportion of all cis-eQTLs. Second, we confined our trans-eQTL analyses to a subset of 10,317 variants previously associated with complex phenotypes. As such, a significant trans-eQTL for a trait-associated variant does not necessarily mean that the same underlying variant affects both the phenotype and gene expression. Third, PGS estimates have been shown to have variable prediction accuracy even when evaluated within the same ancestry. This variability may be caused by differences in sample characteristics (e.g. age, sex, socio-economic status) in the original GWAS as well as the dataset in which the PGS is calculated52. Such variability may therefore have caused either inflation or deflation of our eQTS effect sizes. Although we present several examples that are interpretable in the context of the respective traits, caution is needed when drawing conclusions on higher-level phenotypes. Instead, our resource should serve as a starting point for further in-depth studies that can reliably connect the reported eQTL and eQTS associations to phenotypes.
Although putative biological mechanisms of action could be assigned to half of the identified trans-eQTLs, significant replication in scRNA-seq, purified cell type and cell line datasets was very limited. Such low replication rates suggest two likely causes. First, a number of the distal effects are likely driven by inter-individual cell-type-composition differences, which occur in any bulk tissue. While such effects could be informative in the context of some complex traits (i.e. for autoimmune diseases), the most interesting information lies in the intracellular effects. Furthermore, while we corrected for unknown confounders in our analyses, some residual cell-composition effects remain in the data. Therefore, it was not possible to reliably distinguish cell-type-dependent effects from intracellular ones. Instead, our results should serve as a prioritized list for in-depth functional studies.
Second, our discovery analyses were conducted in a sample >10 times larger than the largest replication datasets available. Because trans-eQTL effects are generally weak, this lack of statistical power likely causes low replication rates. Additionally, trans-eQTL effects are widely considered to be more cell-type- and tissue-specific than local cis-eQTL effects18. Although this belief might be partly caused by variable trans-eQTL strengths in different tissue contexts and the limited power of current trans-eQTL studies, it would also lead to lower replication rates of blood trans-eQTLs in specific cell types.
Compared to the gene expression from bulk tissues, scRNA-seq datasets are less affected by cell-type composition and serve as the best current source for replicating, prioritizing and annotating trans-eQTLs. While we have compiled, to our knowledge, the largest available blood scRNA replication dataset, it was still only 3.6% of the sample size of the discovery study. It is therefore unsurprising that only 35 trans-eQTLs reached the significance threshold (FDR<0.05). None-the-less, 84% of the 729 trans-eQTLs attaining nominal significance (P<0.05) also showed allelic concordance with the discovery analysis, suggesting that there are intracellular effects among our trans-eQTLs, even if case-by-case distinction of cell-type-composition and intracellular effects is not yet possible. Upcoming large-scale single cell eQTL studies53 (e.g. https://www.eqtlgen.org/single-cell.html), as well as highly-powered eQTL analyses in non-blood tissues54 and cell lines, will be instrumental in distinguishing intracellular effects from cell-type composition.
Full summary statistics for our cis-eQTL, trans-eQTL and eQTS analyses (www.eqtlgen.org) can be used to interpret GWAS, to prioritize putative trait-related genes for in-depth functional studies and to develop methods to perform those tasks. We envision that upcoming statistical tools and frameworks that enable federated analyses in consortia will facilitate conducting highly powered global trans-eQTL studies. This will expand the work presented here and enable a better connection between distal effects on gene expression and complex phenotypes.
Online Methods
Cohorts
The eQTLGen Consortium data consist of 31,684 blood and peripheral blood mononuclear cell (PBMC) samples from 37 datasets, pre-processed in a standardized way and analyzed by each cohort analyst. 25,482 (80.4%) of the samples were whole blood samples and 6,202 (19.6%) were PBMCs, and the majority of samples were of European ancestry (Supplementary Table 1). The gene expression levels of the samples were profiled by the Illumina (N=17,421; 55%), Affymetrix U219 (N=2,767; 8.7%), and Affymetrix Hu-Ex v1.0 ST (N=5,075; 16%) expression arrays and by RNA-seq (N=6,422; 20.3%). A summary of each dataset is outlined in Supplementary Table 1. Detailed cohort descriptions can be found in the Supplementary Note. All cohorts participating in this study enrolled participants with informed consent, collected and analyzed data in accordance with ethical and institutional regulations, and provided summary statistics for the meta-analyses. The information about individual institutional review board approvals is available in the original publications for each cohort (Supplementary Note) or in the cohort-specific section in Supplementary Note.
Each of the cohorts carried out genotype and expression data pre-processing, PGS calculation and cis-eQTL-, trans-eQTL- and eQTS-mapping following the steps outlined in the online analysis plans, specific for each platform (https://github.com/molgenis/systemsgenetics/wiki/eQTL-mapping-analysis-cookbook; https://github.com/molgenis/systemsgenetics/wiki/eQTL-mapping-analysis-cookbook-for-RNA-seq-data; https://github.com/molgenis/systemsgenetics/wiki/QTL-mapping-analysis-cookbook-for-Affymetrix-expression-arrays), or with slight alterations as described in Supplementary Table 1 and the Supplementary Note. All but one cohort (Framingham Heart Study), included unrelated individuals into the analysis.
Information about replication datasets is detailed in the Supplementary Note.
Genotype data preprocessing
The primary pre-processing and quality control of genotype data was conducted by each cohort, as specified in the original publications and in the Supplementary Note. The majority of cohorts used genotypes imputed to the 1000 Genomes phase 1 version 3 (1000G p1v3) or a newer reference panel. GenotypeHarmonizer55 v1.4.9 (https://github.com/molgenis/systemsgenetics/wiki/Genotype-Harmonizer) was used to harmonize all genotype datasets to match the GIANT 1000G p1v3 ALL reference panel (ftp://share.sph.umich.edu/1000genomes/fullProject/2012.03.14/GIANT.phase1_release_v3.20101123.snps_indels_svs.genotypes.refpanel.ALL.vcf.gz.tgz) and to fix potential strand issues for A/T and C/G SNPs. Each cohort tested SNPs with minor allele frequency (MAF) >0.01, Hardy-Weinberg P-value >0.0001, call rate >0.95, and MACH r2>0.5. Reported SNP identifiers are in dbSNP v137.
Expression data preprocessing
Illumina arrays
Illumina array expression datasets were profiled by HT-12v3, HT-12v4 and HT-12v4 WGDASL arrays. Before analysis, all the probe sequences from the manifest files of those platforms were re-mapped to the GRCh37.p10 human genome build and transcriptome using SHRiMP v2.2.3 aligner56 (http://compbio.cs.toronto.edu/shrimp/), allowing two mismatches. Probes mapping to multiple locations in the genome were removed from further analyses.
For Illumina arrays, the raw unprocessed expression matrix was exported from GenomeStudio. First, two PCs were calculated on the quantile-normalized and log2 transformed expression data and plotted to identify and exclude outlier samples. The data were normalized in several steps: quantile normalization, log2 transformation, probe centering and scaling by the equation ExpressionProbe,Sample = (ExpressionProbe,Sample - MeanProbe) / Std.Dev.Probe. Genes showing no variance were removed. Next, the first four multidimensional scaling (MDS) components, calculated based on non-imputed and pruned genotypes using plink v1.0757, were regressed out of the expression matrix to account for population stratification. We further removed up to 20 of the first expression-based PCs that were not associated with any SNPs, as these capture non-genetic variation in expression. After regressing out these covariates, the residual gene expression matrix was used for eQTL mapping. Each cohort also ran MixupMapper58 software to identify incorrectly labeled genotype–expression combinations and resolved any sample mix-ups (https://github.com/molgenis/systemsgenetics/wiki/Resolving-mixups).
