

Abstract
Polycomb repressive complexes (PRCs) are a heterogenous collection of dozens, if not hundreds, of protein complexes composed of various combinations of subunits. PRCs are transcriptional repressors important for cell-type specificity during development, and as such, are commonly mis-regulated in cancer. PRCs are broadly characterized as PRC1 with histone ubiquitin ligase activity, or PRC2 with histone methyltransferase activity; however, the mechanism by which individual PRCs, particularly the highly diverse set of PRC1s, alter gene expression has not always been clear. Here we review the current understanding of how PRCs act, both individually and together, to establish and maintain gene repression, the biochemical contribution of individual PRC subunits, the mis-regulation of PRC function in different cancers, and the current strategies for modulating PRC activity. Increased mechanistic understanding of PRC function, as well as cancer-specific roles for individual PRC subunits, will uncover better targets and strategies for cancer therapies.
INTRODUCTION TO POLYCOMB GROUP PROTEINS
The proper regulation of chromatin structure during development is important for establishing and maintaining cell-type specific transcriptional programs. Compared to unicellular organisms, metazoans have an increased number of chromatin regulators, which play essential roles in cell-type specification. One such set of chromatin regulators are the Polycomb group (PcG) proteins, which were originally discovered in Drosophila and named for their roles in leg development (1). Subsequently, PcG proteins were identified as crucial regulators of development in vertebrates, acting primarily to modify histones and reduce DNA accessibility (2,3). PcG proteins fall into two broad complexes: Polycomb Repressive Complex 1 and 2 (PRC1 and PRC2) (Figure 1) (4). PRC1 is defined by a RING subunit, which is capable of H2AK119 monoubiquitination, while PRC2 is defined by an EZH subunit, which is capable of H3K27 methylation. Both complexes incorporate a variety of mutually exclusive subunits giving rise to an array of diverse complexes with distinct biochemical activities (5,6).
Figure 1.

The subunit composition of Polycomb repressive complexes. (A) Subunit composition of canonical PRC1 (cPRC1) and noncanonical PRC1 (ncPRC1). (B) Subunit composition of PRC2.
Polycomb complex composition
All PRC1s contain a RING ubiquitin ligase subunit with one of six possible PCGF paralogs (Figure 1A). PRC1s that contain CBX and PHC subunits are defined as ‘canonical’ PRC1 (cPRC1), and exclusively incorporate PCGF2 or PCGF4, while PRC1s that contain the RYBP/YAF2 subunit are defined as ‘non-canonical’ PRC1 (ncPRC1) and can contain any PCGF paralog (Figure 1A) (7). The incorporation of different PCGF variants defines ncPRC subcomplexes with different accessory subunits (7,8).
All PRC2s contain an EZH methyltransferase subunit, EED, SUZ12 and RbAp46/48 (Figure 1B) and catalyze mono/di/trimethylation of lysine 27 of histone H3 (H3K27me, H3K27me2, H3K27me3) (9,10). PRC2 can be categorized as PRC2.1 and PRC2.2 depending on the accessory subunits PCL1-3 (PRC2.1) or AEBP2 and JARID2 (PRC2.2), which are involved in genome targeting (11–14).
Establishment of Polycomb sites
Mammals lack the so-called Polycomb Response Elements (PREs) found in lower organisms (15,16); instead, ncPRC1 localizes to unmethylated CpG islands via accessory proteins and catalyzes monoubiquitination of H2AK119 (Figure 2A) (17–20). PRC2.2 subunits JARID2/AEBP2 bind a subset of H2AK119ub1 deposited primarily by ncPRC1 (21,22), while PRC2.1 binds unmethylated CpG islands, at least in part, through PCL association with DNA (23,24). The two PRC2 variants cooperate in establishing sites of H3K27me3 across the genome (25), which can repress transcription in part by blocking acetylation of H3K27. Whether H2AK119ub1 can be transcriptionally repressive at sites without PRC2 activity is not known, as ncPRC1 subunits have also been implicated in gene activation (26–33).
Figure 2.

Transcriptional repression by PRCs. (A) Initiation. ncPRC1 targets specific DNA sequences (hypomethylated CpG islands) and ubiquitinates H2AK119. H2AK119ub1 is recognized by JARID2 and AEBP2 of PRC2.2, leading to the recruitment of PRC2 and deposition of trimethylation on H3K27. PRC2.1 also associates with hypomethylated CpG islands via PCL-mediated DNA binding and deposits additional H3K27me3. This leads to an initial H3K27me3-mediated compaction, drawing nucleosomes closer for EED to perform allosteric activation of EZH activity. (B) Propagation/Maintenance. H3K27me3 binds to aromatic cage of EED, and allosterically activates EZH methyltransferase activity, leading to further trimethylation of nearby unmodified H3K27. H3K27me3 is recognized by the CBX subunit within cPRC1. cPRC1 recruitment leads to further chromatin compaction and transcriptional repression.
Polycomb propagation and chromatin compaction
What is sometimes referred to as ‘canonical’ Polycomb function is a mechanism by which Polycomb marks are propagated and maintained (Figure 2B). This is initiated by the association of the PRC2 subunit EED with H3K27me3, which allosterically activates methyltransferase activity of EZH towards unmodified H3K27 at neighboring and non-neighboring nucleosomes (34,35). cPRC1 binds a subset of H3K27me3 via the N-terminal chromodomain of the CBX subunit, monoubiquitinates H2AK119 through the E3 ligase domain of RING1A/B, and oligomerizes via the PHC and CBX subunits to physically compact chromatin (36) into large domains called Polycomb bodies (37). Formation of Polycomb bodies is dependent on PRC2 (38) and cPRC1, in particular the chromodomain of the CBX subunit (39,40) and the sterile alpha motif (SAM) oligomerization domain of PHC (41). These nuclear foci are spatially separated from regions of active transcription as well as regions of constitutive heterochromatin (42). Within these large 3D domains, PcG-bound regions make contacts with other PcG-bound regions several megabases away, resulting in repression across the domain (33). In mammalian cells, Polycomb body distribution changes during differentiation (40,43), as well as transformation (42).
Maintenance of Polycomb sites
After Polycomb binding sites are established, PcG proteins maintain these sites during cell division. The loss of ncPRC1 (RYBP deletion) in mouse embryonic stem cells (mESCs) can be compensated for with elevated EED, H3K27me3, and CBX7 at shared target loci, indicating some redundancy for ncPRC1 by PRC2/cPRC1 in the maintenance of previously established Polycomb binding sites (21). While PRC2/cPRC1 is not required for initiating Polycomb binding sites, it is critical for transcriptional repression (17) and may be sufficient for Polycomb maintenance/memory, at least in some cell types (44). During differentiation, however, dynamic changes in PRC subunit composition and genome localization require establishment of new Polycomb sites (45).
Biochemical functions of individual PRC1 and PRC2 subunits
Advanced proteomic techniques have begun to identify the combinations of individual PRC subunits unique to particular cell types. While structural and functional studies have elucidated the mechanistic contribution of critical core subunits, the biochemical contribution of accessory and substoichiometric subunits, and the unique biochemical contribution of individual paralogs are not always clear. A summary of defined subunit domains and known biochemical functions is provided in Table 1.
Table 1.
Biochemical functions of PRC subunits
| Subunit | Constituent motifs and domains (InterPro and CanSAR database) | PDB structures (including partial structures) | Major biochemical function | Additional structural, allosteric or regulatory functions if any known |
|---|---|---|---|---|
| PRC1 | ||||
| RING1A RING1B (RING2/RNF2) |
|
RING1B |
|
|
| PCGF1 PCGF2 (MEL18) PCGF3 PCGF4 (BMI1) PCGF5 PCGF6 |
|
PCGF1 PCGF4 PCGF5 PCGF6 |
|
|
| CBX2 CBX4 CBX6 CBX7 CBX8 |
|
CBX2 CBX4 CBX6 CBX7 CBX8 |
|
|
| PHC1 PHC2 PHC3 |
|
PHC1 PHC3 |
|
|
| SCMH1 SCML1 SCML2 SCML4 |
|
SCMH1 SCML2 |
||
| RYBP YAF2 |
|
RYBPYAF2 |
|
|
| KDM2B |
|
KDM2B | ||
| BCOR BCORL1 |
|
BCOR BCORL1 |
|
|
| MAX MGA |
|
MAX |
|
|
| E2F6 DP1 DP2 |
|
DP1 DP2 |
|
|
| L3MBTL2 |
|
L3MBTL2 |
|
|
| AUTS2 |
|
|
|
|
| PRC2 | ||||
| EZH1 EZH2 |
|
EZH1 EZH2 |
|
|
| EED |
|
EED |
|
|
| SUZ12 |
|
SUZ12 |
|
|
| RBBP4/RbAp48 RBBP7/RbAp46 |
|
RBBP4 RBBP7 |
|
|
| PCL1/ PHF1 PCL2/ MTF2 PCL3/ PHF19 |
|
PCL1 PCL2 PCL3 |
|
|
| AEBP2 | Zinc finger | AEBP2 |
|
|
| JARID2 |
|
JARID2 |
|
|
| PALI1 PALI2 |
|
PALI1 |
|
|
| EPOP |
|
|
|
|
| EZHIP/CATACOMB |
|
|
|
|
PcG PROTEINS IN CANCER
Tumor genome sequencing has revealed frequent alteration of genes encoding chromatin regulators, including PcG proteins (109). PcG proteins have traditionally been viewed as oncogenic due to their ability to prevent differentiation (110) and bypass senescence (111,112), in part by repressing the INK/ARF tumor suppressor genes (113,114). This function is important for maintaining proliferation of both normal stem cells and cancer stem cells (CSCs) (11,115). Despite a generally oncogenic function for PcGs, the role for individual subunits can vary tremendously, with some subunits acting as both an oncogene and a tumor suppressor depending on the context. Predicting which cancers are dependent on PcG function and then determining the best subunit to target is still challenging, although several trends have emerged based on clinical and experimental data.
Genetic mutation of PcG genes in cancer
From the TCGA mutation data, it is clear that some cancers have a high incidence of PcG gene mutation (Figure 3). The high mutation rates in many of these cancers, such as melanoma (SKCM), stomach cancer (STAD), colon cancer (COAD), uterine cancer (UCEC) and lung cancers (LUAD, LUSC) are more likely due to high mutational burden than any relevant function for PcG proteins (116). However, frequent mutations in some subunits, such as MGA and BCOR/BCORL1, as well as generally high mutation rates in cancers such as diffuse large B-cell lymphoma (DLBCL) and cholangiocarcinoma of the bile duct (CHOL) point to specific roles for Polycomb subunits in tumor suppression.
Figure 3.

Heatmap of PcG subunit mutation frequency in cancer. The number of patient tumors with mutations in PcG genes was determined using TCGA datasets in cBioPortal, and the percentage in each cancer type is displayed as a heatmap. White boxes indicate that no mutational data was available.
MGA
MGA mutations are frequently found in multiple cancer types (Figure 3). The oncogenic role of MGA mutation is likely not due to the loss of PRC1.6 function but instead due to the release of free MAX, which can associate with MYC, and facilitate MYC-mediated oncogenic transcription (117,118). Whether mutations in other PRC1.6 subunits affect MYC function in a similar manner has not been established.
BCOR/BCORL1
BCOR is the member of ncPRC1.1 that directly associates with BCL6 to facilitate repression at BCL6 binding sites. BCOR/BCORL1 mutations are found in an array of cancers (Figure 3), and inactivating BCOR mutations have been identified in various hematological, epithelial and central nervous system (CNS) neoplasms (119–123). Somatic mutations of BCOR/BCORL1 are found in 10% acute myeloid leukemia patients (124–127), as well as in other hematological malignancies (128). BCOR is required for myeloid differentiation (129) and its deletion in mice disrupts H2AK119ub1-mediated repression of Hox genes during differentiation, leading to leukemia when paired with KRAS mutation (130).
DLBCL
Due to a prominent role for Polycomb in both B-cell and T-cell development, blood cancers have frequent genomic alterations in Polycomb genes (34,131). In most lymphoid malignant contexts, PRC subunits are overexpressed or activated; however, loss of function mutations are found in some specific types of lymphoid cancers, typically pediatric cancers, or cancers with mutations that activate Ras signaling (132).
TALL
Loss-of-function mutations in all three PRC2 subunits are found to be paired with Notch mutations in over 40% of early T-cell precursors, leading to T-cell acute lymphocytic leukemia (TALL) (133–138). Loss of PRC2 subunits in this cell type prevents T-cell differentiation by allowing repressed, but primed, promoters marked by H3K27me3 to become fully methylated and constitutively repressed (139). In this setting, it is not the repressive function of PRC2 that is important for tumor suppression, but the reversible nature of PRC2-mediated repression.
MPNST
In conjunction with additional tumor suppressors, the PRC2 subunits EED and SUZ12 (but not EZH2) are mutated in 85% of malignant peripheral nerve sheath tumors (MPNST) that develop in patients with NF1 mutations (140). These cancers have loss of H3K27me2 in addition to H3K27me3, indicating different mechanistic outcomes compared to EZH2 mutant cancers (141).
Other genetic events leading to Polycomb loss-of-function
Oncohistone H3K27M
Lys27Met (K27M) mutations in histone H3.1 and H3.3 genes have been identified as genetic drivers in pediatric cancers (142–145). Even though mutations in a single copy of H3 constitute only a small fraction (3.63%–17.61%) of total H3 expressed, H3K27me3 levels are globally reduced (146–148) due to increased affinity to, and inhibition of, EZH2 by H3K27M (147). The inhibition is specific, however, to allosterically activated EZH2 involved in the spread of H3K27me3 marks, and does not affect EZH2 activity at CpG islands (108). Instead, increased H3K27me3 is observed at this small subset of sites (149) leading to efforts to inhibit EZH2 (150,151) in addition to targeting aberrant H3K27Ac-mediated gene activation (152,153).
EZHIP overexpression
Posterior fossa type A ependymomas have increased expression of EZHIP, which inhibits allosterically activated PRC2 through a motif similar to H3K27M (107,108,152,154). Similar to H3K27M cancers, a global decrease in H3K27me3 is observed in this cancer type; however, therapeutic strategies based on this mechanism have not yet been investigated.
Genetic events leading to Polycomb dependency
Covered more extensively in (155).
EZH2 gain-of-function mutations
EZH2 is critical for the activation of resting B-cells and their entry into the cell cycle during germinal center (GC) development. As such, activation of PcGs through mutation or overexpression is commonly observed in B-cell leukemias and lymphomas. Heterozygous mutations of the Tyr641 (Y641F/N/S/H/C) are found in 22% of follicular lymphomas and GC type DLBCLs (87,156,157) and mutations of A677 and A687 have been described in isolated cases of DLBCLs (88). These mutants are inactive as monomethyltransferases but efficient at the di- and trimethylation, while wild-type EZH2 has the highest activity as a monomethyltransferase. The combined activities of WT and mutant EZH2 augments H3K27 trimethylation, increasing steady state levels of H3K27me3 and downstream gene repression (158). Because of the cooperative nature of WT and mutant EZH2, both forms are good drug targets and EZH2 inhibitors are effective against mutant DLBCL in vivo (158,159), leading to FDA approval of the EZH2 inhibitor tazemetostat for use in patients.
BCOR translocations
In contrast to BCOR’s role as a tumor suppressor in leukemia, activating BCOR alterations and translocations are common in pediatric sarcomas and CNS neuroectodermal tumors (CNS HGNET-BCOR), which have transcriptional profiles reminiscent of BCOR overexpression (160). One of the most common genomic alterations in BCOR is ITD (internal tandem duplication)–BCOR, which has duplications of the N-terminal PCGF1 binding domain. Similarly, translocations BCOR–CCNB3, BCOR–MAML3 and ZC3H7B–BCOR all include the PCGF1 binding domain, implicating a potential role for aberrant ncPRC1.1 function in oncogenesis. Whether BCOR-altered cancers are dependent on other ncPRC1.1 subunits remains to be determined.
Somatic mutation of UTX
UTX is a member of the MLL4 COMPASS complex and is the demethylase responsible for removal of H3K27me3 marks, among others (161). Inactivating mutations in UTX occur in several types of human cancer (162,163) and result in the loss of the JmjC domain essential for demethylase activity (161). Many UTX mutant cancers have increased levels of H3K27me3 and are more sensitive to EZH2 inhibitors (163).
SWI/SNF alterations
Similar to what is observed in Drosophila, Polycomb complexes and SWI/SNF chromatin remodelers are antagonistic in mammalian cells (164,165). As such, several cancers with SWI/SNF subunit alterations have increased H3K27me3 and an increased sensitivity to EZH2 deletion. In particular, pediatric rhabdoid tumors with loss of the SWI/SNF subunit SNF5 are dependent on EZH2 (166) and respond well to EZH2 inhibitors, which are now approved clinically for these cancers (167). Other SWI/SNF altered cancers that are dependent on EZH2 include SS18-SSX translocated synovial sarcoma (168), ARID1A-mutated ovarian cancer (169), SMARCA4-mutated lung (170) and ovarian cancers (171), and PBRM1-mutated renal cancers (172). Although the mechanism is not completely clear, some of the above cancers are dependent only on non-enzymatic functions of EZH2 (171).
MLL translocations
The fusion of the N-terminal portion of MLL with subunits of the super elongation complex, primarily ENL, AF9, or AF4, initiates leukemia through the constitutive activation of HOX and other hematopoietic stem cell maintenance genes. In MLL-rearranged leukemia, PRC2 promotes acute leukemogenesis through repression of general senescence regulators (173), and disruption of EED or both EZH paralogs inhibits growth of these leukemias (174).
In a more specific dependency, PRC1 subunit CBX8 interacts directly with AF9/ENL and is required for MLL-AF9 leukemogenesis in mice (67). Knockdown of CBX8 or inhibition of the CBX8 chromodomain significantly reduces viability and HOX gene expression in MLL-AF9 leukemia cell lines (175). In this context, CBX8 contributes to gene repression via RING1B association, and contributes to gene activation via AF9/ENL association (65,176). An activating role for CBX8 may be a result of CBX8 incorporation into other complexes (67) or a result of selective CBX8 antagonism by MLL-AF9 (62,65). The exact biochemical role of CBX8 in MLL-AF9 leukemia remains to be elucidated.
Cancers with general upregulation of Polycomb group genes
Comparing RNA-Seq data obtained from TCGA tumors to TCGA matched normal tissue, we defined differential gene regulation for all PcG genes across TCGA cancers (Figure 4). Several cancers have increased transcription of almost all PcG genes; these cancers include cholangiocarcinoma of the bile duct (CHOL), diffuse large B-cell lymphoma (DLBCL), pancreatic cancer (PAAD) and thymoma (THYM). The upregulation of Polycomb subunits has been confirmed in cholangiocarcinoma and pancreatic cancer (177,178) but has yet to be investigated in thymomas. While therapeutic targeting of PcG has not been extensively explored in any of these cancers, recent studies in pancreatic cancer indicate that targeting EZH2 or BMI1 may be effective in certain aggressive subtypes or cell populations (179,180).
Figure 4.

