

Abstract
Two billion people worldwide older than 18 years of age, or approximately 30% of the world population, are overweight or obese. In addition, more than 43 million children under the age of 5 are overweight or obese. Among the population in the United States aged 20 and greater, 32.8 percent are overweight and 39.8 percent are obese. Blacks in the United States have the highest age-adjusted prevalence of obesity (49.6%), followed by Hispanics (44.8%), whites (42.2%) and Asians (17.4%). The impact of being overweight or obese on the US economy exceeds $1.7 trillion dollars, which is equivalent to approximately eight percent of the nation’s gross domestic product. Obesity causes chronic inflammation that contributes to atherosclerosis and causes >3.4 million deaths/year. The pathophysiologic mechanisms in obesity that contribute to inflammation and atherosclerosis include activation of adipokines/cytokines and increases in aldosterone in the circulation. The adipokines leptin, resistin, IL-6, and monocyte chemoattractant protein activate and chemoattract monocytes/macrophages into adipose tissue that promote visceral adipose and systemic tissue inflammation, oxidative stress, abnormal lipid metabolism, insulin resistance, endothelial dysfunction, and hypercoagulability that contribute to atherosclerosis. In addition in obesity, the adipokines/cytokines IL-1β, IL-18, and TNF are activated and cause endothelial cell dysfunction and hyperpermeability of vascular endothelial junctions. Increased aldosterone in the circulation not only expands the blood volume but also promotes platelet aggregation, vascular endothelial dysfunction, thrombosis, and fibrosis. In order to reduce obesity and obesity-induced inflammation, therapies including diet, medications, and bariatric surgery are discussed that should be considered in patients with BMIs>35-40 kg/m2 if diet and lifestyle interventions fail to achieve weight loss. In addition, antihypertensive therapy, plasma lipid reduction and glucose lowering therapy should be prescribed in obese patients with hypertension, a 10-year CVD risk >7.5%, or prediabetes or diabetes.
Keywords: Subcutaneous adipose tissue, visceral adipose tissue, body mass index, waist to hip ratio, chronic inflammation, adipokines, insulin resistance, endothelial dysfunction, atherosclerosis, atherosclerotic coronary artery disease, heart failure, weight loss medications, glucose lowering medications, antihypertensive medications
Introduction
Incidence
Obesity is widespread in the industrialized world. The World Health Organization (WHO) and the American Heart Association (AHA) classify obesity based on body mass index (BMI), which is calculated as the body weight in kilograms divided by the height in meters squared [1]. Body mass index categories include: underweight: BMI<18.5 kg/m2, normal weight: BMI 18.5-24.9 kg/m2, overweight: BMI 25.0-29.9 kg/m2, class I obesity: BMI 30.0-34.9 kg/m2, class II obesity: BMI 35.0-39.9 kg/m2, and class III or extreme obesity: BMI≥40.0 kg/m2 [2,3]. For Asians, a BMI greater than 23 kg/m2 is considered overweight and a BMI of 27.5 kg/m2 or more is considered obese. Poor dietary habits, excessive calorie intake, and a sedentary lifestyle contribute to the worldwide development of obesity.
Two billion people worldwide older than 18 years of age, or approximately 30% of the world population, are overweight or obese [4]. In addition, more than 43 million children under the age of 5 are overweight or obese [5,6]. In the United States 180.5 million people ages 2 or greater are overweight or obese. Among the population in the United States aged 20 and greater, 32.8 percent are overweight and 39.8 percent are obese [7]. Blacks in the United States have the highest age-adjusted prevalence of obesity (49.6%), followed by Hispanics (44.8%), whites (42.2%) and Asians (17.4%) [8].
Obesity accounts for 47% of the total cost of chronic disease treatment in the United States [7]. The impact of being overweight or obese on the US economy exceeds $1.7 trillion dollars, which is equivalent to approximately eight percent of the nation’s gross domestic product. The estimate includes $480.7 billion in direct health-care costs and $1.24 trillion in lost productivity from obese individuals [7].
Overweight and obesity cause worldwide more than 3.4 million deaths each year [9]. The years of life lost for obese men and women range from 0.8-0.9 years for those aged 60-79 years to 5.9-6.1 years for those men and women aged 20-39 years. The more an individual weighs and the younger his or her age, the greater the effect of obesity on the risk and complications from cardiovascular disease [10]. The relationship between obesity as measured by BMI and mortality risk, which is primarily atherosclerotic cardiovascular risk, is characterized by a J-shaped curve, with the greatest mortality seen among individuals classified as extremely obese as illustrated in Figure 1. Figure 1 is adapted in part from [11,12].
Figure 1.
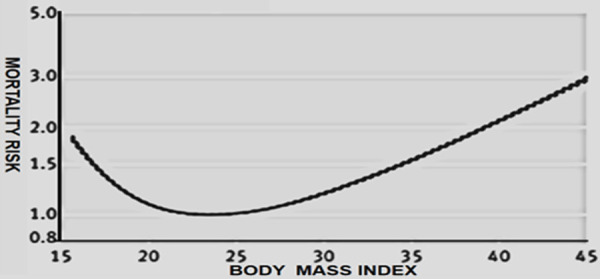
Relative risk of mortality versus body mass index over 20 years. Underweight, overweight and obesity increases the risk of all-cause mortality with a J shaped dose-response relationship. The nadir of the dose-response curve occurs in the BMI range of 23 to 24 among never smokers [11,12].
Obesity is a major risk factor for atherosclerotic cardiovascular disease. Each one-point increase in an individual’s BMI above normal weight causes a ten percent increase in the risk for atherosclerosis and coronary heart disease (CHD) [13]. A 10 kg increase in body weight increases the risk of atherosclerotic coronary artery disease by 12% and increases the systolic blood pressure by approximately 3 mmHg and the diastolic blood pressure by 2.3 mmHg [14]. According to a meta-analysis of 21 studies that included 389,239 individuals and 20,652 coronary heart disease events, men and women who are overweight or obese have a relative risk of 1.32 and 1.81, respectively, for developing atherosclerotic coronary heart disease after adjusting for age, gender, and smoking status [15]. However, mild obesity may be associated with improved survival in patients with established atherosclerosis-a finding that is termed the “obesity paradox”. Whether or not the obesity paradox is a true clinical entity is hotly debated. In this regard, there is a growing body of literature on potential methodological explanations for the obesity paradox, including the “fat but fit” hypothesis ascribed to obese patients with good cardiorespiratory conditioning. Alternatively, opponents of the obesity paradox cite: (1) misclassification bias toward obese patients caused by using BMI as a measure of obesity instead of body fat distribution, (2) unmeasured confounding factors in investigations, such as patient cardiorespiratory fitness, sarcopenia, and tobacco smoking, and (3) a potential for bias toward obese patients in comparison with nonobese patients with severe illness-related weight loss. In addition, the “obesity paradox” often is reported in young obese individuals with few comorbid conditions. Nevertheless, obesity is not a benign disease and individuals frequently termed “metabolically healthy obese” often progress to metabolically unhealthy obese individuals. The important interactions that link body composition, fat distribution, aging, cardiorespiratory fitness and atherosclerotic disease are critical areas of current basic and clinical research.
In addition to diet and lack of physical exercise, obesity is due to a number of contributing factors such as genetics, the microbiome, alterations in circadian rhythms, environmental stressors and confounding factors such as pharmaceutical drugs and chemicals. However, the interaction of these multiple factors across generations and the lifespan of an obese individual make it extremely difficult to determine the precise contribution of these factors to the obesity pandemic in humans. Moreover, despite considerable research efforts devoted to the understanding the biology of obesity, the available knowledge to date has been of little help in curbing the pandemic of obesity. This review interprets and cites the current literature on obesity and obesity-induced atherosclerosis and focuses on the diagnosis and measurement of obesity in patients, the mechanisms whereby adipocytes, primarily from visceral obesity, contribute to atherosclerosis, and the current treatments of obesity in patients.
Measurements of obesity
The determination of BMI is a common clinical measurement for determining the prevalence of obesity. However, BMI gives no information about variations in different ethnic groups or whether body fat is in the subcutaneous or in the visceral depots or both, which is important in determining the risk for atherosclerosis [16,17]. In addition, the BMI does not distinguish between fat mass, fat-free mass, and lean mass and can overestimate the degree of obesity in individuals who are extremely muscular. Some individuals who have a normal weight or who are overweight are at high risk for atherosclerosis if they have an excess of visceral adipose tissue with ectopic fat deposition in the heart, liver, and skeletal muscle. Conversely, individuals who are overweight or obese can be at relatively low atherosclerotic risk if they expand their subcutaneous adipose tissue mass, particularly in the gluteal-femoral area, but not in the visceral organs. These individuals are often termed “metabolically healthy obese” and often have little visceral fat and lack the metabolic syndrome. Consequently, individuals with similar BMIs can have vastly different body compositions and different risks for atherosclerosis.
Measurements of the waist circumference and the waist to hip ratio, as indicators of central adiposity, are better measurements than body mass index, for identifying individuals at increased risk of developing atherosclerotic cardiovascular diseases. A waist to hip ratio greater than 0.9 in men and greater than 0.85 in women indicates central obesity, or abdominal wall plus visceral fat, and an increased risk for atherosclerosis [18]. However, measurements of waist circumference do not distinguish between subcutaneous adipose tissue and the visceral adipose tissues. The distinction between subcutaneous and visceral adipose tissue is important because of the strong association of visceral adiposity with atherosclerosis.
The development of imaging technologies has revolutionized the study of body adiposity and the relationship of obesity with atherosclerotic cardiovascular disease. Because of distinct properties of fat, muscle, and bone tissues, body composition and body fat partitioning phenotypes can be precisely assessed with multidetector computerized axial tomography (CAT), magnetic resonance imaging (MRI), or dual x-ray absorptiometry (DXA). These techniques can characterize in women and men the heterogeneity in abdominal fat distribution and the differences in fat distribution with age [19,20]. However, both CAT and MRI imaging are expensive tests and require a specially trained technologist and physician to administer and interpret the examinations. Dual x-ray absorptiometry (DXA) is currently being used as a low-cost, low-radiation alternative to CAT and MRI and provides measurements of fat mass, fat free mass, and body bone mineral content [20-22]. In general, adult men with a percent body fat >30% and adult women with a percent body fat >41% by DXA are considered obese. The effective radiation dose with DXA is small (1-7 microsievert) [20,21]. Individuals with excess visceral adipose tissue, irrespective of their BMI or total adiposity, have an increased risk for atherosclerotic cardiovascular disease and diabetes. Excess epicardial adipose tissue deposition measured by CAT scan or MRI scan is associated with abnormal myocardial blood flow reserve, coronary plaque vulnerability, coronary artery calcification, and coronary artery disease severity [21A]. Table 1 compares the clinical utility of different noninvasive techniques in the measurement of adiposity and the ability of each technique to estimate visceral adiposity where + = adequate and ++++ = optimal. Table 1 is adapted, in part, from [22].
Table 1.
Utility of different techniques in the measurement of adiposity in obese individuals
| Technique | Clinical utility | Visceral adiposity estimate |
|---|---|---|
| Body mass index | +++ | + |
| Waist circumference | +++ | ++ |
| Waist to hip ratio | +++ | ++ |
| Waist height ratio | +++ | ++ |
| Computrized tomography | ++ | ++++ |
| Magnetic resonance imaging | ++ | ++++ |
| Dual X-ray absorptiometry | +++ | +++ |
This table compares the clinical utility of different noninvasive techniques in the measurement of adiposity and the ability of each technique to estimate visceral adiposity where + = adequate and ++++ = optimal.
Pathological mechanisms in obesity: subcutaneous and visceral obesity are metabolically active tissues that contribute to atherosclerosis
In the human body there are two types of adipose tissue, brown adipose tissue and white adipose tissue. Brown adipose tissue is primarily found in small and discrete regions of newborns and is responsible for the production of heat by thermogenesis. White adipose tissue is the predominant form of adipose tissue found in adults and is responsible for fat storage in the body [23].
Adipose tissue has been thought in the past to act primarily as a storage depot that builds up or breaks down excess triglycerides into free fatty acids and glycerol based on the metabolic needs of the body. More recently, adipose tissue is recognized to be metabolically active and capable of engaging in cross-talk between the organs within the body.
Excessive caloric intake along with a sedentary lifestyle results in an energy imbalance and an increase in subcutaneous adipose tissue in the body. When excessive caloric intake exceeds the normal storage capacity of the subcutaneous adipocytes, with adipocytes exceeding 100-120 μm in size, fatty acids are then stored in visceral adipocytes in the mesentery and omentum and also stored in the pericardium and epicardium, liver, pancreas, and kidneys [24,25].
Distinct adipose tissue depots can be divided among upper body and abdominal subcutaneous adipose depots, visceral depots, and lower body adipose depots. Upper body and visceral adipose tissues are associated with metabolic dysfunction and atherosclerotic disease, whereas lower body gluteofemoral subcutaneous adipose tissue is associated with some protection against diet-induced metabolic derangements. Each adipose depot can function distinctly as an endocrine organ with a different impact on health outcomes.
In general, visceral adipocytes transfer and release fatty acids more extensively than subcutaneous adipocytes, secrete more inflammatory adipokines and less anti-inflammatory adipokines, and have greater responses to glucocorticoids than subcutaneous adipose tissue. Excessive fatty acid accumulation in visceral organs such as the liver results in impairment in liver metabolism and overproduction of apolipoprotein B-containing lipoproteins, increased hepatic glucose production, decreased hepatic degradation of insulin, and systemic hyperinsulinemia [24]. Adipokine overproduction in visceral adipose tissue has an autocrine and paracrine role in crosstalk with endothelial cells, fibroblasts, and immune cells that contribute to obesity-associated inflammation and insulin resistance [26,27]. In addition, adipokines function as circulating cytokines and hormones that communicate with the brain, heart, liver, muscle, and the immune system and are implicated in the genesis of insulin resistance and atherosclerosis. However, abdominal subcutaneous adipose tissue is not protective against atherosclerosis, insulin resistance and diabetes mellitus because a significant accumulation of deep abdominal subcutaneous adipose tissue is associated with dyslipidemia, insulin resistance, glucose intolerance, and the metabolic syndrome [28].
Hypertrophic adipocytes in the viscera release adipokines which induce the infiltration of monocytes and macrophages into the visceral adipose tissue and promote local and systemic inflammation [1,24,29]. The macrophages express CD11c, inducible nitric oxide synthase (iNOS), tumor necrosis factor (TNF), IL-6, monocyte chemoattractant protein (MCP-1), interleukin (IL) IL-1β and IL-12 which contribute to an inflammatory state. These inflammatory mediators activate inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ), c-Jun N-terminal kinase (JNK), and inflammasome pathways, especially the NLR family pyrin domain containing-3 (NLRP3) inflammasome, which negatively regulate insulin signaling, glucose metabolism and fibrinolysis and ultimately contribute to atherosclerosis [29,30].
Visceral adiposity is linked to the deposition of excess triglycerides in the myocardium and localized fat depots in the epicardium and pericardium which are associated with coronary stenosis, myocardial infarction, and mortality [18,31]. In this regard, the 30-day mortality in viscerally obese individuals after hospitalization for myocardial infarction can be as high as 16% in patients without diabetes [30] and 19% in patients with diabetes [31].
