

ABSTRACT
Homologous recombination requires the coordinated effort of several proteins to complete break resection, homologous pairing, and resolution of DNA crossover structures. RecN is a conserved bacterial protein important for double-strand break repair and is a member of the structural maintenance of chromosomes (SMC) protein family. Current models in Bacillus subtilis propose that RecN responds to double-stranded breaks prior to RecA and end processing, suggesting that RecN is among the very first proteins responsible for break detection. Here, we investigate the contribution of RecA and end processing by AddAB to RecN recruitment into repair foci in vivo. Using this approach, we found that recA is required for RecN-green fluorescent protein (GFP) focus formation on the nucleoid during normal growth and in response to DNA damage. In the absence of recA function, RecN foci form in a low percentage of cells, RecN localizes away from the nucleoid, and RecN fails to assemble in response to DNA damage. In contrast, we show that the response of RecA-GFP foci to DNA damage is unchanged in the presence or absence of recN. In further support of RecA activity preceding RecN, we show that ablation of the double-strand break end-processing enzyme addAB results in a failure of RecN to form foci in response to DNA damage. With these results, we conclude that RecA and end processing function prior to RecN, establishing a critical step for the recruitment and participation of RecN during DNA break repair in Bacillus subtilis.
IMPORTANCE Homologous recombination is important for the repair of DNA double-strand breaks. RecN is a highly conserved protein that has been shown to be important for sister chromatid cohesion and for surviving break-inducing clastogens. Here, we show that the assembly of RecN into repair foci on the bacterial nucleoid requires the end-processing enzyme AddAB and the recombinase RecA. In the absence of either recA or end-processing RecN-GFP, foci are no longer DNA damage inducible, and foci form in a subset of cells as large complexes in regions away from the nucleoid. Our results establish the stepwise order of action, where double-strand break end processing and RecA association precede the participation of RecN in break repair in Bacillus subtilis.
KEYWORDS: double-strand break repair, end processing, RecA, RecN, AddAB, Bacillus subtilis
INTRODUCTION
All cells are constantly subject to exogenous and endogenous stressors that result in DNA damage (1). DNA strand breaks are a particularly deleterious type of damage that can result from exogenous sources, including ionizing radiation or endogenous reactive oxygen species, that result in strand breaks when replication forks encounter nicks in the template strand (2). DNA strand breaks are repaired by several different pathways (3–5). The choice between repair pathways depends on a number of factors, including the organism, growth phase, and type of break produced (4, 6). In bacteria, DNA strand breaks are repaired by three pathways, homologous recombination, single-strand annealing (SSA), and nonhomologous end joining (NHEJ) (7–11).
In the Gram-positive bacterium Bacillus subtilis, double-strand break repair (DSBR) occurs by homologous recombination and NHEJ (10, 12). During DSBR by homologous recombination, the break is processed by the dual-function helicase-nuclease enzyme AddAB, resulting in end resection and a 3′ single-strand DNA (ssDNA) extension after AddAB encounters a chi site (13–17). The 3′ overhang provides a substrate for RecA binding and nucleoprotein filament formation. In B. subtilis, RecO helps load RecA in vivo after AddAB-dependent end processing, because AddAB does not have a RecA loading function (9). RecA then catalyzes strand invasion, pairing the 3′ ssDNA with the homologous sequence of a sister chromatid (for a review, see references 18 and 19). During DSBR, a double-crossover intermediate forms, generating a Holliday junction, which is subsequently cleaved by Holliday junction endonuclease RecU or MutS2, repairing and restoring the information lost at the break site (20, 21). Even though there is much more to be learned, each step of homologous recombination is fairly well understood in B. subtilis as well as in many other organisms, ranging from bacteria to mammalian cells (for a review, see references 1, 22, and 23).
Structural maintenance of chromosome (SMC) proteins are conserved throughout biology and function in several processes, including chromosome cohesion, chromosome condensin, dosage compensation, and DSBR (24–33). B. subtilis contains three SMC-like proteins: SMC, involved in organizing and condensing the right and left arms of the chromosome near the replication origin, RecN, a protein involved in DSBR, and YhaN (SbcE), a protein providing a minor contribution to some aspects of DSBR (27, 30, 31, 34). For both RecN and SbcE, their contribution to DSBR is through homologous recombination, and these proteins have no known role in NHEJ. B. subtilis cells deficient for sbcE are sensitive to DNA damage, including a site-specific DSB, but they do not share the same extent of sensitivity as observed with recN-deficient cells (27). Therefore, of the B. subtilis proteins, RecN is the best characterized, although its contribution to DSBR remains unclear.
In Escherichia coli, recN expression is highly induced as part of the SOS regulon to DNA damage, while in B. subtilis RecN is not part of the SOS response, although RecN protein levels do increase slightly following DNA damage (35, 36). Both E. coli and B. subtilis RecN have been shown to assemble into foci in response to DSBs (37, 38). E. coli recN was assayed following expression from an inducible promoter using an N-terminal green fluorescent protein (GFP) fusion (37). This study showed that the plasmid-borne gfp-recN allele was functional when overexpressed and formed large complexes in the cell away from the nucleoid in nearly 100% of cells (37). Following DNA damage with mitomycin C (MMC), approximately 50% of cells showed GFP-RecN foci coincident with the nucleoid (37). Further, the redistribution of RecN from cytoplasmic to nucleoid localized required recA, leading to the conclusion that RecA recruits RecN (37). This work further identified a RecA variant, Q300A, that was proficient for SOS induction but failed to recruit RecN and was as sensitive to MMC as the ΔrecN strain, supporting the notion that RecA recruits RecN through a physical interaction (37). Subsequent genetic characterization of ΔrecN and recAQ300A showed that these alleles are not epistatic and are instead additive for defects in recombination and repair of an I-SceI site-specific DSB, arguing against the conclusion that the recAQ300A allele causes a defect in RecN recruitment (39).
As mentioned above, RecN is an SMC-like family protein (40). It has been shown in eukaryotes that sister chromatid cohesion provides an important contribution to DSBR (41). Recent evidence in E. coli establishes RecN as being important for sister chromatid cohesion during DSBR (42). This work also presented coimmunoprecipitation results showing that RecA physically interacts with RecN and that RecA is required for loading RecN in response to MMC-induced damage (42). The RecAQ300A variant was also found to have no effect on RecN-mediated condensation activity, further indicating that RecAQ300A did not ablate a RecN interaction site on RecA (42). Therefore, in E. coli, RecN functions to maintain sister chromatid cohesion, allowing for efficient homology search by RecA during DSBR. Interestingly, Deinococcus radiodurans RecN has been shown to stimulate RecA-mediated strand exchange to form a D-loop and a nicked circular duplex, while RecA has been shown to stimulate the DNA-dependent ATPase activity of RecN (43). This work has also shown that Deinococcus RecN provides an additional role in promoting DNA strand exchange separate from its function in cohesion (43). Collectively, RecN functions after RecA binding, contributing to DSBR by maintaining sister chromatid cohesion and contributing to strand exchange in E. coli and D. radiodurans.
