

ABSTRACT
The increased transmission of SARS-CoV-2 variants of concern (VOC), which originated in the United Kingdom (B.1.1.7/alpha), South Africa (B1.351/beta), Brazil (P.1/gamma), the United States (B.1.427/429 or epsilon), and India (B.1.617.2/delta), requires a vigorous public health response, including real-time strain surveillance on a global scale. Although genome sequencing is the gold standard for identifying these VOCs, it is time-consuming and expensive. Here, we describe a simple, rapid, and high-throughput reverse transcriptase PCR (RT-PCR) melting-temperature (Tm) screening assay that identifies the first three major VOCs. RT-PCR primers and four sloppy molecular beacon (SMB) probes were designed to amplify and detect the SARS-CoV-2 N501Y (A23063T) and E484K (G23012A) mutations and their corresponding wild-type sequences. After RT-PCR, the VOCs were identified by a characteristic Tm of each SMB. Assay optimization and testing was performed with RNA from SARS-CoV-2 USA WA1/2020 (wild type [WT]), B.1.1.7, and B.1.351 variant strains. The assay was then validated using clinical samples. The limit of detection for both the WT and variants was 4 and 10 genomic copies/reaction for the 501- and 484-codon assays, respectively. The assay was 100% sensitive and 100% specific for identifying the N501Y and E484K mutations in cultured virus and in clinical samples, as confirmed by Sanger sequencing. We have developed an RT-PCR melt screening test for the major VOCs that can be used to rapidly screen large numbers of patient samples, providing an early warning for the emergence of these variants and a simple way to track their spread.
KEYWORDS: SARS-CoV-2, variants, N501Y, E484K, melting temperature, screening test, sloppy molecular beacon
INTRODUCTION
In December 2020, public health officials in the United Kingdom observed a surge of COVID-19 cases in Kent, England, that appeared to be largely due to a specific variant of SARS-CoV-2 (1, 2). The new variant was named a variant of concern (VOC 202012/1) or B.1.1.7 based on its phylogenetic lineage. Early reports suggested that B.1.1.7 was more transmissible and more virulent than previous SARS-CoV-2 strains (3, 4), although the evidence did not suggest that B.1.1.7 caused a decrease in vaccine efficacy (5–8). The B.1.1.7 variant has a number of mutations in the spike protein, including single-nucleotide polymorphisms (SNPs) resulting in N501Y, A570D, D614G, and P681H mutations and deletions at amino acids 69 and 70 and 144Y (6, 9). The N501Y (A23063T) mutation has been identified as an important contributor to the worrisome phenotype (2, 4, 10, 11). Other SARS-CoV-2 strain variants have been implicated in large outbreaks in South Africa (known as 20H/501Y.V2 or B.1.351 lineage) and Brazil (20J/501Y.V3 or P.1 lineage) (12). These variants appear to have decreased the efficiency of some COVID-19 vaccines (2, 13, 14). All three variants, B.1.1.7, B.1.351, and P.1, contain the N501Y mutation, while the South African and Brazilian variants additionally contain mutations in E484K and K417N (1, 12). Thus, the N501Y mutation appears to be an excellent marker for all three strains, while the E484K mutation can be used to differentiate the other two strains from B.1.1.7. New SARS-CoV-2 variants have also been recently reported in the United States (5, 15). Most of the new variants also have mutations at the 501 and/or 484 position, although additional mutations, such as L452R, have also been reported in more recent variants (15, 16); however, the epidemiological and clinical relevance of these new variants are not well understood.
All three of the major SARS-CoV-2 N501Y variants are circulating around the world (1). However, the distribution, extent, and spread of these variants are poorly understood in countries such as the United States, where only a small fraction (0.04 to 3.5%) of COVID-19 cases are analyzed by viral genomic sequencing per the CDC’s national genomic surveillance dashboard (17). Viral genome sequencing is currently the only method available to reliably detect rapidly emerging SARS-CoV-2 variants. Genomic sequencing has the advantage of providing a detailed map of new mutations, which supports new variant discovery as well as monitoring the exact type of the variant. However, genomic sequencing is expensive and difficult to perform in real time. In contrast, reverse transcriptase PCR (RT-PCR) testing for SARS-CoV-2 has become widespread. This diagnostic approach is easy to perform in a high-throughput manner, and rapid turnaround times are possible. However, routine RT-PCR tests do not differentiate among SARS-CoV-2 variants or do so only by producing negative assay results (18), which still require sequence confirmation (19). Furthermore, the potential for additional mutations to appear near key variant-defining alleles may complicate the development of RT-PCR assays for SARS-CoV-2 variants (20). We have previously demonstrated that sloppy molecular beacons (SMBs) combined with melting temperature (Tm) code analysis may be used to specifically detect mutations in short genomic regions where a variety of mutations can exist (21).
Here, we apply this same Tm-based approach to detect and differentiate variant strains of SARS-CoV-2 with high sensitivity and specificity. This approach is flexible and can be used in a high-throughput manner for screening and detection of SARS-CoV-2 variants, easily allowing the addition of new mutation-detecting assays as needed to identify and track new variants as they emerge. Furthermore, this approach can be performed on a wide range of real-time PCR instrumentation as long as they have the capacity to run melting curve analysis. The wide availability of such instruments can allow quick adoption of this assay around the world, increasing access to real-time monitoring of SARS-CoV-2 variant spread.
MATERIALS AND METHODS
Ethical considerations.
The use of deidentified clinical samples from confirmed COVID-19-positive and -negative patients for PCR testing and sequencing was approved by the Rutgers Institutional Review Board under protocol numbers 20170001218 and 2020001541.
Viral cultures and RNA.