Inflammatory adipokines/cytokines in obesity that increase atherosclerosis: leptin, resistin, retinaol binding protein 4, angiopoietin-like protein 2, il-6, and monocyte chemoattractant protein-1
Many studies of the mechanisms of actions of adipokines have been performed in cell cultures and in rodents with widely different approaches and sometimes inconsistent results. Moreover, rodent adipokines are not 100% homogenous with human adipokines. Consequently, not every postulated adipokine mechanism of action reported in the literature can be extrapolated to obese patients. Therefore, this manuscript discusses, and cites the appropriate supporting bibliographic references, the major cardiovascular mechanisms of action of adipokines that are generally accepted by cardiovascular specialists who diagnose and treat obese patients.
Important adipokines that are involved in initiating and promoting inflammation and that are increased in visceral obesity include leptin, resistin, retinol binding protein 4 (RBP4), Angiopoietin Like Proteiin-2 (AngptL-2), IL-6 and MCP-1 [32,33]. These adipokines induce systemic inflammation, endothelial dysfunction, hypercoagulability, and insulin resistance, all of which promote atherosclerosis. In contrast, adiponectin and omentum-1 are anti-inflammatory and anti-atherogenic adipokines.
Leptin
Leptin normally regulates food intake by signaling the hypothalamus to suppress hunger signals. The leptin concentration in the blood increases with body adiposity and is significantly increased in individuals with visceral obesity. However, obese individuals are resistant to the appetite suppressant effects of leptin due to impaired leptin transport across the blood brain barrier, inhibition of leptin signal transduction by suppressor of cyotkine3, defective autophagy, and impairment of the biological function of leptin due to cellular endoplasmic reticulum stress [30,34]. Nevertheless, obese patients are not resistant to the inflammatory actions of leptin, especially in the vasculature and myocardium.
In obese patients, leptin is a potent monocyte/macrophage chemoattractant. Hyperleptinemia in obese individuals stimulates inflammation by increasing monocyte and macrophage secretion of inflammatory molecules TNFα, IL-6, and MCP-1, and increasing the expression of adhesion molecules VCAM-1, ICAM-1, and E-selectin, which cause monocyte attraction to vascular endothelial cell walls, increased permeability of vascular endothelial cell junctions, and atheroma formation [31,35,36]. Leptin also stimulates monocyte production of reactive oxygen species and promotes monocyte/macrophage cell chemoattraction and proliferation in endothelial cells which contributes to endothelial dysfunction and atherosclerosis by impairing nitric oxide (NO) dependent vasorelaxation and causing vasoconstriction and thrombosis [37-39]. In addition, hyperleptinemia can increase the accumulation of cholesterol esters in atherosclerotic foam cells by upregulating acyl-CoA cholesterol acyltransferase and downregulating cholesterol esterase [40]. Leptin can also decrease high density lipoprotein (HDL) concentrations and, in this manner, impede cholesterol removal from tissues by increasing hepatic HDL clearance by hepatic scavenger receptors [41].
In obesity, leptin stimulates vascular endothelial cell and smooth muscle proliferation, hypertrophy, and migration and platelet aggregation, which contributes to vascular stenosis and thrombosis [29,42,43]. In this regard, leptin can upregulate thrombospondin-1, a potent proatherogenic protein, which is an extracellular matrix glycoprotein that facilitates vascular smooth muscle proliferation and migration [44]. In addition, leptin can induce a prothrombotic state by enhancing PAI-1 expression, which is a major endogenous inhibitor of fibrinolysis [45-47]. Increased leptin concentrations positively correlate with the plasma concentration of PAI-1, in men and in women with atherosclerosis [47-49]. Leptin can also play an important role in atheroma plaque instability and rupture by stimulating the expression of matrix metalloproteinase-2 (MMP-2) by human vascular smooth muscle cells with the resultant breakdown of matrix and non-matrix proteins [50,51].
Hyperleptinemia potentiates the deleterious vasoconstrictive actions of angiotensin II and endothelin on blood vessels, while angiotensin II increases leptin synthesis--thereby generating a self-perpetuating cycle of excessive oxidative stress and vasoconstriction [31]. Leptin-mediated aldosterone production contributes to obesity-associated endothelial dysfunction and cardiac fibrosis, which impair myocardial relaxation and thereby contribute to atherosclerotic cardiovascular disease. Clinical studies demonstrate that increased leptin plasma concentrations can foretell acute cardiovascular events, coronary restenosis after coronary angioplasty, and cerebral stroke independently of the traditional cardiovascular risk factors [47]. In this regard, increased plasma leptin concentrations correlate with increased coronary artery calcifications, myocardial infarctions (MI), and carotid artery intima-media thickness [43,47,52]. Consequently, hyperleptinemia can be proatherogenic and prothrombotic in obese patients and, therefore, hyperleptinemia can be an important therapeutic target in the treatment of atherosclerosis in patients.
Resistin
Resistin is a pro-inflammatory adipokine that binds to Toll-like Receptor 4 or Adenylyl Cyclase-Associated Protein 1 (CAP1) in endothelial cells and monocytes and mediates pro-inflammatory signals through nuclear factor kappa beta (NFκB) and mitogen activated protein kinase (MAPK) signaling [53]. Resistin concentrations are positively associated with the concentrations of soluble TNFα receptor-2, interleukin-6, and lipoprotein-associated phospholipase A2. In this regard, resistin significantly upregulates the expression of the inflammatory molecules TNF-α, IL-1, IL-6, IL-12, the adhesion molecules vascular cell adhesion molecule 1 (VCAM1) and intracellular adhesion molecule 1 (ICAM1) via the p38/mitogen-activated protein kinase (MAPK) pathway and the release of endothelin (ET)-1 [53-57]. In addition, resistin induces insulin resistance by decreasing insulin-stimulated glucose uptake by skeletal muscle [53]. Significantly, resistin upregulates lipoprotein-associated phospholipase A2 and free fatty acid expression, thereby increasing intracellular fatty acids in human macrophages and promoting foam cell formation and atherosclerosis [53,58]. In addition, resistin up-regulates MCP-1 to attract monocytes to infiltrate the subendothelium and shifts monocytes or M2 macrophages into inflammatory M1 macrophages [59]. Furthermore, resistin interferes with insulin-sensitizers adiponectin and Fibroblast growth factor 21 by reducing their interaction with membrane receptors [60]. Resistin, also induces human aortic smooth muscle cell proliferation, migration and atheroma production through extracellular-signal-regulated kinase1/2 (ERK1/2) and protein kinase B (Akt) signaling pathways [61,62]. In addition, resistin produces vascular endothelial cell vasoconstriction and predisposes to thrombosis by decreasing endothelial cell nitric oxide synthase (eNOS) and nitric oxide (NO) concentration by activation of p38 and JNK MAPK and by increasing the endothelial cell concentrations of free oxygen radicals [53,63].
Resistin is significantly increased in patients with atherosclerosis and in patients with acute coronary syndromes [54,56,64-66]. A resistin concentration >12.76 ng/ml indicates that a patient is at high risk for acute myocardial ischemia/infarction and positively correlates with the severity of atherosclerotic plaques [67,68]. In addition, resistin is increased in atherosclerotic aneurysms. Increased resistin concentrations also correlate with increased C reactive protein concentrations as well as the number of coronary arteries with calcification and >50% coronary stenosis in patients [69]. Consequently, resistin is emerging as an important biomarker of coronary atherosclerosis and a potentially useful therapeutic target for treatment in patients with coronary artery disease. Preclinical studies indicate that statin drugs can inhibit the upregulation of resistin and decrease resistin concentrations in adipocytes [70,71].
Retinol binding protein 4
Retinol binding protein 4 (RBP4) is expressed in adipocytes, macrophages, and in the liver, and is significantly increased in individuals with excessive visceral fat accumulation. Increased serum RBP4 concentrations are positively associated with atherosclerotic risk factors including increased cholesterol and triglyceride concentrations, decreased HDL concentrations, increased BMI, the metabolic syndrome, and hypertension and is an independent predictor for the incidence of adverse atherosclerotic cardiovascular events after adjustment for traditional risk factors [72-74].
Retinol binding protein stimulates adipose tissue lipolysis and fatty acid release, which subsequently triggers hepatic steatosis. In addition RBP4 can activate toll-like receptors 2/4 (TLR2/4) and stimulate NFκB, JNK, and interleukin 1β proinflammatory cytokine secretion from macrophages [75]. Increased circulating RBP4 limits or inhibits glucose transport in skeletal muscle cells and adipocytes and results in insulin resistance among individuals with obesity, type 2 diabetes and among nonobese, nondiabetic individuals with strong family histories of type 2 diabetes [74-77]. Also RBP4 stimulates vascular endothelial cells to produce pro-inflammatory VCAM1, MCP-1, and IL-6, which results in monocyte/macrophage adherence to vascular endothelium and enhances oxidized low density lipoprotein (LDL) uptake by macrophages and resultant foam cell formation. Conversely, knock down of RBP4 expression can significantly decrease vascular atherosclerotic plaque burden [74]. Consequently, increased RBP4 concentrations in blood correlate with the severity of atherosclerosis and the risk of cardiovascular events and RBP4 is a potential therapeutic clinical target for the treatment of patients with atherosclerosis.
Angiopoietin-like protein
The angiopoietin-like (ANGPTL) family of proteins participate in the pathology of atherosclerosis via inflammation, dyslipidemia, and atherosclerotic foam cell formation. Angiopoietin-like protein 2 (Angptl2) is an adipocyte-derived inflammatory mediator that is overexpressed in obese patients which inhibits lipoprotein lipase, and therefore triglyceride breakdown, and links obesity to inflammation and systemic insulin resistance [78]. Inflammatory cytokines such as TNF-α induce the expression of Angptl2 in adipocytes which, in turn, induces vascular endothelial inflammation, increased vessel permeability through leukocyte attachment to vessel walls, inflammatory vascular remodeling and atherosclerosis [78,79]. Angptl2 concentrations in the blood are positively correlated with the BMI, waist circumference, visceral fat content, CRP concentration, the homeostasis model assessment of insulin resistance (HOMA-R) index, and coronary artery stenosis [80,81]. In contrast, Angptl2 concentrations are negatively correlated with circulating HDL cholesterol [80,81]. ANGPTL8 increases the uptake and decreases the efflux of cholesterol in macrophages and promotes the transition of macrophages to cholesterol accumulating foam cells [82]. Individuals with ANGPTL3 loss-of-function mutations have decreased cholesterol and triglyceride concentrations and decreased risk of atherosclerotic coronary artery disease [81]. Consequently, ANGPTL3 is a potential clinical therapeutic target for the treatment of atherosclerosis. An antisense oligonucleotide inhibitor of ANGPTL3 and a monoclonal antibody against ANGPTL3 are in clinical trials for the treatment of patients with hyperlipidemia and atherosclerosis [83].
Interleukin-6
(IL-6) Interleukin-6 (IL-6) is important in the regulation of inflammation, hematopoiesis, the immune response and host defense mechanisms and has diverse roles on inflammation and insulin sensitivity in diverse tissues which are dependent on different signaling pathways and active or sedentary lifestyles. For example, IL-6 produced in skeletal muscle during physical activity can act as an energy sensor by activating AMP-activated kinase and enhancing glucose disposal, lipolysis and fat oxidation. Conversely, in sedentary individuals with body fat content that is increasing, IL-6 is released from adipose tissue, increases IL-6 plasma concentrations, and contributes to a proinflammatory state and ultimately insulin resistance. In this regard, IL-6 and the IL-6 receptors increase in proportion with increases in visceral, and to a lesser extent subcutaneous fat, and positively correlate with BMI and the percent of body fat. IL-6 expression also correlates with the increased expression of TNF-α, MCP-1, Interferon gamma-induced protein 10 (IP-10) and macrophage infiltration into adipose tissue [84-86]. In obese individuals, IL-6 reduces the expression of the glucose transporter-4 (GLUT-4) and insulin receptor substrate-1 (IRS-1) and in this manner increases insulin resistance [87,88]. In addition, IL-6 decreases lipoprotein lipase activity, which results in increased macrophage uptake of lipids and atherosclerotic foam cell and plaque formation [89]. IL-6 also has pro-thrombotic effects on platelets. In addition, IL-6 inhibits TGF-β-induced regulatory T cell (Treg) differentiation, a subset of CD4+ T cells that normally protect against tissue injury [90]. Of interest is the fact that IL-6 and also TNF are increased in women with normal body weight and body mass index but whose fat mass is >30% of their total body weight and whose risk of developing atherosclerosis is increased [91]. Moreover, in overweight individuals an IL-6 concentration >1 pg/mL is predictive of significant CAD [92]. In contrast, reduction of the IL-6 pathway with decreases of IL 6 to less than 1.65 ng/mL with the interleukin blocker canakinumab produces a 32% reduction in major adverse CV events and decreases the requirement for urgent coronary revascularization [93]. Consequently, pharmacologic reduction of inflammatory IL-6 and also IL-1 in obese patients with normal cholesterol concentrations is an important and promising new area of research in the treatment of atherosclerosis and obesity.
Monocyte chemoattractant protein-1
Monocyte chemoattractant protein-1 (MCP-1) is produced primarily by adipocytes, macrophages, and endothelial cells and the concentration of MCP-1 in adipose tissue increases during obesity [94]. MCP-1 is a potent chemotactic factor for monocytes/macrophages that infiltrate adipose tissue and actively secrete TNFα, IL-6 and MCP-1, which contributes to adipose tissue inflammation, insulin resistance, and atherosclerosis [94-98]. Increased concentrations of MCP-1 in atherosclerotic plaques are associated with large lipid cores, decreased collagen content, decreased smooth muscle cell burden, intraplaque hemorrhage, matrix turnover, and increased major adverse cardiovascular events [99]. In a meta-analysis of seven studies including 21,401 individuals, increased MCP-1 concentrations in the circulation were associated with increased risk of coronary heart disease, nonfatal myocardial infarction and cardiovascular death [100]. Conversely, pharmacological inhibition of MCP-1 pathways in pre-clinical studies significantly limits atherosclerotic lesion burden and produces atherosclerotic plaque stability [101]. Consequently, MCP-1 and MCP-1 signaling antagonism in patients with atherosclerosis could potentially significantly lower the risk of vascular plaque formation, plaque rupture and cardiovascular mortality in patients with atherosclerotic cardiovascular disease.
Anti-inflammatory adipokines that decrease atherosclerosis: adiponectin and omentum-1
Adiponectin
Adiponectin is secreted by adipocytes and blood adiponectin concentrations correlate inversely with visceral fat accumulation [102,103]. The anti-atheromatous effects of adiponectin are mediated, in part, by anti-inflammatory effects on the vascular endothelium through AMP activated protein kinase (AMPK)-mediated modulation of NFκB and Akt [104,105]. Adiponectin inhibits TNF-α production in adipose tissue macrophages through inhibition of NFκB activation and stimulation of anti-inflammatory IL-10 cytokine production [106]. In addition, adiponectin suppresses free oxygen radical generation and attenuates the adhesion of monocytes to vascular endothelial cells and foam cell formation by inhibiting TNF-α and IL-8-induced synthesis of ICAM1, VCAM1, and E-selectin [107-109]. Adiponectin promotes the differentiation of monocytes to anti-inflammatory M2 macrophages and stimulates the phagocytosis by macrophages of apoptotic cells [110]. In addition, adiponectin increases ceramidase production and reduces harmful hepatic ceramide lipid-induced insulin resistance [111,112]. In addition, adiponectin lowers blood glucose concentrations by increasing the expression of hepatic insulin receptor substrate 2 (IRS-2), activating AMPK and increasing glucose utilization in skeletal muscle and fatty-acid oxidation in the liver and skeletal muscle [113]. Conversely, in obese patients with non-insulin dependent diabetes, adiponectin concentrations are decreased. Adiponectin can also directly stimulate NO production in vascular endothelial cells and cause vasodilation via activation of 5-AMPK and enhancement of eNOS activity [114].
