

Abstract
The sequence similarity within the amino-terminal regions of parathyroid hormone (PTH) and PTH-related protein (PTHrP) allow the two to share actions upon a common receptor, PTH1R. A number of biological activities have been ascribed to actions of other domains within PTHrP. PTHrP production by late stage breast cancer has been shown to contribute to bone metastasis formation through promotion of osteoclast formation and bone resorption by action through PTH1R. There is evidence also for a role for PTHrP early in breast cancer that is protective against tumour progression. No signaling pathway has been identified for this effect. PTHrP has also been identified as a factor promoting the emergence of breast cancer cells from dormancy in bone. In that case PTHrP does not function through activation of PTH1R, despite having very substantial effects on transcriptional activity of the breast cancer cells. This indicates actions of PTHrP that are non-canonical, i.e. mediated through domains other than the amino-terminal. It is concluded that PTHrP has several distinct paracrine, autocrine and intracrine actions in the course of breast cancer pathophysiology. Some are mediated through action upon PTH1R, others controlled by other domains within PTHrP.
INTRODUCTION
Humoral hypercalcaemia of malignancy
The discovery of PTHrP arose from interest in the mechanisms by which certain cancers cause hypercalcaemia without necessarily metastasising to the skeleton. Hypercalcaemia was recognised as a complication of cancer in the early 20th century. When Fuller Albright in 1941 [1] was discussing a patient with renal carcinoma, a solitary metastasis and hypercalcemia, he suggested that some tumors might cause hypercalcaemia by secreting parathyroid hormone (PTH) or something very like it. Early studies that implicated PTH in the development of hypercalcaemia seemed to support the concept of “ectopic PTH production” by cancers as the cause of non-metastatic hypercalcemia [2]. In the early 1970s though, some doubt arose regarding the nature of the cancer-derived product that led to this cancer syndrome. In hypercalcaemic patients with non-parathyroid cancer or primary hyperparathyroidism, it was noted that, although the hypercalcaemia was greater in the cancer group, those with primary hyperparathyroidism had lower serum concentrations of an apparently different immunologic identity to PTH itself [3, 4]. Protein extract of a breast cancer from a hypercalcaemic patient also yielded PTH immunoreactivity that was non-parallel to standard [5] and Powell et al [6] showed that immunoreactive PTH could not be detected in a number of hypercalcemic cancers, despite the use of several antisera with a range of epitopes. The term humoral hypercalcaemia of malignancy (HHM) was introduced to describe patients with non-metastatic hypercalcaemia [7]. Ultimately, clinical studies established the biochemical similarity between primary hyperparathyroidism and this syndrome of HHM [8–10], with the weight of evidence being that the responsible factor was chemically different from PTH, although possessing virtually identical biological activities.
Identification of PTHrP
Biological assays of PTH-like activity had been developed by this time [11, 12] that led to the discovery in extracts and culture supernatants of hypercalcemic tumors of activity that promoted PTH-like adenylyl cyclase responses in osteoblast and kidney targets [13] [14] [15]. This paved the way for purification of the active protein from a human lung cancer cell line [16], a breast cancer [17] and a renal cancer cell line [18]. The cloning of its cDNA [19] showed 8 of the first 13 residues of this PTH-related protein (PTHrP) to be identical to those in PTH. The structural requirements for full biological activity of PTHrP were contained within the first 34 amino acids [20], as was known to be the case with PTH [21]. These findings were sufficient to explain the biochemical similarities between syndromes of PTH excess and non-metastatic hypercalcemia in cancer. They signalled the discovery of an evolutionary relationship between these two molecules, most likely derived from a common ancestor and evolving from a gene duplication event.
PTHrP structural domains
PTHrP could be divided into different domains on the basis of its primary amino acid sequence (Figure 1). Intracellular “prepro” and “pro” precursors of the mature peptide, essential for intracellular trafficking, are encoded with the first 36 amino acids (−36 to−1); this domain is cleaved from the molecule when it is secreted. The next domain includes the first 13 residues of the mature protein, of which 8 of this domain are identical with PTH. This domain is critical for most of the agonist effects of PTH and PTHrP on their shared PTH1R receptor [22] [23]. The following residues, PTHrP (14–36), although having almost no homology with PTH, appear to be critical for binding of PTHrP to PTH1R.
Figure 1.
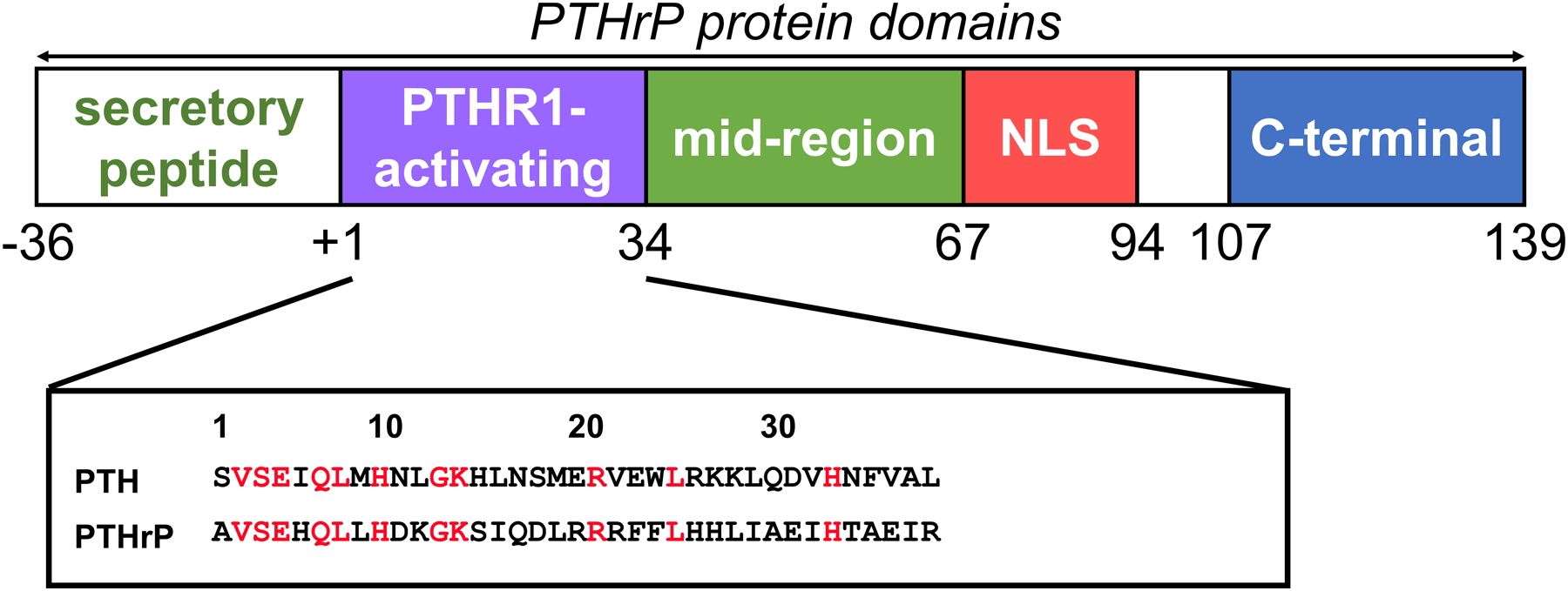
The biological domains of PTHrP (See text).
The marked conservation of the PTHrP amino acid sequence in human, rat, mouse, chicken and canine up to position 111 indicated that important functions are likely to reside in this region. In addition to the actions of PTHrP through PTH1R, there is increasing evidence for other biological activities within the PTHrP molecule that give rise to the concept that PTHrP is a polypeptide precursor of a number of biological activities, analogous with pro-opiomelanocortin [24]. These include data suggesting that PTHrP is an oncofetal hormone, circulating in the fetus and acting on the placenta to promote calcium transport from the mother to the fetus [25–28], an effect mediated by a portion of the PTHrP molecule distinct from the PTH-like region. Of great interest was the discovery that PTHrP is localised either in the nucleus or the cytoplasm of cells, and that its location is cell cycle-dependent [29–31].
Molecular details of the actions of amino acids 36–139/141, including the nuclear localizing sequence (NLS), are not well established. A number of biological actions have been ascribed to the carboxyterminal region of PTHrP, beginning at residue 107 (v infra). The final tail region of PTHrP, amino acids 142–173, is found only in humans and is encoded by only one of the three human PTHrP mRNA isoforms. Its significance in terms of tissue distribution, processing, or function is unknown. Apart from recognition of specific details of the nuclear transport mechanism [30–32], receptors have not yet been identified for any of the non-PTH1R (non-canonical) actions of PTHrP.
ENDOCRINE, AUTOCRINE, AND PARACRINE ROLES OF PTHrP
For the most part, PTHrP has an autocrine or paracrine role. Only three circumstances have been identified postnatally in which PTHrP is convincingly present in the circulation and acting in an endocrine manner. These are: 1) the HHM syndrome, in which PTHrP is secreted by tumors [33] and is targeted to bone and kidney, 2) lactation, in which PTHrP is made in the breast and reaches the circulation [34], and 3) fetal life, where PTHrP regulates maternal-to-fetal placental calcium transport [26].
