

Abstract
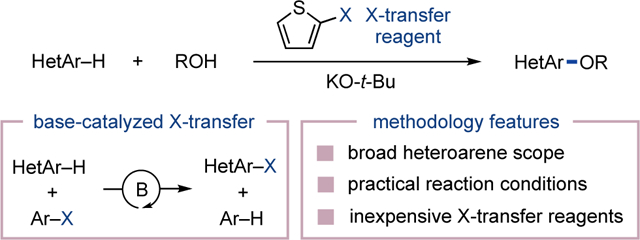
We report a general protocol for the direct C–H etherification of N-heteroarenes. Potassium tert-butoxide catalyzes halogen transfer from 2-halothiophenes to N-heteroarenes to form N-heteroaryl halide intermediates that undergo tandem base-promoted alcohol substitution. Thus, the simple inclusion of inexpensive 2-halothiophenes enables regioselective oxidative coupling of alcohols with 1,3-azoles, pyridines, diazines and polyazines under basic reaction conditions.
Graphical Abstract
The oxidative coupling of aryl C–H bonds with alcohols is an ideal synthetic approach to aromatic ethers that eliminates the need for preinstalled functional groups.1 Such methodology can facilitate late-stage derivatization of complex arenes and resource-efficient large-scale syntheses.2 While significant progress has been made for the C–H etherification of benzene derivatives, there are no direct protocols applicable to a broad range of N-heteroaryl substrates, such as 1,3-azoles and (di)azines.1,3–5 A method that addresses this limitation would streamline access to N-heteroaryl ethers that have wide value, especially in pharmaceuticals and agrochemicals.6
A key challenge for direct C–H etherification is the incompatibility of combining a C–H oxidative process with nucleophilic alcohol coupling partners.7 Thus, existing routes for N-heteroaryl C–H etherification require independent synthesis of a functionalized arene followed by an alcohol substitution reaction. A common strategy in this regard involves heteroaryl N-oxide or N-triflyl formation en route to nitro-, cyano- or phosphonium-substituted azines.8–10 Alternatively, a two-step metalation/halogenation sequence provides heteroaryl halides; however, this often requires irreversible metalation and a directing group for good selectivity.11–13 Thus, a new activation strategy is likely required if N-heteroaryl C–H etherification is to be achieved in a single operation. In addition to increased synthetic efficiency, a new oxidative coupling method could complement the scope, selectivity and practicality of existing routes to aromatic ethers.
In 1951, Nord disclosed the unexpected observation that 2-bromothiophene disproportionates in the presence of strong base (Figure 1a).14 This report represents the first known example of base-catalyzed aromatic halogen transfer, a mechanistic process now widely used in the context of halogen dance methodology.15,16 We realized the intermolecular nature of aromatic halogen transfer could be exploited in a new C–H functionalization strategy as shown in Figure 1b. Here, base-catalyzed halogen transfer from a sacrificial aryl halide (Ar–X) to an N-heteroarene (HetAr–H) is sequenced with a substitution reaction of the N-heteroaryl halide intermediate.
Figure 1.

The use of base-catalyzed halogen transfer (X-transfer) for a new heteroarene C–H etherification reaction.
Merging halogen transfer with a substitution reaction is a critical design feature for an overall high-yielding and selective C–H functionalization reaction.17 In the absence of a thermodynamic driving force, aromatic halogen transfer is expected to be reversible and unselective while generating N-heteroaryl halides prone to undesired side reactions.18–20 A separate challenge for the proposed method is identification of an aryl halide that efficiently transfers a halogen to transient N-heteroaryl anions but does not decompose or undergo nucleophilic substitution under basic conditions. Inspired by Nord’s seminal report, we herein disclose that 2-halothiophenes fulfill this role to enable regioselective N-heteroarene C–H etherification (Figure 1b, bottom).
We first targeted the 2-selective C–H etherification of 1,3-azoles for development as there is currently no general protocol for this coupling process.21,22 Optimization of a model reaction between thiazole (1) and 2-ethyl-1-hexanol (2) is shown in Scheme 1a using KO-t-Bu as base.23 We began by examining 2-bromothiophenes as potential oxidants, which are inert towards nucleophilic aromatic substitution.24 Use of 2-bromothiophene (4) provides 16% etherification yield and a small polybromothiophene screen found that inexpensive 2,5-dibromothiophene (7, <$40 per mole) increases the yield to 74%. Use of common halogenating reagents (e.g. CBr4 and NBS) as oxidants results in no product formation, demonstrating the unique efficacy of 2-bromothiophene reagents under these conditions.25
Scheme 1. Optimization of halogen transfer reagents for the C–H etherification of (a) thiazole and (b) 3-bromopyridine.a.

