

Abstract
Background
Pathway enrichment analysis (PEA) is a well-established methodology for interpreting a list of genes and proteins of interest related to a condition under investigation. This paper aims to extend our previous work in which we introduced a preliminary comparative analysis of pathway enrichment analysis tools. We extended the earlier work by providing more case studies, comparing BiP enrichment performance with other well-known PEA software tools.
Methods
PEA uses pathway information to discover connections between a list of genes and proteins as well as biological mechanisms, helping researchers to overcome the problem of explaining biological entity lists of interest disconnected from the biological context.
Results
We compared the results of BiP with some existing pathway enrichment analysis tools comprising Centrality-based Pathway Enrichment, pathDIP, and Signaling Pathway Impact Analysis, considering three cancer types (colorectal, endometrial, and thyroid), for a total of six datasets (that is, two datasets per cancer type) obtained from the The Cancer Genome Atlas and Gene Expression Omnibus databases. We measured the similarities between the overlap of the enrichment results obtained using each couple of cancer datasets related to the same cancer.
Conclusion
As a result, BiP identified some well-known pathways related to the investigated cancer type, validated by the available literature. We also used the Jaccard and meet-min indices to evaluate the stability and the similarity between the enrichment results obtained from each couple of cancer datasets. The obtained results show that BiP provides more stable enrichment results than other tools.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12859-021-04297-z.
Keywords: Biological pathway, Statistical analysis, Pathway enrichment analysis, Pathway databases
Background
Over the past decade, we have witnessed an increase in the production and availability of omics data aided by the continuous development and refinement of Next Generation Sequencing (NGS), Genome-Wide Association Studies (GWAS), gene expressions, and SNP microarrays platforms, also known as High-Throughput (HT) methodologies. Their ability to produce a massive amount of data has spurred the development of several pipelines for data analysis [1–4]. Several annotation software tools use Gene Ontology (GO) to link HT data analysis results with the affected biological mechanisms [5, 6]. Although these software tools can effectively analyze these vast amounts of available data, the produced results are still not connected to the biological mechanisms they may influence. Common diseases (for example, flu and headaches) and complex conditions (for example, cancer or diabetes) are due to several biological entities’ interactions represented through biological pathways.
Biological pathways are human representations of the existent biomolecules’ interactions regulating cellular functions in healthy and diseased conditions and how cells can interact with the external environment. Biological pathways are networks where nodes represent biomolecules, and edges represent the interaction among them. The pathway representation as a network has contributed to the development of several network alignment [7], and deep learning analysis [8] algorithms.
Biological pathways are fundamental in analyzing, contextualizing and interpreting omics data. The main pathway categories are three: Signalling Pathways, Metabolic Pathways and Regulatory Pathways. Several pathway databases including Kyoto Encyclopedia of Gene and Genome (KEGG) [9], Metabolic Pathway Database (MetaCyc) [10], PantherDB [11], PathwayCommons [12], Pathway Interaction Databse (PID) [13], Reactome [14], SIGnaling Network Open Resource (SIGNOR) [15] and WikiPathways [16] are available online. These databases share different types of pathways. For example, Reactome and KEGG store all three categories of pathways, while SIGNOR includes only signaling pathways and Metacyc showcases only metabolic pathways. Also, databases that contain the same kinds of pathways (like KEGG and Reactome) show minimal overlap on the number of pathways and gene coverage as reported in [17].
Many molecular and cell biologists face a common question in their research: how to link pathways to a specific genes or proteins list? The availability of biological information in a digital format enables the automatic elaboration of these pathway databases for different knowledge discovery tasks.
In particular, Pathway Enrichment Analysis (PEA) is a well-established approach to gain insight into the underlying biological mechanism of a differentially expressed list of genes and proteins of interest. PEA can use the information in pathway databases to elucidate the link between the genes and proteins of interest and the biological mechanisms affected in the biological pathways. The three principal categories of PEA methods are: (1) Over Represented Analysis (ORA); (2) Gene Set Enrichment Analysis (GSEA); and (3) Topological Enrichment Analysis (TEA).
The first two categories of methods perform enrichment analysis using a list of genes, proteins, SNPs or mRNA as input. Moreover, GSEA methods include self-contained or competitive null-hypothesis approaches. Self-contained null hypothesis methods assume that no genes in the gene list are associated with the phenotype. Competitive null hypothesis methods assume that genes in the gene list have a higher probability of being associated with the phenotype than genes outside the gene list. In contrast, the TEA methods need a genes and proteins list along with the network topology information.
PEA methods endeavor to help researchers decipher biological entities of interest disconnected from the biological context, expediting their findings’ validation. Analyzing lists of biological entities at the functional pathway-level can provide more explanatory power than analyzing a list of independent entities.
Several PEA software tools are available, among those: BioPAX-Parser (BiP) [18], Centrality-based Pathway Enrichment (CePa) [19], pathDIP [17] and Signaling Pathway Impact Analysis (SPIA) [20]. BiP and pathDIP belong to the ORA category. BiP employs Hypergeometric function [21] to assess the significance of genes of interest enriched in a specific pathway. Conversely, pathDIP uses a customized version of Fisher’s Test [22]. CePa and SPIA belong to the TEA category. CePa performs TEA by using network centrality measures (node input degree, node output degree, betweenness, input reachability and output reachability) [23]. In contrast, SPIA computes TEA using network measures [24] evaluating a node’s neighbor.
BiP and pathDIP can perform pathways enrichment analysis using a list of genes and proteins of interest without providing any additional information or data manipulation in the phenomena under investigation. In contrast, CePa and SPIA require users to provide additional network information, such as the interactions among the genes and proteins of interest and other network topology information, to perform TEA.
CePa, pathDIP, and SPIA allow performing PEA only using the previously collected and integrated pathway information. Conversely, BiP can perform PEA employing users’ downloaded pathways information from KEGG, Reactome or any other pathway database compliant with the Biological Pathway Exchange (BioPAX) format [25]. In this way, BiP can always provide more accurate and update enrichment results, avoiding losing critical biological features. Authors in [26] and [27] have remarkably highlighted the importance of using updated pathway information along with accurate lists of genes or proteins in performing pathway enrichment analysis. They proved that outdated pathway information negatively influenced the PEA results.
We evaluate the BiP’s pathway enrichment effectiveness using three TCGA cancer data sets and three GEO gene expression data sets, related respectively with colorectal (CC), endometrial (EC), and thyroid (TC) cancer. We used the six gene lists along with KEGG and Reactome pathway databases to evaluate the enrichment results. This way, it is possible to assess if the enrichment results obtained from each tool are similar, when varying the input gene list and keeping the database unchanged.
Although some works, such as [28, 29], use GO in PEA, as reported in Khatri et al. [30], the definition of pathway in certain cases may be misleading or incorrect. For this reason, we did not use GO to perform the experiments.
For each couple of cancer enrichment results we computed the pathway overlap (intersection) and the union between the two enrichments, selecting the first top 10 pathways (p value ) to validate by using published literature. We chose as statistical significance threshold the p value, allowing us to improve the reproducibility of scientific studies as recommended in [31]. We used the Jaccard and meet-min similarities indices to evaluate the tools’ stability (for example, in terms of number and similarity of the enrichment pathways obtained by using different datasets related to the same cancer type). The obtained results proved that BiP is more solid in terms of number and similarity of enriched pathways than other existing tools. Also, BiP can identify some well-known cancer pathways omitted by other compared software frameworks.
This paper aims to extend our previous work [32] in which we introduced a preliminary comparative analysis of pathway enrichment analysis tools. In particular, we performed more case studies and compared BiP enrichment performance with other well-known PEA software tools.
Methods
Related works
PEA software frameworks are available as stand-alone software, web-based applications or program libraries. The first two categories are usually more convenient to use, as they do not require analytical skills or programming abilities. Program libraries are coded in C, Java, R and Python languages, allowing them to automate the process through scripting analysis pipelines. User skills and the cost-benefit ratio of time invested in orchestrating everything necessary to run the analysis may influence selecting software platforms and program libraries. In the following we present a description of some well-known PEA software frameworks.
BiP [18] can perform PEA using pathways encoded in Biological Pathway Exchange (BioPAX) [25] and KGML (KEGG Markup Language) formats. BioPAX is a meta-language defined in OWL (Web Ontology Language) and represented in the RDF/XML (Resource Description Framework / eXtensible Meta Language) format and is the language of choice to store and exchange pathway data. KEGG Markup Language (KGML) is based on an XML-like markup language, providing computational analysis and modeling of gene/protein networks and chemical networks in the KEGG database. BiP has been extended to be compatible with the KGML format used to represent pathways in the KEGG database. BiP is developed in Java programming language, making it platform-independent. PEA in BiP is computed using a list of proteins/genes of interest as input. The genes and proteins enrichment is calculated using a customized version of Hypergeometric Test, along with multiple statistical corrector such as False Discovery Rate (FDR) and Bonferroni. BiP can enrich lists of genes and proteins using KEGG, Reactome and other available pathway databases compliant with the BioPAX format. BiP can be freely downloaded as a stand-alone application at [33].
CePa [19] performs PEA based on topological information in addition to gene-set information. Pathways are collected and integrated from the Pathway Interaction Database (PID) database. PID includes the KEGG database. CePa parses the XML pathway files to obtain pathway data in order to perform the enrichment. To perform PEA, CePa needs a differentially expressed gene list and a background gene list. Several methods are available to produce the differential genes or proteins list, for example, the t-test. The background gene list is the list of genes of a specific microarray platform. The differential gene list and the background gene list must use the same identifiers to represent the gene symbol or protein identifier. CePa is an R application freely available at [34].
pathDIP [17] is an integrated database of pathways in human, model organisms and domesticated animals, comprising core pathways from major curated pathway databases and gene pathway associations predicted using physical protein interactions. pathDIP helps researchers to perform ORA on structured ontology annotations, pathway databases, or set of biological entities. pathDIP is a web-application compatible with all available operating systems, it is available through an Application Program Interface (API) and it is developed in Java, R and Python. pathDIP computes PEA using the Fisher’s Exact Test, providing correction for multiple hypothesis testing through two different methods: Bonferroni and FDR. pathDIP integrates pathway data from 22 databases, including KEGG and Reactome. pathDIP is publicly available at [35].
Signaling Pathway Impact Analysis (SPIA) [20] combines the evidence obtained from the classical enrichment analysis with the measure of the perturbation on a given pathway under a given condition. It allows to calculate a global pathway significance p value, combining the enrichment and perturbation p values. SPIA needs a set of differentially expressed genes and their fold changes and pathways topology to compute PEA from the condition under investigation. Differentially expressed genes need to be in Entrez gene IDs format. SPIA performs PEA by using pathway information from the KEGG database. SPIA is an R program freely available at [36].
The main differences among the surveyed PEA frameworks are summarised as follows. CePa and SPIA are all available as R packages, meaning the user needs some basic programming knowledge. Conversely, BiP efficiently performs PEA through a simple graphical user interface (GUI), that allows loading only a list of genes or proteins, selecting which pathway database use, setting the p value significance threshold and choosing the destination folder where to store the results. The only requirement to use BiP is to have previously installed Java, no additional libraries are necessary. pathDIP is a web application that allows performing enrichment analysis through a graphical interface just by loading a list of protein or gene identifiers and returning the pathways in which those identifiers are involved.
All the PEA software tools are different in terms of pathway enrichment calculation. BiP employs the Hypergeometric Test, along with FDR and Bonferroni correctors to calculate pathway enrichment. CePa computes pathway enrichment using topological information (that are, node input degree, node output degree, betweenness, input reachability and output reachability) and gene-set information. pathDip computes pathway enrichment using the Fisher’s Exact Test, along with Bonferroni and FDR correctors. Finally, SPIA calculates pathway enrichment combining the classical enrichment analysis with the measure of the perturbation on a given pathway under a given condition, evaluating a node’s neighbourhood.
BiP and pathDIP can perform pathways enrichment analysis using a list of genes and proteins of interest without providing any additional information or data manipulation in the phenomena under investigation. In contrast, CePa and SPIA require users to provide additional network information, such as the interactions among the genes and proteins of interest and other network topology information, to perform TEA.
Finally, BiP is the only enrichment analysis tool that allows users to select the pathway database that has to be used to enrich the condition under investigation.
All the PEA software tools and the related algorithms are well established in providing biological context in -omics studies, particularly to analyze the biological molecules, where summarizing the overall biology of a particular disease by pathways enhances interpretability. BiP, CePa, pathDIP and SPIA are some of the available gold standards tools in pathway enrichment analysis.
Most PEA tools evaluate enrichments in a very similar manner; we chose CePa, pathDip and SPIA as representative PEA tools due to their popularity and ability to test a breadth of data sources similar to that of BiP.
The Genomic Regions Enrichment of Annotations Tool (GREAT) [37] is a web-application for ontology enrichments tailored for regions bounding. GREAT requires a set of input genomic regions and an ontology of gene annotations as input, whereas the compared tools require as input a list of proteins or genes and one or more pathway databases. Also, ontology functional enrichment is different from PEA, how stated in [30]. Thus, we have chosen do not to compare GREAT with BiP.
g:Profiler [38] primary purpose is to perform functional enrichment analysis on input genes lists. Only in the last release of g:Profiler, pathway enrichment has been introduced, and it is limited only to Reactome and WikiPathways pathway databases. KEGG, due to licensing reasons, can be used only for terms association and not for enrichment. Conversely, from g:Profiler, all the compared PEA tools use KEGG to perform enrichment analysis, a not negligible difference that would make the comparison between the results obtained using different pathway databases unfair.
Datasets
We downloaded three couples of cancer datasets: colorectal cancer (COAD [39] and GSE41011 [40]), thyroid cancer (THCA [41] and GSE65144 [42]), and endometrial cancer (UCEC [43], and GSE63678 [44]) from the TCGA and GEO databases respectively, to test BiP.
The Gene Expression Omnibus (GEO) database is a public functional genomics database including high-throughput gene expression, chips, and microarrays data.
The Cancer Genome Atlas (TCGA) makes publicly available molecular and clinical information for more than 33 different types of human cancers, including exome (variant analysis), single nucleotide polymorphism (SNP), DNA methylation, transcriptome (mRNA), microRNA (miRNA) and proteome. TCGA data are accessible through the NCI Genomic Data Commons (GDC) data portal, GDC Legacy Archive, and the Broad Institute’s GDAC Firehose.
