

Abstract
Phospholipase C gamma1(PLCγ1) is a member of the PLC family that functions as signal transducer by hydrolyzing membrane lipid to generate second messengers. The unique protein structure of PLCγ1 confers a critical role as a direct effector of VEGFR2 and signaling mediated by other receptor tyrosine kinases. The distinct vascular phenotypes in PLCγ1-deficient animal models and the gain-of-function mutations of PLCγ1 found in human endothelial cancers point to a major physiological role of PLCγ1 in the endothelial system. In this review, we discuss aspects of physiological and molecular function centering around PLCγ1 in the context of endothelial cells and provide a perspective for future investigation.
Introduction
Phospholipase C (PLC) isozymes are a family of enzymes that share a common function of hydrolyzing membrane lipid phosphatidylinositol-4,5-biphosphate (PIP2) to generate inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG), two second messengers commonly involved in membrane receptor signal transduction(1–5). IP3 is released from the plasma membrane and binds IP3 receptors on the endoplasmic reticulum (ER), triggering rapid Ca2+ entry into the cytosol from the ER(2–4, 6). The other PIP2-derived second messenger, DAG, is retained in the plasma membrane, where it can activate protein kinase C (PKC)-dependent signaling cascades (2–4).
The thirteen isozymes comprising the PLC family in mammalian cells are categorized into six subgroups based on structural homology. These are PLCβ (β1, β2, β3, β4), PLCγ (γ1, γ2), PLCδ (δ1, δ3, δ4), PLCε, PLCζ, and PLCη (η1, η2)(3, 4). The unique functional repertoire of each PLC subgroup is determined by distinct protein functional domains and tissue distribution patterns. For instance, the two tandem Src homology 2 (SH2) domains that occur exclusively in PLCγ1 and PLCγ2 confer the unique capability of interacting with receptor tyrosine kinases (RTKs)(3, 4, 7) (Fig.1). The phenotypes in animal models of PLCγ1 deficiency, along with the human diseases associated with aberrant PLCγ1 activation, highlight the major physiological functions of PLCγ1 in endothelial and hematopoietic-immune systems (Table 1). The current review aims to focus on the role of PLCγ1-mediated signaling in endothelial cells. For details of other PLC isozymes and the functions of PLCγ1 in other systems, the reader is referred to recent reviews(3, 4, 7–9).
Fig. 1. Human PLCγ1 protein structure and signal transduction.
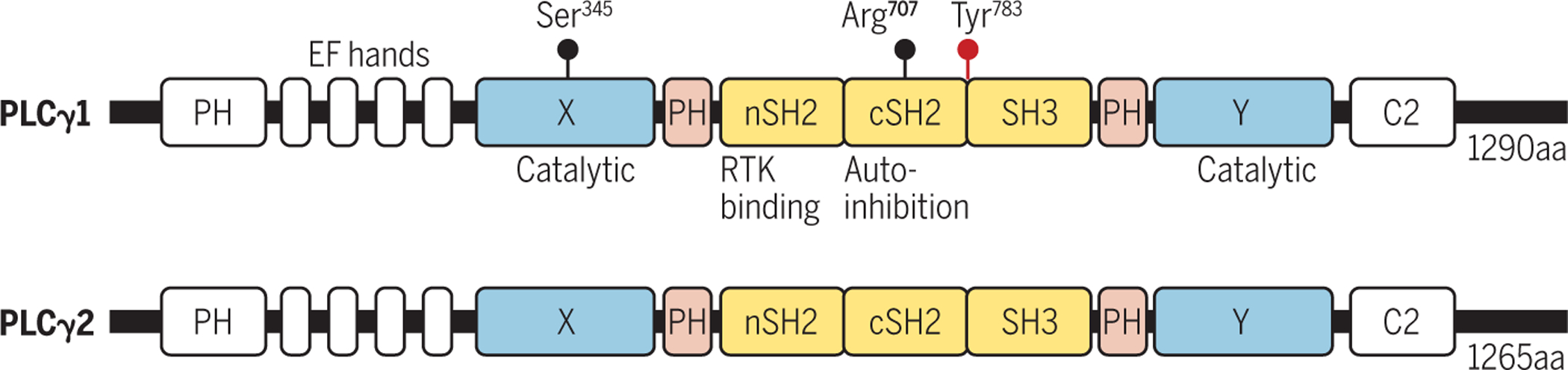
Common domains shared by all PLC isozymes include PH, EF hands, catalytic domains X and Y, and C2. The insertion between X and Y domains, consisting of nSH2, cSH2, SH3 and two split PH domains, is exclusive to PLCγ1/2 and not present in other PLC isozymes. PLCγ1 uses the nSH2 domain to interact with RTKs. The cSH2 domain exerts autoinhibitory function on X/Y domains under basal conditions. Arg707 and Ser345 are mutated in human angiosarcomas and T cell lymphomas, respectively.