Adiponectin suppresses the expression of heparin binding epidermal growth factor, platelet derived growth factor, and basic fibroblastic growth factor in the endothelial cells of injured vascular walls and in this manner inhibits the proliferation and migration of vascular smooth muscle cells and vascular stenosis. Adiponectin also increases the production of tissue inhibitor of metalloproteinase in macrophages through the induction of IL-10 synthesis, and selectively suppresses vascular endothelial cell apoptosis [109]. In this manner, adiponectin can prevent injury-induced vascular intimal thickening and vascular stenosis [115]. Consequently, adiponectin exerts significant anti-inflammatory, anti-atherogenic and vascular protective effects and adiponectin concentrations in blood correlate inversely with development of atherosclerotic cardiovascular disease events [116]. In contrast, hypoadiponectinemia significantly and independently correlates with the presence of atherosclerotic coronary artery disease in males even after adjustment for cigarette smoking, hypertension, dyslipidemia and diabetes mellitus [117-119]. Males with adiponectin concentrations <4.0 μg/mL have a twofold increase in the prevalence of coronary artery disease [119]. However, adiponectin concentrations in blood can significantly increase with weight reduction. For example, a 21% reduction in BMI is associated with a 46% increase in the mean adiponectin blood concentration [105,120]. Patients with the highest concentrations of adiponectin have a greater than 40% decreased 6-year risk of myocardial infarction compared with individuals with the lowest adiponectin concentrations [120,121]. Since adiponectin exerts significant anti-inflammatory, anti-atherogenic and vascular protective effects, measurement of adiponectin concentrations in blood are helpful in the evaluation of a patient’s risk for atherosclerotic coronary artery disease [120,121].
Omentin-1
Omentin-1 is an anti-inflammatory adipokine produced preferentially by stromal vascular cells in visceral adipose tissue. Omentin-1 concentrations in blood correlate inversely with the BMI, body fat mass, and fasting insulin concentrations, and correlate positively with insulin sensitivity, adiponectin concentrations, HDL concentrations, and vascular endothelial cell function [122]. Omentin-1 activates AMPK, phosphatidylinositol 3 kinase (PI3K) and inhibits Nicotinamide Adenine Dinucleotide Phosphate Hydrogen (NADPH) oxidase [123]. These actions ultimately result in insulin sensitization, reduction in TNFα-induced inflammation, and vascular protection. Treatment of human endothelial cells with omentin-1 prevents TNF-α induced cyclooxygenase-2 expression by inhibiting c-Jun N-terminal kinase signaling, suggesting an anti-inflammatory function of omentin-1 on endothelial cells [124,125]. Omentin-1 can also cause vasodilatation and inhibit arterial atheroma and calcification by suppressing vascular inflammatory responses, apoptosis, vascular smooth muscle cell proliferation and neointimal formation, and reducing collagen content [126-133]. In contrast, decreased omentin-1 concentrations are associated with increased arterial stiffness and atherosclerosis, especially in patients with type II diabetes [132].
Table 2 summarizes the source and function of the inflammatory and anti-inflammatory adipokines. Table 2 is adapted, in part, from [30,34,42].
Table 2.
Source and function of adipokines
| Adipokine | Source | Binding site | Function |
|---|---|---|---|
| Leptin | Adipocytes | Leptin Receptor | Appetite Control, Inflammation |
| Resistin | Adipocytes, Monocytes | Adenylyl Cyclase-Associated Protein 1 (CAP1) | Inflammation, Insulin resistance, atherosclerosis |
| RBP4 | Adipocytes, Macrophages, Liver | Retinol, Transthyretin | Insulin resistance |
| AngpL-2 | Adipocytes | Activates signaling adhesion molecules, monocyte chemotaxis | Inflammation, Insulin resistance |
| IL-6 | Adipocytes, Macrophages | IL-6 Receptor | Pro-inflammatory but variable effects |
| MCP-1 | Adipocytes, Stromal Vascular Cells, Macrophages | CCR2 | Recruits monocytes/macrophages. Causes Inflammation |
| Adiponectin | Adipocytes | Adiponectin Receptors 1 & 2 | Anti-inflammatory, insulin sensitization, anti-atherogenic |
| Omentum-1 | Adipocytes, Stromal Vascular Cells | Triggers AKT activation | Anti-inflammatory, insulin sensitization, anti-atherogenic |
Obesity-induced increases in saturated fatty acids contribute to atherosclerosis
Oxidative stress due to increases in circulating saturated fatty acids is an important factor in the pathogenesis of vascular endothelial cell dysfunction and atherosclerosis [134]. Accumulation of toxic fatty acids, including diacylglycerides and ceramides, contributes to endoplasmic reticulum (ER) stress, mitochondrial dysfunction and the generation of reactive oxygen species (ROS), which together result in inflammation, insulin resistance, endothelial cell apoptosis and atherosclerosis [134]. Increased concentrations of fatty acids and an impaired expression in adipose tissues of anti-oxidative enzymes, such as superoxide dismutase, glutathione peroxidase, and catalase in obese individuals, cause increased activation of NADPH oxidase and induce the production of H2O2, free oxygen radicals, and hydroxyl radicals [135]. Moreover, free fatty acid activation of the NF-κB pathway increases inflammatory TNF-α, IL-1β, IL-6, and MCP-1, inhibits eNOS and decreases nitric oxide production and, in this manner, cause vascular endothelial cell inflammation, vasoconstriction and thrombosis [136,137]. In addition, excess free fatty acids have direct effects on vascular endothelial cells with the expression of cell-surface adhesion molecules, myeloperoxidases and plasminogen activator inhibitor-1 (PAI-1) which promote macrophage infiltration, vascular endothelial cell inflammation, thrombosis, and atherosclerosis [138]. Moreover, excess free fatty acids can impair cellular mitochondria and lysosome integrity and cause endoplasmic reticulum (ER) stress and, in this manner, inactivate the serine-threonine kinase AMPK and decrease intracellular energy [136,139]. Furthermore, excess free fatty acids activate innate immune system cells in adipose tissue and trigger NLRP3 inflammasome activation, which stimulates caspase-1, IL-1, IL-18 and an inflammatory cascade that contributes to chronic inflammation in arteries and tissues [140,141].
In the Framingham Health Study, obese individuals, especially individuals with visceral obesity and increased free fatty acids, had a 50 to 60 percent increase in the risk for atherosclerotic cardiovascular disease in comparison with non-obese individuals [142-144]. In contrast, individuals who habitually consume monounsaturated fatty acids in a Mediterranean diet or individuals who chronically consume statin drugs, have increased adiponectin concentrations, decreased free fatty acids and inflammatory markers TNF-α, MCP-1, IFN-γ, CRP, IL-18, and IL-6, and decreases in atherosclerotic vascular disease [145,146].
Adipose immune cells in chronic inflammation and atherosclerosis
Innate and adaptive immune cells are present in adipose tissue. Innate immune cells include macrophages, neutrophils, eosinophils, and mast cells, whereas adaptive immune cells include T cells and B cells [147]. Adipocytes express Major Histocompatibility Complex (MHC) class II molecules and T cell co-stimulators and can act as antigen-presenting cells and induce the activation of CD4+ T cells in visceral adipose tissue [148]. Adipocytes also secrete inflammatory adipokines that modulate B cell generation.
In lean body weight individuals with healthy adipose tissue, anti-inflammatory M2 macrophages, CD4Th2 cells, regulatory T cells (Tregs), and eosinophils play important roles in the removal of apoptotic cells and help maintain a homeostatic and tolerogenic environment. These immune cells secrete anti-inflammatory IL-4, IL-10, IL-13, and IL-1Ra [147,148]. In obesity, the number and activity of macrophages, mast cells, neutrophils, and T- and B lymphocytes in visceral adipose tissue are increased due to chemoattraction by adipokines while the numbers of eosinophils, T helper 2 (Th2), Treg, and Invariant natural killer T (iNKT) cells are decreased [149]. M1 macrophages, Th1 cells, CD8 T cells, and mast cells augment the production of adipose tissue-derived proinflammatory cytokines and produce insulin resistance, and endothelial cell dysfunction [147]. CD8 T cells induce macrophage M1 differentiation from monocytes and their cytokine secretion [150]. CD4Th1 cell secretion of interferon (IFN)-γ also drives monocyte polarization toward M1 type macrophages and promotes the secretion of inflammatory TNF-α, IL-1β, IL-6, IL-12, and MCP-1 [151]. In addition, neutrophils infiltrate visceral adipose tissue where they stimulate M1 type macrophage infiltration, pro-inflammatory cytokine secretion and chronic inflammation. Mast cells, upon receiving stimulatory signals from T cells, rapidly release histamine, serotonin, heparin, serine protease, and eicosanoids which significantly contribute to the inflammatory responses in obesity [152]. Consequently, M1 macrophages, CD4Th1, CD8 and mast cells play critical roles in chronic inflammation, insulin resistance, and ultimately atherosclerosis in obese patients. Table 3 lists the changes in immune cells in the visceral adipose tissue of obese individuals. Table 3 is adapted, in part from [147-152].
Table 3.
Proinflammatory and anti-inflammatory immune cells in adipose tissue of obese individuals
| Immune cell | Obese adipose tissue changes | Adipose tissue response | Insulin sensitivity |
|---|---|---|---|
| Neutrophils | Increased | Pro-inflammatory | Decreased |
| CD8 T cells | Increased | Pro-inflammatory | Decreased |
| CD4 TH1 cells | Increased | Pro-inflammatory | Decreased |
| CD4 T reg | Decreased | Anti-inflammatory | Increased |
| M1 macrophages | Increased | Pro-inflammatory | Decreased |
| M2 macrophages | Variable | Anti-inflammatory | Increased |
| MAST cells | Increased | Pro-inflammatory | Decreased |
The abnormalities in oxidative stress, abnormal lipid metabolism with excess free fatty acids, and endothelial dysfunction that contributes significantly to obesity-induced atherosclerosis are summarized in Figure 2: obesity induced atherosclerosis.
Figure 2.
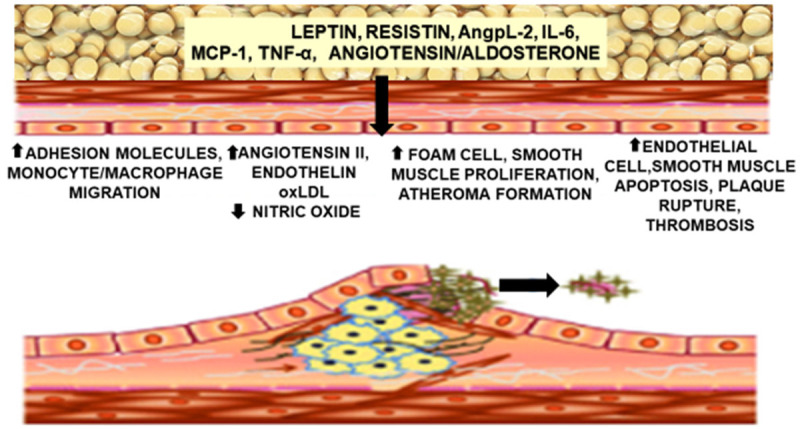
Obesity is characterized by decreases in anti-inflammatory/atheroprotective adipokines (adiponectin and omentum-1) and increases in proinflammatory/atherogenic adipokines [leptin, resistin, rbp4, angp1, IL-6, macrophage chemoattractant protein (MCP)-1] as well as increases in free fatty acids, and inflammatory cells. These factors participate in vascular inflammation and endothelial cell dysfunction which contribute to the inception and progression of atherosclerosis.
Obesity: increased sympathetic nerve activation and hypertension promote atherosclerosis
Obesity is associated with an increase in sympathetic nerve activation which is triggered by increases in pro-inflammatory adipokines and fatty acids, resistance to insulin, and impairment in arterial baroreceptor function. Vascular tone is also increased in obese individuals due to increased endothelin-1 (ET-1) in the blood, which augments the vasoconstrictive actions of angiotensin-II, norepinephrine, and serotonin. Angiotensin II promotes vascular endothelial cell dysfunction through activation of NADPH oxidase, formation of free oxygen radicals, and production of H2O2 [153,154]. Angiotensin II also impairs phosphorylation of the insulin receptor substrate 1 (IRS-1), with resultant impairment in insulin signaling and insulin resistance. Moreover, angiotensin II participates actively in leukocyte and platelet activation and vascular thrombosis in obese individuals [154].
Activation of the sympathetic nervous system and the renin-angiotensin-aldosterone system is also promoted by increased circulating leptin and leptin receptor signaling in obesity. Increased aldosterone in the blood of obese individuals promotes plasma volume expansion [155]. Leptin also augments the release of neprilysin from adipocytes and the kidneys, which causes a deficiency of endogenous natriuretic peptides in obese individuals with resultant additional sodium retention and plasma volume expansion [155]. Each of these actions of aldosterone, neprilysin and leptin work in concert with increased sympathetic nerve activity to promote sodium retention, plasma volume expansion, hypertension, vascular endothelial dysfunction and ultimately atherosclerosis.
Obesity treatment to reduce inflammation and slow or prevent the progression of atherosclerosis
Medications or bariatric surgery should be considered in patients with BMIs>35-40 kg/m2 if diet and lifestyle interventions fail to achieve weight loss. In addition, lipid reduction, glucose lowering therapy and antihypertensive medications should be considered in obese patients with a 10-year cardiovascular disease risk >7.5%, prediabetes or diabetes, and hypertension. The following sections present specific recommendations by the Author for the treatment of obese patients with appropriate supporting bibliographic medical references.
Weight loss, exercise, medications, lipid and glucose lowering medications, antihypertensive medications in the treatment of obesity
Weight loss and exercise
Sugars and saturated fatty acids can induce the synthesis and activation of inflammasomes and activate pro-inflammatory IL-1 and IL-18 cytokines that can cause endothelial cell dysfunction, increase the permeability of the endothelial junctions in vessels, and cause vascular smooth muscle proliferation, migration, and ultimately vascular stenosis. In contrast, consumption of a Mediterranean diet with increased concentrations of omega three fatty acids is correlated with increased secretion of adiponectin, decreases secretion of TNF-α, MCP-1, IFN-γ, CRP, IL-18, and IL-6, and decreases in atherosclerosis [156]. Diets that restrict sugars and saturated fats and induce mild to moderate weight loss of body fat and not lean body mass are beneficial and improve the quality of life in obese patients. As little as five percent weight loss by dietary intervention is associated with a reduction in triglycerides and free fatty acids, an increase in HDL cholesterol, and a decrease in the requirements for lipid lowering medications in obese patients [157].