Heatmap of transcriptional changes in PcG subunits in cancer. Transcriptional fold change of both noncanonical and canonical Polycomb genes in TCGA tumor samples is normalized to TCGA normal, which is obtained from normal tissues near the tumors (T/N) using GEPIA2. Increases (red) or decreases (green) in cancer are displayed as Log2 fold change.
DLBCL
The oncogenic role of PcG upregulation in B-cell lymphoma is well-established and is related to a requirement for Polycomb in germinal center activation (see Section 2.1) (181). While EZH2 inhibitors are not as cytotoxic in DLBCL with increased EZH2 as they are in DLBCL with activating mutations in EZH2 (see Section 2.4), they still inhibit growth and may be effective in combination therapies (182).
BMI1 has been studied extensively in the context of leukemia, and was one of the first PcG proteins identified as oncogenic in hematological diseases (183,184). Ectopic expression of BMI1 gives rise to B- and T-cell lymphomas (185,186), and deletion of BMI1 delays primary leukemia and blocks secondary leukemia (187–189). These results implicate cPRC1 as a potential target; however, the best therapeutic strategy to modulate cPRC1 function in this setting is unclear.
The upregulation of ncPRC1 subunits is also important in DLBCL, particularly subunits of ncPRC1.1, which is important for silencing B-cell differentiation genes during the development of DLBCL (190). This is likely related to the mechanism by which ncPRC1.1 restricts myeloid differentiation by cooperating with BCL6 and PRC2 to repress myeloid regulator genes (129,191). BCL6 inhibitors and degraders are under development (192–194), and ncPRC1.1-specific inhibition may be similarly effective. KDM2B has two potentially targetable domains including the JmjC demethylase domain and the Zinc Finger (ZF)-CXXC DNA binding domain. Inhibitors targeting other JmjC domains have been reported; however, selectivity has been challenging (195). If resolved, selective inhibitors to either domain could be extended to the development of KDM2B-specific inhibitors or degraders for DLBCL treatment.
PRC paralogs as oncogene/tumor suppressor pairs
While many cancer-specific alterations in PcG subunits are related to specific developmental pathways or cancer-specific phenotypes, some sets of paralogs are universally up and downregulated across multiple cancers, including EZH2/EZH1, RING1B/RING1A and (CBX2/CBX8)/(CBX6/CBX7) (Figure 4).
EZH
EZH2 upregulation paired with EZH1 downregulation in tumors is likely related to the fact that EZH2 is expressed only in actively dividing cells and plays a vital role in cellular proliferation, while EZH1 is expressed in both dividing and differentiated cells. While some cancers are dependent on both paralogs, others are dependent only on EZH2, leading to the development of both selective and nonselective catalytic inhibitors (196). In some cancers with EZH2 upregulation, inhibition of the catalytic activity alone is insufficient to stall tumor growth, implicating catalytic-independent roles for EZH2 in cancer progression. This may include non-enzymatic roles for EZH2 in gene activation (197), as well as non-transcriptional functions, such as in cytosolic actin polymerization during metastasis (198). To modulate catalytic-independent functions of EZH2, selective degraders of EZH2 may be more generally effective as a therapeutic strategy (199).
RING
RING1B is the more commonly upregulated RING paralog in cancer, and as the catalytic subunit of PRC1, represents a good therapeutic target (Figure 4); in contrast, RING1A is commonly downregulated in cancers and low expression correlates with poor prognosis, indicating that it may act as a tumor suppressor (200). Considering this, as well as the central importance of RING activity in many normal cells, the development of RING1B-specific inhibitors may prove more selective for cancer.
CBX
CBX paralogs are frequently misregulated in cancer, with CBX2 and CBX8 as the most commonly upregulated paralogs, and CBX7 and CBX6 as the most commonly downregulated (Figure 4). Supporting generally oncogenic roles, CBX2 and CBX8 have emerged as targets in lymphoma (190), hepatocellular carcinoma (201), breast cancer (202,203), prostate cancer (204) and ovarian cancer (205), while CBX6 and CBX7 have been implicated as tumor suppressors in thyroid cancer (206) lung cancer (207), glioblastoma (208), bladder cancer (209), cervical cancer (210) and breast cancer (211). There are exceptions, however; CBX7 acts as an oncogene in lymphoma and prostate cancer (212,213), and CBX6 acts as an oncogene in hepatocellular carcinoma (214). A correlation to any known role for CBX2 and CBX8 in development or cell cycle regulation is not apparent; however, the recently uncovered biochemical role for CBX2, and to a lesser degree for CBX8, in phase separation, may be related to oncogenic function (215,216). Whether selective inhibition of these paralogs has general utility for cancer therapy remains to be seen.
Cancer subpopulations with dependencies on PcG proteins
In many cancers, PcG proteins are not involved in the development of tumors but are instead involved in cancer progression or resistance to therapy, either through facilitating therapy-resistant subtypes or therapy-resistant cell populations.
Metastatic prostate cancer
Multiple PcG proteins are potential targets in prostate cancer, including EZH2, CBX2 and BMI1 (217–223). The most established target is the PRC2 subunit EZH2, which is overexpressed in metastatic prostate cancer and is strongly correlated with cell cycle and DNA repair genes. EZH2’s role in prostate cancer may be at least partially separate from its function in PRC2, as it promotes androgen receptor (AR)-dependent gene activation and DNA damage repair in a manner independent of H3K27me3 (224,225). EZH2 is also upregulated in therapy-resistant neuroendocrine prostate cancer, where it functions in conjunction with CBX2 to repress AR target gene expression and promote resistance to AR-targeted therapies (226,227). EZH2 inhibition across a variety of therapy-resistant prostate cancers shows promising results, particularly to reverse chemotherapy and radiotherapy resistance (228,229). The oncogenic functions of RING1B and BMI1 along with the fusion BMI1-COMMD3 (230–233) may be additional targets for treating advanced prostate cancer.
Glioblastoma multiforme
Glioblastoma multiforme (GBM) is malignant Grade IV brain tumor, composed of heterogeneous cell populations, predominantly abnormal astrocytic cells. Within this population are glioma stem-like cell (GSC) with high expression of PcG genes (234). This population is responsible for resistance to radiation or chemotherapy and tumor repopulation after treatment (196,235,236). Short-term EZH2 depletion or inhibition stalls tumor growth, while prolonged EZH2 depletion drastically alters tumor cell identity and enhances tumor progression (196). The expression of other PcG subunits, such as BMI1, is also enriched in GBM cancer stem cells (237), although in a different GSC population, indicating that dual targeting of multiple PRC complexes may be necessary (196).
Colorectal cancer
Similar to glioblastoma, targeting cancer-initiating cells (CICs) in colorectal cancer is a promising strategy to prevent tumor recurrence after therapy. Human colorectal CIC function is dependent on BMI-1 (PCGF4), a subcomponent of PRC1 (238).
POLYCOMB GROUP PROTEIN MODULATORS: INHIBITORS/ DEGRADERS/ ACTIVATORS
Considering the importance of PcG proteins in gene regulation and cancer progression, many chemical approaches have been developed to target different aspects of Polycomb function. An overview of the different subunits targeted and the strategies for modulation are described in Figure 5 and summarized with chemical structures in Table 2. Additional derivatives of similar scaffolds are not all included.
Figure 5.

Mechanism of Action (MoA) for PcG protein modulators. Representative modulators targeting (A) PRC2 and (B) PRC1 are shown with illustrations of their MoA. PPI: Protein–protein interaction.
Table 2.
Mechanism of action and structure of Polycomb modulators
| Target subunit | Ligand | Mechanism of action | Structure | Reference | Associated Clinical Trials (NCT number) |
|---|---|---|---|---|---|
| PRC2 | |||||
| EZH2 | 3-Deazane-planocin A | Inhibits the histone methyltransferase activity of EZH2 while inducing degradation of the PRC2 core subunits EZH2, EED, and SUZ12. In immunocompromised mice, this compound reduced the time of formation of tumors originating from prostate cancer cells |

|
(240) |
|
| EZH2 | GSK926 | SAM-e competitive inhibitor discovered from a high-throughput screening of the GSK compound collection. Reduces H3K27me3 levels in a breast cancer cell line and inhibits cell proliferation in breast and prostate cancer cell-based models |

|
(241) |
|
| EZH2 | GSK126 | In a similar fashion as GSK926, GSK126 was discovered from a high-throughput screening of the GSK compound collection. Highly selective for EZH2 over other methyltransferases. Inhibits cell proliferation in B-cell lymphoma cell-based and murine models that contain an EZH2-activating mutation |

|
(159) | NCT02082977 |
| EZH2 | EPZ005678 | Selectively reduces H3K27 methylation by EZH2 in vitro and in lymphoma cell-based models. Treatment of lymphoma cells bearing a mutant EZH2, leads to antiproliferative effects, indicating that these cancers are critically dependent on mutant EZH2 |

|
(158) |
|
| EZH2 | EPZ-6438 (tazemetostat) | Discovered along with EPZ005678 but shows better potency and oral bioavailability in animals. Treatment of mice bearing a lymphoma xenograft with mutant EZH2 reduces cell growth in a concentration dependent manner. FDA-approved for follicular lymphoma and epithelioid sarcoma with SNF5 deletions |
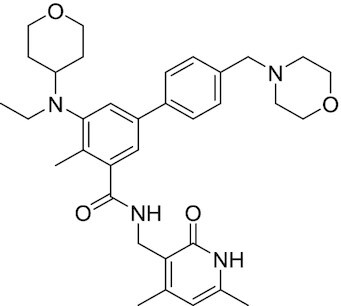
|
(243) |
NCT03456726 NCT02601950 NCT01897571 NCT03213665 NCT02860286 NCT02601937 NCT04557956 NCT04762160 NCT03010982 NCT03874455 NCT02875548 NCT04179864 NCT03854474 NCT03028103 NCT04917042 |
| EZH2 | EPZ011989 | Inhibits EZH2 in a mouse xenograft model of DLBCL, resulting in tumor growth inhibition while showing oral bioavailability |
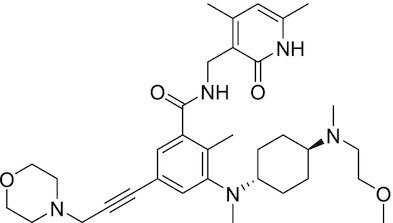
|
(247) |
|
| EZH2 | EI1 | Inhibits the methyl-transferase activity of EZH2/PRC2 leading to reduction of H3K27 methylation over other H3 methylation marks. EI1 shows antiproliferative effects and down-regulates the proliferation gene signature in DLBCL |
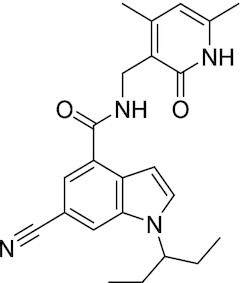
|
(248) |
|
| EZH2 | UNC1999 | Inhibits the methyl-transferase activity of EZH2 and EZH1 by acting as competitor of SAM-e. UNC1999 is orally bioavailable and shows no adverse effects in Swiss albino mice |

|
(249) |
|
| EZH2 | CPI-360 | Competes with SAM-e for the EZH2 SET domain, leading to reduction of H3K27 trimethylation levels without affecting the protein levels of EZH2, SUZ12, and EED. CPI-360 has antiproliferative effects in different lymphoma cell-based models as well as in a human B-cell non-Hodgkin lymphoma murine model |

|
(251) |
|
| EZH2 | CPI-1205 | Binds to the EZH2 catalytic domain. CPI-1205 proved to be efficacious, well-tolerated and highly bioavailable in a lymphoma xenograft model. Currently under clinical trials |

|
(291) |
NCT02395601 NCT03480646 NCT03525795 |
| EZH2 | PF06821497 | EZH2 catalytic inhibitor effective in mouse xenograft model of DLBCL |

|
(253) | NCT03460977 |
| EZH1/2 | Valemetostat (DS-3201b) | EZH1/EZH2 dual inhibitor with activity in DLBCL, as well as AML, TAL and urogenital cancers |

|
(255) |
NCT04703192 NCT04842877 NCT04102150 NCT04388852 |
| EZH2 | MS1943 | First-in-class EZH2 degrader, selective for EZH2 over other methyltransferases. Induces EZH2 degradation and cytotoxicity in triple-negative breast cancer cell-based models |

|
(199) |
|
| EZH2 | Ebastine | Initially discovered as an antihistamine drug, repurposed as an EZH2 inhibitor by decreasing EZH2 expression and reducing the levels of H3K27me3 in breast cancer and prostate cancer cells. Also active in a triple-negative breast cancer murine model |

|
(229) |
|
| EZH2-EED | Astemizole | Disrupts EZH2-EED protein-protein interaction, which results in inhibition of the methyltransferase activity of PRC2. Astemizole inhibits proliferation of DLBCL cells |

|
(258) |
|
| EZH2-EED | SAH-EZH2 | Peptidomimetic of stabilized alpha-helix of EZH2 which disrupt the EZH2-EED interaction leading to reduced H3K27me3 and EZH2 protein levels. SAH-EZH2 is capable of inducing growth arrest in leukemia cells and shows antiproliferative effects in B-cell lymphoma cell lines. | FSSNRQKILERTEILNQEWKQRRIQPV | (259) |
|
| EZH2-EED | DC-PRC2in-01 | Inhibits EZH2-EED interaction leading to reduced H3K27me3, as well as degradation of PRC2 core subunits. DC-PRC2in-01 inhibits PRC2-driven lymphomas cell growth and demonstrates cell cycle arrest at G0/G1 phase |

|
(260) |
|
| EED | A-395 | Inhibits EED H3K27me3 recognition by binding to the H3K27me3 binding pocket. Inhibits growth in DLBCL cell lines that have acquired resistance to EZH2 inhibitors. Also active in a xenograft murine model |

|
(292) |
|
| EED | EED226 | Binds to the EED binding pocket that recognizes H3K27me3. Reduces H3K27 methylation in a human B-cell non-Hodgkin lymphoma cell line and inhibits tumor proliferation in a human B-cell non-Hodgkin lymphoma murine model |

|
(263) |
|
| EED | MAK-683 | Currently under Phase I/II study to be evaluated as an anti-tumor agent in DLBCL, nasopharyngeal carcinoma (NPC) or other advanced solid tumors for whom no further effective standard treatment is available | Unknown | (265) | NCT02900651 |
| EED | UNC5115 | Discovered with UNC5114. Binds to the H3K27me3 binding pocket and inhibits the catalytic activity of PRC2 |

|
(266) |
|
| EED | Compound 19 | Acts as a competitor for the H3K27me3 binding pocket in EED, leading to reduction in the methyltransferase activity of PRC2. Inhibits growth in a DLBCL cell line. |

|
(267) |
|
| EED | UNC6852 | Bivalent chemical degrader that binds to EED and leads to degradation of PRC2. Derived from EED226 and a VHL ligand. Decreases H3K27me3 levels in DLBCL cell lines |

|
(268) |
|
| EED | PROTAC 2 | Degrades EED along with EZH2 and SUZ12. PROTAC 2 is a more potent degrader than its analogue PROTAC 1. Both molecules inhibit growth in a DLBCL cell line as well as a rhabdoid cancer cell line |