Genomic RNA from SARS-CoV-2 USA WA1/2020 (wild type [WT]) and viral culture stocks of SARS-CoV-2 hCoV-19/England/204820464/2020 (B.1.1.7 variant, 501-MT [mutant]), SARS-CoV-2 isolate hCoV-19/South Africa/KRISP-K005325/2020 (B.1.351 variant, 484-MT), and SARS-CoV-2 isolate hCoV-19/Japan/TY7-503/2021 (P.1, 484-MT) were obtained from BEI Resources, NIAID (Manassas, VA). RNA was isolated from both variant strains in a biosafety level 3 (BSL3) laboratory, using an RNAdvance viral RNA extraction kit (Beckman Coulter, Indianapolis, IN). RNA extracted from all three variant strains was quantified against a standard curve generated with the N1 gene-specific real-time RT-PCR assay (22, 23), and the concentration was determined to be 4 × 105 GE/μl for B.1.1.7, 7 × 106 GE/μl for B.1.351, and 8.5 × 106 GE/μl for the P.1 variant strains.
Genome sequence analysis for assay design.
For initial analysis of the mutations in SARS-CoV-2, a total of 330,132 high-quality viral genome sequences deposited in GISAID (24) as of 12 January 2021, were analyzed. Publicly available data sets were analyzed in this study. These data can be found at https://www.gisaid.org/. A 250-nucleotide region around the N501Y (A23063T) and E484K (G23012A) positions in the reference strain (GenBank accession number MN908947) was selected and used to identify the corresponding regions in the GISAID data set using BLAST (25). These matching sequences were condensed into a set of unique sequences and aligned using a multiple-sequence alignment program, MAFFT (26). Candidate amplification primers and probes were identified on the basis of sequence conservation and predicted Tm using the algorithm of SantaLucia (27), and a final set of primers was designed with the help of the Primer3 program (28). SMB probe design was performed using the web servers DNA mfold (http://www.unafold.org/mfold/applications/dna-folding-form.php) and DINAmelt (http://www.unafold.org/hybrid2.php) to predict the probe folding structures and probe-target hybrid Tm values, respectively.
Primers and probes.
The list of primers and probes used for the 501 variant (SMB-501) and the 484 variant (SMB-484) assays are shown in Table 1. Both assays were run as two separate reactions. For the SMB-501 assay, an 89-bp region surrounding the 23063 position was amplified using an asymmetric PCR. For SMB-484 assay, a 76-bp region surrounding the 23012 position was amplified using an asymmetric PCR. SMB were designed targeting both the wild-type 501N/484E (23063A/23012A) and the mutant 501Y/484K (23063T/23012G) sequences (SMB-501-MT). Primers were obtained from Millipore Sigma (The Woodlands, TX), and SMBs were synthesized by LGC Biosearch Technologies (Petaluma, CA). An internal control (IC) assay developed by the CDC (22, 23), targeting the human RNaseP gene, was simultaneously performed for each extracted RNA specimen as a separate reaction in a separate well, using the TaqMan real-time PCR assay probe tagged with 6-carboxyfluorescein (FAM) at the 5′ end and Dabcyl quencher at the 3′ end.
TABLE 1.
Primers and probesa
| Assay | Primer/probe | 5′ End | Sequence | 3′ End | Amplicon size (bp) |
|---|---|---|---|---|---|
| SMB-501 | 501-F | ggttttaattgttactttcctttacaa | 89 | ||
| 501-R | gaaagtactactactctgtatggttgg | ||||
| 501-WT | Quasar 570 | CCGCgtt[pdU]ccatcccactaatgctg[pdU]tggttaccaacGCGG | BHQ-2 | ||
| 501-MT | Quasar 670 | CGCGgtt[pdu]ccatcccacttatgctg[pdU]tggttaccaacCGCG | BHQ-2 | ||
| SMB-484 | 484-F | ctatcaggccggtagcacac | 76 | ||
| 484-R | gaaaccatatgattgtaaaggaaag | ||||
| 484-WT | Quasar 570 | CCGCGccttgtaatggtgttgaaggttttaattgttacGCGCGG | BHQ-2 | ||
| 484-MT | Quasar 670 | CCGCGccttgtaatggtgttaaaggttttaattgttacGCGCGG | BHQ-2 |
The lowercase letters indicate the SMB probe region, and the uppercase letters indicate the SMB stem region. [pdU], C5 propynyl-deoxyuridine. BHQ, black hole quencher.
SMB assay formulation and procedure.
TaqPath 1-step RT-quantitative PCR (qPCR) master mix CG (ThermoFisher Scientific, Waltham, MA) was used for the RT-PCR. Each one-step reaction mix was supplemented with 0.2 μM the forward primer 501-F/484-F and 2 μM/4 μM the reverse primer 501-R/484-R, 0.4 μM each of the SMB probes (SMB-501-WT and SMB-501-MT/SMB-484-WT and SMB-484-MT), and 1 μl of the template RNA (note that the concentration of the forward primers did not match the concentration of the reverse primers so as to create an asymmetric PCR). The internal control contained primers and a probe specific for human RNaseP as described previously (22, 23). Each reaction was run in replicates of 4 in 384-well plates in a Roche LightCycler 480 (Roche, Indianapolis, IN). The one-step RT-PCR amplification was performed with the following thermocycling conditions: uracil DNA glucosylase incubation for 2 min at 37°C and reverse transcription (RT) for 15 min at 50°C, followed by asymmetric PCR for 45 cycles (denaturation at 95°C for 1 s, annealing/extension at 55°C for 30 s). The post-PCR melt was performed with the following conditions: denaturation at 95°C for 30 s, followed by cooling down to 45°C and gradual heating to 85°C, with continuous monitoring of fluorescence at a rate of 2 acquisitions per degree Celsius. The total assay time was 77 min. Automated Tm calls were performed by the LC480 Tm detection software at the end of the PCR. The resulting Tm for each probe was identified and matched with the Tm signature code defined for the wild type or the mutant variants.
Analytical sensitivity.