Table 1:
PLCγ1 in animal models and human diseases
| Genotype | Functional impact | Disease or phenotype | Reference |
|---|---|---|---|
| PLCγ1 R707Q (cSH2 domain) | Loss of autoinhibitory function, constitutive activation. | A prevalence of 9% in angiosarcomas from various organs. | (28), (29), (46), (47) |
| PLCγ1 S345F (catalytic domain X) | Constitutive activation. | T-cell lymphoma. 19% prevalence in cutaneous T-cell lymphoma | (30), (49) |
| PLCγ1 −/− mice | Global loss of PLCγ1 | Embryonic lethality between E9.5–10.5; defective hematopoiesis and likely defective angiogenesis. | (32), (33), (37) |
| Chimeric PLCγ1 (−/− and +/+) mice | Global chimeras | Impaired hematopoiesis, multicystic kidneys. | (37) |
| Zebrafish PLCγ1 y10 | Loss-of-function mutant | Defective angiogenesis and segmental vessel formation. Reduced expression of arterial markers in the dorsal aorta. The dorsal aorta is formed with compromised lumen. | (38) |
| Flk1 Y1173F knock-in mouse | Loss of VERGFR2 binding and activation of PLCγ1 | Embryonic lethality at E8.5–9.5; defective vasculogenesis and hematopoiesis | (51) |
| Fgfr1 Y776F knock-in mouse | Loss of FGFR1 binding and activation of PLCγ1 | Minor defects in posterior thoracic vertebrae | (104) |
| PLCγ1 T-cell specific KO (CD4-cre) | Loss of T-cells PLCγ1expression | Defective T cell selection, inflammatory and autoimmune symptoms | (44) |
| PLCγ1 f/f; PLCg2−/−; Mx-cre mice, | PLCγ1/γ2 double KO in B cells | Defective B cell development | (45) |
Protein structure of PLCγ1
Several structural domains are commonly shared by all PLC family isozymes (Fig.1). The X and Y domains, which fold together and resemble a triose phosphate isomerase (TIM) α/β barrel in their tertiary structure, constitute the catalytic region in all PLCs(3, 7, 10). The catalytic site responsible for PIP2 hydrolysis is highly conserved in all PLCs (10), as is the pleckstrin homology (PH) domain shared by most PLC isozymes with the exception of PLCε and PLCζ (3, 4, 7). The PH domain of PLCγ1 is thought to be the binding site for phosphatidylinositol-3,4,5-trisphosphate (PIP3) (11–13). Finally, EF-hand and C2 domains are involved in Ca2+ binding.