The COAD dataset contains 750 cases and 21, 224 probes, THCA includes 681 cases and 13, 564 probes, UCEC dataset comprises 560 cases and 22, 162 probes. The GSE41011 dataset contains 30, 968 probes and 19 cases, GSE65144 includes 12 cases and 54, 675 probes and, finally, GSE63678 comprises 7 cases and 22, 277 probes. Here cases indicate the positive samples, while probes indicate the dimension of the microarray (that is, the number of rows).
Table 1 summarises the information about the six dowloaded datasets.
Table 1.
Summary of the downloaded datasets
| Dataset Name | Data Source | Cancer Type | Cases | MGenes |
|---|---|---|---|---|
| COAD | TCGA | Colorectal Cancer (CC) | 750 | 5913 |
| THCA | TCGA | Thyroid Cancer (TC) | 681 | 6270 |
| UCEC | TCGA | Endometrial Cancer (EC) | 560 | 6574 |
| GSE41011 | GEO | Colorectal Cancer (CC) | 19 | 791 |
| GSE65144 | GEO | Thyroid Cancer (TC) | 12 | 6202 |
| GSE63678 | GEO | Endometrial Cancer (EC) | 7 | 1024 |
refers to the number of mutated/deregulated genes involved with the various cancer types
We downloaded biological pathways database in BioPAX format for each available organism from Reactome [45], but we used Homo-sapiens to perform pathway enrichment analysis. Furthermore, we downloaded from KEGG [46] database all the Homo-sapiens pathways.
Reactome is an open-source freely available curated relational database of signaling and metabolic molecules and their relations organized into biological pathways and processes. Reactome uses pathway steps that indicate any event in biology that changes the state of a biological molecule. Molecules such as nucleic acids, proteins, complexes, and small molecules participating in reactions form a network of biological interactions called pathways.
KEGG is a database that integrates genomic, chemical and systemic functional information to analyze gene functions. KEGG contains various types of data classified as systems information, genomic information, chemical information and health information. The central KEGG element is the molecular network, representing systemic functions of the cell and the organism.
Mutated genes (MGenes) were downloaded from cBioPortal [47, 48] for the TCGA datasets (mutated + (Copy-number alterations) CNAs), while deregulated genes were obtained using the GEO2R web portal [49] for GEO datasets.
The cBioPortal for Cancer Genomics provides a Web resource for graphically analyzing multidimensional cancer genomics data. The portal reduces molecular profiling data from cancer tissues and cell lines into readily understandable genetic, gene expression, etc data. The cBioPortal allows to integrate multiple data types at the gene level and then query for the presence of specific biological events in each sample (for example, genetic mutation, gene amplification, and increased mRNA or miRNA expression). Data integration includes CNAs, mRNA and microRNA (miRNA) expression, and so on.
GEO2R is an online framework with which users can separate the samples into groups and select the differential expressed genes (DEG). We used the False Discovery Rate (FDR) corrector to adjust the p value by reducing the type I error in the null hypothesis due to multiple comparisons.
Both downloaded genes and pathways data sets have been used as input for the four PEA frameworks to get new biological insights.
We evaluated each tool’s capability to enrich the same relevant pathways using different data sets related to the same condition. Thus, it was not needed to perform any batch effect removal.
Additional file 1 contains the pathway enrichment analysis user guide illustrating how to download genes or proteins data sets from GEO and TCGA databses and highlighting how to use the downloaded data sets with the surveyed PEA software tools.
Pathways enrichment computation
Equation 1 defines the Hypergeometric function implemented in BiP to compute the pathway enrichment score.
| 1 |
In Eq. 1 is the number of proteins into the pathway under enrichment analysis, refers to the number of proteins with which to perform the enrichment and represents the intersection between the proteins in the pathway and the input proteins.
To balance the errors due to multiple tests, we implemented the False Discovery Rate (FDR) corrector along with the Bonferroni’s corrector. Equations 2 and 3 define FDR and Bonferroni correctors.
| 2 |
In Eq. 2, is the i-th smallest p value out the total number of the p value for the performed experiment, N represents the number of performed tests, and i is the number of accepted p value using the i-th p value threshold.
| 3 |
In Eq. 3, is the corrected significance level, is the chosen significance level, and k is the number of performed individual tests.
Similarity indices computation
To assess the BiP abilities to detect essential pathways influenced by the genes under investigation, we compared the pathway enrichment results by using the two gene lists from the same disease obtained by BiP with respect to those provided by CePa, pathDIP, and SPIA, using the following similarity indices:
(1) Jaccard similarity index (JI) measures the percentage of similarity between the two enrichment sets. Equation 4 defines the Jaccard index.
| 4 |
In Eq. 4, and represent the total number of pathways in the two enrichments respectively. JI index range between 0 and 1, indicating respectively low and high similarity between the two enrichments.
(2) meet-min index (mi) [50] evaluates the similarity between the two sets, in terms of set containment. The definition of the meet-min index is reported in Eq. 5:
| 5 |
In Eq. 5, and represent the total number of pathways respectively in the two enrichments sets. mi index ranges between 0, that indicates no containment between the two sets, and 1 that indicates a perfect containment of a set into another.
We chose to use Jaccard and meet-min indices as similarity measures for the following reasons. The Jaccard index is a common trend to compare populations by determining what percent of objects identified were present in both populations, which means that the Jaccard index evaluates the similarity of two sets. Meet-min is a measure that can assess the similarity (the common objects into the two sets). The containment between two sets or one set contains the other, which means that the meet-min can evaluate both similarity and containment between two sets.
Results
This section compares the BiP, CePa, pathDIP and SPIA capability to perform pathway enrichment analysis using the six gene lists obtained from the cancer datasets in Table 1, using the Reactome and KEGG databases.
We compared BiP with pathDIP [17], CePa [19] and SPIA [20] based on the KEGG database. We also compared BiP and pathDIP using the Reactome database because CePa and SPIA do not support pathway data coming from this database. Then, we validated the identified pathways by using the available literature.
Colorectal cancer enrichment using KEGG database
In this subsection, we present the pathway enrichment results obtained by all the software tools by analyzing the two gene lists related to Colorectal Cancer (CC) -COAD and GSE41011- and using the KEGG database.
BiP was able to identify 274 and 53 significant pathways (p value ) respectively from the COAD and GSE41011 genes lists (let see Additional file 2 for the whole enriched pathway list). Table 2 shows the intersection of the first top 10 (The first top ten pathways are the first ten ones ordered by p value from lower to higher values) pathways (p value ) obtained from the two CC gene lists using the KEGG database.
Table 2.
The intersection of the first 10 pathways (sorted by the relevance of p value ) obtained from BiP by performing PEA using CC gene lists and the KEGG database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Metabolic pathways | 4.33 × 10 | 1.26 × 10 | 1.26 × 10 |
| (2) Pathways in cancer | 2.95 × 10 | 2.14 × 10 | 8.58 × 10 |
| (3) Transcriptional misregulation in cancer | 2.44 × 10 | 1.18 × 10 | 7.10 × 10 |
| (4) MAPK signaling pathway | 2.04 × 10 | 8.50 × 10 | 5.95 × 10 |
| (5) Salmonella infection | 2.75 × 10 | 8.90 × 10 | 8.01 × 10 |
| (6) Neuroactive ligand-receptor interaction | 1.00 × 10 | 2.65 × 10 | 2.91 × 10 |
| (7) Herpes simplex virus 1 infection | 1.85 × 10 | 3.85 × 10 | 5.39 × 10 |
| (8) Proteoglycans in cancer | 3.08 × 10 | 5.60 × 10 | 8.95 × 10 |
| (9) Epstein-Barr virus infection | 5.75 × 10 | 9.29 × 10 | 1.67 × 10 |
| (10) Human papillomavirus infection | 1.48 × 10 | 2.06 × 10 | 4.32 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
The first BiP’s enriched pathway is the “Metabolic pathway” that in a recent manuscript [51] is cited as pathway that regulates the colorectal cancer initiation and progression. In [52] authors identified how the “Transcriptional misregulation in cancer” pathway correlates with some outcomes of colorectal cancer. The “MAPK signaling pathway” regulates many cellular function including cell proliferation and apoptosis in colorectal cancer as reported in [53]. In [54] authors assessed the functional role of “salmonella infection” in downregulating Wnt1 in the inflammatory response and colorectal cancer progression. The functional implication of “Neuroactive ligand-receptor interaction pathway” in colorectal cancer has been described in [55]. In [56] the implication of “Herpes virus” in human colorectal polyps and adenocarcinomas, inducing important oncogenic pathways in colon-cancer cells, is reported. In the manuscript published by Vicente et al. [57] the role of the “proteoglycans molecules” in colorectal cancer progression is described. In the review of Fernandes et al. [58] the role of “Epstein-Barr virus infection” and “Human papillomavirus infection” in the onset and progression of colorectal cancer, is assessed.
CePa was able to identify 24 and 22 significant pathways respectively from the COAD and GSE41011 genes lists (let see Additional file 3 for the whole enriched pathway list).
Table 3 shows the CePa’s top 10 pathways obtained from the intersection of the pathway enrichment results for the two CC genes lists and the KEGG database.
Table 3.
The intersection of the first 10 pathways obtained from CePa by performing pathway enrichment analysis by using CC genes lists and the KEGG database. The table summarises the values obtained for each topology measure employed by CePa
| Name | Weight | In degree | Out degree | Betweenness | In reachability | Out reachability |
|---|---|---|---|---|---|---|
| (1) Pyrimidine biosynthesis | 0.034 | 0.031 | 0.188 | 0.031 | 0.037 | 0.037 |
| (2) Inactivation of APC C via direct inhibition of the APC C complex | 0.031 | 0.047 | 0.047 | 0.037 | 0.037 | 0.037 |
| (3) Removal of licensing factors from origins | 0.031 | 0.034 | 0.031 | 0.037 | 0.037 | 0.037 |
| (4) CDT1 association with the CDC6 ORC origin complex | 0.031 | 0.0059 | 0.047 | 0.0058 | 0.037 | 0.037 |
| (5) NCAM signaling for neurite out growth | 0.031 | 0.034 | 0.098 | 0.083 | 0.037 | 0.037 |
| (6) FGFR1c and Klotho ligand binding and activation | 0.044 | 0.169 | 0.10 | 0.066 | 0.180 | 0.180 |
| (7) Switching of origins to a post replicative state | 0.031 | 0.034 | 0.031 | 0.037 | 0.037 | 0.037 |
| (8) Fanconi Anemia pathway | 0.031 | 0.034 | 0.031 | 0.037 | 0.037 | 0.037 |
| (9) ChREBP activates metabolic gene expression | 0.044 | 0.321 | 0.400 | 0.0058 | 0.338 | 0.338 |
| (10) Telomere C strand Lagging Strand Synthesis | 0.065 | 0.098 | 0.075 | 0.071 | 0.047 | 0.047 |
CePa enrichment results include the following KEGG pathways. Authors show in [59] that Klotho family members are associated with “FGFRs” to adjust “FGF” binding to “FGFRs”, important molecules in CC [59, 60]. Satoh et al. [61] describes the importance of “phyrymidine biosynthesis” in colorectal cancer, while [62] highlights that loss of “APC expression” in tumor tissue may be related with the risk for recurrence and a poor survival rate for patients with colorectal cancer. Tsaniras et al. [63] reviews the link between pre-replicative complex and cancer (pathways “Removal of licensing factors from origin”, “CDT1 association with the CDC6 ORC origin complex”). Bernal et al. [64] evaluates the role of dysfunctional “telomeres” in contributing to genomic instability in cancer. Tong et al. [65] shows that “ChREBP” plays a critical role in redirecting glucose metabolism to anabolic pathways as well as suppressing p53 activity. Esteban-Jurado et al. [66] evaluates how the “Fanconi anemia DNA damage repair pathway” performs an important role in germline predisposition to colorectal cancer. Fernández-Briera et al. [67] describes how “NCAM” (but not the neurite outgrowth) is responsible of worse prognosis and lymph nodes metastasis in colorectal cancer. To the best of our knowledge, we could not find any evidence for the “Switching of origins to a post-replicative state” pathway.
pathDIP in the colorectal cancer enrichment was able to identify 53 and 291 significant pathways (p value ) respectively from the COAD and GSE41011 genes lists (let see Additional file 4 for the whole enriched pathway list). Table 4 shows the intersection of the first top 10 pathways (p value ) obtained from the two CC gene lists using the KEGG database.
Table 4.
The intersection of the first 10 pathways (sorted by the relevance of p value ) obtained from pathDIP by performing PEA using CC genes lists and the KEGG database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Pathways in cancer | 1.93 × 10 | 6.16 × 10 | 6.16 × 10 |
| (2) Human papillomavirus infection | 6.34 × 10 | 1.01 × 10 | 2.03 × 10 |
| (3) Endocytosis | 4.28 × 10 | 4.56 × 10 | 1.37 × 10 |
| (4) Lysosome | 8.40 × 10 | 6.72 × 10 | 2.69 × 10 |
| (5) MicroRNAs in cancer | 8.88 × 10 | 5.68 × 10 | 2.84 × 10 |
| (6) Human T-cell leukemia virus 1 infection | 1.42 × 10 | 7.59 × 10 | 4.56 × 10 |
| (7) MAPK signaling | 1.94 × 10 | 8.88 × 10 | 6.22 × 10 |
| (8) PI3K-Akt signaling | 5.31 × 10 | 1.54 × 10 | 1.70 × 10 |
| (9) Cell cycle | 1.47 × 10 | 3.92 × 10 | 4.70 × 10 |
| (10) Autophagy-animal | 1.92 × 10 | 3.07 × 10 | 6.13 × 10 |
In the table FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value by using Bonferroni corrector
The pathDIP enrichment results include the following KEGG pathways. The “Pathways in cancer” is too general, making its role trivial in colorectal cancer. Fernandes et al. [58] assesses the role of “Human papillomavirus infection” in the onset and progression of colorectal cancer. Park et al. [68] describes the role of “Endocytosis” in colorectal cancer cells. Sun et al. [69] describes how the dichloroacetate attenuates the stemness of colorectal cancer cells via triggering ferroptosis through sequestering iron in “lysosomes” pathway. Grady et al. [70] describes a possible tumor suppressor role of “microRNA” in colorectal cancer due to the epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL. We couldn’t find a link with the “Human T-cell leukemia virus 1 infection” and colorectal cancer. Slattery et al. [53] describes the involvement of “MAPK signaling pathway” in colorectal cancer. Agarwal et al. [71] assesses the role of “PI3K-Akt signaling” in cell survival and metastasis regulation in colorectal cancer. Mita et al. [72], Kuerbitz et al. [73] describe how “Cell Cycle” pathway is related to colorectal cancer. We did not investigate “Autophagy - animal” pathway because it is not related to the human species.