The prequantitated genomic RNA from the SARS-CoV-2 USA WA1/2020 (WT), B.1.1.7 (alpha/UK variant), and B.1.351 (beta/SA variant), obtained from BEI Resources, were diluted in Tris-EDTA (TE) buffer. Total RNA was extracted from a SARS-CoV-2 negative nasopharyngeal (NP) specimen (confirmed negative by Xpert Xpress SARS-CoV-2 test). The diluted SARS-CoV-2 RNA from the WT and the variants was spiked into the extracted matrix at final concentrations of 1,000, 500, 100, 50, 10, 5, and 1 GE/μl and mixed well. A 1-μl aliquot of the RNA spiked into the NP matrix was added to 19 μl of the one-step RT-PCR mix containing the primers and probes and was evaluated in the SMB-501 assay or the SMB-484 assay; 1,000 GE/μl of the quantified RNA from P.1 was also validated with both assays (Table 2).
TABLE 2.
Tm profile for SARS-CoV-2 VOC tested by SMB-501 and SMB-484 assays
| SARS-CoV-2 strain | SMB-501 assay |
SMB-484 assay |
||
|---|---|---|---|---|
| 501-WT probe (°C) | 501-MT probe (°C) | 484-WT probe (°C) | 484-MT probe (°C) | |
| WA1 USA/2020 (WT strain) | 59.8 ± 0.4 | 58.2 ± 1 | 64 ± 0.1 | 58.7 ± 0.4 |
| B.1.1.7 (alpha, MT strain) | 55.2 ± 0.4 | 62.2 ± 0.6 | 64.5 ± 0.05 | 59.2 ± 0.03 |
| B.1.351 (beta, MT strain) | 56.2 ± 0.01 | 63.1 ± 0.01 | 59.7 ± 0.5 | 63 ± 0.6 |
| P.1 (gamma, MT strain) | 56.3 ± 0.07 | 63 ± 0.16 | 60.7 ± 0.03 | 63.5 ± 0.05 |
Validation with patient samples.
Deidentified NP swabs obtained from patients undergoing routine COVID-19 clinical testing using the Xpert Xpress SARS-CoV-2 or Xpert Xpress SARS-CoV-2/Flu/RSV (4-plex) test (Cepheid, Sunnyvale CA) assay in the CLIA- and CAP-certified laboratory at the Public Health Research Institute (PHRI), Newark, NJ, were selected for this study. NP swabs were collected in 3 ml viral transport medium (VTM) from Hardy Diagnostics (Santa Maria, CA) or Labscoop (Little Rock, AR) and were banked under either refrigerated conditions (for specimens first tested within the previous 2 weeks) or frozen (for specimens stored longer than 2 weeks). The samples consisted of 46 randomly selected COVID-19-positive samples of unknown genotype and an RT-PCR cycle threshold (CT) of <42 with either Xpert Xpress SARS-CoV-2 or Xpert Xpress CoV-2/Flu/RSV test. Nine of the samples had been banked between October 2020 and 31 December 2020, 16 in the month of January 2021, 15 in February 2021, and 6 in March 2021. Thirty COVID-19-negative specimens were randomly selected from samples banked between October 2020 and February 2021. RNA was extracted from each specimen using a QiaAmp viral RNA isolation kit by following the manufacturer’s instructions (Qiagen, Valencia, CA) in a BSL2 laboratory. A 5-μl volume of RNA was added to the one-step RT-PCR mix containing the primers and probes. A subset of these clinical samples consisting of 26/46 of the COVID-19-positive samples and 19/30 of the COVID-19-negative samples used the SMB-484 assay, mainly based on the continued availability of the sample for testing. The SMB-501, SMB-484, and IC assays were tested in separate wells in replicates of two. The samples that failed the IC assay were repeated starting with the RNA extraction. A subset of samples that tested positive either for 501N/484E wild type or 501Y/484K mutant was confirmed by Sanger sequencing using the primer pair F, 5′CTATCAGGCCGGTAGCACAC3′, and R, 5′CTTTCTTTTGAACTTCTACATG3′, which amplifies a 143-bp segment of the S-gene, inclusive of amino acid positions 484 and 501. Sequencing chromatograms were analyzed using Ugene (ver 37) compared to the known WT and MT sequences using MegAlign Pro software (DNAStar, ver16).
Statistical analysis.
Standard statistical analyses (averages and standard deviations [SD]) and graphing were performed using Microsoft Excel (ver 2102) and GraphPad Prism 8.4.3 for Windows.
RESULTS
Limit of detection.
The one-step RT mix containing asymmetric PCR primers and both wild-type and MT-specific probes was added with either WT RNA or MT RNA at different concentrations, ranging from 1,000 to 1 GE/reaction for both SMB-501 and SMB-484 assays. After Tm analysis was performed, the Tm peak heights were highest in the assays with the largest added number of GE, and the peak heights progressively decreased as the number of GEs present in the reactions decreased. However, Tm peak heights produced by both SMB 501-WT/SMB 484-WT and SMB 501-MT/SMB 484-MT could still be reproducibly detected at concentrations down to 10 GE/reaction when tested against both WT and N501Y mutant strains, defining the assay limit of detection as ≤10 GE per reaction for both the SMB-501 and SMB-484 assays (Fig. 1; see also Fig. S1 in the supplemental material).
FIG 1.

Analytical limit of detection and Tm values generated by the SMB-501 (A and B) and SMB-484 (C and D) assays tested against SARS-CoV-2 RNA in the presence of the nasopharyngeal (NP) matrix. Shown are SARS-CoV-2 wild type (WT) RNA (A and C), B.1.1.7 mutant (MT) RNA (B), and B.351 mutant (MT) RNA (D) at the indicated number of genomic equivalents (GEs).
Tm code definition.