|
(269) |
|
| EED | UNC5636 | Peptidomimetic compound that selectively activates EED bearing a I363M mutation. This promotes PRC2 catalytic activity shown by the incorporation of a methyl group to lysine 27 of H3 peptide |

|
(270) |
|
| PRC1 | |||||
| RING | PRT4165 | Inhibits H2A ubiquitination of topoisomerase Top2α at double-strand break sites in cells |

|
(275) |
|
| RING | RB-3 | Binds RING1B and alters protein conformation, preventing association with histones and subsequent H2A119Ub |

|
(276) |
|
| PCGF4 (BMI-1) | PTC-209 | Inhibits colorectal cancer-initiating cells by reducing the protein levels of PCGF4 (BMI-1) |

|
(238) |
|
| PCGF4 (BMI-1) | PTC-596 | Reduces the levels of functional BMI-1 by inducing its hyper-phosphorylation. Currently under Phase 1 clinical trials |

|
(277) |
NCT03206645 NCT03761095 NCT03605550 NCT02404480 |
| PCGF4 (BMI-1) | QW24 | Induces BMI-1 protein degradation through the autophagy-lysosome pathway, leading to inhibition of colorectal CICs’ self-renewal |
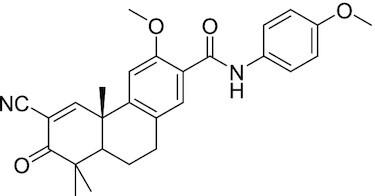
|
(278) |
|
| CBX4/7 | UNC3866 | Binds to the ChD of the CBX paralogs, preventing them from binding methyllysine. UNC3866 inhibits proliferation of PC3 prostate cancer cells |

|
(282) |
|
| CBX7 | MS37452 | Displaces CBX7 from the INK4A/ARF locus in prostate cancer cells, hence de-repressing the transcription of p16/CDKN2A |

|
(283) |
|
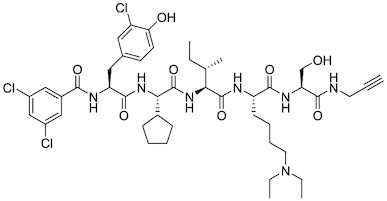
| CBX7 | MS351 | Discovered via structure-guided drug design, MS351 inhibits CBX7 binding to H3K27me3 when it is bound to RNA. It also derepresses CBX7 target genes in both mouse embryonic stem cells and PC3 prostate cancer cells |
|
(284) |
|
| CBX7 | Compound 33F | Developed using rational design to modify a L3MBTL1 methyllysine binding inhibitor |

|
(285) |
|
| CBX4/7 | UNC4976 | Allosteric modulator of CBX7, abrogating its function as reader of H3K27me3 marks and increasing its non-specific binding to DNA |

|
(286) |
|
| CBX6 | Ligand 5 | Binds to the beta groove of CBX6, which includes the lysine trimethylation binding pocket along with a (−2) pocket and a hydrophobic cleft extending from the binding site |

|
(287) |
|
| CBX6/8 | Ligand 22 | Selectively binds to both CBX6 and CBX8 over other CBX ChDs |

|
(288) |
|
| CBX8 | SW2_110A | Binds to the ChD of CBX8 and prevents its association with chromatin, leading to inhibition of proliferation and deactivation of MLL-AF9 target genes in THP1 leukemia cells |

|
(175) |
|
| CBX8 | UNC7040 | Allosteric modulator of CBX8, abrogating its function as reader of H3K27me3 marks and increasing its non-specific binding to DNA, leading to inhibition of proliferation in lymphoma cells |