aFor all reactions, 1 equiv of heteroarene and 1.5 equiv of alcohol used. Yields determined by 1H NMR spectroscopy of the crude reaction mixture. See Supporting Information for full optimization details. b65% yield without 18-crown-6 additive.
We reasoned this functionalization strategy could operate on other distinct classes of N-heteroarenes, including pyridine derivatives (Scheme 1b). Targeting the C–H etherification of 3-bromopyridine (8) as a model reaction, use of 2,5-dibromothiophene (7) provides low yields under the previously optimized conditions (12% at 60 °C and 8% at 25 °C). Since pyridines are less acidic than 1,3-azoles, we speculated the lower concentration of pyridyl anions in this reaction would require faster halogen transfer for efficient reactivity.23 In this regard, we expected iodothiophenes to be more active halogen transfer reagents due to iodine’s increased polarizability and decreased electronegativity.26 While 2,5-diiodothiophene (10) does increase the yield to 82% at 25 °C, we also found inexpensive 2-iodothiophene (11, <$100 per mole) to be equally effective (84% yield).27,28 When 2-iodothiophene (11) is used, 3-iodothiophene is observed as a major byproduct that likely forms via disproportionation processes. This led us to develop 2,3-diiodobenzothiophene (12, prepared in three steps on 100 mmol scale)29 as an alternative oxidant that is stable to the reaction conditions but still enables high C–H etherification yield (86%).30 We note 18-crown-6 is used as an additive for reactions in Scheme 1b, although useful yields are obtained without it (65% of 9) and many azine substrates reported below do not require its use.
Table 1 provides a scope of heteroaryl ethers that can be accessed using the three 2-halothiophene reagents identified in our optimization studies. High regio- and chemoselectivity is observed for C–H etherification (>10:1 for shown product, unless noted) as judged by examination of crude reaction mixtures. Primary, secondary and benzylic alcohols are effective coupling partners, including amino-alcohols, alkenols and complex alcohols such as the pharmaceutical perphenazine (36).
Table 1.
Substrate scope for heteroarene C–H etherification using optimized halogen transfer reagents.a

|
Reactions conducted on 0.5 to 1 mmol scale of heteroarene; yields are of isolated products; reaction conducted at 60 °C for 3 h for Table 1a and at 25 °C for 20 h for Table 1b and 1c.
18-crown-6 (1.5 equiv) additive used.
Side products (20% total) from 2-substitution of the starting material observed.
2 equiv of alcohol used.
DMF used as solvent.
Reaction conducted at 60 °C.
Reaction conducted with 2 equiv pyridazine and 1 equiv alcohol; product forms in 4:1 ratio to other aryl ethers.
Product forms in 6:1 ratio to other aryl ethers. See Supporting Information for full details.
Table 1a shows the use of 2,5-dibromothiophene (7) for the 2-etherification of 1,3-azole compounds, including thiazoles, oxazoles, imidazoles and benzofused variants (13–21). 4,5-Dimethylthiazole (13) undergoes 2-etherification without competing methyl oxidation. Substrates with halogen substituents (15, 18) and additional aryl groups (15, 17, 21) favor 1,3-azole etherification over potential substitution or unselective halogenation side reactions.
Table 1b shows the use of 2-iodothiophene (11) for the etherification of 6-membered N-heteroarenes.31 3-Halopyridines (9, 24, 36), including 3,5-dichloropyridine (22), produce 4-etherified products without competing 2-etherification or 3-substitution. The selective etherification of 5-bromo-2-iodopyridine (23) and 2-chloro-5-methoxypyridine (25) illustrates the preferred 4-selectivity over 2-halogen substitution.32 3-(Phenylthio)pyridine (26) reacts smoothly as do pyridines without electron-withdrawing groups in the 3-position, including 2,6-disubstituted pyridines (27, 28) and pyridine 29 that contains two acidic difluoromethyl groups.33
Structurally diverse azines and diazines also participate in this method. Selective etherification of a furo[3,2-b]pyridine (30), quinoline (31) and pyrazolo[1,5-a]pyrimidine (35) occurs without competing halogenation of other positions. Pyridazine (32) and derivatives with 3,6-dialkoxy (33) and amide (34) substituents undergo 4-selective C–H etherification.
For certain azines, use of 2,3-diiodobenzothiophene (12) in place of 2-iodothiophene (11) improves the etherification yield (Table 1c).34 Pyridines with phenylsulfonyl (37) and sulfonamide (38) groups produce 4-substituted ethers in high yield under these conditions. More diverse arenes such as 5-chloro-2-phenylpyrimidine (39), imidazo[1,2-b]pyridazine (40) and phthalazine (41) also react.