Current models in B. subtilis suggest that RecN is the first protein to act during the process of DSBR (44). In B. subtilis, RecN has been assayed when expressed from its native promoters at its normal chromosomal location with a C-terminal fusion to yellow fluorescent protein (YFP) (38, 45). In these studies, RecN-YFP localization was shown to be independent of end processing and recA (38, 45). This work leads to the conclusion that RecN organizes into a single repair complex gathering DSBs before end processing by AddAB or RecA binding, suggesting that RecN is the first known protein to act in this process (38, 44, 45). Further, this work showed that RecN-YFP foci were faint and formed foci in a small subset of cells, a result that disagrees with the observations using E. coli GFP-RecN (37, 38, 45). The differences observed could be due to a number of factors, including N- versus C-terminal fusions, plasmid relative to chromosomal expression, and certain species-specific differences in biochemical activity between RecA and RecN from the different organisms studied.
To understand how RecN organizes during DSBR in B. subtilis, we constructed N- and C-terminal fluorescent fusions to RecN expressed from its normal chromosomal position under the control of its native promoters. We show that the N-terminal RecN fusion is nonfunctional, while the C-terminal RecN fusion behaves similarly to the wild type. Through imaging repair complex formation, we show that RecA and end processing are required for the formation of DNA damage-inducible RecN-GFP foci, demonstrating the concerted order of participation during DSBR. Further, our work demonstrates that in the absence of recA and end processing, RecN-GFP foci form larger inactive complexes away from the nucleoid, suggesting that in the absence of RecA, the RecN protein is rendered unresponsive to DNA damage and unable to load onto DNA. Our work underscores the sequential order of action during DSBR and establishes RecA and end processing as critical components important for the recruitment of RecN in response to DNA damage in live cells.
RESULTS
AddAB is responsible for the majority of end resection in vivo.
During DSBR, the ends are resected, generating 3′ ssDNA tails at chi sites during a procedure referred to as end processing or end resection (46, 47). The 3′ ssDNA ends serve as suitable substrates for RecA binding and nucleoprotein filament formation (48). It has previously been shown in B. subtilis that AddAB is critical for end resection in vitro and in vivo (13, 46, 49). Other evidence in B. subtilis suggests that recJ exonuclease, in combination with DNA helicase RecQ, provides end resection at stalled replication forks (38). To understand the contribution of AddAB and RecJ to end processing, we performed an assessment of cell survival following exposure to mitomycin C (MMC). MMC was chosen because it indirectly results in double-strand breaks (DSBs) and causes replication fork stalling, providing a broad and effective method to sample the importance of AddAB and RecJ to end resection in vivo (9, 50).
We show that cells disrupted for addA or addB showed the same percent survival, which was expected given their gene products form an active enzyme (16, 17) (Fig. 1). Importantly, we show that, in an otherwise wild-type background, recJ provides only a minor contribution to cell survival following MMC exposure. In B. subtilis, RecO is responsible for loading RecA regardless of the type of lesion encountered (9). In support of this, we show that cells deficient for recO are highly sensitive to DNA damage, with a sensitivity greater than that of a single end-processing mutant (9) (Fig. 1). Further, we show that combining an addA deficiency with a recJ deletion further sensitized B. subtilis cells, providing evidence that in the absence of addA, recJ provides some overlapping function (Fig. 1). Importantly, cells deficient for addA and recJ showed approximately the same percent survival as a recO deficiency, further supporting the model that, in B. subtilis, RecO is required for RecA loading following end resection during DSBR (9). The recA-deficient strain was even more sensitive, likely due to its role in SOS induction (Fig. 1). We conclude that in B. subtilis, AddAB is responsible for the vast majority of end resection with RecJ, providing a minor overlapping contribution.
FIG 1.

AddAB is responsible for the majority of end processing in vivo. Shown is a killing curve of various recombination deficient mutants in B. subtilis to mitomycin C. Survival is relative to untreated wild-type cells. The error bars indicate standard errors of the means (SEM) from at least three independent measurements.
RecN is functional with a C-terminal fluorescent fusion.
Current models in B. subtilis suggest that RecN is the earliest acting protein during DSBR and that RecN functions before RecA and end processing (44, 45). Further, it has been shown biochemically that B. subtilis RecN can bind and protect 3′ ssDNA tails, supporting the model that RecN acts prior to RecA (51). The subcellular localization of RecN fused to GFP or YFP has been examined in E. coli and B. subtilis (37, 38, 45). In E. coli, a functional N-terminal fusion to plasmid-expressed GFP-RecN showed RecN forms large complexes away from the nucleoid (37). In B. subtilis, RecN-YFP showed small complexes in a subset of cells that were concluded to form independent of RecA and end processing (38).
To understand the subcellular localization of RecN and choreography during DSBR, we began by building N- and C-terminal GFP fusions to assess their functionality. Each fusion was expressed from the recN native chromosomal location under the control of the native promoters to prevent any localization patterns that could be symptomatic of plasmid-borne expression. Our fusion constructs include monomeric gfpmut3 with a 22- or 23-amino-acid flexible linker for the N-terminal and C-terminal fusions, respectively. We tested strains with gfp-recN, recN-gfp, and an recN-deficient strain as a control in spot titer assays following UV challenge, the break-inducing peptide phleomycin, or the cross-linking agent MMC (Fig. 2). We found that recN-deficient cells were sensitive to phleomycin and MMC but not UV, further supporting the model that RecN is important for DSBR. Interestingly, we found that the gfp-recN allele was indistinguishable from the recN-deficient control (recN::tet). Further, the recN-gfp allele showed the same survival as the wild-type control on MMC and phleomycin (Fig. 2). With these data, we conclude that gfp-recN is nonfunctional and recN-gfp shows nearly wild-type levels of survival to DNA damage, providing an appropriate fusion to determine the genetic requirements supporting RecN-GFP focus formation.
FIG 2.
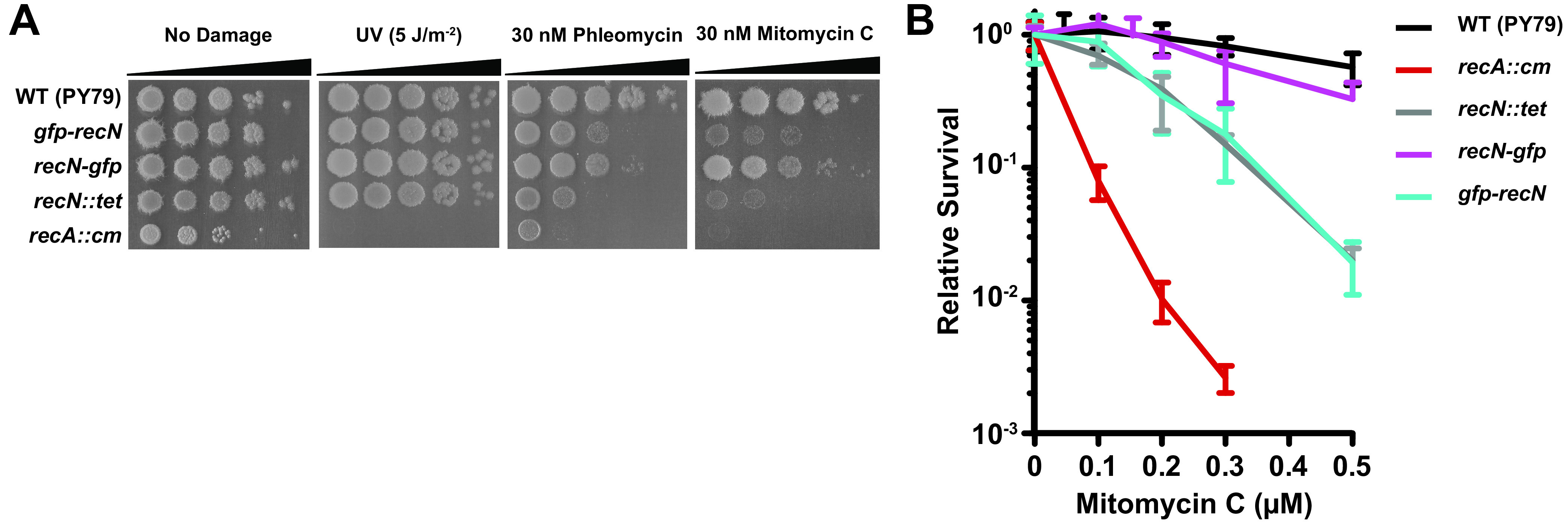
RecN is functional with a C-terminal GFP fusion. (A) Spot titer plates with the indicated strains challenged with UV irradiation, phleomycin, or mitomycin C. Each spot was serially diluted 10-fold. (B) Killing curve of the indicated strain to increasing concentrations of mitomycin C. Relative survival indicates survival compared to untreated wild type. Error bars represent SEM from at least three independent measurements.