|
(290) |
|
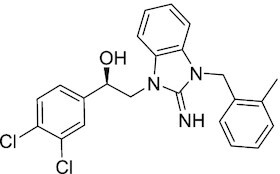
| CBX2 | SW2_152F | Selective CBX2 chromodomain inhibitor. Prevents and reverts neuroendocrine differentiation in prostate cancer cells |
|
(226) |
|
PRC2 modulators
Overview of this section is depicted in Figure 5A.
EZH2 catalytic inhibitors
Given the central role for H3K27me3 in Polycomb-mediated repression, most inhibitors to date target the catalytic activity of EZH2. Within this category, most developed inhibitors bind to the SET domain of EZH2 and compete with S-adenosyl-L-methionine (SAM-e) cofactor binding. EZH2 enzymatic inhibitors have impressive efficacy in certain preclinical and clinical cancer models. A more detailed description of the medicinal chemistry is highlighted in (239).
3-Deazaneplanocin A (DZNep)
DZNep is an inhibitor of the enzyme S-adenosylhomocysteine hydrolase. Inhibition of this hydrolase results in a reduction of cellular SAM-e, thereby inhibiting all SAM-e-dependent enzymes, including EZH2. DZNep effectively decreases the expression of PRC2 proteins EZH2, SUZ12, and EED, as well as global H3K27me3 levels, leading to the reactivation of the PRC2-repressed genes and apoptosis in breast cancer cell line MCF-7 and colorectal cancer cell line HCT116, but not in normal cells (240). However, the lack of specificity for EZH2 inhibition makes DZNep unsuitable as a probe of EZH2 dependency, leading to further efforts to elucidate its contribution to EZH2-independent processes.
GSK926 and GSK126
GSK926 was the first highly potent, SAM-e competitive and selective EZH2 inhibitor developed by GSK (241). GSK126 has improved properties and activity against cell migration and angiogenesis (159).
EPZ005687
EPZ005687 is a selective inhibitor of EZH2 (Ki = 24 nM) and therefore blocks H3K27 methylation (158). EPZ005687 displays 50-fold selectivity for EZH2 over the closest methyltransferase EZH1, and >500-fold selectivity over 15 other methyltransferases. EPZ005687 inhibits the growth of EZH2-activating-mutant lymphoma cells, with lesser effect on wild-type lymphoma, making it the first-in-class chemical probe for evaluating the effect of inhibiting EZH2 enzymatic activity in cancer. EPZ005687 is also effective in inhibiting the proliferation and migration of synovial sarcoma cells driven by a translocation in SWI/SNF subunit SS18 (242).
EPZ-6438 (Tazemetostat)
EPZ-6438 is a potent (Ki = 2.5 nM) and selective (35-fold over EZH1 and 4500-fold to other HMTs) SAM-e competitive small molecule inhibitor of EZH2. Although EPZ-6438 has similar MoA and selectivity in comparison to EPZ005678, EPZ-6438 has superior potency and drug-like properties. EPZ-6438 treatment of mice bearing EZH2-mutant non-Hodgkin lymphoma xenografts demonstrated complete and sustained tumor regressions with concurrent diminution of H3K27me3 levels in tumors. EPZ-6438 is currently FDA-approved for follicular lymphoma, B-cell non-Hodgkin lymphoma and epithelioid sarcoma (243–246).
EPZ011989
Optimized from EPZ-6438, EPZ011989 has a lower amine pKa, while the selectivity for EZH2 and potency against EZH2-mutant lymphoma are maintained (Ki < 3 nM). In addition, it reduces H3K27me3 levels in tumors and exhibits good bioavailability, metabolic stability, PK/PD profile, and in vivo activity (247).
EI1
The SAM-e competitive EZH2 inhibitor, EI1, was identified from a high-throughput screening (248). EI1 demonstrates potent inhibition of the enzymatic activity of both wildtype EZH2 and Y641F mutant with IC50 of 15 nM and 13 nM, respectively. DLBCL cells treated with EI1 exhibit genome-wide loss of H3K27 methylation and activation of PRC2 target genes, leading to decreased proliferation, increased cell cycle arrest, and apoptosis.
UNC1999
UNC1999 is an orally available and selective SAM-e competitive inhibitor with high potency for both EZH2 and EZH1 (249). UNC1999 suppresses global H3K27 trimethylation/dimethylation with concurrent gain of H3K27ac, leading to growth inhibition of mixed-lineage leukemia (MLL) cells (250). UNC1999 displays in vivo activity in a well-defined murine MLL-AF9 leukemia model, delaying MLL-AF9-induced leukemogenicity and prolonging survival (250).
CPI-1205 and CPI-360
CPI-360 is a highly potent (Ki = 0.5 nM) EZH2 inhibitor developed at Constellation Pharmaceuticals from a pyridone-based high-throughput screen hit. It displays activity in both mutant EZH2-containing lymphomas as well as lymphomas with WT EZH2 (251). Optimized compound CPI-1205 is effective in vivo and well-tolerated in clinical trials (252). Further modifications to improve residence time and metabolism are being pursued.
PF068721497
A catalytic inhibitor developed at Pfizer from a focused library of dimethylpyridones. It is potent against both WT and mutant EZH2 and induces tumor regression in DLBCL mouse xenograft models (253).
Valemostat
A catalytic inhibitor of both EZH1 and EZH2 developed by Daiichi Sankyo Co. (254) It is active against EZH2-mutant DLBCL as well as AML (255). It is in clinical trials for these cancers, as well as for TALL and urogenital cancers.
EZH2 degraders and downregulators
MS1943
Although EZH2 inhibitors are effective in treating sarcoma and lymphoma, other cancers that are dependent on EZH2, such as triple negative breast cancer (TNBC), do not respond to catalytic inhibition. For these cancers, chemical approaches to degrade EZH2 are being explored. MS1943 is the first-in-class EZH2-selective degrader that effectively reduces EZH2 levels in cells (199). It is a bivalent compound consisting of an EZH2 inhibitor linked to an adamantane hydrophobic tag. The hydrophobic tag interacts with greasy core residues of the protein and destabilizes the protein folding, resulting in a partially folded protein that is subsequently degraded by the proteasome (256,257). MS1943 has profound cytotoxic effect in multiple TNBC cells in vitro and in vivo, with minimal effect on normal cells.
Ebastine
Ebastine, a marketed antihistamine drug, reduces EZH2 transcript levels, leading to decreased H3K27me3 levels in breast and prostate cancer cell lines. It decreases cancer cell growth and reduces tumor growth and progression in xenograft mice models (229). Ebastine demonstrates promise as an anticancer medication considering its potency as EZH2 inhibitor and its safety as an antihistamine that is already available on the market.
EZH2-EED interaction disruptors
Since EZH2 enzymatic function depends on its interaction with EED, disruption of this interaction provides another route to modulate EZH2 activity. Disruption of EZH2-EED interaction also reduces the protein levels of EZH2 and may have broader therapeutic implications for PRC2-addicted cancers that are either dependent or independent of its HMT activity (172,199).
Astemizole
This antihistamine drug has been recently identified as a small molecule inhibitor of EZH2-EED interaction. It inhibits PRC2 activity by depleting the PRC2 subunits and decreasing H3K27me3 levels, resulting in anti-proliferation activity in lymphomas (258).
SAH-EZH2
SAH-EZH2 is a stabilized alpha-helix mimetic of the EZH2 peptide. It selectively inhibits H3K27 trimethylation of PRC2 in vitro and selectively suppresses the growth of leukemia and lymphoma cells by disrupting the EZH2-EED interaction (259). Stapled peptides have sub-optimal pharmacokinetics, reducing their utility as single dose agents; however, they may have therapeutic potential administered along with enzymatic inhibitors to boost efficacy and decrease resistance.
DC-PRC2in-01
The co-crystal structure of astemizole in complex with EED was used for the structure-guided design of DC-PRC2in-01, a novel EZH2-EED interaction inhibitor (260). With a Kd of 4.56 μM, it causes degradation of PRC2 core subunits and a decrease in global H3K27me3 levels in lymphoma cancer cells.
EED inhibitors
The PRC2 subunit EED binds to the H3K27me3 mark and augments EZH2 enzymatic activity. As an alternative strategy to inhibiting the PRC2 methylation function, targeting EED can reduce PRC2 activity and prevent PRC2 binding to histones. EED inhibitors may also be advantageous in being capable of targeting both wildtype and mutant EZH2-dependent functions.
A-395
A-395 binds to the WD40 domain of EED, competing with H3K27me3 in the aromatic binding pocket with an IC50 of 7 nM. Inhibition of H3K27me3 binding prevents allosteric activation of PRC2; this resembles the results achieved with EZH2 inhibitors, but expands with activity against cell lines with acquired resistance to EZH2 inhibitors (261). Further structure-activity relationship studies on this compound found that 2,6-disubstitution of the N-benzyl improved the binding and cellular activities (262).
EED226
EED226 is a potent and selective PRC2 inhibitor developed from a high throughput screen hit that directly binds to the WD40 domain of EED. EED226 binding to EED induces a conformational change in the aromatic cage leading to the destabilization of EED-H3K27me3 interaction and a loss of PRC2 enzymatic activity. EED226 is effective in treating cancers susceptible to EZH2 inhibition, as well as cancers with a mutant EZH2 protein resistant to SAM-e competitive inhibitors (263). Several additional small molecules (EED666, EED162, EED210, and EED709) bind to EED in a similar manner, defining a common mechanism of inhibition and providing a starting point for further inhibitor development (263,264).
MAK683
MAK683 is an allosteric inhibitor of PRC2 developed by Novartis (265). Similar to EED226, it binds EED and alters the conformation of the EED-H3K27me3 binding pocket, preventing EED-H3K27me3 interaction and EZH2 activation. This drug is being tested in clinical trials for patients with DLBCL, nasopharyngeal carcinoma, gastric cancer, ovarian cancer, prostate cancer and sarcomas.
Peptidomimetic ligand UNC5114 and UNC5115
Using JARID2-K116me3 peptides as a starting point, smaller, more potent peptidomimetic ligands for the EED aromatic cage were developed. UNC5114 (Kd = 0.68 ± 0.05 μM) and UNC5115 (Kd = 1.14 ± 0.14 μM) exhibit 10-fold improvement in affinity and physicochemical properties compared with JARID2-K116me3 (114–118,266) and inhibit allosteric activation of PRC2 catalytic activity by EED.
Compound 19
A set of fragment-sized small molecules (compounds 14, 16, and 19) were discovered using structure-guided inhibitor design. The optimized compound 19 inhibits PRC2 with IC50 = 1.3 μM and inhibits rhabdoid cell growth with IC50 = 2.9 μM (267).
EED proteolysis targeting chimeras (PROTACs)
UNC6852
An EED-targeted bivalent chemical degrader UNC6852 selectively degrades EED and other PRC2 components through recruitment of the VHL ubiquitin ligase. UNC6852 inhibits PRC2 catalytic activity, decreases H3K27me3 levels, and inhibits proliferation of DLBCL cell lines. PRC2-targeted degraders have the potential to overcome acquired resistance to EZH2 inhibitors. More importantly, UNC6852 provides a useful tool for identifying and treating cancers dependent on non-catalytic functions for PRC2 (268).
PROTAC 1 and PROTAC 2
Simultaneously to UNC6852, a team at AstraZeneca reported different EED-based PROTAC molecules that utilize VHL recruitment to degrade PRC2 subunits and reduce PRC2 activity (269).
EED activators
UNC5636
Loss-of-function (LOFs) mutations in cancer, unlike gain-of-function mutations, are bigger challenges for targeted therapeutics. A subset of myeloid disorders has a I363M LOF mutation of EED that leads to a loss in EZH2 enzymatic activity. UNC5636 was developed using computational simulations and structure-based design to selectively stimulate the catalytic activity of PRC2-EED-I363M and not WT PRC2. Although the compound lacks cell permeability, this work demonstrates the feasibility of developing targeted agonists for reversing the LOF mutant phenotype (270), which can also be a promising direction for other PcG LOF mutations.
Limitations of PRC2 inhibitors
Although EZH2 demonstrates a remarkable level of substrate specificity in catalyzing methylation of only one lysine (H3K27) among all histone peptide substrates, various in vitro and cell-based studies indicate that EZH2 has additional substrates such as JARID2, H2BK120, talin, and transcription factors such as STAT3, which may or may not be desirable targets for cancer (246,271–274). In addition, the cytotoxic effects of EZH2 inhibition on adult stem cells remain a major concern in therapeutical strategies.
PRC1 inhibitors
An overview of this section is summarized in Figure 5B.
RING inhibitors
PRT4165
PRT4165 inhibits RING1A and RING1B E3 ubiquitin ligase activity in vitro. While inhibition of PRC1-mediated H2A monoubiquitination in cells has not been shown, it does inhibit polyubiquitination at double stranded breaks, which is proposed to be downstream of PRC1 activity (275). This molecule is the first chemical inhibitor of PRC1 E3 ubiquitin ligase activity, although its low potency and structural similarity to pan assay interference compounds raises concerns over cellular activity and selectivity.
RB-3
Fragment-based screening and subsequent optimization of molecules that bind to the BMI1/RING1B dimer recently produced the first selective inhibitor of PRC1 RING ubiquitin ligase activity (276). This inhibitor binds RING1B with 3.6 μM affinity and alters the conformation to block ubiquitination activity. It is selective for PRC1 and effectively blocks H2AK119ub1 in cells, but it is not selective for RING1B over RING1A or BMI1 over PCGF1, indicating that it likely blocks all PRC1-mediated ubiquitination. While complete inhibition of PRC1 may be toxic to some cell types, the reduction of PRC1 ubiquitin ligase activity, or subsequent development of inhibitors selective for individual PRC1 subcomplexes, may prove therapeutically useful.
BMI-1 inhibitors
PTC209 and PTC596
PTC209 induces BMI-1 degradation and reduces PRC1 activity. Treatment of primary colorectal cancer xenografts with PTC-209 resulted in loss of colorectal CICs with irreversible tumor growth impairment and little cytotoxicity (238). Additional efforts from PTC Therapeutics resulted in PTC596, an orally bioavailable molecule currently in clinical trials (NCT02404480) for glioblastoma and fibrosarcoma. PTC596 inhibits APC/CCDC20 activity resulting in the persistent activation of CDK1 and CDK2, leading to hyperphosphorylation of BMI-1 and its degradation (277).
QW24
BMI-1 inhibitor QW24 decreases BMI-1 protein expression through the autophagy-lysosomal degradation pathway. Based on cell viability and protein regulation, QW24 is more potent than previously reported BMI-1 inhibitor PTC209. QW24 significantly inhibits stem-like properties in colorectal cancer cell lines, leading to attenuation of proliferation and metastasis. In animal studies, QW24 exhibits little toxicity in the subcutaneous xenograft model, with reduced tumor metastasis and increased mice survival (278).
CBX inhibitors
Five CBX proteins are incorporated into PRC1, which bind H3K27me3 via their chromodomains (ChDs). The flexibility of the ChD and high homology among the CBX ChD provide as major obstacles for developing potent, selective, and cell permeable CBX inhibitors.
CBX4/7
Peptidic inhibitors
CBX7 promotes proliferation of prostate cancer cells (279) and malignant hematopoietic progenitor cells (280). The first CBX ChD ligands were peptidic in nature with the highest affinity for CBX7, with 0.28 ±0.05 μM, and five-fold selectivity over CBX8 (281). UNC3866 was then developed with similar specificity for CBX7 and CBX4, but higher affinity and, most importantly, utilized a modification of the trimethyllysine to diethyllysine to allow for cell permeability. UNC3866 induces a senescence-like phenotype in PC3 prostate cancer cells and inhibits their proliferation at IC50 = 7.6 μM (282).
Small molecule inhibitors MS37452 and MS351
MS37452 and MS351 are small molecule CBX ChD inhibitors identified using high-throughput screening. Although the affinity to CBX7 ChD is low, they are selective for CBX7 over the other paralogs. Both molecules inhibit H3K27me3 binding; however, MS351 only inhibits H3K27me3 binding when CBX7 is bound to RNA (283,284). In PC3 prostate cancer cells, MS37452 and MS351 can inhibit CBX7 binding at the INK4A/ARF locus to induce transcriptional de-repression of p16/CDKN2A and inhibit PC3 growth.
Compound 33F
A set of small molecule inhibitors with low potency developed by rational adaption of inhibitors of L3MBTL1, a methyllysine-binding protein (285). This work identified multiple small-molecule inhibitors with modest to low potency (IC50: 257–500 μM).
Positive allosteric modulator UNC4976
UNC4976 is a modified version of UNC3866 with a large norcamphor group on the lysine instead of diethyls. The bulky modification does not change in vitro binding properties but does induce a conformational change that increases the affinity of CBX7 ChD to DNA/RNA. The enhanced efficacy of UNC4976 results from simultaneously antagonizing H3K27me3-specific binding while increasing non-specific DNA and RNA binding, shifting the equilibrium of CBX7-containing PRC1 from H3K27me3 target regions (286).
CBX6
Peptidic ligand 5
This inhibitor selectively inhibits the CBX6 ChD by occupying a small hydrophobic pocket adjacent to the aromatic cage. It displays good affinity (Kd = 900 nM) with in vitro selectivity >5-fold over other CBX paralogs (287).
CBX6/CBX8
Peptidic ligands 22 and 23
The first dual-selective inhibitors for CBX6 and CBX8 demonstrate high affinity to both CBX6 and CBX8 (288). Lead compounds from this study demonstrated efficacy in rhabdoid tumor cell line.
CBX8
Peptidic inhibitor SW2_110A
The selection of directed DNA-encoded libraries (DELs) against multiple ChDs led to the development of SW2_110A, a selective, cell-permeable inhibitor of the CBX8 ChD with a Kd of ∼800 nM, and minimal 5-fold selectivity (no binding to CBX4 and CBX6, 20-fold over CBX7, 5-fold over CBX2) for CBX8 ChD over all other CBX paralogs in vitro. SW2_110A specifically inhibits the association of CBX8 with chromatin in cells and inhibits the proliferation of THP1 leukemia cells driven by the MLL-AF9 translocation (175,289).
Allosteric modulator UNC7040
A potent positive allosteric modulator of CBX8, UNC7040, can antagonize H3K27me3 binding to CBX8 while increasing interactions with nucleic acids and participation in variant PRC1 (290). It has increased potency against CBX8 binding in DLBCL cell compared to similar inhibitors that only inhibit H3K27me3 binding.
CBX2
Peptidic inhibitor SW2_152F
The CBX2 ChD inhibitor SW2_152F has a Kd of ∼80 nM, and 24 to 1000-fold selectivity for CBX2 over the other CBX paralogs in vitro. This inhibitor blocks CBX2-facilitated neuroendocrine differentiation in prostate cancer, mainly through de-repressing AR signaling in neuroendocrine differentiated prostate cancer cells (226).
CONCLUSIONS AND FUTURE OPPORTUNITIES