This etherification protocol enables several benefits over existing routes to N-heteroaryl ethers. For example, practical alkoxide bases are employed under ambient conditions, requiring no strong directing groups to achieve regioselective C–H etherification in a single step. This contrasts with existing methods for net heteroaryl C–H etherification, including a three-step metalation, halogenation and substitution sequence. We expect these features to be especially valuable for the selective functionalization of polyazines. For example, Scheme 2a shows a regioselective C–H etherification reaction on a bipyridine (42) that lacks directing groups required for a 4-selective metalation/halogenation protocol.35 This selectivity also complements the scope of (pseudo)halogenation methods reliant on pyridine N-activation that typically functionalize the most basic and unencumbered N-heteroarene.36
Scheme 2. Direct C–H etherification of polyazines.a.

aIsolated yields using conditions from Table 1b and 1c. bShown product forms in 7:1 ratio to other ether side products. c18-Crown-6 additive used. See Supporting Information for details.
Scheme 2b shows additional polyazines that exhibit high regioselectivity using conditions from Table 1. Here, a pyridine-containing alcohol selectively couples with a chlorinated 2,3’-bipyridine substrate (44). An imidazo[1,2-b]pyridazine with an 8-quinolinyl substituent undergoes selective etherification on the pyridazine ring (45) while 4-selective etherification of a 3,6-disubstituted pyridazine occurs in the presence of two pendant pyridines (46). In sum, numerous C–H etherification products in Table 1, Scheme 2 and Equation 1 (vide infra) are accessed directly from readily available heteroarenes where a corresponding heteroaryl (pseudo)halide is not commercially available and is challenging to access.
Equation 1 demonstrates how this base-promoted method can be incorporated into cascade substitution sequences to access products of greater complexity. Thus, reaction of 5-bromo-2-(trifluoromethyl)-pyridine (47) with L-prolinol (48) produces morpholine-fused pyridine 49 in 70% yield through tandem C–H etherification and C–Br amination reactions.37
 |
(1) |