SPIA did not enrich any pathway from both CC genes lists (let see Additional file 5 for the whole enriched pathway list).
Endometrial cancer enrichment using KEGG database
Analyzing with BiP the two datasets related to the Endometrial Cancer, that is UCEC and GSE63678, we have been able to identify 276 and 252 significant pathways (p value ) respectively. Both enrichment results have been able to detect pathways linked to endometrial cancer (let see Additional file 2 for the whole enriched pathway list). Table 5 shows the BiP’s top 10 enriched pathways using both EC genes lists and the KEGG database.
Table 5.
The intersection of the first 10 pathways (sorted by the relevance of p value ) obtained from BiP by performing PEA using EC genes lists and the KEGG database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Metabolic pathways | 1.18 × 10 | 3.21 × 10 | 3.21 × 10 |
| (2) Cytokine-cytokine receptor interaction | 3.14 × 10 | 4.27 × 10 | 8.53 × 10 |
| (3) Pathways in cancer | 1.12 × 10 | 1.01 × 10 | 3.04 × 10 |
| (4) Neuroactive ligand-receptor interaction | 5.60 × 10 | 3.81 × 10 | 1.52 × 10 |
| (5) Pathways of neurodegeneration-multiple diseases | 2.09 × 10 | 1.14 × 10 | 5.69 × 10 |
| (6) Transcriptional misregulation in cancer | 7.91 × 10 | 3.59 × 10 | 2.15 × 10 |
| (7) MAPK signaling pathway | 4.41 × 10 | 1.71 × 10 | 1.20 × 10 |
| (8) NOD-like receptor signaling pathway | 2.64 × 10 | 8.98 × 10 | 7.18 × 10 |
| (9) Shigellosis | 9.48 × 10 | 2.86 × 10 | 2.58 × 10 |
| (10) Proteoglycans in cancer | 1.94 × 10 | 5.29 × 10 | 5.29 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
The BiP enrichment results comprise the “Metabolic” pathway whose role in endometrial cancer is described in [74]. Liu et al. [75] shows the involvement of the “Cytokine-cytokine receptor interaction” in the development of endometrial cancer. The Pathways in cancer is too general, making its role obvious in endometrial cancer. Ouyang et al. [76] describes the role of “Neuroactive ligand-receptor interaction” with the endometrial cancer developing. Wu et al. [77] assesses the role of “Transcriptional misregulation in cancer” pathway in metastatic endometrial cancers development. Wang et al. [78] aimed to assess the potential role of long non-coding RNA BANCR promoting endometrial cancer cell proliferation and invasion by regulating MMP2 and MMP1 via ERK/“MAPK signaling” pathway. Yang et al. [79] describes how “NOD-like receptors signaling pathway” through Hydrogen could contribute to inhibit endometrial cancer growth. Kodati et al. [80] propose an hypothesis that “shigellosis” may be the trigger for the endometriosis. Winship et al. [81] describes the role of Chondroitin sulfate “proteoglycan” protein that stimulated by interleukin 11 promotes endometrial epithelial cancer cell proliferation and migration.
Table 6 shows the top 10 pathways obtained from CePa (let see Additional file 3 for the whole enriched pathway list). As shown, the first eight enriched pathways in Table 6 are identical to the ones obtained in colorectal cancer. Wang et al. [82] shows that reprogrammed branch chain amino-acid metabolism could promote endometrial cancer cells’ proliferation. While, for the Removal of DNA patch containing an essential residue pathway there are only few evidence of its involvement in the endometrial cancer [83, 84].
Table 6.
The intersection of the first 10 pathways obtained from CePa by performing TEA using EC genes lists and the KEGG database. The table summarises the values obtained for each topology measure employed by CePa
| Name | weight | in degree | out degree | betweenness | in reachability | out reachability |
|---|---|---|---|---|---|---|
| (1) Pyrimidine biosynthesis | 0.031 | 0.029 | 0.146 | 0.037 | 0.0056 | 0.034 |
| (2) Inactivation of APC C via direct inhibition of the APC C complex | 0.044 | 0.0290 | 0.034 | 0.037 | 0.0056 | 0.034 |
| (3) Removal of licensing factors from origins | 0.031 | 0.029 | 0.034 | 0.037 | 0.047 | 0.034 |
| (4) CDT1 association with the CDC6 ORC origin complex | 0.031 | 0.029 | 0.066 | 0.062 | 0.047 | 0.034 |
| (5) NCAM signaling for neurite out growth | 0.031 | 0.029 | 0.034 | 0.085 | 0.121 | 0.034 |
| (6) Switching of origins to a post replicative state | 0.031 | 0.029 | 0.034 | 0.037 | 0.047 | 0.034 |
| (7) Fanconi Anemia pathway | 0.031 | 0.029 | 0.034 | 0.0377 | 0.047 | 0.041 |
| (8) Telomere C strand Lagging Strand Synthesis | 0.0056 | 0.062 | 0.066 | 0.0053 | 0.0056 | 0.041 |
| (9) Branched chain amino acid catabolism | 0.031 | 0.029 | 0.089 | 0.0053 | 0.047 | 0.034 |
| (10) Removal of DNA patch containing a basic residue | 0.031 | 0.062 | 0.034 | 0.085 | 0.087 | 0.034 |
Analyzing with pathDIP the two EC genes lists, that is, UCEC and GSE63678, we have been able to identify 133 and 60 significant pathways (p value ) respectively. Both enrichment results have been able to detect pathways linked to endometrial cancer. Table 7 shows the pathDIP’s top 10 enriched pathways (p value ) in both endometrial cancer genes lists (let see Additional file 4 for the whole enriched pathway list).
Table 7.
The intersection of the first 10 pathways (sorted by the relevance of p value ) obtained from pathDIP performing PEA using EC genes lists and the KEGG database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Alzheimer disease | 2.47 × 10 | 3.46 × 10 | 6.93 × 10 |
| (2) Parkinson disease | 7.46 × 10 | 5.24 × 10 | 2.10 × 10 |
| (3) Non-alcoholic fatty liver disease (NAFLD) | 9.14 × 10 | 5.13 × 10 | 2.57 × 10 |
| (4) Cellular senescence | 1.31 × 10 | 4.60 × 10 | 3.68 × 10 |
| (5) Huntington disease | 4.98 × 10 | 1.40 × 10 | 1.40 × 10 |
| (6) Human T-cell leukemia virus 1 infection | 8.80 × 10 | 2.25 × 10 | 2.47 × 10 |
| (7) Oxidative phosphorylation | 9.73 × 10 | 2.28 × 10 | 2.73 × 10 |
| (8) Apoptosis | 1.28 × 10 | 2.76 × 10 | 3.59 × 10 |
| (9) Mineral absorption | 1.69 × 10 | 3.17 × 10 | 4.75 × 10 |
| (10) Cardiac muscle contraction | 1.84 × 10 | 3.24 × 10 | 5.18 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
Among the pathways enriched by pathDIP we could not find any link for the “Alzheimer diseases”, “Parkinson diseases”, “Non-alcoholic fatty liver disease (NAFLD)”, “Human T-cell leukemia virus 1 infection”, “Cardiac muscle contraction” pathways and the endometrial cancer. Konno et al. [85] delineates the role of “Cellular senescence” pathway in suppressing proliferating, and stem cell-like phenotype of aggressive endometrial cancer cells. Nevadunsky et al. [86] assesses the effects of such “oxidative phosphorylation” in both endometrial and non-endometrial cancer types. Wong et al. [87] demonstrate that treatment based on Dichloroacetate promotes “apoptosis” in endometrial cancer. Huo et al. [88] describes the involvement of “Mineral absorption” pathway in the underlying biological mechanisms driving the tumorgenesis of endometrial cancer.
SPIA did not enrich any pathway from both EC genes lists (let see Additional file 5 for the whole enriched pathway list).
Thyroid cancer enrichment using KEGG database
Analyzing with BiP the two thyroid cancer genes lists, that is, THCA and GSE65144, we have been able to identify 280 and 54 significant pathways (p value ) respectively. Both BiP’s enrichment results have been able to detect thyroid cancer-associated pathways. Table 8 shows the BiP’s top 10 overlapping pathways (p value ) obtained from the two TC genes lists and the KEGG database (let see Additional file 2 for the whole enriched pathway list).
Table 8.
The intersection of the first 10 pathways (sorted by the relevance of p value ) obtained from BiP, performing PEA using TC genes lists and the KEGG database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Metabolic pathways | 1.59 × 10 | 4.98 × 10 | 4.98 × 10 |
| (2) Cytokine-cytokine receptor interaction | 1.02 × 10 | 1.60 × 10 | 3.21 × 10 |
| (3) Pathways in cancer | 2.50 × 10 | 2.62 × 10 | 7.85 × 10 |
| (4) Pathways of neurodegeneration-multiple diseases | 7.56 × 10 | 5.93 × 10 | 2.37 × 10 |
| (5) Ubiquitin mediated proteolysis | 1.75 × 10 | 3.44 × 10 | 5.50 × 10 |
| (6) Neuroactive ligand-receptor interaction | 2.72 × 10 | 4.75 × 10 | 8.56 × 10 |
| (7) Oxidative phosphorylation | 7.94 × 10 | 1.25 × 10 | 2.49 × 10 |
| (8) Ribosome | 2.02 × 10 | 2.89 × 10 | 6.35 × 10 |
| (9) Protein processing in endoplasmic reticulum | 1.39 × 10 | 1.62 × 10 | 4.37 × 10 |
| (10) Human immunodeficiency virus 1 infection | 2.06 × 10 | 2.16 × 10 | 6.47 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
Searching the literature, we found the following evidence for the enriched pathways listed in Table 8. Filetti et al. [89] shows that the intrathyroidal iodine “metabolism pathway” represents one of the most peculiar abnormalities present in neoplastic thyroid tissue. Feng et al. [90] describes the role of “Cytokine-cytokine receptor interaction” with thyroid cancer. The “Pathways in cancer” is too general, making its role obvious in thyroid cancer. To the best of our knowledge, we could not find any link between “Pathways of neurodegeneration - multiple diseases”, “Human immunodeficiency virus 1 infection” and thyroid cancer. Han et al. [91] delineates the link between “Ubiquitin mediated proteolysis” pathway and thyroid cancer. Xu et al. [92] describes the involvement of “neuroactive ligand-receptor interaction” in thyroid cancer. Bonara et al. [93] describe the defective “oxidative phosphorylation” in thyroid cancer associated with pathogenic mitochondrial DNA mutations. [94] describes the link between and ribosome activity to develop new diagnostic and therapeutic targets in thyroid cancers. Zhao et al. [95] presents how the tunicamycin promotes metastasis through up-regulating “endoplasmic reticulum” in thyroid carcinoma.
CePa enrichment shows nine pathways in common between colorectal and endometrial cancer. The number of shared genes among the six analyzed cancer data sets is equal to 30, a value that is not enough to support this overlap in the pathways enrichment results, obtained using these different genes lists. Table 9 shows the CePa’s top 10 pathways obtained from the intersection of the pathway enrichment results for the two TC genes lists and the KEGG database (let see Additional file 3 for the whole enriched pathway list). The remaining, “Generation of second messenger molecules” in [96] describes as the PIP3 acts as the second messenger into the thyroid cancer.
Table 9.
The 10 enriched pathways with CePa using the two TC genes lists and the KEGG database. The table summarises the values obtained for each topology measure employed by CePa
| Name | Weight | In degree | Out degree | Betweenness | In reachability | Out reachability |
|---|---|---|---|---|---|---|
| (1) Pyrimidine biosynthesis | 0.034 | 0.034 | 0.180 | 0.041 | 0.034 | 0.029 |
| (2) Inactivation of APC C via direct inhibition of the APC C complex | 0.047 | 0.0050 | 0.029 | 0.041 | 0.034 | 0.047 |
| (3) Generation of second messenger molecules | 0.034 | 0.097 | 0.0059 | 0.118 | 0.091 | 0.118 |
| (4) Removal of licensing factors from origins | 0.034 | 0.034 | 0.029 | 0.041 | 0.034 | 0.029 |
| (5) CDT1 association with the CDC6 ORC origin complex | 0.047 | 0.034 | 0.041 | 0.0058 | 0.071 | 0.029 |
| (6) Association of licensing factors with the pre replicative complex | 0.205 | 0.034 | 0.188 | 1 | 0.066 | 0.233 |
| (7) NCAM signaling for neurite out growth | 0.034 | 0.062 | 0.029 | 0.068 | 0.091 | 0.029 |
| (8) Switching of origins to a post replicative state | 0.034 | 0.034 | 0.029 | 0.041 | 0.034 | 0.029 |
| (9) Fanconi Anemia pathway | 0.034 | 0.034 | 0.029 | 0.041 | 0.034 | 0.029 |
| (10) Telomere C strand Lagging Strand Synthesis | 0.0051 | 0.065 | 0.041 | 0.082 | 0.075 | 0.029 |
Analyzing with pathDIP the two thyroid cancer genes lists, that is, THCA and GSE65144, we have been able to identifying 71 and 223 significant pathways (p value ) respectively (let see Additional file 4 for the whole enriched pathway list). Both pathDIP’s enrichment results, have been able to detect thyroid cancer associated pathways. Table 10 shows the pathDIP’s top 10 overlapping pathways (p value ) from the two TC genes lists and the KEGG database.
Table 10.