There are many cancers dependent on PcG proteins for initiation, progression, and chemotherapy resistance; however, the most effective method for targeting Polycomb function in a specific cancer is not always clear. Due to a central role for H3K27me3 in Polycomb-mediated repression and the relative homogeneity of PRC2 subunit composition, the majority of inhibitors developed to date target PRC2-mediated H3K27 trimethylation. Targeting the enzymatic SET domain of EZH1/2 is the most straightforward approach; however, drug-resistant mutant cancers, as well as cancers dependent on non-enzymatic PRC2 function, have necessitated alternate approaches to inhibit PRC2. Allosteric EED inhibitors, PRC2 disrupters, and EZH2/EED degraders can inhibit the growth of cancers insensitive to EZH2 enzymatic inhibitors, although the full utility of these compounds remains to be explored. Additional investigation of how this class of inhibitors affects both normal cells and cancers will reveal additional therapeutic utility.
Targeting PRC2 is effective against many Polycomb-dependent cancers; however, the complete loss of H3K27me3 may be detrimental to many normal cells, particularly adult stem cells. In addition, it is likely that not all PRC2 function in a particular cancer is oncogenic, and some PRC2 activity may actually be tumor suppressive, as has been shown in certain cancer types. Therefore, inhibiting a subset of Polycomb targets or inhibiting cancer-specific PRCs may be more effective in some situations. For instance, in many cancers CBX2/CBX8 are oncogenic while CBX6/CBX7 are tumor suppressive. Inhibitors for cPRC1, specifically CBX ChD inhibitors, can define the utility of more specifically targeting the subset of oncogenic cPRC1s that act downstream of PRC2. Current CBX ChD inhibitors are limited by poor pharmacological properties; however, they are useful for defining individual CBX paralog(s) to focus on as drug targets. The development of more potent CBX inhibitors will potentially require novel strategies, such as allosteric modulation or even degradation.
Targeting ncPRC1s is another strategy that may be more selective for certain cancers. ncPRC1 has higher ubiquitin ligase activity than cPRC1; however, if nonspecific RING enzymatic inhibitors prove toxic, a better strategy may be to target specific ncPRC1 variant subunits. ncPRC1.1 and ncPRC1.6 bind different regions of the genome but are functionally redundant in ESCs. In contrast, ncPRC1.1 is essential in B-cell lymphomas, while ncPRC1.6 acts as a tumor suppressor. Degraders provide an opportunity to target subcomplex-specific ncPRC1 subunits containing non-essential enzymatic or binding domains; for instance, targeting ncPRC1.1 through KDM2B or targeting ncPRC1.6 through L3MBTL2.
The functional redundancy for many Polycomb subunits along with the cell-type specific expression of PcG genes indicates that dual inhibition of PRC subunits may be a more effective pharmacological strategy. In probe development, selectivity is the highest priority; however, for drug development targeting of multiple functionally redundant PcG targets might be more effective in increasing cancer death and decreasing resistance. Even so, identifying and inhibiting just the subset of PcG targets important in cancer would reduce toxicity to normal cells. Defining the redundant and specific biological functions for individual PcG proteins in cancer is required for this polypharmacology approach; however, using genetic screens for multiple targets is highly challenging. Instead, small molecule inhibitors can be rapidly screened in combination to identify dual targeting approaches that selectively inhibit Polycomb activity in a cancer-specific manner. Increasing the diversity of Polycomb subunit inhibitors will significantly facilitate this approach. An increased understanding of how specific PRCs/subunits biochemically contribute to Polycomb activity will help with prioritizing targets, as well as developing effective strategies for inhibition.
ACKNOWLEDGEMENTS
Figures created with BioRender.com.
Contributor Information
Sijie Wang, Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University and Purdue University Center for Cancer Research, 201 S. University St., West Lafayette, IN 47907 USA.
Sandra C. Ordonez-Rubiano, Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University and Purdue University Center for Cancer Research, 201 S. University St., West Lafayette, IN 47907 USA.
Alisha Dhiman, Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University and Purdue University Center for Cancer Research, 201 S. University St., West Lafayette, IN 47907 USA.
Guanming Jiao, Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University and Purdue University Center for Cancer Research, 201 S. University St., West Lafayette, IN 47907 USA.
Brayden P Strohmier, Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University and Purdue University Center for Cancer Research, 201 S. University St., West Lafayette, IN 47907 USA.
Casey J Krusemark, Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University and Purdue University Center for Cancer Research, 201 S. University St., West Lafayette, IN 47907 USA.
Emily C Dykhuizen, Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University and Purdue University Center for Cancer Research, 201 S. University St., West Lafayette, IN 47907 USA.
FUNDING
Lilly Graduate Research Award from the Purdue College of Pharmacy (to S.W., in part); NIH [R35GM128894 to C.J.K.]; NIH [U01CA207532 to E.C.D.]; ACS [RSG-21–012-01-DMC].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kennison J.A. The polycomb and trithorax group proteins of Drosophila: Trans-regulators of homeotic gene function. Annu. Rev. Genet. 1995; 29:289–303. [DOI] [PubMed] [Google Scholar]
- 2. Morey L., Santanach A., Blanco E., Aloia L., Nora E.P., Bruneau B.G., Di Croce L. Polycomb regulates mesoderm cell fate-specification in embryonic stem cells through activation and repression mechanisms. Cell Stem Cell. 2015; 17:300–315. [DOI] [PubMed] [Google Scholar]
- 3. Müller J., Verrijzer P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr. Opin. Genet. Dev. 2009; 19:150–158. [DOI] [PubMed] [Google Scholar]
- 4. Hauri S., Comoglio F., Seimiya M., Gerstung M., Glatter T., Hansen K., Aebersold R., Paro R., Gstaiger M., Beisel C. A High-Density map for navigating the human polycomb complexome. Cell Rep. 2016; 17:583–595. [DOI] [PubMed] [Google Scholar]
- 5. Schuettengruber B., Bourbon H.M., Di Croce L., Cavalli G. Genome regulation by polycomb and trithorax: 70 Years and Counting. Cell. 2017; 171:34–57. [DOI] [PubMed] [Google Scholar]
- 6. Piunti A., Shilatifard A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat. Rev. Mol. Cell Biol. 2021; 22:326–345. [DOI] [PubMed] [Google Scholar]
- 7. Gao Z., Zhang J., Bonasio R., Strino F., Sawai A., Parisi F., Kluger Y., Reinberg D. PCGF Homologs, CBX Proteins, and RYBP Define Functionally Distinct PRC1 Family Complexes. Mol. Cell. 2012; 45:344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tavares L., Dimitrova E., Oxley D., Webster J., Poot R., Demmers J., Bezstarosti K., Taylor S., Ura H., Koide H. et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. 2012; 148:664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiao L., Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science. 2015; 350:aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mierlo G., Veenstra G.J.C., Vermeulen M., Marks H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019; 29:660–671. [DOI] [PubMed] [Google Scholar]
- 11. Ciferri C., Lander G.C., Maiolica A., Herzog F., Aebersold R., Nogales E. Molecular architecture of human polycomb repressive complex 2. Elife. 2012; 1:e00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holoch D., Margueron R. Mechanisms regulating PRC2 recruitment and enzymatic activity. Trends Biochem. Sci. 2017; 42:531–542. [DOI] [PubMed] [Google Scholar]
- 13. Chammas P., Mocavini I., Croce L.Di Engaging chromatin: PRC2 structure meets function. Br. J. Cancer. 2019; 122:315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laugesen A., Højfeldt J.W., Helin K. Molecular mechanisms directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell. 2019; 74:8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bauer M., Trupke J., Ringrose L. The quest for mammalian Polycomb response elements: are we there yet?. Chromosoma. 2015; 125:471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanay A., O’Donnell A.H., Damelin M., Bestor T.H. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl. Acad. Sci. 2007; 104:5521–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kahn T.G., Dorafshan E., Schultheis D., Zare A., Stenberg P., Reim I., Pirrotta V., Schwartz Y.B. Interdependence of PRC1 and PRC2 for recruitment to Polycomb Response Elements. Nucleic Acids Res. 2016; 44:10132–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fursova N.A., Blackledge N.P., Nakayama M., Ito S., Koseki Y., Farcas A.M., King H.W., Koseki H., Klose R.J. Synergy between Variant PRC1 complexes defines polycomb-mediated gene repression. Mol. Cell. 2019; 74:1020–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blackledge N.P., Fursova N.A., Kelley J.R., Huseyin M.K., Feldmann A., Klose R.J. PRC1 catalytic activity is central to polycomb system function. Mol. Cell. 2020; 77:857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tamburri S., Lavarone E., Fernández-Pérez D., Conway E., Zanotti M., Manganaro D., Pasini D. Histone H2AK119 mono-ubiquitination is essential for polycomb-mediated transcriptional repression. Mol. Cell. 2020; 77:840–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zepeda-Martinez J.A., Pribitzer C., Wang J., Bsteh D., Golumbeanu S., Zhao Q., Burkard T.R., Reichholf B., Rhie S.K., Jude J. et al. Parallel PRC2/cPRC1 and vPRC1 pathways silence lineage-specific genes and maintain self-renewal in mouse embryonic stem cells. Sci. Adv. 2020; 6:eaax5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kasinath V., Beck C., Sauer P., Poepsel S., Kosmatka J., Faini M., Toso D., Aebersold R., Nogales E. JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science. 2021; 371:eabc3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Højfeldt J.W., Hedehus L., Laugesen A., Tatar T., Wiehle L., Helin K. Non-core Subunits of the PRC2 complex are collectively required for its target-site specificity. Mol. Cell. 2019; 76:423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H., Liefke R., Jiang J., Kurland J.V., Tian W., Deng P., Zhang W., He Q., Patel D.J., Bulyk M.L. et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature. 2017; 549:287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Healy E., Mucha M., Glancy E., Fitzpatrick D.J., Conway E., Neikes H.K., Monger C., Mierlo G.V, Baltissen M.P., Koseki Y. et al. PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol. Cell. 2019; 76:437–452. [DOI] [PubMed] [Google Scholar]
- 26. Scelfo A., Fernández-Pérez D., Tamburri S., Zanotti M., Lavarone E., Soldi M., Bonaldi T., Ferrari K.J., Pasini D. Functional landscape of PCGF proteins reveals both RING1A/B-dependent-and RING1A/B-independent-specific activities. Mol. Cell. 2019; 74:1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Endoh M., Endo T.A., Endoh T., Isono K., Sharif J., Ohara O., Toyoda T., Ito T., Eskeland R., Bickmore W.A. et al. Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain cell identity. PLOS Genet. 2012; 8:e1002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao Z., Lee P., Stafford J.M., Schimmelmann M., Schaefer A., Reinberg D. An AUTS2–Polycomb complex activates gene expression in the CNS. Nature. 2014; 516:349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao W., Huang Y., Zhang J., Liu M., Ji H., Wang C., Cao N., Li C., Xia Y., Jiang Q. et al. Polycomb group RING finger proteins 3/5 activate transcription via an interaction with the pluripotency factor Tex10 in embryonic stem cells. J. Biol. Chem. 2017; 292:21527–21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Q., Geng Z., Gong Y., Warren K., Zheng H., Imamura Y., Gao Z. WDR68 is essential for the transcriptional activation of the PRC1-AUTS2 complex and neuronal differentiation of mouse embryonic stem cells. Stem Cell Res. 2018; 33:206–214. [DOI] [PubMed] [Google Scholar]
- 31. Frangini A., Sjöberg M., Roman-Trufero M., Dharmalingam G., Haberle V., Bartke T., Lenhard B., Malumbres M., Vidal M., Dillon N. The Aurora B Kinase and the Polycomb Protein Ring1B Combine to Regulate Active Promoters in Quiescent Lymphocytes. Mol. Cell. 2013; 51:647–661. [DOI] [PubMed] [Google Scholar]
- 32. Maezawa S., Hasegawa K., Yukawa M., Sakashita A., Alavattam K.G., Andreassen P.R., Vidal M., Koseki H., Barski A., Namekawa S.H. Polycomb directs timely activation of germline genes in spermatogenesis. Genes Dev. 2017; 31:1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Loubiere V., Papadopoulos G.L., Szabo Q., Martinez A.-M., Cavalli G. Widespread activation of developmental gene expression characterized by PRC1-dependent chromatin looping. Sci. Adv. 2020; 6:eaax4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leicher R., Ge E.J., Lin X., Reynolds M.J., Xie W., Walz T., Zhang B., Muir T.W., Liu S. Single-molecule and in silico dissection of the interaction between Polycomb repressive complex 2 and chromatin. Proc. Natl. Acad. Sci. 2020; 117:30465–30475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Poepsel S., Kasinath V., Nogales E. Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat. Struct. Mol. Biol. 2018; 25:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grau D.J., Chapman B.A., Garlick J.D., Borowsky M., Francis N.J., Kingston R.E. Compaction of chromatin by diverse polycomb group proteins requires localized regions of high charge. Genes Dev. 2011; 25:2210–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matheson L., Elderkin S. Polycomb Bodies. Nuclear Architecture and Dynamics. 2018; Elsevier; 297–320. [Google Scholar]
- 38. Hernández-Muñoz I., Taghavi P., Kuijl C., Neefjes J., van Lohuizen M. Association of BMI1 with Polycomb Bodies Is Dynamic and Requires PRC2/EZH2 and the Maintenance DNA Methyltransferase DNMT1. Mol. Cell. Biol. 2005; 25:11047–11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Messmer S., Franke A., Paro R. Analysis of the functional role of the Polycomb chromo domain in Drosophila melanogaster. Genes Dev. 1992; 6:1241–1254. [DOI] [PubMed] [Google Scholar]
- 40. Ren X., Vincenz C., Kerppola T.K. Changes in the Distributions and Dynamics of Polycomb Repressive Complexes during Embryonic Stem Cell Differentiation. Mol. Cell. Biol. 2008; 28:2884–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Isono K., Endo T.A., Ku M., Yamada D., Suzuki R., Sharif J., Ishikura T., Toyoda T., Bernstein B.E., Koseki H. SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell. 2013; 26:565–577. [DOI] [PubMed] [Google Scholar]
- 42. Saurin A.J., Shiels C., Williamson J., Satijn D.P.E., Otte A.P., Sheer D., Freemont P.S. The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J. Cell Biol. 1998; 142:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buchenau P., Hodgson J., Strutt H., Arndt-Jovin D.J. The distribution of polycomb-group proteins during cell division and development in Drosophila embryos: Impact on models for silencing. J. Cell Biol. 1998; 141:469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moussa H.F., Bsteh D., Yelagandula R., Pribitzer C., Stecher K., Bartalska K., Michetti L., Wang J., Zepeda-Martinez J.A., Elling U. et al. Canonical PRC1 controls sequence-independent propagation of Polycomb-mediated gene silencing. Nat. Commun. 2019; 10:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kloet S.L., Makowski M.M., Baymaz H.I., Van Voorthuijsen L., Karemaker I.D., Santanach A., Jansen P.W.T.C., Di Croce L., Vermeulen M. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol. 2016; 23:682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Endoh M., Endo T.A., Endoh T., Fujimura Y.I., Ohara O., Toyoda T., Otte A.P., Okano M., Brockdorff N., Vidal M. et al. Polycomb group proteins Ring1A/B are functionally linked to the core transcriptional regulatory circuitry to maintain ES cell identity. Development. 2008; 135:1513–1524. [DOI] [PubMed] [Google Scholar]
- 47. Cao R., Tsukada Y.I., Zhang Y. Role of Bmi-1 and Ring1A in H2A Ubiquitylation and Hox Gene Silencing. Mol. Cell. 2005; 20:845–854. [DOI] [PubMed] [Google Scholar]
- 48. Li Z., Cao R., Wang M., Myers M.P., Zhang Y., Xu R.-M. Structure of a Bmi-1-Ring1B Polycomb Group Ubiquitin Ligase Complex *. J. Biol. Chem. 2006; 281:20643–20649. [DOI] [PubMed] [Google Scholar]
- 49. Buchwald G., Stoop P., Weichenrieder O., Perrakis A., Lohuizen M., Sixma T.K. Structure and E3-ligase activity of the Ring–Ring complex of Polycomb proteins Bmi1 and Ring1b. EMBO J. 2006; 25:2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McGinty R.K., Henrici R.C., Tan S. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature. 2014; 514:591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bezsonova I., Walker J.R., Bacik J.P., Duan S., Dhe-Paganon S., Arrowsmith C.H. Ring1B Contains a Ubiquitin-Like Docking Module for Interaction with Cbx Proteins. Biochemistry. 2009; 48:10542–10548. [DOI] [PubMed] [Google Scholar]
- 52. Chittock E.C., Latwiel S., Miller T.C.R., Müller C.W. Molecular architecture of polycomb repressive complexes. Biochem. Soc. Trans. 2017; 45:193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang R., Taylor A.B., Leal B.Z., Chadwell L.V., Ilangovan U., Robinson A.K., Schirf V., Hart P.J., Lafer E.M., Demeler B. et al. Polycomb group targeting through different binding partners of RING1B C-terminal domain. Structure. 2010; 18:966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kaustov L., Ouyang H., Amaya M., Lemak A., Nady N., Duan S., Wasney G.A., Li Z., Vedadi M., Schapira M. et al. Recognition and Specificity Determinants of the Human Cbx Chromodomains *. J. Biol. Chem. 2011; 286:521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Völkel P., le Faou P., Vandamme J., Pira D., Angrand P.O. A human polycomb isoform lacking the Pc box does not participate to PRC1 complexes BUT forms protein assemblies and represses transcription. Epigenetics. 2012; 7:482–491. [DOI] [PubMed] [Google Scholar]
- 56. Bernstein E., Duncan E.M., Masui O., Gil J., Heard E., Allis C.D. Mouse Polycomb Proteins Bind Differentially to Methylated Histone H3 and RNA and Are Enriched in Facultative Heterochromatin. Mol. Cell. Biol. 2006; 26:2560–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Connelly K.E., Weaver T.M., Alpsoy A., Gu B.X., Musselman C.A., Dykhuizen E.C. Engagement of DNA and H3K27me3 by the CBX8 chromodomain drives chromatin association. Nucleic Acids Res. 2019; 47:2289–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Plys A.J., Davis C.P., Kim J., Rizki G., Keenen M.M., Marr S.K., Kingston R.E. Phase separation of polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019; 33:799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kagey M.H., Melhuish T.A., Wotton D. The polycomb protein Pc2 is a SUMO E3. Cell. 2003; 113:127–137. [DOI] [PubMed] [Google Scholar]
- 60. Li B., Zhou J., Liu P., Hu J., Jin H., Shimono Y., Takahashi M., Xu G. Polycomb protein Cbx4 promotes SUMO modification of de novo DNA methyltransferase Dnmt3a. Biochem. J. 2007; 405:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ismail I.H., Gagné J.P., Caron M.C., McDonald D., Xu Z., Masson J.Y., Poirier G.G., Hendzel M.J. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 2012; 40:5497–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hemenway C.S., De Erkenez A.C., Gould G.C.D. The polycomb protein MPc3 interacts with AF9, an MLL fusion partner in t(9;11)(p22;q23) acute leukemias. Oncogene. 2001; 20:3798–3805. [DOI] [PubMed] [Google Scholar]