Affymetrix arrays
Affymetrix-array-based datasets used expression data previously pre-processed and quality controlled as indicated in the Supplementary Note.
RNA-seq
Alignment, initial quality control and quantification differed slightly across datasets, as described in the Supplementary Note. Each cohort removed outliers as described above, and then used Trimmed Mean of M-values normalization59 and a counts per million (CPM) filter to include genes with >0.5 CPM in at least 1% of the samples. Subsequent steps were identical to the Illumina processing, with some exceptions for the BIOS Consortium datasets (Supplementary Note).
Empirical probe matching
To integrate the different expression platforms for the purpose of meta-analysis, we developed an empirical probe-matching approach. We used the pruned SNPs to conduct per-platform meta-analyses for all Illumina arrays, for all RNA-seq datasets, and for each Affymetrix dataset separately, using summary statistics from analyses without correction for PCs. For each platform, this yielded an empirical trans-eQTL Z-score matrix, as well as 10 permuted Z-score matrices in which links between genotype and expression files were shuffled. These permuted Z-score matrices reflect the gene–gene or probe–probe correlation structure.
We then used RNA-seq permuted Z-score matrices as a gold standard reference and calculated, for each gene, the Pearson correlation coefficients with all the other genes, yielding a correlation profile for each gene. We then repeated the same analysis for the Illumina meta-analysis and the two different Affymetrix platforms. Finally, we correlated the correlation profiles from each array platform with the correlation profiles from RNA-seq. If there were multiple probes detecting the expression of one gene, we selected the probe showing the highest Pearson correlation with the corresponding gene in the RNA-seq data and treated those as matching expression features in the combined meta-analyses. This yielded 19,942 genes that were detected in RNA-seq datasets and tested in the combined meta-analyses. Genes and probes were matched to Ensembl v7160 (ftp://ftp.ensembl.org/pub/release-71/gtf/homo_sapiens/Homo_sapiens.GRCh37.71.gtf.gz) stable gene IDs and HGNC symbols in all the analyses.
Meta-analysis procedure
The results presented in this study were meta-analyzed using a weighted Z-score method61, where the Z-scores are weighted by the square root of the sample size of the cohort. For cis-eQTL and trans-eQTL meta-analyses, this resulted in a final sample size of up to 31,684. The combined eQTS meta-analysis included only unrelated individuals from the Framingham Heart Study, resulting in a combined sample size of up to 28,158. Considering that our analysis contained many different gene expression and genotyping platforms, we limited our meta-analysis to associations present in at least two cohorts in order to reduce platform-specific effects. Specifics for each meta-analysis (cis-eQTL, trans-eQTL, eQTS) are detailed below.
Cross-platform replications
To test the performance of the empirical probe-matching approach, we conducted discovery cis-, trans- and eQTS meta-analyses for each expression platform (RNA-seq, Illumina, Affymetrix U219 and Affymetrix Hu-Ex v1.0 ST arrays; array probes matched to 19,942 genes by empirical probe matching). For each discovery analysis, we conducted replication analyses in the three remaining platforms.
Cis-eQTL mapping
Cis-eQTL mapping was performed in each cohort using a pipeline described previously5. In brief, the pipeline takes a window of 1 Mb upstream and 1 Mb downstream around each SNP to select genes or expression probes to test, based on the center position of the gene or probe. The associations between these SNP–gene combinations are calculated using Spearman correlation. Next, every cohort performed 10 permutations. In each permutation, the links between genotype and expression identifiers were shuffled prior to re-calculating all associations. Both the non-permuted results and each round of permuted results were meta-analyzed across cohorts.
Multiple testing correction for cis-eQTL mapping
For our multiple testing procedure, we used the meta-analyzed permutations to calculate the overall FDR, as previously described5. In short, we reasoned that the large numbers of correlated SNPs and genes present in the cis-eQTL results might cause inflated estimates (i.e. highly correlated SNPs associated with a specific gene would result in equal permuted P-values for that particular gene). To circumvent this issue, we first selected the lowest association P-value per gene in both the permuted and non-permuted meta-analyses. The resulting lists of P-values were sorted and, per given P-value in the non-permuted data, we determined the proportion of P-values equal to or below this value in both the permuted and non-permuted data. We then determined our FDR estimate as the proportion of permuted P-values over the proportion of non-permuted P-values. If a specific eQTL from the full set was not among the set of per-gene lowest association P-values, this eQTL was assigned the higher FDR value corresponding to the next eQTL available among the set of lead variants per gene. We refer to this procedure as ‘gene-level’ FDR, but note that the FDR estimates should be evaluated as ‘analysis-wide’, since the ultimate distribution of permuted P-values used to calculate our FDR estimates was derived for all tested genes, rather than per-gene. Cis-eQTLs with a gene-level FDR<0.05 (corresponding to P<2.02×10−5) that were tested in more than one cohort were deemed significant.
Trans-eQTL mapping
Trans-eQTL mapping was performed using a previously described pipeline5 while testing a subset of 10,317 SNPs associated with complex traits. We required the distance between the SNP and the center of the gene to be >5 Mb. To maximize the power to identify trans-eQTL effects, the expression matrices were corrected for the results of summary-statistics-based or iterative conditional cis-eQTL mapping analyses (Supplementary Note) before trans-eQTL mapping. For that, lead SNPs for significant (FDR<0.05) conditional cis-eQTLs were regressed out from the expression matrix. Finally, we removed potential false positive trans-eQTLs caused by reads cross-mapping with cis regions (Supplementary Note).
Selection of SNPs for trans-eQTL mapping
Genetic risk factors were downloaded from three public repositories: the EBI GWAS Catalog73 (https://www.ebi.ac.uk/gwas/; downloaded 21 November 2016), the NIH GWAS Catalogue and Immunobase (www.immunobase.org; accessed 26 April 2016), applying a significance threshold of P≤5×10−8. Additionally, we added 2,706 genome-wide significant GWAS SNPs from a blood trait GWAS23. SNP coordinates were lifted to hg19 using the liftOver command from R package rtracklayer v1.34.174 and subsequently standardized to match the GIANT 1000G p1v3 ALL reference panel. This yielded 10,562 SNPs (Supplementary Table 4). We tested associations between all risk factors and genes that were at least 5 Mb away to ensure that that they did not tag a cis-eQTL effect. In total, 10,317 trait-associated SNPs were tested in trans-eQTL analyses.
eQTS mapping
PGS trait inclusion
Full association summary statistics were downloaded from several publicly available resources (Supplementary Table 13). PGSs are most predictive when individuals in the original GWAS are from similar ancestry as individuals for whom the PGS is calculated. Because most of the individuals in our meta-analyses were from European ancestry, we investigated the information presented on the web sites or abstracts of corresponding publications and omitted GWASs performed exclusively in non-European cohorts. Filters applied to the separate data sources are indicated in the Supplementary Note. All the dbSNP rs numbers and directions of effects were standardized to match GIANT 1000G p1v3 identifiers and minor alleles. SNPs with opposite alleles compared to GIANT alleles were flipped. SNPs with A/T and C/G alleles, tri-allelic SNPs, indels, SNPs with unknown or different alleles to GIANT 1000G p1v3 were removed from the analysis. Genomic control was applied to all the P-values for the datasets not genotyped by Immunochip or Metabochip. Additionally, genomic control was skipped for one dataset that did not have full associations available75 and for all the datasets from the GIANT consortium because genomic control had already been applied for these. In total, 1,263 summary statistics files were added to the analysis. Information about the summary statistics files can be found in the Supplementary Note and Supplementary Table 13.