The intersection of the first 10 pathways (sorted by the relevance of p value ) obtained from pathDIP performing PEA using TC genes lists and the KEGG database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) MicroRNAs in cancer | 1.95 × 10 | 6.18 × 10 | 6.18 × 10 |
| (2)Endocytosis | 2.82 × 10 | 4.47 × 10 | 8.94 × 10 |
| (3)Pathways in cancer | 2.71 × 10 | 2.86 × 10 | 8.59 × 10 |
| (4)PI3K-Akt signaling | 4.67 × 10 | 3.70 × 10 | 1.48 × 10 |
| (5)Proteoglycans in cancer | 1.04 × 10 | 6.58 × 10 | 3.29 × 10 |
| (6)Human papillomavirus infection | 5.76 × 10 | 2.61 × 10 | 1.83 × 10 |
| (7)Focal adhesion | 5.79 × 10 | 2.29 × 10 | 1.84 × 10 |
| (8)MAPK signaling | 7.28 × 10 | 2.31 × 10 | 2.31 × 10 |
| (9)Phagosome | 2.29 × 10 | 6.59 × 10 | 7.25 × 10 |
| (10)Regulation of actin cytoskeleton | 8.49 × 10 | 1.92 × 10 | 2.69 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
To the best of our knowledge we could not find any study directly linking “MicroRNAs in cancer”, “Proteoglycans in cancer”, “Human papillomavirus infection”, “phagosome”, “Regulation of actin cytoskeleton” and the thyroid cancer. The “Pathways in cancer” is too general, making its role obvious in thyroid cancer. Theret et al. [97] describes the identification of LRP-1 as an “endocytosis” and recycling receptor for 1-integrin in thyroid cancer cells. Liu et al. [98] describes how the LncRNA modulates the cell proliferation and cancer growth of thyroid cancer through “PI3K-Akt signaling” pathway. Owens et al. [99] explained the possible role of “Focal adhesion” in a mechanism for metastasis of thyroid cancer. Eissing et al. [100] describes how Notch pathway activation by the “MAPK signaling” is responsible for thyroid cancer proliferation.
SPIA did not enrich any pathway from both TC genes lists (let see Additional file 5 for the whole enriched pathway list).
Colorectal cancer enrichment using Reactome database
Analyzing with BiP the two genes lists related to colorectal cancer -COAD and GSE41011 - and the Reactome database we identified 586 and 896 significant pathways (p value ) respectively. The enrichment results obtained by BiP point out its capability to identify some well-known colon cancer risk pathways. Table 11 shows the top 10 pathways (p value ) obtained by the intersection results from the two CC genes lists and the Reactome database.
Table 11.
The intersection of the first 10 pathways (sorted by the relevance of p value ) obtained from BiP performing PEA using CC genes lists and the Reactome database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Metabolism of proteins | 2.32 × 10 | 4.66 × 10 | 4.66 × 10 |
| (2) Post-translational protein modification | 2.13 × 10 | 2.14 × 10 | 4.29 × 10 |
| (3) Cellular responses to external stimuli | 5.70 × 10 | 3.82 × 10 | 1.14 × 10 |
| (4) Cell Cycle, Mitotic | 1.90 × 10 | 9.57 × 10 | 3.83 × 10 |
| (5) Mitotic G1-G1/S phases | 1.90 × 10 | 7.65 × 10 | 3.83 × 10 |
| (6) Cellular responses to stress | 1.98 × 10 | 6.63 × 10 | 3.98 × 10 |
| (7) Cell Cycle | 3.03 × 10 | 8.71 × 10 | 6.10 × 10 |
| (8) Mitotic G2-G2/M phases | 9.07 × 10 | 2.28 × 10 | 1.82 × 10 |
| (9) DNA Repair | 6.35 × 10 | 1.42 × 10 | 1.28 × 10 |
| (10) Cell Cycle Checkpoints | 6.55 × 10 | 1.32 × 10 | 1.32 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
Hughes et al. [101] describes the associations between “Metabolism of Proteins” pathway and the colorectal cancer. Jaén et al. [102], Tomonaga et al. [103] describe the role of “The Post-translational protein modification pathway” in the colorectal cancer. Fang et al. [104] describes the “Cellular responses to external stimuli” as a general pathway that regulates how a single cell detects and responds to external molecular and physical signals, comprising the mitogen-activated protein kinases (MAPK), the extracellular-signal-regulated kinases in colorectal cancer. Mita et al. [72], Kuerbitz et al. [73] desribe how the “Cell Cycle, Mitotic”, “Cell Cycle” and “Cell Cycle Checkpoints” pathways, are related to colorectal cancer. Furthermore, survivin-transcription is controlled by specific sequences in the promoter region, and it increases during “Mitotic G1-G1/S phases” [72, 105], and reaches a peak in “Mitotic G2-G2/M phases” [72, 106], other two pathways enriched by BiP placed in the first 10 positions by p value relevance. A recent manuscript published by Reilly et al. [107] describes as the alterations in DNA repair genes could provide new therapeutic opportunities for colorectal cancer, that is a further evidence for the “DNA repair” pathway in colorectal cancer enriched by BiP. Finally, [108] describes the role of “Cellular responses to stress” pathway in colorectal cancer.
Analyzing with pathDIP the two genes lists related to colorectal cancer, that is, COAD and GSE65144, we identified 59 and 704 significant pathways (p value ) respectively. Both pathDIP’s enrichment results, have been able to detect colorectal cancer associated pathways. Table 12 shows the top 10 overlapping pathways (p value ) obtained from the two enrichment analysis using Reactome database.
Table 12.
The intersection of the first 10 pathways (sorted by the relevance of p value ) obtained from pathDIP performing PEA using CC genes lists and the Reactome database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Metabolism of proteins | 3.18 × 10 | 6.31 × 10 | 6.31 × 10 |
| (2) Immune System | 1.46 × 10 | 9.64 × 10 | 2.89 × 10 |
| (3) Post-translational protein modification | 6.40 × 10 | 3.17 × 10 | 1.27 × 10 |
| (4) Disease | 4.02 × 10 | 1.33 × 10 | 7.96 × 10 |
| (5) Gene expression (Transcription) | 1.34 × 10 | 3.79 × 10 | 2.66 × 10 |
| (6) RNA Polymerase II Transcription | 1.01 × 10 | 2.00 × 10 | 2.00 × 10 |
| (7) Innate Immune System | 4.51 × 10 | 8.13 × 10 | 8.95 × 10 |
| (8) Cell Cycle | 2.07 × 10 | 2.93 × 10 | 4.11 × 10 |
| (9) Generic Transcription Pathway | 2.94 × 10 | 3.88 × 10 | 5.83 × 10 |
| (10) Transcriptional Regulation by TP53 | 3.27 × 10 | 3.09 × 10 | 6.48 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
Hughes et al. [101] delineates the associations between “Metabolism of Proteins” pathway and the colorectal cancer. [109] shows how the “immune system” pathway plays an integral role in preventing and promoting the development of colorectal cancer. Jaén et al. [102] and Tomonaga et al. [103] show the role of “The Post-translational protein modification pathway” in colorectal cancer. At the best of our knowledge, it was not possible to identify any link between “disease”, “Generic Transcription Pathway”, “Transcriptional Regulation by TP53” pathways and colorectal cancer. [110] investigates the effect of PI3K pathway in regulating colorectal cancer cell lines and the “gene expression” pathway. Wang et al. [111] shows that “RNA polymerase II transcription” through BTF3 contributes to primary colorectal cancer or metastasis. [112] discusses the mechanisms of colitis and colitis-associated colorectal cancer used by the innate immune system in the intestine. Mita et al. [72], Kuerbitz et al. [73] describes how the “Cell Cycle” pathway is related to colorectal cancer.
Endometrial cancer enrichment using Reactome database
Analyzing with BiP the two genes lists related to the Endometrial Cancer , that is, UCEC and GSE63678, we have been able to identify 566 and 1, 173 significant pathways (p value ) respectively. Both enrichment results have been able to detect pathways linked to endometrial cancer. Table 13 shows the top 10 enriched pathways (p value ) obtained using both EC genes lists and the Reactome database.
Table 13.
The intersection of the first 10 pathways (sorted by the relevance of p value ) obtained from BiP performing PEA using EC genes lists and the Reactome database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Metabolism of proteins | 2.35 × 10 | 4.83 × 10 | 4.83 × 10 |
| (2) Metabolism | 3.89 × 10 | 4.00 × 10 | 8.00 × 10 |
| (3) Cellular responses to external stimuli | 5.22 × 10 | 3.57 × 10 | 1.07 × 10 |
| (4) Signaling by Receptor Tyrosine Kinases | 2.82 × 10 | 1.45 × 10 | 5.80 × 10 |
| (5) Cellular responses to stress | 5.82 × 10 | 2.39 × 10 | 1.20 × 10 |
| (6) Cell Cycle | 1.30 × 10 | 4.46 × 10 | 2.67 × 10 |
| (7) Post-translational protein modification | 3.30 × 10 | 9.68 × 10 | 6.77 × 10 |
| (8) Cell Cycle, Mitotic | 7.04 × 10 | 1.81 × 10 | 1.44 × 10 |
| (9) Mitotic G1-G1/S phases | 7.04 × 10 | 1.61 × 10 | 1.44 × 10 |
| (10) Cellular Senescence | 3.54 × 10 | 7.27 × 10 | 7.27 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
Mehlen et al. [113] describes the connections between “Axon guidance” and endometrial cancer. Baylin et al. [114], Mäkinen et al. [115] report the relationships between transcription pathways (“Gene expression (Transcription)”, “RNA Polymerase II Transcription” and “Generic Transcription Pathway”) with endometrial cancer. Takai et al. [116] shows the connection between “Signaling by Receptor Tyrosine Kinases” and endometrial cancer. Yu et al. [117], Change et al. [118, 119] prove the link between “ERBB2 and ERBB4 signaling pathways” and the endometrial cancer, while [120] reviews the role of “Rho GTPases signaling” in cancer. Deregulation of the cell cycle is famously linked to cancer development [121], while the bi-directional correlation between the neural factors and cancer progression and metastasis is more recent [122].
Analyzing with pathDIP the two genes lists related to the Endometrial Cancer, that is, UCEC and GSE63678, we identified 22 and 215 significant pathways (p value ) respectively. The pathway enrichment intersection obtained from pathDIP using the two EC genes lists and the Reactome database, contains only 7 pathways in common. Table 14 shows the top 7 enriched pathways (p value ) obtained using EC genes lists and Reactome database.
Table 14.
The intersection of the first 7 pathways (sorted by the relevance of p value ) obtained from pathDIP performing PEA using EC genes lists and the Reactome database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) RNA Polymerase II Transcription | 5.04 × 10 | 7.43 × 10 | 5.94 × 10 |
| (2) Gene expression (Transcription) | 6.83 × 10 | 8.95 × 10 | 8.06 × 10 |
| (3) Generic Transcription Pathway | 1.67 × 10 | 1.51 × 10 | 1.97 × 10 |
| (4) Metabolism of proteins | 6.64 × 10 | 2.70 × 10 | 7.83 × 10 |
| (5) Immune System | 6.85 × 10 | 2.07 × 10 | 8.09 × 10 |
| (6) Innate Immune System | 4.79 × 10 | 3.25 × 10 | 1.00 |
| (7) Translation | 7.01 × 10 | 4.31 × 10 | 1.00 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
Baylin et al. [114], and Mäkinen et al. [115] delineate the relationships between transcription pathways (“Gene expression Transcription”, “RNA Polymerase II Transcription” and “Generic Transcription Pathway”) with endometrial cancer. At the best of our knowledge, it was not possible to identify any link between “Metabolism of proteins”, “Innate Immune System”, and “Translation” pathways with endometrial cancer. [123] delineates the implications of “immune system” in endometrial cancer development.
Thyroid cancer enrichment using Reactome database
Analyzing with BiP the two thyroid cancer genes lists that is, THCA and GSE65144, we identified 374 and 921 relevant pathways (p value ) respectively. Both enrichment results, have been able to detect thyroid cancer associated pathways. Table 15 shows the top 10 overlapping pathways obtained enriching the two TC genes lists and using Reactome database.
Table 15.
The first 10 enriched pathways (sorted by the relevance of p value ) obtained from BiP performing PEA using TC genes lists and the Reactome database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Mitotic Prometaphase | 7.74 × 10 | 2.52 × 10 | 1.51 × 10 |
| (2) Membrane Trafficking | 3.61 × 10 | 8.80 × 10 | 7.04 × 10 |
| (3) Signaling by Rho GTPases | 7.67 × 10 | 1.15 × 10 | 1.50 × 10 |
| (4) Post-translational protein modification | 1.49 × 10 | 1.94 × 10 | 2.91 × 10 |
| (5) Extracellular matrix organization | 1.40 × 10 | 1.36 × 10 | 2.72 × 10 |
| (6) Metabolism of proteins | 2.48 × 10 | 2.11 × 10 | 4.85 × 10 |
| (7) M Phase | 3.18 × 10 | 2.48 × 10 | 6.20 × 10 |
| (8) Cell Cycle, Mitotic | 1.17 × 10 | 7.86 × 10 | 2.28 × 10 |
| (9) Mitotic G1-G1/S phases | 1.17 × 10 | 7.59 × 10 | 2.28 × 10 |
| (10) Cell Cycle | 1.58 × 10 | 9.64 × 10 | 3.08 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
Nucera et al. [124] reports the link between “Extracellular matrix organization” and thyroid cancer. Zhong et al. [125] describes the involvement of “RHO GTPases” in thyroid cancer, while [126] describes the connection between “Membrane Trafficking” pathway and thyroid cancer. Interestingly, few pathways are linked to cell cycle mentioned above, that is well known to be deregulated in cancer (pathways: “Mitotic Prometaphase”, “M Phase”, “Cell Cycle, Mitotic”, “Mitotic G1-G1/S phase”, “Cell Cycle”). Ząbczyńska et al. [127] describes the link between Changes in the glycosylation profile (a popular type of post-translational modification (PTM) ) pathway with thyroid cancer. While we couldn’t find a link with the metabolism of proteins pathway.
Analyzing with pathDIP the two genes lists related to thyroid cancer, that is, THCA and GSE65144, we identified 28 and 405 relevant pathways (p value ) respectively. Both enrichment results, have been able to detect thyroid cancer associated pathways. Table 16 shows the top 10 overlapping pathways (p value ) using the two TC genes lists and Reactome database.
Table 16.