- 63. Connelly K.E., Dykhuizen E.C. Compositional and functional diversity of canonical PRC1 complexes in mammals. Biochim. Biophys. Acta - Gene Regul. Mech. 2017; 1860:233–245. [DOI] [PubMed] [Google Scholar]
- 64. Kawaguchi T., Machida S., Kurumizaka H., Tagami H., Nakayama J. Phosphorylation of CBX2 controls its nucleosome-binding specificity. J. Biochem. 2017; 162:343–355. [DOI] [PubMed] [Google Scholar]
- 65. Maethner E., Garcia-Cuellar M.P., Breitinger C., Takacova S., Divoky V., Hess J.L., Slany R.K. MLL-ENL Inhibits Polycomb Repressive Complex 1 to Achieve Efficient Transformation of Hematopoietic Cells. Cell Rep. 2013; 3:1553–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Malik B., Hemenway C.S. CBX8, a component of the Polycomb PRC1 complex, modulates DOT1L-mediated gene expression through AF9/MLLT3. FEBS Lett. 2013; 587:3038–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tan J., Jones M., Koseki H., Nakayama M., Muntean A.G., Maillard I., Hess J.L. CBX8, a Polycomb Group Protein, Is Essential for MLL-AF9-Induced Leukemogenesis. Cancer Cell. 2011; 20:563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Seif E., Kang J.J., Sasseville C., Senkovich O., Kaltashov A., Boulier E.L., Kapur I., Kim C.A., Francis N.J. Phase separation by the polyhomeotic sterile alpha motif compartmentalizes Polycomb Group proteins and enhances their activity. Nat. Commun. 2020; 11:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wani A.H., Boettiger A.N., Schorderet P., Ergun A., Münger C., Sadreyev R.I., Zhuang X., Kingston R.E., Francis N.J. Chromatin topology is coupled to Polycomb group protein subnuclear organization. Nat. Commun. 2016; 7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Robinson A.K., Leal B.Z., Chadwell L.V., Wang R., Ilangovan U., Kaur Y., Junco S.E., Schirf V., Osmulski P.A., Gaczynska M. et al. The growth-suppressive function of the polycomb group protein polyhomeotic is mediated by polymerization of its sterile alpha motif (SAM) domain. J. Biol. Chem. 2012; 287:8702–8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim C.A., Francis N.J. Chromatin: Polycomb Group SAMs Unite. Curr. Biol. 2016; 26:R710–R712. [DOI] [PubMed] [Google Scholar]
- 72. Saurin A.J., Shao Z., Erdjument-Bromage H., Tempst P., Kingston R.E. A drosophila polycomb group complex includes zeste and dTAFII proteins. Nature. 2001; 412:655–660. [DOI] [PubMed] [Google Scholar]
- 73. Levine S.S., Weiss A., Erdjument-Bromage H., Shao Z., Tempst P., Kingston R.E. The Core of the Polycomb Repressive Complex Is Compositionally and Functionally Conserved in Flies and Humans. Mol. Cell. Biol. 2002; 22:6070–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li M., Zhang S., Zhao W., Hou C., Ma X., Li X., Huang B., Chen H., Chen D. RYBP modulates stability and function of Ring1B through targeting UBE3A. FASEB J. 2019; 33:683–695. [DOI] [PubMed] [Google Scholar]
- 75. Rose N.R., King H.W., Blackledge N.P., Fursova N.A., Ember K.J., Fischer R., Kessler B.M., Klose R.J. RYBP stimulates PRC1 to shape chromatin-based communication between polycomb repressive complexes. Elife. 2016; 5:e18591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhao J., Wang M., Chang L., Yu J., Song A., Liu C., Huang W., Zhang T., Wu X., Shen X. et al. RYBP/YAF2-PRC1 complexes and histone H1-dependent chromatin compaction mediate propagation of H2AK119ub1 during cell division. Nat. Cell Biol. 2020; 22:439–452. [DOI] [PubMed] [Google Scholar]
- 77. Blackledge N.P., Farcas A.M., Kondo T., King H.W., McGouran J.F., Hanssen L.L.P., Ito S., Cooper S., Kondo K., Koseki Y. et al. Variant PRC1 Complex-Dependent H2A Ubiquitylation Drives PRC2 recruitment and polycomb domain formation. Cell. 2014; 157:1445–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Frescas D., Guardavaccaro D., Bassermann F., Koyama-Nasu R., Pagano M. JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature. 2007; 450:309–313. [DOI] [PubMed] [Google Scholar]
- 79. Tsukada Y.I., Fang J., Erdjument-Bromage H., Warren M.E., Borchers C.H., Tempst P., Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006; 439:811–816. [DOI] [PubMed] [Google Scholar]
- 80. Wang Z., Gearhart M.D., Lee Y.W., Kumar I., Ramazanov B., Zhang Y., Hernandez C., Lu A.Y., Neuenkirchen N., Deng J. et al. A Non-canonical BCOR-PRC1.1 Complex Represses Differentiation Programs in Human ESCs. Cell Stem Cell. 2018; 22:235–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wong S.J., Senkovich O., Artigas J.A., Gearhart M.D., Ilangovan U., Graham D.W., Abel K.N., Yu T., Hinck A.P., Bardwell V.J. et al. Structure and Role of BCOR PUFD in Noncanonical PRC1 Assembly and Disease. Biochemistry. 2020; 59:2718–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cascoń A., Robledo M. MAX and MYC: A heritable breakup. Cancer Res. 2012; 72:3119–3124. [DOI] [PubMed] [Google Scholar]
- 83. Hurlin P.J., Steingrìmsson E., Copeland N.G., Jenkins N.A., Eisenman R.N. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J. 2000; 19:3841–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Stielow B., Finkernagel F., Stiewe T., Nist A., Suske G. MGA, L3MBTL2 and E2F6 determine genomic binding of the non-canonical Polycomb repressive complex PRC1.6. PLoS Genet. 2018; 14:e1007193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Trojer P., Cao A.R., Gao Z., Li Y., Zhang J., Xu X., Li G., Losson R., Erdjument-Bromage H., Tempst P. et al. L3MBTL2 Protein Acts in Concert with PcG Protein-Mediated Monoubiquitination of H2A to Establish a Repressive Chromatin Structure. Mol. Cell. 2011; 42:438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of zeste protein. Genes Dev. 2002; 16:2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Morin R.D., Johnson N.A., Severson T.M., Mungall A.J., An J., Goya R., Paul J.E., Boyle M., Woolcock B.W., Kuchenbauer F. et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010; 42:181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sneeringer C.J., Scott M.P., Kuntz K.W., Knutson S.K., Pollock R.M., Richon V.M., Copeland R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. 2010; 107:20980–20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. McCabe M.T., Graves A.P., Ganji G., Diaz E., Halsey W.S., Jiang Y., Smitheman K.N., Ott H.M., Pappalardi M.B., Allen K.E. et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. 2012; 109:2989–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Majer C.R., Jin L., Scott M.P., Knutson S.K., Kuntz K.W., Keilhack H., Smith J.J., Moyer M.P., Richon V.M., Copeland R.A. et al. A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 2012; 586:3448–3451. [DOI] [PubMed] [Google Scholar]
- 91. Cao R., Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell. 2004; 15:57–67. [DOI] [PubMed] [Google Scholar]
- 92. Zhang Q., Vo N., Goodman R.H. Histone Binding Protein RbAp48 Interacts with a Complex of CREB Binding Protein and Phosphorylated CREB. Mol. Cell. Biol. 2000; 20:4970–4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sun J.M., Hou Y.C., Davie J.R. Differential distribution of unmodified and phosphorylated histone deacetylase 2 in chromatin. J. Biol. Chem. 2007; 282:33227–33236. [DOI] [PubMed] [Google Scholar]
- 94. Mahajan M.C., Narlikar G.J., Boyapaty G., Kingston R.E., Weissman S.M. Heterogeneous nuclear ribonucleoprotein C1/C2, MeCP1, and SWI/SNF form a chromatin remodeling complex at the β-globin locus control region. Proc. Natl. Acad. Sci. 2005; 102:15012–15017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ballaré C., Lange M., Lapinaite A., Martin G.M., Morey L., Pascual G., Liefke R., Simon B., Shi Y., Gozani O. et al. Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat. Struct. Mol. Biol. 2012; 19:1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Boulay G., Rosnoblet C., Guérardel C., Angrand P.O., Leprince D. Functional characterization of human Polycomb-like 3 isoforms identifies them as components of distinct EZH2 protein complexes. Biochem. J. 2011; 434:333–342. [DOI] [PubMed] [Google Scholar]
- 97. Kalb R., Latwiel S., Baymaz H.I., Jansen P.W.T.C., Müller C.W., Vermeulen M., Müller J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014; 21:569–571. [DOI] [PubMed] [Google Scholar]
- 98. Kim H., Kang K., Kim J. AEBP2 as a potential targeting protein for Polycomb Repression Complex PRC2. Nucleic Acids Res. 2009; 37:2940–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Grijzenhout A., Godwin J., Koseki H., Gdula M.R., Szumska D., McGouran J.F., Bhattacharya S., Kessler B.M., Brockdorff N., Cooper S. Functional analysis of AEBP2, a PRC2 Polycomb protein, reveals a Trithorax phenotype in embryonic development and in ESCs. Development. 2016; 143:2716–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. He G.P., Kim S., Ro H.S. Cloning and characterization of a novel zinc finger transcriptional repressor. A direct role of the zinc finger motif in repression. J. Biol. Chem. 1999; 274:14678–14684. [DOI] [PubMed] [Google Scholar]
- 101. Li G., Margueron R., Ku M., Chambon P., Bernstein B.E., Reinberg D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010; 24:368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sanulli S., Justin N., Teissandier A., Ancelin K., Portoso M., Caron M., Michaud A., Lombard B., da Rocha S.T., Offer J. et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol. Cell. 2015; 57:769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Conway E., Jerman E., Healy E., Ito S., Holoch D., Oliviero G., Deevy O., Glancy E., Fitzpatrick D.J., Mucha M. et al. A Family of Vertebrate-Specific Polycombs Encoded by the LCOR/LCORL Genes Balance PRC2 Subtype Activities. Mol. Cell. 2018; 70:408–421. [DOI] [PubMed] [Google Scholar]
- 104. Zhang Q., Agius S.C., Flanigan S.F., Uckelmann M., Levina V., Owen B.M., Davidovich C. PALI1 facilitates DNA and nucleosome binding by PRC2 and triggers an allosteric activation of catalysis. Nat. Commun. 2021; 12:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Malte Beringer A., Pisano P., Di Carlo V., Payer B., Wierer M., Di Croce Correspondence L. EPOP functionally links elongin and polycomb in pluripotent stem cells. Mol. Cell. 2016; 64:645–658. [DOI] [PubMed] [Google Scholar]
- 106. Piunti A., Smith E.R., Morgan M.A.J., Ugarenko M., Khaltyan N., Helmin K.A., Ryan C.A., Murray D.C., Rickels R.A., Yilmaz B.D. et al. CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci. Adv. 2019; 5:eaax2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ragazzini R., Pérez-Palacios R., Baymaz I.H., Diop S., Ancelin K., Zielinski D., Michaud A., Givelet M., Borsos M., Aflaki S. et al. EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat. Commun. 2019; 10:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jain S.U., Rashoff A.Q., Krabbenhoft S.D., Hoelper D., Do T.J., Gibson T.J., Lundgren S.M., Bondra E.R., Deshmukh S., Harutyunyan A.S. et al. H3 K27M and EZHIP Impede H3K27-Methylation spreading by inhibiting allosterically stimulated PRC2. Mol. Cell. 2020; 80:726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Baylin S.B., Jones P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016; 8:a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lee T.I., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K. et al. Control of developmental regulators by polycomb in human embryonic stem cells. Cell. 2006; 125:301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kamminga L.M., Bystrykh L.V., De Boer A., Houwer S., Douma J., Weersing E., Dontje B., De Haan G. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006; 107:2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wen Y., Cai J., Hou Y., Huang Z., Wang Z. Role of EZH2 in cancer stem cells: From biological insight to a therapeutic target. Oncotarget. 2017; 8:37974–37990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Popov N., Gil J. Epigenetic regulation of the INK4B-ARF-INK4a locus: In sickness and in health. Epigenetics. 2010; 5:685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bracken A.P., Helin K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer. 2009; 9:773–784. [DOI] [PubMed] [Google Scholar]
- 115. Wang M.C., Li C.L., Cui J., Jiao M., Wu T., Jing L., Nan K.J. BMI-1, a promising therapeutic target for human cancer (Review). Oncol. Lett. 2015; 10:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wu H.-X., Wang Z.-X., Zhao Q., Chen D.-L., He M.-M., Yang L.-P., Wang Y.-N., Jin Y., Ren C., Luo H.-Y. et al. Tumor mutational and indel burden: a systematic pan-cancer evaluation as prognostic biomarkers. Ann. Transl. Med. 2019; 7:640–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Llabata P., Mitsuishi Y., Choi P.S., Cai D., Francis J.M., Torres-Diz M., Udeshi N.D., Golomb L., Wu Z., Zhou J. et al. Multi-Omics Analysis Identifies MGA as a Negative Regulator of the MYC Pathway in Lung Adenocarcinoma. Mol. Cancer Res. 2020; 18:574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mathsyaraja H., Catchpole J., Freie B., Eastwood E., Babaeva E., Geuenich M., Cheng P.F., Ayers J., Yu M., Wu N. et al. Loss of MGA repression mediated by an atypical polycomb complex promotes tumor progression and invasiveness. Elife. 2021; 10:e64212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Astolfi A., Fiore M., Melchionda F., Indio V., Bertuccio S.N., Pession A. BCOR involvement in cancer. Epigenomics. 2019; 11:835–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wamstad J.A., Bardwell V.J. Characterization of Bcor expression in mouse development. Gene Expr. Patterns. 2007; 7:550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hoffman L.M., DeWire M., Ryall S., Buczkowicz P., Leach J., Miles L., Ramani A., Brudno M., Kumar S.S., Drissi R. et al. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol. Commun. 2016; 4:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Argani P., Kao Y.C., Zhang L., Bacchi C., Matoso A., Alaggio R., Epstein J.I., Antonescu C.R. Primary Renal Sarcomas with BCOR-CCNB3 Gene Fusion. Am. J. Surg. Pathol. 2017; 41:1702–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. McEvoy J., Nagahawatte P., Finkelstein D., Richards-Yutz J., Valentine M., Ma J., Mullighan C., Song G., Chen X., Wilson M. et al. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget. 2014; 5:438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Grossmann V., Tiacci E., Holmes A.B., Kohlmann A., Martelli M.P., Kern W., Spanhol-Rosseto A., Klein H.U., Dugas M., Schindela S. et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011; 118:6153–6163. [DOI] [PubMed] [Google Scholar]
- 125. Lindsley R.C., Mar B.G., Mazzola E., Grauman P.V., Shareef S., Allen S.L., Pigneux A., Wetzler M., Stuart R.K., Erba H.P. et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015; 125:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Damm F., Chesnais V., Nagata Y., Yoshida K., Scourzic L., Okuno Y., Itzykson R., Sanada M., Shiraishi Y., Gelsi-Boyer V. et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013; 122:3169–3177. [DOI] [PubMed] [Google Scholar]
- 127. Dobashi A., Tsuyama N., Asaka R., Togashi Y., Ueda K., Sakata S., Baba S., Sakamoto K., Hatake K., Takeuchi K. Frequent BCOR aberrations in extranodal NK/T-Cell lymphoma, nasal type. Genes Chromosom. Cancer. 2016; 55:460–471. [DOI] [PubMed] [Google Scholar]
- 128. Isshiki Y., Iwama A. Emerging role of noncanonical polycomb repressive complexes in normal and malignant hematopoiesis. Exp. Hematol. 2018; 68:10–14. [DOI] [PubMed] [Google Scholar]
- 129. Cao Q., Gearhart M.D., Gery S., Shojaee S., Yang H., Sun H., Lin D.C., Bai J.W., Mead M., Zhao Z. et al. BCOR regulates myeloid cell proliferation and differentiation. Leukemia. 2016; 30:1155–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kolla L., Kelly M.C., Mann Z.F., Anaya-Rocha A., Ellis K., Lemons A., Palermo A.T., So K.S., Mays J.C., Orvis J. et al. Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat. Commun. 2020; 11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Iwama A. Polycomb repressive complexes in hematological malignancies. Blood. 2017; 130:23–29. [DOI] [PubMed] [Google Scholar]
- 132. Raedt T.De, Beert E., Pasmant E., Luscan A., Brems H., Ortonne N., Helin K., Hornick J.L., Mautner V., Kehrer-Sawatzki H. et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014; 514:247–251. [DOI] [PubMed] [Google Scholar]
- 133. Ernst T., Chase A.J., Score J., Hidalgo-Curtis C.E., Bryant C., Jones A.V, Waghorn K., Zoi K., Ross F.M., Reiter A. et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010; 42:722–726. [DOI] [PubMed] [Google Scholar]
- 134. Nikoloski G., Langemeijer S.M.C., Kuiper R.P., Knops R., Massop M., Tönnissen E.R.L.T.M., Heijden A., Scheele T.N., Vandenberghe P., Witte T. et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010; 42:665–667. [DOI] [PubMed] [Google Scholar]
- 135. Bejar R., Stevenson K., Abdel-Wahab O., Galili N., Nilsson B., Garcia-Manero G., Kantarjian H., Raza A., Levine R.L., Neuberg D. et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011; 364:2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mochizuki-Kashio M., Aoyama K., Sashida G., Oshima M., Tomioka T., Muto T., Wang C., Iwama A. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1-dependent manner. Blood. 2015; 126:1172–1183. [DOI] [PubMed] [Google Scholar]
- 137. Shirahata-Adachi M., Iriyama C., Tomita A., Suzuki Y., Shimada K., Kiyoi H. Altered EZH2 splicing and expression is associated with impaired histone H3 lysine 27 tri-Methylation in myelodysplastic syndrome. Leuk. Res. 2017; 63:90–97. [DOI] [PubMed] [Google Scholar]
- 138. Gangat N., Mudireddy M., Lasho T.L., Finke C.M., Nicolosi M., Szuber N., Patnaik M.M., Pardanani A., Hanson C.A., Ketterling R.P. et al. Mutations and prognosis in myelodysplastic syndromes: karyotype-adjusted analysis of targeted sequencing in 300 consecutive cases and development of a genetic risk model. Am. J. Hematol. 2018; 93:691–697. [DOI] [PubMed] [Google Scholar]
- 139. Wang C., Mochel N.S.R., Christenson S.A., Cassandras M., Moon R., Brumwell A.N., Byrnes L.E., Li A., Yokosaki Y., Shan P. et al. Expansion of hedgehog disrupts mesenchymal identity and induces emphysema phenotype. J. Clin. Invest. 2018; 128:4343–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Korfhage J., Lombard D.B. Malignant Peripheral Nerve Sheath Tumors: From Epigenome to Bedside. Mol. Cancer Res. 2019; 17:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Marchione D.M., Lisby A., Viaene A.N., Santi M., Nasrallah M., Wang L.-P., Williams E.A., Larque A.B., Chebib I., Garcia B.A. et al. Histone H3K27 dimethyl loss is highly specific for malignant peripheral nerve sheath tumor and distinguishes true PRC2 loss from isolated H3K27 trimethyl loss. Mod. Pathol. 2019; 32:1434–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Khuong-Quang D.-A., Buczkowicz P., Rakopoulos P., Liu X.-Y., Fontebasso A.M., Bouffet E., Bartels U., Albrecht S., Schwartzentruber J., Letourneau L. et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012; 124:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Schwartzentruber J., Korshunov A., Liu X.-Y., Jones D.T.W., Pfaff E., Jacob K., Sturm D., Fontebasso A.M., Quang D.-A.K., Tönjes M. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012; 482:226–231. [DOI] [PubMed] [Google Scholar]