RecN-GFP forms damage-inducible foci on the bacterial nucleoid.
To understand the subcellular localization of RecN and to provide insight into the differences that were observed between the E. coli GFP-RecN and the B. subtilis RecN-YFP fusions, we assayed and quantified the subcellular localization of the B. subtilis GFP-RecN and RecN-GFP fusions. For the GFP-RecN fusion in untreated cells, we observed robust foci that formed in approximately 38.8% of cells with 4.3% of foci coincident with the nucleoid (n = 616) (Fig. 3A). In cells challenged with MMC, we found that GFP-RecN formed foci in 37.6% of cells, with 4.7% (n = 782) of foci coincident with the nucleoid.
FIG 3.

RecN-GFP foci are damage inducible and form foci on the nucleoid. (A) Shown are representative micrographs of GFP-RecN or RecN-GFP foci untreated or following challenge with mitomycin C. The cell membrane is stained with FM 4-64 and pseudocolored blue, the nucleoid is stained with DAPI and pseudocolored magenta. Bars, 4 μm. (B) The graph shows the percentage of cells with foci in each category. The cyan color reflects the fraction of foci that were nucleoid associated, while the black bars represent the fraction of foci away from the nucleoid. Conditions with mitomycin C (MMC) are indicated, and the error bars reflect the 95% confidence interval. For RecN-GFP, n = 958 and n = 1,077 cells scored untreated or challenged with MMC, respectively. For GFP-RecN, n = 616 and n = 782 cells were scored for untreated or cells challenged with MMC, respectively. (C) The focus intensity was determined using an automated program in Fiji with the MicrobeJ plugin. For RecN-GFP and GFP-RecN, the number of foci analyzed was the following: RecN-GFP, n = 798; GFP-RecN, n = 495. The distribution of focus intensity was graphed as a violin plot with the dotted center line representing the median and the solid lines representing the 25th and 75th percentiles. The difference between the medians was significant, with a P value of <0.001 using a two-tailed t test. The foci analyzed were from at least two independent experiments.
These results show that GFP-RecN foci assembled in areas of the cell away from the nucleoid. Further, the foci were not damage inducible, and we did not observe GFP-RecN foci redistribute to the nucleoid following DNA damage (Fig. 3B; see also Fig. S1 in the supplemental material). Most of the results we observe with the GFP-RecN fusion were similar to those for the E. coli GFP-RecN fusion, with the exception that the E. coli protein showed damage-inducible relocalization (37). It should be noted that the B. subtilis gfp-recN strain was indistinguishable from an recN-deficient strain for growth in the presence of DNA damage and that the gfp-recN allele assayed in E. coli was overexpressed from a plasmid using an inducible promoter (37) as opposed to the fusion allele used here. Based on our genetic and subcellular localization results, we conclude that GFP-RecN in B. subtilis forms a nonfunctional inactive complex that is unable to participate in DSBR.
In contrast to GFP-RecN, RecN-GFP formed foci in about 7.3% of cells, with 1.6% (n = 958) of foci coincident with the nucleoid when grown in the absence of DNA damage. RecN-GFP then formed foci in 56.9% of cells, with 50.6% (n = 1,077) of foci coincident with the nucleoid following DNA damage with MMC (Fig. 3). This result shows that the vast majority of DNA damage-inducible foci were coincident with the nucleoid (Fig. 3), suggesting that the RecN-GFP foci are participating in DSBR. The foci that formed were qualitatively smaller, and the fluorescence intensity was significantly reduced compared with that of the GFP-RecN foci (Fig. 3A and C; Fig. S1). Prior studies characterizing RecN-YFP reported that 0.05% of undamaged cells showed foci while 70% of cells showed foci following challenge with MMC (45). The percentage of cells we observe to have RecN-GFP foci in untreated cells is inconsistent with the prior report; however, the percentage of cells we observe with RecN-GFP foci is similar to that of the prior report following damage with MMC (45).
During the DNA damage response, RecN forms multiple repair centers on the nucleoid.
Prior work characterizing B. subtilis RecN-YFP suggested that RecN forms a limited number of “repair centers” per cell regardless of the DNA damage dose used (45). We quantified the number of RecN-GFP foci per cell and found that following DNA damage, RecN-GFP forms as many as 4 foci (repair centers) per cell, with most cells forming one or two foci per cell (Fig. 4). Prior work has suggested that RecN is capable of gathering several DSBs into one repair center (45). Our data are more supportive of a model where RecN can assemble at several sites of DNA damage to help facilitate chromosome cohesion and DSBR at multiple points along the nucleoid. Further, based on our results, we suggest that RecN is unlikely to coalesce several DSBs into one large repair center within B. subtilis; instead, RecN is more likely to help facilitate repair after it is recruited to DNA breaks.
FIG 4.
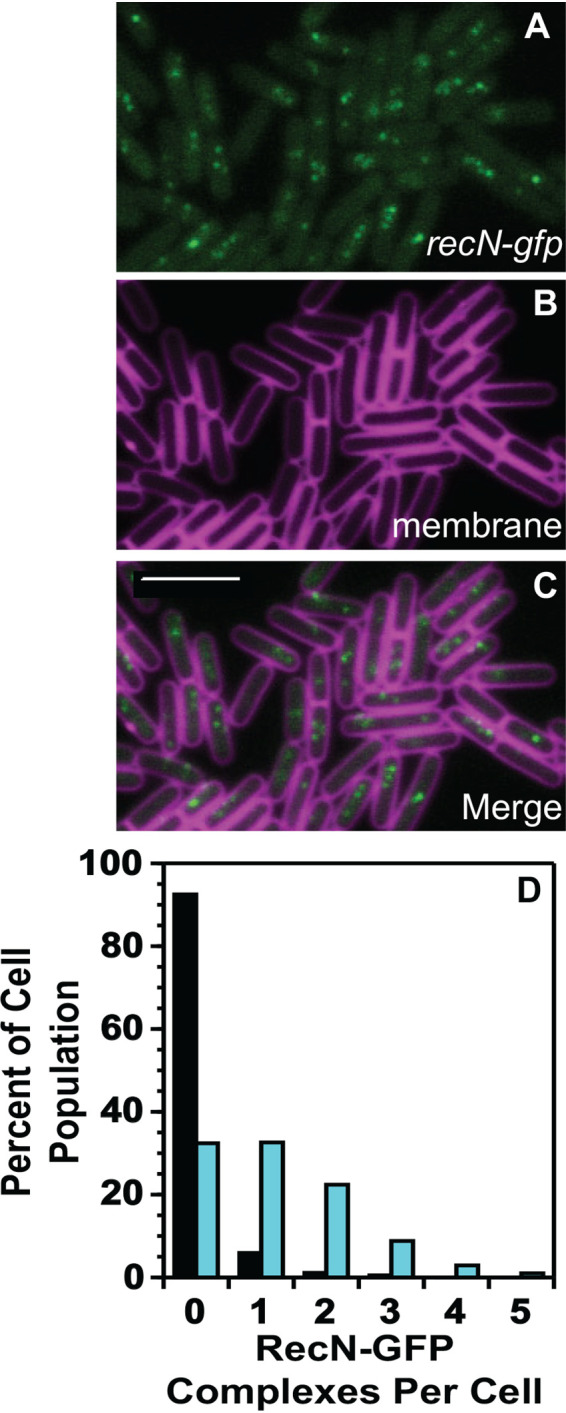
RecN-GFP forms multiple repair complexes on the nucleoid per cell. (A to C) Representative images of RecN-GFP foci, the cell membrane imaged with TMA-DPH pseudocolored magenta, and the merged imaged. The bar indicates 4 μm. (D) Number of foci per cell for untreated cells (black bars) and cells challenged with mitomycin C (cyan bars). The number of cells scored for untreated and mitomycin C treatment were over 900 cells per condition.