The insertion domains between the catalytic domains X and Y, composed of nSH2, cSH2 and SH3 domains flanked by the two split halves of the PH domain (spPH), which exists exclusively in PLCγ1 and PLCγ2 but not in other PLC isozymes, are essential for responsiveness to growth factors (3, 4, 7, 14) (Fig.1). The nSH2 domain mediates interactions with RTK growth factor receptors, whereas the cSH2 domain serves an auto-inhibitory role by masking the catalytic active site in the X and Y domains(8, 14–17). The activation of PLCγ1 by an RTK is driven by a phosphorylation-induced conformational change(8, 15). Binding of a growth factor ligand to an RTK triggers its dimerization and activation resulting in autophosphorylation of the cytoplasmic tyrosine kinase domains of the RTK. PLCγ is then recruited to a phosphotyrosine in the RTK and becomes phosphorylated at Tyr783 between cSH2 and SH3 domains(17–20). This triggers a cascade of conformational changes resulting in the auto-inhibitory cSH2 domain moving away from the catalytic X/Y domains to become associated with phosphorylated Tyr783, thereby unmasking the catalytic active sites in the X/Y domains and allowing substrate PIP2 binding and subsequent hydrolysis (18, 19, 21)(Fig.1–2). Mutations in cSH2 tyrosine binding sites in PLCγ can disable auto-inhibition and consequently result in a state of constitutive activation(17, 22).
Fig. 2. PLCγ1 in regulation of VEGFR2 and FGFR1 signaling.
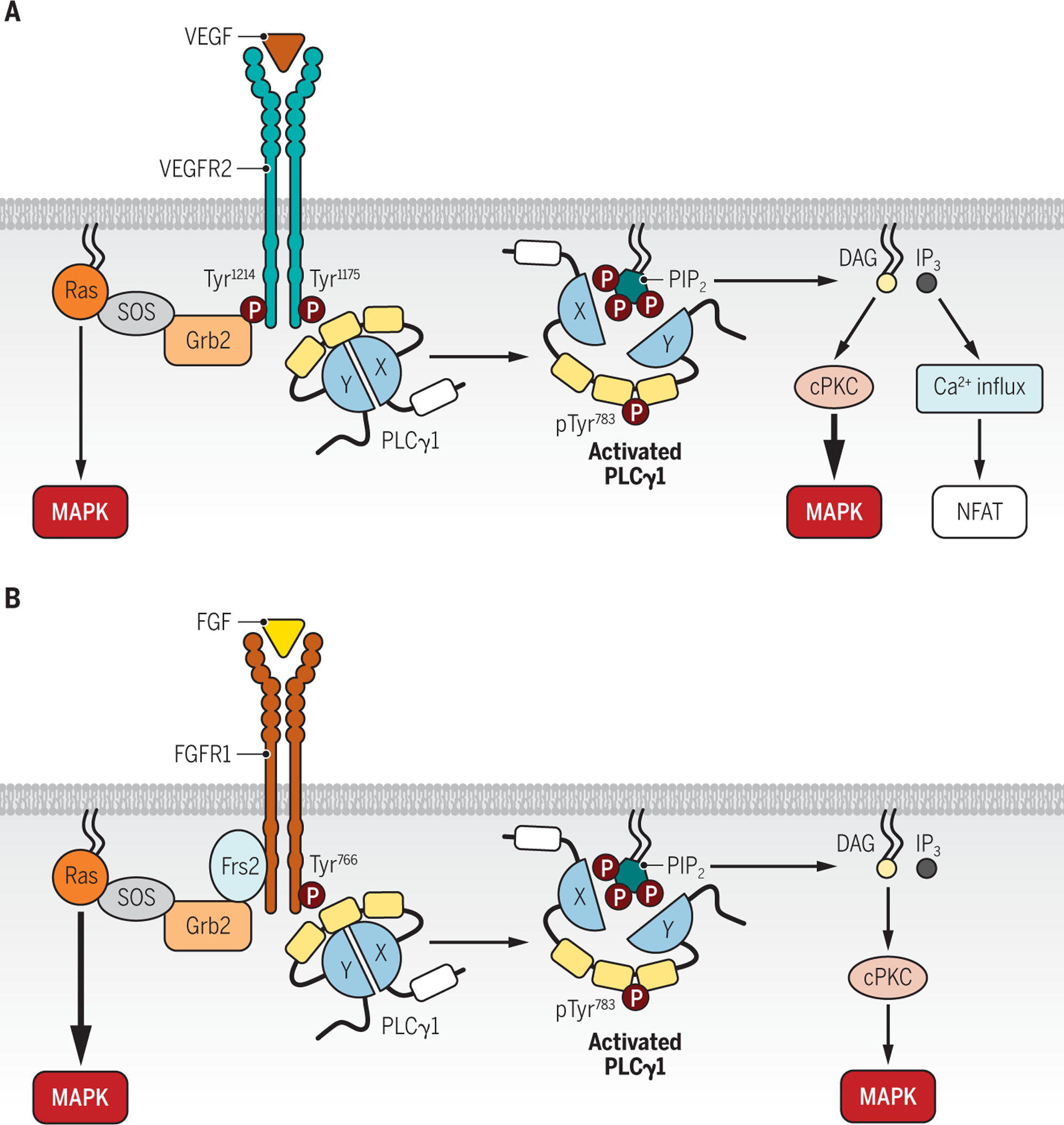
A. PLCγ1 is autoinhibited under basal conditions. VEGF induces the recruitment of PLCγ1 to Tyr1175 in VEGFR2, which activates PLCγ1 by phosphorylating Tyr783, thereby initiating a conformational change to unmask the catalytic X and Y domains for PIP2 access. PIP2 is hydrolyzed into the second messengers DAG and IP3, which trigger downstream PKC-MAPK and Ca2+-NFAT pathways, respectively. pTyr1212-VEGFR2-Grb2-Ras could play an additive role to pTyr1175-VEGFR2-PLCγ1 to activate MAPK signaling.