Individualized nutrition therapy is recommended for each obese patient. In general, energy goals of ≤800 to 1200 kcal/d should be considered for patients weighing <114 kg and approximately 1200 to 1800 kcal/d for those patients weighing ≥114 kg. Diets of 200-800 kcal/day are not recommended unless there is a clinical need for rapid weight loss. Additional goals include restricting fat in the diet to <30% of total calories with <10% from saturated fats. Recommendations should include one to two glasses of water with each meal, carbohydrate monitoring, consumption of fruits, legumes, vegetables, whole grains and the substitution of polyunsaturated fatty acids for saturated and trans fats [158]. Long term weight loss is dependent on caloric restriction and diet adherence. However, dietary effects on weight loss can decrease and plateau with time due to compensatory body adaptation.
Each patient should undergo a physical examination and receive an exercise prescription, which should include moderate intensity walking, cycling or swimming, five times per week [158]. In order to promote clinically significant weight loss, 225 to 420 minutes of physical exercise/week are required. In addition, 200 to 300 minutes of physical exercise/week are required to prevent weight gain after weight loss. A “warm-up” and “cool-down” component to the exercise program minimizes any possible muscular injury. The goal should be to achieve and maintain a 7 to 10% loss of initial body weight. In addition, a rehabilitation program with aerobic and resistance training is helpful in improving patient cardiorespiratory fitness. Weight loss achieved through dietary intervention and exercise results in decreased visceral adipose tissue and adipose tissue macrophages and reduces circulating IL-6, CRP, PAI-1, TNF-α, soluble TNF receptor, P-selectin, intercellular adhesion molecule-1 (ICAM-1), VCAM-1, and IL-18 in obese men and women [159,160]. Physical activity improves glucose tolerance and sensitivity by improving non-insulin-dependent glucose uptake. In addition, physical activity decreases the plasma triglycerides, improves the ratio between HDL and LDL cholesterol, increases fibrinolysis, decreases platelet aggregation, improves oxygen uptake in the heart and in the peripheral tissues, lowers the resting heart rate by increasing vagal tone, and decreases the blood pressure [109]. Each consistent 1 hour metabolic equivalent increase in physical activity is associated with an 8% decrease in cardiovascular disease risk [109].
Weight loss medications
If lifestyle interventions fail to achieve the desired weight loss goals, medications for weight loss should be considered in addition to a diet and an exercise program. Pharmacological therapy should be considered for individuals with a BMI of 25 to 30 kg/m2 with comorbidities or a BMI>30 kg/m2 with or without comorbidities [158,161]. See Table 4. The effect of weight loss medications on cardiovascular morbidity and mortality has not been definitively determined and is under investigation. Table 4 is Adapted from [161].
Table 4.
Weight loss medications
| Generic drug | Drug dosage | Average weight loss | Side effects | Dea schedule |
|---|---|---|---|---|
| Pancreatic lipase inhibitor | ||||
| Orlistat | 120 mg tid before meal | 3.4 KG | Steatorrhea, flatulence, fecal discharge | Not scheduled |
| Orlistat | 60 mg tid before meal | 2.5 KG | Steatorrhea, flatulence, fecal discharge | Not scheduled |
| Seotonin receptor agonist | ||||
| Lorcaserin | 10 mg bid for 12 months | 3.2 KG | Headache, nausea, blurred vision, hypoglycemia | Schedule IV |
| Phentermine-topiramate | ||||
| Phentermine-topiramate | 3.75/23 mg, 7.5/46 mg, 15/92 mg for 12 months | 6.7-8.9 KG | Insomnia, confusion, dry mouth, nausea, paresthesias | Schedule IV |
| Adrenergic drugs | ||||
| Diethlypropion | 25 mg tid or 75 mg in am for <12 weeks | 3 KG | Insomnia, headache, nausea | Schedule IV |
| Phentermine | 15-30 mg/day for <12 weeks | 3.6 KG | Headache, dry mouth, itching | Schedule IV |
| Benzphetamine | 25-50 mg tid for <12 weeks | 3-4 KG | Insomina, dizziness, hyperactivity | Schedule III |
| Phendimetrazine | 17.5-70 mg tid for <12 weeks | 3.8 KG | Dizziness, flushing, sweating | Schedule III |
| Glucagon like peptide analogs | ||||
| Liraglutide | 0.6-3.0 mg sq <12 months | 5.85 KG | Pancreatitis, hypoglycemia, nausea, vomiting, fatigue, contraindicated in medullary thyroid cancer | Not scheduled |
| Semaglutide | 1 to 2.4 mg sq weekly for ≥20 weeks | Nausea, diarrhea, vomiting, constipation, abdominal pain, headache, fatigue, dyspepsia, dizziness, abdominal distension, eructation, flatulence, gastroenteritis and gastroesophageal reflux disease | Not scheduled | |
| Opioid receptor antagonist-adrenergic reuptake inhibitor | ||||
| Naltrexone-bupropion | 8/90-16/180 mg bid<12 months | 2.0-4 KG | Mood changes, blurred vision, dry mouth, constipation, seizures | Not scheduled |
This table contains weight loss medications currently available. Full drug information should be carefully reviewed with and individualized for each patient prior to prescription and administration. Weight loss medication should not be administered to pregnant females or females who breast feed their infants. Liraglutide or Semaglutide should not be prescribed in individuals with medullary thyroid cancer or family history of medullary thyroid cancer. The effect of weight loss medications on cardiovascular morbidity and mortality has not been definitively established. MEN2 = Multiple Endocrine Neoplasia syndrome type 2.
Surgery for weight loss
In patients who are motivated to lose weight but who fail to lose weight despite diet, exercise, and weight loss medications, the AHA/ACC/Obesity Society guidelines recommend that adults with BMI>35-40 kg/m2 or >32.4-35 kg in Asians and significant obesity-related comorbidities, such as atherosclerotic vascular disease and/or diabetes mellitus, be considered for bariatric surgery in a high volume bariatric surgery center [158,162]. Both gastric bypass and gastric sleeve surgery can significantly reduce visceral fat by approximately 40% to 50% with a more modest reduction in abdominal subcutaneous fat [163]. In addition, bariatric surgery results in an improvement in insulin sensitization and glucose utilization in skeletal muscle and adipose tissue. In the Swedish Obese Subjects (SOS) Bariatric Surgery study, weight-loss surgery led to a 30% reduction in the incidence of cardiovascular events and a 50% reduction in cardiovascular deaths in individuals with severe obesity compared with those who received usual care after 15 years of follow-up [164]. In this study the remission rate for patients with obesity and type 2 diabetes mellitus was 72% at 2 years and 36% at 10 years compared with 21% and 13%, respectively, for the nonsurgical control obese patients [164]. Bariatric surgery was also markedly more effective than medical treatment alone in the prevention of type 2 diabetes mellitus, with a relative risk reduction of 78% [165]. Consequently, bariatric surgery is emerging as an important cost effective management option for obese patients with atherosclerotic cardiovascular disease and other obesity related co-morbidities [166]. Micronutrient and nutritional status must be carefully monitored in all patients after bariatric surgery.
Laparoscopic adjustable gastric banding, in which an inflatable silicone band is placed around the upper part of the stomach to narrow its lumen, restrict food passage, and form a small proximal pouch of stomach, can limit the quantity of food that can be ingested [166]. This procedure can promote as much as 55% weight loss at two years after surgery and promotes the remission of hypertension in as many as 54% of patients, dyslipidemia in 40%, and diabetes in as many as 72% [164,166].
Although bariatric surgery is capable of reversing many of the deleterious effects of obesity, removal of large amounts of abdominal subcutaneous fat by liposuction does not appear to improve an obese patient’s insulin sensitivity or glucose utilization in muscle, liver or adipose tissues and does not significantly decrease the plasma concentrations of C-reactive protein, IL-6, and TNF-α [167]. This observation may be explained by the fact that liposuction reduces subcutaneous adipose mass without decreasing visceral fat or major organ fat.
Lipid lowering therapy
Obese patients with pre-diabetes or diabetes mellitus who are less than age 40 are advised to take a high-intensity statin if they have a LDL-c greater than 100 mg/dl or clinical evidence of atherosclerotic cardiovascular disease. In addition, obese patients greater than age 40 with an estimated 10-year ASCVD risk greater than 7.5% should be treated with a high-intensity statin [168]. Due to anti-inflammatory and lipid lowering effects, statins can ameliorate the CAD prognosis in obese pre-diabetic or diabetic patients [169]. Fibrates, such as gemfibrozil or fenofibrate, also decrease circulating systemic inflammatory mediators such as endothelin-1, IL-6, CRP, TNF-α, and IFN-γ and are effective in lowering serum triglyceride and total cholesterol concentrations in obese pre-diabetic and diabetic patients [170].
Glucose lowering therapy in obese patients with pre-diabetes
Patients with obesity have an increased prevalence of impaired glucose tolerance that contribute to their high risk for diabetes and cardiovascular disease. The duration of glycemic burden in obese patients is a strong predictor of adverse outcomes such as atherosclerotic cardiovascular disease. For obese patients with prediabetes, especially for those with BMI≥35 kg/m2, those aged <60 years, and women with a prior history of gestational diabetes mellitus, metformin therapy should be considered [171]. Metformin has the strongest evidence base, has demonstrated long-term safety as pharmacologic therapy for diabetes prevention and is recommended by the World Health Organization [168,171]. In a substudy of the United Kingdom Prospective Diabetes Study, metformin, compared with lifestyle modifications alone, resulted in a 32% reduction in microvascular and macrovascular diabetes-related outcomes, a 39% reduction in myocardial infarction, and a 36% reduction in all-cause mortality rate [158]. Metformin, however, is contraindicated in individuals with renal disease with a glomerular filtration rate <30 ml/min/1.73 m2.
The 2021 US treatment guidelines for diabetes mellitus incorporate sodium-glucose co-transporter-2 (SGLT2) inhibitors as second-line glucose-lowering agents after metformin especially in patients with high cardiovascular risk and in patients with established atherosclerotic cardiovascular disease [172]. Postulated mechanisms of the beneficial effects of SGLT2 inhibitors include a reduction in adipokine and cytokine production, as well as reductions in cardiac cells necrosis and cardiac fibrosis, improved cardiac cell metabolism, improved ventricular loading conditions secondary to a reduction in preload due to diuretic and natriuretic effects, and inhibition of the Na+/H+ exchange in myocardial and renal tubular cells [173]. Two recent meta-analyses of SGLT2 inhibitors have reported that SGLT2 inhibitors can significantly reduce major adverse cardiovascular events (myocardial infarction, stroke, heart failure hospitalization) and the progression of kidney disease [173,174]. SGLT2 inhibitors are contraindicated when the glomerular filtration rate is <30 ml/in/1.73 m2. Head-to-head randomized control trials that compare SGLT2 inhibitors with metformin are needed to clarify whether SGLT2 inhibitors can safely and cost-effectively replace metformin as a first-line agent in the management of obese patients with prediabetes or non-insulin dependent diabetes mellitus.
Antihypertensive therapy
The Framingham Heart Study and the Nurses’ Health Study have identified a direct, continuous and almost linear relationship between patient obesity and the magnitude of blood pressure elevation [175,176]. In addition, the percentage of patients with blood pressure control in a therapeutic range decreases with increasing obesity. The ACC/AHA guidelines recommend treating patients with obesity with cardiovascular risk factors to a goal blood pressure of less than 130/80 mmHg, whereas the American Diabetes Association (ADA) guidelines recommend a blood pressure target of less than 140/90 mmHg [172]. The ADA suggests reserving a blood pressure target of less than 130/80 mmHg for patients at high risk of cardiovascular disease if the blood pressure can be safely reduced [159,177]. Two randomized clinical trials demonstrate in individuals with obesity and confirmed hypertension that a reduction of atherosclerotic cardiovascular disease events occurs with a decrease in blood pressures to <140 mmHg systolic and <90 mmHg diastolic [178,179]. In adult obese patients at high risk for atherosclerotic cardiovascular disease, especially stroke, a systolic and diastolic blood pressure ≤130/80 mmHg, is appropriate only when such a lower blood pressure can be achieved without hypotension, syncope, or kidney injury [177,180].
Diet, weight reduction, sodium restriction and an angiotensin converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB) are the recommended first line treatments for obese patients with hypertension [181,182]. A diet rich in fresh fruits, vegetables, and low-fat dairy products (the Dietary Approaches to Stop Hypertension or DASH diet) lowers systolic and diastolic blood pressure as much as 11.6 and 5.3 mmHg, respectively, independent of weight reduction [183]. Angiotensin converting enzyme inhibitors and angiotensin receptor blockers cause decreases in CRP, IL-6, MMP-9, and platelet aggregation by the blockade of angiotensin effects on the direct activation of inflammatory cells, vascular permeability, adhesion molecules and chemokines [184,185]. In the Treatment in Obese Patients with Hypertension study, which compared the efficacy and safety of Lisinopril with hydrochlorothiazide in in 232 obese hypertensive patients, the number of blood pressure responders was greater with Lisinopril (40% versus 33%) and plasma glucose decreased by -0.21±0.71 mmol/L with Lisinopril but increased +0.31±0.99 mmol/L with hydrochlorothiazide treatment [186]. In the Candesartan Role on Obesity and on Sympathetic System study, blood pressure reduction was similar in patients treated with either candesartan or hydrochlorothiazide, but insulin sensitivity increased and sympathetic nerve activity decreased in candesartan-treated patients [187].
An angiotensin converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB) significantly reduces the risk of obese patients with albuminuria progressing to advanced renal disease [188]. Serum creatinine, estimated glomerular filtration rate and serum potassium concentrations should be monitored in these patients. The use of ACE inhibitors and ARBs in combination is not recommended given the lack of added atherosclerotic cardiovascular benefits and the increased rate of hyperkalemia, syncope, and acute kidney injury. Among patients without albuminuria for whom cardiovascular disease prevention is the primary goal of blood pressure control and in obese black patients with hypertension, a dihydropyridine calcium channel blocker (CCB), a thiazide-type diuretic, or both can be considered in place of or in addition to an ACE inhibitor or an ARB for blood pressure control [172,180]. Since obese patients with hypertension often require two or more antihypertensive drugs, ACE+CCB or ACE+CCB+ thiazide diuretic can play an important role in blood pressure control. Spironolactone, or eplerenone, can be added to the treatment regimen if the blood pressure is not controlled. Beta-adrenergic receptor blocking drugs are utilized for blood pressure control in patients with atherosclerotic vascular disease, heart failure, atrial fibrillation or in young hypertensive obese women who are planning pregnancy or are pregnant. Table 5 lists antihypertensive medications used in the treatment of obesity induced hypertension, their anatomic/physiologic target(s) and their metabolic effects. Table 5 is adapted, in part, from [189-191].
Table 5.
Antihypertensive drugs and their effects in treatment of obesity induced hypertension

|
Summary and future perspective
Visceral adiposity is a serious chronic inflammatory condition that contributes to vascular atherosclerosis. In obese patients, the adipokines leptin, resistin, IL-6, and monocyte chemoattractant protein 1, chemoattract monocytes and macrophages into visceral adipose tissue and promote local and systemic inflammation, oxidative stress, abnormal lipid metabolism, vascular endothelial hyperpermeability and dysfunction, hypercoaguability, insulin resistance, and atherosclerotic cardiovascular disease. Obesity also increases the number of mineralocorticoid receptors and circulating aldosterone, which increase the plasma volume and cause platelet aggregation, endothelial dysfunction, and vascular and myocardial interstitial fibrosis. Weight loss medications and bariatric surgery should be considered if lifestyle interventions fail to achieve weight reduction. Substantial weight loss from diet and exercise, medications, and bariatric surgery is capable of reversing many of the increases in the inflammatory adipokines, the oxidative stress and the abnormal lipid metabolism due to obesity. All physicians must employ a multidisciplinary approach with obese patients that involves lifestyle modification and pharmacological and/or surgical interventions in order to combat the pandemic of obesity and increase patient longevity. Continued intensive research into the inflammatory and anti-inflammatory adipokines will facilitate our understanding of the important contribution of obesity adipokines to atherosclerosis and will ultimately lead to treatments that contribute to healthier patients that are not limited by atherosclerotic cardiovascular disease.