The fact that PTHrP cannot be detected in the circulation of postnatal animals, together with the widespread expression of PTHrP in the developing embryo and adult tissues, supported the hypothesis that PTHrP is a cellular cytokine whose actions involve both cell growth and differentiation. PTHrP mRNA or protein are detected in the following human tissues: adrenal, bone, brain, heart, intestine, kidney, liver, lung, mammary gland, ovary, parathyroid, placenta, prostate, skeletal muscle, skin, spleen, stomach, and smooth muscle (reviewed in [35]). The spatial and temporal distribution of PTHrP correlates highly with that of the PTH1R [36, 37], which can be detected in the parietal endoderm from day 5.5 in the mouse and at sites of epithelial/mesenchymal interactions in the rat embryo from day 9.5 [36]. The relative expression levels of PTHrP and its receptor are often inversely correlated within a tissue or in certain locales along a border of apposition. Such a tight inverse coupling of expression seemed to imply either feedback downregulation of the receptor or a precise coordinate regulation of the two genes during the course of fetal development [37].
INTRACRINE AND AUTOCRINE ACTIONS OF OTHER PTHrP DOMAINS
In addition to its actions through the PTH1R, PTHrP translocates to the nucleus through a specific transport process. This localization through a defined sequence in the mid-region was found to be essential for the ability of PTHrP to confer enhanced survival on chondrocytes following serum starvation [38]. The mechanism of import of PTHrP requires interaction with importin β, which structural analysis revealed binds to PTHrP (67–94) [39]. A nuclear targeting sequence inhibiting apoptosis exists at PTHrP (87–107) [40]), and PTHrP (109–139) is involved in its nuclear export. In quiescent cells, nuclear/nucleolar location is evident, with predominant cytoplasmic location and increased production and secretion as cells move towards mitosis [30]. The nuclear transport of PTHrP is carried out by specific binding to importin β, and phosphorylation of Thre85 of PTHrP by the cyclin dependent protein kinases, CDK2 and CDC2, which favours extrusion of PTHrP from the nucleus [30]. The nuclear/nucleolar location, its phosphorylation control, cell cycle dependence, and specific nuclear import mechanism all suggest that the protein exerts important functions(s) in the nucleus, the nature of which remain to be determined.
In vascular smooth muscle cells, PTHrP localized to the nucleus increases cell proliferation, whereas extracellular PTHrP treatment decreases cell proliferation and enhances muscle relaxation in the same cells by acting through PTH1R [41, 42]. Remarkably, in the vascular smooth muscle experiments, the increased mitogenesis resulting from PTHrP transfection was found to require not only the NLS, but also the C-terminal (108–139) domain of the molecule [42], suggesting that additional non-nuclear actions are involved in the intracrine action of PTHrP. The C-terminal domain of PTHrP has had many biological actions ascribed to it in pharmacological experiments carried out in vitro and in vivo.
Interest in the C-terminal domain began with the finding that PTHrP (107–139) inhibited osteoclast activity and bone resorption by isolated rat osteoclasts in vitro, an effect exerted by the pentapeptide TRSAW (residues 107–111), that was then named “osteostatin” [43]. Although injection of PTHrP (107–139) over the calvariae in mice was found to inhibit bone resorption [44], the anti-resorptive effect of TRSAW in organ culture has been controversial, with some investigators not finding this effect in vitro [45]. The peptide was found to be mitogenic for osteoblasts in vitro [46].
Although no receptor has yet been identified, both TRSAW and PTHrP (107–139) increased protein kinase C activity in rat splenocytes at low picomolar concentrations [47], with similar actions in ROS 17.2/8 osteosarcoma cells [48]. The same group of authors reported protein kinase C activation in osteosarcoma cells by PTH (28–34) and PTHrP (28–34) [49]. The C-terminal domain is the least conserved among species, with only PTHrP (107–111) (TRSAW) being conserved among mammals. It should be noted that in many of the cited studies, the TRSAW peptide reproduced faithfully the effects of PTHrP (107–139). Thus, this short sequence seems likely to be the most important contributor to the host of pharmacologic effects reported, in which case it could provide a pathway to receptor identification. Although there can be no certainty of any physiological implications from these pharmacological studies, possible roles for the C-terminal domain should continue to be sought, and this would include studies in bone.
An interesting further insight into cancer-derived PTHrP action comes from the finding that PTHrP is a substrate of matrix metalloproteinases (MMPs), which can generate PTHrP (1–17) [50]. This peptide fragment promoted pre-osteoblast motility and differentiation, signalling through PTH1R to increase Ca2+ flux and ERK phosphorylation. PTHrP (1–17) had no effect on cAMP production or osteoclast formation through RANKL, unlike the PTHrP (1–36) effect (v infra). Such an effect of this short peptide, favouring a bone-forming effect of PTHrP, will be of interest in further work on its formation in cancers and its actions in context with other products of PTHrP proteolysis.
PTHrP IN BREAST CANCER AND BONE METASTASIS: PRECLINICAL DATA
Paracrine and autocrine mechanisms extend to the roles of PTHrP in cancer, with much of the evidence coming from breast cancer. Some PTHrP sequence data was obtained from a breast cancer extract in early work [51], and PTHrP was detected in lactating breast [52] as well as in breast milk [53]. In addition to circulating and acting as hormonal mediator of HHM, PTHrP is produced by two thirds of primary breast cancers [54], and plasma levels of PTHrP were elevated in 60% of patients with hypercalcaemia in breast cancer associated with bone metastases [33]. Furthermore, immunohistochemical staining of PTHrP was detected in 90% of bone metastases but less than 20% of metastases in soft tissue sites [55], prompting the suggestion that the local production of PTHrP in the bone marrow by breast cancer cells could promote the bone resorption process, providing a niche for tumour establishment and subsequent growth and expansion.
Indeed the view was commonly expressed that the single most important property required of cancer cells to establish and grow in bone is the ability to promote bone resorption [56–58]. An experiment that seemed to support this was one in which human MCF7 breast cancer cells, which normally do not grow in bone after intracardiac injection in nude mice, did so prolifically with substantial lytic deposits when PTHrP was overexpressed in the cells [59]. In that same work, cancer cell-derived PTHrP was shown to promote production of receptor activator of NFκB ligand (RANKL) by host osteoblastic cells, resulting in osteoclast formation and the resorption required for the lytic deposits (Figure 2). In addition to the evidence from animal studies there is ample histological evidence that human breast tumor deposits in bone are surrounded by active osteoclasts [60, 61].
Figure 2. PTHrP effects on osteoclasts and bone resorption.
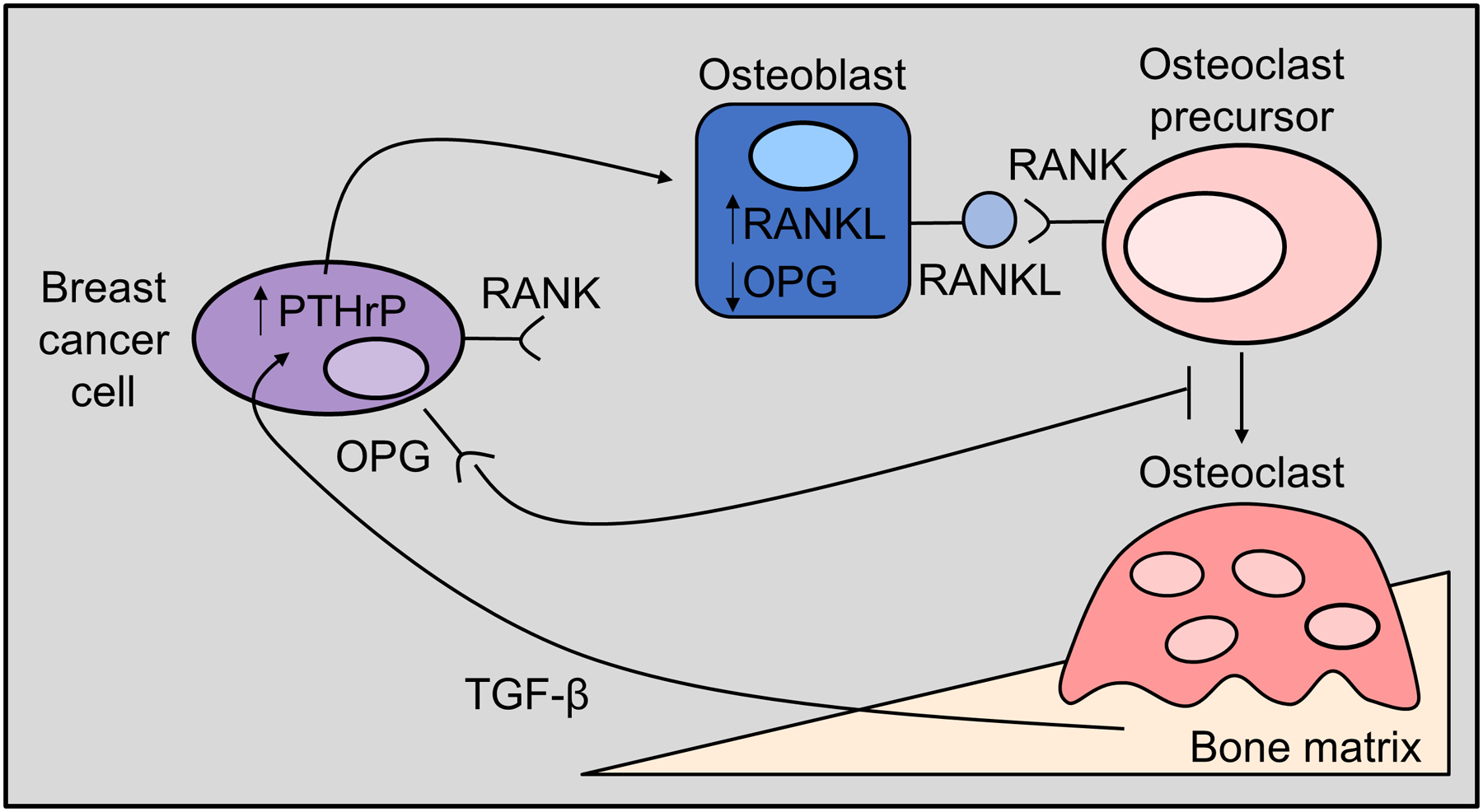
Tumour-derived PTHrP stimulates RANKL production by host osteoblast lineage cells, resulting in osteoclast formation [108] and the resorption required for the formation of osteolytic lesions and tumor colonisation of the bone marrow. Increased resorption releases growth factors from the bone matrix, such as transforming growth factor beta (TGF-β) [108], which signal back to the tumour cells to enhance PTHrP production and further drive tumour growth in the bone marrow.