The first 10 enriched pathways (sorted by the relevance of p value ) obtained from pathDIP performing PEA using TC genes lists and the Reactome database
| Pathway Name | p value | FDRc | Bc |
|---|---|---|---|
| (1) Immune System | 1.56 × 10 | 2.90 × 10 | 2.90 × 10 |
| (2) Metabolism | 9.59 × 10 | 5.94 × 10 | 1.78 × 10 |
| (3) Metabolism of proteins | 6.89 × 10 | 3.20 × 10 | 1.28 × 10 |
| (4) Cell Cycle, Mitotic | 5.72 × 10 | 1.52 × 10 | 1.06 × 10 |
| (5) Post-translational protein modification | 3.01 × 10 | 6.20 × 10 | 5.58 × 10 |
| (6) Membrane Trafficking | 1.37 × 10 | 2.54 × 10 | 2.54 × 10 |
| (7) Neutrophil degranulation | 3.17 × 10 | 4.91 × 10 | 5.90 × 10 |
| (8) M Phase | 5.68 × 10 | 4.59 × 10 | 1.06 × 10 |
| (9) Disease | 7.65 × 10 | 5.07 × 10 | 1.42 × 10 |
| (10) Signaling by Receptor Tyrosine Kinases | 4.96 × 10 | 2.88 × 10 | 9.21 × 10 |
In the table, FDRc represents the corrected p value using FDR corrector, and Bc refers to the corrected p value using Bonferroni corrector
[126] describes the connection between “Membrane Trafficking” pathway and thyroid cancer. Interestingly, few pathways are linked to “cell cycle” mentioned above, that is well known to be deregulated in cancer (pathways: “Mitotic Prometaphase”, “M Phase”, “Cell Cycle, Mitotic”, “Mitotic G1-G1/S phase”, “Cell Cycle”). We couldn’t find a link between the “Metabolism”, “metabolism of proteins”, “Immune System”, “Post-translational protein modification”, “Neutrophil degranulation”, “Disease” pathways and thyroid cancer.
Pathway enrichment similarity assessment
To compare the pathway enrichment results obtained from BiP with CePa, pathDIP, and SPIA, we used the same six gene lists obtained from the cancer datasets listed in Table 1, along with the KEGG and Reactome databases.
For each couple of enrichments, we measured the total number of the enriched pathway with p value , and the number of enriched pathways that belong to the union, and the intersection between the enriched results, concerning the used database. These information have been summarized in Tables 17 and 18.
Table 17.
The number of enriched pathways obtained by using the six genes datasets and the KEGG database
| Tool | CC | EC | TC | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TCGA | GEO | TCGA | GEO | TCGA | GEO | |||||||
| NP | NP | U | I | NP | NP | U | I | NP | NP | U | I | |
| BiP | 274 | 53 | 274 | 53 | 276 | 252 | 295 | 233 | 280 | 54 | 284 | 50 |
| CePa | 23 | 25 | 27 | 21 | 23 | 25 | 27 | 21 | 23 | 25 | 28 | 20 |
| pathDIP | 52 | 290 | 291 | 51 | 133 | 60 | 152 | 41 | 71 | 233 | 226 | 68 |
| SPIA | 4 | 1 | 5 | 0 | 1 | 6 | 7 | 0 | 0 | 19 | 19 | 0 |
NP stands for Number of Enriched Pathways, U is short for Union, and I is short for Intersection
Table 18.
The number of enriched pathways obtained by using the six genes datasets and the Reactome database
| Tool | CC | EC | TC | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TCGA | GEO | TCGA | GEO | TCGA | GEO | |||||||
| NP | NP | U | I | NP | NP | U | I | NP | NP | U | I | |
| BiP | 586 | 896 | 936 | 546 | 566 | 1173 | 1184 | 555 | 374 | 921 | 930 | 365 |
| pathDIP | 59 | 703 | 730 | 32 | 22 | 215 | 230 | 7 | 28 | 405 | 414 | 19 |
NP stands for Number of Enriched Pathways, U is short for Union, and I is short for Intersection
To measure the similarity between the pairs of pathway enrichments obtained from the investigated cancer genes lists, we adopted the Jaccard index able to measure the similarity between two sets and the meet-min index that can assess the containment property between two groups. High Jaccard index values indicate that the two enrichments are similar, while high values of meet-min index suggest that the smallest set is contained in the biggest. Figures 1, 2, 3, 4, 5 and 6 show the Jaccard and meet-min indices values respectively for each software tool applied using KEGG or Reactome, to each cancer data set.
Fig. 1.

The similarity score obtained by each tool analyzing both CC gene lists using the KEGG database. The histograms show the score reached by each tool in percentage. In the Figure, JI indicates the Jaccard Index, which measures the similarity of two classes of samples. mi corresponds to the meet-min index used to quantify the containment between two sets
Fig. 2.

The similarity score obtained by each tool analyzing both EC gene lists using the KEGG database. The histograms show the score reached by each tool in percentage. In the Figure, JI indicates the Jaccard Index, which measures the similarity of two classes of samples. mi corresponds to the meet-min index used to quantify the containment between two sets
Fig. 3.

The similarity score, obtained by each tool analyzing both TC gene lists using the KEGG database. The histograms show the score reached by each tool in percentage. In the Figure, JI indicates the Jaccard Index, which measures the similarity of two classes of samples. mi corresponds to the meet-min index used to quantify the containment between two sets
Fig. 4.
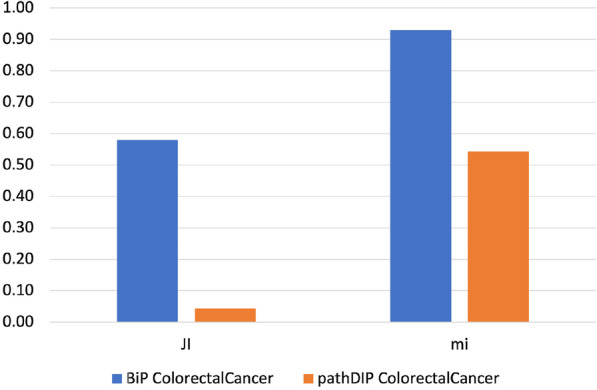
The similarity score obtained by each tool analyzing both CC gene lists using the Reactome database. The histograms show the score reached by each tool in percentage. In the Figure, JI indicates the Jaccard Index, which measures the similarity of two classes of samples. mi corresponds to the meet-min index used to quantify the containment between two sets
Fig. 5.
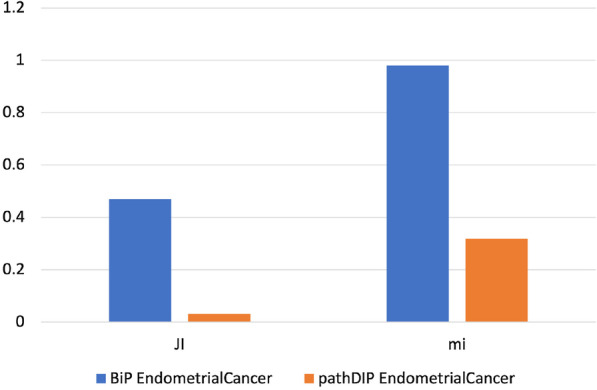
The similarity score obtained by each tool analyzing both EC gene lists using the Reactome database. The histograms show the score reached by each tool in percentage. In the Figure, JI indicates the Jaccard Index, which measures the similarity of two classes of samples. mi corresponds to the meet-min index used to quantify the containment between two sets
Fig. 6.
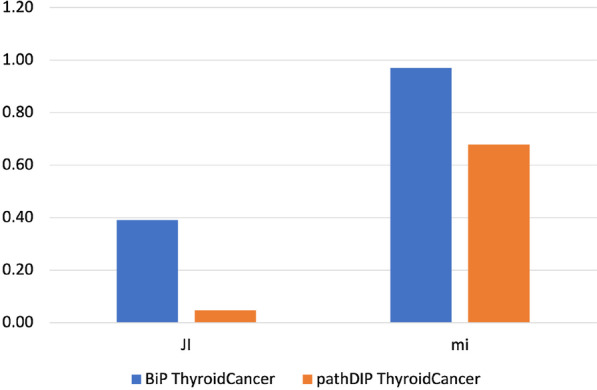
The similarity score obtained by each tool analyzing both TC gene lists using the Reactome database. The histograms show the score reached by each tool in percentage. In the Figure, JI indicates the Jaccard Index, which measures the similarity of two classes of samples. mi corresponds to the meet-min index used to quantify the containment between two set
Results show that BiP achieves the highest Jaccard and meet-min indexes values in almost all comparisons, revealing that BiP is more stable than the other methods. Only in two cases, CePa obtains better Jaccard values when enriching CC and TC gene lists. Result due to the relatively similar number of enriched pathways in the two enrichment results (see Table 17), indicating that the two sets are only in the overlap. Conversely, BiP obtains higher values of meet-min, a most suitable measure of containment between two groups.
The better enrichment stability yielded by BiP may be due to the use of updated pathway information. Pathway enrichment analysis is influenced by outdated pathway information, and the used database [26]. Outdated pathway information negatively affects pathway enrichment analysis. To use updated pathway information, BiP allows users to load and use the last version of pathway data available from Reactome, KEGG, and the other web databases compliant with the BioPAX or KGML data formats. We will add in BiP software framework a KEGG downloader module to directly download the last version of KEGG pathways, making it possible to perform pathway enrichment analysis by using pathway data coded in KGML. BiP [33] is freely available, and we are working to make the updated version of BiP available as soon as possible. Thus, updated pathway information avoids to underrate the functional significance of relevant genes, preventing the lost of some possible significant results. A consideration that could explain the different pathway enrichment results obtained by CePa and SPIA is due to the use of an outdated version of KEGG (containing only 108 pathways). Whereas BiP and pathDIP used the last version of KEGG containing 139 pathways (at the time of writing), allowing them to enrich more pathways and providing more consistent pathways enrichment concerning the investigated cancer genes list.
Discussion
To better understand the impact of the PEA on a gene or protein list of interest, we performed PEA employing the three couple of gene lists obtained from TCGA and GEO databases, respectively, related to the colorectal (CC), endometrial (EC), and thyroid (TC) cancers. The goal of PEA was to evaluate the capability of BiP to identify relevant pathways for the three types of cancer under investigation with respect to the other tools.
For the CC genes list and KEGG database, BiP enriched 274 pathways from COAD data set, and 53 pathways from GSE41011 with a p value . BiP enriched insulin signaling pathway (p value = ), Notch signaling pathway (p value = ), and apoptosis (p value = 0.002), all of which are well recognized pathways whose dysregulation can contribute in accelerate the risk of CC development and progression. It is worth to note that, in the intersection between the top 10 enriched pathway from both COAD and GSE41011 data sets, only BiP identified as novel candidate risk pathways the Metabolic pathway (p value = ) which was disregarded by the other tools, the Transcriptional misregulation in cancer (p value = ), and proteoglycans in cancer (p value = ) pathways, which were ignored by CePa and SPIA tools. The enrichment of the three pathways related to CC is also described in [128, 129]. Zhao et al. [129] illustrates a protocol to figure out some critical biomarkers associated with colorectal cancer. The authors obtain the CC differential expressed genes from the GEO database (GSE32323 data set), whereas the KEGG pathway database was used to enrich those genes. [128] shows a pipeline to investigate possible competing endogenous RNA (ceRNA) networks in CC. The authors obtain the CC differential expressed genes from the TCGA database, whereas the KEGG pathway database was used to enrich those genes. The overlap between the pathway enrichment results obtained by BiP and those in [128, 129] confirms the effectiveness of BiP in identifying pathways that play a relevant role in complex diseases.
For the EC genes list and KEGG database, BiP enriched 276 pathways from UCEC data set, and 252 pathways from GSE63678 data set with a (p value ). BiP enriched p53 signaling pathway (p value = ), VEGF signaling pathway (p value = ), and Ras signaling pathway (p value = ), all of which are well recognized pathways whose dysregulation can contribute in EC development. Noticeably, in the intersection between the top 10 enriched pathway from both data sets UCEC and GSE63678, only BIP identified as novel candidate risk pathways Metabolic pathway (p value = ), MAPK signaling pathway (p value = ) in endometrial cancer (see [130], and [131]), which were ignored by the other tools.
For the TC genes list and KEGG database, BiP enriched 280 pathways from THCA data set, and 54 pathways from GSE65144 with a ( p value ). BiP enriched Thyroid hormone pathway (p value = ), Rap1 signaling pathway (p value = ), and Parathyroid hormone synthesis, secretion and action pathway (p value = ), all of which are well recognized pathways whose dysregulation can contribute to TC development. Noticeably, in the intersection between the top 10 enriched pathways from both data sets UCEC and GSE63678, only BIP identified as novel candidate risk pathways Ubiquitin mediated proteolysis pathway (p value = ) and Oxidative phosphorylation pathway (p value = ) and reticulum pathway. [132–134] and [95] describe the involvement of those pathways in TC, which were ignored by the other tools.
Finally, we compared BiP enrichment results with those obtained by Yang et al. [135] by using the same gene lists and pathway database. It is worthy to note that BiP enriches the same relevant pathways of Yang et al. This result confirms the effectiveness of BiP in identifying pertinent pathways in the condition under investigation, even if different data sets related to the same disease, are used. The production of very different pathway enrichment results for each investigated dataset associated with the same disease may limit the usefulness of those results in clinical and therapeutic scenarios. The production of very different pathway enrichment results for each investigated dataset associated with the same disease may limit the usefulness of those results in clinical and therapeutic scenarios. We used the Jaccard and meet-min indices to better prove the stability and the similarity between the enrichment results obtained from each couple of cancer datasets. The obtained results show that BiP provides more stable enrichment results than other tools, when analyzing different genes or proteins data sets related to the same diseases.