Acknowledgements
The Author thanks the librarians of the James A. Haley Hospital/University of South Florida for their assistance in providing many of the articles cited in this manuscript. Work in the cardiovascular research laboratory of the Author is supported by a grant from the Children’s Cardiomyopathy Foundation.
Disclosure of conflict of interest
None.
References
- 1.Alpert M, Agrawal H, Kumar S, Kumar A. Heart failure and obesity in adults: pathophysiology, clinical manifestations and management. Curr Heart Fail Rep. 2014;11:156–165. doi: 10.1007/s11897-014-0197-5. [DOI] [PubMed] [Google Scholar]
- 2.Obesity: preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser. 2000;894:1–253. [PubMed] [Google Scholar]
- 3.Poirier P, Alpert MA, Fleisher LA, Thompson PD, Sugerman HJ, Burke LE, Marceau P, Franklin BA American Heart Association Obesity Committee of Council on Nutrition, Physical Activity and Metabolism, Council on Cardiopulmonary Perioperative and Critical Care, Council on Cardiovascular Surgery and Anesthesia and Council on Cardiovas. Cardiovascular evaluation and management of severely obese patients undergoing surgery: a science advisory from the American Heart Association. Circulation. 2009;120:86–95. doi: 10.1161/CIRCULATIONAHA.109.192575. [DOI] [PubMed] [Google Scholar]
- 4.Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu-Rmeileh NM, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, Anwari P, Banerjee A, Barquera S, Basu S, Bennett DA, Bhutta Z, Blore J, Cabral N, Nonato IC, Chang JC, Chowdhury R, Courville KJ, Criqui MH, Cundiff DK, Dabhadkar KC, Dandona L, Davis A, Dayama A, Dharmaratne SD, Ding EL, Durrani AM, Esteghamati A, Farzadfar F, Fay DF, Feigin VL, Flaxman A, Forouzanfar MH, Goto A, Green MA, Gupta R, Hafezi-Nejad N, Hankey GJ, Harewood HC, Havmoeller R, Hay S, Hernandez L, Husseini A, Idrisov BT, Ikeda N, Islami F, Jahangir E, Jassal SK, Jee SH, Jeffreys M, Jonas JB, Kabagambe EK, Khalifa SE, Kengne AP, Khader YS, Khang YH, Kim D, Kimokoti RW, Kinge JM, Kokubo Y, Kosen S, Kwan G, Lai T, Leinsalu M, Li Y, Liang X, Liu S, Logroscino G, Lotufo PA, Lu Y, Ma J, Mainoo NK, Mensah GA, Merriman TR, Mokdad AH, Moschandreas J, Naghavi M, Naheed A, Nand D, Narayan KM, Nelson EL, Neuhouser ML, Nisar MI, Ohkubo T, Oti SO, Pedroza A, Prabhakaran D, Roy N, Sampson U, Seo H, Sepanlou SG, Shibuya K, Shiri R, Shiue I, Singh GM, Singh JA, Skirbekk V, Stapelberg NJ, Sturua L, Sykes BL, Tobias M, Tran BX, Trasande L, Toyoshima H, van de Vijver S, Vasankari TJ, Veerman JL, Velasquez-Melendez G, Vlassov VV, Vollset SE, Vos T, Wang C, Wang X, Weiderpass E, Werdecker A, Wright JL, Yang YC, Yatsuya H, Yoon J, Yoon SJ, Zhao Y, Zhou M, Zhu S, Lopez AD, Murray CJ, Gakidou E. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study. Lancet. 2013;384:766–81. doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. WHO Fact Sheet, March 3, 2020.
- 6.De Onis M, Blossner M, Borghi E. Global prevalence and trends of overweight and obesity among preschool children. Am J Clin Nutr. 2010;92:1257–1264. doi: 10.3945/ajcn.2010.29786. [DOI] [PubMed] [Google Scholar]
- 7.Waters H, Graf M. America’s obesity crisis: the health and economic impact of excess weight. Miliken Institute. 2018:1–24. [Google Scholar]
- 8.Hales C, Carroll M, Fryar C, Ogden C. Prevalence of obesity and severe obesity among adults: United States, 2017-2018. NCHS Data Brief. 2020;360:1–6. [PubMed] [Google Scholar]
- 9.GBD 2015 Obesity Collaborators. Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, Marczak L, Mokdad AH, Moradi-Lakeh M, Naghavi M, Salama JS, Vos T, Abate KH, Abbafati C, Ahmed MB, Al-Aly Z, Alkerwi A, Al-Raddadi R, Amare AT, Amberbir A, Amegah AK, Amini E, Amrock SM, Anjana RM, Ärnlöv J, Asayesh H, Banerjee A, Barac A, Baye E, Bennett DA, Beyene AS, Biadgilign S, Biryukov S, Bjertness E, Boneya DJ, Campos-Nonato I, Carrero JJ, Cecilio P, Cercy K, Ciobanu LG, Cornaby L, Damtew SA, Dandona L, Dandona R, Dharmaratne SD, Duncan BB, Eshrati B, Esteghamati A, Feigin VL, Fernandes JC, Fürst T, Gebrehiwot TT, Gold A, Gona PN, Goto A, Habtewold TD, Hadush KT, Hafezi-Nejad N, Hay SI, Horino M, Islami F, Kamal R, Kasaeian A, Katikireddi SV, Kengne AP, Kesavachandran CN, Khader YS, Khang YH, Khubchandani J, Kim D, Kim YJ, Kinfu Y, Kosen S, Ku T, Defo BK, Kumar GA, Larson HJ, Leinsalu M, Liang X, Lim SS, Liu P, Lopez AD, Lozano R, Majeed A, Malekzadeh R, Malta DC, Mazidi M, McAlinden C, McGarvey ST, Mengistu DT, Mensah GA, Mensink GBM, Mezgebe HB, Mirrakhimov EM, Mueller UO, Noubiap JJ, Obermeyer CM, Ogbo FA, Owolabi MO, Patton GC, Pourmalek F, Qorbani M, Rafay A, Rai RK, Ranabhat CL, Reinig N, Safiri S, Salomon JA, Sanabria JR, Santos IS, Sartorius B, Sawhney M, Schmidhuber J, Schutte AE, Schmidt MI, Sepanlou SG, Shamsizadeh M, Sheikhbahaei S, Shin MJ, Shiri R, Shiue I, Roba HS, Silva DAS, Silverberg JI, Singh JA, Stranges S, Swaminathan S, Tabarés-Seisdedos R, Tadese F, Tedla BA, Tegegne BS, Terkawi AS, Thakur JS, Tonelli M, Topor-Madry R, Tyrovolas S, Ukwaja KN, Uthman OA, Vaezghasemi M, Vasankari T, Vlassov VV, Vollset SE, Weiderpass E, Werdecker A, Wesana J, Westerman R, Yano Y, Yonemoto N, Yonga G, Zaidi Z, Zenebe ZM, Zipkin B, Murray CJL. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377:13–27. doi: 10.1056/NEJMoa1614362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grover SA, Kaouache M, Rempel P, Joseph L, Dawes M, Lau DC, Lowensteyn I. Years of life lost and healthy life-years lost from diabetes and cardiovascular disease in overweight and obese people: a modelling study. Lancet Diabetes Endocrinol. 2015;3:114–22. doi: 10.1016/S2213-8587(14)70229-3. [DOI] [PubMed] [Google Scholar]
- 11.Aune D, Sen A, Prasad M, Norat T, Janszky I, Tonstad S, Romundstad P, Vatten LJ. BMI and all- cause mortality: systematic review and non-linear dose-response meta-analysis of 230 cohort studies with 3.74 million deaths among 30.3 million participants. BMJ. 2016;353:i2156. doi: 10.1136/bmj.i2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. 2005;28:1769–1778. doi: 10.2337/diacare.28.7.1769. [DOI] [PubMed] [Google Scholar]
- 13.Hu F. Obesity epidemiology. Oxford University Press, Incorporated; 2008. pp. 1–384. [Google Scholar]
- 14.Din-Dzietham R, Liu Y, Bielo M, Shamsa F. High blood pressure trends in children and adolescents in national surveys, 1963 to 2002. Circulation. 2007;116:1488–1496. doi: 10.1161/CIRCULATIONAHA.106.683243. [DOI] [PubMed] [Google Scholar]
- 15.Bogers RP, Bemelmans WJ, Hoogenveen RT, Boshuizen HC, Woodward M, Knekt P, van Dam RM, Hu FB, Visscher TL, Menotti A, Thorpe RJ Jr, Jamrozik K, Calling S, Strand BH, Shipley MJ BMI-CHD Collaboration Investigators. Association of overweight with increased risk of coronary heart disease partly independent of blood pressure and cholesterol levels: a meta-analysis of 21 cohort studies including more than 300,000 persons. Arch Intern Med. 2007;167:1720–1728. doi: 10.1001/archinte.167.16.1720. [DOI] [PubMed] [Google Scholar]
- 16.Batsis JA, Mackenzie TA, Bartels SJ, Sahakyan KR, Somers VK, Lopez-Jimenez F. Diagnostic accuracy of body mass index to identify obesity in older adults: NHANES 1999-2004. Int J Obes (Lond) 2016;40:761–767. doi: 10.1038/ijo.2015.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prado C, Gonzalez M, Heymsfi eld S. Body composition phenotypes and obesity paradox. Curr Opin Clin Nutr Metab Care. 2015;18:535–551. doi: 10.1097/MCO.0000000000000216. [DOI] [PubMed] [Google Scholar]
- 18.Zeller M, Steg PG, Ravisy J, Lorgis L, Laurent Y, Sicard P, Janin-Manificat L, Beer JC, Makki H, Lagrost AC, Rochette L, Cottin Y RICO Survey Working Group. Relation between body mass index, waist circumference, and death after acute myocardial infarction. Circulation. 2008;118:482–490. doi: 10.1161/CIRCULATIONAHA.107.753483. [DOI] [PubMed] [Google Scholar]
- 19.Maurovich-Horvat P, Massaro J, Fox CS, Moselewski F, O’Donnell CJ, Hoffmann U. Comparison of anthropometric, area- and volume-based assessment of abdominal subcutaneous and visceral adipose tissue volumes using multi-detector computed tomography. Int J Obes (Lond) 2007;31:500–506. doi: 10.1038/sj.ijo.0803454. [DOI] [PubMed] [Google Scholar]
- 20.De Lorenzo A, Gratteri S, Gualtieri P, Cammarano A, Bertucci P, Di Renzo L. Why primary obesity is a disease? J Transl Med. 2019;17:169. doi: 10.1186/s12967-019-1919-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marra M, Sammarco R, De Lorenzo A, Iellamo F, Siervo M, Pietrobelli A, Donini LM, Santarpia L, Cataldi M, Pasanisi F, Contaldo F. Assessment of body composition in health and disease using Bioelectrical Impedance Analysis (BIA) and Dual Energy X-Ray Absorptiometry (DXA): a critical overview. Contrast Media Mol Imaging. 2019;2019:3548284. doi: 10.1155/2019/3548284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21A.Packer M. Epicardial adipose tissue may mediate deleterious effects of obesity and inflammation on the myocardium. J Am Coll Cardiol. 2018;71:2360–2372. doi: 10.1016/j.jacc.2018.03.509. [DOI] [PubMed] [Google Scholar]
- 22.Neeland IJ, Poirier P, Despres JP. Cardiovascular and metabolic heterogeneity of obesity: clinical challenges and implications for management. Circulation. 2018;37:1391–1406. doi: 10.1161/CIRCULATIONAHA.117.029617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saely CH, Geiger K, Drexel H. Brown versus white adipose tissue: a mini-review. Gerontology. 2012;58:15–23. doi: 10.1159/000321319. [DOI] [PubMed] [Google Scholar]
- 24.Tchernof A, Després JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2011;93:359–404. doi: 10.1152/physrev.00033.2011. [DOI] [PubMed] [Google Scholar]
- 25.Varlamov O, Somwar R, Cornea A, Kievit P, Grove KL, Roberts CT Jr. Single-cell analysis of insulin-regulated fatty acid uptake in adipocytes. Am J Physiol Endocrinol Metab. 2010;299:E486–E496. doi: 10.1152/ajpendo.00330.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuzawa Y, Funahashi T, Nakamura T. Molecular mechanism of metabolic syndrome X: contribution of apocytokines adipocyte-derived bioactive substances. Ann N Y Acad Sci. 1999;892:146–54. doi: 10.1111/j.1749-6632.1999.tb07793.x. [DOI] [PubMed] [Google Scholar]
- 27.Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–43. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 28.Hamdy O, Porramatikul S, Al-Ozari E. Metabolic obesity: the paradox between visceral and subcutaneous fat. Curr Diabetes Rev. 2006;2:367–73. doi: 10.2174/1573399810602040367. [DOI] [PubMed] [Google Scholar]
- 29.Esser N, L’homme l, De Roover A, Kohnen L, Scheen A, Moutschen M, Piette J, Legrand-Poels S, Pauot N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia. 2013;56:2487–97. doi: 10.1007/s00125-013-3023-9. [DOI] [PubMed] [Google Scholar]
- 30.Kwon H, Pessin J. Adipokines mediate inflammation and insulin resistance. Front Endocrinol (Lausanne) 2013;4:71. doi: 10.3389/fendo.2013.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koh K, Park S, Quon M. Leptin and cardiovascular diseases: response to therapeutic interventions. Circulation. 2012;117:3238–3249. doi: 10.1161/CIRCULATIONAHA.107.741645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blüher M. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes. 2009;117:241–250. doi: 10.1055/s-0029-1192044. [DOI] [PubMed] [Google Scholar]
- 33.Van de Voorde J, Pauwels B, Boydens C, Decaluwé K. Adipocytokines in relation to cardiovascular disease. Metabolism. 2013;62:1513–21. doi: 10.1016/j.metabol.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 34.Ouchi N, Parker J, Lugus J, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adya R, Tan B, Randeva H. Differential effects of leptin and adiponectin in endothelial angiogenesis. J Diabetes Res. 2015;2015:648239. doi: 10.1155/2015/648239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porreca E, Di Febbo C, Fusco L, Moretta V, Di Nisio M, Cuccurullo F. Soluble thrombomodulin and vascular adhesion molecule-1 are associated to leptin plasma levels in obese women. Atherosclero. 2004;172:175–180. doi: 10.1016/j.atherosclerosis.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 37.Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab. 2008;4:619–626. doi: 10.1038/ncpendmet0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bouloumié A, Curat CA, Segenes C, Lomede K, Miranville A, Busse R. Role of macrophage tissue infiltration in metabolic diseases. Curr Opin Clin Nutr Metab Care. 2005;8:347–354. doi: 10.1097/01.mco.0000172571.41149.52. [DOI] [PubMed] [Google Scholar]
- 39.Bełtowski J, Wojcicka G, Jamroz-Wiśniewska A, Borkowska E, Marciniak A. Antioxidant treatment normalizes nitric oxide production, renal sodium handling and blood pressure in experimental hyperleptinemia. Life Sci. 2005;77:1855–68. doi: 10.1016/j.lfs.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Li RS, Zhou SH, Wang ZP, Du YY, Pan HW. Effects of leptin on acyl-CoA: cholesterol O-acyltransferase-1 expression and activity during foam cell differentiation in human mononuclear THP-1 cells. Int J Clin Exp Pathol. 2016;9:3417–3423. [Google Scholar]