The hypothesis that bone-disseminated tumour cells produce PTHrP at localised sites to enhance their growth in bone is further supported by bioengineering studies that demonstrate PTHrP is specifically up-regulated when breast or lung cancer cells are cultured on substrates with rigidities similar to those of bone [62, 63]. Furthermore, these effects are reversed when the cells are treated with neutralising antibodies against Roc or integrin β3 to block mechanotransduction signals.
The idea that PTHrP provides a favorable niche for tumour cells to grow in bone was supported by studies of Guise et al [64] in which MDA-MB-231 human breast cancer cells are injected into the left cardiac ventricle of nude mice, and examined by radiology and histology to quantitate lytic bone lesions that develop after approximately 3 weeks. Importantly, treatment of tumour-injected animals with a neutralising monoclonal antibody against PTHrP largely prevented tumour growth and histological evidence of bone invasion. In these studies, tumours growing in bone were marked by prolific osteoclast appearance at the tumour-bone interface, an effect that was lost by treatment with bisphosphonates, or with neutralising monoclonal antibodies against PTHrP [64, 65], [66]. This efficacy of anti-PTHrP against tumour growth in bone recapitulated the success of polyclonal [67] and monoclonal [68] antibodies in treating the hypercalcaemia in mouse models of HHM. A fully humanised anti-PTHrP monoclonal antibody was developed that was shown to be fully effective in these experimental models [69], but there is no published data on the application of such an antibody in human studies. Informative as these studies have been, a mouse model of a mammary carcinoma that spontaneously metastasises to bone and represents the entire metastasis pathway from the primary site to bone would be desirable. Such a tumour, the 4T1 mouse mammary carcinoma model, has proven useful in defining the roles of PTHrP in the early invasive processes as well as in the establishment of tumor cells at secondary bone sites [70]; however, the frequency of spontaneous bone metastases in this model varies so widely that it has been of limited use [71]. Other bone-tropic variants of the 4T1 model (e.g. 4T1.2, 4T1BM2) have been employed to study tumour dissemination to bone in vivo [72, 73]. These cell line variants are detectable in bone by histology and qPCR for genomic DNA but have not been characterised for PTHrP production.
These experiments do not exclude contributions from other cytokines, e.g. IL-1, IL-6 [74], IL-8 [66], TNFα or cyclo-oxygenase products [75], which could be produced by tumor or host cells in response to the tumor [66] [74, 75]. Indeed, IL-6 in particular is well-established to promote tumour growth in the bone marrow through osteoclast activation [74]. As is the case with PTHrP [59], these cytokines promote osteoclast formation and resorption either by increasing RANKL production by host osteoblast lineage cells, or as in the case of IL-8, by direct action upon haemopoietic precursors, independent of RANKL [66, 76]. Together these data seemed to indicate that bone metastatic growth of tumours required not just the general invasive properties that are expected of cancer cells, but specifically, the a ability of the cells to promote bone resorption.
For some years, tumour production of PTHrP has been proposed to be linked to the cachexia that often accompanies hypercalcaemic and bone metastatic cancers. Evidence first came from hypercalcaemic tumours grown in immune-deficient mice. Cachexia was closely associated with high circulating PTHrP and calcium levels and could be corrected by anti-PTHrP treatment [68, 69], but in these studies it was not possible to establish PTHrP as the definitive cause of cachexia as distinct from a hypercalcaemic cause. This question was pursued further in studies of the Lewis lung carcinoma model of cancer cachexia in nude mice [77], which suggested PTHrP has a role in wasting by promoting expression of genes involved in thermogenesis in adipose tissues. Antibody neutralisation of PTHrP blocked cachexia development in these mice that had elevated PTHrP levels, but surprisingly, the mice were normocalcaemic. A possible related link to muscle effects has also come from work showing that muscle weakness in nude mice bearing osteolytic human cancers likely resulted from resorption-induced TGFβ, promoting a series of effects that resulted in decreased Ca2+-induced muscle force production [78]. A conclusion from that work was that muscle weakness preceded the lost muscle mass of cachexia. Release of TGFβ by PTHrP - stimulated bone resorption had been established previously by this group [59, 64, 79]. The questions raised by these studies concerning PTHrP involvement in processes of muscle weakness and wasting clearly merit further study.
PTHrP IN BREAST CANCER AND BONE METASTASIS: CLINICAL DATA
There has been considerable conflation between the role for PTHrP in tumor-induced bone disease and its potential effects on spontaneous metastasis to the bone marrow. Preclinical data has primarily focused on the mechanism by which PTHrP promotes osteolysis in the tumor-bone microenvironment. In contrast, clinical studies have focused on evaluating PTHrP expression in the primary tumor and bone metastatic site. The incidence of 60 to 70% of positive staining for PTHrP in primary breast cancers has been amply confirmed at the protein [54, 80–82] and mRNA level [81, 83]. Several of these studies concluded that PTHrP expression in primary breast cancers is related to subsequent bone metastasis development [80–82]. However, all of these studies had limitations from a number of points of view. The patients were selected subjects with advanced disease and there were limited numbers of patients, with limited follow-up and retrospective accrual.
The only long-term prospective study of consecutively accrued patients has been carried out on 526 consecutive patients treated by surgery at one centre, and analysed after evaluation over 5 years [84] and 10 years [85]. Importantly, these studies demonstrate that PTHrP in the primary tumor site may in fact provide protection against the formation of bone metastases. With an incidence of 79% of patients with PTHrP positive breast cancers at the time of operation, and PTHrP positive staining associated significantly with ER, PR and menopausal status, the analyses at 5 [84] and 10 years [85] showed that patients with PTHrP positive tumours had significantly improved survival (p < 0.001), and had significantly fewer metastases at all sites, including bone (p=.04) (Figure 3). Although this finding was at odds with the starting hypothesis, which was that expression of PTHrP in primary breast cancers would correlate with subsequent development of bone metastases, it is by no means inconsistent with a role for PTHrP in bone metastasis development. This clinical study suggested that PTHrP can have effects on breast cancer behaviour that differ from the distinct ability to promote bone resorption. The latter remains likely to be an important contributor to resorption and bone metastasis in late stages of the disease, but the outcome of the prospective clinical study indicates that PTHrP might confer upon cancer cells a less invasive phenotype, most likely through actions exerted earlier in tumour development. Consistent with the findings of Henderson et al in their prospective study [84], a recent analysis by PTHrP immunostaining in two cohorts of patients demonstrated that loss of PTHrP nuclear, but not cytoplasmic, staining was associated with unfavourable prognosis [84].
Figure 3. PTHrP is associated with increased breast cancer patient survival.
