

Abstract
Summary: A comprehensive genomic analysis of structural variants in multiple myeloma in this issue highlights the key role of these events, involving primarily the immunoglobulin heavy chain locus in disease initiation and the MYC locus in disease progression. However, the current study reveals the large number of genomic hotspots, oncogenes, tumor suppressor genes, and recombination mechanisms that contribute to multiple myeloma heterogeneity.
See related article by Rustad et al., p. 258.
The study by Rustad and colleagues (1) includes samples from 752 newly diagnosed tumors and provides the first comprehensive analysis of structural variants in multiple myeloma. The analysis is based on several datasets: low coverage (median, 4×–8×) paired-end sequences of long whole-genome inserts (median, 852 bp), whole-exome sequences, and also RNA sequencing for 79% of the tumors. Simple structural variants were classified as deletion, tandem duplication, inversion, reciprocal translocation, unbalanced translocation, and insertion with a single templated insert (2). Complex structural variants included chromothripsis, chromoplexy, insertions at a translocation breakpoint, multiple templated inserts, and unspecified complex structural variant. Chromothripsis, which appears to result from a single catastrophic event that includes copy-number gains and losses, was defined by the presence of 10 or more interconnected structural variant breakpoint pairs associated with: (i) clustering of breakpoints, (ii) randomness of DNA fragment joins, and (iii) randomness of DNA fragment order across one or more chromosomes (3). Chromoplexy was defined by interconnected structural variant breakpoints across >2 chromosomes associated with copy-number loss (4). Multiple myeloma tumors had fewer structural variants (median of 16) compared with solid tumors. The authors appropriately suggest that the prevalence of structural variants probably is significantly higher, but the low coverage of long inserts likely misses some structural variants, particularly those that are subclonal. As reported previously, by far the most common loci involved in recurrent structural variant were immunoglobulin (5) and MYC (6, 7), implicated in disease initiation and progression, respectively.
One of the most surprising results from this study was that chromothripsis was present in 24% of multiple myeloma tumors, substantially lower than in most solid tumors, but much higher than in previous studies, which found a prevalence as low as 3% of multiple myeloma tumors. The authors explain that the higher prevalence of chromothripsis in their study includes both the improved resolution of whole-genome sequences and evolving criteria to identify chromothripsis. Chromothripsis has a particularly strong association with biallelic inactivation of TP53 (HR, 6.6). Despite its higher prevalence, chromothripsis continues to be a strong independent negative predictor for both progression-free survival (HR, 1.42) and overall survival (HR, 1.81).
Templated insertions are present in 19% of the multiple myeloma tumors. Previous studies (8) suggest that there are two types of templated insertions: (i) duplicated sequences (median of 100 kb but sometimes >1,000 kb) flanking putative double-stranded breakpoints and (ii) insertions of one or more unrelated chromosomal fragments into a recipient chromosomal site or at the breakpoint junction of a translocation or inversion breakpoint. Figure 1A hypothesizes that a yet to be defined mechanism generates a double-strand break with duplicated sequences flanking the breakpoint; this breakpoint can be repaired to generate a tandem duplication, a translocation or inversion with the duplicated sequences found on both derivative chromosomes, or a templated insertion(s) flanked by the duplicated breakpoint sequences. Both kinds of templated insertions have been found in many kinds of tumors. The strong correlation of both tandem duplications and templated insertions associated with MYC and other sites as reported in the current article provides additional support for the proposed model. Both kinds of duplicated sequences often contain super-enhancers or oncogenes. There also is limited data suggesting that there is a correlation of the presence of both types of templated insertion occurring together (8).
Figure 1.