- 41.Silver D, Wang N, Tall A. Defective HDL particle uptake in ob/ob hepatocytes causes decreased recycling, degradation, and selective lipid uptake. J Clin Invest. 2000;105:151–159. doi: 10.1172/JCI8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blüher M. Adipokines-removing road blocks to obesity and diabetes therapy. Mol Metab. 2014;3:230–240. doi: 10.1016/j.molmet.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beltowski J. Leptin and atherosclerosis. Atherosclerosis. 2006;189:47–60. doi: 10.1016/j.atherosclerosis.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Chavez R, Haney R, Cuadra R, Ganguly R, Adapala R, Thodeti C, Raman P. Upregulation of thrombospondin-1 expression by leptin in vascular smooth muscle cells via JAK2- and MAPK-dependent pathways. Am J Physiol Cell Physiol. 2012;303:C179–C191. doi: 10.1152/ajpcell.00008.2012. [DOI] [PubMed] [Google Scholar]
- 45.Singh P, Peterson TE, Barber KR, Kuniyoshi FS, Jensen A, Hoffmann M, Shamsuzzaman AS, Somers VK. Leptin upregulates the expression of plasminogen activator inhibitor-1 in human vascular endothelial cells. Biochem Biophys Res Commun. 2010;392:47–52. doi: 10.1016/j.bbrc.2009.12.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elbatarny H, Maurice D. Leptin-mediated activation of human platelets: involvement of a leptin receptor and phosphodiesterase 3A-containing cellular signaling complex. Am J Physiol Endocrinol Metab. 2005;289:E695–702. doi: 10.1152/ajpendo.00125.2005. [DOI] [PubMed] [Google Scholar]
- 47.Thøgersen AM, Söderberg S, Jansson JH, Dahlén G, Boman K, Nilsson TK, Lindahl B, Weinehall L, Stenlund H, Lundberg V, Johnson O, Ahrén B, Hallmans G. Interactions between fibrinolysis, lipoproteins and leptin related to a first myocardial infarction. Eur J Cardiovasc Prev Rehabil. 2004;11:33–40. doi: 10.1097/01.hjr.0000116824.84388.a2. [DOI] [PubMed] [Google Scholar]
- 48.Soderberg S, Olsson T, Eliasson M, Johnson O, Ahren B. Plasma leptin levels are associated with abnormal fibrinolysis in men and postmenopausal women. J Intern Med. 1999;245:533–43. doi: 10.1046/j.1365-2796.1999.00472.x. [DOI] [PubMed] [Google Scholar]
- 49.De Mitrio V, De Pergola G, Vettor R, Marino R, Sciaraffia M, Pagano C, Scaraggi FA, Di Lorenzo L, Giorgino R. Plasma plasminogen activator inhibitor-I is associated with plasma leptin irrespective of body mass index, body fat mass, and plasma insulin and metabolic parameters in premenopausal women. Metabolism. 1999;48:960–4. doi: 10.1016/s0026-0495(99)90190-7. [DOI] [PubMed] [Google Scholar]
- 50.Li L, Mamputu JC, Wiernsperger N, Renier G. Signaling pathways involved in human vascular smooth muscle cell proliferation and matrix metalloproteinase-2 expression induced by leptin: inhibitory effect of metformin. Diabetes. 2005;54:2227–2234. doi: 10.2337/diabetes.54.7.2227. [DOI] [PubMed] [Google Scholar]
- 51.Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE, Jang Y, Cho SY, Kim HS. Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med. 2001;33:95–102. doi: 10.1038/emm.2001.17. [DOI] [PubMed] [Google Scholar]
- 52.Wallace AM, McMahon AD, Packard CJ, Kelly A, Shepherd J, Gaw A, Sattar N. Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS) Circulation. 2001;104:3052–3056. doi: 10.1161/hc5001.101061. [DOI] [PubMed] [Google Scholar]
- 53.Jamaluddin M, Weakley S, Yao Q, Chen C. Resistin: functional roles and therapeutic considerations for cardiovascular disease. British J Pharmacol. 2012;165:622–632. doi: 10.1111/j.1476-5381.2011.01369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chait A, den Hartigh LJ. Adipose tissue distribution, inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Front Cardiovasc Med. 2020;7:22. doi: 10.3389/fcvm.2020.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ. Human resistin stimulates the proinflammatory cytokines TNF-alpha and IL-12 in macrophages by NF kappaB-dependent pathway. Biochem Biophys Res Comm. 2005;334:1092–1101. doi: 10.1016/j.bbrc.2005.06.202. [DOI] [PubMed] [Google Scholar]
- 56.Verma S, Li SH, Wang CH, Fedak PW, Li RK, Weisel RD, Mickle DA. Resistin promotes endothelial cell activation: further evidence of adipokine-endothelial interaction. Circulation. 2003;108:736–740. doi: 10.1161/01.CIR.0000084503.91330.49. [DOI] [PubMed] [Google Scholar]
- 57.Rae C, Robertson S, Taylor J, Graham A. Resistin induces lipolysis and re-esterification of triacylglycerol stores, and increases cholesteryl ester deposition, in human macrophages. FEBS Lett. 2007;581:4877–4883. doi: 10.1016/j.febslet.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 58.Lee TS, Lin CY, Tsai JY, Wu YL, Su KH, Lu KY, Hsiao SH, Pan CC, Kou YR, Hsu YP, Ho LT. Resistin increases lipid accumulation by affecting class A scavenger receptor, CD36 and ATP-binding cassette transporter-A1 in macrophages. Life Sci. 2009;84:97–104. doi: 10.1016/j.lfs.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Chen W, Wang S, Lin C, Tsai C, Fong Y, Lin T, Weng S, Huang H, Liao K, Tang C. Resistin enhances monocyte chemoattractant protein-1 production in human synovial fibroblasts and facilitates monocyte migration. Cell Physiol Biochem. 2019;52:408–420. doi: 10.33594/000000029. [DOI] [PubMed] [Google Scholar]
- 60.Benomar Y, Amine H, Crepin D, Al Rifai S, Riffault L, Gertler A, Taouis M. Central Resistin/TLR4 impairs adiponectin signaling, contributing to insulin and fgf21 resistance. Diabetes. 2016;65:913–926. doi: 10.2337/db15-1029. [DOI] [PubMed] [Google Scholar]
- 61.Calabro P, Samudio I, Willerson JT, Yeh ET. Resistin promotes smooth muscle cell proliferation through activation of extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase pathways. Circualtion. 2004;110:3335–3340. doi: 10.1161/01.CIR.0000147825.97879.E7. [DOI] [PubMed] [Google Scholar]
- 62.Jiang C, Zhang H, Zhang W, Kong W, Zhu Y, Zhang H, Xu Q, Li Y, Wang X. Homocysteine promotes vascular smooth muscle cell migration by induction of the adipokine resistin. Am J Physiol Cell Physiol. 2009;297:C1466–C1476. doi: 10.1152/ajpcell.00304.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen C, Jiang J, Lü JM, Chai H, Wang X, Lin PH, Yao Q. Resistin decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2010;299:H193–H201. doi: 10.1152/ajpheart.00431.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohmori R, Momiyama Y, Kato R, Taniguchi H, Ogura M, Ayaori M, Nakamura H, Ohsuzu F. Associations between serum resistin levels and insulin resistance, inflammation, and coronary artery disease. J Am Coll Cardiol. 2005;46:379–380. doi: 10.1016/j.jacc.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 65.Reilly M, Lehrke M, Wolfe M, Rohatgi A, Lazar M, Rader D. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111:932–939. doi: 10.1161/01.CIR.0000155620.10387.43. [DOI] [PubMed] [Google Scholar]
- 66.Chu S, Ding W, Li K, Pang Y, Tang C. Plasma resistin is associated with myocardium injury in patients with acute coronary syndrome. Circ J. 2008;72:1249–1253. doi: 10.1253/circj.72.1249. [DOI] [PubMed] [Google Scholar]
- 67.de Leon A, Gonzalez A, Hernandez G, Sanchez J, Diaz B, Coello D, Rodriguez M, Garcia G, Jaime A, Mdel C. The association of resistin with coronary disease in the general population. J Atheroscler Thromb. 2014;21:273–281. doi: 10.5551/jat.19273. [DOI] [PubMed] [Google Scholar]
- 68.Gencer B, Auer R, de Rekeneire N, Butler J, Kalogeropoulos A, Bauer D, Kritchevsky B, Miljkovic L, Vittinghoff E, Harris T, Rodondi N. Association between resistin levels and cardiovascular disease events in older adults: the health, aging and body composition study. Atherosclero. 2016;245:181–186. doi: 10.1016/j.atherosclerosis.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang H, Chen DY, Cao J, He ZY, Zhu BP, Long M. High serum resistin level may be an indicator of the severity of coronary disease in acute coronary syndrome. Chin Med Sci J. 2009;24:161–166. doi: 10.1016/s1001-9294(09)60082-1. [DOI] [PubMed] [Google Scholar]
- 70.Hu W, Qiao S, Li J. Decreased C-reactive protein-induced resistin production in human monocytes by simvastatin. Cytokine. 2007;40:201–206. doi: 10.1016/j.cyto.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 71.Ichida Y, Hasegawa G, Fukui M, Obayashi H, Ohta M, Fujinami A, Ohta K, Nakano K, Yoshikawa T, Nakamura N. Effect of atorvastatin on in vitro expression of resistin in adipocytes and monocytes/macrophages and effect of atorvastatin treatment on serum resistin levels in patients with type 2 diabetes. Pharmacol. 2006;76:34–39. doi: 10.1159/000088948. [DOI] [PubMed] [Google Scholar]
- 72.Graham TE, Yang Q, Blüher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson PA, Smith U, Kahn BB. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. 2006;354:2552–2563. doi: 10.1056/NEJMoa054862. [DOI] [PubMed] [Google Scholar]
- 73.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–62. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 74.Liu Y, Zhong Y, Chen H, Wang D, Wang M, Ou JS, Xia M. Retinol-binding protein-dependent cholesterol uptake regulates macrophage foam cell formation and promotes atherosclerosis. Circulation. 2017;135:1339–1354. doi: 10.1161/CIRCULATIONAHA.116.024503. [DOI] [PubMed] [Google Scholar]
- 75.Moraes-Vieira PM, Yore MM, Sontheimer-Phelps A, Castoldi A, Norseen J, Aryal P, Simonyté Sjödin K, Kahn BB. Retinol binding protein 4 primes the NLRP3 inflammasome by signaling through Toll-like receptors 2 and 4. Proc Natl Acad Sci U S A. 2020;117:31309–31318. doi: 10.1073/pnas.2013877117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rychter AM, Skrzypczak-Zielińska M, Zielińska A, Eder P, Souto EB, Zawada A, Ratajczak AE, Dobrowolska A, Krela-Kaźmierczak I. Is the retinol-binding protein 4 a possible risk factor for cardiovascular diseases in obesity? Int J Mol Sci. 2020;21:5229–5249. doi: 10.3390/ijms21155229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Majerczyk M, Olszanecka-Glinianowicz M, Purianowska-Kuznicka M, Chudek J. Retinol-binding protein 4 (RBP4) as the causative factor and marker of vascular injury related to insulin resistance. Postepy Hig Med Dosw. 2016;70:1267–1275. [PubMed] [Google Scholar]
- 78.Zheng JY, Zou JJ, Wang WZ, Feng XY, Shi YY, Zhao Y, Jin G, Liu ZM. Tumor necrosis factor-alpha increases angiopoietin-like protein 2 gene expression by activating Foxo1 in 3T3-L1 adipocytes. Mol Cell Endocrinol. 2011;339:120–129. doi: 10.1016/j.mce.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 79.Tabata M, Kadomatsu T, Fukuhara S, Miyata K, Ito Y, Endo M, Urano T, Zhu HJ, Tsukano H, Tazume H, Kaikita K, Miyashita K, Iwawaki T, Shimabukuro M, Sakaguchi K, Ito T, Nakagata N, Yamada T, Katagiri H, Kasuga M, Ando Y, Ogawa H, Mochizuki N, Itoh H, Suda T, Oike Y. Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell Metab. 2009;10:178–188. doi: 10.1016/j.cmet.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 80.Aryal B, Price N, Suarez Y, Fernández-Hernando C. ANGPTL4 in metabolic and cardiovascular disease. Trends Mol Med. 2019;25:723–734. doi: 10.1016/j.molmed.2019.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cao Y, Li R, Zhang F, Guo Z, Tuo S, Li Y. Correlation between angiopoietin-like proteins in inflammatory mediators in peripheral blood and severity of coronary arterial lesion in patients with acute myocardial infarction. Exp Ther Med. 2019;17:3495–3500. doi: 10.3892/etm.2019.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiao X, Yang Y, Li L, Yu H, Yang Y, Li J, Du Y, Zhang J, Hu C, Qin Y. Angiopoietin-like protein 8 accelerates atherosclerosis in ApoE-/- mice. Atherosclero. 2020;307:63–71. doi: 10.1016/j.atherosclerosis.2020.06.014. [DOI] [PubMed] [Google Scholar]
- 83.Wang X, Musunuru K. Angiopoietin-like 3: from discovery to therapeutic gene editing. JACC Basic Transl Sci. 2019;4:755–762. doi: 10.1016/j.jacbts.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sindhu S, Thomas R, Shihab P, Sriraman D, Behbehani K, Ahmad R. Obesity is a positive modulator of IL-6R and IL-6 expression in the subcutaneous adipose tissue: significance for metabolic inflammation. PLoS One. 2010;10:e0133494. doi: 10.1371/journal.pone.0133494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lukic L, Lalic NM, Rajkovic N, Jotic A, Lalic K, Milicic T, Seferovic JP, Macesic M, Gajovic JS. Hypertension in obese type 2 diabetes patients is associated with increases in insulin resistance and IL-6 cytokine levels: potential targets for an efficient preventive intervention. Int J Environ Res Public Health. 2014;11:3586–3598. doi: 10.3390/ijerph110403586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Serrano-Marco L, Barroso E, El Kochairi I, Palomer X, Michalik L, Wahli W, Vázquez-Carrera M. The peroxisome proliferator-activated receptor (PPAR) β/δ agonist GW501516 inhibits IL-6-induced signal transducer and activator of transcription 3 (STAT3) activation and insulin resistance in human liver cells. Diabetologia. 2012;55:743–751. doi: 10.1007/s00125-011-2401-4. [DOI] [PubMed] [Google Scholar]
- 87.Dou L, Zhao T, Wang L, Huang X, Jiao J, Gao D, Zhang H, Shen T, Man Y, Wang S, Li J. miR-200s contribute to interleukin-6 (IL-6)-induced insulin resistance in hepatocytes. J Biol Chem. 2013;288:22596–606. doi: 10.1074/jbc.M112.423145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yin J, Hao Z, Ma Y, Liao S, Li X, Fu J, Wu Y, Shen J, Zhang P, Li X, Wang H. Concomitant activation of the PI3K/Akt and ERK1/2 signalling is involved in cyclic compressive force-induced IL-6 secretion in MLO-Y4 cells. Cell Biol Int. 2014;38:591–598. doi: 10.1002/cbin.10235. [DOI] [PubMed] [Google Scholar]