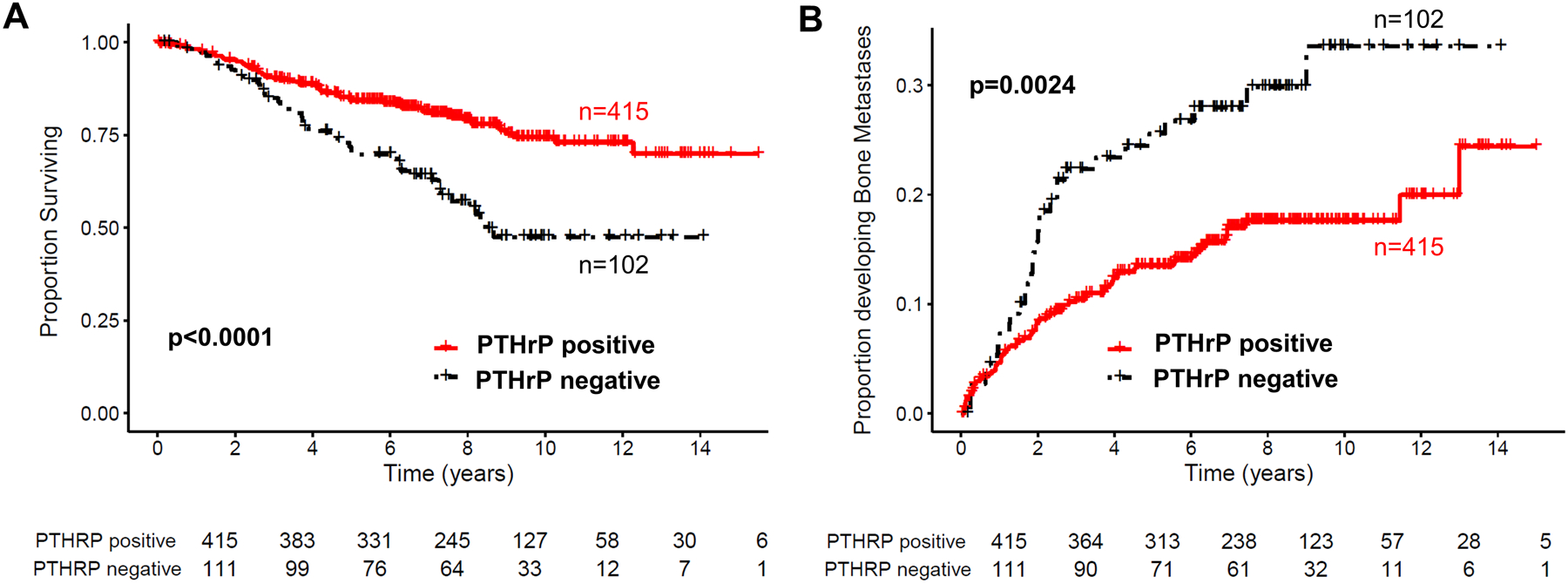
(A, B) Data sourced from [108]. PTHrP protein by immunohistochemistry in N=526 breast cancer patients with mean follow-up of 10.8 years. PTHrP in the primary breast tumour was an independent predictor of improved survival and decreased risk of developing bone metastases.
Such a protective effect of PTHrP might be ascribed to an action early in tumour progression of any of the several domains of the molecule apart from the amino-terminal region that acts upon PTH1R to mediate the bone resorptive effect. Another example of a protein with a divergent effect in cancer is transforming growth factor beta (TGFβ), which acts early as a tumor suppressor by inhibiting proliferation of epithelial, endothelial and hemopoietic cells. Refractoriness to these effects develops later, and overexpression of TGFβ leads to a microenvironment conducive to tumor growth (reviewed in [86–89]).
Surprising though the outcome of the clinical trial was, support for it comes from a publicly available data base of independent transcript analyses of PTHLH (n = 3549 patients) [90–92]. These data showed association between low PTHLH transcript levels and unfavourable prognosis that applied to both estrogen receptor (ER) positive and ER negative breast cancer patients.
Mouse genetic studies
The questions arising from the finding of a “protective” effect of PTHrP in breast cancer pathogenesis remain unresolved. They were addressed in two independent studies using genetically induced spontaneous mouse mammary carcinoma models. When PTHrP was conditionally deleted in mice with MMTV-Neu-induced carcinogenesis, loss of PTHrP resulted in a higher tumour incidence, suggesting that PTHrP prevents tumor progression. The gene expression signature associated with loss of PTHrP in vivo correlated with poorer outcome in breast cancer. The conclusion was that loss of PTHrP accelerates mammary tumourogenesis by a non-cell-autonomous tumour suppressor pathway [93]. Quite the opposite conclusion was reached when PTHrP was ablated in the MMTV-PyMT mouse mammary carcinoma model [94], where there was a delay in primary tumour initiation, reduced tumour progression, and reduced metastases to all sites when PTHrP was deleted. Thus the role for PTHrP in primary tumor progression has not been clarified by these studies.
BONE METASTASIS MECHANISMS BEYOND RESORPTION
The unexpected outcome of the prospective clinical study [84, 85] indicating that PTHrP may be protective against tumor progression and bone metastasis had no easy explanation, and was further complicated by the two studies carried out in genetically manipulated mice with differing outcomes [93, 94]. The contribution of cancer-derived PTHrP to the skeletal complications of late stage breast cancer had been supplemented by the possibility of an entirely separate action. This would be one in which PTHrP exerts an effect upon an earlier stage of tumourigenesis or upon aspects of invasion. Any thinking about mechanisms for such effects would need to include the possibility that PTHrP could exert actions through domains of the molecule other than that acting through PTH1R.
An experimental model that seemed to offer a way to examine other possible actions of PTHrP was the estrogen receptor positive human MCF7 breast cancer cell line. As discussed above, these cells lay dormant in bone after intracardiac injection into nude mice, but colonized bone and grew aggressively as lytic deposits when PTHrP was overexpressed [59]. In the course of examining the possible role of the leukemia inhibitory factor receptor (LIFR) in colonization and growth of tumour in bone, LIFR expression was found to be lower in primary breast cancers of patients with bone metastases [95], and was correlated with patient outcome. Using MCF7 cells as an experimental model of dormancy, knockdown of LIFR in the MCF7 cells conferred upon them the ability to colonise and grow in bone in vivo in a similar manner to the PTHrP-overexpressing cells, thus overcoming the dormancy behaviour of MCF7 cells [95]. In taking this further, gene expression analysis of these cells revealed that overexpression of PTHrP resulted in the down-regulation of several pro-dormancy genes [95]. Among these genes were LIFR and its downstream signaling target SOCS3.
Subsequent RNAseq analysis of MCF7 PTHrP over-expressing cells, which had previously been shown to aggressively colonize the bone in vivo [59], identified more than 2500 genes differentially regulated with a log 2-fold change >1 and p<0.05 in MCF7 PTHrP-overexpressing vs MCF7 control cells [96] (Figure 4A).This was of particular interest since it had long been known that although MCF7 cells expressed functional receptors for calcitonin and prostaglandin E2 linked to adenylyl cyclase activation, no such activation could be detected in response to PTH (1–34) [97]. This lack of activation through the PTH1R in MCF7 cells was confirmed [96], in addition to finding no effect on activation of a CREB reporter construct that is readily activated by either salmon calcitonin or PGE2 (Figure 4B&C). The latter two agonists, unlike PTH and PTHrP, also promoted expression of genes known to be regulated by the protein kinase A (PKA) – CREB pathway [96]. Consistent with a lack of regulation through PTH1R in MCF7 cells, RNAseq analysis confirmed that only 2 of a previously described panel of 32 CREB-responsive genes [98] were significantly up-regulated in MCF7 PTHrP-overexpressing cells. Three CREB-responsive genes were significantly down-regulated, and the remaining 27 were not altered by PTHrP over-expression, confirming that even long-term overexpression of PTHrP does not induce genes that result from cAMP signaling in MCF7 cells.
Figure 4. PTHrP induces gene expression independent of cAMP signaling in MCF7 cells.
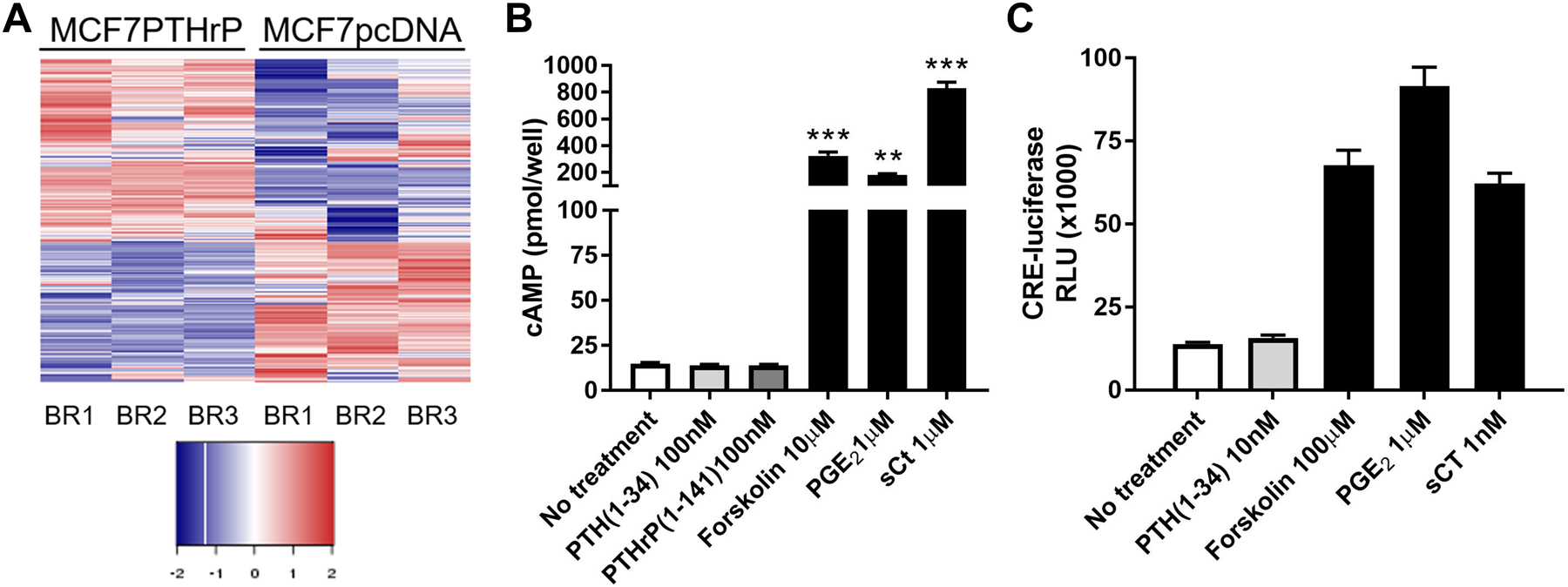
(A) >2500 genes were significantly changed by log 2-fold change >1 and p<0.05 when PTHrP was over-expressed in MCF7 cells. Data sourced from [108]. (B) Neither PTH not PTHrP are able to induce cAMP production in MCF7 cells, but positive controls forskolin, prostaglandin E2 (PGE2), and salmon calcitonin (sCT), which do not use the PTH1R, are able to induce cAMP. Data sourced from [108]. (C) PTH does not activate a cAMP-responsive element (CRE)-luciferase reporter construct in MCF7 cells, but positive controls forskolin, prostaglandin E2 (PGE2), and salmon calcitonin (sCT) activate the CRE luciferase reporter. Data sourced from [108].