RecA is required for efficient RecN-GFP damage-inducible foci on the nucleoid.
Current models for DSBR in B. subtilis suggest that RecN acts early in repair and upstream of RecA and end processing (38, 44, 45). Cytological studies in E. coli show that the redistribution of GFP-RecN foci to the nucleoid is dependent on the recA gene (37), yet genetic studies show that mutations in recN and recA are genetically separable (39). Therefore, we asked if RecN-GFP foci were altered in cells with a recA-deficient allele (recA::spc) (Fig. 5). We found that although the percentage of cells with RecN-GFP foci increased slightly after DNA damage (8.4% [n = 1,008 cells scored] to 13.2% [n = 1,336 cells scored]) in the absence of recA, the RecN-GFP subcellular localization response to DNA damage was almost entirely lost compared with a wild-type recA strain. Further, we find that the foci that do form are located away from the nucleoid, showing the same result as our findings with the nonfunctional GFP-RecN fusion (compare Fig. 3B and 5B).
FIG 5.

RecA is required for the formation of RecN-GFP foci on the nucleoid in response to DNA damage. (A) Representative micrographs of B. subtilis cells with RecN-GFP and in cells deficient for recA left untreated or challenged with mitomycin C. Cell membranes were stained with FM 4-64 (pseudocolored blue), and the nucleoid was imaged with DAPI (pseudocolored magenta). (B) Bar graph shows the percentage of cells with foci that were nucleoid associated (cyan) or off the nucleoid (black) in the presence or absence of DNA damage in cells deficient for recA. Error bars represent the 95% confidence interval. For the untreated condition, n = 1,008, and for MMC treatment, n = 1,336 for cells scored. (C) Shown is a graph with the percentage of cells with RecA-GFP foci challenged with mitomycin C or phleomycin that were wild type for recN or carrying the recN::cm allele. The number of cells scored for each condition are the following: for recA-GFP and recN::cm untreated, n = 1,211; for 40 nM MMC, n = 924; for 80 nM MMC, n = 301; for 300 nM phleomycin, n = 446; for recA-GFP and wild-type recN, untreated n = 479; 40 nM MMC, n = 547; for 80 nM MMC, n = 340; for 300 nM phleomycin, n = 347. Error bars represent the 95% confidence intervals.
The simplest explanation for our results is that the organization of RecN-GFP into foci on the nucleoid is recA dependent. If RecA acts before RecN, then we would expect the percentage of cells with RecA-GFP foci to remain unchanged in the presence or absence of a functional recN gene. Indeed, we show that the percentage of cells with RecA-GFP foci were unchanged between wild-type and recN-deficient cells, demonstrating that the organization of RecA-GFP into foci is independent of recN, yet the organization of RecN-GFP into foci on the nucleoid is strictly dependent on recA. We conclude that RecN acts downstream of RecA, providing their order of action in B. subtilis.
End processing is required for RecN-GFP to form DNA damage-inducible repair centers.
Our data show that recA is required for RecN to form damage-inducible foci that are coincident with the nucleoid (Fig. 3B). Because we find that RecN-GFP foci are recA dependent, we hypothesize that RecN foci will also be dependent on end resection, because end resection is necessary for RecA to form nucleoprotein filaments on ssDNA (52). Therefore, we examined RecN-GFP foci in cells deficient for AddAB by testing the effect of addA::erm or addB::erm, separately ablating the major end-processing pathway in B. subtilis (13, 38) (Fig. 1). We find that cells lacking addA or addB showed RecN-GFP focus formation in ∼11% (n = 416) and 13% (n = 444) of untreated cells, respectively. Thus, the percentage of cells with RecN-GFP foci was initially elevated in cells lacking addA or addB relative to the wild type. Importantly, however, RecN-GFP foci were no longer DNA damage inducible in the absence of end processing by AddAB, showing the same overall result we find in the absence of recA (Table 1, bottom two rows). In the absence of recA, RecN-GFP foci only formed in 13% of damaged cells (Fig. 5), the same effect we observe following DNA damage in the absence of end processing using a deficient addA or addB allele with foci forming in ∼12.7% (n = 431) and ∼12.0% (n = 516) of cells, respectively (Table 1). Qualitatively, we did notice that the RecN-GFP foci that formed in the absence of end processing were larger and formed away from the cell center (Fig. S2). Because the larger RecN-GFP foci are not damage inducible, these results indicate that the ∼12% of RecN-GFP complexes that form in cells lacking AddAB appear to represent inactive complexes, mirroring the localization pattern observed for RecN-GFP in the absence of recA or when using the nonfunctional N-terminal GFP fusion. With these results, we conclude that RecN-GFP focus formation in the absence of end processing represents coalesced RecN protein that does not contribute to DSBR in B. subtilis.
TABLE 1.
RecA and AddAB are important for RecN to form DNA repair complexes
| Genotype | % foci formed (no. of cells) |
|
|---|---|---|
| Endogenous | Mitomycin C treated | |
| recA-gfp | 10.6 (795) | 67.3 (800) |
| recA-gfp addA::erm | 3.5 (202) | 32.6 (306) |
| recA-gfp recJ::erm | 6.4 (287) | 40.7 (226) |
| recA-gfp addA::erm ΔrecJ | 2.6 (267) | 25.4 (193) |
| recN-gfp | 7.7 (456) | 28.7 (324) |
| recN-gfp addA::erm | 11.3 (416) | 12.8 (431) |
| recN-gfp addB::erm | 13.7 (444) | 12.0 (516) |
DISCUSSION
RecN is a highly conserved SMC-like bacterial protein that is critical for homologous recombination (39, 42, 49, 53–55). Discovered nearly 40 years ago (49, 55–57), the contribution of RecN to DSBR has been clear, although the mechanistic contribution and the step when RecN contributes has remained under investigation. In E. coli, recN-deficient cells are sensitive to ionizing radiation, MMC, and a DSB induced by the I-SceI homing endonuclease (39, 42, 54). Further, E. coli recN-deficient strains show wild-type levels of DNA degradation during DSBR, suggesting normal function of RecBCD (54). Importantly, cells lacking recN are more strongly sensitized when two or three I-SceI-induced breaks occur in the chromosome (54). In B. subtilis, similar results have been shown, as recN-deficient cells are sensitive to MMC (45) (Fig. 1), the break-inducing peptide phleomycin, and 4-nitroquinoline 1 oxide (4-NQO) (49) but not UV-induced damage (Fig. 1). Further, recN-deficient B. subtilis cells show no defect in transformation with plasmid DNA and are 80% as efficiently transformed with chromosomal DNA as wild-type cells (49). Taken together, the current sensitivity data in E. coli, Bacillus, D. radiodurans, and C. crescentus suggest that RecN has a conserved contribution to DSBR (39, 54, 58, 59).