- 89.Berg AH, Scherer P. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–949. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- 90.Bettelli E, Carrier Y, Gao W, Korn T, Strom T, Oukka M, Weiner H, Kuchroo V. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 91.De Lorenzo A, Del Gobbo V, Premrov MG, Bigioni M, Galvano F, Di Renzo L. Normal-weight obese syndrome: early inflammation? Am J Clin Nutr. 2007;85:40–45. doi: 10.1093/ajcn/85.1.40. [DOI] [PubMed] [Google Scholar]
- 92.Wainstein MV, Mossmann M, Araujo GN, Gonçalves SC, Gravina GL, Sangalli M, Veadrigo F, Matte R, Reich R, Costa FG, Andrades M, da Silva AMV, Bertoluci MC. Elevated serum interleukin-6 is predictive of coronary artery disease in intermediate risk overweight patients referred for coronary angiography. Diabetol Metab Syndr. 2017;9:67. doi: 10.1186/s13098-017-0266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ridker P, Libby P, MacFadyen J, Thuren T, Ballantyne C, Fonseca F, Koenig W, Shimokawa H, Everett B, Glynn R. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) Eur Heart J. 2018;39:3499–350. doi: 10.1093/eurheartj/ehy310. [DOI] [PubMed] [Google Scholar]
- 94.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Uchida Y, Takeshita K, Yamamoto K, Kikuchi R, Nakayama T, Nomura M, Cheng XW, Egashira K, Matsushita T, Nakamura H, Murohara T. Stress augments insulin resistance and prothrombotic state: role of visceral adipose-derived monocyte chemoattractant protein-1. Diabetes. 2012;61:1552–1561. doi: 10.2337/db11-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Simeoni E, Hoffmann MM, Winkelmann BR, Ruiz J, Fleury S, Boehm BO, März W, Vassalli G. Association between the A-2518G polymorphism in the monocyte chemoattractant protein-1 gene and insulin resistance and type 2 diabetes mellitus. Diabetologia. 2004;47:1574–1580. doi: 10.1007/s00125-004-1494-4. [DOI] [PubMed] [Google Scholar]
- 97.Yadav A, Saini V, Arora S. MCP-1: chemoattractant with a role beyond immunity: a review. Clin Chim Acta. 2010;411:1570–1579. doi: 10.1016/j.cca.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 98.Bruun J, Lihn A, Pedersen S, Richelsen B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): implication of macrophages resident in the AT. J Clin Endocrinol Metab. 2005;90:2282–2289. doi: 10.1210/jc.2004-1696. [DOI] [PubMed] [Google Scholar]
- 99.Georgakis M, van der Laan S, Asare Y, Mekke J, Haitjema S, Schoneveld A, de Jager S, Nurmohamed N, Kroon J, Stroes E, de Kleijn D, de Borst G, Maeqdefessel L. Monocyte-chemoattractant protein-1 levels in human atherosclerosis associate with plaque vulnerability. Arterioscler Thromb Vasc Biol. 2021;41:2038–2048. doi: 10.1161/ATVBAHA.121.316091. [DOI] [PubMed] [Google Scholar]
- 100.Georgakis MK, de Lemos JA, Ayers C, Wang B, Björkbacka H, Pana TA, Thorand B, Sun C, Fani L, Malik R, Dupuis J, Engström G, Orho-Melander M, Melander O, Boekholdt SM, Zierer A, Elhadad MA, Koenig W, Herder C, Hoogeveen RC, Kavousi M, Ballantyne CM, Peters A, Myint PK, Nilsson J, Benjamin EJ, Dichgans M. Association of circulating monocyte chemoattractant protein-1 levels with cardiovascular mortality: a meta-analysis of population-based studies. JAMA Cardiol. 2021;6:587–592. doi: 10.1001/jamacardio.2020.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Živković L, Asare Y, Bernhagen J, Dichgans M, Georgakis M. CCL2/CCR2 inhibition in atherosclerosis: a meta-analysis of preclinical studies. bioRxiv. 2021 [Google Scholar]
- 102.Ryo M, Nakamura T, Kihara S, Kumada M, Shibazaki S, Takahashi M, Nagai M, Matsuzawa Y, Funahashi T. Adiponectin as a biomarker of the metabolic syndrome. Circ J. 2004;68:975–981. doi: 10.1253/circj.68.975. [DOI] [PubMed] [Google Scholar]
- 103.Silva TE, Colombo G, Schiavon L. Adiponectin: a multitasking player in the field of liver diseases. Diabetes Metab. 2014;40:95–107. doi: 10.1016/j.diabet.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 104.Ouchi N, Kihara S, Arita Y. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation. 2001;103:1057–1063. doi: 10.1161/01.cir.103.8.1057. [DOI] [PubMed] [Google Scholar]
- 105.Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, Yano W, Froguel P, Nagai R, Kimura S, Kadowaki T, Noda T. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277:25863–25866. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- 106.Kumada M, Kihara S, Ouchi N, Kobayashi H, Okamoto Y, Ohashi K, Maeda K, Nagaretani H, Kishida K, Maeda N, Nagasawa A, Funahashi T, Matsuzawa Y. Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation. 2004;109:2046–2049. doi: 10.1161/01.CIR.0000127953.98131.ED. [DOI] [PubMed] [Google Scholar]
- 107.Elmekci H, Ekmekci O. The role of adiponectin in atherosclerosis. Clin Appl Thromb Hemost. 2006;12:163–168. doi: 10.1177/107602960601200203. [DOI] [PubMed] [Google Scholar]
- 108.Mathis D. Immunological goings-on in visceral adipose tissue. Cell Metab. 2013;17:851–859. doi: 10.1016/j.cmet.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Van Gaal L, Mertens I, DeBlock C. Mechanisms linking obesity with cardiovascular disease. Nature. 2008;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 110.Takemura Y, Ouchi N, Shibata R, Aprahamian T, Kirber MT, Summer RS, Kihara S, Walsh K. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest. 2007;117:375–386. doi: 10.1172/JCI29709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Luo L, Liu M. Adipose tissue in control of metabolism. J Endocrinol. 2016;231:R77–R99. doi: 10.1530/JOE-16-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yamauchi T, Kadowaki T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013;17:185–96. doi: 10.1016/j.cmet.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 113.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 114.Chen H, Montagnani M, Funahashi T, Shimomura I, Quon M. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem. 2003;278:45021–45026. doi: 10.1074/jbc.M307878200. [DOI] [PubMed] [Google Scholar]
- 115.Matsuda M, Shimomura I, Sata M, Arita Y, Nishida M, Maeda N, Kumada M, Okamoto Y, Nagaretani H, Nishizawa H, Kishida K, Komuro R, Ouchi N, Kihara S, Nagai R, Funahashi T, Matsuzawa Y. Role of adiponectin in preventing vascular stenosis: the missing link of adipo-vascular axis. J Biol Chem. 2002;277:37487–37491. doi: 10.1074/jbc.M206083200. [DOI] [PubMed] [Google Scholar]
- 116.Okamoto Y, Kihara S, Funahashi T, Matsuzawa Y, Libby P. Adiponectin: a key adipocytokine in metabolic syndrome. Clin Sci (Lond) 2006;110:267–278. doi: 10.1042/CS20050182. [DOI] [PubMed] [Google Scholar]
- 117.Fisman E, Tenenbaum A. Adiponectin: a manifold therapeutic target for metabolic syndrome, diabetes, and coronary disease? Cardiovasc Diabetol. 2014;13:103. doi: 10.1186/1475-2840-13-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kumada M, Kihara S, Sumitsuji S, Kawamoto T, Matsumoto S, Ouchi N, Arita Y, Okamoto Y, Shimomura I, Hiraoka H, Nakamura T, Funahashi T, Matsuzawa Y Osaka CAD Study Group. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol. 2003;23:85–89. doi: 10.1161/01.atv.0000048856.22331.50. [DOI] [PubMed] [Google Scholar]
- 119.Nakamura Y, Shimada K, Fukuda D, Shimada Y, Ehara S, Hirose M, Kataoka T, Kamimori K, Shimodozono S, Kobayashi Y, Yoshiyama M, Takeuchi K, Yoshikawa J. Implications of plasma concentrations of adiponectin in patients with coronary artery disease. Heart. 2004;90:528–533. doi: 10.1136/hrt.2003.011114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang WS, Lee WJ, Funahashi T, Tanaka S, Matsuzawa Y, Chao CL, Chen CL, Tai TY, Chuang LM. Weight reduction increases plasma levels of an adipose-derived anti-inflammatory protein, adiponectin. J Clin Endocrinol Metab. 2001;86:3815–3819. doi: 10.1210/jcem.86.8.7741. [DOI] [PubMed] [Google Scholar]
- 121.Pischon T, Girman C, Hotamisligil G, Rifai N, Hu F, Rimm E. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004;291:1730–1737. doi: 10.1001/jama.291.14.1730. [DOI] [PubMed] [Google Scholar]
- 122.Greulich S, Chen WJ, Maxhera B, Rijzewijk LJ, van der Meer RW, Jonker JT, Mueller H, de Wiza DH, Floerke RR, Smiris K, Lamb HJ, de Roos A, Bax JJ, Romijn JA, Smit JW, Akhyari P, Lichtenberg A, Eckel J, Diamant M, Ouwens DM. Cardioprotective properties of omentin-1 in type 2 diabetes: evidence from clinical and in vitro studies. PLoS One. 2013;8:e59697. doi: 10.1371/journal.pone.0059697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhou Y, Zhang B, Hao C, Huang X, Li X, Huang Y, Luo Z. Omentin-A novel adipokine in respiratory diseases. Int J Mol Sci. 2017;19:1–16. doi: 10.3390/ijms19010073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tan BK, Adya R, Farhatullah S, Lewandowski KC, O’Hare P, Lehnert H, Randeva HS. Omentin-1, a novel adipokine, is decreased in overweight insulin-resistant women with polycystic ovary syndrome. Ex vivo and in vivo regulation of omentin-1 by insulin and glucose. Diabetes. 2008;57:801–808. doi: 10.2337/db07-0990. [DOI] [PubMed] [Google Scholar]
- 125.Yamawaki H, Kuramoto J, Kameshima S, Usui T, Okada M, Hara Y. Omentin a novel adipocytokine inhibits TNF-induced vascular inflammation in human endothelial cells. Biochem Biophys Res Commun. 2011;408:339–343. doi: 10.1016/j.bbrc.2011.04.039. [DOI] [PubMed] [Google Scholar]
- 126.Yamawaki H, Tsubaki N, Mukohda M, Okada M, Hara Y. Omentin, a novel adipokine, induces vasodilatation in rat isolated blood vessels. Biochem Biophys Res Commun. 2010;393:668–672. doi: 10.1016/j.bbrc.2010.02.053. [DOI] [PubMed] [Google Scholar]
- 127.Duan X, Xie P, Ma Y, Tang S. Omentin inhibits osteoblastic differentiation of calcifying vascular smooth muscle cells through the PI3K/Akt pathway. Amino Acids. 2011;41:1223–1231. doi: 10.1007/s00726-010-0800-3. [DOI] [PubMed] [Google Scholar]
- 128.Yamawaki H, Kuramoto J, Kameshima S, Usui T, Okada M, Hara Y. Omentin, a novel adipocytokine inhibits TNF-induced vascular inflammation in human endothelial cells. Biochem Biophys Res Commun. 2011;408:339–343. doi: 10.1016/j.bbrc.2011.04.039. [DOI] [PubMed] [Google Scholar]
- 129.Uemura Y, Shibata R, Kanemura N, Ohashi K, Kambara T, Hiramatsu-Ito M, Enomoto T, Yuasa D, Joki Y, Matsuo K, Ito M, Hayakawa S, Ogawa H, Murohara T, Ouchi N. Adipose-derived protein omentin prevents neointimal formation after arterial injury. FASEB J. 2015;29:141–151. doi: 10.1096/fj.14-258129. [DOI] [PubMed] [Google Scholar]
- 130.Jager C, Pasterkamp G. Atheroprotective properties of human Omentin-1 in experimental atherosclerosis. Cardiovasc Res. 2016;110:1–3. doi: 10.1093/cvr/cvw040. [DOI] [PubMed] [Google Scholar]
- 131.Watanabe K, Watanabe R, Konii H, Shirai R, Sato K, Matsuyama TA, Ishibashi-Ueda H, Koba S, Kobayashi Y, Hirano T, Watanabe T. Counteractive effects of omentin-1 against atherogenesis. Cardiovasc Res. 2016;110:118–128. doi: 10.1093/cvr/cvw016. [DOI] [PubMed] [Google Scholar]
- 132.Yoo HJ, Hwang SY, Hong HC, Choi HY, Yang SJ, Seo JA, Kim SG, Kim NH, Choi KM, Choi DS, Baik SH. Association of circulating omentin-1 level with arterial stiffness and carotid plaque in type 2 diabetes. Cardiovasc Diabetol. 2011;10:103. doi: 10.1186/1475-2840-10-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ouerghi N, Ben Fradj MK, Bezrati I, Feki M, Kaabachi N, Bouassida A. Effect of high-intensity interval training on plasma omentin-1 concentration in overweight/obese and normal-weight youth. Obes Facts. 2017;10:323–331. doi: 10.1159/000471882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sobczak A, Blindauer C, Stewart A. Changes in plasma free fatty acids associated with type-2 diabetes. Nutrients. 2019;11:2022–2064. doi: 10.3390/nu11092022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ghosh A, Gao L, Thakur A, Siu PM, Lai CWK. Role of free fatty acids in endothelial dysfunction. J Biomed Sci. 2017;24:50. doi: 10.1186/s12929-017-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kobayasi R, Akamine EH, Davel AP, Rodrigues MA, Carvalho CR, Rossoni LV. Oxidative stress and inflammatory mediators contribute to endothelial dysfunction in high-fat diet-induced obesity in mice. J Hypertens. 2010;28:2111–2119. doi: 10.1097/HJH.0b013e32833ca68c. [DOI] [PubMed] [Google Scholar]
- 138.Mathew M, Tay E, Cusi K. Elevated plasma free fatty acids increase cardiovascular risk by inducing plasma biomarkers of endothelial activation, myeloperoxidase and PAI-1 in healthy subjects. Cardiovasc Diabetol. 2010;9:9. doi: 10.1186/1475-2840-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.García-Prieto CF, Hernández-Nuño F, Rio DD, Ruiz-Hurtado G, Aránguez I, Ruiz-Gayo M, Somoza B, Fernández-Alfonso MS. High-fat diet induces endothelial dysfunction through a down-regulation of the endothelial AMPK-PI3K-Akt-eNOS pathway. Mol Nutr Food Res. 2015;59:520–532. doi: 10.1002/mnfr.201400539. [DOI] [PubMed] [Google Scholar]
- 140.Traupe T, Lang M, Goettsch W, Münter K, Morawietz H, Vetter W, Barton M. Obesity increases prostanoid mediated vasoconstriction and vascular thromboxane receptor gene expression. J Hypertens. 2002;20:2239–2245. doi: 10.1097/00004872-200211000-00024. [DOI] [PubMed] [Google Scholar]
- 141.Grassi G, Seravalle G, Scopelliti F, Dell’Oro R, Fattori L, Quarti-Trevano F, Brambilla G, Schiffrin EL, Mancia G. Structural and functional alterations of subcutaneous small resistance arteries in severe human obesity. Obesity. 2010;18:92–98. doi: 10.1038/oby.2009.195. [DOI] [PubMed] [Google Scholar]