The Tm values produced by both SMBs against the reference WT and the MT SARS-CoV-2 strains are listed in Table 2. The means and standard deviations shown were derived from at least 4 replicates. When reference RNA (1,000 GE/reaction) was tested with the SMB 501-WT probe, WT-RNA produced a Tm of 59.8 ± 0.4°C, while MT N501Y RNA from B.1.1.7/B.1.351/P.1 variants produced Tm values of 55.2 ± 0.4°C, 56.2 ± 0.01°C, and 56.3 ± 0.07°C, respectively. Similarly, when RNA was tested with the SMB 501-MT probe, WT-RNA produced a Tm of 58.2 ± 1°C, while MT N501Y RNA from B.1.1.7/B.1.351/P.1 variants produced Tm values of 62.2 ± 0.6°C/63.1 ± 0.01°C/63 ± 0.16°C, respectively. Given that Tm values can vary slightly between clinical samples, we specified a two-temperature Tm code that identified either 501N or 501Y alleles within a Tm range of approximately 1.5 to 4 times the experimentally verified ±SD values for each probe Tm. Thus, the 501N (WT) Tm code was defined as an SMB-501-WT Tm of 59.8 ± 1.5°C and an SMB-501-MT Tm of 58.2 ± 1.5°C, and a 501Y (MT) Tm code was defined as an SMB-501-WT Tm of 55.2 ± 1.5°C and an SMB-501-MT Tm of 62.2 ± 1.5°C. Similarly, for SMB-484 assay, the reference Tm code for 484E (WT) was 64 ± 0.1 (with SMB-484-WT probe) and 58.7 ± 0.4 (with SMB-484-MT probe), and the reference Tm code for 484K (MT, B.1.351 and P.1) was 59.7 ± 0.5 (WT probe) and 63 ± 0.6 (MT probe). Any samples that failed to produce a Tm value for either of the SMBs or produced Tm values outside the range defined for the Tm codes would have been defined as indeterminate and the assay repeated. Using this code definition, we retested our reference RNA samples 20 times, and the correct 501/484 allele was detected in each case, achieving an analytic sensitivity and specificity of 100%. These results clearly indicate that the combination of both SMB 501-WT and SMB 501-MT probes can specifically detect and differentiate the N501Y variants from the wild-type strains with high confidence.
Validation with patient samples.
A total of 76 patient samples, 46 confirmed COVID-19 positive and 30 confirmed COVID-19 negative, were tested using the SMB-501 assay. A subset of 45 of these same samples (26 COVID-19 positive and 19 COVID negative) were additionally tested with the SMB-484 assay. None of the COVID-19-negative samples produced any measurable Tm values from either of the assays, yielding a specificity of 100%. Positive samples were selected based on a wide range of N2-CT values between 16 and 41 based on the Xpert Xpress SARS-CoV-2 assay. As shown in Table 3, 40/45 (89%) of the COVID-19-positive samples produced measurable Tm values for both SMB probes in the SMB-501 assay in at least one of the two replicates. Two samples were identified as indeterminate based on the inability of either the WT or MT probe to generate a measurable Tm and/or due to the presence of a failed internal control. Five clinical samples that had previously tested positive for SARS-CoV-2 generated negative results in our variant assay, in that neither probe produced a measurable Tm. All of the samples with negative assay results had previously shown initial Xpert CT values of ≥35. Although many of the samples with Xpert CT values as late as 39 could still be detected by our assay, we found that the samples with a CT of ≥35 using the Xpert assay yielded relatively stunted melting peak heights of ≤0.3 with WT-SMB and ≤0.1 with MT-SMB, similar to that observed when testing <10 GE in our limit-of-detection studies, which is indicative of very low viral loads in these samples. However, the Tm could still be identified for at least one of the 501 SMB probes in all samples, except the negatives. Overall, 27/45 (60%) of the COVID-19-positive samples had SMB 501-WT and SMB 501-MT Tm values consistent with the WT N501 allele, and 11/45 (24.4%) of the samples had Tm values consistent with the mutant Y501 allele within ±1 standard deviation of our reference standards (Table 3, Fig. 2). Twelve representative WT and mutant samples, as predicted by the SMB assay, underwent Sanger sequencing of the PCR products for confirmation of the PCR results. In all cases, the WT or mutant sequences identified by the SMB N501Y assay were confirmed by the sequencing result. Using the confirmed sequencing results as a gold standard, 4/4 of the wild-type clinical samples were detected as WT by the SMB N501Y assay, and 8/8 N501Y mutant results were detected as mutant by the assay, demonstrating a clinical sensitivity and specificity of 100%. Similarly, with the SMB-484 assay, 19/26 (73%) of the samples tested were wild type and 4/26 (15.3%) were variants. Out of the 4 variants, 4/4 were sequenced and confirmed to harbor the G-to-A mutation in the 484th codon, yielding an assay specificity of 100% compared to sequencing. It should be noted that one of the specimens (SMBP-27) contained an E484K mutation but was found to have a WT sequence at codon 501. Considering that the major three VOCs contain either N501Y alone (B.1.1.7) or both N501Y and E484K mutations (B.1.351 and P.1), the presence of this single 484K mutant indicates that this sample contains the variant of interest, B.1.525/B.1.526, which originated in New York City. Thus, the combination of our SMB-501 and SMB-484 assays should help capture most of the variants in circulation other than those with 452 mutations. We also observed that the frequency of N501Y and E484K variants increased substantially between the clinical samples obtained in October 2020 and the samples obtained in February and March 2021 (Fig. 3).
TABLE 3.
