

Abstract
Many organs experience a loss of tissue mass and a decline in regenerative capacity during aging. In contrast, the prostate continues to grow in volume. In fact, age is the most important risk factor for prostate cancer. However, the age-related factors that influence the composition, morphology and molecular features of prostate epithelial progenitor cells, the cells-of-origin for prostate cancer, are poorly understood. Here, we review the evidence that prostate luminal progenitor cells are expanded with age. We explore the age-related changes to the microenvironment that may influence prostate epithelial cells and risk of transformation. Finally, we raise a series of questions about models of aging and regulators of prostate aging which need to be addressed. A fundamental understanding of aging in the prostate will yield critical insights into mechanisms that promote the development of age-related prostatic disease.
1. Introduction
Aging is a significant risk factor for disease in many tissues, including the prostate. Unlike tissues that atrophy with age, the prostate gland undergoes expansion. Prostatic enlargement or benign prostatic hyperplasia (BPH) causes lower urinary tract symptoms such as increased urinary frequency, urinary incontinence and, more rarely, renal failure[1]. BPH is the most common benign neoplasm of aging men. Autopsy studies have revealed that approximately 20% of men in their 40s and 50-60% of men in their 60s have histological evidence of BPH[2]. Furthermore, roughly 80% of men in their 70s exhibit symptoms of BPH[3]. Risk of prostate cancer also increases with age. Prostate cancer incidence increases from 1 in 20,000 for men younger than 39 to 1 in 45 for men aged 40-59 and to 1 in 7 for men aged 60-79[4]. As 64% of prostate cancer diagnoses are made in men over the age of 65 and the number of men in this age cohort is predicted to increase 4-fold by 2050, there will be a growing population requiring management[5]. Understanding the molecular and cellular mechanisms that underlie age-associated changes in the prostate will be essential to combat disease risk.
Recent DNA sequencing studies have established mutational landscapes in normal adult tissues, including somatic mutations in cancer-associated genes that increase in frequency with age[6–9]. These findings suggest an evolutionary process from normal cells to morphologically indistinguishable precancerous cells toward rare clones that become cancers. Epigenetic profiling has also revealed age-related changes in multiple human tissues, suggesting that epigenetic and genetic changes accumulate simultaneously and jointly contribute to aging[10–12]. Clonality is increased with age in blood and other adult tissues[13, 14], suggesting that aged tissues are maintained by fewer progenitor cells. The process of cell competition that enables normal epithelial cells to replace neighboring mutated cells[15], also termed epithelial defense against cancer, declines with age[14]. Similarly, age-related immune dysfunction may reduce efficiency of immune surveillance[16, 17], enabling the survival and expansion of pre-malignant cells.
2. Age-related accumulation of mutations in the prostate
Many studies have demonstrated that aging, but histologically-normal, cells in adult tissues gradually accumulate mutations[18]. Indeed, we and others discovered mutagenic fields in histologically-normal prostate tissue from patients diagnosed with localized prostate tumors[19]. These fields contain comparable numbers of single nucleotide variants (SNVs) to frank prostate cancers, along with significant numbers of copy number aberrations (CNAs) and genomic rearrangements (GRs). This, along with phylogenetic evidence of similar mutational histories of prostate cancers of differing grades[20–22], suggests that prostate cancers emerge from specific subclones in a broader mutagenic field. If this hypothesis were correct, it would suggest significant age-associated variability in prostate cancer mutational and evolutionary profiles. Two broad types of strategies have been used to search for this variability. First, there have been systematic genomic studies of age-related outliers: those rare prostate cancers that arise in men under the age of 55. Second, there have been studies of age-related trends in prostate cancer molecular features across large populations of sporadic disease. These have each revealed intriguing features.
Studies of early-onset prostate cancer (EOPC) have been relatively few compared to the multiple large cohorts of typical late onset prostate cancer. The largest study of EOPC evaluated 203 distinct tumors with germline and tumor whole-genome sequencing (WGS), along with methylome and RNA-seq profiling of tumor tissue[23]. Whilst many mutational features were shared between early and late onset prostate cancers, intriguing differences occurred. EOPCs were preferentially monoclonal, and showed less evolutionary diversification – consistent with a shorter lifespan. EOPCs showed similar frequencies of many driver events, but with a few intriguing differences. For example, chromosome 3p14 deletion (centered at FOXP1) was common in EOPC, but is relatively rare in later-onset prostate cancer. By contrast, while point mutations in SPOP were moderately prevalent (~10%) in late-onset cancers, they are rare in EOPC. Further, as anticipated EOPC showed a lower total burden of point mutations, and the mutational processes generating them showed an apparent age-association. Thus prostate cancers in very young men share many mutational features with those in older men, but with a reduction in mutations and evolutionary diversity consistent with a shorter natural history, and a small number of age-divergent driver mutations hinting at novel mechanisms of tumorigenesis.
In a similar way, several groups have considered how the number of mutations of each type present in a prostate tumor vary as a function of age. Remarkably, a linear relationship has been observed for effectively all types of mutations studied. For example, the sensitivity of point mutations in the nuclear genome (measured as point mutations per Mbp sequenced) rises by about 0.01 for each year of age at diagnosis[24]. For a 3,000 Mbp genome, this corresponds to 30 extra point mutations. Considering that a typical prostate cancer has a mutation rate of ~0.5 per Mbp sequenced[25], this corresponds to an annual 2% rise the year of diagnosis alone. A smaller, but still linear, increase with age is seen in the ~16 kbp mitochondrial genome, and linear increases in the numbers of translocations, inversions, deletions and gains have all been described[23, 24]. Point mutations in particular are biased towards classic deamination trinucleotide signatures that have been associated with age in several other cancer types[21, 23].
Besides age, several other factors are associated with the accumulation of mutations in the prostate. Strongest of these is the association between the germline genome and prostate cancer evolution. Rare deleterious germline variants in DNA Damage Repair (DDR) genes like BRCA2 are associated with both an increased rate of evolution[26] and with adverse clinical outcomes[27]. Similarly, common germline variants are strongly associated with both risk of a prostate cancer diagnosis[28, 29] and with tumor evolution[30, 31]. Similarly, ancestry is strongly associated with prostate cancer phenotype[32–35]. It is wholly unknown how these features relate to prostatic aging, and it is unclear if the observations in large cohorts of Caucasian men will fully generalize to different ancestry groups. Nevertheless, accumulating evidence suggests that localized prostates are continually accumulating somatic mutations, and it is unclear how this links to their clinical manifestations. A pair of recent pan-cancer surveys have recapitulated several of these observations in diverse cancer types[36, 37].