- 142.Rahmani A, Ahmadi M, Misgavam S, Farhadi F, Shariatpanahi Z. Body composition and abdominal obesity in patients with and without coronary heart disease. Cardiol Res. 2014;5:68–71. doi: 10.14740/cr324w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bastien M, Poirier P, Lemieux I, Despres J. Overview of epidemiology and contribution of obesity to cardiovascular disease. Prog Cardiovasc Dis. 2014;56:369–381. doi: 10.1016/j.pcad.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 144.Wilson P, D’Agostino R, Sullivan L, Parise H, Kannel W. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Arch Int Med. 2002;162:1867–1872. doi: 10.1001/archinte.162.16.1867. [DOI] [PubMed] [Google Scholar]
- 145.Koushki K, Shahbaz SK, Mashayekhi K, Sadeghi M, Zayeri ZD, Taba MY, Banach M, Al-Rasadi K, Johnston TP, Sahebkar A. Anti-inflammatory action of statins in cardiovascular disease: the role of inflammasome and toll-like receptor pathways. Clinic Rev Allerg Immunol. 2021;60:175–199. doi: 10.1007/s12016-020-08791-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sahebkar A, Simental-Mendía LE, Pedone C, Ferretti G, Nachtigal P, Bo S, Derosa G, Maffioli P, Watts GF. Statin therapy and plasma free fatty acids: a systematic review and meta-analysis of controlled clinical trials. Br J Clin Pharmacol. 2018;81:807–818. doi: 10.1111/bcp.12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Huh J, Park Y, Ham M, Kim J. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells. 2014;37:365–371. doi: 10.14348/molcells.2014.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Song J, Deng T. The adipocyte and adaptive immunity. Front Immunol. 2020;11:593058. doi: 10.3389/fimmu.2020.593058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cildir G, Akincilar S, Tergaonkar V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol Med. 2013;19:487–500. doi: 10.1016/j.molmed.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 150.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 151.Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, Libby P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. 2008;103:467–476. doi: 10.1161/CIRCRESAHA.108.177105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu J, Divoux A, Sun J, Zhang J, Clément K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ, Doria A, Libby P, Blumberg RS, Kahn BB, Hotamisligil GS, Shi GP. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15:940–945. doi: 10.1038/nm.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Weil BR, Westby CM, Van Guilder GP, Greiner JJ, Stauffer BL, DeSouza CA. Enhanced endothelin-1 system activity with overweight and obesity. Am J Physiol Heart Circ Physiol. 2011;301:H689–695. doi: 10.1152/ajpheart.00206.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Folli F, Kahn C, Hansen H, Bouchie J, Feener E. Angiotensin II inhibits insulin signaling in aortic smooth muscle cells at multiple levels. A potential role for serine phosphorylation in insulin/angiotensin II crosstalk. J Clin Invest. 1997;100:2158–2169. doi: 10.1172/JCI119752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Packer M. Leptin-aldosterone-neprilysin axis: identification of its distinctive role in the pathogenesis of the three phenotypes of heart failure in people with obesity. Circulation. 2018;137:1614–1631. doi: 10.1161/CIRCULATIONAHA.117.032474. [DOI] [PubMed] [Google Scholar]
- 156.Casas R, Urpi-Sardà M, Sacanella E, Arranz S, Corella D, Castañer O, Lamuela-Raventós RM, Salas-Salvadó J, Lapetra J, Portillo MP, Estruch R. Anti-inflammatory effects of the mediterranean diet in the early and late stages of atheroma plaque development. Mediators Infl amm. 2017;2017:3674390. doi: 10.1155/2017/3674390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jensen MD, Ryan DH, Apovian CM, Ard JD, Comuzzie AG, Donato KA, Hu FB, Hubbard VS, Jakicic JM, Kushner RF, Loria CM, Millen BE, Nonas CA, Pi-Sunyer FX, Stevens J, Stevens VJ, Wadden TA, Wolfe BM, Yanovski SZ, Jordan HS, Kendall KA, Lux LJ, Mentor-Marcel R, Morgan LC, Trisolini MG, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Smith SC Jr, Tomaselli GF American College of Cardiology/American Heart Association Task Force on Practice Guidelines; The Obesity Society. Guideline for the management of overweight and obesity in adults. Circulation. 2014;129(Suppl 2):S102–S138. doi: 10.1161/01.cir.0000437739.71477.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, Himmelfarb CD, Khera A, Lloyd-Jones D, McEvoy JW, Michos ED, Miedema MD, Muñoz D, Smith SC Jr, Virani SS, Williams KA Sr, Yeboah J, Ziaeian B. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Circulation. 2019;140:e596–e646. doi: 10.1161/CIR.0000000000000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nicoletti G, Giugliano G, Pontillo A, Cioffi M, D’Andrea F, Giugliano D, Esposito K. Effect of a multidisciplinary program of weight reduction on endothelial functions in obese women. J Endocrinol Invest. 2003;26:RC5–RC8. doi: 10.1007/BF03345154. [DOI] [PubMed] [Google Scholar]
- 160.Heilbronn LK, Noakes M, Clifton PM. Energy restriction and weight loss on very-low-fat diets reduce C-reactive protein concentrations in obese, healthy women. Arterioscler Thromb Vasc Biol. 2001;21:968–970. doi: 10.1161/01.atv.21.6.968. [DOI] [PubMed] [Google Scholar]
- 161.Henning RJ. Diabetes mellitus and cardiovascular disease. Future Cardiol. 2018;14:491–509. doi: 10.2217/fca-2018-0045. [DOI] [PubMed] [Google Scholar]
- 162.Rubino F, Nathan DM, Eckel RH, Schauer PR, Alberti KG, Zimmet PZ, Del Prato S, Ji L, Sadikot SM, Herman WH, Amiel SA, Kaplan LM, Taroncher-Oldenburg G, Cummings DE Delegates of the 2nd Diabetes Surgery Summit. Metabolic surgery in the treatment algorithm for type 2 diabetes: a joint statement by International Diabetes Organizations. Diabetes Care. 2016;39:861–877. doi: 10.2337/dc16-0236. [DOI] [PubMed] [Google Scholar]
- 163.Dadson P, Landini L, Helmiö M, Hannukainen JC, Immonen H, Honka MJ, Bucci M, Savisto N, Soinio M, Salminen P, Parkkola R, Pihlajamäki J, Iozzo P, Ferrannini E, Nuutila P. Effect of bariatric surgery on adipose tissue glucose metabolism in different depots in patients with or without type 2 diabetes. Diabetes Care. 2016;39:292–299. doi: 10.2337/dc15-1447. [DOI] [PubMed] [Google Scholar]
- 164.Sjöström L, Lindroos AK, Peltonen M, Torgerson J, Bouchard C, Carlsson B, Dahlgren S, Larsson B, Narbro K, Sjöström CD, Sullivan M, Wedel H Swedish Obese Subjects Study Scientific Group. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med. 2004;351:2683–2693. doi: 10.1056/NEJMoa035622. [DOI] [PubMed] [Google Scholar]
- 165.Carlsson LM, Peltonen M, Ahlin S, Anveden Å, Bouchard C, Carlsson B, Jacobson P, Lönroth H, Maglio C, Näslund I, Pirazzi C, Romeo S, Sjöholm K, Sjöström E, Wedel H, Svensson PA, Sjöström L. Bariatric surgery and prevention of type 2 diabetes in Swedish obese subjects. N Engl J Med. 2012;367:695–704. doi: 10.1056/NEJMoa1112082. [DOI] [PubMed] [Google Scholar]
- 166.Ruban A, Stoenchev K, Ashrafian H, Teare J. Current treatments for obesity. Clin Med (Lond) 2019;19:205–212. doi: 10.7861/clinmedicine.19-3-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Klein S, Fontana L, Young VL, Coggan AR, Kilo C, Patterson BW, Mohammed BS. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med. 2004;350:2549–2557. doi: 10.1056/NEJMoa033179. [DOI] [PubMed] [Google Scholar]
- 168.Herman W, Kalyani R, Cherrington A. American Diabetes Association Standards of medical care in diabetes: 2017. Diabetes Care. 2017;40(Suppl 1):S1–S132. doi: 10.2337/dci17-0007. [DOI] [PubMed] [Google Scholar]
- 169.Diamantis E, Kyriakos G, Quiles-Sanchez LV, Farmaki P, Troupis T. The anti-inflammatory effects of statins on coronary artery disease: an updated review of the literature. Curr Cardiol Rev. 2017;13:209–216. doi: 10.2174/1573403X13666170426104611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Madej A, Okopien B, Kowalski J, Zielinski M, Wysocki J, Szygula B, Kalina Z, Herman ZS. Effects of fenofibrate on plasma cytokine concentrations in patients with atherosclerosis and hyperlipoproteinemia IIb. Int J Clin Pharmacol Ther. 1998;36:345–349. [PubMed] [Google Scholar]
- 171.World Health Organization. HEARTS D: diagnosis and management of type 2 diabetes. 2020 [Google Scholar]
- 172.American Diabetes Association. Standards of medical care in diabetes-2021. Diabetes Care. 2021;44:S1–S222. doi: 10.2337/dc21-Sint. [DOI] [PubMed] [Google Scholar]
- 173.Tentolouris A, Vlachakis P, Tzeravini E, Eleftheriadou I, Tentolouris N. SGLT2 inhibitors: a review of their antidiabetic and cardioprotective effects. Int J Environ Res Public Health. 2019;16:2965. doi: 10.3390/ijerph16162965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.McGuire DK, Shih WJ, Cosentino F, Charbonnel B, Cherney D, Dagogo-Jack S, Pratley R, Greenberg M, Wang S, Huyck s, Gantz I, Terra S, Masiukiewicz U, Cannon C. Association of SGLT2 inhibitors with cardiovascular and kidney outcomes in patients with type 2 diabetes: a meta-analysis. JAMA Cardiol. 2021;6:148–158. doi: 10.1001/jamacardio.2020.4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hall J. The kidney, hypertension, and obesity. Hypertension. 2003;41:625–633. doi: 10.1161/01.HYP.0000052314.95497.78. [DOI] [PubMed] [Google Scholar]
- 176.Mertens I, Van Gaal L. Overweight, obesity, and blood pressure: the effects of modest weight reduction. Obes Res. 2000;8:270–278. doi: 10.1038/oby.2000.32. [DOI] [PubMed] [Google Scholar]
- 177.American Diabetes Association. Classification and diagnosis of diabetes: standards of medical care in diabetes-2021. Diabetes Care. 2021;44(Suppl 1):S15–S33. doi: 10.2337/dc21-S002. [DOI] [PubMed] [Google Scholar]
- 178.Lewington S, Clarke R, Qizilbash N, Peto R, Collins R Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–1913. doi: 10.1016/s0140-6736(02)11911-8. [DOI] [PubMed] [Google Scholar]
- 179.Arguedas J, Leiva V, Wright J. Blood pressure targets for hypertension in people with diabetes mellitus. Cochrane Database Syst Rev. 2013;10:CD008277. doi: 10.1002/14651858.CD008277.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC Jr, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA Sr, Williamson JD, Wright JT Jr. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. J Am Coll Cardiol. 2018;71:2199–2269. [Google Scholar]
- 181.Sharma A, Pischon T, Engeli S, Scholze J. Choice of drug treatment for obesity-related hypertension: where is the evidence? J Hypertens. 2001;19:667–674. doi: 10.1097/00004872-200104000-00001. [DOI] [PubMed] [Google Scholar]
- 182.Wenzel U, Benndor R, Lange S. Treatment of arterial hypertension in obese patients. Semin Nephrol. 2013;33:66–74. doi: 10.1016/j.semnephrol.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 183.Appel LJ, Brands MW, Daniels SR, Karanja N, Elmer PJ, Sacks FM American Heart Association. Dietary approaches to prevent and treat hypertension: a scientific statement for the American Heart Association. Hypertension. 2006;47:296–308. doi: 10.1161/01.HYP.0000202568.01167.B6. [DOI] [PubMed] [Google Scholar]
- 184.Di Napoli M, Papa F. Angiotensin-converting enzyme inhibitor use is associated with reduced plasma concentration of C-reactive protein in patients with first-ever ischemic stroke. Stroke. 2003;34:2922–2929. doi: 10.1161/01.STR.0000099124.84425.BB. [DOI] [PubMed] [Google Scholar]
- 185.Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35:881–900. doi: 10.1016/s1357-2725(02)00271-6. [DOI] [PubMed] [Google Scholar]
- 186.Reisin E, Weir MR, Falkner B, Hutchinson HG, Anzalone DA, Tuck ML. Lisinopril versus hydrochlorothiazide in obese hypertensive patients: a multicenter placebo-controlled trial. Treatment in Obese Patients with Hypertension (TROPHY) Study Group. Hypertension. 1997;30:140–145. doi: 10.1161/01.hyp.30.1.140. [DOI] [PubMed] [Google Scholar]
- 187.Grassi G, Seravalle G, Dell’Oro R, Trevano FQ, Bombelli M, Scopelliti F, Facchini A, Mancia G. Comparative effects of candesartan and hydrochlorothiazide on blood pressure, insulin sensitivity, and sympathetic drive in obese hypertensive individuals: results of the CROSS study. J Hypertension. 2003;21:1761–1769. doi: 10.1097/00004872-200309000-00027. [DOI] [PubMed] [Google Scholar]
- 188.Palmer SC, Mavridis D, Navarese E, Craig JC, Tonelli M, Salanti G, Wiebe N, Ruospo M, Wheeler DC, Strippoli GF. Comparative efficacy and safety of blood pressure lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet. 2015;385:2047–2056. doi: 10.1016/S0140-6736(14)62459-4. [DOI] [PubMed] [Google Scholar]
- 189.Owen J, Reisin E. Anti-hypertensive drug treatment of patients with and the metabolic syndrome and obesity: a review of evidence, meta-analysis, post hoc and guidelines publications. Curr Hypertens Rep. 2015;17:558. doi: 10.1007/s11906-015-0558-9. [DOI] [PubMed] [Google Scholar]
- 190.Wofford M, Smith G, Minor S. The treatment of hypertension in obese patients. Curr Hypertens Rep. 2008;10:143–150. doi: 10.1007/s11906-008-0027-9. [DOI] [PubMed] [Google Scholar]
- 191.Unger T, Borghi C, Charchar F, Khan NA, Poulter NR, Prabhakaran D, Ramirez A, Schlaich M, Stergiou GS, Tomaszewski M, Wainford RD, Williams B, Schutte AE. International society of hypertension global hypertension practice guidelines. Hypertension. 2020;75:1334–1357. doi: 10.1161/HYPERTENSIONAHA.120.15026. [DOI] [PubMed] [Google Scholar]