We next performed studies to gain insight into the mechanism of C–H etherification. When alcohol is excluded from the optimized reaction conditions with the model substrates thiazole (1) and 3-bromopyridine (8), 2-bromothiazole and 3-bromo-4-iodopyridine are observed, respectively (see Supporting Information for details). In these control reactions, only low quantities of N-heteroaryl halides accumulate with the reactions ultimately resulting in low mass balances. These observations are consistent with the need to sequence halogen transfer with a substitution reaction for a high C–H functionalization yield.
Another insightful observation came during our substrate scope studies. When the reaction of 3-fluoropyridine (50) and methanol using 2-iodothiophene (11) was attempted, 4-iodo-3-methoxypyridine (51) was obtained as the major product (Scheme 3a). Subjection of 3-fluoro-4-iodopyridine (52) to methanol and KO-t-Bu results in a 3-selective nucleophilic aromatic substitution reaction (SNAr, Scheme 3b).38 A control experiment in Scheme 3c shows that 3-methoxypyridine (53) does not undergo 4-iodination under these conditions. These results are consistent with halogen transfer and subsequent SNAr as the operative mechanism for C–H etherification.39,40 Studies on the halogen transfer process, including further development of halogen transfer reagents, are ongoing.41,42
Scheme 3. Observation of a halogen transfer adduct.a.
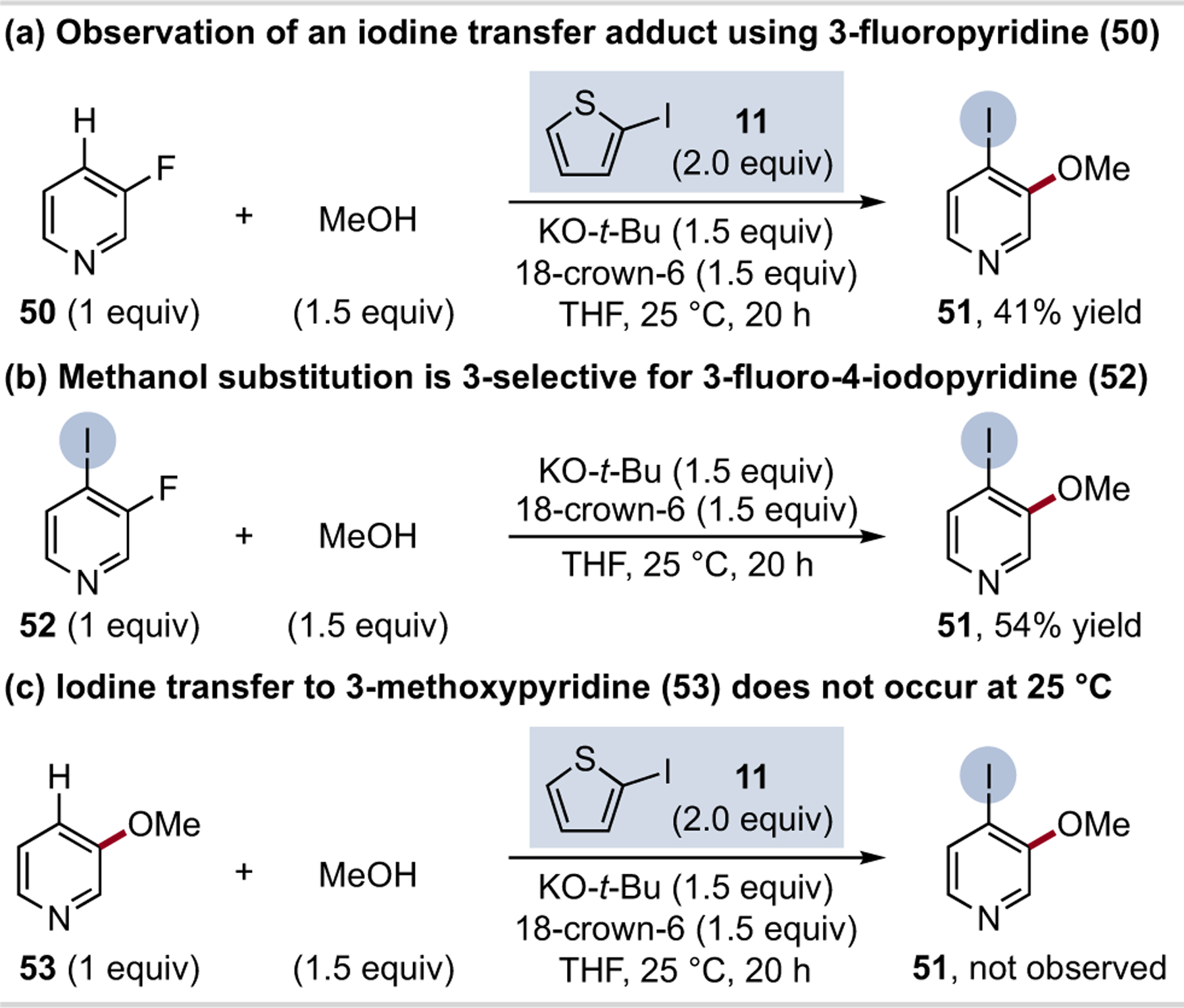
aYields determined by 1H NMR spectroscopy of crude reaction mixture; see Supporting Information for details.
Key to development of this C–H etherification protocol was the discovery that 2-halothiophenes can serve as base- and nucleophile-tolerant halogen oxidants. Thus, heteroarene C–H etherification can be accomplished using typical SNAr reaction conditions with the simple inclusion of a 2-halothiophene. The underlying mechanistic steps of this process represent a general framework for C–H functionalization methodology that we are currently expanding upon.
Supplementary Material
ACKNOWLEDGMENT
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number 1R35GM138350. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. Acknowledgment is also made to the Donors of the American Chemical Society Petroleum Research Fund for support of this research (ACS PRF #60171-DNI1). We thank Professors Andrew McNally (CSU) and Yiming Wang (University of Pittsburgh) for input on this manuscript.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Detailed experimental procedures, characterization data, and NMR spectra for all compounds (PDF).