We would highlight the key role of the metabolic pathway in reprogramming cancer cells, that might play an important role in the progress of several types of cancers. Studies on cancer metabolism show that metabolic pathways aberrations and reprogramming are necessary to sustain rapid cell proliferation, tumor progression and cell death resistance [136]. Cancer metabolic reprogramming represents a fundamental trait of most cancer cells. Several works have evidenced that this metabolic reprogramming is an active process governed by oncogenes and cancer suppressors, which provides energy to the cancer cells [137]. Indeed, La Vecchia et al. in [138] provide a review of key findings in cancer metabolism pathway as a significant contributor of tumor initiation, growth, and metastatic dissemination in CC. Also, in [139] many findings show that metabolic pathway contributes to endometrial cancer cell survival and tumor growth are provided. Finally, in [140] a recent molecular study in thyroid cancer revealed that oncogenes and tumor suppressor genes not only control growth and apoptotic phenotypes of thyroid carcinomas but also directly affect the outcome of treatment and or disease progression in thyroid cancer. Thus, it appears clear that metabolic pathway dysregulation is a crucial factor in regulating tumor progression and survival in all the investigated tumor types. Thus, the association of this pathway with all three cancers types in our study is not surprising and confirms the effectiveness of BiP in identifying pathways that play a relevant role in complex diseases.
To the best of our knowledge, we were not able to find any evidence in literature proving the involvement of Branched chain amino acid catabolism pathway in developing, sustaining or growing EC. In [141] Sweatt et al. describe the implication of Branched chain amino acid catabolism pathway to regulate anabolic signals in digestive systems, as well as in neurons of the peripheral nervous system. To the best of our knowledge, we were not able to find any evidence in literature proving the involvement of Removal of DNA patch containing a basic residue pathway with EC. We didn’t get any results even looking for the Removal of DNA patch containing a basic residue pathway in KEGG. This result point outs that CePa to perform pathway enrichment used an older version of KEGG, that impact negatively on the enrichment results, since it provide an obsolete pathway name. In the current version of KEGG the Removal of DNA patch containing a basic residue pathway may have been replaced by the DNA base excision repair pathway. Furthermore, this can contribute to explain why even using different gene lists related to different cancer types, CePa provides the same first 8 pathway in both CC and EC enrichment results. Notability, the number of genes in common among the CC and EC cancer data sets is equal to 30; value that does not justify this overlap in the pathways enrichment results using different genes data sets. We also investigated the neuroactive ligand receptor interaction pathway that is a collection of receptors located on the plasma membranes, involved in the transduction of signals from the extracellular environment into cells [142]. The neuroactive ligand-receptor interaction pathway contains a very large gene set, consisting of more than 300 genes representing a variety of signalling molecules including many types of neuroreceptors. Many of these neuroreceptor genes have been found to be associated with multiple cancers, as well as playing an important role in the development of CC [143, 144]. Thus, the association of this gene set with CC observed in our study is not surprising, and confirms the effectiveness of BiP in identifying pathways that play a key role in complex diseases.
Conclusion
In this work, we presented a comparative study of pathway enrichment analysis tools, conducted by using BiP, CePa, pathDIP and SPIA software tools. We have shown that the used database version influences the pathway enrichment results; the updated versions do not underrate the functional significance of relevant genes and proteins and do not omit the significant effects. This conclusion was specifically pronounced for CePa and SPIA because they are topology-based enrichment approaches and consequently expected to be most sensitive to the definition of a pathway available in the adopted database. At the same time, we observed that using an updated version of pathway databases (BiP) or an integrative pathway approach (pathDIP) led to more biologically consistent results.
The results obtained by BiP show good performance in terms of the number of relevant enriched pathways, especially compared to SPIA. BiP shows higher Jaccard and meet-min indexes values in almost all similarity comparisons results obtained from different gene lists of the same cancer types than CePa, pathDIP, and SPIA, suggesting BiP’s efficacy to achieve more reliable results. Of course, part of the boost in performance may be due to using more recently update versions of Reactome and KEGG databases, considering the difference in the size of the two databases.
As future works, we are improving the enrichment approach to limit the number of enriched pathways, ensuring at the same time the best possible result accuracy and improving their effectiveness. Finally, we will extend the BiP parsing capability to make it compatible with as many pathway representation formats as possible, such as SBML, GMTL, and XML-like.
Supplementary Information
Additional file 1. Pathway Enrichment Analysisuser guide illustrating how to download genes or proteins data setsfrom GEO and TCGA databases and highlighting how to use thedownloaded data sets with the surveyed PEA software tools.
Additional file 2. The list of relevant enriched pathways(p value ) obtained by BiP using each cancergene list and the related intersection between the two lists ofenriched pathways referring to the same cancer type.
Additional file 3. The list of relevant enriched pathways(p value ) obtained by CePa using each cancergene list and the related intersection between the two lists ofenriched pathways referring to the same cancer type.
Additional file 4. The list of relevant enriched pathways(p value ) obtained by pathDip using eachcancer gene list and the related intersection between the two listsof enriched pathways referring to the same cancer type.
Additional file 5. The list of relevant enriched pathways(p value ) obtained by SPIA using each cancergene list and the related intersection between the two lists ofenriched pathways referring to the same cancer type.
Acknowledgements
This work is the extended version of “Using BioPAX-Parser (BiP) to annotate lists of biological entities with pathway data” accepted at the “1st International Workshop on Conceptual Modeling for Life Sciences (CMLS 2020)”, in conjunction with the “39th International Conference on Conceptual Modeling (ER 2020)”, and published on the Lecture Notes on Computer Science (LNCS 12584) proceedings, Springer Nature.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 22, Supplement 13 2021: Selected articles from the 1st International Workshop on Conceptual Modeling for Life Sciences (CMLS 2020) - Part 1. The full contents of the supplement are available at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-22-supplement-13.
Abbreviations
- API
Application Program Interface
- BiP
BioPAX-Parser
- CC
Colorectal cancer
- CePa
Centrality-based Pathway Enrichment
- CNA
Copy-number alteration
- DEG
Differential expressed gene
- EC
Endometrial cancer
- FDR
False discovery rate
- GEO
Gene Expression Omnibus
- GSEA
Gene Set Enrichment Analysis
- GWAS
Genome-Wide Association Studies
- HT
High Throughput
- JI
Jaccard similarity Index
- KEGG
Kyoto Encyclopaedia of Gene and Genome
- mi
meet-min index
- miRNA
micro RNA
- NGS
Next Generation Sequencing
- ORA
Over Represented Analysis
- PEA
Pathway enrichment analysis
- SNPs
Single nucleotide polymorphism
- SPIA
Signaling Pathway Impact Analysis
- TC
Thyroid cancer
- TCGA
The Cancer Genome Atlas
- TEA
Topological Enrichment Analysis
Authors’ contributions
G.A. and M.C. contributed to the conceptual idea of the study. G.A. directed the writing of the manuscript. G.A. conceived and designed the experiments. G.A. performed the experimental work and analyzed the results. G.A. and M.C. wrote the paper. All authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
The datasets used and analyzed in this study are freely available in TCGA and GEO. TCGA datasets links: https://portal.gdc.cancer.gov/projects/TCGA-COAD, https://portal.gdc.cancer.gov/projects/TCGA-THCA, https://portal.gdc.cancer.gov/projects/TCGA-UCEC. GEO datasets links: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41011, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65144, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63678. Reactome database link: https://reactome.org/download-data, KEGG database link: https://www.kegg.jp, BiP software tool link: https://gitlab.com/giuseppeagapito/bip, CePa software tool link: http://cran.r-project.org/web/packages/CePa/, pathDIP software tool link: http://ophid.utoronto.ca/pathDIP, SPIA software tool link: http://bioconductor.org/packages/SPIA/. Also, all the links to the datasets and materials have been provided through the manuscript.
Declarations
Ethics approval and consent to participate
No ethics approval was required for the study.
Consent for publication
All the authors contributed to manuscript read, and approved the submitted version.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Giuseppe Agapito, Email: agapito@unicz.it.
Mario Cannataro, Email: cannataro@unicz.it.
References
- 1.Agapito G, Guzzi PH, Cannataro M. DMET-Miner: Efficient discovery of association rules from pharmacogenomic data. J Biomed Inform. 2015;56:273–283. doi: 10.1016/j.jbi.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Guzzi PH, Agapito G, Cannataro M. coresnp: Parallel processing of microarray data. IEEE Trans Comput. 2013;63(12):2961–2974. doi: 10.1109/TC.2013.176. [DOI] [Google Scholar]
- 3.Agapito G, Guzzi PH, Cannataro M. Parallel extraction of association rules from genomics data. Appl Math Comput. 2019;350:434–446. [Google Scholar]
- 4.Agapito G, Guzzi PH, Cannataro M. An efficient and scalable SPARK preprocessing methodology for Genome Wide Association Studies. In: 2020 28th Euromicro International Conference on Parallel, Distributed and Network-Based Processing (PDP); 2020. p. 369–375.
- 5.Agapito G, Milano M, Guzzi PH, Cannataro M. Extracting cross-ontology weighted association rules from gene ontology annotations. IEEE/ACM Trans Comput Biol Bioinf. 2015;13(2):197–208. doi: 10.1109/TCBB.2015.2462348. [DOI] [PubMed] [Google Scholar]
- 6.Agapito G, Cannataro M, Guzzi PH, Milano M. Using GO-WAR for mining cross-ontology weighted association rules. Comput Methods Programs Biomed. 2015;120(2):113–122. doi: 10.1016/j.cmpb.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Milano M, Milenković T, Cannataro M, Guzzi PH. L-HetnetAligner: a novel algorithm for Local Alignment of Heterogeneous Biological networks. Sci Rep. 2020;10(1):1–20. doi: 10.1038/s41598-019-56847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calimeri F, Cauteruccio F, Marzullo A, Stamile C, Terracina G. Mixing logic programming and neural networks to support neurological disorders analysis. In: International joint conference on rules and reasoning. Springer; 2018. p. 33–47.
- 9.Ogata H, Goto S, Fujibuchi W, Kanehisa M. Computation with the KEGG pathway database. Biosystems. 1998;47(1–2):119–128. doi: 10.1016/S0303-2647(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 10.Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2007;36(suppl-1):D623–D631. doi: 10.1093/nar/gkm900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33(suppl-1):D284–D288. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur Ö, Anwar N, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2010;39(suppl-1):D685–D690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, et al. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37(Database issue):D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joshi-Tope G, Gillespie M, Vastrik I, D’Eustachio P, Schmidt E, de Bono B, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33(suppl-1):D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Licata L, Lo Surdo P, Iannuccelli M, Palma A, Micarelli E, Perfetto L, et al. SIGNOR 2.0, the SIGnaling network open resource 2.0: 2019 update. Nucleic acids research. 2020;48(D1):D504–D510. [DOI] [PMC free article] [PubMed]
- 16.Pico AR, Kelder T, Van Iersel MP, Hanspers K, Conklin BR, Evelo C. WikiPathways: pathway editing for the people. PLoS Biol. 2008;6(7):e184. doi: 10.1371/journal.pbio.0060184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahmati S, Abovsky M, Pastrello C, Jurisica I. pathDIP: an annotated resource for known and predicted human gene-pathway associations and pathway enrichment analysis. Nucleic Acids Res. 2016;45(D1):D419–D426. doi: 10.1093/nar/gkw1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agapito G, Pastrello C, Guzzi PH, Jurisica I, Cannataro M. BioPAX-Parser: parsing and enrichment analysis of BioPAX pathways. Bioinformatics. 2020;36(15):4377–8. 10.1093/bioinformatics/btaa529. [DOI] [PubMed]
- 19.Gu Z, Wang J. CePa: an R package for finding significant pathways weighted by multiple network centralities. Bioinformatics. 2013;29(5):658–660. doi: 10.1093/bioinformatics/btt008. [DOI] [PubMed] [Google Scholar]
- 20.Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, et al. A novel signaling pathway impact analysis. Bioinformatics (Oxford, England) 2009;25(1):75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nørlund NE. Hypergeometric functions. Acta Mathematica. 1955;94(1):289–349. doi: 10.1007/BF02392494. [DOI] [Google Scholar]
- 22.Upton GJ. Fisher’s exact test. J R Stat Soc Ser A (Stat Soc) 1992;155(3):395–402. doi: 10.2307/2982890. [DOI] [Google Scholar]
- 23.Friedkin NE. Theoretical foundations for centrality measures. Am J Sociol. 1991;96(6):1478–1504. doi: 10.1086/229694. [DOI] [Google Scholar]
- 24.Pavlopoulos GA, Secrier M, Moschopoulos CN, Soldatos TG, Kossida S, Aerts J, et al. Using graph theory to analyze biological networks. BioData Min. 2011;4(1):1–27. doi: 10.1186/1756-0381-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demir E, Cary MP, Paley S, Fukuda K, Lemer C, Vastrik I, et al. The BioPAX community standard for pathway data sharing. Nat Biotechnol. 2010;28(9):935. doi: 10.1038/nbt.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mubeen S, Hoyt CT, Gemünd A, Hofmann-Apitius M, Fröhlich H, Domingo-Fernández D. The impact of pathway database choice on statistical enrichment analysis and predictive modeling. Front Genet. 2019;10:1203. doi: 10.3389/fgene.2019.01203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wadi L, Meyer M, Weiser J, Stein LD, Reimand J. Impact of outdated gene annotations on pathway enrichment analysis. Nat Methods. 2016;13(9):705–706. doi: 10.1038/nmeth.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le NQK, Yapp EKY, Nagasundaram N, Chua MCH, Yeh HY. Computational identification of vesicular transport proteins from sequences using deep gated recurrent units architecture. Comput Struct Biotechnol J. 2019;17:1245–1254. doi: 10.1016/j.csbj.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le NQK, Yapp EKY, Yeh HY. ET-GRU: using multi-layer gated recurrent units to identify electron transport proteins. BMC Bioinform. 2019;20(1):1–12. doi: 10.1186/s12859-019-2972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012;8(2):e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benjamin DJ, Berger JO, Johannesson M, Nosek BA, Wagenmakers EJ, Berk R, et al. Redefine statistical significance. Nat Human Behav. 2018;2(1):6–10. doi: 10.1038/s41562-017-0189-z. [DOI] [PubMed] [Google Scholar]
- 32.Agapito G, Cannataro M. Using BioPAX-Parser (BiP) to annotate lists of biological entities with pathway data. In: International conference on conceptual modeling. Springer; 2020. p. 92–101.
- 33.The BioPAX Parser Framework. https://gitlab.com/giuseppeagapito/bip. Accessed 11 Feb 2021.
- 34.The Centrality-based Pathway Enrichment (CePa) Framework. Available from: http://cran.r-project.org/web/packages/CePa/. Accessed 11 Feb 2021.