B. FGFR1 primarily utilizes Frs2-Grb2-SOS-Ras, but not PLCγ1, to activate MAPK pathway. In a similar manner to the activation of PLCγ1 by VEGFR2, FGFR1 phosphorylated at Tyr766 recruits and activates PLCγ1 by phosphorylating Tyr783 in PLCγ1, leading to a conformational change that exposes the catalytic X/Y domains for PIP2. The magnitude and downstream signaling of FGF-induced PLCγ1 activation are likely cell type-specific and context-dependent.
The PLC SH3 domain can function as a GEF by interacting with GTPases for dynamin-1 in the cytoplasm(23) or PIKE (phosphatidylinositol-3-OH kinase (PI3K) enhancer) within the nucleus(24), though such mechanisms could depend on specific ligand and/or cell type. The spPH domains interacts with the cSH2 domain to enhance its autoinhibitory function(21, 25). Surprisingly, the spPH domains of PLCγ2, but not PLCγ1, accounts for GTPase Rac-mediated activation, but the reason for such selective interaction is unknown(26). In addition, a study reports that the C-terminal half of spPH directly interacts with the ion channel TRPC3 to induce Ca2+ entry(27).
Several gain-of-function point mutations in PLCγ1 have been discovered in human endothelial and hematopoietic cancers(9) (Table 1). The angiosarcoma-associated mutation R707Q in the cSH2 domain disrupts the stability of its autoinhibitory function(28, 29) (Fig.1, table1). S345F in the catalytic domain X is a mutation found in T-cell lymphoma(17, 30) (Fig.1, table1). In both cases, these mutations render PLCγ1 constitutively active as a causative association with tumorigenesis.
PLCγ1 in mouse and zebrafish models
Though structurally highly homologous isozymes, PLCγ1 and PLCγ2, are encoded by distinct genomic regions(31). The PLCγ1 gene maps to chromosomes 20q12 in human and 2 H2 in mouse genomes, whereas the PLCγ2 gene is located in the human chromosome 16q23.3 and mouse chromosome 8 E1 (NCBI gene database). Studies using LacZ reporter incorporated in the endogenous PLCγ1 gene show ubiquitous expression in all embryonic tissues, with a particularly high level in the endothelium of the dorsal aorta, the heart, the gut, and the yolk sac(32, 33). The overall expression pattern of PLCγ2 has not been satisfactorily examined, despite some evidence of expression reported in hematopoietic lineages, particularly B cells, and central nervous system(34–36).