REFERENCES
- (1).For reviews on oxidative alcohol coupling reactions, see:; (a) Krylov IB; Vil’ VA; Terent’ev AO Cross-dehydrogenative coupling for the intermolecular C–O bond formation. Beilstein J. Org. Chem 2015, 11, 92–146. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu B; Shi B-F Transition-metal-catalyzed etherification of unactivated C–H bonds. Tetrahedron Lett 2015, 56, 15–22. [Google Scholar]; (c) Borpatra PJ; Deka B; Deb ML; Baruah PK Recent advances in intramolecular C–O/C–N/C–S bond formation via C–H functionalization. Org. Chem. Front 2019, 6, 3445–3489. [Google Scholar]; (d) Zheng Q; Chen J; Rao G-W Recent Advances in C-O Bond Construction via C-H Activation. Russ. J. Org. Chem 2019, 55, 569–586. [Google Scholar]
- (2).For selected reviews on the utility of C–H functionalization, see:; (a) Dalton T; Faber T; Glorious F C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci 2021, 7, 245–261. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cernak T; Dykstra KD; Vachal P; Krska SW The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]; (c) Yamaguchi J; Yamaguchi AD; Itami K C–H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed 2012, 51, 8960–9009. [DOI] [PubMed] [Google Scholar]; (d) Abrams DJ; Provencher PA; Sorensen EJ Recent applications of C–H functionalization in complex natural product synthesis. Chem. Soc. Rev 2018, 47, 8925–8967. [DOI] [PubMed] [Google Scholar]
- (3).For a selection of recent approaches, see:; (a) Huang H; Lambert TH Electrophotocatalytic C–H Heterofunctionalization of Arenes. Angew. Chem. Int. Ed 2021, 60, 11163–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bhadra S; Matheis C; Katayev D; Gooßen LJ Copper-Catalyzed Dehydrogenative Coupling of Arenes with Alcohols. Angew. Chem. Int. Ed 2013, 52, 9279–9283. [DOI] [PubMed] [Google Scholar]; (c) Roane J; Daugulis O Copper-Catalyzed Etherification of Arene C–H Bonds. Org. Lett 2013, 15, 5842–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For reviews on N-heteroarene C–H functionalization, see:; (a) Murakami K; Yamada S; Kaneda T; Itami K C–H Functionalization of Azines. Chem. Rev 2017, 117, 9302–9332. [DOI] [PubMed] [Google Scholar]; (b) Stephens DE; Larionov OV Recent advances in the C–H-functionalization of the distal positions in pyridines and quinolines. Tetrahedron 2015, 71, 8683–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Verbitskiy EV; Rusinov GL; Chupakhin ON; Charushin VN Recent Advances in Direct C–H Functionalization of Pyrimidines. Synthesis 2018, 50, 193–210. [Google Scholar]; (d) kosza M; Wojciechowski K Nucleophilic Substitution of Hydrogen in Heterocyclic Chemistry. Chem. Rev 2004, 104, 2631–2666. [DOI] [PubMed] [Google Scholar]
- (5). References 8–12 review multistep approaches for N-heteroaryl C–H etherification. For examples of direct alcohol C–H coupling reactions that are limited to specific substrate classes, see:; (a) Takemura N; Kuninobu Y; Kanai M Copper-Catalyzed C–H Alkoxylation of Azoles. Org. Lett 2013, 15, 844–847. [DOI] [PubMed] [Google Scholar]; (b) Sugimoto T; Pfleiderer W Pteridines CIV. Regioselective Alkoxylation of Pteridines at the 6-Position by N-Bromo-succinimide and Alcohol. Heterocycl 1995, 41, 781–788. [Google Scholar]; (c) Kibriya G; Samanta S; Jana S; Mondal S; Hajra A Visible Light Organic Photoredox-Catalyzed C–H Alkoxylation of Imidazopyridine with Alcohol. J. Org. Chem 2017, 82, 13722–13727. [DOI] [PubMed] [Google Scholar]; (d) Heinisch G; Huber T Pyridazines LX Telesubstitution and Dismutation Reactions in the Series of Phenyl-3-pyridazinylmethane Derivatives. Liebigs Ann. Chem 1992, 1992, 19–22. [Google Scholar]; For a representative example of a directing group-assisted benzene C–H methoxylation method that includes N-heteroarenes in the substrate scope, see:; (e) Yin X-S; Li Y-C; Yuan J; Gu W-J; Shi B-F Copper(II)-catalyzed methoxylation of unactivated (hetero)aryl C–H bonds using a removable bidentate auxiliary. Org. Chem. Front 2015, 2, 119–123. [Google Scholar]
- (6).(a) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (b) Baumann M; Baxendale IR; Ley SV; Nikbin N An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals. Beilstein J. Org. Chem 2011, 7, 442–495. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Umetsu N; Shirai Y Development of novel pesticides in the 21st century. J. Pestic. Sci 2020, 45, 54–74. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jeschke P Progress of modern agricultural chemistry and future prospects. Pest Manag. Sci 2016, 72, 433–455. [DOI] [PubMed] [Google Scholar]
- (7). In addition to the incompatibility of many oxidants with alkoxides due to competing alcohol oxidation and elimination reactions, metal-catalyzed C–H etherification is difficult as metal-alkoxide intermediates are prone to β-hydride elimination and C–O reductive elimination can be difficult. See reference 1 for discussions.