3. Aging of prostate progenitor cells
3.1. Prostate stem and progenitor cells as the cells-of-origin for prostate cancer
As prostate cancers arise from epithelial cells that accumulate mutations with age, it is essential to understand which cell-types are proficient for transformation. The epithelium of the prostate is a double layer comprised of basal cells adjacent to the basement membrane and luminal cells adjacent to the fluid-filled lumen, in addition to a rare population of neuroendocrine cells[38]. Postnatal mouse prostate development is mediated by multipotent basal stem and progenitor cells as well as unipotent luminal progenitor cells[39, 40]. In contrast, post-pubertal adult mouse prostate epithelium is maintained predominantly by distinct pools of unipotent basal progenitor cells and unipotent luminal progenitors[41, 42]. In response to inflammation[43], high fat diet[44], or luminal cell death[45], basal cells can exhibit multipotency. Multipotency has also been demonstrated in a subset of castration-resistant luminal cells[46] in mouse prostate using lineage tracing approaches. Using mitochondrial mutations to track clonality in human prostate, Moad et al. demonstrated that multipotent basal stem and progenitor cells maintain epithelial glands, with a minor contribution from rare unipotent luminal progenitor cells[47].
Mouse models of prostate cancer have demonstrated roles for both basal and luminal cells in prostate cancer initiation. Upon loss of the tumor-suppressor PTEN, adult prostate basal cells undergo luminal differentiation[41, 48, 49], giving rise to phenotypically luminal cancer cells. Recent studies suggest that distinct subpopulations of luminal cells residing in distinct prostate microenvironments can initiate prostate cancer with different functional activity[50, 51]. For example, prostate cancer arising from proximally-located progenitor-enriched Sca-1+ luminal cells exhibit a greater capacity for neuroendocrine differentiation following androgen ablation than distally-located Sca-1− luminal cells[52]. Single cell RNA sequencing studies have better defined luminal heterogeneity, revealing a progenitor-like luminal subset expressing stem/progenitor genes including Sca-1, Trop2, Prostate stem cell antigen (Psca) and Keratin 4 (Krt4)[53–56]. Lineage tracing using Krt4 or Psca promoters confirms the progenitor features of self-renewal and differentiation in progenitor-like luminal cells[56]. Pten deletion in Krt4+ cells confirms that prostate cancer can arise in transformed luminal progenitor cells[56].
We and others have utilized an approach to transform primary benign human prostate epithelial cells and evaluate the capacity of distinct cell-types to initiate cancer in immune-deficient mice[57, 58]. Basal cells from primary naïve human prostate tissue can be transformed by multiple combinations of oncogenes to initiate prostate adenocarcinoma[58, 59] and small cell prostate cancer[60, 61]. In contrast, luminal cells from primary naïve human prostate tissue are not easily transformed. To enhance luminal cell survival and expansion prior to transformation, 3D organoid culture has been utilized, revealing that oncogene-expressing luminal cell-derived organoids can initiate human prostate cancer[62]. Further studies identified a subset of human prostate luminal cells characterized by low protein expression of CD38 and high protein expression of PSCA that are enriched for progenitor-like organoid-forming capacity and can serve as cells-of-origin for human prostate cancer[63]. These luminal progenitor cells are expanded in regions with inflammation and share features with Proliferative Inflammatory Atrophy, a proposed precursor for prostate cancer[64]. Collectively these studies using mouse and human tissue show that basal cells and luminal progenitor cells are likely to be the cells most at risk for developing into prostate cancer. It should be noted that a wide range of approaches and markers have been used to identify and characterize luminal progenitor cells in mouse and human prostate, reviewed in detail by Zhang et al[65], underscoring the importance of this subset in development, homeostasis and disease.
3.2. Age-related changes in the prostate epithelial compartment defined by an increase in luminal progenitor cells
As stem and progenitor cells are preferred cells-of-origin for prostate cancer, it is critical to evaluate the effects of aging on progenitor activity. We defined age-related changes to prostate epithelium by profiling epithelial cells isolated from 3-month old and 24-month old mouse prostate. Unlike in muscle[66] and brain[67], aging in the prostate is associated with maintenance of progenitor activity. Aging does not diminish the progenitor-like primary organoid forming capacity of basal and luminal cells. Furthermore, aging does not affect the self-renewing capacity of basal and luminal cells upon re-plating into secondary organoid culture[68]. Luminal cells from aged mice form larger organoids on average and generate a greater proportion of large organoids. In addition, these cells share features with human luminal progenitor cells defined by low CD38 expression including elevated mRNA expression of Bcl2, CD74, Pigr, and Psca, and low Cd38 mRNA expression[68].
Recent studies have better specified luminal heterogeneity, defining a progenitor enriched subset identified by Trop2, Psca, and Krt4[53, 54, 56]. These luminal cells exhibit increased capacity to generate organoids compared to other luminal subsets and can be cells-of-origin for prostate cancer[56]. Since aged luminal cells exhibit increased mRNA expression of Trop2, Psca, and Krt4, we evaluated whether the luminal progenitor signature in aged mouse prostate arises from uniform upregulation of progenitor markers, or an age-related expansion of a pre-existing progenitor population. Flow cytometry analysis revealed the mean percentage of Trop2+ luminal cells increased from 6% in adult prostates to 21% in old prostates[68]. This finding supports a model where the luminal aging signature arises from an age-related expansion of Trop2+ luminal progenitor cells (Figure 1). A recent study identified an age-related expansion of progenitor-like luminal cells in the mouse mammary gland[69]. These findings suggest that age-related changes to prostate epithelium may be conserved in other epithelial tissues.
Figure 1. Features of aging in the prostate.