- 35.The pathDIP Framework. http://ophid.utoronto.ca/pathDIP. Accessed 11 Feb 2021.
- 36.The Signaling Pathway Impact Analysis (SPIA) Framework. Available from: http://bioconductor.org/packages/SPIA/. Accessed 11 Feb 2021.
- 37.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47(W1):W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The TCGA Colorectal Cancer (COAD) data set. Available from: https://portal.gdc.cancer.gov/projects/TCGA-COAD. Accessed 11 Feb 2021.
- 40.The GEO Colorectal Cancer (GSE41011) data set. Available from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41011. Accessed 11 Feb 2021.
- 41.The TCGA Thyroid Cancer (THCA) data set. Available from: https://portal.gdc.cancer.gov/projects/TCGA-THCA. Accessed 11 Feb 2021.
- 42.The GEO Thyroid Cancer (GSE65144) data set. Available from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65144. Accessed 11 Feb 2021.
- 43.The TCGA Endometrial Cancer (UCEC) data set. Available from: https://portal.gdc.cancer.gov/projects/TCGA-UCEC. Accessed 11 Feb 2021.
- 44.The GEO Endometrial Cancer (GSE63678) data set. Available from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63678. Accessed 11 Feb 2021.
- 45.The Reactome Pathway Database. Available from: https://reactome.org/download-data. Accessed 11 Feb 2021.
- 46.The Kyoto Encyclopedia of Genes and Genomes Pathway Database. Available from: https://www.kegg.jp. Accessed 11 Feb 2021.
- 47.The cBioPortal for Cancer Genomics. Available from: https://www.cbioportal.org/. Accessed 11 Feb 2021.
- 48.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1–pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.The GEO2R Portal. Available from: https://www.ncbi.nlm.nih.gov/geo/geo2r/. Accessed 11 Feb 2021.
- 50.Meng F, Dai E, Yu X, Zhang Y, Chen X, Liu X, et al. Constructing and characterizing a bioactive small molecule and microRNA association network for Alzheimer’s disease. J R Soc Interface. 2014;11(92):20131057. doi: 10.1098/rsif.2013.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.La Vecchia S, Sebastián C. Metabolic pathways regulating colorectal cancer initiation and progression. In: Seminars in cell and developmental biology. vol. 98. Elsevier; 2020. p. 63–70. [DOI] [PubMed]
- 52.Andersen CL, Christensen LL, Thorsen K, Schepeler T, Sørensen FB, Verspaget HW, et al. Dysregulation of the transcription factors SOX4, CBFB and SMARCC1 correlates with outcome of colorectal cancer. Br J Cancer. 2009;100(3):511–523. doi: 10.1038/sj.bjc.6604884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Slattery ML, Mullany LE, Sakoda LC, Wolff RK, Samowitz WS, Herrick JS. The MAPK-signaling pathway in colorectal cancer: dysregulated genes and their association with micrornas. Cancer Inform. 2018;17:1176935118766522. doi: 10.1177/1176935118766522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Lu R, Fu X, Dan Z, Zhang YG, Chang X, et al. Novel regulatory roles of Wnt1 in infection-associated colorectal cancer. Neoplasia. 2018;20(5):499–509. doi: 10.1016/j.neo.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang QR, Pan XB. Prognostic lncRNAs, miRNAs, and mRNAs form a competing endogenous RNA network in colon cancer. Front Oncol. 2019;9:712. doi: 10.3389/fonc.2019.00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harkins L, Volk AL, Samanta M, Mikolaenko I, Britt WJ, Bland KI, et al. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet. 2002;360(9345):1557–1563. doi: 10.1016/S0140-6736(02)11524-8. [DOI] [PubMed] [Google Scholar]
- 57.Vicente CM, da Silva DA, Sartorio PV, Silva TD, Saad SS, Nader HB, et al. Heparan sulfate proteoglycans in human colorectal cancer. Anal Cell Pathol. 2018. 10.1155/2018/8389595. [DOI] [PMC free article] [PubMed]
- 58.Fernandes Q, Gupta I, Vranic S, Al Moustafa AE. Human papillomaviruses and Epstein-Barr virus interactions in colorectal cancer: a brief review. Pathogens. 2020;9(4):300. doi: 10.3390/pathogens9040300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katoh M, Nakagama H. FGF receptors: cancer biology and therapeutics. Med Res Rev. 2014;34(2):280–300. doi: 10.1002/med.21288. [DOI] [PubMed] [Google Scholar]
- 60.Chaffer CL, Dopheide B, Savagner P, Thompson EW, Williams ED. Aberrant fibroblast growth factor receptor signaling in bladder and other cancers. Differentiation. 2007;75(9):831–842. doi: 10.1111/j.1432-0436.2007.00210.x. [DOI] [PubMed] [Google Scholar]
- 61.Satoh K, Yachida S, Sugimoto M, Oshima M, Nakagawa T, Akamoto S, et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Nat Acad Sci. 2017;114(37):E7697–E7706. doi: 10.1073/pnas.1710366114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim IY, Kwon HY, Park KH, Kim DS. Anaphase-promoting complex 7 is a prognostic factor in human colorectal cancer. Ann Coloproctol. 2017;33(4):139. doi: 10.3393/ac.2017.33.4.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsaniras SC, Kanellakis N, Symeonidou I, Nikolopoulou P, Lygerou Z, Taraviras S. Licensing of DNA replication, cancer, pluripotency and differentiation: an interlinked world? In: Seminars in cell and developmental biology. vol. 30. Elsevier; 2014. p. 174–180. [DOI] [PubMed]
- 64.Bernal A, Tusell L. Telomeres: implications for cancer development. Int J Mol Sci. 2018;19(1):294. doi: 10.3390/ijms19010294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tong X, Zhao F, Mancuso A, Gruber JJ, Thompson CB. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc Nat Acad Sci. 2009;106(51):21660–21665. doi: 10.1073/pnas.0911316106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Esteban-Jurado C, Franch-Expósito S, Muñoz J, Ocaña T, Carballal S, López-Cerón M, et al. The Fanconi anemia DNA damage repair pathway in the spotlight for germline predisposition to colorectal cancer. Eur J Human Genet. 2016;24(10):1501–5. 10.1038/ejhg.2016.44. [DOI] [PMC free article] [PubMed]
- 67.Fernández-Briera A, García-Parceiro I, Cuevas E, Gil-Martín E. Effect of human colorectal carcinogenesis on the neural cell adhesion molecule expression and polysialylation. Oncology. 2010;78(3–4):196–204. doi: 10.1159/000313699. [DOI] [PubMed] [Google Scholar]
- 68.Park BG, Kim YJ, Min JH, Cheong TC, Nam SH, Cho NH, et al. Assessment of cellular uptake efficiency according to multiple inhibitors of Fe 3 O 4-Au core-shell nanoparticles: possibility to control specific endocytosis in colorectal cancer cells. Nanoscale Res Lett. 2020;15(1):1–10. doi: 10.1186/s11671-020-03395-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun J, Cheng X, Pan S, Wang L, Dou W, Liu J, et al. Dichloroacetate attenuates the stemness of colorectal cancer cells via trigerring ferroptosis through sequestering iron in lysosomes. Environ Toxicol. 2020;36(4):520–9. 10.1002/tox.23057. [DOI] [PubMed]
- 70.Grady W, Parkin R, Mitchell P, Lee J, Kim Y, Tsuchiya K, et al. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene. 2008;27(27):3880–3888. doi: 10.1038/onc.2008.10. [DOI] [PubMed] [Google Scholar]
- 71.Agarwal E, Brattain MG, Chowdhury S. Cell survival and metastasis regulation by Akt signaling in colorectal cancer. Cell Signal. 2013;25(8):1711–1719. doi: 10.1016/j.cellsig.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14(16):5000–5005. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- 73.Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci. 1992;89(16):7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang HP, Gonzalez Bosquet J, Li Q, Platz EA, Brinton LA, Sherman ME, et al. Common genetic variation in the sex hormone metabolic pathway and endometrial cancer risk: pathway-based evaluation of candidate genes. Carcinogenesis. 2010;31(5):827–833. doi: 10.1093/carcin/bgp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu A, Zhang D, Yang X, Song Y. Estrogen receptor alpha activates MAPK signaling pathway to promote the development of endometrial cancer. J Cell Biochem. 2019;120(10):17593–17601. doi: 10.1002/jcb.29027. [DOI] [PubMed] [Google Scholar]
- 76.Ouyang D, Li R, Li Y, Zhu X. A 7-lncRNA signature predict prognosis of Uterine corpus endometrial carcinoma. J Cell Biochem. 2019;120(10):18465–18477. doi: 10.1002/jcb.29164. [DOI] [PubMed] [Google Scholar]
- 77.Wu X, Miao J, Jiang J, Liu F. Analysis of methylation profiling data of hyperplasia and primary and metastatic endometrial cancers. Eur J Obstetr Gynecol Reprod Biol. 2017;217:161–166. doi: 10.1016/j.ejogrb.2017.08.036. [DOI] [PubMed] [Google Scholar]
- 78.Wang D, Wang D, Wang N, Long Z, Ren X. Long non-coding RNA BANCR promotes endometrial cancer cell proliferation and invasion by regulating MMP2 and MMP1 via ERK/MAPK signaling pathway. Cell Physiol Biochem. 2016;40(3–4):644–656. doi: 10.1159/000452577. [DOI] [PubMed] [Google Scholar]
- 79.Yang Y, Liu PY, Bao W, Chen SJ, Wu FS, Zhu PY. Hydrogen inhibits endometrial cancer growth via a ROS/NLRP3/caspase-1/GSDMD-mediated pyroptotic pathway. BMC Cancer. 2020;20(1):28. doi: 10.1186/s12885-019-6491-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kodati V, Govindan S, Movva S, Ponnala S, Hasan Q. Role of Shigella infection in endometriosis: a novel hypothesis. Med Hypotheses. 2008;70(2):239–243. doi: 10.1016/j.mehy.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 81.Winship A, Van Sinderen M, Heffernan-Marks A, Dimitriadis E. Chondroitin sulfate proteoglycan protein is stimulated by interleukin 11 and promotes endometrial epithelial cancer cell proliferation and migration. Int J Oncol. 2017;50(3):798–804. doi: 10.3892/ijo.2017.3848. [DOI] [PubMed] [Google Scholar]
- 82.Wang P, Wu S, Zeng X, Zhang Y, Zhou Y, Su L, et al. BCAT1 promotes proliferation of endometrial cancer cells through reprogrammed BCAA metabolism. Int J Clin Exp Pathol. 2018;11(12):5536–+. [PMC free article] [PubMed]
- 83.Hosono S, Matsuo K, Ito H, Oze I, Hirose K, Watanabe M, et al. Polymorphisms in base excision repair genes are associated with endometrial cancer risk among postmenopausal Japanese women. Int J Gynecol Cancer. 2013;23(9):1561–1568. doi: 10.1097/IGC.0b013e3182a80a7e. [DOI] [PubMed] [Google Scholar]
- 84.Krupa R, Sobczuk A, Poplawski T, Wozniak K, Blasiak J. DNA damage and repair in endometrial cancer in correlation with the hOGG1 and RAD51 genes polymorphism. Mol Biol Rep. 2011;38(2):1163–1170. doi: 10.1007/s11033-010-0214-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Konno Y, Dong P, Xiong Y, Suzuki F, Lu J, Cai M, et al. MicroRNA-101 targets EZH2, MCL-1 and FOS to suppress proliferation, invasion and stem cell-like phenotype of aggressive endometrial cancer cells. Oncotarget. 2014;5(15):6049. doi: 10.18632/oncotarget.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nevadunsky NS, Van Arsdale A, Strickler HD, Moadel A, Kaur G, Frimer M, et al. Metformin use and endometrial cancer survival. Gynecol Oncol. 2014;132(1):236–240. doi: 10.1016/j.ygyno.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008;109(3):394–402. doi: 10.1016/j.ygyno.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huo X, Sun H, Liu Q, Ma X, Peng P, Yu M, et al. Clinical and expression significance of AKT1 by co-expression network analysis in endometrial cancer. Front Oncol. 2019;9:1147. doi: 10.3389/fonc.2019.01147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Filetti S, Bidart JM, Arturi F, Caillou B, Russo D, Schlumberger M, et al. Sodium/iodide symporter: a key transport system in thyroid cancer cell metabolism. Eur J Endocrinol. 1999;141(5):443–457. doi: 10.1530/eje.0.1410443. [DOI] [PubMed] [Google Scholar]
- 90.Feng X, Li T, Liu Z, Shi Y, Peng Y. HOXC10 up-regulation contributes to human thyroid cancer and indicates poor survival outcome. Mol BioSyst. 2015;11(11):2946–2954. doi: 10.1039/C5MB00253B. [DOI] [PubMed] [Google Scholar]
- 91.Han CG, Huang Y, Qin L. Long non-coding RNA ZFAS1 as a novel potential biomarker for predicting the prognosis of thyroid cancer. Med Sci Monit Int Med J Exp Clin Res. 2019;25:2984. doi: 10.12659/MSM.912921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu Y, Chen J, Yang Z, Xu L. Identification of RNA expression profiles in thyroid cancer to construct a competing endogenous RNA (ceRNA) network of mRNAs, long noncoding RNAs (lncRNAs), and microRNAs (miRNAs) Med Sci Monit Int Med J Exp Clin Res. 2019;25:1140. doi: 10.12659/MSM.912450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bonora E, Porcelli AM, Gasparre G, Biondi A, Ghelli A, Carelli V, et al. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res. 2006;66(12):6087–6096. doi: 10.1158/0008-5472.CAN-06-0171. [DOI] [PubMed] [Google Scholar]
- 94.Jeong S, Kim IK, Kim H, Choi MJ, Lee J, Jo YS. Liver X receptor related to tumor progression and ribosome gene expression in papillary thyroid cancer. Endocrinol Metabol. 2020;35(3):656. doi: 10.3803/EnM.2020.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao G, Kang J, Xu G, Wei J, Wang X, Jing X, et al. Tunicamycin promotes metastasis through upregulating endoplasmic reticulum stress induced GRP78 expression in thyroid carcinoma. Cell Biosci. 2020;10(1):1–10. doi: 10.1186/s13578-019-0370-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alsina J, Alsina R, Gulec S. A concise atlas of thyroid cancer next-generation sequencing panel ThyroSeq v.2. Mol Imaging Radionuclide Therapy. 2017;26(Suppl 1):102–117. [DOI] [PMC free article] [PubMed]