- (8).For reviews on heteroaryl N-oxides, see:; (a) Kutasevich AV; Perevalov VP; Mityanov VS Recent Progress in Non-Catalytic C–H Functionalization of Heterocyclic N-Oxides. Eur. J. Org. Chem 2021, 357–373. [Google Scholar]; (b) Baykov SV; Boyarskiy VP Metal-free functionalization of azine N-oxides with electrophilic reagents. Chem. Heterocycl. Compd 2020, 56, 814–823. [Google Scholar]; (c) Wang Y; Zhang L Recent Developments in the Chemistry of Heteroaromatic N-Oxides. Synthesis 2015, 47, 289–305. [Google Scholar]; For etherification via activated N-oxides:; (d) Lian Y; Coffey SB; Li Q; Londregan AT Preparation of Heteroaryl Ethers from Azine N-Oxides and Alcohols. Org. Lett 2016, 18, 1362–1365. [DOI] [PubMed] [Google Scholar]
- (9).For the activation of N-heteroarenes with triflic anhydride en route to phosphonium salts, haloarenes and cyanoarenes, see:; (a) Hilton MC; Dolewski RD; McNally A Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts. J. Am. Chem. Soc 2016, 138, 13806–13809. [DOI] [PubMed] [Google Scholar]; (b) Dolewski RD; Hilton MC; McNally A 4-Selective Pyridine Functionalization Reactions via Heterocyclic Phosphonium Salts. Synlett 2018, 29, 8–14. [Google Scholar]; (c) Levy JN; Alegre-Requena JV; Liu R; Paton RS; McNally A Selective Halogenation of Pyridines Using Designed Phosphine Reagents. J. Am. Chem. Soc 2020, 142, 11295–11305. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zi Y; Schömberg F; Wagner K; Vilotijevic I C–H Functionalization of Benzothiazoles via Thiazol-2-yl-phosphonium Intermediates. Org. Lett 2020, 22, 3407–3411. [DOI] [PubMed] [Google Scholar]; (e) Elbert BL; Farley AJM; Gorman TW; Johnson TC; Genicot C; Lallemand B; Pasau P; Flasz J; Casro JL; MacCoss M; Paton RS; Schofield CJ; Smith MD; Willis MC; Dixon DJ C–H Cyanation of 6-Ring N-Containing Heteroaromatics. Chem. Eur. J 2017, 23, 14733–14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For other methods to install functional groups that undergo substitution, see:; (a) Fier SP; Hartwig JF Synthesis and Late-Stage Functionalization of Complex Molecules through C–H Fluorination and Nucleophilic Aromatic Substitution. J. Am. Chem. Soc 2014, 136, 10139–10147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fier PS A Bifunctional Reagent Designed for the Mild, Nucleophilic Functionalization of Pyridines. J. Am. Chem. Soc 2017, 139, 9499–9502. [DOI] [PubMed] [Google Scholar]; (c) Kim M; You E; Park S; Hong S Divergent reactivity of sulfinates with pyridinium salts based on one- versus two-electron pathways. Chem. Sci 2021, 12, 6629–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For selected reviews and discussions on heteroarene metalation, see:; (a) Queguiner G; Marsais F; Snieckus V; Epsztajn J Directed Metalation of Pi-Deficient Azaaromatics: Strategies of Functionalization of Pyridines, Quinolines, and Diazines. Adv. Heterocycl. Chem 1991, 52, 187–304. [Google Scholar]; (b) Schlosser M The 2×3 Toolbox of Organometallic Methods for Regiochemically Exhaustive Functionalization. Angew. Chem. Int. Ed 2005, 44, 376–393. [DOI] [PubMed] [Google Scholar]; (c) Balkenhohl M; Knochel P Regioselective C–H Activation of Substituted Pyridines and other Azines using Mg- and Zn-TMP-Bases. SynOpen 2018, 2, 78–95. [Google Scholar]; (d) Haag B; Mosrin M; Ila H; Malakhov V; Knochel P Regio- and Chemoselective Metalation of Arenes and Heteroarenes Using Hindered Metal Amide Bases. Angew. Chem. Int. Ed 2011, 50, 9794–9824. [DOI] [PubMed] [Google Scholar]; (e) Balkenhohl M; Jangra H; Makarov IS; Yang S-M; Zipse H; Knochel P A Predictive Model Towards Site-Selective Metalations of Functionalized Heterocycles, Arenes, Olefins, and Alkanes using TMPZnCl·LiCl. Angew. Chem. Int. Ed 2020, 59, 14992–14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For examples of in situ base-promoted halogenation of heteroarenes under noncryogenic conditions, see:; (a) Do H-Q; Daugulis O A Simple Base-Mediated Halogenation of Acidic sp2 C-H Bonds under Noncryogenic Conditions. Org. Lett 2009, 11, 421–423. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu X; Zhao X; Liang F; Ren B t-BuONa-mediated direct C–H halogenation of electron-deficient (hetero)arenes. Org. Biomol. Chem 2018, 16, 886–890. [DOI] [PubMed] [Google Scholar]; We note these methods were examined in the context of this C–H etherification reaction to no effect; see reference 25.