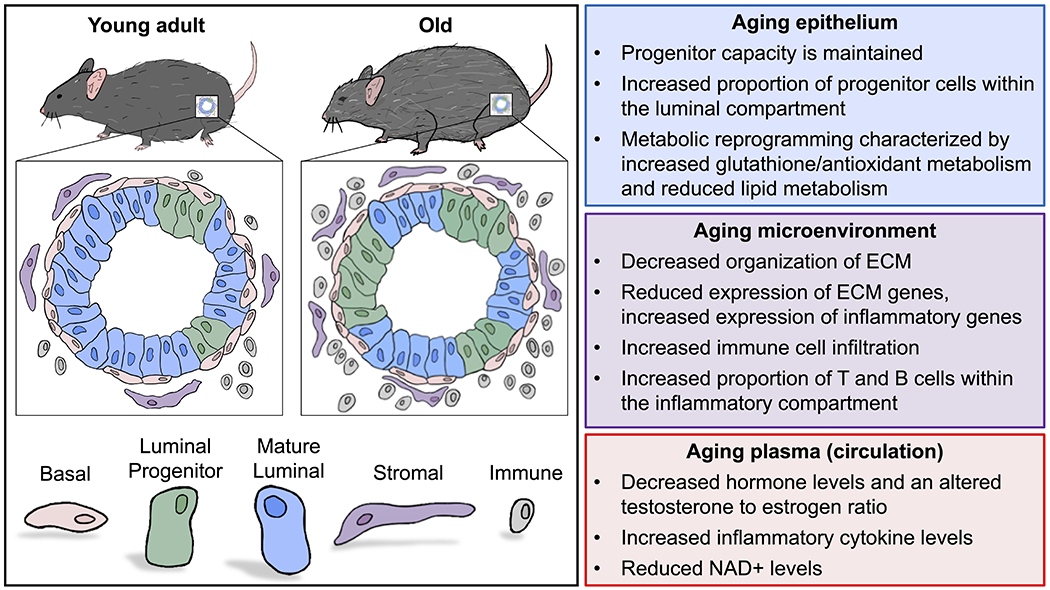
Aging in the prostate is characterized by an increased proportion of progenitor-like luminal cells, which are phenotypically defined by high expression of Trop2 and Psca (left). Reported age-related changes in the prostate epithelium, the prostate microenvironment, and plasma (circulation) are listed (right).
As the proportion of Trop2+ luminal progenitors increases during aging, we investigated whether luminal cells exhibit a heterogeneous response to aging. Gene expression analysis revealed that the adult Trop2+ signature is largely retained in old Trop2+ luminal cells, whereas less overlap was observed between the adult and old Trop2− cells[68]. Organoid assays revealed that while the organoid-forming capacity of Trop2+ cells is retained with age, the organoid-forming capacity of Trop2− cells is significantly diminished. Consistent with diminished organoid capacity, gene ontology analysis of genes downregulated in old Trop2− cells returned terms related to cell cycle and cell division[68]. These findings suggest that distinct luminal subsets exhibit altered responses to aging. Whereas progenitor-like luminal cells retain their capacity, mature luminal cells exhibit diminished capacity resulting in a dramatic age-related shift in the luminal compartment. Similar findings have recently been reported in the mouse brain where microglial subsets have a heterogeneous response to aging, resulting in the expansion of pro-inflammatory microglial cells[70].
Since we observed significant overlap in gene expression between mouse Trop2+ luminal progenitor cells and human luminal progenitor cells defined by low CD38 and high PSCA[63], we explored whether human luminal progenitor cells are expanded with age. Using normal prostates obtained from organ donors through the Southwest Transplant Alliance and UT Southwestern, we found a significant correlation between age and the percentage of PSCA+ luminal progenitor cells[68]. These findings suggest that the expansion of luminal progenitors during aging is a conserved feature across species.
When transcriptional profiles of three distinct epithelial cell-types (basal, Trop2+ luminal progenitor, and Trop2− luminal cells) were evaluated, each cell-type exhibited common age-related changes reflective of metabolic reprogramming and an altered microenvironment[71]. Increased glutathione and antioxidant metabolism in aging epithelial cells suggests a response to tissue hypoxia, while increased inflammatory signaling likely reflects a response to prostatic inflammation. Further evaluation of the aged prostate microenvironment may provide insight into the extrinsic factors regulating epithelial aging.
4. Aging of the prostate microenvironment
4.1. Age-related reprogramming of prostate stromal cells and the extracellular matrix
Prostate aging and disease pathogenesis is associated with the expansion of stromal cells[72], which is an important factor contributing to tissue growth and dysregulation of aging epithelial progenitor cells. Numerous studies have defined crosstalk between the prostate stroma and adjacent epithelium[73]. Furthermore, there is emerging evidence that age-related changes to the prostatic stroma influence the growth of the prostatic epithelium[74]. Conditioned media from fibroblasts isolated from younger prostates, but not from older prostates, has been shown to suppress prostate epithelial cell growth[75], suggesting that young fibroblasts secrete different factors than aged fibroblasts. Aging has also been shown to influence the prostate extracellular matrix (ECM), which is primarily comprised of fibrillar collagen[73]. Collagen abundance is relatively conserved between younger and older prostates, whereas an age-related decrease in ECM organization occurs in addition to emergence of rough and fragmented fibrils[74]. RNA profiling of the aging prostatic stroma revealed a reduction in several collagen genes including Col1a1 and Col3a1[74]. Similar age-related changes to the ECM have been observed in other tissues, such as the skin, where a reduction in mechanical stimulation leads to reduced collagen synthesis[76].
Consistent with earlier work[74], we have defined age-related changes in prostate stromal cell morphology and gene expression. Dissociated cells from the aged stromal compartment exhibit reduced forward scatter levels as measured by Fluorescence Activated Cell Sorting (FACS), indicative of a reduction in cell size[68]. In addition to reduced collagen gene expression, aged stromal cells also exhibit reduced expression of the elastin gene (Eln), which is critical for the formation of ECM elastic fibers[71]. While ECM-related genes exhibit reduced mRNA expression levels in aged stromal cells, inflammatory-related genes are increased with age[71, 77–79].
4.2. Increased infiltration of lymphocytes in aged prostate
A loss of ECM integrity in the aged prostate may enable increased infiltration of inflammatory cells into the prostate microenvironment, leading to increased inflammatory cytokines that influence both stromal and epithelial cells. Alternatively, aged stromal or epithelial cells may upregulate pro-inflammatory signals to enhance immune cell infiltration into the old prostate. Using immunohistochemistry, Bianchi-Frias et al., demonstrated increased infiltration of myeloid and lymphocyte cells into aged mouse prostate[74]. More recently, we have utilized mass cytometry to comprehensively characterize age-related changes to the prostate immune compartment, revealing a significant increase in the proportion of B and T lymphocytes in aged prostates[68]. Upon further investigating the timeline where the shift within the immune compartment takes place, we found a significant increase in lymphocyte infiltration arising between 6 and 12 months of age[80]. This age-related phenotype occurs much earlier than previously reported, indicating that “inflamm-aging” may contribute to other age-related processes in the prostate including expansion of the pool of luminal progenitor cells[68]. The presence of chronic inflammation in the benign human prostate nearly doubles the risk for developing prostate cancer[81]. Furthermore, experimentally promoting inflammation in rodent models can increase prostatic epithelial cell proliferation[43, 44, 82]. These findings suggest that an age-related increase in inflammatory cells may contribute to disease pathogenesis.