The major goal of this investigation is determining the concerted order of action between RecA, end processing (AddAB), and RecN. Current models of B. subtilis suggest that RecN-YFP foci form independently of both recA and DSB end processing (60), while in D. radiodurans RecN functions in vitro after RecA (43). The important findings from our work in B. subtilis are that an N-terminal fusion to RecN inactivates the protein, while a functional C-terminal fusion requires both RecA and end processing to assemble into multiple foci on the nucleoid in response to DNA damage. We provide quantitative evidence showing that in the absence of recA or end processing by AddAB, RecN-GFP foci are no longer DNA damage inducible. Based on our results, we conclude that during DSBR, RecN functions after end processing and RecA nucleoprotein filament formation on ssDNA.
The results we present here with RecN-GFP differ, in part, from prior studies using a RecN-YFP fusion protein (38). The RecN-GFP fusion used in this study is monomeric gfpmut3, which has been shown to be one of the most monomeric fluorescent proteins available (61). In contrast, YFP has a strong tendency to form dimers (62). In addition, we used an extended flexible linker, which we have shown previously contributes to the functionality of fusion proteins (63). Previous work showing that RecN-YFP forms foci in the absence of recA or end processing in part agrees with our findings, as we also show RecN-GFP is capable of forming foci in the absence of recA and addA or addB. The difference is that in our experiments, RecN-GFP foci fail to respond to DNA damage and generally form near the cell periphery when recA or end processing (addAB) are nonfunctional. The differences between our work and the prior study (38) could be a reflection of the fluorescent proteins used in each study.
Does RecA bind RecN and recruit it during DSBR? One study identified a recA mutant (recAQ300A) proposed to define the site of interaction between RecA and RecN (37). More recent studies have shown that recAQ300A has no effect on RecN function in E. coli (39, 42). RecN-Flag has been shown to coimmunoprecipitate E. coli RecA and vice versa following MMC challenge, suggesting a physical interaction occurs between these proteins during repair. Further, in D. radiodurans it was shown that RecA and RecN can stimulate activity of the other protein in a defined system (42, 43). It was also shown that D. radiodurans RecA and RecN interact directly in the presence or absence of DNA and that the stimulatory effect of RecN on RecA required D. radiodurans RecA, suggesting a species-specific interaction (43). Therefore, there is indeed strong evidence that RecA physically interacts with RecN, although a discrete site on either protein has yet to be identified to mediate their interaction and facilitate the recruitment of RecN in vitro or in vivo. Given that RecN is critical for repair, we speculate that RecN also recognizes a DNA structure that forms during repair, contributing to its recruitment to break sites by RecA.
An important difference between E. coli and B. subtilis is the regulation of recN expression. In E. coli, recN is part of the SOS regulon (57, 64). Expression of recN is low, with high expression induced following SOS induction (64). In contrast, B. subtilis recN is not under SOS control (35, 65). B. subtilis RecN protein levels do increase slightly following DNA damage, although the increase in RecN is independent of SOS regulation (36). Our work here shows that RecN forms foci in a small percentage of untreated cells away from the nucleoid. In otherwise wild-type cells, DNA damage causes RecN to increase focus formation on the nucleoid, indicating a damage-dependent relocalization of RecN. Importantly, we show that recA and addAB are required for the DNA damage-dependent induction of RecN-GFP focus formation to occur. We suggest that RecN is not under SOS control in B. subtilis because it is able to undergo a redistribution to the nucleoid in response to DNA damage and does not require much of an increase in protein abundance to participate in DSBR. We further suggest that the large RecN-GFP complexes we observe at the cell periphery in the absence of recA or addAB represent inactive RecN complexes. Our data show that RecN is unable to redistribute to the bacterial nucleoid in response to DNA damage and participate in homologous recombination in the absence of recA and addAB, suggesting we have ablated a RecN loading pathway yielding “dead end” RecN complexes.
In E. coli, following DNA replication, sister loci remain coincident and do not separate for 10 to 20 min (66). Sister chromatid cohesion in E. coli is regulated by topoisomerase IV (Topo IV) (66). Recombination in E. coli cells deficient for recN can be rescued by inactivation of Topo IV using a temperature-sensitive allele (parEts) (42). This work showed that by conditionally inactivating Topo IV to maintain precatenanes and, thus, sister chromatid cohesion, recombination in a recN mutant can be restored (42). This work considered with the finding that recN mutants are error-prone in recombination suggests that sister loci could misalign, leading to errors and reduced fidelity during DSBR (54). These studies, considered with our own findings showing that RecA and AddAB are required for RecN to assemble on the nucleoid, suggest that RecN contributes to DSBR after RecA by helping to maintain sister chromatid cohesion. Such a role by RecN would allow for an efficient homology search during DSBR. In the absence of recN, sister chromatids would segregate early, impairing RecA-mediated homology search and reducing the fidelity of DSBR. Such an effect might not be as critical for repair of a single DSB; however, repair of multiple site-specific DSBs or multiple breaks caused by chemical damage would reveal the importance of RecN in mediating cohesion (42, 54). Thus, just like DSBR in eukaryotes, a critical feature in bacterial repair is the involvement of an SMC-like condensin to aid in the homology search of the intact sister contributing to repair and the overall fidelity of the process.
MATERIALS AND METHODS
Basic bacteriology.
All strains used in this study were derivates of the laboratory strain PY79 (67) and are listed in Table S1 in the supplemental material. The media used were lysogeny broth (68) (LB) (10 g/liter tryptone, 10 g/liter NaCl, and 5 g/liter yeast extract) and S750 minimal medium (1× S750 salts from 10× S750 salts stock [104.7 g/liter morpholinepropanesulfonic acid, 13.2 g/liter, ammonium sulfate, 6.8 g/liter monobasic potassium phosphate, pH 7.0, adjusted with potassium hydroxide], 1× metals diluted from 100× metals stock [0.2 M MgCl2, 70 mM CaCl2, 5 mM MnCl2, 0.1 mM ZnCl2, 100 μg/ml thiamine-HCl, 2 mM HCl, 0.5 mM FeCl3], 0.1% potassium glutamate, 40 μg/ml phenylalanine, 40 μg/ml tryptophan), and 2% glucose was added as a carbon source as described previously (69). Unless otherwise stated, all B. subtilis strains were grown at 30°C in a shaking water bath or on plates in an incubator. The final concentrations of antibiotics used were 12.5 μg/ml tetracycline, 100 μg/ml spectinomycin, 5 μg/ml chloramphenicol, and 0.5 μg/ml erythromycin. Each was diluted from a 10× stock. For DNA damage mitomycin C (Fisher Bioreagents) and phleomycin (Sigma) were used at several different concentrations as indicated in the figure legends.
Microscopy.