- (13).Electrophilic aromatic halogenation represents an alternative route, although N-heteroarenes do not typically undergo electrophilic halogenation under practical conditions unless an electron-donating group is present.; Joule JA and Mills K Heterocyclic Chemistry, 5th ed. Wiley: West Sussex, 2011. [Google Scholar]
- (14).Vaitiekunas A; Nord FF Tetrabromothiophene from 2-Bromothiophene by means of Sodium Acetylide in Liquid Ammonia. Nature 1951, 168, 875–876. [Google Scholar]
- (15).Bunnett JF Base-catalyzed halogen dance, and other reactions of aryl halides. Acc. Chem. Res 1972, 5, 139–147. [Google Scholar]
- (16).For reviews on halogen dance methods, see:; (a) Schnürch M; Spina M; Khan AF; Mihovilovic MD; Stanetty P Halogen dance reactions—A review. Chem. Soc. Rev 2007, 36, 1046–1057. [DOI] [PubMed] [Google Scholar]; (b) Erb W; Mongin F Halogen ‘dance’: a way to extend the boundaries of arene deprotolithiation. Tetrahedron 2016, 72, 4973–4988. [Google Scholar]
- (17).For application of this strategy in the context of aryl halide isomerization, see:; Puleo TR; Bandar JS Base-catalyzed aryl halide isomerization enables the 4-selective substitution of 3-bromopyridines. Chem. Sci 2020, 11, 10517–10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18). Base-promoted “halogen dance” reactions result in complex product mixtures in the absence of a strong thermodynamic driving force, typically an acidity gradient; see references 15–16.
- (19).For examples of the instability of N-heteroaryl halides under basic conditions, see:; (a) Smith AJ; Poole DL; Murphy JA The role of organic electron donors in the initiation of BHAS base-induced coupling reactions between haloarenes and arenes. Sci. China Chem 2019, 62, 1425–1438. [Google Scholar]; (b) Dong Y; Lipschutz MI; Tilley TD Regioselective, Transition Metal-Free C–O Coupling Reactions Involving Aryne Intermediates. Org. Lett 2016, 18, 1530–1533. [DOI] [PubMed] [Google Scholar]
- (20).For an aromatic halogenation method proposed to occur via several mechanisms, including aromatic iodine transfer, see:; (a) Shi Q; Zhang S; Zhang J; Oswald VF; Amassian A; Marder SR; Blakey SB KOtBu-Initiated Aryl C–H Iodination: A Powerful Tool for the Synthesis of High Electron Affinity Compounds. J. Am. Chem. Soc 2016, 138, 3946–3949. [DOI] [PubMed] [Google Scholar]; (b) Donham LL; Gronert S Substitution Reactions on Iodine and Bromine: Mechanisms for Facile Halogenations of Heterocycles. J. Org. Chem 2019, 84, 5757–5762. [DOI] [PubMed] [Google Scholar]
- (21). 1,3-Azole C–H etherification is limited primarily to intramolecular examples described in reference 5a. For additional work, see:; (a) Yu M; Wang Z; Hu J; Li S; Du H Copper-Catalyzed Intramolecular Alkoxylation of Purine Nucleosides: One-Step Synthesis of 5′-O,8-Cyclopurine Nucleosides. J. Org. Chem 2015, 80, 9446–9453. [DOI] [PubMed] [Google Scholar]; (b) Dörr M; Röckl JL; Rein J; Schollmeyer D; Waldvogel SR Electrochemical C–H Functionalization of (Hetero)Arenes—Optimized by DoE. Chem. Eur. J 2020, 26, 10195–10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).For a sequential base-promoted iodination/Cu-catalyzed cyanation of N-heteroarenes, see:; Do H-Q; Daugulis O Copper-Catalyzed Cyanation of Heterocycle Carbon–Hydrogen Bonds. Org. Lett 2010, 12, 2517–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).For acidity measurements relevant to this method, see:; (a) Shen K; Fu Y; Li J-N; Liu L; Guo Q-X What are the pKa values of C–H bonds in aromatic heterocyclic compounds in DMSO? Tetrahedron 2007, 63, 1568–1576. [Google Scholar]; (b) Bordwell FG Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res 1988, 21, 456–463. [Google Scholar]
- (24).For the use of 2-bromothiophenes as oxidants in Ni-catalyzed dehydrogenation reactions, see:; Zhang P; Huang D; Newhouse TR Aryl-Nickel-Catalyzed Benzylic Dehydrogenation of Electron-Deficient Heteroarenes. J. Am. Chem. Soc 2020, 142, 1757–1762. [DOI] [PubMed] [Google Scholar]
- (25).See Supporting Information for a full list of halogenating reagents examined. Typically, no N-heteroarene etherification was observed and side reactions occurred between the base, alcohol, and halogenating reagent. 1-Iodoperfluorobutane was the only halogenating reagent to provide C–H etherification yield, producing 2% of 9.