Assay and Sanger sequencing results from COVID-positive clinical samplesa
| Sample | Date of 1st test | PCR CTb | SMB-501 assay |
SMB-484 assay |
Confirmation by sequencingc |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Tm (°C) |
ID |
Tm (°C) |
ID | Wild type | N501Y mutant (AAT-TAT) | E484K mutant (GAA-AAA) | |||||
| WT probe (Cy3) | MT probe (Cy5) | WT probe (Cy3) | MT probe (Cy5) | ||||||||
| WT-reference | 59.8 ± 0.4 | 58.2 ± 1 | WT | 64 ± 0.1 | 58.7 ± 0.4 | WT | Yes | No | No | ||
| MT-reference | 55.2 ± 0.4 | 62.2 ± 0.6 | MT | 59.7 ± 0.5 | 63 ± 0.6 | MT | No | Yes | No | ||
| SMBP-1 | Oct–Nov 2020 | 29.1 | 59.9 | 58.4 | WT | 63.9 | 58.2 | WT | Yes | No | No |
| SMBP-2 | 34.8 | NP | NP | Neg | |||||||
| SMBP-3 | 34.5 | NP | NP | Neg | 64.0 | 58.7 | WT | ||||
| SMBP-4 | 35.4 | NP | NP | Neg | |||||||
| SMBP-5 | 39.2 | 59.5 | 58.2 | WT | |||||||
| SMBP-6 | 36.0 | 59.1 | 58.2 | WT | Yes | No | No | ||||
| SMBP-7 | 33.7 | 58.9 | 58.1 | WT | 64.0 | NP | Ind | ||||
| SMBP-8 | 36.8 | 59.5 | 58.2 | WT | |||||||
| SMBP-9 | 27.9 | 59.4 | 58.3 | WT | |||||||
| SMBP-10 | Jan 2021 | 29.4 | 59.3 | 58.3 | WT | 63.8 | 58.2 | WT | Yes | No | No |
| SMBP-11 | 39.1 | 59.9 | 58.6 | WT | |||||||
| SMBP-12 | 33.8 | 59.0 | 58.2 | WT | 64.3 | 58.8 | WT | Yes | No | No | |
| SMBP-13 | 17.7 | 59.3 | 58.5 | WT | 64.2 | 58.9 | WT | Yes | No | No | |
| SMBP-14 | 21.3 | 59.4 | 58.5 | WT | 64.2 | 58.8 | WT | Yes | No | No | |
| SMBP-15 | 17.5 | 59.1 | 58.3 | WT | 64.0 | 58.7 | WT | Yes | No | No | |
| SMBP-16 | 30.0 | 55.5 | 62.6 | MT | 64.2 | 58.5 | WT | No | Yes | No | |
| SMBP-17 | 41.2 | NP | NP | Neg | |||||||
| SMBP-18 | 23.2 | 59.8 | 58.9 | WT | 64.0 | 58.7 | WT | ||||
| SMBP-19 | 28.3 | 59.3 | 58.1 | WT | 63.8 | 58.2 | WT | Yes | No | No | |
| SMBP-20 | 17.1 | 59.2 | 58.3 | WT | 64.0 | 58.8 | WT | Yes | No | No | |
| SMBP-21 | 16.4 | 59.2 | 58.4 | WT | 64.2 | 58.9 | WT | Yes | No | No | |
| SMBP-22 | 18.2 | 58.9 | 58.1 | WT | Yes | No | No | ||||
| SMBP-23 | 21.1 | 58.9 | 58.1 | WT | 64.1 | 58.6 | WT | Yes | No | No | |
| SMBP-24 | 39.0 | 59.5 | 58.2 | WT | |||||||
| SMBP-25 | 28.4 | 54.5 | 61.6 | MT | 64.1 | NP | Ind | No | Yes | No | |
| SMBP-26 | Feb 2021 | 27.8 | 54.5 | 61.6 | MT | 63.9 | 58.3 | NP | No | Yes | No |
| SMBP-27 | 33.5 | 59.3 | 58.9 | WT | 60.1 | 63.0 | MT | Yes | No | Yes | |
| SMBP-28 | 38.3 | 59.7 | 58.5 | WT | 64.1 | NP | Ind | ND | ND | ND | |
| SMBP-29 | 18.1 | 58.9 | 58.1 | WT | 64.1 | 58.8 | WT | Yes | No | No | |
| SMBP-30 | 27.8 | 59.2 | 58.2 | WT | |||||||
| SMBP-31a | 17.6 | 55.1 | 61.9 | MT | 60.4 | 63.3 | MT | No | Yes | Yes | |
| SMBP-32a | 17.4 | 55.1 | 61.9 | MT | 64.0 | 58.7 | WT | No | Yes | No | |
| SMBP-33 | 32.8 | 55.0 | 62.1 | MT | |||||||
| SMBP-34 | 25.2 | 55.5 | 62.7 | MT | 60.2 | 63.3 | MT | No | Yes | Yes | |
| SMBP-35 | 18.4 | 55.6 | 62.5 | MT | 60.5 | 63.4 | MT | No | Yes | Yes | |
| SMBP-36 | 18.8 | 54.9 | 61.9 | MT | No | Yes | No | ||||
| SMBP-37 | 28.4 | 54.9 | 62.2 | MT | |||||||
| SMBP-38 | 16.4 | 59.8 | 58.5 | WT | 64.1 | 58.6 | WT | ||||
| SMBP-39 | 32.2 | NP | NP | Neg | |||||||
| SMBP-40 | 32.3 | 58.9 | 57.8 | WT | Yes | No | No | ||||
| SMBP-41 | Mar 2021 | 32.6 | 55.4 | 62.6 | MT | 63.9 | 58.3 | WT | No | Yes | No |
| SMBP-42 | 36.8 | 59.7 | 58.4 | WT | |||||||
| SMBP-43 | 30.1 | 59.8 | 58.9 | WT | Yes | No | No | ||||
| SMBP-44 | 33.3 | NP | NP | Inv | |||||||
| SMBP-45 | 34.6 | NP | 61.3 | Ind | |||||||
| SMBP-46 | 23.3 | 59.2 | 58.285 | WT | 64.0 | 58.6 | WT | Yes | No | No | |
IC, internal control; NP, no Tm peak; Neg, negative; ND, not determined; Inv, invalid; Ind, indeterminate. Gray cells indicate corresponding samples that were not tested by the SMB-484 assay or by sequencing due to insufficient clinical sample remaining.
Xpert Xpress SARS-CoV-2 or Xpert Xpress SARS-CoV-2/Flu/RSV tests.
Representative strains were sequenced. Sequencing was repeated twice to confirm the identified mutations.
FIG 2.