4.3. Local hypoxia may influence the aging prostate
Aging in the prostate is associated with increased prostatic mass. Therefore, we hypothesized that aged prostates may feature localized hypoxic regions that emerge due to lack of sufficient vasculature. Consistent with this hypothesis, motif analysis on genes upregulated in aged prostate epithelial cells identified transcriptional regulators of hypoxia[71]. Exposure to hypoxia has been shown to modulate several processes that may contribute to the prostate epithelial aging phenotype. For example, hypoxia increases normal prostate epithelial cell resistance to apoptosis[83]. Hypoxia can alter the differentiation capacity of tissue progenitor cells[84] and promote local tissue regeneration[85]. Importantly, exposure to hypoxia can promote antioxidant metabolism and recent studies demonstrate that upregulation of antioxidant capacity is sufficient for oncogenic transformation[86]. In cancer cells, hypoxia has been shown to facilitate survival and propagation of tumor cells by inducing intracellular signaling pathways including PI3K/AKT/mTOR, MAPK/ERK and NFĸB[87]. Studies comparing hypoxic and normoxic tumors suggest that hypoxic tumors preferentially accumulate specific types of mutations[88, 89], such as disruption of p53 or over-expression of Bcl-2 which impairs the apoptotic response[90]. As normal epithelial cells accumulate mutations with age, a hypoxic microenvironment in the aging prostate may similarly select for certain subclones or cell populations with intrinsic resistance to hypoxic stress. In support of this, hypoxic prostate cancers preferentially form specific sub-histologies[91] and are more often visible to modern MRI protocols[92]. Future studies should define cellular mechanisms or somatic mutations that may provide a fitness advantage for aging prostate epithelial cells under hypoxic conditions.
4.4. Age-related changes in circulating factors that may influence the prostate
During aging, changes to the concentration of various circulating factors including hormones, metabolites and inflammatory cytokines have been reported[93]. In males, an age-related decrease in testosterone[94] likely alters the estrogen to testosterone ratio. Estrogen can modulate the prostate ECM[95] as well as the pool of prostate progenitor cells[96], both of which are altered with age. Upon analyzing circulating metabolites, Gomes et al., identified higher levels of methylmalonic acid, a known mediator of tumor progression, in the serum of older patients[97]. Conversely, NAD+: NADH and NADP+: NADPH plasma ratios, which are crucial to a range of cellular processes, are decreased with age[98]. A decrease in NAD+ has been correlated with reduced mitochondrial- and nuclear-function and increased age-associated pathogenesis[99]. NAD+ supplementation may counter age-related phenotypes in the prostate as has been shown in other organs[100–102]. Age-related changes in hormones and other factors may be caused in part by increased body weight of older mice, as obesity in humans is associated with reduced testosterone levels[103]. Body weight and conditions of obesity should be considered when evaluating circulating factors in old mice. A deeper understanding of the factors that regulate prostate aging will require investigation into how epithelial and non-epithelial cells of the prostate are regulated by circulating hormones, metabolites and cytokines that change in abundance with age.
5. Unanswered questions in the area of prostate epithelial aging and cancer risk
Despite the importance of age in risk of cancer incidence, our knowledge of prostate epithelial aging is limited and many important questions remain unanswered. In the following section, we will raise open questions that need to be addressed to better resolve the role of aging in progenitor cell activity and to elucidate the factors that mediate age-related prostate cancer risk.
1. Do aging progenitor cells accumulate somatic mutations in a different manner than mature prostate cells?
Recent studies utilizing deep sequencing of non-cancerous tissues including the skin, esophagus and colon during aging have revealed an accumulation of mutations with age[7–9, 104]. The rate of accumulation of somatic mutations varies between individuals and, in the liver, between stem cells and differentiated cells[18]. This suggests that different cells in the same tissue can accumulate mutations at different rates. Based on our recent findings that distinct luminal subsets in the mouse prostate age differently, distinct cell-types in the aging prostate may accumulate mutations at different rates, or even accumulate different types of mutations as a function of their differential epigenetic context, chromatin architecture and capacities for repairing specific types of DNA damage.
2. What are the origins of increased luminal progenitor cells with age?
Luminal progenitor cells in the human prostate, defined by high PSCA expression and low CD38 expression[63, 105] exhibit increased organoid-forming activity and are capable of responding to oncogenic insults to initiate human prostate cancer[63]. Importantly, the percentage of luminal progenitor cells increases with age in human prostate. High Trop2 protein expression marks Psca+ luminal cells in the mouse prostate with enhanced organoid-forming progenitor activity[56, 68]. The proportion of luminal cells with a Trop2+ progenitor phenotype was elevated three- to four-fold in old prostates[68], confirming expansion of the population of luminal progenitor cells in aging mouse prostate. Deletion of Pten in luminal progenitor cells leads to tumor initiation[56], further demonstrating that luminal progenitor cells are cells-of-origin for prostate cancer. Collectively, these studies suggest age-related dysregulation of epithelial heterogeneity increases the number of cells at risk for transformation. How luminal progenitor cells are expanded in aging prostates has not been established. Aging luminal progenitor cells may arise as a result of enhanced self-renewal from pre-existing luminal progenitor cells, or through increased differentiation of aging basal cells. Alternatively, microenvironmental cues may direct the dedifferentiation of mature luminal cells into luminal progenitor cells. Lineage tracing experiments would be necessary to distinguish these possibilities.