- 144. Fontebasso A.M., Papillon-Cavanagh S., Schwartzentruber J., Nikbakht H., Gerges N., Fiset P.-O., Bechet D., Faury D., Jay N.De, Ramkissoon L.A. et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014; 46:462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Taylor K.R., Mackay A., Truffaux N., Butterfield Y.S., Morozova O., Philippe C., Castel D., Grasso C.S., Vinci M., Carvalho D. et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014; 46:457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Bender S., Tang Y., Lindroth A.M., Hovestadt V., Jones D.T.W., Kool M., Zapatka M., Northcott P.A., Sturm D., Wang W. et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013; 24:660–672. [DOI] [PubMed] [Google Scholar]
- 147. Lewis P.W., Müller M.M., Koletsky M.S., Cordero F., Lin S., Banaszynski L.A., Garcia B.A., Muir T.W., Becher O.J., Allis C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science. 2013; 340:857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Venneti S., Garimella M.T., Sullivan L.M., Martinez D., Huse J.T., Heguy A., Santi M., Thompson C.B., Judkins A.R. Evaluation of Histone 3 Lysine 27 Trimethylation (H3K27me3) and Enhancer of Zest 2 (EZH2) in Pediatric Glial and Glioneuronal Tumors Shows Decreased H3K27me3 in H3F3A K27M Mutant Glioblastomas. Brain Pathol. 2013; 23:558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Chan K.-M., Fang D., Gan H., Hashizume R., Yu C., Schroeder M., Gupta N., Mueller S., James C.D., Jenkins R. et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013; 27:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Mohammad F., Weissmann S., Leblanc B., Pandey D.P., Højfeldt J.W., Comet I., Zheng C., Johansen J.V., Rapin N., Porse B.T. et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017; 23:483–492. [DOI] [PubMed] [Google Scholar]
- 151. Brien G.L., Bressan R.B., Monger C., Gannon D., Lagan E., Doherty A.M., Healy E., Neikes H., Fitzpatrick D.J., Deevy O. et al. Simultaneous disruption of PRC2 and enhancer function underlies histone H3.3-K27M oncogenic activity in human hindbrain neural stem cells. Nat. Genet. 2021; 53:1221–1232. [DOI] [PubMed] [Google Scholar]
- 152. Piunti A., Hashizume R., Morgan M.A., Bartom E.T., Horbinski C.M., Marshall S.A., Rendleman E.J., Ma Q., Takahashi Y., Woodfin A.R. et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017; 23:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wiese M., Hamdan F.H., Kubiak K., Diederichs C., Gielen G.H., Nussbaumer G., Carcaboso A.M., Hulleman E., Johnsen S.A., Kramm C.M. Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. 2020; 11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Jain S.U., Do T.J., Lund P.J., Rashoff A.Q., Diehl K.L., Cieslik M., Bajic A., Juretic N., Deshmukh S., Venneti S. et al. PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat. Commun. 2019; 10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Xu B., Konze K.D., Jin J., Wang G.G. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 2015; 43:698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Caganova M., Carrisi C., Varano G., Mainoldi F., Zanardi F., Germain P.-L., George L., Alberghini F., Ferrarini L., Talukder A.K. et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J. Clin. Invest. 2013; 123:5009–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Okosun J., Bödör C., Wang J., Araf S., Yang C.-Y., Pan C., Boller S., Cittaro D., Bozek M., Iqbal S. et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2013; 46:176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Knutson S.K., Wigle T.J., Warholic N.M., Sneeringer C.J., Allain C.J., Klaus C.R., Sacks J.D., Raimondi A., Majer C.R., Song J. et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012; 8:890–896. [DOI] [PubMed] [Google Scholar]
- 159. McCabe M.T., Ott H.M., Ganji G., Korenchuk S., Thompson C., Van Aller G.S., Liu Y., Pietra A.D, LaFrance L.V., Mellinger M. et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012; 492:108–112. [DOI] [PubMed] [Google Scholar]
- 160. Paret C., Theruvath J., Russo A., Kron B., Malki K.El, Lehmann N., Wingerter A., Neu M.A., Gerhold-Ay A., Wagner W. et al. Activation of the basal cell carcinoma pathway in a patient with CNS HGNET-BCOR diagnosis: consequences for personalized targeted therapy. Oncotarget. 2016; 7:83378–83391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Hong S., Cho Y.-W., Yu L.-R., Yu H., Veenstra T.D., Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. 2007; 104:18439–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kim J.-H., Sharma A., Dhar S.S., Lee S.-H., Gu B., Chan C.-H., Lin H.-K., Lee M.G. UTX and MLL4 Coordinately Regulate Transcriptional Programs for Cell Proliferation and Invasiveness in Breast Cancer Cells. Cancer Res. 2014; 74:1705–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Wang L., Shilatifard A. UTX Mutations in Human Cancer. Cancer Cell. 2019; 35:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Bracken A.P., Brien G.L., Verrijzer C.P. Dangerous liaisons: interplay between SWI/SNF, NuRD, and Polycomb in chromatin regulation and cancer. Genes Dev. 2019; 33:936–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kadoch C., Crabtree G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015; 1:e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Wilson B.G., Wang X., Shen X., McKenna E.S., Lemieux M.E., Cho Y.-J., Koellhoffer E.C., Pomeroy S.L., Orkin S.H., Roberts C.W.M. Epigenetic Antagonism between Polycomb and SWI/SNF Complexes during Oncogenic Transformation. Cancer Cell. 2010; 18:316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Kim K.H., Roberts C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016; 22:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. McBride M.J., Pulice J.L., Beird H.C., Ingram D.R., D’Avino A.R., Shern J.F., Charville G.W., Hornick J.L., Nakayama R.T., Garcia-Rivera E.M. et al. The SS18-SSX fusion oncoprotein hijacks BAF complex targeting and function to drive synovial sarcoma. Cancer Cell. 2018; 33:1128–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Bitler B.G., Aird K.M., Garipov A., Li H., Amatangelo M., Kossenkov A.V, Schultz D.C., Liu Q., Shih I.-M., Conejo-Garcia J.R. et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A -mutated cancers. Nat. Med. 2015; 21:231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Fillmore C.M., Xu C., Desai P.T., Berry J.M., Rowbotham S.P., Lin Y.-J., Zhang H., Marquez V.E., Hammerman P.S., Wong K.-K. et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015; 520:239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Wang Y., Chen S.Y., Karnezis A.N., Colborne S., Santos N.D, Lang J.D., Hendricks W.P., Orlando K.A., Yap D., Kommoss F. et al. The histone methyltransferase EZH2 is a therapeutic target in small cell carcinoma of the ovary, hypercalcaemic type. J. Pathol. 2017; 242:371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Kim K.H., Kim W., Howard T.P., Vazquez F., Tsherniak A., Wu J.N., Wang W., Haswell J.R., Walensky L.D., Hahn W.C. et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015; 21:1491–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Shi J., Wang E., Zuber J., Rappaport A., Taylor M., Johns C., Lowe S.W., Vakoc C.R. The Polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9;NrasG12D acute myeloid leukemia. Oncogene. 2013; 32:930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Neff T., Sinha A.U., Kluk M.J., Zhu N., Khattab M.H., Stein L., Xie H., Orkin S.H., Armstrong S.A. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc. Natl. Acad. Sci. 2012; 109:5028–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Wang S., Denton K.E., Hobbs K.F., Weaver T., McFarlane J.M.B., Connelly K.E., Gignac M.C., Milosevich N., Hof F., Paci I. et al. Optimization of ligands using focused DNA-Encoded Libraries To Develop a Selective, Cell-Permeable CBX8 chromodomain inhibitor. ACS Chem. Biol. 2019; 15:112–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Mueller D., Bach C., Zeisig D., Garcia-Cuellar M.-P., Monroe S., Sreekumar A., Zhou R., Nesvizhskii A., Chinnaiyan A., Hess J.L. et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007; 110:4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Neureiter D., Kiesslich T., Ritter M., Mayr C. Update on the role and therapeutic potential of polycomb repressive complexes in (biliary tract) cancer. Expert Opin Ther Targets. 2017; 22:1–3. [DOI] [PubMed] [Google Scholar]
- 178. Grzenda A., Ordog T., Urrutia R. Polycomb and the Emerging Epigenetics of Pancreatic Cancer. J. Gastrointest. Cancer. 2011; 42:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Patil S., Steuber B., Kopp W., Kari V., Urbach L., Wang X., Küffer S., Bohnenberger H., Spyropoulou D., Zhang Z. et al. EZH2 regulates pancreatic cancer subtype identity and tumor progression via transcriptional repression of GATA6. Cancer Res. 2020; 80:4620–4632. [DOI] [PubMed] [Google Scholar]
- 180. Wang M.-C., Jiao M., Wu T., Jing L., Cui J., Guo H., Tian T., Ruan Z., Wei Y.-C., Jiang L.-L. et al. Polycomb complex protein BMI-1 promotes invasion and metastasis of pancreatic cancer stem cells by activating PI3K/AKT signaling, an ex vivo, in vitro, and in vivo study. Oncotarget. 2016; 7:9586–9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Pasqualucci L., Dalla-Favera R. Genetics of diffuse large b-cell lymphoma. Blood. 2018; 131:2307–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Brach D., Johnston-Blackwell D., Drew A., Lingaraj T., Motwani V., Warholic N.M., Feldman I., Plescia C., Smith J.J., Copeland R.A. et al. EZH2 inhibition by tazemetostat results in altered dependency on B-cell activation signaling in DLBCL. Mol. Cancer Ther. 2017; 16:2586–2597. [DOI] [PubMed] [Google Scholar]
- 183. Haupt Y., Alexander W.S., Barri G., Klinken S.P., Adams J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in Eμ-myc transgenic mice. Cell. 1991; 65:753–763. [DOI] [PubMed] [Google Scholar]
- 184. Lohuizen M., Frasch M., Wientjens E., Berns A. Sequence similarity between the mammalian bmi-1 proto-oncogene and the Drosophila regulatory genes Psc and Su(z)2. Nature. 1991; 353:353–355. [DOI] [PubMed] [Google Scholar]
- 185. Lessard J., Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003; 423:255–260. [DOI] [PubMed] [Google Scholar]
- 186. Alkema M.J., Jacobs H., Lohuizen M., Berns A. Perturbation of B and T cell development and predisposition to lymphomagenesis in Eμ Bmi 1 transgenic mice require the Bmi1 RING finger. Oncogene. 1997; 15:899–910. [DOI] [PubMed] [Google Scholar]
- 187. Jacobs J.J.L., Scheijen B., Voncken J.W., Kieboom K., Berns A., Van Lohuizen M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc- induced apoptosis via INK4a/ARF. Genes Dev. 1999; 13:2678–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Park I., Qian D., Kiel M., Becker M.W., Pihalja M., Weissman I.L., Morrison S.J., Clarke M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003; 423:302–305. [DOI] [PubMed] [Google Scholar]
- 189. Rizo A., Olthof S., Han L., Vellenga E., de Haan G., Schuringa J.J. Repression of BMI1 in normal and leukemic human CD34+ cells impairs self-renewal and induces apoptosis. Blood. 2009; 114:1498–1505. [DOI] [PubMed] [Google Scholar]
- 190. Béguelin W., Teater M., Gearhart M.D., Calvo Fernández M.T., Goldstein R.L., Cárdenas M.G., Hatzi K., Rosen M., Shen H., Corcoran C.M. et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell. 2016; 30:197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Tara S., Isshiki Y., Nakajima-Takagi Y., Oshima M., Aoyama K., Tanaka T., Shinoda D., Koide S., Saraya A., Miyagi S. et al. Bcor insufficiency promotes initiation and progression of myelodysplastic syndrome. Blood. 2018; 132:2470–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Słabicki M., Yoon H., Koeppel J., Nitsch L., Roy Burman S.S., Di Genua C., Donovan K.A., Sperling A.S., Hunkeler M., Tsai J.M. et al. Small-molecule-induced polymerization triggers degradation of BCL6. Nature. 2020; 588:164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Kerres N., Steurer S., Schlager S., Bader G., Berger H., Caligiuri M., Dank C., Engen J.R., Ettmayer P., Fischerauer B. et al. Chemically Induced Degradation of the Oncogenic Transcription Factor BCL6. Cell Rep. 2017; 20:2860–2875. [DOI] [PubMed] [Google Scholar]
- 194. McCoull W., Cheung T., Anderson E., Barton P., Burgess J., Byth K., Cao Q., Castaldi M.P., Chen H., Chiarparin E. et al. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC to Provide Insight into Small Molecule Targeting of BCL6. ACS Chem. Biol. 2018; 13:3131–3141. [DOI] [PubMed] [Google Scholar]
- 195. McAllister T.E., England K.S., Hopkinson R.J., Brennan P.E., Kawamura A., Schofield C.J. Recent Progress in Histone Demethylase Inhibitors. J. Med. Chem. 2016; 59:1308–1329. [DOI] [PubMed] [Google Scholar]
- 196. Lee J., Kotliarova S., Kotliarov Y., Li A., Su Q., Donin N.M., Pastorino S., Purow B.W., Christopher N., Zhang W. et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006; 9:391–403. [DOI] [PubMed] [Google Scholar]
- 197. Shi B., Liang J., Yang X., Wang Y., Zhao Y., Wu H., Sun L., Zhang Y., Chen Y., Li R. et al. Integration of Estrogen and Wnt Signaling Circuits by the Polycomb Group Protein EZH2 in Breast Cancer Cells. Mol. Cell. Biol. 2007; 27:5105–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Su I.H., Dobenecker M.W., Dickinson E., Oser M., Basavaraj A., Marqueron R., Viale A., Reinberg D., Wülfing C., Tarakhovsky A. Polycomb group protein Ezh2 controls actin polymerization and cell signaling. Cell. 2005; 121:425–436. [DOI] [PubMed] [Google Scholar]
- 199. Ma A., Stratikopoulos E., Park K.S., Wei J., Martin T.C., Yang X., Schwarz M., Leshchenko V., Rialdi A., Dale B. et al. Discovery of a first-in-class EZH2 selective degrader. Nat. Chem. Biol. 2020; 16:214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Gao S., Wang S.-Y., Zhang X.-D., Wu H., Pang D. Low expression of the polycomb protein RING1 predicts poor prognosis in human breast cancer. Front. Oncol. 2021; 10:3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Zhang C.Z., Chen S.L., Wang C.H., He Y.F., Yang X., Xie D., Yun J.P. CBX8 exhibits oncogenic activity via AKT/b-catenin activation in hepatocellular carcinoma. Cancer Res. 2018; 78:51–63. [DOI] [PubMed] [Google Scholar]
- 202. Chung C.-Y., Sun Z., Mullokandov G., Bosch A., Qadeer Z.A., Cihan E., Rapp Z., Parsons R., Aguirre-Ghiso J.A., Farias E.F. et al. Cbx8 Acts Non-canonically with Wdr5 to Promote Mammary Tumorigenesis. Cell Rep. 2016; 16:472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Zheng S., Lv P., Su J., Miao K., Xu H., Li M. Overexpression of CBX2 in breast cancer promotes tumor progression through the PI3K/AKT signaling pathway. Am. J. Transl. Res. 2019; 11:1668–1682. [PMC free article] [PubMed] [Google Scholar]
- 204. Clermont P.L., Crea F., Chiang Y.T., Lin D., Zhang A., Wang J.Z.L., Parolia A., Wu R., Xue H., Wang Y. et al. Identification of the epigenetic reader CBX2 as a potential drug target in advanced prostate cancer. Clin. Epigenetics. 2016; 8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Wheeler L.J., Watson Z.L., Qamar L., Yamamoto T.M., Post M.D., Berning A.A., Spillman M.A., Behbakht K., Bitler B.G. CBX2 identified as driver of anoikis escape and dissemination in high grade serous ovarian cancer. Oncogenesis. 2018; 7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Pallante P., Federico A., Berlingieri M.T., Bianco M., Ferraro A., Forzati F., Iaccarino A., Russo M., Pierantoni G.M., Leone V. et al. Loss of the CBX7 Gene Expression Correlates with a Highly Malignant Phenotype in Thyroid Cancer. Cancer Res. 2008; 68:6770–6778. [DOI] [PubMed] [Google Scholar]
- 207. Forzati F., Federico A., Pallante P., Abbate A., Esposito F., Malapelle U., Sepe R., Palma G., Troncone G., Scarfò M. et al. CBX7 is a tumor suppressor in mice and humans. J. Clin. Invest. 2013; 123:934–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Li G., Warden C., Zou Z., Neman J., Krueger J.S., Jain A., Jandial R., Chen M. Altered expression of polycomb group genes in glioblastoma multiforme. PLoS One. 2013; 8:e80970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Huang Z., Yan Y., Zhu Z., Liu J., He X., Dalangood S., Li M., Tan M., Cai J., Tang P. et al. CBX7 suppresses urinary bladder cancer progression via modulating AKR1B10–ERK signaling. Cell Death Dis. 2021; 12:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Li R., Yan Q., Tian P., Wang Y., Wang J., Tao N., Ning L., Lin X., Ding L., Liu J. et al. CBX7 inhibits cell growth and motility and induces apoptosis in cervical cancer cells. Mol. Ther. - Oncolytics. 2019; 15:108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Deng H., Guan X., Gong L., Zeng J., Zhang H., Chen M.Y., Li G. CBX6 is negatively regulated by EZH2 and plays a potential tumor suppressor role in breast cancer. Sci. Rep. 2019; 9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Scott C.L., Gil J., Hernando E., Teruya-Feldstein J., Narita M., Martínez D., Visakorpi T., Mu D., Cordon-Cardo C., Peters G. et al. Role of the chromobox protein CBX7 in lymphomagenesis. Proc. Natl. Acad. Sci. 2007; 104:5389–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Bernard D., Martinez-Leal J.F., Rizzo S., Martinez D., Hudson D., Visakorpi T., Peters G., Carnero A., Beach D., Gil J. CBX7 controls the growth of normal and tumor-derived prostate cells by repressing the Ink4a/Arf locus. Oncogene. 2005; 24:5543–5551. [DOI] [PubMed] [Google Scholar]
- 214. Zheng H., Jiang W.H., Tian T., Tan H.S., Chen Y., Qiao G.L., Han J., Huang S.Y., Yang Y., Li S. et al. CBX6 overexpression contributes to tumor progression and is predictive of a poor prognosis in hepatocellular carcinoma. Oncotarget. 2017; 8:18872–18884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Plys A.J., Davis C.P., Kim J., Rizki G., Keenen M.M., Marr S.K., Kingston R.E. Phase separation of polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019; 33:799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Kundu S., Ji F., Sunwoo H., Jain G., Lee J.T., Sadreyev R.I., Dekker J., Kingston R.E. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol. Cell. 2017; 65:432–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Varambally S., Dhanasekaran S.M., Zhou M., Barrette T.R., Kumar-Sinha C., Sanda M.G., Ghosh D., Pienta K.J., Sewalt R.G.A.B., Rubin M.A. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002; 419:624–629. [DOI] [PubMed] [Google Scholar]
- 218. van Leenders G.J.L.H., Dukers D., Hessels D., van den Kieboom S.W.M., Hulsbergen C.A., Witjes J.A., Otte A.P., Meijer C.J., Raaphorst F.M. Polycomb-Group Oncogenes EZH2, BMI1, and RING1 Are Overexpressed in Prostate Cancer With Adverse Pathologic and Clinical Features. Eur. Urol. 2007; 52:455–463. [DOI] [PubMed] [Google Scholar]