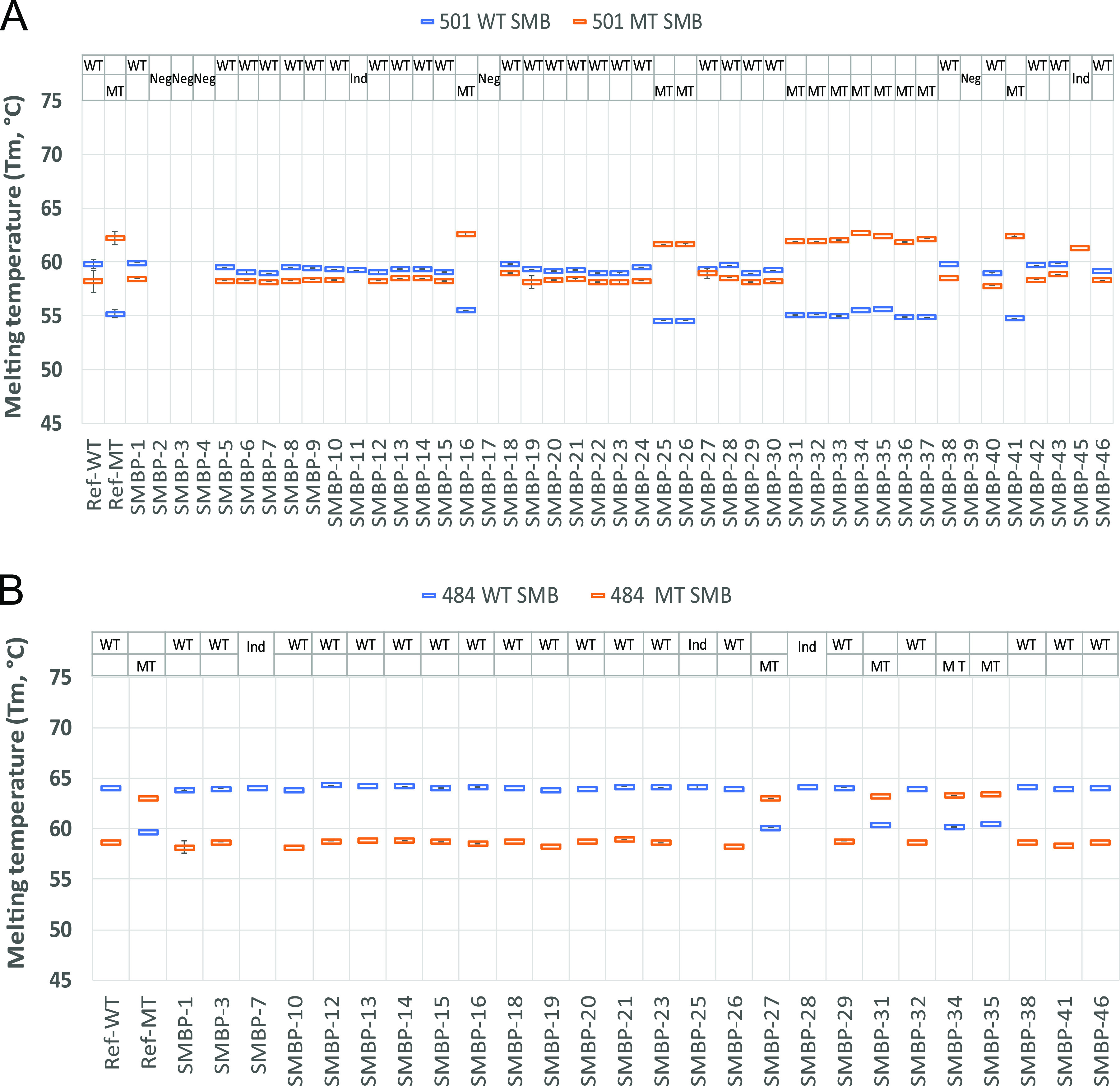
Sloppy molecular beacon (SMB) Tm profile of positive clinical nasopharyngeal (NP) samples tested using the SMB-501 and SMB-484 Tm assay. Tm signatures consisting of the Tm values for both the WT probe (blue) and MT probe (orange) are shown for the SMB-501 assay (A) and the SMB-484 assay (B). Ref WT indicates the Tm profile of the reference WT SARS-CoV-2 strain, and Ref MT indicates the Tm profile of the reference MT SARS-CoV-2 B.1.1.7 strain (A) and B.351 (B). SMBP1 to SMBP46 indicate that Tm profiles of the 46 COVID-positive clinical samples tested in this study with SMB-501 assay and the 26 COVID-positive samples tested by SMB-484 assay. Error bars show ± one standard deviation.
FIG 3.

Prevalence of N501Y variant strains among the tested sample set over time. The proportion of N501Y and E484K variants is shown for samples obtained during the periods of October to November (n = 9 tested with SMB-501/n = 3 tested with SMB-484), January (n = 16/n = 12), February (n = 15/n = 9), and the first week of March (n = 6/n = 2). No samples from December were tested in our study.
DISCUSSION
The emergence of SARS-CoV-2 VOC with the potential for increased transmission, disease severity, and resistance to vaccine-induced immunity is of grave concern (14, 29). A simple screening assay to monitor the emergence and spread of these strains may be helpful for implementing public health strategies to counter these and future strains. Our study demonstrates that our assay is simple, rapid, and sensitive and specific for detecting key variant-identifying mutations using a high-throughput PCR assay platform. Thus, this assay has the potential for relatively inexpensive high-throughput testing for rapid identification of N501Y, E484K variants. In designing this assay, we took advantage of the fact that the N501Y and E484K mutations are common in the major SARS-CoV-2 variants. Since this mutation also appears to be responsible for the increased infectivity and possibly the other adverse manifestations of these strains (4), assays that detect this mutation may also prove useful to detect any future strain that evolves to have increased transmission potential.