PGS calculation
A custom Java program, GeneticRiskScoreCalculator-v0.1.0c (https://github.com/molgenis/systemsgenetics/tree/master/GeneticRiskScoreCalculator), was used for calculating several PGSs in parallel. Independent effect SNPs for each summary statistics file were identified by double-clumping, first using a 250kb distance and subsequently a 5Mb distance with a linkage disequilibrium (LD) threshold R2=0.1. Weighted PGSs were calculated by summing the risk alleles for each independent SNP weighted by its GWAS effect size (beta or log(OR) from the GWAS study). Five GWAS P-value thresholds (P<5×10−8, 1×10−5, 1×10−4, 1×10−3 and 1×10−2) were used for constructing PGSs for each summary statistics file. HLA region (chr6:25,000,000–35,000,000) was omitted from calculations and PGSs were scaled to fall between 0 and 2, for compatibility with the QTL mapping pipeline.
Pruning SNPs and PGSs
To identify a set of independent SNPs, we conducted LD-based pruning as implemented in PLINK 1.976 with the setting --indep-pairwise 50 5 0.1. This yielded 4,586 uncorrelated SNPs (R2<0.1, GIANT 1000G p1v3 ALL).
To identify the set of uncorrelated PGSs, 10 permuted trans-eQTL Z-score matrices from the combined trans-eQTL analysis were first confined to the pruned set of SNPs. Those matrices were then used to identify 3,042 uncorrelated genes based on Z-score correlations (absolute Pearson R<0.05). Next, permuted eQTS Z-score matrices were confined to uncorrelated genes and used to calculate pairwise correlations between all genetic risk scores to define a set of 1,873 uncorrelated PGSs (Pearson R2<0.1).
Multiple testing correction in trans-eQTL and eQTS mapping
To calculate FDR estimates for trans-eQTLs and eQTS, we compared each P-value from the non-permuted meta-analysis with all P-values from 10 meta-analyzed permutation rounds. We note that this differs from the permutation strategy used in the cis-eQTL analysis, because here we used the P-values from all SNP-gene combinations, not just the smallest P-value for each gene. Nevertheless, the 10,317 SNPs tested for trans-eQTLs contained many linked variants. To establish a conservative FDR estimate, we therefore used the pruned set of 4,586 SNPs to perform a meta-analysis for both the non-permuted and permuted datasets. We derived FDR estimates from these limited meta-analyses by sorting the lists of P-values and determining the proportion of P-values in the non-permuted and permuted datasets for each given P-value in the non-permuted dataset. We then applied these FDR estimates to the trans-eQTL results from all 10,317 genetic trait-associated SNPs. If a specific eQTL from the full set was not tested in the meta-analysis conducted on the pruned set, this eQTL was assigned the higher FDR value corresponding to the next eQTL tested in the pruned set. We used an FDR threshold of 0.05 (corresponding to P<8.3×10−6) to declare a trans-eQTL effect significant. Similarly, in the eQTS analysis, we used a set of 1,873 uncorrelated (Pearson R2<0.1) PGSs and performed an analogous FDR calculation. The FDR threshold for eQTS corresponded to P<3.02×10−6. We analyzed only SNP/PGS–gene pairs tested in at least two cohorts.
Replication of trans-eQTLs and eQTSs in bulk datasets
Information about replication cohorts and their respective settings for replication analyses is outlined in the Supplementary Note. If applicable, summary statistics from different replication datasets for the same cell type or tissue were meta-analyzed using a weighted Z-score method61. Benjamini-Hochberg FDR17 was used to adjust replication analysis P-values for multiple testing. We required FDR<0.05 and the same effect direction with discovery to declare effect replicating. R package pwr (https://cran.r-project.org/web/packages/pwr/index.html) was used to conduct power analyses for replication datasets.
scRNA-seq analyses
scRNA-seq cohorts and data
For the replication of trans-eQTLs in scRNA-seq, we used unpublished data of PBMCs from 1,139 unrelated individuals in two cohorts generated using the 10X Chromium platform: OneK1K (N=982) and 1M-scBloodNL (N=157). The data were processed using the Cell Ranger Single Cell Software Suite v3.0.2 (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger) and aligned using STAR62 implementation within Cell Ranger Single Cell Software Suite . Cells were demultiplexed and doublets removed before performing cell type classification. We combined the data in a meta-analysis within each of the eight available cell types: B-cells, CD4+ T-cells, CD8+ T-cells, classical monocytes, non-classical monocytes, dendritic cells, natural killer cells and plasma cells. See Supplementary Note for methodological details.
Replication of trans-eQTL effects
We tested the replication of the 59,786 discovery trans-eQTLs only if the trans-eQTL gene was sufficiently expressed (i.e. had a missing sample fraction that was at most 20% in the large OneK1K dataset), leaving between 1,917 and 27,582 eQTLs to be studied, depending on cell type. We estimated the inflation of signal by calculating the lambda inflation relative to the inverse chi-square cumulative distribution function of 0.5. Trans-eQTLs with FDR<0.05 in any cell type were deemed significantly replicating. To get a better idea of replication across cell types, we calculated the average Z-score across cell types. We selected effects with an absolute average Z-score>1.96 (equivalent to P<0.05) to calculate the allelic concordance with the discovery trans-eQTLs.
Correlation of trans-eQTL effects
To test the correlation between trans-eQTL effects in the discovery and replication datasets, we used the rb approach24, which accounts for the errors in the estimated eQTL effects so that the estimate of correlation is less dependent on sample sizes. First, we derived the estimate of the trans-eQTL effect (beta) and the standard error of the beta (SE(beta)) from the Z-score and the MAF of the significant trans-eQTLs, using the following formulae from Zhu et al. 201663
where p is the MAF, n is the sample size and z is the meta-analysis Z-score. MAF was computed from 26,609 eQTLGen samples (excluding FHS) for discovery analysis and from 1,139 replication samples for scRNA-seq replication analyses. For analyses in purified cell types and cell lines (LCL, iPSC) where allele frequencies were not available, we used the MAF as observed in eQTLGen instead.
In order to include independent effects in the analysis, for each trans-eQTL gene, we included only the strongest significant discovery effect in each 2 Mb window. Statistics of rb and SE(rb) were calculated as detailed in Qi et al. 201824 assuming no sample overlap between discovery and replication datasets. Because we were only seeking to correlate the effects of identified trans-eQTLs, we did not use any reference discovery dataset for selecting trans-eQTLs to estimate rb, and hence did not consider potential ascertainment bias, although such bias is likely to be small. To calculate a P-value, the Z-score was first calculated by dividing rb by SE(rb) and then squared to calculate the χ2 statistic. The P-value was then derived from the χ2 distribution with one degree of freedom.