- 97.Theret L, Jeanne A, Langlois B, Hachet C, David M, Khrestchatisky M, et al. Identification of LRP-1 as an endocytosis and recycling receptor for 1-integrin in thyroid cancer cells. Oncotarget. 2017;8(45):78614. doi: 10.18632/oncotarget.20201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu H, Deng H, Zhao Y, Li C, Liang Y. LncRNA XIST/miR-34a axis modulates the cell proliferation and tumor growth of thyroid cancer through MET-PI3K-AKT signaling. J Exp Clin Cancer Res. 2018;37(1):1–12. doi: 10.1186/s13046-017-0664-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Owens LV, Xu L, Dent GA, Yang X, Sturge GC, Craven RJ, et al. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Ann Surg Oncol. 1996;3(1):100–105. doi: 10.1007/BF02409059. [DOI] [PubMed] [Google Scholar]
- 100.Eissing L, Scherer T, Todter K, Knippschild U, Greve JW, Buurman WA, et al. De novo lipogenesis in human fat and liver is linked to ChREBP- Band metabolic health. Nat Commun. 2013;4:1528 EP. 10.1038/ncomms2537. [DOI] [PMC free article] [PubMed]
- 101.Hughes R, Magee E, Bingham S, et al. Protein degradation in the large intestine: relevance to colorectal cancer. Curr Issues Intestinal Microbiol. 2000;1(2):51–58. [PubMed] [Google Scholar]
- 102.Jaén RI, Prieto P, Casado M, Martín-Sanz P, Boscá L. Post-translational modifications of prostaglandin-endoperoxide synthase 2 in colorectal cancer: an update. World J Gastroenterol. 2018;24(48):5454–5461. doi: 10.3748/wjg.v24.i48.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tomonaga T, Matsushita K, Yamaguchi S, Oh-Ishi M, Kodera Y, Maeda T, et al. Identification of altered protein expression and post-translational modifications in primary colorectal cancer by using agarose two-dimensional gel electrophoresis. Clin Cancer Res. 2004;10(6):2007–2014. doi: 10.1158/1078-0432.CCR-03-0321. [DOI] [PubMed] [Google Scholar]
- 104.Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6(5):322–327. doi: 10.1016/S1470-2045(05)70168-6. [DOI] [PubMed] [Google Scholar]
- 105.Park J, Cartwright CA. Src activity increases and Yes activity decreases during mitosis of human colon carcinoma cells. Mol Cell Biol. 1995;15(5):2374–2382. doi: 10.1128/MCB.15.5.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Simão ÉM, Sinigaglia M, Bugs CA, Castro MAA, Librelotto GR, Alves R, et al. Induced genome maintenance pathways in pre-cancer tissues describe an anti-cancer barrier in tumor development. Mol BioSyst. 2012;8(11):3003–3009. doi: 10.1039/c2mb25242b. [DOI] [PubMed] [Google Scholar]
- 107.Reilly NM, Novara L, Di Nicolantonio F, Bardelli A. Exploiting DNA repair defects in colorectal cancer. Mol Oncol. 2019 doi: 10.1002/1878-0261.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fung KY, Brierley GV, Henderson S, Hoffmann P, McColl SR, Lockett T, et al. Butyrate-induced apoptosis in HCT116 colorectal cancer cells includes induction of a cell stress response. J Proteome Res. 2011;10(4):1860–1869. doi: 10.1021/pr1011125. [DOI] [PubMed] [Google Scholar]
- 109.Markman JL, Shiao SL. Impact of the immune system and immunotherapy in colorectal cancer. J Gastrointest Oncol. 2015;6(2):208. doi: 10.3978/j.issn.2078-6891.2014.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Agarwal A, Das K, Lerner N, Sathe S, Cicek M, Casey G, et al. The AKT/I B kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor- B and -catenin. Oncogene. 2005;24(6):1021–1031. doi: 10.1038/sj.onc.1208296. [DOI] [PubMed] [Google Scholar]
- 111.Wang CJ, Frånbergh-Karlson H, Wang DW, Arbman G, Zhang H, Sun XF. Clinicopathological significance of BTF3 expression in colorectal cancer. Tumor Biol. 2013;34(4):2141–2146. doi: 10.1007/s13277-013-0745-8. [DOI] [PubMed] [Google Scholar]
- 112.Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11(1):9–20. doi: 10.1038/nri2891. [DOI] [PubMed] [Google Scholar]
- 113.Mehlen P, Llambi F. Role of netrin-1 and netrin-1 dependence receptors in colorectal cancers. Br J Cancer. 2005;93(1):1. doi: 10.1038/sj.bjc.6602656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001;10(7):687–692. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- 115.Mäkinen N, Mehine M, Tolvanen J, Kaasinen E, Li Y, Lehtonen HJ, et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science. 2011;334(6053):252–255. doi: 10.1126/science.1208930. [DOI] [PubMed] [Google Scholar]
- 116.Takai N, Miyazaki T, Fujisawa K, Nasu K, Miyakawa I. Expression of receptor tyrosine kinase EphB4 and its ligand ephrin-B2 is associated with malignant potential in endometrial cancer. Oncol Rep. 2001;8(3):567–573. doi: 10.3892/or.8.3.567. [DOI] [PubMed] [Google Scholar]
- 117.Yu T, Li J, Yan M, Liu L, Lin H, Zhao F, et al. MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4, PIK3R3, mTOR, S6K2 signaling pathway. Oncogene 2014;34:413 EP. 10.1038/onc.2013.574. [DOI] [PubMed]
- 118.Chang JL, Tsao YP, Liu DW, Han CP, Lee WH, Chen SL. The expression of type I growth factor receptors in the squamous neoplastic changes of uterine cervix. Gynecologic Oncol. 1999;73(1):62–71. doi: 10.1006/gyno.1998.5301. [DOI] [PubMed] [Google Scholar]
- 119.The activation of an extracellular signal-regulated kinase by oestradiol interferes with the effects of trastuzumab on HER2 signalling in endometrial adenocarcinoma cell lines. Eur J Cancer. 2003;39(9):1302–1309. [DOI] [PubMed]
- 120.Porter AP, Papaioannou A, Malliri A. Deregulation of Rho GTPases in cancer. Small GTPases. 2016;7(3):123–138. doi: 10.1080/21541248.2016.1173767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res. 2017;7(5):1016–1036. [PMC free article] [PubMed] [Google Scholar]
- 122.Kuol N, Stojanovska L, Apostolopoulos V, Nurgali K. Role of the nervous system in cancer metastasis. J Exp Clin Cancer Res. 2018;37(1):5. doi: 10.1186/s13046-018-0674-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vanderstraeten A, Tuyaerts S, Amant F. The immune system in the normal endometrium and implications for endometrial cancer development. J Reprod Immunol. 2015;109:7–16. doi: 10.1016/j.jri.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 124.Nucera C, Lawler J, Hodin R, Parangi S. The BRAFV600E mutation: what is it really orchestrating in thyroid cancer? Oncotarget. 2010;1(8):751–756. doi: 10.18632/oncotarget.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhong WB, Liang Y, Wang CY, Chang TC, Lee W. Lovastatin suppresses invasiveness of anaplastic thyroid cancer cells by inhibiting Rho geranylgeranylation and RhoA/ROCK signaling. Endocr Relat Cancer. 2005;12(3):615–629. doi: 10.1677/erc.1.01012. [DOI] [PubMed] [Google Scholar]
- 126.Jung EJ, Moon HG, Park ST, Cho BI, Lee SM, Jeong CY, et al. Decreased annexin A3 expression correlates with tumor progression in papillary thyroid cancer. PROTEOMICS-Clin Appl. 2010;4(5):528–537. doi: 10.1002/prca.200900063. [DOI] [PubMed] [Google Scholar]
- 127.Ząbczyńska M, Kozłowska K, Pocheć E. Glycosylation in the thyroid gland: vital aspects of glycoprotein function in thyrocyte physiology and thyroid disorders. Int J Mol Sci. 2018;19(9):2792. doi: 10.3390/ijms19092792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liang Y, Zhang C, Ma MH, Dai DQ. Identification and prediction of novel non-coding and coding RNA-associated competing endogenous RNA networks in colorectal cancer. World J Gastroenterol. 2018;24(46):5259. doi: 10.3748/wjg.v24.i46.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao ZW, Fan XX, Yang LL, Song JJ, Fang SJ, Tu JF, et al. The identification of a common different gene expression signature in patients with colorectal cancer. Math Biosci Eng. 2019;16(4):2942–2958. doi: 10.3934/mbe.2019145. [DOI] [PubMed] [Google Scholar]
- 130.Guo JL, Tang T, Li JH, Yang YH, Zhang L, Quan Y. LncRNA HEIH enhances paclitaxel-tolerance of endometrial cancer cells via activation of MAPK signaling pathway. Pathol Oncol Res. 2020;26(3):1757–1766. doi: 10.1007/s12253-019-00718-w. [DOI] [PubMed] [Google Scholar]
- 131.Shen J, Chen L, Cheng J, Jin X, Mu Y, Li Q, et al. Circular RNA sequencing reveals the molecular mechanism of the effects of acupuncture and moxibustion on endometrial receptivity in patients undergoing infertility treatment. Mol Med Rep. 2019;20(2):1959–1965. doi: 10.3892/mmr.2019.10386. [DOI] [PubMed] [Google Scholar]
- 132.Ding YG, Ren YL, Xu YS, Wei CS, Zhang YB, Zhang SK, et al. Identification of key candidate genes and pathways in anaplastic thyroid cancer by bioinformatics analysis. Am J Otolaryngol. 2020;41(3):102434. doi: 10.1016/j.amjoto.2020.102434. [DOI] [PubMed] [Google Scholar]
- 133.Thakur S, Daley B, Gaskins K, Vasko VV, Boufraqech M, Patel D, et al. Metformin targets mitochondrial glycerophosphate dehydrogenase to control rate of oxidative phosphorylation and growth of thyroid cancer in vitro and in vivo. Clin Cancer Res. 2018;24(16):4030–4043. doi: 10.1158/1078-0432.CCR-17-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kumari S, Adewale R, Klubo-Gwiezdzinska J. The molecular landscape of Hürthle cell thyroid cancer is associated with altered mitochondrial function-a comprehensive review. Cells. 2020;9(7):1570. doi: 10.3390/cells9071570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang Q, Wang S, Dai E, Zhou S, Liu D, Liu H, et al. Pathway enrichment analysis approach based on topological structure and updated annotation of pathway. Briefings Bioinform. 2019;20(1):168–177. doi: 10.1093/bib/bbx091. [DOI] [PubMed] [Google Scholar]
- 136.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. 2017;168(4):657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.La Vecchia S, Sebastián C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin Cell Dev Biolo. 2020;98:63–70. SI: Cancer Cells and Therapeutic Targets. [DOI] [PubMed]
- 139.Byrne FL, Poon IKH, Modesitt SC, Tomsig JL, Chow JDY, Healy ME, et al. Metabolic vulnerabilities in endometrial cancer. Cancer Res. 2014;74(20):5832–5845. doi: 10.1158/0008-5472.CAN-14-0254. [DOI] [PubMed] [Google Scholar]
- 140.Coelho RG, Fortunato RS, Carvalho DP. Metabolic reprogramming in thyroid carcinoma. Front Oncol. 2018;8:82. doi: 10.3389/fonc.2018.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sweatt AJ, Wood M, Suryawan A, Wallin R, Willingham MC, Hutson SM. Branched-chain amino acid catabolism: unique segregation of pathway enzymes in organ systems and peripheral nerves. Am J Physiol Endocrinol Metabol. 2004;286(1):E64–E76. doi: 10.1152/ajpendo.00276.2003. [DOI] [PubMed] [Google Scholar]
- 142.Lauss M, Kriegner A, Vierlinger K, Noehammer C. Characterization of the drugged human genome. Pharmacogenomics. 2007;8(8):1063–1073. doi: 10.2217/14622416.8.8.1063. [DOI] [PubMed] [Google Scholar]
- 143.Fu T, Song W, Ren J, Wang C, Ge Y. Analysis of circular RNA-related competing endogenous RNA identifies the immune-related risk signature for colorectal cancer. Front Genet. 2020;11:505. doi: 10.3389/fgene.2020.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu BX, Huang GJ, Cheng HB. Comprehensive analysis of core genes and potential mechanisms in rectal cancer. J Comput Biol. 2019;26(11):1262–1277. doi: 10.1089/cmb.2019.0073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Pathway Enrichment Analysisuser guide illustrating how to download genes or proteins data setsfrom GEO and TCGA databases and highlighting how to use thedownloaded data sets with the surveyed PEA software tools.
Additional file 2. The list of relevant enriched pathways(p value ) obtained by BiP using each cancergene list and the related intersection between the two lists ofenriched pathways referring to the same cancer type.
Additional file 3. The list of relevant enriched pathways(p value ) obtained by CePa using each cancergene list and the related intersection between the two lists ofenriched pathways referring to the same cancer type.
Additional file 4. The list of relevant enriched pathways(p value ) obtained by pathDip using eachcancer gene list and the related intersection between the two listsof enriched pathways referring to the same cancer type.
Additional file 5. The list of relevant enriched pathways(p value ) obtained by SPIA using each cancergene list and the related intersection between the two lists ofenriched pathways referring to the same cancer type.
Data Availability Statement
The datasets used and analyzed in this study are freely available in TCGA and GEO. TCGA datasets links: https://portal.gdc.cancer.gov/projects/TCGA-COAD, https://portal.gdc.cancer.gov/projects/TCGA-THCA, https://portal.gdc.cancer.gov/projects/TCGA-UCEC. GEO datasets links: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41011, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65144, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63678. Reactome database link: https://reactome.org/download-data, KEGG database link: https://www.kegg.jp, BiP software tool link: https://gitlab.com/giuseppeagapito/bip, CePa software tool link: http://cran.r-project.org/web/packages/CePa/, pathDIP software tool link: http://ophid.utoronto.ca/pathDIP, SPIA software tool link: http://bioconductor.org/packages/SPIA/. Also, all the links to the datasets and materials have been provided through the manuscript.