Taken together, this provides evidence that substantial effects of PTHrP overexpression on gene expression in MCF7 cells are unrelated to PTH1R-mediated actions through the cAMP/PKA/CREB activation pathway (Figure 5). Thus, the other (non-canonical) domains of PTHrP need to be considered, acting either in an intracrine or autocrine/paracrine manner. Exploration of these domains may reveal novel mechanisms by which PTHrP acts to promote tumor cell exit from dormancy in the bone marrow, and may begin to provide some insight into the yet undefined role for PTHrP in primary tumor progression.
Figure 5. PTHrP can regulate gene expression through intracrine actions.
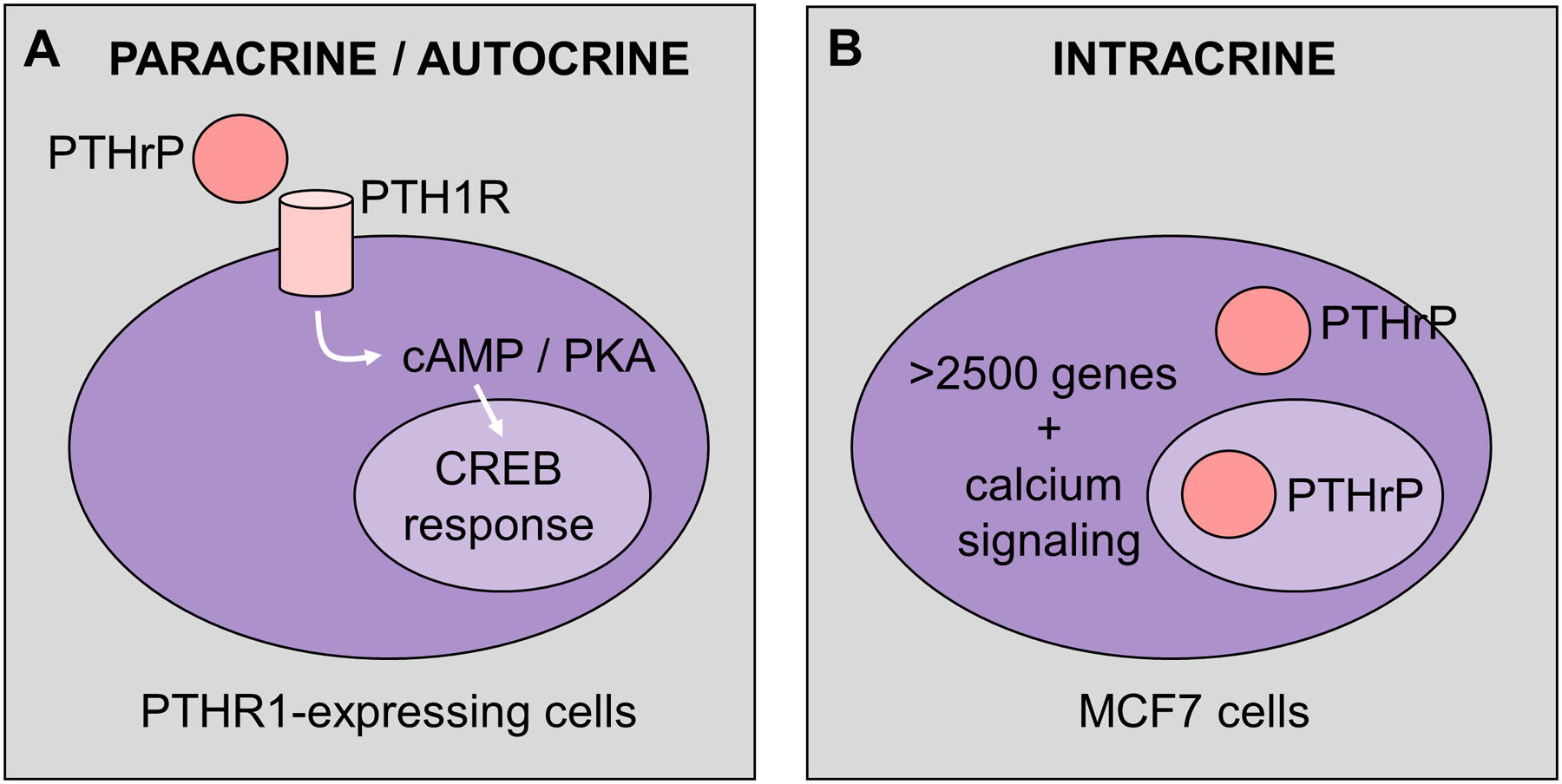
(A) In cells that express PTH1R, PTHrP binds to PTH1R to induce cAMP / CREB signaling, through a known CREB-responsive gene signature [108]. (B) The MCF7 breast cancer cells do not express a functional PTH1R, but are able to induce >2500 genes when PTHrP is overexpressed. This is not through induction of cAMP signaling, but rather activation of alternative pathways, including calcium signaling. It remains unknown if these intracrine actions are mediated through the cytosolic or nuclear actions of PTHrP.
LESSONS FROM THERAPEUTICS: THE AZURE AND RELATED CLINICAL STUDIES
The PTHrP action on osteoclastogenesis was influential in generating the view that bone resorption is the most important property that cancer cells must have in order to grow in bone (v supra). However, this might prove to be an over-simplification, with evidence gathering of other PTHrP actions that are potentially important. Recent clinical studies provide information that could be helpful in this regard.
With bisphosphonates emerging in the 1990s as powerful inhibitors of bone resorption, it was natural that they be approached as potential therapies to prevent metastasis, and possibly improving survival as a result. Inhibition of osteoclast-mediated bone resorption was shown to be effective in reducing skeletal complications from metastatic bone disease in early clinical studies, without effects on disease progression, time to progression or survival [99, 100].
In the later AZURE study of over 3,000 women with Stage II or III breast cancer who were treated with adjuvant zoledronate, no effect on disease-free survival was found with a median follow-up period of 59 months [101], but when a prespecified subgroup analysis was carried out in this extended trial, with a mean follow-up period of 84 months, it revealed significant improvements in disease-free survival in those women in the trial who had passed through the menopause at the time of study entry [102].
These benefits from the use of adjuvant bisphosphonates provided the impetus for a major study investigating the efficacy of denosumab, a human monoclonal antibody against RANKL, which inhibits osteoclast formation and has been shown to be a very powerful inhibitor of bone resorption in clinical studies of osteoporosis [103]. In a randomized comparator study, denosumab was found to be significantly more effective than zoledronate in delaying skeletal-related events in metastatic breast cancer [104], with similar findings in other studies [105]. Subsequently a 5-year international phase III clinical study of denosumab was carried out in 4,500 patients, half treated with placebo and half with denosumab. The outcomes, presented in 2018 and published in abstract form [106] included that there was a significant reduction in time to bone metastasis as a site of first recurrence (p=0.024). Crucially though, there was no discernible effect on the primary endpoint of bone metastasis-free survival, nor on the key secondary endpoints of disease-free survival and distant disease recurrence. Furthermore, unlike the AZURE trial, menopause status had no effect on the outcomes.
This lack of an effect on survival by denosumab was surprising in view of the concept of the primacy of bone resorption in bone metastasis development and progression. It might suggest that inhibition of osteoclastic resorption is not sufficient alone to reduce tumour spread and increase survival in breast cancer.
Such a concept of factors other than osteoclastic resorption contributing to invasion and metastasis of breast cancers is relevant when considering the possibility that PTHrP might influence breast cancer invasive capacity by non-canonical pathways other than through PTH1R and osteoclastogenesis.
Summary and conclusions.
We conclude that there are a number of ways in which tumour-derived PTHrP can influence breast cancer behavior, as depicted in Figure 6. These include (i) action(s) to suppress tumour development and invasion. This would likely be at an early cancer stage. The signaling pathways need to be determined and should include the possibility of actions in the nucleus or through another PTHrP domain. (ii) Action(s) that contribute to cancer cells in bone emerging from a dormant state. Based on current evidence, we hypothesize that the action favouring emergence from dormancy is mediated by non-canonical domains, i.e. distinct from the amino-terminal region that acts through PTH1R on the cAMP-PKA pathway. Such a mechanism would complement rather than exclude the likelihood that a changed microenvironment resulting from increased bone resorption facilitates reactivation of dormant cancer cells in bone [107]. (iii) Action(s) to promote osteoclast formation and activity in the tumour host. This makes use of the PTH1R-PKA-Creb pathway and would be regarded as an action late in tumour pathogenesis (Figure 6).