3. Does prostatic “inflamm-aging” modulate progenitor expansion and transformation?
Chronic inflammation increases with age and correlates with disease risk[81]. In human prostate tissue, progenitor cells are often found in close proximity to immune cells and exhibit an inflammatory signature including NFkB activation[63], suggesting an interaction between immune cells and epithelial progenitor cells. We and others have defined age-related changes to the inflammatory compartment characterized by increasing immune cells as well as a shift toward an increased proportion of T and B lymphocytes[68, 80]. Signatures of old prostate epithelial and stromal cells reveal elevated inflammatory signaling[71], suggesting that inflammatory cues likely influence aging epithelial cells. However, it is not known whether age-related inflammation plays a role in protecting against or promoting the risk of transformation. Immune cell-secreted cytokines can increase epithelial proliferation and reduce apoptosis[43, 44, 106], which may contribute to epithelial cell survival and risk of transformation through activation of Toll-like receptors, NFkB or JAK/STAT pathways[107–109]. Conversely, immune cell surveillance might eliminate cells that accumulate somatic mutations. Functional studies will be required to delineate the precise role of immune cells in the aging prostate and how anti-inflammatory approaches would alter prostate epithelial aging and transformation.
4. What is the role of antioxidant/glutathione metabolism in aging prostate epithelial cells?
The antioxidant response emerged as a common signature of prostate aging across multiple distinct cell-types[71]. Nuclear factor E2-related factor 2 (Nrf2) is considered a master regulator of the antioxidant response to counteract oxidative stress and prevent DNA damage through the activation of enzymes that coordinate glutathione metabolism[110]. Importantly, modulation of Nrf2 is sufficient to alter lifespan in model organisms[111]. In several tissues including liver and muscle, Nrf2-mediated glutathione metabolism declines with age and is predicted to contribute to an age-related decline in tissue function[112, 113]. The elevated glutathione metabolism signature in aging prostate epithelial cells may simply be an indicator of oxidative stress and the natural cellular response to this threat. The activation of Nrf2 signaling may protect aging epithelial cells from DNA damage and mutagenesis, reducing the risk of transformation. Alternatively, the antioxidant response may promote survival of mutated epithelial cells and sustain progenitor capacity in the aging prostate. Loss-of-function or gain-of-function studies will be necessary to determine the precise role of Nrf2 and glutathione metabolism during prostate epithelial aging.
5. Is the wild-type mouse prostate a good model of prostatic aging?
Prostate cancer does not spontaneously develop in aging mice as it does in aging humans. In fact, the commonly used C57BL/6 mouse strain is generally refractory to tumorigenesis. Therefore, it is important to evaluate whether the mouse prostate can be used effectively to study prostatic aging. Our recent findings that both luminal progenitor cells and CD45+ immune cells accumulate in aging mouse and human prostate tissue suggest that certain features of aging are conserved across species[68]. These results support the use of mouse prostate to study factors that regulate these conserved aging phenotypes. For example, could interventions that delay aging and extend lifespan in model organisms, such as caloric restriction or treatment with metformin[114, 115], alter the trajectory of prostate aging phenotypes? It will be critical to understand why aging mouse prostates exhibit progenitor expansion without transformation while aging human prostates exhibit frequent BPH and are at risk for tumor initiation. Several important distinctions can be made between mouse and human prostate. Basal cells form a continuous layer around luminal cells in human prostate forming a stratified epithelium, while the basal cell layer is more sporadic in mouse prostate consistent with a pseudostratified epithelium[116]. Differences in telomere length between inbred mouse strains and humans may also contribute to aging and transformation[117]. Additionally, while the human prostate is separated into distinct zones, the mouse prostate is made up of distinct lobes with varying transcriptional profiles between lobes[54]. In mouse models of prostate cancer, cancer lesions can initiate in distinct lobes depending on the genetic event, construct used, or mouse strain[118]. As recent studies used total mouse prostate and did not distinguish between lobes[68, 71], future studies should address whether progenitor cells and inflammatory cells vary between aging prostate lobes. Determining whether prostate aging can be best modeled in one or another lobe of the mouse prostate will enable better inter-species comparisons.
6. How does aging in the prostate compare to other adult tissues?
A recent study on age-related changes to the mammary gland defined several aging phenotypes that we have previously reported in the prostate[68, 71]: (1) an increase in the proportion of progenitor-like luminal cells within the epithelium, (2) reduced expression of ECM genes in aging epithelial and stromal cells, and (3) an increase in the lymphocyte proportion within the immune compartment[69]. Similar to prostate cancer, age is a risk factor for breast cancer[119]. These findings suggest that studies of aging in one tissue may inform mechanisms driving age-related changes in other tissues. We have reported changes in gene expression that reflect metabolic reprogramming in aging epithelial cells[71], consistent with reports of age-related metabolic reprogramming in several tissues contributing to aging phenotypes[120–122]. However, not all aging phenotypes in the prostate are shared by other tissues. While we have found that old prostates maintain their progenitor capacity[68], stem and progenitor activity is reduced in other aging tissues[123, 124]. Comparing distinct tissues with different phenotypic responses to aging may aid in our understanding of the cell-intrinsic and extrinsic factors that maintain progenitor capacity in the aging prostate.
6. Summary
Fundamental to understanding prostate disease pathogenesis is a deeper understanding of age-related prostate biology. To a certain extent, the field of prostate aging research is still in its infancy, with an initial focus on characterizing age-related phenotypes before defining important mechanisms that drive those age-related changes. The prostate is a heterogeneous tissue, with multiple epithelial and non-epithelial components, each of which is likely to exhibit distinct aging phenotypes. In this review article, we have focused on the epithelial progenitor cells that are cells-of-origin for prostate cancer and the influences on those cells in particular. Moving forward, researchers should continue to address the ways in which heterogeneous cell-types age individually and collectively within a shared aging microenvironment. Defining interventions to interfere with or delay prostate aging may enable significant progress in reducing disease pathogenesis in the prostate.
Acknowledgements:
The authors are supported by the American Cancer Society (RSG-17-068-01-TBG), NIH/NCI (R01CA237191, P30CA016042, U01CA214194, UCLA Prostate SPORE P50CA0912131), Rose Hills Foundation, Gill endowment dedicated to cancer research and the Spitzer Family Foundation Fund, as well as UCLA’s Jonsson Comprehensive Cancer Center, Broad Stem Cell Research Center, Clinical and Translational Science Institute, and Institute of Urologic Oncology. PDC acknowledges the support of the UCLA Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research Rose Hills Foundation Graduate Scholarship Training Program. JMG is supported by a Ruth L. Kirschstein National Research Service Award GM007185. PCB was supported by a Prostate Cancer Foundation Special Challenge Award to PCB (Award ID #: 20CHAS01) made possible by the generosity of Mr. Larry Ruvo.
Footnotes
Conflicts of Interest: None declared.