Our assay is the first, to our knowledge, that uses post-PCR Tm-based analysis to detect and differentiate SARS-CoV-2 variants using SMB probes demonstrated in clinical samples. This assay format has the benefit of producing a measurable Tm result irrespective of whether the SMB probe is fully complementary to its target nucleic acid sequence. Instead of detecting a mutation by either producing or not producing a signal, SMBs detect mutations by producing a Tm shift. Failure to produce a Tm signal indicates an invalid assay rather than the absence of a mutation. The robustness of our assay is further increased by our use of two different SMBs, one complementary to the WT sequence and one complementary to the mutant sequence. The pattern of Tm values or Tm signature produced by the combined Tm values of each SMB probe provides an unequivocal identification of a WT or MT sequence. We have also shown that Tm signatures can be used to detect mixtures of mutant and WT sequences and to identify numerous mutations present in an assay’s target region (29). Thus, we expect that our assay should continue to be able to identify N501Y and E484K variants even if additional mutations develop near this primary mutation within the probe-footprint region once the specific Tm signatures of each new genotype are characterized. Our assay is meant to be a screening assay that will identify samples likely to contain SARS-CoV-2 variants of concern. Although in this study we have proposed the use of a combination of two probes per assay for increased accuracy, we expect that most SARS CoV-2 variants can be identified using only the MT probe alone, and future versions of our assay are likely to use a single SMB to identify mutations that are less critical than N501Y and E484K. We suggest that a useful public health strategy would be to screen COVID-19-positive samples in near real time with our assay and then to perform genomic sequencing on a subset of screen-positive samples. This sequencing would confirm the presence of the expected variants and, in some cases, lead to the discovery of new variants. Ongoing sequencing of a subset of all SARS-CoV-2 samples will also be required to identify completely novel variants or to investigate the epidemiology and clinical characteristics of variants such as those recently reported in the United States. Fortunately, our assay is easily extensible, and additional tests for new key mutations can be added in a modular format to a screening panel when new mutations associated with critical new variants are discovered. In fact, it is our intention to continuously update this assay until our work is superseded by a better approach or much more widespread genome sequencing becomes commonplace. At the time of this assay development and manuscript preparation, additional strains originated in United States (epsilon) and India (delta) were not considered VOCs. We are currently optimizing and screening samples for these additional mutations. Key updates will also be posted on a preprint journal. In the meantime, we hope that our current test will help increase surveillance and potentially help control the spread of the new emerging variants of concern.
ACKNOWLEDGMENTS
This study was funded by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01 AI131617. D.A. receives research support and royalty payments from Cepheid, which sells the Xpert Xpress SARS-CoV-2 and Xpert Xpress SARS-CoV-2/Flu/RSV tests. S.C. is an employee of Cepheid, and R.J. is a paid consultant for Cepheid.
The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: genomic RNA from SARS-related coronavirus 2, isolate USA-WA1/2020, NR-52285. The following reagent was obtained through BEI Resources, NIAID, NIH: severe acute respiratory syndrome-related coronavirus 2, isolate hCoV-19/England/204820464/20200, NR-54000, contributed by Bassam Hallis.
We thank all the laboratories that contributed sequence data to the GISAID EpiCoV database. A GISAID acknowledgment table reporting the geographic origin and contributions of genomes analyzed in this study is included in the supplemental material as Table S1.
Footnotes
Supplemental material is available online only.
Contributor Information
Padmapriya Banada, Email: banadapp@njms.rutgers.edu.
David Alland, Email: allandda@njms.rutgers.edu.
Melissa B. Miller, UNC School of Medicine
REFERENCES
- 1.Galloway SE, Paul P, MacCannell DR, Johansson MA, Brooks JT, MacNeil A, Slayton RB, Tong S, Silk BJ, Armstrong GL, Biggerstaff M, Dugan VG. 2021. Emergence of SARS-CoV-2 B.1.1.7 lineage–United States, December 29, 2020–January 12, 2021. MMWR Morb Mortal Wkly Rep 70:95–99. doi: 10.15585/mmwr.mm7003e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lauring AS, Hodcroft EB. 2021. Genetic variants of SARS-CoV-2—what do they mean? JAMA 325:529–531. doi: 10.1001/jama.2020.27124. [DOI] [PubMed] [Google Scholar]
- 3.Lacobucci G. 2021. Covid-19: new UK variant may be linked to increased death rate, early data indicate. BMJ 372:n230. doi: 10.1136/bmj.n230. [DOI] [PubMed] [Google Scholar]
- 4.Santos JC, Passos GA. 2021. The high infectivity of SARS-CoV-2 B.1.1.7 is associated with increased interaction force between Spike-ACE2 caused by the viral N501Y mutation. bioRxiv doi: 10.1101/2020.12.29.424708. [DOI]
- 5.Callaway E, Mallapaty S. 2021. Novavax offers first evidence that COVID vaccines protect people against variants. Nature 590:17. doi: 10.1038/d41586-021-00268-9. [DOI] [PubMed] [Google Scholar]
- 6.CDC. 2021. Emerging SARS-CoV-2 variants. https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html.