Comparison of pLI metrics over gene expression bins
All genes were divided into 10 bins based on the average expression value of the TMM-normalized and log2-transformed expression matrix from BIOS cohort. If the number of tested genes did not divide by 10, the lowest bin was set to include more genes than the rest of the bins. ExAC pLI metrics19 were acquired from ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.3.1/functional_gene_constraint/fordist_cleaned_exac_r03_march16_z_pli_rec_null_data.txt. For every expression bin, genes for which pLI metric was available were tested for difference by two-sided Wilcoxon rank sum test.
TF and tissue enrichment analyses for REST locus
We downloaded curated sets of known TF-targets and tissue-expressed genes from the Enrichr64,65 website (http://amp.pharm.mssm.edu/Enrichr/). TF-target gene sets were assayed by ChIP-X experiments from the ChEA66 and ENCODE67,68 projects. Tissue-expressed genes were based on the ARCHS4 database69. Gene sets were processed, mapped to entrez IDs with R package ClusterProfiler v3.10.170 (http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html) and tested for over-representation by one-sided Fisher’s exact test as implemented in the R package GeneOverlap v1.18.0 (https://www.bioconductor.org/packages/release/bioc/html/GeneOverlap.html), by using 19,942 genes tested in trans-eQTL analysis as background. Multiple testing correction was conducted using Benjamini-Hochberg method17.
Biological mechanisms explaining trans-eQTLs
To better understand the biological mechanisms underlying the trans-eQTLs, we performed a number of enrichment analyses. We converted trans-eQTLs to a gene-by-gene matrix via three methods: using Pascal27, using cis-eQTL information and combining both (Supplementary Note). For the enrichments, we calculated whether there was significant overlap with known TF–target pairs25 (www.RegulatoryCircuits.org), gene co-regulation patterns (Supplementary Note), PPIs26 (https://www.intomics.com/inbio/map/api/get_data?file=InBio_Map_core_2016_09_12.tar.gz) and Hi-C contacts in LCL cells28 using a two-sided Fisher’s exact test.
Capture Hi-C overlap for cis-eQTLs
To assess whether cis-eQTL lead SNPs overlapped with chromosomal contact as measured by Hi-C data, we used promoter capture Hi-C (CHi-C) data71 downloaded from CHiCP72 (https://www.chicp.org/). We took the lead eQTL SNPs, overlapped these with the CHi-C data, and studied the 10,428 cis-eQTL genes for which this data was available. We tested whether the CHi-C target maps within 5kb of the lead SNP. Of the 803 cis-eQTL genes for which the lead SNP mapped more than 100 kb away from the TSS or TES, 223 overlapped with the CHi-C data (27.8%). Of 9,625 cis-eQTL genes for which the lead SNP mapped within 100kb from the TSS or TES, 1,641 overlapped with the CHi-C data (17.0%). To test if these observed overlaps were not happening by chance, we performed the same analysis while flipping the location of the CHi-C target relative to the location of the bait and tested the difference with a two-tailed two-sample test of equal proportions.
Data availability
Primary genotype and gene expression data was analyzed by individual cohorts participating in the study and our study analyzed summary statistics. Full summary statistics of the eQTLGen cis-eQTL, trans-eQTL and eQTS meta-analyses are available on the eQTLGen website, www.eqtlgen.org, which was built using the MOLGENIS framework73. We also provide cis-eQTL files formatted for use in SMR, MAFs, and replication statistics for cis-eQTLs, trans-eQTLs and eQTSs. Per-cohort summary statistics for discovery cohorts can be made available after approval of an analysis proposal in eQTLGen and with agreement of the cohort PIs, contact corresponding authors for further information. Trait-associated variants were collected from EBI GWAS Catalogue (https://www.ebi.ac.uk/gwas/; accessed on 21 November 2016), NIH GWAS Catalogue (now hosted by EBI GWAS Catalogue: https://www.ebi.ac.uk/gwas/) and Immunobase (www.immunobase.org; accessed 26 April 2016; now hosted by Open Targets: https://genetics.opentargets.org/immunobase). Sources of numerous GWAS summary statistics used for eQTS analyses are outlined in Supplementary Note and Supplementary Table 13. ExAC pLI scores used for Figure 2 originate from: ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.3.1/functional_gene_constraint/fordist_cleaned_exac_r03_march16_z_pli_rec_null_data.txt. Genotype reference files used for harmonizing discovery datasets for meta-analysis originate from here: ftp://share.sph.umich.edu/1000genomes/fullProject/2012.03.14/GIANT.phase1_release_v3.20101123.snps_indels_svs.genotypes.refpanel.ALL.vcf.gz.tgz. Gene model used for gene annotations originates from ENSEMBL v71 (ftp://ftp.ensembl.org/pub/release-71/gtf/homo_sapiens/Homo_sapiens.GRCh37.71.gtf.gz). FANTOM TF annotations used for eQTS enrichment analyses originate from: http://fantom.gsc.riken.jp/5/sstar/Browse_Transcription_Factors_hg19. CHIP-seq data used for cis-eQTL overlap originates from https://www.chicp.org/. Protein-protein interaction data used for trans-eQTL mechanism enrichment analyses originate from: https://www.intomics.com/inbio/map/api/get_data?file=InBio_Map_core_2016_09_12.tar.gz. Hi-C data used for trans-eQTL mechanism enrichment is deposited in GEO (GM12878, GEO accession GSE63525). Curated gene sets used for enrichment analyses (Gene Ontology sets, ENCODE CHiP-X and CheA CHiP-X TF targets, TRANSFAC and JASPAR PWMs, ARCHS4 tissue expression, TargetScan miRNA target predictions, Tarbase miRNA validated targets) were downloaded from Enrichr web site (https://maayanlab.cloud/Enrichr/#stats). Gene expression summaries and meta-data from GTEx v7 originate from: https://gtexportal.org/home/. Gene expression summaries from BIOS are available in Source Data 1 and also in BIOS Omics Atlas (http://bbmri.researchlumc.nl/atlas/#data). Mean cell counts for eight immune cell types from 1M-scBloodNL cohort are available in Source Data 2. Per-cohort individual-level genotype and gene expression data is governed by respective biobanks and access can be requested according to procedures established by each biobank, with relevant restrictions applying as imposed by IRB or local legislation. Data access procedures established for BIOS Consortium are available on https://www.bbmri.nl/acquisition-use-analyze/bios.