- (26). Both the halogen identity and relative stability of the aryl anion nucleofuge contribute to the efficiency of halogenophilic substitution reactions. For approximations of aryl anion stabilities, see reference 23a for aryl C–H pKa values. For a review of halogenophilic reactions, see:; Sazonov PK; Artamkina GA; Beletskaya IP Nucleophilic substitution at the halogen atom (halogenophilic reactions). Russ. Chem. Rev 2012, 81, 317–335. [Google Scholar]
- (27).2,5-Dibromothiophene ($76/500g) and 2-iodothiophene ($470/kg) are inexpensive commercial reagents. 2,5-Diiodothiophene is also commercially available but more expensive ($266/25g). Prices obtained from www.emolecules.com on 4/19/2021.
- (28).Use of 2-iodothiophenes do not enable thiazole (1) C–H etherification, likely due to the fact that 2-iodothiazole does not undergo alcohol substitution under conditions shown in Scheme 1.
- (29).Mehta S; Lacrock RC Iodine/Palladium Approaches to the Synthesis of Polyheterocyclic Compounds. J. Org. Chem 2010, 75, 1652–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).When 2,3-diidobenzothiophene (12) is subjected to the standard reaction conditions in the absence of N-heteroarene, 8% of 3-iodobenzothiophene forms with 92% of 12 remaining after 24 h.
- (31).We note pyridines without an inductive electron-withdrawing group are incompatible substrates under the optimized conditions as are variants with unsubstituted methyl substituents.
- (32).For an examination of selective substitution of a 2-chloro-4-iodopyridine derivative, see:; Seto S; Convenient synthesis of 7-aryl-3,4,5,6-tetrahydro-2H-pyrido[4,5-b]-and [2,3-b]-1,5-oxazocine-6-ones. Tetrahedron Lett 2004, 45, 8475–8478. [Google Scholar]
- (33).Huang L; Liu W; Zhao L-L; Zhang Z; Yan X Base-Catalyzed H/D Exchange Reaction of Difluoromethylarenes. J. Org. Chem 2021, 86, 3981–3988. [DOI] [PubMed] [Google Scholar]
- (34).Use of 2,3-diiodobenzothiophene (12) in place of 2-iodothiophene (11) for substrates shown in Table 1c provides increased yields of 20–60%; see Supporting Information.
- (35). Attempted metalation/iodination of 42 using LiTMP base resulted in an approximate 1:1 ratio of 3- and 4-iodination of the 2-trifluoromethylpyridine ring in 49% yield; see Supporting Information and:; Schlosser M; Marull M The Direct Metalation and Subsequent Functionalization of Trifluoromethyl-Substituted Pyridines and Quinolines. Eur. J. Org. Chem 2003, 2003, 1569–1575. [Google Scholar]
- (36). For methods likely to functionalize other positions of 42, see references 8–10. For a discussion of site-selective control in polyazine C–H functionalization, see:; Dolewski RD; Fricke PJ; McNally A Site-Selective Switching Strategies to Functionalize Polyazines. J. Am. Chem. Soc 2018, 140, 8020–8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).The structural assignment of 49 was confirmed by a multistep synthesis of an authentic sample as described in the Supporting Information.
- (38).This selectivity is not consistent with a transition-metal catalyzed etherification pathway that would likely be selective for 4-substitution of iodine; for example, see:; Chen Z; Jiang Y; Guo Y; Ma D Oxalic Diamides and tert-Butoxide: Two Types of Ligands Enabling Practical Access to Alkyl Aryl Ethers via Cu-Catalyzed Coupling Reaction. J. Am. Chem. Soc 2019, 141, 3541–3549. [DOI] [PubMed] [Google Scholar]
- (39).Formation of an aryne intermediate is a potential alternative alcohol substitution pathway of the in situ generated N-heteroaryl halide; however, the high regioselectivities observed suggest this is not a major competing pathway.
- (40).Another mechanism considered is alkoxide addition to the N-heteroarene with subsequent oxidation of the dearomatized intermediate by a 2-halothiophene. The observation of N-heteroaryl halide intermediates suggests this is not a dominant pathway. See also reference 20b.
- (41). Halogen transfer may occur through rearranged halothiophenes formed in situ; for relevant discussions, see references 15, 16 and:; Yamane Y; Sunahara K; Okana K; Mori A Magnesium Bisamide-Mediated Halogen Dance of Bromothiophenes. Org. Lett 2018, 20, 1688–1691. [DOI] [PubMed] [Google Scholar]
- (42). Aromatic halogen transfer typically involves aryl anion attack on an active haloarene transfer reagent, although hypohalite intermediate have also been proposed; see reference 12 and:; Mach MH; Bunnett JF Duality of mechanism in the electrophilic bromination of aromatic compounds. J. Am. Chem. Soc 1974, 96, 936–938. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.