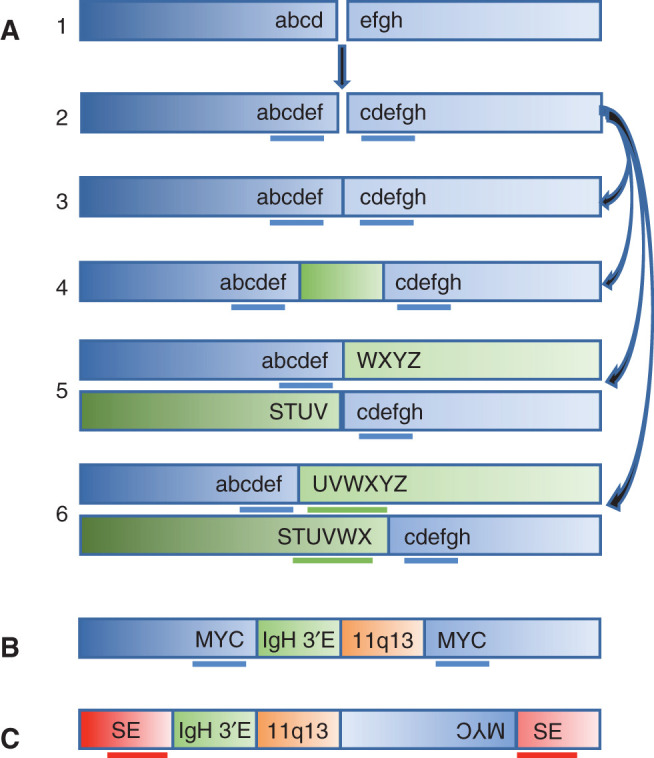
A, Model for possible resolutions of a double-strand break with duplicated sequences. A double strand break (1), with generation of duplicated sequences “cdef” on both sides of the breakpoint (2), can be repaired with generation of tandem duplicated sequence (3), insertion of an unrelated piece of DNA (4), and reciprocal translocation without (5) or with duplication “UVWX” on the partner chromosome (6). B, Insertion of an 11;14 fragment containing 3′ IGH enhancer (3′E) 11q13 breakpoint, but not CCND1 gene, in the KMS12 multiple myeloma cell line. C, Insertion of an 11;14 fragment containing the 3′ IGH enhancer 11q13 breakpoint, but not CCND1 gene, and also a chromosome 8 fragment containing the MYC gene between duplicated sequences on chromosome 4. For B and C, chromosome 8 is blue, chromosome 14 is green, chromosome 11 is orange, and chromosome 4 is red with duplicated sequences underlined and a possible super-enhancer (SE) indicated.
The abstract to the current article states, “Importantly, in 31% of patients, two or more seemingly independent putative driver events were caused by a single structural event.” However, a previous article (8) shows four examples of multiple myeloma cell lines in which a primary t(11;14) translocation provided the template for a secondary templated insertion of an 11;14 sequence containing the 3′ IGH super-enhancer, but not the CCND1 gene. For the KMS12 (Fig. 1B) and MM.M1 (8) cell lines, there was a der14 t(11;14) presumptive primary translocation and insertion of an 11;14 fragment between duplicated chromosome 8 sequences including MYC; for each cell line, precisely the same unique chr11;14 breakpoint was found on the der14 t(11;14) chromosome and the chromosome 8 with the inserted fragment. The MOLP8 cell line was more complex with an 11;14 fragment from the der14 t(11;14) and a chromosome 8 fragment containing MYC inserted between duplicated chromosome 4 sequences (Fig. 1C). For the Karpas620 (8) cell line, a translocation between der14 t(11;14) and chromosome 8, with duplicated sequences flanking both breakpoints, resulted in translocation products containing 3′ IGH super-enhancer and MYC sequences on both translocation products. We also note that others have used an IGH capture procedure that identified 8;11 breakpoints in two primary multiple myeloma tumors with t(11;14) translocations (9). The current article shows that templated sequences often are associated with MYC or CCND1 sequences, but not NSD2 sequences. Therefore, it would be interesting to know whether the structures of templated sequences associated with the CCND1 locus in the current article are similar to the four multiple myeloma cell lines described above (8).