Global deletion of PLCγ1 in mouse embryos leads to developmental retardation and death between embryonic days (E) 9.5 and 10.5 (32, 33, 37). Critically, there is a lack of erythrocyte progenitors in the yolk sac and no detectable blood island (32, 33, 37). Although a general reduction of endothelial marker expression has been reported in PLCγ1 null embryo at E9.5 (33) (Table 1 and 2), histological assessment demonstrates generally normal anatomy of the heart and the paired dorsal aortae despite smaller size of the embryo body (32) (Table2). In particular, the dorsal aortae are formed with conspicuous lumens as visualized in transverse sections (32) (Table2).
Table 2:
VEGFR2-MAPK pathway mutants
| Model | Species | Survival | Cardiovascular phenotype | Reference |
|---|---|---|---|---|
| Flk1 −/− | Mouse | Embryonic lethality between E8.5–9.5 | Defective vasculogenesis and hematopoiesis: no blood vessel formation; absence of yolk-sac blood island. | (50), (51) |
| Flk1 -/Y1173F | Mouse | Embryonic lethality between E8.5–9.5 | Defective vasculogenesis and hematopoiesis; no blood vessel formation; absence of yolk-sac blood island. | (51) |
| Flk1-/Y1212F and Flk1Y1212F/Y1212F | Mouse | Viable and fertile | Neonatal retina angiogenesis is normal in C57Bl/6 background but has mild defect in FVB background | (51), (53) |
| Flk1 f/f ; Cdh5cre ERT2 | Mouse | N/A | Postnatally induced deletion of Flk1 leads to compromised angiogenesis in neonatal retina. | (60), (61) |
| PLCγ1 −/− | Mouse | Embryonic lethality between E9.5–10.5 | Small embryo body. Defective hematopoiesis and possible defective angiogenesis. The paired dorsal aortae form and have conspicuous lumens. | Fig. 3D in (32); Figs. 4A and 5A in (33), (37) |
| PLCγ1Y10 mutation | Zebrafish | N/A | Defective angiogenesis and segmental vessel formation. Reduced expression of arterial markers in the dorsal aorta. The cardinal vein is formed normally; the dorsal aorta is formed with smaller-than-normal lumen. | Fig. 1 in (38) |
| PKCβ −/− | Mouse | Viable, fertile and normal | No cardiovascular anomaly. Impaired immunity in adult. |
(144) |
| PKCα −/− | Mouse | Viable, fertile and normal | No cardiovascular anomaly. | (143) |
| ERK1 −/− | Mouse | Viable, fertile and normal | No cardiovascular anomaly. | (57) |
| ERK2 −/− | Mouse | Embryonic lethality by E11.5 | Placental defect. | (58) |
| ERK1 −/− ; ERK2 f/f ; Tie2cre | Mouse | Embryonic lethality by E10.5 | Defective angiogenesis | (56) |
Fish embryos bearing a loss-of-function (LOF) mutation y10 that affects the PH domain of PLCγ1, develop various vascular abnormalities (38, 39) including a compromised development of segmental vessels from the cardinal vein (38, 39). These mutants exhibit a reduction in mRNA expression of the arterial markers EphB2 and Notch5 in the dorsal aorta and are nonresponsive to ectopic VEGFA stimulation (38). However, the dorsal aorta and the cardinal vein in PLCγ1-deficient fish are morphologically visible (38) (Table1 and 2). Although the dorsal aorta appears smaller in the mutant than wild type fish (38), morphology and size of the cardinal vein are normal (38, 39) (Table1 and 2).