Code availability
Individual cohorts participating in the study followed the analysis plans as specified in our analysis cookbooks (https://github.com/molgenis/systemsgenetics/wiki/eQTL-mapping-analysis-cookbook-(eQTLGen); https://github.com/molgenis/systemsgenetics/wiki/eQTL-mapping-analysis-cookbook-for-RNA-seq-data; https://github.com/molgenis/systemsgenetics/wiki/QTL-mapping-analysis-cookbook-for-Affymetrix-expression-arrays) or with slight alterations as described in the Methods and Supplementary Note. Tools and source codes used for genotype harmonization, identification of sample mix-ups, eQTL mapping, meta-analyses and calculation of PGSs are available at https://github.com/molgenis/systemsgenetics/. Tools used for primary analyses were written in Java (v6, v7, v8; www.java.com). Plink v1.0.7 (https://zzz.bwh.harvard.edu/plink/) and v1.90 (https://www.cog-genomics.org/plink/1.9/) was used for clumping and pruning. Downstream analyses and plots were done with R (v3.4.4, v3.6.1, v4.0.0; https://cran.r-project.org/) using packages data.table v1.12 (https://cran.r-project.org/web/packages/data.table/), tidyverse v1.2.1 (https://cran.r-project.org/web/packages/tidyverse/), broom v0.5.1 package (https://cran.r-project.org/web/packages/broom/), pheatmap v1.0.12 package (https://cran.r-project.org/web/packages/pheatmap/), GeneOverlap v1.18.0 (https://bioconductor.org/packages/release/bioc/html/GeneOverlap.html). Power analyses were conducted by R package pwr v1.3–0 (https://cran.r-project.org/web/packages/pwr/). scRNA-seq analyses made use of Cell Ranger Single Cell Software Suite v3.0.2 (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger) and its implementation of STAR aligner. ToppGene web tool (https://toppgene.cchmc.org/) was used for some interpretative enrichment analyses, as well as GeneNetwork web tool (https://genenetwork.nl/). Decon2 framework was (https://github.com/molgenis/systemsgenetics/tree/master/Decon2) used for predicting cell counts in BIOS data. We formatted our cis-eQTLs into the BESD format using SMR (https://cnsgenomics.com/software/smr/#Overview).
Extended Data
Extended Data Fig. 1. Cis-eQTL replication in GTEx v7 tissues.
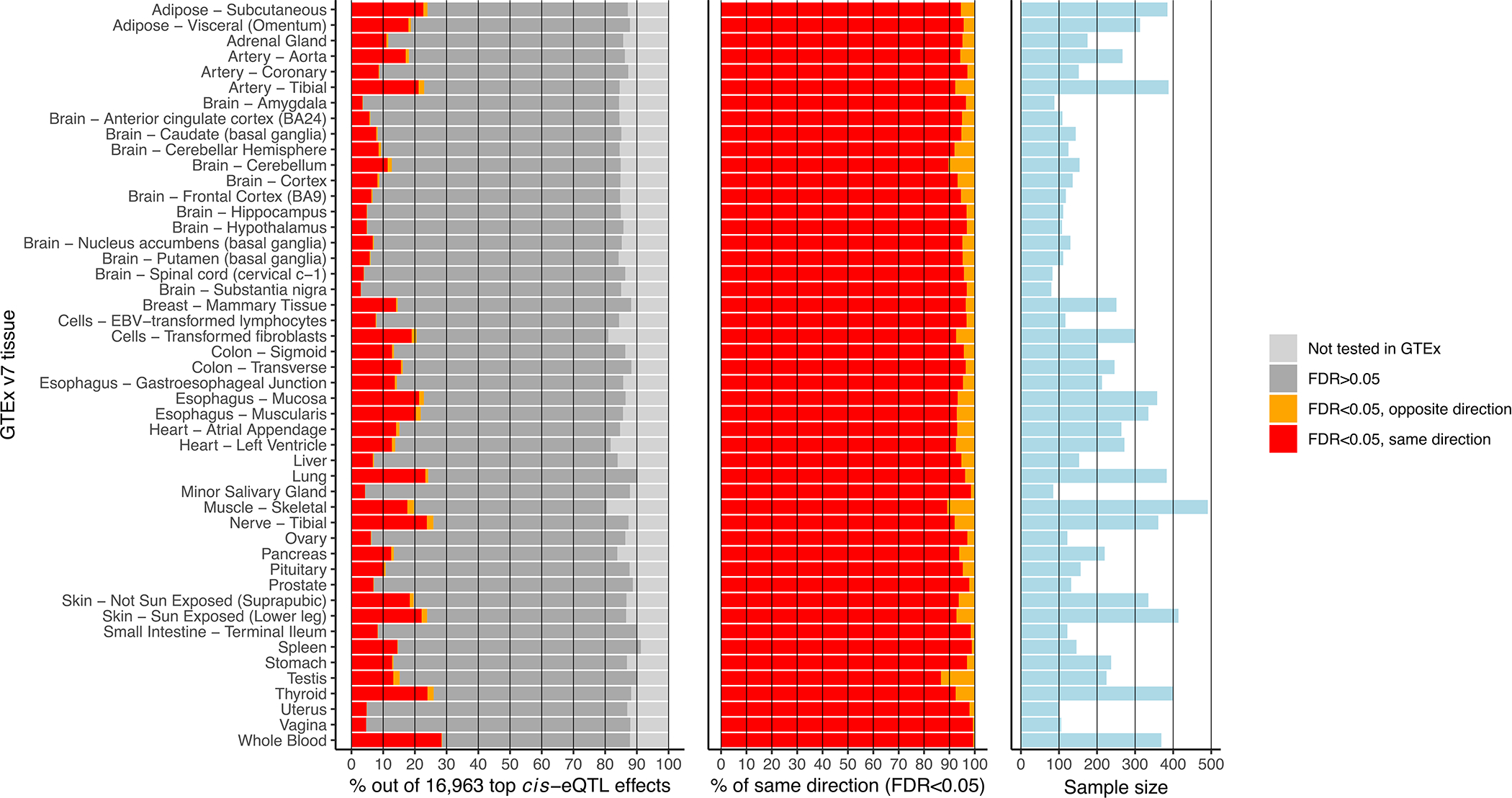
Cis-eQTL replication in GTEx v7 tissues. For this analysis, the most significant cis-eQTL SNP for each gene was tested in the available post-mortem tissues in GTEx v7. Since GTEx was part of our discovery meta-analysis, the cis-eQTL discovery analysis was repeated while excluding GTEx whole blood, identifying 16,963 lead cis-eQTL effects that were subsequently replicated in each GTEx tissue. Left: while the majority of the 16,963 cis-eQTLs were tested in the GTEx replication study, a relatively small fraction had an FDR<0.05. Middle: of those ciseQTLs showing a replication FDR<0.05, allelic directions were highly consistent with the discovery meta-analysis. Right: sample sizes of GTEx tissues. Limited replication rates at FDR<0.05 were probably due to the relatively small sample size per GTEx tissue.
Extended Data Fig. 2. Dot-plot showing the locations of the trans-eQTL effects identified in discovery meta-analysis and their association P-values (-log10 scale).