Figure 6. PTHrP has different actions at different stages of disease progression.
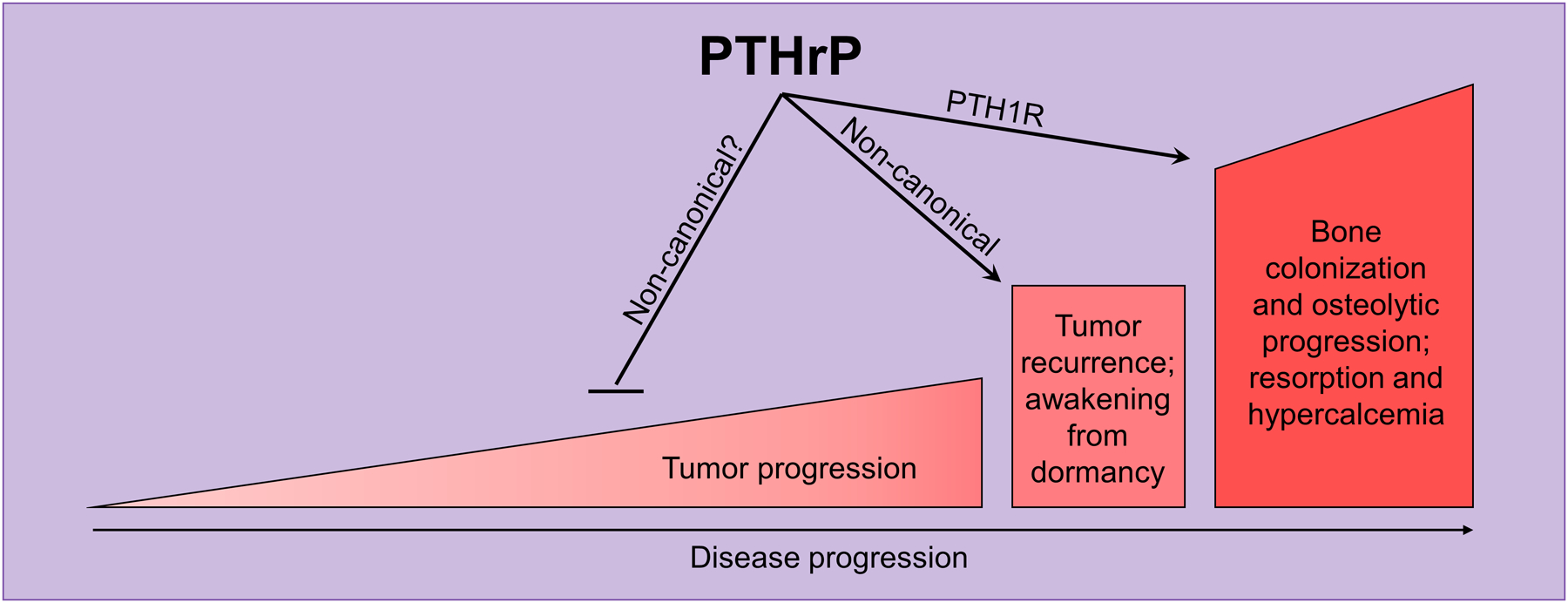
Early in tumour progression, the clinical data suggest that PTHrP inhibits tumour progression since breast cancer patients with PTHrP staining in the primary tumour have better overall survival and reduced risk of developing bone metastases [108]. Once tumour cells have disseminated to the bone marrow, increased PTHrP reduces pro-dormancy genes and drives tumour cells out of a quiescent state [108]. At this stage, high intratumoural PTHrP levels promote osteoclast activation and increased resorption, as well as increased hypercalcemia [108].
Acknowledgements
TJM acknowledges research support from the National Health and Medical Research Council (Australia) and the Victorian Government OIS Program. RWJ acknowledges
NIH award R00CA194198 (RWJ) and DoD CDMRP award W81XWH-18-1-0029 (RWJ). The authors acknowledge the helpful advice of Michael A Henderson.
Abbreviations
- PTH
parathyroid hormone
- PTHrP
PTH-related protein
- PTH1R
PTH/PTHrP receptor1
- HHM
humoral hypercalcaemia of malignancy
- RANKL
receptor activator of NFκB ligand
- IL-6, IL-8, IL-11
interleukins 6, 8 and 11
- TNFα
tumour necrosis factor α
- TGFβ
transforming growth factor β
- SOCS3
suppressor of cytokine signaling 3
- cAMP
cyclic adenosine 3,5-monophosphate
- PKA
protein kinase A
- CREB
cyclic AMP response element binding protein
- LIFR
leukemia inducing factor receptor
- sCT
salmon calcitonin
- PGE2
prostaglandin E2
Footnotes
Competing interests statement.
The authors declare no competing interests.
REFERENCES
- 1.Albright F, Smith PH, and Richardson AM, Postmenopausal osteoporosis. Journal of the American Medical Association, 1941. 116: p. 2465–2474. [Google Scholar]
- 2.Omenn GS, Roth SI, and Baker WH, Hyperparathyroidism associated with malignant tumors of nonparathyroid origin. Cancer, 1969. 24(5): p. 1004–11. [DOI] [PubMed] [Google Scholar]
- 3.Benson RC Jr., et al. , Immunoreactive forms of circulating parathyroid hormone in primary and ectopic hyperparathyroidism. J Clin Invest, 1974. 54(1): p. 175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riggs BL, et al. , Immunologic differentiation of primary hyperparathyroidism from hyperparathyroidism due to nonparathyroid cancer. J Clin Invest, 1971. 50(10): p. 2079–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melick RA, Martin TJ, and Hicks JD, Parathyroid hormone production and malignancy. Br Med J, 1972. 2(5807): p. 204–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powell D, et al. , Nonparathyroid humoral hypercalcemia in patients with neoplastic diseases. N Engl J Med, 1973. 289(4): p. 176–81. [DOI] [PubMed] [Google Scholar]
- 7.Martin TJ and Atkins D, Biochemical regulaors of bone resorption and their significance in cancer. Essays in Medical Biochemistry, 1979. 4: p. 49 – 82. [Google Scholar]
- 8.Stewart AF, et al. , Biochemical evaluation of patients with cancer-associated hypercalcemia: evidence for humoral and nonhumoral groups. N Engl J Med, 1980. 303(24): p. 1377–83. [DOI] [PubMed] [Google Scholar]
- 9.Kukreja SC, et al. , Elevated nephrogenous cyclic AMP with normal serum parathyroid hormone levels in patients with lung cancer. J Clin Endocrinol Metab, 1980. 51(1): p. 167–9. [DOI] [PubMed] [Google Scholar]
- 10.Rude RK, et al. , Urinary and nephrogenous adenosine 3’,5’-monophosphate in the hypercalcemia of malignancy. J Clin Endocrinol Metab, 1981. 52(4): p. 765–71. [DOI] [PubMed] [Google Scholar]
- 11.Atkins D, et al. , Rat osteogenic sarcoma cells: isolation and effects of hormones on the production of cyclic AMP and cyclic GMP. Endocrinology, 1977. 101(2): p. 555–61. [DOI] [PubMed] [Google Scholar]
- 12.Majeska RJ, Rodan SB, and Rodan GA, Parathyroid hormone-responsive clonal cell lines from rat osteosarcoma. Endocrinology, 1980. 107(5): p. 1494–503. [DOI] [PubMed] [Google Scholar]
- 13.Rodan SB, et al. , Factors associated with humoral hypercalcemia of malignancy stimulate adenylate cyclase in osteoblastic cells. J Clin Invest, 1983. 72(4): p. 1511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart AF, et al. , Identification of adenylate cyclase-stimulating activity and cytochemical glucose-6-phosphate dehydrogenase-stimulating activity in extracts of tumors from patients with humoral hypercalcemia of malignancy. Proc Natl Acad Sci U S A, 1983. 80(5): p. 1454–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strewler GJ, Williams RD, and Nissenson RA, Human renal carcinoma cells produce hypercalcemia in the nude mouse and a novel protein recognized by parathyroid hormone receptors. J Clin Invest, 1983. 71(3): p. 769–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moseley JM, et al. , Parathyroid hormone-related protein purified from a human lung cancer cell line. Proc Natl Acad Sci U S A, 1987. 84(14): p. 5048–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burtis WJ, et al. , Identification of a novel 17,000-dalton parathyroid hormone-like adenylate cyclase-stimulating protein from a tumor associated with humoral hypercalcemia of malignancy. J Biol Chem, 1987. 262(15): p. 7151–6. [PubMed] [Google Scholar]
- 18.Thiede MA, et al. , Human renal carcinoma expresses two messages encoding a parathyroid hormone-like peptide: evidence for the alternative splicing of a single-copy gene. Proc Natl Acad Sci U S A, 1988. 85(13): p. 4605–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suva LJ, et al. , A parathyroid hormone-related protein implicated in malignant hypercalcemia: cloning and expression. Science, 1987. 237(4817): p. 893–6. [DOI] [PubMed] [Google Scholar]
- 20.Kemp BE, et al. , Parathyroid hormone-related protein of malignancy: active synthetic fragments. Science, 1987. 238(4833): p. 1568–70. [DOI] [PubMed] [Google Scholar]
- 21.Tregear GW and Potts JT Jr., Synthetic analogues of residues 1–34 of human parathyroid hormone: influence of residue number 1 on biological potency in vitro. Endocr Res Commun, 1975. 2(8): p. 561–70. [DOI] [PubMed] [Google Scholar]
- 22.Juppner H, et al. , A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science, 1991. 254(5034): p. 1024–6. [DOI] [PubMed] [Google Scholar]
- 23.Martin TJ, Parathyroid Hormone-Related Protein, Its Regulation of Cartilage and Bone Development, and Role in Treating Bone Diseases. Physiol Rev, 2016. 96(3): p. 831–71. [DOI] [PubMed] [Google Scholar]
- 24.Philbrick WM, et al. , Defining the roles of parathyroid hormone-related protein in normal physiology. Physiol Rev, 1996. 76(1): p. 127–73. [DOI] [PubMed] [Google Scholar]
- 25.Rodda CP, et al. , Evidence for a novel parathyroid hormone-related protein in fetal lamb parathyroid glands and sheep placenta: comparisons with a similar protein implicated in humoral hypercalcaemia of malignancy. J Endocrinol, 1988. 117(2): p. 261–71. [DOI] [PubMed] [Google Scholar]
- 26.Kovacs CS, et al. , Parathyroid hormone-related peptide (PTHrP) regulates fetal-placental calcium transport through a receptor distinct from the PTH/PTHrP receptor. Proc Natl Acad Sci U S A, 1996. 93(26): p. 15233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abbas SK, et al. , Stimulation of ovine placental calcium transport by purified natural and recombinant parathyroid hormone-related protein (PTHrP) preparations. Q J Exp Physiol, 1989. 74(4): p. 549–52. [DOI] [PubMed] [Google Scholar]
- 28.Wu TL, et al. , Structural and physiologic characterization of the mid-region secretory species of parathyroid hormone-related protein. J Biol Chem, 1996. 271(40): p. 24371–81. [DOI] [PubMed] [Google Scholar]
- 29.Lam MH, et al. , PTHrP and cell division: expression and localization of PTHrP in a keratinocyte cell line (HaCaT) during the cell cycle. J Cell Physiol, 1997. 173(3): p. 433–46. [DOI] [PubMed] [Google Scholar]