References
- 1.Chughtai B, et al. Benign prostatic hyperplasia. Nat Rev Dis Primers, 2016. 2: p. 16031. [DOI] [PubMed] [Google Scholar]
- 2.Roehrborn CG, Benign prostatic hyperplasia: an overview. Rev Urol, 2005. 7 Suppl 9: p. S3–S14. [PMC free article] [PubMed] [Google Scholar]
- 3.Liu D, et al. Integrative multiplatform molecular profiling of benign prostatic hyperplasia identifies distinct subtypes. Nat Commun, 2020. 11(1): p. 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crawford ED, Epidemiology of prostate cancer. Urology, 2003. 62(6 Suppl 1): p. 3–12. [DOI] [PubMed] [Google Scholar]
- 5.Bechis SK, Carroll PR, and Cooperberg MR, Impact of age at diagnosis on prostate cancer treatment and survival. J Clin Oncol, 2011. 29(2): p. 235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yizhak K, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science, 2019. 364(6444). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee-Six H, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature, 2019. 574(7779): p. 532–537. [DOI] [PubMed] [Google Scholar]
- 8.Martincorena I, et al. Somatic mutant clones colonize the human esophagus with age. Science, 2018. 362(6417): p. 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martincorena I, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science, 2015. 348(6237): p. 880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Florath I, et al. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet, 2014. 23(5): p. 1186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tajuddin SM, et al. Novel age-associated DNA methylation changes and epigenetic age acceleration in middle-aged African Americans and whites. Clin Epigenetics, 2019. 11(1): p. 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benayoun BA, et al. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res, 2019. 29(4): p. 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jan M, Ebert BL, and Jaiswal S, Clonal hematopoiesis. Semin Hematol, 2017. 54(1): p. 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu N, et al. Stem cell competition orchestrates skin homeostasis and ageing. Nature, 2019. 568(7752): p. 344–350. [DOI] [PubMed] [Google Scholar]
- 15.Kon S, et al. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol, 2017. 19(5): p. 530–541. [DOI] [PubMed] [Google Scholar]
- 16.Montecino-Rodriguez E, Berent-Maoz B, and Dorshkind K, Causes, consequences, and reversal of immune system aging. J Clin Invest, 2013. 123(3): p. 958–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ovadya Y, et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun, 2018. 9(1): p. 5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brazhnik K, et al. Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci Adv, 2020. 6(5): p. eaax2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper CS, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet, 2015. 47(4): p. 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu LY, et al. Quantifying the influence of mutation detection on tumour subclonal reconstruction. Nat Commun, 2020. 11(1): p. 6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Espiritu SMG, et al. The Evolutionary Landscape of Localized Prostate Cancers Drives Clinical Aggression. Cell, 2018. 173(4): p. 1003–1013 e15. [DOI] [PubMed] [Google Scholar]
- 22.Wedge DC, et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat Genet, 2018. 50(5): p. 682–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerhauser C, et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell, 2018. 34(6): p. 996–1011 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopkins JF, et al. Mitochondrial mutations drive prostate cancer aggression. Nat Commun, 2017. 8(1): p. 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraser M, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature, 2017. 541(7637): p. 359–364. [DOI] [PubMed] [Google Scholar]
- 26.Taylor RA, et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat Commun, 2017. 8: p. 13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castro E, et al. Effect of BRCA Mutations on Metastatic Relapse and Cause-specific Survival After Radical Treatment for Localised Prostate Cancer. Eur Urol, 2015. 68(2): p. 186–93. [DOI] [PubMed] [Google Scholar]
- 28.Huynh-Le MP, et al. Polygenic hazard score is associated with prostate cancer in multi-ethnic populations. Nat Commun, 2021. 12(1): p. 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conti DV, et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat Genet, 2021. 53(1): p. 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Briollais L, et al. Germline Mutations in the Kallikrein 6 Region and Predisposition for Aggressive Prostate Cancer. J Natl Cancer Inst, 2017. 109(4). [DOI] [PubMed] [Google Scholar]
- 31.Houlahan KE, et al. Genome-wide germline correlates of the epigenetic landscape of prostate cancer. Nat Med, 2019. 25(10): p. 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang FW, et al. Exome Sequencing of African-American Prostate Cancer Reveals Loss-of-Function ERF Mutations. Cancer Discov, 2017. 7(9): p. 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaratlerdsiri W, et al. Whole-Genome Sequencing Reveals Elevated Tumor Mutational Burden and Initiating Driver Mutations in African Men with Treatment-Naive, High-Risk Prostate Cancer. Cancer Res, 2018. 78(24): p. 6736–6746. [DOI] [PubMed] [Google Scholar]
- 34.Ren S, et al. Whole-genome and Transcriptome Sequencing of Prostate Cancer Identify New Genetic Alterations Driving Disease Progression. Eur Urol, 2018. 73(3): p. 322–339. [DOI] [PubMed] [Google Scholar]
- 35.Li J, et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature, 2020. 580(7801): p. 93–99. [DOI] [PubMed] [Google Scholar]
- 36.Chatsirisupachai K PL, Van Loo P, de Magalhães JP, An Integrative Analysis of the Age-Associated Genomic, Transcriptomic and Epigenetic Landscape across Cancers. bioRxiv, 2020. 10.1101/2020.07.07.192237. [DOI] [Google Scholar]
- 37.Li CH HS, Boutros PC, Age Influences on the Molecular Presentation of Tumours. bioRxiv, 2020. 10.1101/2020.07.07.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen MM and Abate-Shen C, Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev, 2010. 24(18): p. 1967–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ousset M, et al. Multipotent and unipotent progenitors contribute to prostate postnatal development. Nat Cell Biol, 2012. 14(11): p. 1131–8. [DOI] [PubMed] [Google Scholar]
- 40.Tika E, et al. Spatiotemporal regulation of multipotency during prostate development. Development, 2019. 146(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi N, et al. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell, 2012. 21(2): p. 253–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J, et al. Regenerated luminal epithelial cells are derived from preexisting luminal epithelial cells in adult mouse prostate. Mol Endocrinol, 2011. 25(11): p. 1849–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwon OJ, et al. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc Natl Acad Sci U S A, 2014. 111(5): p. E592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwon OJ, et al. High fat diet promotes prostatic basal-to-luminal differentiation and accelerates initiation of prostate epithelial hyperplasia originated from basal cells. Stem Cell Res, 2016. 16(3): p. 682–91. [DOI] [PubMed] [Google Scholar]