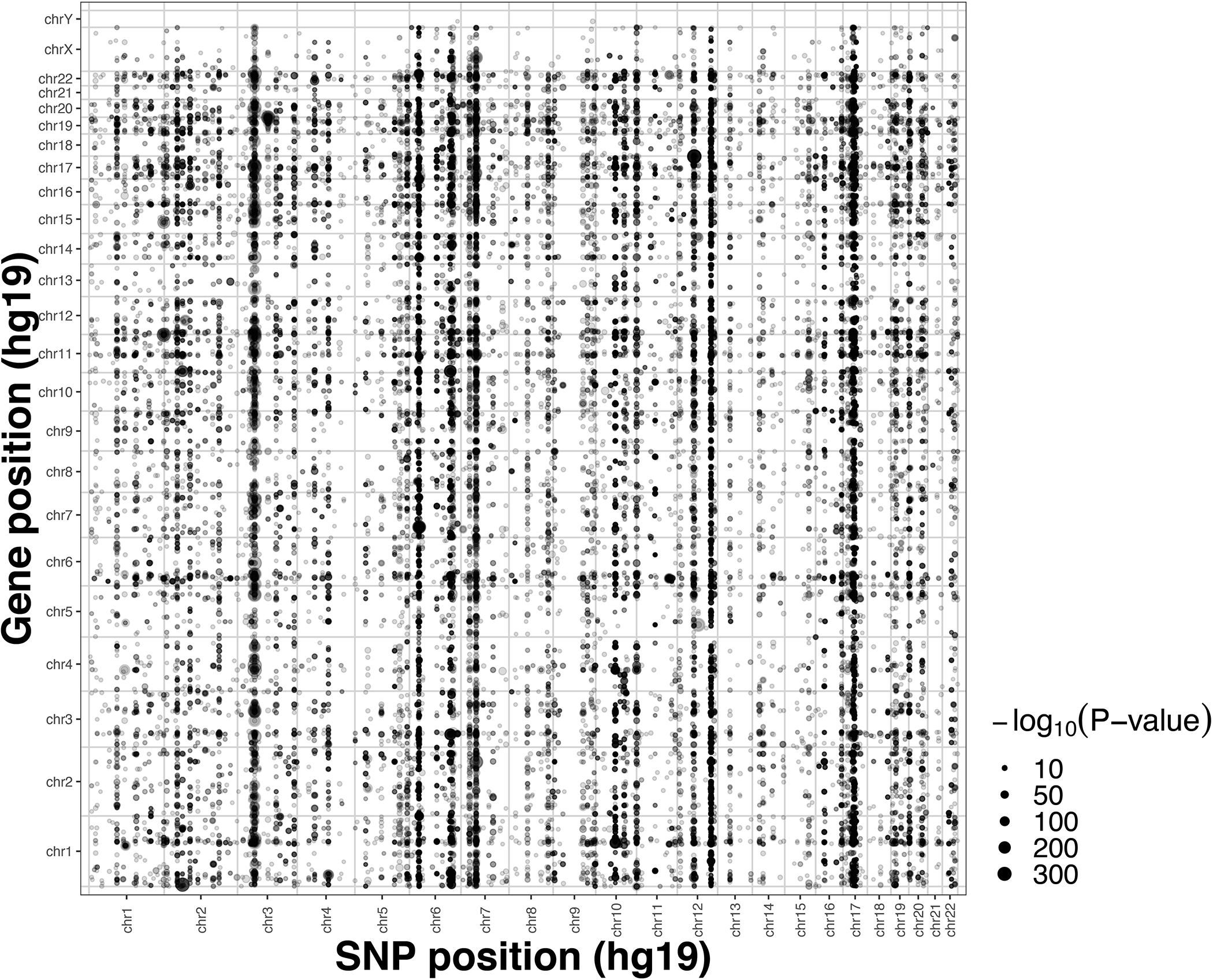
Dot-plot showing the locations of the trans-eQTL effects identified in discovery meta-analysis (weighted Z-score meta-analysis on Spearman correlation) and their respective two-sided association P-values in -log10 scale. SNP positions are shown on the x-axis and gene locations on the y-axis, each dot shows one significant trans-eQTL effect (FDR<0.05). Vertical bands appear where a single genomic locus affects many genes in trans, while horizontal bands illustrate genes affected by many SNPs.
Extended Data Fig. 3. Overview of tested and significant (FDR<0.05) GWAS trait classes in eQTS analysis.
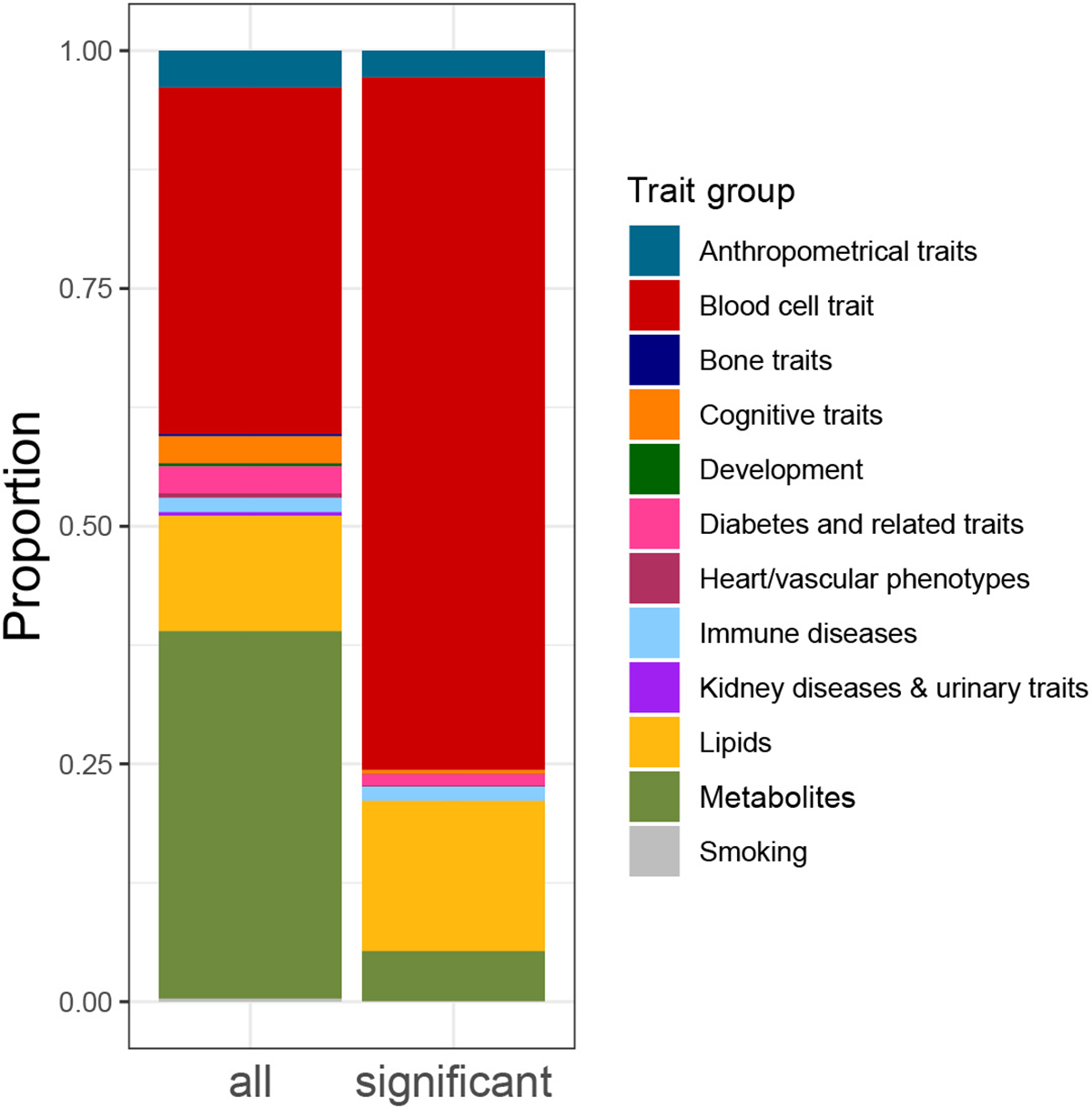
Overview of tested and significant (FDR<0.05) GWAS trait classes in eQTS analysis.
Supplementary Material
Acknowledgments
The cohorts participating in this study list their acknowledgments in the cohort-specific sections of Supplementary Note.