Cells were plated from frozen stocks onto LB agar plates, with the appropriate antibiotics to select for markers listed in Table 1. Cells were then grown either at room temperature or 30°C, generating a light lawn of confluent growth with very few discernible colonies. Plates were then washed using prewarmed minimal medium (S750 supplemented with 2% glucose). The culture was inoculated at a starting optical density at 600 nm (OD600) of 0.05 to 0.1 in a final volume of 12.5 ml in a 125-ml flask. The culture was wrapped with foil to block light and incubated in a shaking water bath at 30°C at 250 rpm. Cultures were then grown for a few hours to achieve 3 doublings, reaching an OD600 of 0.4 to 0.8. Cultures were then challenged with DNA-damaging agents for 60 min or vehicle control to remain untreated prior to imaging. Cells were then placed on agarose pads and imaged as described previously (9, 70, 71). Focus intensity was analyzed using ImageJ with the MicrobeJ plugin to identify focus intensity using the point function. The intensity was determined for the number of foci indicated in the figure legend from at least two independent experiments. The data were graphed and statistics completed using Prism 9.
Cell membranes were visualized with the fluorescent dye TMA-DPH at a working concentration of 10 μM or FM 4-64 as described previously (9, 63, 71–75). TMA-DPH and FM 4-64 were added about 10 min before imaging, followed by incubating the sample for a few minutes in a shaking water bath at 30°C or at room temperature for FM 4-64. TMA-DPH images were captured after GFP for 65 ms. Following membrane staining slides were prepared for imaging on 1% agarose pads as described previously (9, 63, 71–75).
Killing curves.
For killing curves, cells were grown to mid-exponential phase at an OD600 of 0.5 to 0.8, followed by harvesting cells by centrifugation and resuspension in 0.85% saline. Cells were challenged with the indicated damaging agent for 30 min, followed by centrifugation and serial dilution. Cells were then plated on LB agar for viable cells, followed by enumeration (9, 76). The relative survival was normalized to the survival of the untreated wild-type control.
Spot plates.
Cells were grown to mid-exponential phase (OD600 of 0.5 to 0.8), followed by harvesting 1.5 ml of cells by centrifugation and resuspension in 0.85% saline. Cells were then serially diluted in 0.85% saline and followed by spotting 10 μl of resuspended cells on LB agar plates or LB agar plates with the indicated DNA-damaging agent. For UV treatment, cells were serially diluted and then challenged on the plate with 5 J/m2 UV-C. Plates were incubated at 30°C overnight and imaged the following day after 12 to 16 h of growth at 30°C.
The recA-gfp allele used was recA23-mGFPmut2, as described previously (9, 65, 73, 77). The recN-gfp allele used was recN23-mGFPmut3. Twenty-three denotes a 23-amino-acid flexible linker between RecN and mGFPmut3, which has been described previously (61). In Table 1, the recN-GFP strain used is EKM19, which is the same background as JSL486 with the same recN-gfp construct. All data presented in Table 1 were completed independent of the data presented in Fig. 3B and 5B. MMC was added to 20 ng/ml for 1 h prior to image acquisition.
ACKNOWLEDGMENTS
We thank members of the Simmons lab for comments on the work. We thank Pusparanee Hakim for her help and expertise in quantifying RecN foci and Caroline Lowder for technical help with experiments.
This work was supported by NIH grant R35 GM131772-01 to L.A.S. and in part by a Donald R. Shepherd Scholarship to E.K.M. J.S.L. was also supported in part by a Rackham Predoctoral Fellowship.
We have no conflict of interest to declare.
J.S.L., E.K.M., and L.A.S. conceived this study. J.S.L. and E.K.M. performed all experiments. J.S.L. performed the experiments shown in Fig. 1 through 5 and Fig. S1. E.K.M. completed all data shown in Table 1 and Fig. S2. E.K.M. and L.A.S. assembled and wrote the manuscript. All authors have read and edited the final manuscript and agree to the contents.
Footnotes
Supplemental material is available online only.
Contributor Information
Lyle A. Simmons, Email: lasimm@umich.edu.
Thomas J. Silhavy, Princeton University
REFERENCES
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2006. DNA repair and mutagenesis: second edition, p 9–57. American Society for Microbiology, Washington, DC. [Google Scholar]
- 2.Charbon G, Bjorn L, Mendoza-Chamizo B, Frimodt-Moller J, Lobner-Olesen A. 2014. Oxidative DNA damage is instrumental in hyperreplication stress-induced inviability of Escherichia coli. Nucleic Acids Res 42:13228–13241. 10.1093/nar/gku1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox MM. 1999. Recombinational DNA repair in bacteria and the RecA protein. Prog Nucleic Acids Res Mol Biol 63:311–366. 10.1016/s0079-6603(08)60726-6. [DOI] [PubMed] [Google Scholar]
- 4.Lenhart JS, Schroeder JW, Walsh BW, Simmons LA. 2012. DNA repair and genome maintenance in Bacillus subtilis. Microbiol Mol Biol Rev 76:530–564. 10.1128/MMBR.05020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthews LA, Simmons LA. 2014. Bacterial non-homologous end joining requires teamwork. J Bacteriol 196:3363–3365. 10.1128/JB.02042-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shuman S, Glickman MS. 2007. Bacterial DNA repair by non-homologous end joining. Nat Rev Microbiol 5:852–861. 10.1038/nrmicro1768. [DOI] [PubMed] [Google Scholar]
- 7.Gupta R, Barkan D, Redelman-Sidi G, Shuman S, Glickman MS. 2011. Mycobacteria exploit three genetically distinct DNA double-strand break repair pathways. Mol Microbiol 79:316–330. 10.1111/j.1365-2958.2010.07463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta R, Ryzhikov M, Koroleva O, Unciuleac M, Shuman S, Korolev S, Glickman MS. 2013. A dual role for mycobacterial RecO in RecA-dependent homologous recombination and RecA-independent single-strand annealing. Nucleic Acids Res 41:2284–2295. 10.1093/nar/gks1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenhart JS, Brandes ER, Schroeder JW, Sorenson RJ, Showalter HD, Simmons LA. 2014. RecO and RecR are necessary for RecA loading in response to DNA damage and replication fork stress. J Bacteriol 196:2851–2860. 10.1128/JB.01494-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weller GR, Kysela B, Roy R, Tonkin LM, Scanlan E, Della M, Devine SK, Day JP, Wilkinson A, d'Adda di Fagagna F, Devine KM, Bowater RP, Jeggo PA, Jackson SP, Doherty AJ. 2002. Identification of a DNA nonhomologous end-joining complex in bacteria. Science 297:1686–1689. 10.1126/science.1074584. [DOI] [PubMed] [Google Scholar]
- 11.Aniukwu J, Glickman MS, Shuman S. 2008. The pathways and outcomes of mycobacterial NHEJ depend on the structure of the broken DNA ends. Genes Dev 22:512–527. 10.1101/gad.1631908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hadden CT. 1977. Repair and subsequent fragmentation of deoxyribonucleic acid in ultraviolet-irradiated Bacillus subtilis recA. J Bacteriol 132:856–861. 10.1128/jb.132.3.856-861.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chedin F, Handa N, Dillingham MS, Kowalczykowski SC. 2006. The AddAB helicase/nuclease forms a stable complex with its cognate chi sequence during translocation. J Biol Chem 281:18610–18617. 10.1074/jbc.M600882200. [DOI] [PubMed] [Google Scholar]