- 30.Lam MH, et al. , Phosphorylation at the cyclin-dependent kinases site (Thr85) of parathyroid hormone-related protein negatively regulates its nuclear localization. J Biol Chem, 1999. 274(26): p. 18559–66. [DOI] [PubMed] [Google Scholar]
- 31.Lam MH, et al. , Importin beta recognizes parathyroid hormone-related protein with high affinity and mediates its nuclear import in the absence of importin alpha. J Biol Chem, 1999. 274(11): p. 7391–8. [DOI] [PubMed] [Google Scholar]
- 32.Lam MH, et al. , Nuclear and nucleolar localization of parathyroid hormone-related protein. Immunol Cell Biol, 2000. 78(4): p. 395–402. [DOI] [PubMed] [Google Scholar]
- 33.Grill V, et al. , Parathyroid hormone-related protein: elevated levels in both humoral hypercalcemia of malignancy and hypercalcemia complicating metastatic breast cancer. J Clin Endocrinol Metab, 1991. 73(6): p. 1309–15. [DOI] [PubMed] [Google Scholar]
- 34.Grill V, et al. , Parathyroid hormone-related protein: a possible endocrine function in lactation. Clin Endocrinol (Oxf), 1992. 37(5): p. 405–10. [DOI] [PubMed] [Google Scholar]
- 35.Martin TJ, Moseley JM, and Williams ED, Parathyroid hormone-related protein: hormone and cytokine. J Endocrinol, 1997. 154Suppl: p. S23–37. [PubMed] [Google Scholar]
- 36.Karperien M, et al. , Expression pattern of parathyroid hormone/parathyroid hormone related peptide receptor mRNA in mouse postimplantation embryos indicates involvement in multiple developmental processes. Mech Dev, 1994. 47(1): p. 29–42. [DOI] [PubMed] [Google Scholar]
- 37.Lee K, et al. , Parathyroid hormone-related peptide delays terminal differentiation of chondrocytes during endochondral bone development. Endocrinology, 1996. 137(11): p. 5109–18. [DOI] [PubMed] [Google Scholar]
- 38.Henderson JE, et al. , Nucleolar localization of parathyroid hormone-related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol Cell Biol, 1995. 15(8): p. 4064–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cingolani G, et al. , Molecular basis for the recognition of a nonclassical nuclear localization signal by importin beta. Mol Cell, 2002. 10(6): p. 1345–53. [DOI] [PubMed] [Google Scholar]
- 40.Jans DA, Thomas RJ, and Gillespie MT, Parathyroid hormone-related protein (PTHrP): a nucleocytoplasmic shuttling protein with distinct paracrine and intracrine roles. Vitam Horm, 2003. 66: p. 345–84. [DOI] [PubMed] [Google Scholar]
- 41.Massfelder T, et al. , Parathyroid hormone-related peptide--a smooth muscle tone and proliferation regulatory protein. Curr Opin Nephrol Hypertens, 1998. 7(1): p. 27–32. [DOI] [PubMed] [Google Scholar]
- 42.Massfelder T, et al. , Opposing mitogenic and anti-mitogenic actions of parathyroid hormone-related protein in vascular smooth muscle cells: a critical role for nuclear targeting. Proc Natl Acad Sci U S A, 1997. 94(25): p. 13630–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fenton AJ, et al. , A carboxyl-terminal peptide from the parathyroid hormone-related protein inhibits bone resorption by osteoclasts. Endocrinology, 1991. 129(4): p. 1762–8. [DOI] [PubMed] [Google Scholar]
- 44.Cornish J, et al. , Parathyroid hormone-related protein-(107–139) inhibits bone resorption in vivo. Endocrinology, 1997. 138(3): p. 1299–304. [DOI] [PubMed] [Google Scholar]
- 45.Murrills RJ, Stein LS, and Dempster DW, Lack of significant effect of carboxyl-terminal parathyroid hormone-related peptide fragments on isolated rat and chick osteoclasts. Calcif Tissue Int, 1995. 57(1): p. 47–51. [DOI] [PubMed] [Google Scholar]
- 46.Cornish J, et al. , Interleukin-18 is a novel mitogen of osteogenic and chondrogenic cells. Endocrinology, 2003. 144(4): p. 1194–201. [DOI] [PubMed] [Google Scholar]
- 47.Whitfield JF, et al. , C-terminal fragments of parathyroid hormone-related protein, PTHrP-(107–111) and (107–139), and the N-terminal PTHrP-(1–40) fragment stimulate membrane-associated protein kinase C activity in rat spleen lymphocytes. J Cell Physiol, 1994. 158(3): p. 518–22. [DOI] [PubMed] [Google Scholar]
- 48.Gagnon L, et al. , Protein kinase C-activating domains of parathyroid hormone-related protein. J Bone Miner Res, 1993. 8(4): p. 497–503. [DOI] [PubMed] [Google Scholar]
- 49.Jouishomme H, et al. , The protein kinase-C activation domain of the parathyroid hormone. Endocrinology, 1992. 130(1): p. 53–60. [DOI] [PubMed] [Google Scholar]
- 50.Frieling JS, et al. , Matrix metalloproteinase processing of PTHrP yields a selective regulator of osteogenesis, PTHrP1-17. Oncogene, 2017. 36(31): p. 4498–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stewart AF, et al. , N-terminal amino acid sequence of two novel tumor-derived adenylate cyclase-stimulating proteins: identification of parathyroid hormone-like and parathyroid hormone-unlike domains. Biochem Biophys Res Commun, 1987. 146(2): p. 672–8. [DOI] [PubMed] [Google Scholar]
- 52.Thiede MA and Rodan GA, Expression of a calcium-mobilizing parathyroid hormone-like peptide in lactating mammary tissue. Science, 1988. 242(4876): p. 278–80. [DOI] [PubMed] [Google Scholar]
- 53.Budayr AA, et al. , High levels of a parathyroid hormone-like protein in milk. Proc Natl Acad Sci U S A, 1989. 86(18): p. 7183–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Southby J, et al. , Immunohistochemical localization of parathyroid hormone-related protein in human breast cancer. Cancer Res, 1990. 50(23): p. 7710–6. [PubMed] [Google Scholar]
- 55.Powell GJ, et al. , Localization of parathyroid hormone-related protein in breast cancer metastases: increased incidence in bone compared with other sites. Cancer Res, 1991. 51(11): p. 3059–61. [PubMed] [Google Scholar]
- 56.Mundy GR and Martin TJ, Pathophysiology of Skeletal Complications of Cancer. Handbook of Experimental Pharmacology, 1993. 107: p. 617 – 640. [Google Scholar]
- 57.Mundy GR, Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer, 2002. 2(8): p. 584–93. [DOI] [PubMed] [Google Scholar]
- 58.Martin TJ and Moseley JM, Mechanisms in the skeletal complications of breast cancer. Endocr Relat Cancer, 2000. 7(4): p. 271–84. [DOI] [PubMed] [Google Scholar]
- 59.Thomas RJ, et al. , Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology, 1999. 140(10): p. 4451–8. [DOI] [PubMed] [Google Scholar]
- 60.Mundy GR and Martin TJ, The hypercalcemia of malignancy: pathogenesis and management. Metabolism, 1982. 31(12): p. 1247–77. [DOI] [PubMed] [Google Scholar]
- 61.Clohisy DR and Ramnaraine ML, Osteoclasts are required for bone tumors to grow and destroy bone. J Orthop Res, 1998. 16(6): p. 660–6. [DOI] [PubMed] [Google Scholar]
- 62.Ruppender NS, et al. , Matrix rigidity induces osteolytic gene expression of metastatic breast cancer cells. PLoS One, 2010. 5(11): p. e15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Page JM, et al. , Matrix rigidity regulates the transition of tumor cells to a bone-destructive phenotype through integrin beta3 and TGF-beta receptor type II. Biomaterials, 2015. 64: p. 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guise TA, et al. , Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest, 1996. 98(7): p. 1544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoneda T, et al. , Actions of bisphosphonate on bone metastasis in animal models of breast carcinoma. Cancer, 2000. 88(12 Suppl): p. 2979–88. [DOI] [PubMed] [Google Scholar]
- 66.Bendre MS, et al. , Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer cells correlates with bone metastasis in vivo. Cancer Res, 2002. 62(19): p. 5571–9. [PubMed] [Google Scholar]