- 45.Toivanen R, Mohan A, and Shen MM, Basal Progenitors Contribute to Repair of the Prostate Epithelium Following Induced Luminal Anoikis. Stem Cell Reports, 2016. 6(5): p. 660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature, 2009. 461(7263): p. 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moad M, et al. Multipotent Basal Stem Cells, Maintained in Localized Proximal Niches, Support Directed Long-Ranging Epithelial Flows in Human Prostates. Cell Rep, 2017. 20(7): p. 1609–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang ZA, et al. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol, 2013. 15(3): p. 274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu TL, et al. Conditionally ablated Pten in prostate basal cells promotes basal-to-luminal differentiation and causes invasive prostate cancer in mice. Am J Pathol, 2013. 182(3): p. 975–91. [DOI] [PubMed] [Google Scholar]
- 50.Kwon OJ, Zhang L, and Xin L, Stem Cell Antigen-1 Identifies a Distinct Androgen-Independent Murine Prostatic Luminal Cell Lineage with Bipotent Potential. Stem Cells, 2016. 34(1): p. 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kwon OJ, et al. The Sca-1(+) and Sca-1(−) mouse prostatic luminal cell lineages are independently sustained. Stem Cells, 2020. 38(11): p. 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwon OJ, et al. Sox2 is necessary for androgen ablation-induced neuroendocrine differentiation from Pten null Sca-1(+) prostate luminal cells. Oncogene, 2021. 40(1): p. 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karthaus WR, et al. Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science, 2020. 368(6490): p. 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crowley L, et al. A single-cell atlas of the mouse and human prostate reveals heterogeneity and conservation of epithelial progenitors. Elife, 2020. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joseph DB, et al. Urethral luminal epithelia are castration-insensitive cells of the proximal prostate. Prostate, 2020. 80(11): p. 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo W, et al. Single-cell transcriptomics identifies a distinct luminal progenitor cell type in distal prostate invagination tips. Nat Genet, 2020. 52(9): p. 908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldstein AS, et al. Purification and direct transformation of epithelial progenitor cells from primary human prostate. Nat Protoc, 2011. 6(5): p. 656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldstein AS, et al. Identification of a cell of origin for human prostate cancer. Science, 2010. 329(5991): p. 568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stoyanova T, et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc Natl Acad Sci U S A, 2013. 110(50): p. 20111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JK, et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell, 2016. 29(4): p. 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park JW, et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science, 2018. 362(6410): p. 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park JW, et al. Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proc Natl Acad Sci U S A, 2016. 113(16): p. 4482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu X, et al. Low CD38 Identifies Progenitor-like Inflammation-Associated Luminal Cells that Can Initiate Human Prostate Cancer and Predict Poor Outcome. Cell Rep, 2016. 17(10): p. 2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sfanos KS, et al. The inflammatory microenvironment and microbiome in prostate cancer development. Nat Rev Urol, 2018. 15(1): p. 11–24. [DOI] [PubMed] [Google Scholar]
- 65.Zhang D, et al. Prostate Luminal Progenitor Cells in Development and Cancer. Trends Cancer, 2018. 4(11): p. 769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baumgartner RN, et al. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol, 1998. 147(8): p. 755–63. [DOI] [PubMed] [Google Scholar]
- 67.Meier-Ruge W, et al. Age-related white matter atrophy in the human brain. Ann N Y Acad Sci, 1992. 673: p. 260–9. [DOI] [PubMed] [Google Scholar]
- 68.Crowell PD, et al. Expansion of Luminal Progenitor Cells in the Aging Mouse and Human Prostate. Cell Rep, 2019. 28(6): p. 1499–1510 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li CM, et al. Aging-Associated Alterations in Mammary Epithelia and Stroma Revealed by Single-Cell RNA Sequencing. Cell Rep, 2020. 33(13): p. 108566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tabula Muris C, A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature, 2020. 583(7817): p. 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crowell PD, et al. Distinct cell-types in the prostate share an aging signature suggestive of metabolic reprogramming. Am J Clin Exp Urol, 2020. 8(4): p. 140–151. [PMC free article] [PubMed] [Google Scholar]
- 72.Untergasser G, Madersbacher S, and Berger P, Benign prostatic hyperplasia: age-related tissue-remodeling. Exp Gerontol, 2005. 40(3): p. 121–8. [DOI] [PubMed] [Google Scholar]
- 73.Levesque C and Nelson PS, Cellular Constituents of the Prostate Stroma: Key Contributors to Prostate Cancer Progression and Therapy Resistance. Cold Spring Harb Perspect Med, 2018. 8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bianchi-Frias D, et al. The effects of aging on the molecular and cellular composition of the prostate microenvironment. PLoS One, 2010. 5(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Begley L, et al. CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell, 2005. 4(6): p. 291–8. [DOI] [PubMed] [Google Scholar]
- 76.Varani J, et al. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol, 2006. 168(6): p. 1861–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reyes I, et al. Aging-associated changes in gene expression in the ACI rat prostate: Implications for carcinogenesis. Prostate, 2005. 63(2): p. 169–86. [DOI] [PubMed] [Google Scholar]
- 78.Lau KM, et al. Age-associated changes in histology and gene-expression profile in the rat ventral prostate. Lab Invest, 2003. 83(5): p. 743–57. [DOI] [PubMed] [Google Scholar]
- 79.de Magalhaes JP, Curado J, and Church GM, Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics, 2009. 25(7): p. 875–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fox JJ HT, Navarro HI, Garcia AJ, Shou BL, Goldstein AS, Highly multiplexed immune profiling throughout adulthood reveals kinetics of lymphocyte infiltration in the aging mouse prostate. bioRxiv, 2020. 10.1101/2020.06.18.160556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gurel B, et al. Chronic inflammation in benign prostate tissue is associated with high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer Epidemiol Biomarkers Prev, 2014. 23(5): p. 847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kessler OJ, et al. Role of chronic inflammation in the promotion of prostatic hyperplasia in rats. J Urol, 1998. 159(3): p. 1049–53. [PubMed] [Google Scholar]
- 83.Walsh S, et al. Hypoxia increases normal prostate epithelial cell resistance to receptor-mediated apoptosis via AKT activation. Int J Cancer, 2009. 124(8): p. 1871–8. [DOI] [PubMed] [Google Scholar]