- 14.Chedin F, Kowalczykowski SC. 2002. A novel family of regulated helicases/nucleases from Gram-positive bacteria: insights into the initiation of DNA recombination. Mol Microbiol 43:823–834. 10.1046/j.1365-2958.2002.02785.x. [DOI] [PubMed] [Google Scholar]
- 15.Chedin F, Noirot P, Biaudet V, Ehrlich SD. 1998. A five-nucleotide sequence protects DNA from exonucleolytic degradation by AddAB, the RecBCD analogue of Bacillus subtilis. Mol Microbiol 29:1369–1377. 10.1046/j.1365-2958.1998.01018.x. [DOI] [PubMed] [Google Scholar]
- 16.Saikrishnan K, Yeeles JT, Gilhooly NS, Krajewski WW, Dillingham MS, Wigley DB. 2012. Insights into Chi recognition from the structure of an AddAB-type helicase-nuclease complex. EMBO J 31:1568–1578. 10.1038/emboj.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeeles JT, Gwynn EJ, Webb MR, Dillingham MS. 2011. The AddAB helicase-nuclease catalyses rapid and processive DNA unwinding using a single Superfamily 1A motor domain. Nucleic Acids Res 39:2271–2285. 10.1093/nar/gkq1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. 1994. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev 58:401–465. 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roca A, Cox M. 1990. The RecA protein: structure and function. Crit Rev Biochem Mol Biol 25:415–456. 10.3109/10409239009090617. [DOI] [PubMed] [Google Scholar]
- 20.Ayora S, Carrasco B, Doncel-Perez E, Doncel E, Lurz R, Alonso JC. 2004. Bacillus subtilis RecU protein cleaves Holliday junctions and anneals single-stranded DNA. Proc Natl Acad Sci USA 101:452–457. 10.1073/pnas.2533829100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burby PE, Simmons LA. 2017. MutS2 promotes homologous recombination in Bacillus subtilis. J Bacteriol 199:e00682-16. [PMC] 10.1128/JB.00682-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lusetti SL, Cox MM. 2002. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem 71:71–100. 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- 23.Jasin M, Rothstein R. 2013. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5:a012740. 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. 2004. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell 16:991–1002. 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 25.Ampatzidou E, Irmisch A, O'Connell MJ, Murray JM. 2006. Smc5/6 is required for repair at collapsed replication forks. Mol Cell Biol 26:9387–9401. 10.1128/MCB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Britton RA, Lin DC, Grossman AD. 1998. Characterization of a prokaryotic SMC protein involved in chromosome partitioning. Genes Dev 12:1254–1259. 10.1101/gad.12.9.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krishnamurthy M, Tadesse S, Rothmaier K, Graumann PL. 2010. A novel SMC-like protein, SbcE (YhaN), is involved in DNA double-strand break repair and competence in Bacillus subtilis. Nucleic Acids Res 38:455–466. 10.1093/nar/gkp909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nasmyth K, Haering CH. 2005. The structure and function of SMC and kleisin complexes. Annu Rev Biochem 74:595–648. 10.1146/annurev.biochem.74.082803.133219. [DOI] [PubMed] [Google Scholar]
- 29.Stephan AK, Kliszczak M, Dodson H, Cooley C, Morrison CG. 2011. Roles of vertebrate Smc5 in sister chromatid cohesion and homologous recombinational repair. Mol Cell Biol 31:1369–1381. 10.1128/MCB.00786-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sullivan NL, Marquis KA, Rudner DZ. 2009. Recruitment of SMC by ParB-parS organizes the origin region and promotes efficient chromosome segregation. Cell 137:697–707. 10.1016/j.cell.2009.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Tang OW, Riley EP, Rudner DZ. 2014. The SMC condensin complex is required for origin segregation in Bacillus subtilis. Curr Biol 24:287–292. 10.1016/j.cub.2013.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Csankovszki G, McDonel P, Meyer BJ. 2004. Recruitment and spreading of the C. elegans dosage compensation complex along X chromosomes. Science 303:1182–1185. 10.1126/science.1092938. [DOI] [PubMed] [Google Scholar]
- 33.Jessberger R. 2002. The many functions of SMC proteins in chromosome dynamics. Nat Rev Mol Cell Biol 3:767–778. 10.1038/nrm930. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Le TB, Lajoie BR, Dekker J, Laub MT, Rudner DZ. 2015. Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev 29:1661–1675. 10.1101/gad.265876.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Au N, Kuester-Schoeck E, Mandava V, Bothwell LE, Canny SP, Chachu K, Colavito SA, Fuller SN, Groban ES, Hensley LA, O'Brien TC, Shah A, Tierney JT, Tomm LL, O'Gara TM, Goranov AI, Grossman AD, Lovett CM. 2005. Genetic composition of the Bacillus subtilis SOS system. J Bacteriol 187:7655–7666. 10.1128/JB.187.22.7655-7666.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cardenas PP, Gandara C, Alonso JC. 2014. DNA double strand break end-processing and RecA induce RecN expression levels in Bacillus subtilis. DNA Repair 14:1–8. 10.1016/j.dnarep.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Keyamura K, Sakaguchi C, Kubota Y, Niki H, Hishida T. 2013. RecA protein recruits structural maintenance of chromosomes (SMC)-like RecN protein to DNA double-strand breaks. J Biol Chem 288:29229–29237. 10.1074/jbc.M113.485474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanchez H, Kidane D, Castillo Cozar M, Graumann PL, Alonso JC. 2006. Recruitment of Bacillus subtilis RecN to DNA double-strand breaks in the absence of DNA end processing. J Bacteriol 188:353–360. 10.1128/JB.188.2.353-360.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klimova AN, Sandler SJ. 2020. An epistasis analysis of recA and recN in Escherichia coli K-12. Genetics 216:381–393. 10.1534/genetics.120.303476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pellegrino S, Radzimanowski J, de Sanctis D, Boeri Erba E, McSweeney S, Timmins J. 2012. Structural and functional characterization of an SMC-like protein RecN: new insights into double-strand break repair. Structure 20:2076–2089. 10.1016/j.str.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 41.Strom L, Lindroos HB, Shirahige K, Sjogren C. 2004. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell 16:1003–1015. 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 42.Vickridge E, Planchenault C, Cockram C, Junceda IG, Espeli O. 2017. Management of E. coli sister chromatid cohesion in response to genotoxic stress. Nat Commun 8:14618. 10.1038/ncomms14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uranga LA, Reyes ED, Patidar PL, Redman LN, Lusetti SL. 2017. The cohesin-like RecN protein stimulates RecA-mediated recombinational repair of DNA double-strand breaks. Nat Commun 8:15282. 10.1038/ncomms15282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosch TC, Altenburger S, Oviedo-Bocanegra L, Pediaditakis M, Najjar NE, Fritz G, Graumann PL. 2018. Single molecule tracking reveals spatio-temporal dynamics of bacterial DNA repair centres. Sci Rep 8:16450. 10.1038/s41598-018-34572-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kidane D, Sanchez H, Alonso JC, Graumann PL. 2004. Visualization of DNA double-strand break repair in live bacteria reveals dynamic recruitment of Bacillus subtilis RecF, RecO and RecN proteins to distinct sites on the nucleoids. Mol Microbiol 52:1627–1639. 10.1111/j.1365-2958.2004.04102.x. [DOI] [PubMed] [Google Scholar]