- 7.Wu K, Werner AP, Moliva JI, Koch M, Choi A, Stewart-Jones GBE, Bennett H, Boyoglu-Barnum S, Shi W, Graham BS, Carfi A, Corbett KS, Seder RA, Edwards DK. 2021. mRNA-1273 vaccine induces neutralizing antibodies against spike mutants from global SARS-CoV-2 variants. bioRxiv doi: 10.1101/2021.01.25.427948. [DOI]
- 8.Greaney AJ, Loes AN, Crawford KHD, Starr TN, Malone KD, Chu HY, Bloom JD. 2021. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 29:463–476. doi: 10.1016/j.chom.2021.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grubaugh ND, Hodcroft EB, Fauver JR, Phelan AL, Cevik M. 2021. Public health actions to control new SARS-CoV-2 variants. Cell 184:1127–1132. doi: 10.1016/j.cell.2021.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golubchik T, Lythgoe KA, Hall M, Ferretti L, Fryer HR, MacIntyre-Cockett G, de Cesare M, Trebes A, Piazza P, Buck D, Todd JA, Fraser C, Bonsall D. 2021. Early analysis of a potential link between viral load and the N501Y mutation in the SARS-COV-2 spike protein. medRxiv doi: 10.1101/2021.01.12.20249080. [DOI] [Google Scholar]
- 11.Leung K, Shum MH, Leung GM, Lam TT, Wu JT. 2021. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance 26:2002106. doi: 10.2807/1560-7917.ES.2020.26.1.2002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nonaka CKV, Franco MM, Gräf T, de Lorenzo Barcia CA, de Ávila Mendonça RN, de Sousa KAF, Neiva LMC, Fosenca V, Mendes AVA, de Aguiar RS, Giovanetti M, de Freitas Souza BS. 2021. Genomic evidence of SARS-CoV-2 reinfection involving E484K spike mutation, Brazil. Emerg Infect Dis 27:1522–1524. doi: 10.3201/eid2705.210191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JC, Muecksch F, Rutkowska M, Hoffmann HH, Michailidis E, Gaebler C, Agudelo M, Cho A, Wang Z, Gazumyan A, Cipolla M, Luchsinger L, Hillyer CD, Caskey M, Robbiani DF, Rice CM, Nussenzweig MC, Hatziioannou T, Bieniasz PD. 2020. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 9:e61312. doi: 10.7554/eLife.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubin R. 2021. COVID-19 vaccines vs variants—determining how much immunity is enough. JAMA 325:1241–1243. doi: 10.1001/jama.2021.3370. [DOI] [PubMed] [Google Scholar]
- 15.Zhang W, Davis BD, Chen SS, Sincuir Martinez JM, Plummer JT, Vail E. 2021. Emergence of a novel SARS-CoV-2 variant in southern California. JAMA 325:1324–1326. doi: 10.1001/jama.2021.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, Planchais C, Porrot F, Robillard N, Puech J, Prot M, Gallais F, Gantner P, Velay A, Le Guen J, Kassis-Chikhani N, Edriss D, Belec L, Seve A, Péré H, Courtellemont L, Hocqueloux L, Fafi-Kremer S, Prazuck T, Mouquet H, Bruel T, Simon-Lorière E, Rey FA, Schwartz O. 2021. Reduced sensitivity of infectious SARS-CoV-2 variant B.1.617.2 to monoclonal antibodies and sera from convalescent and vaccinated individuals. bioRxiv doi: 10.1101/2021.05.26.445838. [DOI]
- 17.CDC. 2021. National genomic surveillance dashboard. CDC, Atlanta, GA. [Google Scholar]
- 18.Vogels CBF, Alpert T, Breban M, Fauver JR, Grubaugh ND. 2021. Multiplexed RT-qPCR to screen for SARS-COV-2 B.1.1.7 variants: preliminary results. https://virological.org/t/multiplexed-rt-qpcr-to-screen-for-sars-cov-2-b-1-1-7-variants-preliminary-results/588.
- 19.Ozsolak F, Milos PM. 2011. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Rincon A, Tonda A, Mendoza-Maldonado L, Mulders D, Molenkamp R, Perez-Romero CA, Claassen E, Garssen J, Kraneveld AD. 2021. Classification and specific primer design for accurate detection of SARS-CoV-2 using deep learning. Sci Rep 11:947. doi: 10.1038/s41598-020-80363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roh SS, Smith LE, Lee JS, Via LE, BarryCE, III, Alland D, Chakravorty S. 2015. Comparative evaluation of sloppy molecular beacon and dual-labeled probe melting temperature assays to identify mutations in Mycobacterium tuberculosis resulting in rifampin, fluoroquinolone and aminoglycoside resistance. PLoS One 10:e0126257. doi: 10.1371/journal.pone.0126257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emery SL, Erdman DD, Bowen MD, Newton BR, Winchell JM, Meyer RF, Tong S, Cook BT, Holloway BP, McCaustland KA, Rota PA, Bankamp B, Lowe LE, Ksiazek TG, Bellini WJ, Anderson LJ. 2004. Real-time reverse transcription-polymerase chain reaction assay for SARS-associated coronavirus. Emerg Infect Dis 10:311–316. doi: 10.3201/eid1002.030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wyllie AL, Fournier J, Casanovas-Massana A, Campbell M, Tokuyama M, Vijayakumar P, Warren JL, Geng B, Muenker MC, Moore AJ, Vogels CBF, Petrone ME, Ott IM, Lu P, Venkataraman A, Lu-Culligan A, Klein J, Earnest R, Simonov M, Datta R, Handoko R, Naushad N, Sewanan LR, Valdez J, White EB, Lapidus S, Kalinich CC, Jiang X, Kim DJ, Kudo E, Linehan M, Mao T, Moriyama M, Oh JE, Park A, Silva J, Song E, Takahashi T, Taura M, Weizman OE, Wong P, Yang Y, Bermejo S, Odio CD, Omer SB, Dela Cruz CS, Farhadian S, Martinello RA, Iwasaki A, Grubaugh ND, et al. 2020. Saliva or nasopharyngeal swab specimens for detection of SARS-CoV-2. N Engl J Med 383:1283–1286. doi: 10.1056/NEJMc2016359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elbe S, Buckland-Merrett G. 2017. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall 1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.SantaLuciaJ, Jr.1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci U S A 95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3–new capabilities and interfaces. Nucleic Acids Res 40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson G, Buzko O, Spilman P, Niazi K, Rabizadeh S, Soon-Shiong P. 2021. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y.V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv doi: 10.1101/2021.01.13.426558. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download JCM.00845-21-s0001.xlsx, XLSX file, 11.8 MB (11.8MB, xlsx)
Fig. S1. Download JCM.00845-21-s0002.pdf, PDF file, 0.6 MB (624.7KB, pdf)