Among the 752 multiple myeloma tumors, 31 (5%) had a translocation with a noncanonical immunoglobulin partner, including 19 IGH, 11 IGL, and one IGK. The authors suggest that 15 of the 19 IGH translocations are in tumors that lack a canonical IGH translocation, but that the noncanonical partner might be an initiating event. In support of this speculation, they state that the seven IgH translocations involving MAP3K14 have IGH breakpoints similar to canonical IGH translocations. This is an interesting but not totally convincing suggestion. However, if this is correct, then other mechanisms that increase MAP3K14 activity, for example, deletions of BIRC2 and BIRC3 or TRAF3 might also be initiating events.
Recurrent intrachromosomal copy-number abnormalities (CNA) represent an important oncogenic event in multiple myeloma, and they reported that 83% of CNAs are attributed to a specific structural variant. The authors conclude that at least one driver CNA is present in 47% of all chromothripsis events, 42% of chromoplexy events, and 28% of all templated insertions involving two or more chromosomes. As expected, chromothripsis was associated with copy-number gain and copy-number loss, chromoplexy with copy-number loss, and templated insertions with copy-number gains. Excluding structural events involving immunoglobulin loci, structural variants that affected copy number had the strongest average effect on expression, with an increase or decrease in expression for each gain or loss of a gene copy.
The authors identified 68 recurrent structural variant hotspots not involving immunoglobulin loci or known fragile sites. Hotspots were defined using a piecewise constant algorithm comparing local structural variant breakpoint density with an empirical background model. Gain-of-function hotspots (n = 49), defined by the presence of copy-number gains and translocation-type events, were associated with templated insertions and tandem duplication. These included well-defined multiple myeloma oncogenes (MYC, CCND1, NSD2/FGFR3, IRF4, and MAP3K14) and novel putative driver genes that are targets of therapy (TNFRSF17, SLAMF7, and MCL1). In addition, several gain-of-function hotspots lacking an oncogene were located near enhancers, many of which have been reported to be hijacked by MYC (FAM46C, FBXW7, FOXO3, TXNDC5, NSMCE2, and FCHSD2). They also identified 19 loss-of-function hotspots, defined by copy-number loss, typically resulting from simple deletion of tumor suppressor genes (CDKN2C, SP3, SP140, RPL5, CDKN2A, CYLD, RB1, CDKN1B, MAX, TRAF3, TP53, and KDM6A).
The current study highlights the importance of whole-genome sequencing for the detection and classification of structural variants in multiple myeloma. As the authors note, future studies should use greater sequencing coverage to identify subclonal mutations and follow their course over time in patients that have not yet progressed to multiple myeloma, and in those in stable remission, at risk for relapse. When whole-genome sequencing structural variant analysis is combined with RNA-sequencing expression analysis, it becomes evident that structural variants are major contributors to dysregulated gene expression in multiple myeloma and a critical piece of the puzzle to understand disease pathogenesis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported by grant numbers CA186781, CA195688, and CA224018 (to P.L. Bergsagel) and the Intramural Research Program of the NIH, NCI and Center for Cancer Research (to W.M. Kuehl).
Footnotes
Blood Cancer Discov 2020;1:221–3
References
- 1.Rustad EH, Yellapantula VD, Glodzik D, Maclachlan KH, Diamond B, Boyle EM, et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov 2020;1:258–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020;578:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell 2013;152:1226–36. [DOI] [PubMed] [Google Scholar]
- 4.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell 2013;153:666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL, Kuehl WM.Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc Natl Acad Sci U S A 1996;93:13931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A 2000;97:228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014;28:1725–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demchenko Y, Roschke A, Chen WD, Asmann Y, Bergsagel PL, Kuehl WM. Frequent occurrence of large duplications at reciprocal genomic rearrangement breakpoints in multiple myeloma and other tumors. Nucleic Acids Res 2016;44:8189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker BA, Wardell CP, Johnson DC, Kaiser MF, Begum DB, Dahir NB, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood 2013;121:3413–9. [DOI] [PubMed] [Google Scholar]