- 46.Chedin F, Ehrlich SD, Kowalczykowski SC. 2000. The Bacillus subtilis AddAB helicase/nuclease is regulated by its cognate Chi sequence in vitro. J Mol Biol 298:7–20. 10.1006/jmbi.2000.3556. [DOI] [PubMed] [Google Scholar]
- 47.Yeeles JT, Dillingham MS. 2007. A dual-nuclease mechanism for DNA break processing by AddAB-type helicase-nucleases. J Mol Biol 371:66–78. 10.1016/j.jmb.2007.05.053. [DOI] [PubMed] [Google Scholar]
- 48.Shan Q, Cox MM. 1997. RecA filament dynamics during DNA strand exchange reactions. J Biol Chem 272:11063–11073. 10.1074/jbc.272.17.11063. [DOI] [PubMed] [Google Scholar]
- 49.Alonso JC, Stiege AC, Luder G. 1993. Genetic recombination in Bacillus subtilis 168: effect of recN, recF, recH and addAB mutations on DNA repair and recombination. Mol Gen Genet 239:129–136. 10.1007/BF00281611. [DOI] [PubMed] [Google Scholar]
- 50.Goranov AI, Kuester-Schoeck E, Wang JD, Grossman AD. 2006. Characterization of the global transcriptional responses to different types of DNA damage and disruption of replication in Bacillus subtilis. J Bacteriol 188:5595–5605. 10.1128/JB.00342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanchez H, Alonso JC. 2005. Bacillus subtilis RecN binds and protects 3'-single-stranded DNA extensions in the presence of ATP. Nucleic Acids Res 33:2343–2350. 10.1093/nar/gki533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bell JC, Plank JL, Dombrowski CC, Kowalczykowski SC. 2012. Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature 491:274–278. 10.1038/nature11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dunman PM, Ren L, Rahman MS, Palejwala VA, Murphy HS, Volkert MR, Humayun MZ. 2000. Escherichia coli cells defective for the recN gene display constitutive elevation of mutagenesis at 3,N(4)-ethenocytosine via an SOS-induced mechanism. Mol Microbiol 37:680–686. 10.1046/j.1365-2958.2000.02045.x. [DOI] [PubMed] [Google Scholar]
- 54.Meddows TR, Savory AP, Grove JI, Moore T, Lloyd RG. 2005. RecN protein and transcription factor DksA combine to promote faithful recombinational repair of DNA double-strand breaks. Mol Microbiol 57:97–110. 10.1111/j.1365-2958.2005.04677.x. [DOI] [PubMed] [Google Scholar]
- 55.Picksley SM, Attfield PV, Lloyd RG. 1984. Repair of double-strand breaks in Escherichia coli requires a functional recN gene product. Mol Gen Genet 195:267–274. 10.1007/BF00332758. [DOI] [PubMed] [Google Scholar]
- 56.Sargentini NJ, Smith KC. 1983. Characterization of an Escherichia coli mutant (radB101) sensitive to gamma and uv radiation, and methyl methanesulfonate. Radiat Res 93:461–478. 10.2307/3576026. [DOI] [PubMed] [Google Scholar]
- 57.Lloyd RG, Picksley SM, Prescott C. 1983. Inducible expression of a gene specific to the recF pathway for recombination in Escherichia coli K12. Mol Gen Genet 190:162–167. 10.1007/BF00330340. [DOI] [PubMed] [Google Scholar]
- 58.Badrinarayanan A, Le TB, Laub MT. 2015. Rapid pairing and resegregation of distant homologous loci enables double-strand break repair in bacteria. J Cell Biol 210:385–400. 10.1083/jcb.201505019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Funayama T, Narumi I, Kikuchi M, Kitayama S, Watanabe H, Yamamoto K. 1999. Identification and disruption analysis of the recN gene in the extremely radioresistant bacterium Deinococcus radiodurans. Mutat Res 435:151–161. 10.1016/s0921-8777(99)00044-0. [DOI] [PubMed] [Google Scholar]
- 60.Sanchez H, Cardenas PP, Yoshimura SH, Takeyasu K, Alonso JC. 2008. Dynamic structures of Bacillus subtilis RecN-DNA complexes. Nucleic Acids Res 36:110–120. 10.1093/nar/gkm759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. 2012. Segregation of molecules at cell division reveals native protein localization. Nat Methods 9:480–482. 10.1038/nmeth.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zacharias DA, Violin JD, Newton AC, Tsien RY. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916. 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 63.Simmons LA, Davies BW, Grossman AD, Walker GC. 2008. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol Cell 29:291–301. 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41–64. 10.1093/genetics/158.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simmons LA, Goranov AI, Kobayashi H, Davies BW, Yuan DS, Grossman AD, Walker GC. 2009. Comparison of responses to double-strand breaks between Escherichia coli and Bacillus subtilis reveals different requirements for SOS induction. J Bacteriol 191:1152–1161. 10.1128/JB.01292-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lesterlin C, Gigant E, Boccard F, Espeli O. 2012. Sister chromatid interactions in bacteria revealed by a site-specific recombination assay. EMBO J 31:3468–3479. 10.1038/emboj.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Youngman P, Perkins JB, Losick R. 1984. Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid 12:1–9. 10.1016/0147-619x(84)90061-1. [DOI] [PubMed] [Google Scholar]
- 68.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. 10.1128/jb.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burby PE, Simmons ZW, Simmons LA. 2019. DdcA antagonizes a bacterial DNA damage checkpoint. Mol Microbiol 111:237–253. 10.1111/mmi.14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nye TM, McLean EK, Burrage AM, Dennison DD, Kearns DB, Simmons LA. 2021. RnhP is a plasmid-borne RNase HI that contributes to genome maintenance in the ancestral strain Bacillus subtilis NCIB 3610. Mol Microbiol 115:99–115. 10.1111/mmi.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lenhart JS, Sharma A, Hingorani MM, Simmons LA. 2013. DnaN clamp zones provide a platform for spatiotemporal coupling of mismatch detection to DNA replication. Mol Microbiol 87:553–568. 10.1111/mmi.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lenhart JS, Pillon MC, Guarne A, Simmons LA. 2013. Trapping and visualizing intermediate steps in the mismatch repair pathway in vivo. Mol Microbiol 90:680–698. 10.1111/mmi.12389. [DOI] [PubMed] [Google Scholar]
- 73.Simmons LA, Grossman AD, Walker GC. 2007. Replication is required for the RecA localization response to DNA damage in Bacillus subtilis. Proc Natl Acad Sci USA 104:1360–1365. 10.1073/pnas.0607123104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Simmons LA, Grossman AD, Walker GC. 2008. Clp and Lon proteases occupy distinct subcellular positions in Bacillus subtilis. J Bacteriol 190:6758–6768. 10.1128/JB.00590-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith BT, Grossman AD, Walker GC. 2002. Localization of UvrA and effect of DNA damage on the chromosome of Bacillus subtilis. J Bacteriol 184:488–493. 10.1128/JB.184.2.488-493.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simmons LA, Kaguni JM. 2003. The DnaAcos allele of Escherichia coli: hyperactive initiation is caused by substitution of A184V and Y271H, resulting in defective ATP binding and aberrant DNA replication control. Mol Microbiol 47:755–765. 10.1046/j.1365-2958.2003.03333.x. [DOI] [PubMed] [Google Scholar]
- 77.Walsh BW, Bolz SA, Wessel SR, Schroeder JW, Keck JL, Simmons LA. 2014. RecD2 helicase limits replication fork stress in Bacillus subtilis. J Bacteriol 196:1359–1368. 10.1128/JB.01475-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 and S2 and Table S1. Download JB.00240-21-s0001.pdf, PDF file, 1.9 MB (1.9MB, pdf)