- 67.Kukreja SC, et al. , Antibodies to parathyroid hormone-related protein lower serum calcium in athymic mouse models of malignancy-associated hypercalcemia due to human tumors. J Clin Invest, 1988. 82(5): p. 1798–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iguchi H, et al. , Effects of anti-parathyroid hormone-related protein monoclonal antibody and osteoprotegerin on PTHrP-producing tumor-induced cachexia in nude mice. J Bone Miner Metab, 2006. 24(1): p. 16–9. [DOI] [PubMed] [Google Scholar]
- 69.Onuma E, et al. , Generation of a humanized monoclonal antibody against human parathyroid hormone-related protein and its efficacy against humoral hypercalcemia of malignancy. Anticancer Res, 2004. 24(5A): p. 2665–73. [PubMed] [Google Scholar]
- 70.Lelekakis M, et al. , A novel orthotopic model of breast cancer metastasis to bone. Clin Exp Metastasis, 1999. 17(2): p. 163–70. [DOI] [PubMed] [Google Scholar]
- 71.Campbell JP, et al. , Models of bone metastasis. J Vis Exp, 2012(67): p. e4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sloan EK, Stanley KL, and Anderson RL, Caveolin-1 inhibits breast cancer growth and metastasis. Oncogene, 2004. 23(47): p. 7893–7. [DOI] [PubMed] [Google Scholar]
- 73.Kusuma N, et al. , Integrin-dependent response to laminin-511 regulates breast tumor cell invasion and metastasis. Int J Cancer, 2012. 130(3): p. 555–66. [DOI] [PubMed] [Google Scholar]
- 74.Zheng Y, et al. , Targeting IL-6 and RANKL signaling inhibits prostate cancer growth in bone. Clin Exp Metastasis, 2014. 31(8): p. 921–33. [DOI] [PubMed] [Google Scholar]
- 75.Luo X, et al. , Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep, 2016. 14(1): p. 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bendre MS, et al. , Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res, 2005. 65(23): p. 11001–9. [DOI] [PubMed] [Google Scholar]
- 77.Kir S, et al. , Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature, 2014. 513(7516): p. 100–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waning DL, et al. , Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat Med, 2015. 21(11): p. 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yin JJ, et al. , TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest, 1999. 103(2): p. 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bundred NJ, et al. , Parathyroid hormone related protein and skeletal morbidity in breast cancer. Eur J Cancer, 1992. 28(2–3): p. 690–2. [DOI] [PubMed] [Google Scholar]
- 81.Bouizar Z, Spyratos F, and De vernejoul MC, The parathyroid hormone-related protein (PTHrP) gene: use of downstream TATA promotor and PTHrP 1–139 coding pathways in primary breast cancers vary with the occurrence of bone metastasis. J Bone Miner Res, 1999. 14(3): p. 406–14. [DOI] [PubMed] [Google Scholar]
- 82.Kissin MW, et al. , Parathyroid hormone related protein in breast cancers of widely varying prognosis. Eur J Surg Oncol, 1993. 19(2): p. 134–42. [PubMed] [Google Scholar]
- 83.Vargas SJ, et al. , Localization of parathyroid hormone-related protein mRNA expression in breast cancer and metastatic lesions by in situ hybridization. J Bone Miner Res, 1992. 7(8): p. 971–9. [DOI] [PubMed] [Google Scholar]
- 84.Henderson M, et al. , Parathyroid hormone-related protein production by breast cancers, improved survival, and reduced bone metastases. J Natl Cancer Inst, 2001. 93(3): p. 234–7. [DOI] [PubMed] [Google Scholar]
- 85.Henderson MA, et al. , Parathyroid hormone-related protein localization in breast cancers predict improved prognosis. Cancer Res, 2006. 66(4): p. 2250–6. [DOI] [PubMed] [Google Scholar]
- 86.Siegel PM and Massague J, Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer, 2003. 3(11): p. 807–21. [DOI] [PubMed] [Google Scholar]
- 87.Wakefield LM and Roberts AB, TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev, 2002. 12(1): p. 22–9. [DOI] [PubMed] [Google Scholar]
- 88.Dumont N and Arteaga CL, Targeting the TGF beta signaling network in human neoplasia. Cancer Cell, 2003. 3(6): p. 531–6. [DOI] [PubMed] [Google Scholar]
- 89.Tang B, et al. , TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest, 2003. 112(7): p. 1116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lanczky A, et al. , miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat, 2016. 160(3): p. 439–446. [DOI] [PubMed] [Google Scholar]
- 91.Tran TH, et al. , Loss of Nuclear Localized Parathyroid Hormone-Related Protein in Primary Breast Cancer Predicts Poor Clinical Outcome and Correlates with Suppressed Stat5 Signaling. Clin Cancer Res, 2018. [DOI] [PubMed] [Google Scholar]
- 92.Freeman AN, PhD thesis:The contribution of genetic variations in the region of PTHLH to breast cancer susceptibility. 2016, University of Melbourne: Melbourne. p. 350. [Google Scholar]
- 93.Fleming NI, et al. , Parathyroid hormone-related protein protects against mammary tumor emergence and is associated with monocyte infiltration in ductal carcinoma in situ. Cancer Res, 2009. 69(18): p. 7473–9. [DOI] [PubMed] [Google Scholar]
- 94.Li J, et al. , PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J Clin Invest, 2011. 121(12): p. 4655–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Johnson RW, et al. , Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol, 2016. 18(10): p. 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Johnson RW, et al. , Parathyroid Hormone-Related Protein Negatively Regulates Tumor Cell Dormancy Genes in a PTHR1/Cyclic AMP-Independent Manner. Front Endocrinol (Lausanne), 2018. 9: p. 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martin TJ, et al. , Calcitonin receptors in a cloned human breast cancer cell line (MCF 7). Biochem Biophys Res Commun, 1980. 96(1): p. 150–6. [DOI] [PubMed] [Google Scholar]
- 98.Walia MK, et al. , Activation of PTHrP-cAMP-CREB1 signaling following p53 loss is essential for osteosarcoma initiation and maintenance. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lipton A, et al. , Pamidronate prevents skeletal complications and is effective palliative treatment in women with breast carcinoma and osteolytic bone metastases: long term follow-up of two randomized, placebo-controlled trials. Cancer, 2000. 88(5): p. 1082–90. [DOI] [PubMed] [Google Scholar]
- 100.Hillner BE, The role of bisphosphonates in metastatic breast cancer. Semin Radiat Oncol, 2000. 10(3): p. 250–3. [DOI] [PubMed] [Google Scholar]
- 101.Coleman RE, et al. , Breast-cancer adjuvant therapy with zoledronic acid. N Engl J Med, 2011. 365(15): p. 1396–405. [DOI] [PubMed] [Google Scholar]
- 102.Coleman R, et al. , Adjuvant zoledronic acid in patients with early breast cancer: final efficacy analysis of the AZURE (BIG 01/04) randomised open-label phase 3 trial. Lancet Oncol, 2014. 15(9): p. 997–1006. [DOI] [PubMed] [Google Scholar]
- 103.Cummings SR, et al. , Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med, 2009. 361(8): p. 756–65. [DOI] [PubMed] [Google Scholar]
- 104.Stopeck AT, et al. , Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol, 2010. 28(35): p. 5132–9. [DOI] [PubMed] [Google Scholar]
- 105.Martin M, et al. , Bone-related complications and quality of life in advanced breast cancer: results from a randomized phase III trial of denosumab versus zoledronic acid. Clin Cancer Res, 2012. 18(17): p. 4841–9. [DOI] [PubMed] [Google Scholar]
- 106.Coleman RE, et al. , American Society for Clinical Oncology, in 2018 ASCIO Annual Meeting. 2018, Journal of Clinical Oncology: USA. [Google Scholar]
- 107.Croucher PI, McDonald MM, and Martin TJ, Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer, 2016. 16(6): p. 373–86. [DOI] [PubMed] [Google Scholar]
- 108.Boissier S, et al. , Bisphosphonates inhibit breast and prostate carcinoma cell invasion, an early event in the formation of bone metastases. Cancer Res, 2000. 60(11): p. 2949–54. [PubMed] [Google Scholar]