This work is supported by a grant from the European Research Council (ERC, ERC Starting Grant agreement number 637640 ImmRisk), a VIDI grant (917.14.374) and VICI grant from the Netherlands Organisation for Scientific Research (NWO) to L.F. This work has been supported by the European Regional Development Fund and the programme Mobilitas Pluss (MOBTP108) to U.V. The project was supported by Foundation “De Drie Lichten” in the Netherlands with a grant to A.C. M.G.N. is supported by ZonMw grants 849200011 and 531003014 from The Netherlands Organisation for Health Research and Development, a VENI grant from NWO (VI.Veni.191G.030) and a Jacobs foundation research fellowship. H.Y. is funded by a Diabetes UK RD Lawrence fellowship (17/0005594). This project received funding from the ERC under the European Union’s Horizon 2020 research and innovation programme (grant agreement n° 772376 - EScORIAL) to J.H.V. T.E. and A.K. were supported by the Estonian Research Council grant PRG (PRG1291). A.Ba. was supported by NIH grant 1R01MH109905, NIH grant R01HG008150 (NHGRI; Non-Coding Variants Program) and NIH grant R01MH101814 (NIH Common Fund; GTEx Program). M.v.d.W was funded by Nederlandse Organisatie voor Wetenschappelijk onderzoek, NWO-Veni 192.029. This work was supported by National Institutes of Health grants R21ES024834 (B.P. and M.A.), R01ES020506 (B.P.), R01ES023834 (B.P.), R35ES028379 (B.P.), R01 GM108711 (L.C.), and R01CA107431 (H.A.). This work was supported through The Sigrid Juselius Foundation (J.Ke.) and funds from the Academy of Finland [grant numbers 297338 and 307247] (J.Ke.) and Novo Nordisk Foundation [grant number NNF17OC0026062] (J.Ke.). S.Ri. was supported by the Academy of Finland Centre of Excellence in Complex Disease Genetics (Grant No. 312062). M.G. was supported by EU Horizon 2020 (grant 733100 for SYSCID); and grant from EOS excellence of Science (FNRS and FWO) (gnaf N° 30770923). We acknowledge support from BBMRI–NL (Biobanking and Biomolecular Resources Research Infrastructure 184.021.007 and 184.033.111); Spinozapremie (NWO- 56–464-14192), the European Research Council (ERC Advanced 230374) and KNAW Academy Professor Award (PAH/6635) to D.I.B. G.H. works in a unit that receives funding from the UK MRC (MC_UU_12013/1&2&5) and the University of Bristol. S.B. was supported by the Swiss National Science Foundation (310030–152724). This work was supported by the German Federal Ministry of Education and Research (BMBF) within the framework of the e:Med research and funding concept (grant # 01ZX1906B), and by LIFE – Leipzig Research Center for Civilization Diseases, Universität Leipzig (which is funded by means of the European Union, by the European Regional Development Fund (ERDF) and by means of the Free State of Saxony within the framework of the excellence initiative to H.K. and M.Sc.
We thank the UMCG Genomics Coordination Center, the MOLGENIS team, the UG Center for Information Technology, and the UMCG research IT program and their sponsors in particular BBMRI-NL for data storage, high performance computing and web hosting infrastructure. BBMRI-NL is a research infrastructure financed by the Netherlands Organization for Scientific Research (NWO) [grant number 184.033.111].
We thank Kate McIntyre for editing the manuscript text.
Competing Interests
B.M.P. serves on a DSMB for a clinical trial of a device funded by Zoll LifeCor and on the Steering Committee for the Yale Open Data Access Project funded by Johnson & Johnson. Both activities are unrelated to this work. Rest of the authors declare no competing interests.
Footnotes
Consortium Authors
BIOS Consortium (Biobank-based Integrative Omics Study)
Bastiaan T. Heijmans43, Peter A.C. ‘t Hoen69, Joyce van Meurs27, Rick Jansen19, Lude Franke1,3, Dorret I. Boomsma71, Jenny van Dongen71, Coen D.A. Stehouwer66, Cisca Wijmenga1, Eline P. Slagboom43, Jan H. Veldink72, Hailang Mei56, Maarten van Iterson43, Patrick Deelen1,3,6,7, Marc Jan Bonder1,5, Morris A. Swertz6, Wibowo Arindrarto43
A full list of members and their affiliations appears in the Supplementary Note.
i2QTL Consortium
Marc Jan Bonder1,5, Oliver Stegle5,14,85
A full list of members and their affiliations appears in the Supplementary Note.
References for main text
- 1.Yao C et al. Dynamic Role of trans Regulation of Gene Expression in Relation to Complex Traits. American Journal of Human Genetics 100, 571–580 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Connor LJ et al. Extreme Polygenicity of Complex Traits Is Explained by Negative Selection. American Journal of Human Genetics 105, 456–476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng J et al. Signatures of negative selection in the genetic architecture of human complex traits. Nature Genetics 50, 746–753 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Yao DW, O’Connor LJ, Price AL & Gusev A Quantifying genetic effects on disease mediated by assayed gene expression levels. Nature Genetics 52, 626–633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westra HJ et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature Genetics 45, 1238–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirsten H et al. Dissecting the genetics of the human transcriptome identifies novel trait-related trans-eQTLs and corroborates the regulatory relevance of non-protein coding loci. Human Molecular Genetics 24, 4746–4763 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lloyd-Jones LR et al. The Genetic Architecture of Gene Expression in Peripheral Blood. American Journal of Human Genetics 100, 228–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen R et al. Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Human Molecular Genetics 26, 1444–1451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joehanes R et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biology 18, 16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brynedal B et al. Large-Scale trans-eQTLs Affect Hundreds of Transcripts and Mediate Patterns of Transcriptional Co-regulation. American Journal of Human Genetics 100, 581–591 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis CM & Vassos E Prospects for using risk scores in polygenic medicine. Genome Medicine 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Natarajan P et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 135, 2091–2101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyle EA, Li YI & Pritchard JK An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Li YI & Pritchard JK Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell 177, 1022–1034.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhernakova DV et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nature Genetics 49, 139–145 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Bonder MJ et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nature Genetics 49, 131–138 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289–300 (1995). [Google Scholar]
- 18.Aguet F et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glassberg EC, Gao Z, Harpak A, Lan X & Pritchard JK Evidence for weak selective constraint on human gene expression. Genetics 211, 757–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, Zheng Z, Visscher PM & Yang J Quantifying the mapping precision of genome-wide association studies using whole-genome sequencing data. Genome Biology 18, 86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Astle WJ et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 167, 1415–1429.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melé M et al. The human transcriptome across tissues and individuals. Science 348, 660–665 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi T et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nature Communications 9, 2282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marbach D et al. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nature Methods 13, 366–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li T et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nature Methods 14, 61–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamparter D, Marbach D, Rueedi R, Kutalik Z & Bergmann S Fast and Rigorous Computation of Gene and Pathway Scores from SNP-Based Summary Statistics. PLoS Computational Biology 12, 1–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao SSP et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikpay M et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nature Genetics 47, 1121–1130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bentham J et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nature Genetics 47, 1457–1464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davenport EE et al. Discovering in vivo cytokine-eQTL interactions from a lupus clinical trial. Genome Biology 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McBride JM et al. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: Results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis and Rheumatism 64, 3666–3676 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Yao Y et al. Development of Potential Pharmacodynamic and Diagnostic Markers for Anti-IFN-α Monoclonal Antibody Trials in Systemic Lupus Erythematosus. Human Genomics and Proteomics 1, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perry JRB et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature 514, 92–97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemaitre RN et al. Genetic loci associated with plasma phospholipid N-3 fatty acids: A Meta-Analysis of Genome-Wide association studies from the charge consortium. PLoS Genetics 7, e1002193 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu JZ et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nature Genetics 47, 979–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gateva V et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nature Genetics 41, 1228–1233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moffatt MF et al. A large-scale, consortium-based genomewide association study of asthma. New England Journal of Medicine 363, 1211–1221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wood AR et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nature Genetics 46, 1173–1186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Der Harst P et al. Seventy-five genetic loci influencing the human red blood cell. Nature 492, 369–375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teslovich TM et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willer CJ et al. Discovery and refinement of loci associated with lipid levels. Nature Genetics 45, 1274–1285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. Journal of Clinical Investigation 117, 2216–2224 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldstein JL & Brown MS Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. Journal of Biological Chemistry 249, 5153–5162 (1974). [PubMed] [Google Scholar]
- 45.Singh AB, Kan CFK, Shende V, Dong B & Liu J A novel posttranscriptional mechanism for dietary cholesterol-mediated suppression of liver LDL receptor expression. Journal of Lipid Research 55, 1397–1407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El-Hattab AW Serine biosynthesis and transport defects. Molecular Genetics and Metabolism 118, 153–159 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Leuzzi V, Alessandrì MG, Casarano M, Battini R & Cioni G Arginine and glycine stimulate creatine synthesis in creatine transporter 1-deficient lymphoblasts. Analytical Biochemistry 375, 153–155 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Hart CE et al. Phosphoserine aminotransferase deficiency: A novel disorder of the serine biosynthesis pathway. American Journal of Human Genetics 80, 931–937 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klomp LWJ et al. Molecular characterization of 3-phosphoglycerate dehydrogenase deficiency - A neurometabolic disorder associated with reduced L-serine biosynthesis. American Journal of Human Genetics 67, 1389–1399 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shaheen R et al. Neu-laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH. American Journal of Human Genetics 94, 898–904 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Price AL et al. Single-tissue and cross-tissue heritability of gene expression via identity-by-descent in related or unrelated individuals. PLoS Genetics 7, e1001317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mostafavi H et al. Variable prediction accuracy of polygenic scores within an ancestry group. eLife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van der Wijst M et al. The single-cell eQTLGen consortium. eLife 9, e52155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang D et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 55.Deelen P et al. Genotype harmonizer: Automatic strand alignment and format conversion for genotype data integration. BMC Research Notes 7, 901 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rumble SM et al. SHRiMP: Accurate mapping of short color-space reads. PLoS Computational Biology 5, e1000386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Purcell S et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Westra HJ et al. MixupMapper: Correcting sample mix-ups in genome-wide datasets increases power to detect small genetic effects. Bioinformatics 27, 2104–2111 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zerbino DR et al. Ensembl 2018. Nucleic Acids Research 46, D754–D761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zaykin DV Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. Journal of Evolutionary Biology 24, 1836–1841 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobin A et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nature Genetics 48, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Chen EY et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lachmann A et al. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feingold EA et al. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 306, 636–640 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Myers RM et al. A user’s guide to the Encyclopedia of DNA elements (ENCODE). PLoS Biology 9, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lachmann A et al. Massive mining of publicly available RNA-seq data from human and mouse. Nature Communications 9, 1366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu G, Wang LG, Han Y & He QY ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A Journal of Integrative Biology 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Javierre BM et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 167, 1369–1384.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schofield EC et al. CHiCP: A web-based tool for the integrative and interactive visualization of promoter capture Hi-C datasets. Bioinformatics 32, 2511–2513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Swertz MA et al. The MOLGENIS toolkit: Rapid prototyping of biosoftware at the push of a button. BMC Bioinformatics 11, S12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Primary genotype and gene expression data was analyzed by individual cohorts participating in the study and our study analyzed summary statistics. Full summary statistics of the eQTLGen cis-eQTL, trans-eQTL and eQTS meta-analyses are available on the eQTLGen website, www.eqtlgen.org, which was built using the MOLGENIS framework73. We also provide cis-eQTL files formatted for use in SMR, MAFs, and replication statistics for cis-eQTLs, trans-eQTLs and eQTSs. Per-cohort summary statistics for discovery cohorts can be made available after approval of an analysis proposal in eQTLGen and with agreement of the cohort PIs, contact corresponding authors for further information. Trait-associated variants were collected from EBI GWAS Catalogue (https://www.ebi.ac.uk/gwas/; accessed on 21 November 2016), NIH GWAS Catalogue (now hosted by EBI GWAS Catalogue: https://www.ebi.ac.uk/gwas/) and Immunobase (www.immunobase.org; accessed 26 April 2016; now hosted by Open Targets: https://genetics.opentargets.org/immunobase). Sources of numerous GWAS summary statistics used for eQTS analyses are outlined in Supplementary Note and Supplementary Table 13. ExAC pLI scores used for Figure 2 originate from: ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.3.1/functional_gene_constraint/fordist_cleaned_exac_r03_march16_z_pli_rec_null_data.txt. Genotype reference files used for harmonizing discovery datasets for meta-analysis originate from here: ftp://share.sph.umich.edu/1000genomes/fullProject/2012.03.14/GIANT.phase1_release_v3.20101123.snps_indels_svs.genotypes.refpanel.ALL.vcf.gz.tgz. Gene model used for gene annotations originates from ENSEMBL v71 (ftp://ftp.ensembl.org/pub/release-71/gtf/homo_sapiens/Homo_sapiens.GRCh37.71.gtf.gz). FANTOM TF annotations used for eQTS enrichment analyses originate from: http://fantom.gsc.riken.jp/5/sstar/Browse_Transcription_Factors_hg19. CHIP-seq data used for cis-eQTL overlap originates from https://www.chicp.org/. Protein-protein interaction data used for trans-eQTL mechanism enrichment analyses originate from: https://www.intomics.com/inbio/map/api/get_data?file=InBio_Map_core_2016_09_12.tar.gz. Hi-C data used for trans-eQTL mechanism enrichment is deposited in GEO (GM12878, GEO accession GSE63525). Curated gene sets used for enrichment analyses (Gene Ontology sets, ENCODE CHiP-X and CheA CHiP-X TF targets, TRANSFAC and JASPAR PWMs, ARCHS4 tissue expression, TargetScan miRNA target predictions, Tarbase miRNA validated targets) were downloaded from Enrichr web site (https://maayanlab.cloud/Enrichr/#stats). Gene expression summaries and meta-data from GTEx v7 originate from: https://gtexportal.org/home/. Gene expression summaries from BIOS are available in Source Data 1 and also in BIOS Omics Atlas (http://bbmri.researchlumc.nl/atlas/#data). Mean cell counts for eight immune cell types from 1M-scBloodNL cohort are available in Source Data 2. Per-cohort individual-level genotype and gene expression data is governed by respective biobanks and access can be requested according to procedures established by each biobank, with relevant restrictions applying as imposed by IRB or local legislation. Data access procedures established for BIOS Consortium are available on https://www.bbmri.nl/acquisition-use-analyze/bios.