- 219. Clermont P.L., Lin D., Crea F., Wu R., Xue H., Wang Y., Thu K.L., Lam W.L., Collins C.C., Wang Y. et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin. Epigenetics. 2015; 7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Pickl J.M.A., Tichy D., Kuryshev V.Y., Tolstov Y., Falkenstein M., Schüler J., Reidenbach D., Hotz-Wagenblatt A., Kristiansen G., Roth W. et al. Ago-RIP-Seq identifies Polycomb repressive complex I member CBX7 as a major target of miR-375 in prostate cancer progression. Oncotarget. 2016; 7:59589–59603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Zacharopoulou N., Tsapara A., Kallergi G., Schmid E., Tsichlis P.N., Kampranis S.C., Stournaras C. The epigenetic factor KDM2B regulates cell adhesion, small rho GTPases, actin cytoskeleton and migration in prostate cancer cells. Biochim. Biophys. Acta - Mol. Cell Res. 2018; 1865:587–597. [DOI] [PubMed] [Google Scholar]
- 222. Liu Q., Wang G., Li Q., Jiang W., Kim J.-S., Wang R., Zhu S., Wang X., Yan L., Yi Y. et al. Polycomb group proteins EZH2 and EED directly regulate androgen receptor in advanced prostate cancer. Int. J. Cancer. 2019; 145:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Jain P., Ballare C., Blanco E., Vizan P., Di Croce L. PHF19 mediated regulation of proliferation and invasiveness in prostate cancer cells. Elife. 2020; 9:e51373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Zhu J., Jin L., Zhang A., Gao P., Dai G., Xu M., Xu L., Yang D. Coexpression analysis of the EZH2 gene using the cancer genome atlas and oncomine databases identifies coexpressed genes involved in biological networks in breast cancer, glioblastoma, and prostate cancer. Med. Sci. Monit. 2020; 26:e922346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Xu K., Wu Z.J., Groner A.C., He H.H., Cai C., Lis R.T., Wu X., Stack E.C., Loda M., Liu T. et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science. 2012; 338:1465–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Wang S., Alpsoy A., Sood S., Ordonez-Rubiano S.C., Dhiman A., Sun Y., Jiao G., Krusemark C.J., Dykhuizen E.C. A potent, selective CBX2 chromodomain ligand and its cellular activity during prostate cancer neuroendocrine differentiation. ChemBioChem. 2021; 22:2335–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Dardenne E., Beltran H., Benelli M., Gayvert K., Berger A., Puca L., Cyrta J., Sboner A., Noorzad Z., MacDonald T. et al. N-Myc induces an EZH2-Mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell. 2016; 30:563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Bai Y., Zhang Z., Cheng L., Wang R., Chen X., Kong Y., Feng F., Ahmad N., Li L., Liu X. Inhibition of enhancer of zeste homolog 2 (EZH2) overcomes enzalutamide resistance in castration-resistant prostate cancer. J. Biol. Chem. 2019; 294:9911–9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Li Q., Liu K.Y., Liu Q., Wang G., Jiang W., Meng Q., Yi Y., Yang Y., Wang R., Zhu S. et al. Antihistamine drug ebastine inhibits cancer growth by targeting polycomb group protein EZH2. Mol. Cancer Ther. 2020; 19:2023–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Zhu S., Zhao D., Yan L., Jiang W., Kim J.-S., Gu B., Liu Q., Wang R., Xia B., Zhao J.C. et al. BMI1 regulates androgen receptor in prostate cancer independently of the polycomb repressive complex 1. Nat. Commun. 2018; 9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Zhu S., Zhao D., Li C., Li Q., Jiang W., Liu Q., Wang R., Fazli L., Li Y., Zhang L. et al. BMI1 is directly regulated by androgen receptor to promote castration-resistance in prostate cancer. Oncogene. 2019; 39:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Umbreen S., Banday M.M., Jamroze A., Mansini A.P., Ganaie A.A., Ferrari M.G., Maqbool R., Beigh F.H., Murugan P., Morrissey C. et al. COMMD3:BMI1 Fusion and COMMD3 Protein Regulate C-MYC Transcription: Novel therapeutic target for metastatic prostate cancer. Mol. Cancer Ther. 2019; 18:2111–2123. [DOI] [PubMed] [Google Scholar]
- 233. Su W., Han H.H., Wang Y., Zhang B., Zhou B., Cheng Y., Rumandla A., Gurrapu S., Chakraborty G., Su J. et al. The polycomb repressor complex 1 Drives Double-Negative prostate cancer metastasis by coordinating stemness and immune suppression. Cancer Cell. 2019; 36:139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Yi Y., Hsieh I.-Y., Huang X., Li J., Zhao W. Glioblastoma stem-like cells: characteristics, microenvironment, and therapy. Front. Pharmacol. 2016; 7:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Chen J., Li Y., Yu T.-S., McKay R.M., Burns D.K., Kernie S.G., Parada L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012; 488:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Singh S.K., Hawkins C., Clarke I.D., Squire J.A., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B. Identification of human brain tumour initiating cells. Nature. 2004; 432:396–401. [DOI] [PubMed] [Google Scholar]
- 237. Abdouh M., Facchino S., Chatoo W., Balasingam V., Ferreira J., Bernier G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J. Neurosci. 2009; 29:8884–8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Kreso A., Galen P., Pedley N.M., Lima-Fernandes E., Frelin C., Davis T., Cao L., Baiazitov R., Du W., Sydorenko N. et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 2013; 20:29–36. [DOI] [PubMed] [Google Scholar]
- 239. Martin M.C., Zeng G., Yu J., Schiltz G.E. Small molecule approaches for targeting the polycomb repressive complex 2 (PRC2) in Cancer. J. Med. Chem. 2020; 63:15344–15370. [DOI] [PubMed] [Google Scholar]
- 240. Tan J., Yang X., Zhuang L., Jiang X., Chen W., Puay L.L., Karuturi R.K.M., Tan P.B.O., Liu E.T., Yu Q. Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007; 21:1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Verma S.K., Tian X., Lafrance L.V., Duquenne C., Suarez D.P., Newlander K.A., Romeril S.P., Burgess J.L., Grant S.W., Brackley J.A. et al. Identification of potent, selective, cell-Active inhibitors of the histone lysine methyltransferase EZH2. ACS Med. Chem. Lett. 2012; 3:1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Shen J.K., Cote G.M., Gao Y., Choy E., Mankin H.J., Hornicek F.J., Duan Z. Targeting EZH2-mediated methylation of H3K27 inhibits proliferation and migration of Synovial Sarcoma in vitro. Sci. Rep. 2016; 6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Knutson S.K., Kawano S., Minoshima Y., Warholic N.M., Huang K.-C., Xiao Y., Kadowaki T., Uesugi M., Kuznetsov G., Kumar N. et al. Selective Inhibition of EZH2 by EPZ-6438 Leads to Potent Antitumor Activity in EZH2-Mutant Non-Hodgkin Lymphoma. Mol. Cancer Ther. 2014; 13:842–854. [DOI] [PubMed] [Google Scholar]
- 244. Morschhauser F., Tilly H., Chaidos A., McKay P., Phillips T., Assouline S., Batlevi C.L., Campbell P., Ribrag V., Damaj G.L. et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020; 21:1433–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Italiano A., Soria J.-C., Toulmonde M., Michot J.-M., Lucchesi C., Varga A., Coindre J.-M., Blakemore S.J., Clawson A., Suttle B. et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018; 19:649–659. [DOI] [PubMed] [Google Scholar]
- 246. Gounder M., Schöffski P., Jones R.L., Agulnik M., Cote G.M., Villalobos V.M., Attia S., Chugh R., Chen T.W.W., Jahan T. et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol. 2020; 21:1423–1432. [DOI] [PubMed] [Google Scholar]
- 247. Campbell J.E., Kuntz K.W., Knutson S.K., Warholic N.M., Keilhack H., Wigle T.J., Raimondi A., Klaus C.R., Rioux N., Yokoi A. et al. EPZ011989, A potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS Med. Chem. Lett. 2015; 6:491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Qi W., Chan H., Teng L., Li L., Chuai S., Zhang R., Zeng J., Li M., Fan H., Lin Y. et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. 2012; 109:21360–21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Konze K.D., Ma A., Li F., Barsyte-Lovejoy D., Parton T., MacNevin C.J., Liu F., Gao C., Huang X.-P., Kuznetsova E. et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013; 8:1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Xu B., On D.M., Ma A., Parton T., Konze K.D., Pattenden S.G., Allison D.F., Cai L., Rockowitz S., Liu S. et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015; 125:346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Bradley W.D., Arora S., Busby J., Balasubramanian S., Gehling V.S., Nasveschuk C.G., Vaswani R.G., Yuan C.C., Hatton C., Zhao F. et al. EZH2 inhibitor efficacy in Non-Hodgkin's lymphoma does not require suppression of H3K27 monomethylation. Chem. Biol. 2014; 21:1463–1475. [DOI] [PubMed] [Google Scholar]
- 252. Vaswani R.G., Gehling V.S., Dakin L.A., Cook A.S., Nasveschuk C.G., Duplessis M., Iyer P., Balasubramanian S., Zhao F., Good A.C. et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J. Med. Chem. 2016; 59:9928–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Kung P.-P., Bingham P., Brooun A., Collins M., Deng Y.-L., Dinh D., Fan C., Gajiwala K.S., Grantner R., Gukasyan H.J. et al. Optimization of Orally Bioavailable Enhancer of Zeste Homolog 2 (EZH2) Inhibitors Using Ligand and Property-Based Design Strategies: Identification of Development Candidate (R)-5,8-Dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497). J. Med. Chem. 2017; 61:650–665. [DOI] [PubMed] [Google Scholar]
- 254. Honma D., Kanno O., Watanabe J., Kinoshita J., Hirasawa M., Nosaka E., Shiroishi M., Takizawa T., Yasumatsu I., Horiuchi T. et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017; 108:2069–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Fujita S., Honma D., Adachi N., Araki K., Takamatsu E., Katsumoto T., Yamagata K., Akashi K., Aoyama K., Iwama A. et al. Dual inhibition of EZH1/2 breaks the quiescence of leukemia stem cells in acute myeloid leukemia. Leukemia. 2017; 32:855–864. [DOI] [PubMed] [Google Scholar]
- 256. Neklesa T.K., Crews C.M. Chemical biology: Greasy tags for protein removal. Nature. 2012; 487:308–309. [DOI] [PubMed] [Google Scholar]
- 257. Burslem G.M., Crews C.M. Small-molecule modulation of protein homeostasis. Chem. Rev. 2017; 117:11269–11301. [DOI] [PubMed] [Google Scholar]
- 258. Kong X., Chen L., Jiao L., Jiang X., Lian F., Lu J., Zhu K., Du D., Liu J., Ding H. et al. Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2. J. Med. Chem. 2014; 57:9512–9521. [DOI] [PubMed] [Google Scholar]
- 259. Kim W., Bird G.H., Neff T., Guo G., Kerenyi M.A., Walensky L.D., Orkin S.H. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol. 2013; 9:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Du D., Xu D., Zhu L., Stazi G., Zwergel C., Liu Y., Luo Z., Li Y., Zhang Y., Zhu K. et al. Structure-Guided Development of Small-Molecule PRC2 Inhibitors Targeting EZH2–EED Interaction. J. Med. Chem. 2021; 64:8194–8207. [DOI] [PubMed] [Google Scholar]
- 261. He Y., Selvaraju S., Curtin M.L., Jakob C.G., Zhu H., Comess K.M., Shaw B., The J., Lima-Fernandes E., Szewczyk M.M. et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 2017; 13:389–395. [DOI] [PubMed] [Google Scholar]
- 262. Curtin M.L., Pliushchev M.A., Li H.Q., Torrent M., Dietrich J.D., Jakob C.G., Zhu H., Zhao H., Wang Y., Ji Z. et al. SAR of amino pyrrolidines as potent and novel protein-protein interaction inhibitors of the PRC2 complex through EED binding. Bioorganic Med. Chem. Lett. 2017; 27:1576–1583. [DOI] [PubMed] [Google Scholar]
- 263. Qi W., Zhao K., Gu J., Huang Y., Wang Y., Zhang H., Zhang M., Zhang J., Yu Z., Li L. et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017; 13:381–388. [DOI] [PubMed] [Google Scholar]
- 264. Huang Y., Zhang J., Yu Z., Zhang H., Wang Y., Lingel A., Qi W., Gu J., Zhao K., Shultz M.D. et al. Discovery of First-in-Class, potent, and orally bioavailable embryonic ectoderm development (EED) inhibitor with robust anticancer efficacy. J. Med. Chem. 2017; 60:2215–2226. [DOI] [PubMed] [Google Scholar]
- 265. US20160176882A1 - Triazolopyrimidine compounds and uses thereof - Google Patents.
- 266. Barnash K.D., The J., Norris-Drouin J.L., Cholensky S.H., Worley B.M., Li F., Stuckey J.I., Brown P.J., Vedadi M., Arrowsmith C.H. et al. Discovery of peptidomimetic ligands of EED as allosteric inhibitors of PRC2. ACS Comb. Sci. 2017; 19:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Lingel A., Sendzik M., Huang Y., Shultz M.D., Cantwell J., Dillon M.P., Fu X., Fuller J., Gabriel T., Gu J. et al. Structure-guided design of EED binders allosterically inhibiting the epigenetic polycomb repressive complex 2 (PRC2) methyltransferase. J. Med. Chem. 2017; 60:415–427. [DOI] [PubMed] [Google Scholar]
- 268. Potjewyd F., Turner A.-M.W., Beri J., Rectenwald J.M., Norris-Drouin J.L., Cholensky S.H., Margolis D.M., Pearce K.H., Herring L.E., James L.I. Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem. Biol. 2020; 27:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Hsu J.H., Rasmusson T., Robinson J., Pachl F., Read J., Kawatkar S., Donovan D.H.O., Bagal S., Code E., Rawlins P. et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem. Biol. 2020; 27:41–46. [DOI] [PubMed] [Google Scholar]
- 270. Suh J.L., Barnash K.D., Abramyan T.M., Li F., The J., Engelberg I.A., Vedadi M., Brown P.J., Kireev D.B., Arrowsmith C.H. et al. Discovery of selective activators of PRC2 mutant EED-I363M. Sci. Rep. 2019; 9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Kadoch C., Copeland R.A., Keilhack H. PRC2 and SWI/SNF chromatin remodeling complexes in health and disease. Biochemistry. 2016; 55:1600–1614. [DOI] [PubMed] [Google Scholar]
- 272. Kogure M., Takawa M., Saloura V., Sone K., Piao L., Ueda K., Ibrahim R., Tsunoda T., Sugiyama M., Atomi Y. et al. The oncogenic polycomb histone methyltransferase EZH2Methylates Lysine 120 on Histone H2B and competes ubiquitination. Neoplasia. 2013; 15:1251–IN10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Gunawan M., Venkatesan N., Loh J.T., Wong J.F., Berger H., Neo W.H., Li L.Y.J., La Win M.K., Yau Y.H., Guo T. et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat. Immunol. 2015; 16:505–516. [DOI] [PubMed] [Google Scholar]
- 274. Kim E., Kim M., Woo D.H., Shin Y., Shin J., Chang N., Oh Y.T., Kim H., Rheey J., Nakano I. et al. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma Stem-like Cells. Cancer Cell. 2013; 23:839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Ismail I.H., McDonald D., Strickfaden H., Xu Z., Hendzel M.J. A small molecule inhibitor of polycomb repressive complex 1 inhibits ubiquitin signaling at DNA double-strand breaks. J. Biol. Chem. 2013; 288:26944–26954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Shukla S., Ying W., Gray F., Yao Y., Simes M.L., Zhao Q., Miao H., Cho H.J., González-Alonso P., Winkler A. et al. Small-molecule inhibitors targeting Polycomb repressive complex 1 RING domain. Nat. Chem. Biol. 2021; 17:784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Kim M.J., Cao L., Sheedy J., Risher N., Dumble M., Lee C.-S., Sydorenko N., Baiazitov R., Du W., Moon Y.-C. et al. Abstract 5517: PTC596-induced Bmi1 hyper-phosphorylation via Cdk1/2 activation resulting in tumor stem cell depletion. Cancer Res. 2014; 74:5517–5517. [Google Scholar]
- 278. Wang J., Xing Y., Wang Y., He Y., Wang L., Peng S., Yang L., Xie J., Li X., Qiu W. et al. A novel BMI-1 inhibitor QW24 for the treatment of stem-like colorectal cancer. J. Exp. Clin. Cancer Res. 2019; 38:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Gil J., Bernard D., Martínez D., Beach D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat. Cell Biol. 2004; 6:67–72. [DOI] [PubMed] [Google Scholar]
- 280. Jung J., Buisman S.C., Weersing E., Dethmers-Ausema A., Zwart E., Schepers H., Dekker M.R., Lazare S.S., Hammerl F., Skokova Y. et al. CBX7 Induces Self-Renewal of human normal and malignant hematopoietic stem and progenitor cells by canonical and Non-canonical interactions. Cell Rep. 2019; 26:1906–1918. [DOI] [PubMed] [Google Scholar]
- 281. Simhadri C., Daze K.D., Douglas S.F., Quon T.T.H., Dev A., Gignac M.C., Peng F., Heller M., Boulanger M.J., Wulff J.E. et al. Chromodomain antagonists that target the polycomb-group methyllysine reader protein chromobox homolog 7 (CBX7). J. Med. Chem. 2014; 57:2874–2883. [DOI] [PubMed] [Google Scholar]
- 282. Stuckey J.I., Dickson B.M., Cheng N., Liu Y., Norris J.L., Cholensky S.H., Tempel W., Qin S., Huber K.G., Sagum C. et al. A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat. Chem. Biol. 2016; 12:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Ren C., Morohashi K., Walsh M., Correspondence M.-M.Z. Small-Molecule Modulators of Methyl-Lysine Binding for the CBX7 Chromodomain Chemical inhibition of CBX7 chromodomain derepresses p16/CDKN2A in PC3 cells. Chem. Biol. 2015; 22:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Ren C., Smith S.G., Yap K., Li S., Li J., Mezei M., Rodriguez Y., Vincek A., Aguilo F., Walsh M.J. et al. Structure-Guided discovery of selective antagonists for the chromodomain of polycomb repressive protein CBX7. ACS Med. Chem. Lett. 2016; 7:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Simhadri C., Daze K.D., Douglas S.F., Milosevich N., Monjas L., Dev A., Brown T.M., Hirsch A.K.H., Wulff J.E., Hof F. Rational adaptation of L3MBTL1 inhibitors to create Small-Molecule Cbx7 antagonists. Chem. Med. Chem. 2019; 14:1444–1456. [DOI] [PubMed] [Google Scholar]
- 286. Lamb K.N., Bsteh D., Dishman S.N., Moussa H.F., Fan H., Stuckey J.I., Norris J.L., Cholensky S.H., Li D., Wang J. et al. Discovery and characterization of a cellular potent positive allosteric modulator of the polycomb repressive complex 1 chromodomain, CBX7. Cell Chem. Biol. 2019; 26:1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Milosevich N., Gignac M.C., McFarlane J., Simhadri C., Horvath S., Daze K.D., Croft C.S., Dheri A., Quon T.T.H., Douglas S.F. et al. Selective inhibition of CBX6: A methyllysine reader protein in the polycomb family. ACS Med. Chem. Lett. 2015; 7:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Milosevich N., Wilson C.R., Brown T.M., Alpsoy A., Wang S., Connelly K.E., Sinclair K.A.D., Ponio F.R., Hof R., Dykhuizen E.C. et al. Polycomb paralog chromodomain inhibitors active against both CBX6 and CBX8. ChemMedChem. 2021; 16:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Denton K.E., Wang S., Gignac M.C., Milosevich N., Hof F., Dykhuizen E.C., Krusemark C.J. Robustness of in vitro selection assays of DNA-Encoded peptidomimetic ligands to CBX7 and CBX8. SLAS Discov. 2018; 23:417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Suh J.L., Bsteh D., Si Y., Hart B., Weaver T.M., Pribitzer C., Lau R., Soni S., Ogana H., Rectenwald J.M. et al. Reprogramming CBX8-PRC1 function with a positive allosteric modulator. 2021; bioRxiv doi23 February 2021, preprint: not peer-reviewed 10.1101/2021.02.23.432388. [DOI] [PMC free article] [PubMed]
- 291. Khanna A., Côté A., Arora S., Moine L., Gehling V.S., Brenneman J., Cantone N., Stuckey J.I., Apte S., Ramakrishnan A. et al. Design, synthesis, and pharmacological evaluation of second generation EZH2 inhibitors with long residence time. ACS Med. Chem. Lett. 2020; 11:1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. He Y., Selvaraju S., Curtin M.L., Jakob C.G., Zhu H., Comess K.M., Shaw B., The J., Lima-Fernandes E., Szewczyk M.M. et al. The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 2017; 13:389–395. [DOI] [PubMed] [Google Scholar]