- 84.Shivaraju M, et al. Airway stem cells sense hypoxia and differentiate into protective solitary neuroendocrine cells. Science, 2021. 371(6524): p. 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xi Y, et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat Cell Biol, 2017. 19(8): p. 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Y, et al. Upregulation of Antioxidant Capacity and Nucleotide Precursor Availability Suffices for Oncogenic Transformation. Cell Metab, 2021. 33(1): p. 94–109 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muz B, et al. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl), 2015. 3: p. 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bhandari V, et al. Molecular landmarks of tumor hypoxia across cancer types. Nat Genet, 2019. 51(2): p. 308–318. [DOI] [PubMed] [Google Scholar]
- 89.Bhandari V, et al. Divergent mutational processes distinguish hypoxic and normoxic tumours. Nat Commun, 2020. 11(1): p. 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Graeber TG, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature, 1996. 379(6560): p. 88–91. [DOI] [PubMed] [Google Scholar]
- 91.Chua MLK, et al. A Prostate Cancer “Nimbosus”: Genomic Instability and SChLAP1 Dysregulation Underpin Aggression of Intraductal and Cribriform Subpathologies. Eur Urol, 2017. 72(5): p. 665–674. [DOI] [PubMed] [Google Scholar]
- 92.Houlahan KE, et al. Molecular Hallmarks of Multiparametric Magnetic Resonance Imaging Visibility in Prostate Cancer. Eur Urol, 2019. 76(1): p. 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ershler WB and Keller ET, Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu Rev Med, 2000. 51: p. 245–70. [DOI] [PubMed] [Google Scholar]
- 94.McBride JA, Carson CC 3rd, and Coward RM, Testosterone deficiency in the aging male. Ther Adv Urol, 2016. 8(1): p. 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Suzuki H and Nakada T, Alteration of collagen biosynthesis and analysis of type I and type III collagens of prostate in young rats following sex hormone treatments. Arch Androl, 1996. 36(3): p. 205–16. [DOI] [PubMed] [Google Scholar]
- 96.Hu WY, et al. Estrogen-initiated transformation of prostate epithelium derived from normal human prostate stem-progenitor cells. Endocrinology, 2011. 152(6): p. 2150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gomes AP, et al. Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature, 2020. 585(7824): p. 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Clement J, et al. The Plasma NAD(+) Metabolome Is Dysregulated in “Normal” Aging. Rejuvenation Res, 2019. 22(2): p. 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Imai S and Guarente L, NAD+ and sirtuins in aging and disease. Trends Cell Biol, 2014. 24(8): p. 464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Elhassan YS, et al. Nicotinamide Riboside Augments the Aged Human Skeletal Muscle NAD(+) Metabolome and Induces Transcriptomic and Anti-inflammatory Signatures. Cell Rep, 2019. 28(7): p. 1717–1728 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Martens CR, et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat Commun, 2018. 9(1): p. 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang Q, et al. Increasing ovarian NAD(+) levels improve mitochondrial functions and reverse ovarian aging. Free Radic Biol Med, 2020. 156: p. 1–10. [DOI] [PubMed] [Google Scholar]
- 103.Fui MN, Dupuis P, and Grossmann M, Lowered testosterone in male obesity: mechanisms, morbidity and management. Asian J Androl, 2014. 16(2): p. 223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yokoyama A, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature, 2019. 565(7739): p. 312–317. [DOI] [PubMed] [Google Scholar]
- 105.Henry GH, et al. A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra. Cell Rep, 2018. 25(12): p. 3530–3542 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dang T and Liou GY, Macrophage Cytokines Enhance Cell Proliferation of Normal Prostate Epithelial Cells through Activation of ERK and Akt. Sci Rep, 2018. 8(1): p. 7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin R, et al. Activation of NF-kappa B signaling promotes growth of prostate cancer cells in bone. PLoS One, 2013. 8(4): p. e60983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kroon P, et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res, 2013. 73(16): p. 5288–98. [DOI] [PubMed] [Google Scholar]
- 109.Yeh DW, et al. Interplay between Inflammation and Stemness in Cancer Cells: The Role of Toll-Like Receptor Signaling. J Immunol Res, 2016. 2016: p. 4368101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vomund S, et al. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int J Mol Sci, 2017. 18(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bruns DR, et al. Nrf2 Signaling and the Slowed Aging Phenotype: Evidence from Long-Lived Models. Oxid Med Cell Longev, 2015. 2015: p. 732596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Suh JH, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A, 2004. 101(10): p. 3381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bose C, et al. Sulforaphane prevents age-associated cardiac and muscular dysfunction through Nrf2 signaling. Aging Cell, 2020. 19(11): p. e13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fontana L, Partridge L, and Longo VD, Extending healthy life span--from yeast to humans. Science, 2010. 328(5976): p. 321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Martin-Montalvo A, et al. Metformin improves healthspan and lifespan in mice. Nat Commun, 2013. 4: p. 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.El-Alfy M, et al. Unique features of the basal cells of human prostate epithelium. Microsc Res Tech, 2000. 51(5): p. 436–46. [DOI] [PubMed] [Google Scholar]
- 117.Artandi SE and DePinho RA, Mice without telomerase: what can they teach us about human cancer? Nat Med, 2000. 6(8): p. 852–5. [DOI] [PubMed] [Google Scholar]
- 118.Ittmann M, et al. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res, 2013. 73(9): p. 2718–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jenkins EO, et al. Age-specific changes in intrinsic breast cancer subtypes: a focus on older women. Oncologist, 2014. 19(10): p. 1076–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ren R, et al. Regulation of Stem Cell Aging by Metabolism and Epigenetics. Cell Metab, 2017. 26(3): p. 460–474. [DOI] [PubMed] [Google Scholar]
- 121.Mansell E, et al. Mitochondrial Potentiation Ameliorates Age-Related Heterogeneity in Hematopoietic Stem Cell Function. Cell Stem Cell, 2021. 28(2): p. 241–256 e6. [DOI] [PubMed] [Google Scholar]
- 122.Morris O, et al. Warburg-like Metabolic Reprogramming in Aging Intestinal Stem Cells Contributes to Tissue Hyperplasia. Cell Rep, 2020. 33(8): p. 108423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Conboy IM, et al. Notch-mediated restoration of regenerative potential to aged muscle. Science, 2003. 302(5650): p. 1575–7. [DOI] [PubMed] [Google Scholar]
- 124.Molofsky AV, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature, 2006. 443(7110): p. 448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]