In vertebrates, morphogenesis of the dorsal aortae and the cardinal veins, the first blood vessels formed during embryogenesis, relies completely on de novo vasculogenesis from mesodermal progenitors (40–43). Although the original reports (32, 33) suggested that PLCγ1 deletion results in defective vasculogenesis, the presence of lumenized dorsal aorta and cardinal vein in both mouse and fish embryos indicates that vasculogenesis is completed in the absence of PLCγ1. Moreover, mouse models with a true vasculogenic defect and consequently no blood vasculature of any kind (such as Flk1−/− mice and mice expressing the nonphosphorylatable VEGFR2 Y1173F mutant) typically cannot survive beyond E9.5 when primordial blood circulation is supposed to be established. The lethality of PLCγ1 null embryos at a later time point (E9.5–10.5) demonstrates that they die after the completion of vasculogenesis. It is most likely that PLCγ1 null embryos die due to defective angiogenesis that, by E9.5, leads to generalized developmental retardation. In agreement with this hypothesis, the observation of reduced endothelial markers in PLCγ1-null embryo body at E9.5 is likely the consequence of a decrease in endothelial cells population size due to defective angiogenesis.
Together, these observations implicate the critical roles of PLCγ1 primarily in VEGF-driven angiogenesis as well as hematopoiesis (Tables 1 and 2). The early embryonic mortality following a global PLCγ1 knockout prevented studies of its function in later stages of development and adult animals. Investigations employing a conditional inducible knockout of PLCγ1 indicate its critical functions in T and B cell development and immunity(44, 45). To date, no studies of endothelial-specific inducible PLCγ1 knockouts have been reported.
PLCγ1 mutations in endothelial and hematopoietic malignancies
Although PLCγ1-deficient animals primarily feature vascular and hematopoietic abnormalities, gain-of-function (GOF) PLCγ1 mutations are found in human vascular and hematopoietic malignancies (Table 1). Angiosarcomas are malignant tumors of endothelial origin that arise due to uncontrolled endothelial growth. PLCγ1 activating mutations have been identified in multiple angiosarcomas, including Kaposi’s sarcoma, as well as hepatic and cardiac angiosarcomas (28, 29, 46, 47). The most common is a missense mutation R707Q in the highly conserved cSH2 domain that has a reported prevalence of 9% in angiosarcomas (28, 46). It impairs the auto-inhibitory function of the cSH2 domain, resulting in a constitutive activation of PLCγ1. Ectopic expression of R707Q PLCγ1 in HUVECs activates MAPK and NFAT signaling independently of VEGF stimulation and phosphorylation at Tyr783 (the activation site of PLCγ1)(46). Thus transformed, HUVECs display increased invasiveness and migration capability similar to angiosarcoma cells(46). A patient with hepatic angiosarcoma with wild-type PLCγ1 in the primary tumor developed a PLCγ1R707Q mutation in metastatic foci during treatment with the RTK inhibitor sunitinib (48). This raises the possibility that PLCγ1 activation due to R707Q mutation confers enhanced migratory ability on tumor cells leading to metastatic spread (48).
In T lymphocytes, PLCγ1 is an important effector of T cell receptor (TCR) signaling(44). The activating mutation S345F in the catalytic domain X of PLCγ1 is prevalent in T-cell lymphomas (30, 49). Mechanistically, calcineurin-NFAT signaling is increased in PLCγ1-S345F cutaneous T-cell lymphoma cells(30), similar to the hyperactive signaling caused by the R707Q mutation in PLCγ1 in angiosarcomas(46).
PLCγ1 and vascular endothelial growth factor receptors (VEGFRs)
VEGFR2-ERK activation: PLCγ1 or Ras?
VEGF signaling is a key feature of endothelial biology. VEGF receptor 2 (VEGFR2, encoded by Flk1) is expressed as early as E7.0 in the yolk sac blood islands and by E8.5, in the endocardium, dorsal aorta, capillaries and the yolk sac vasculature(50, 51). VEGFR2 is a typical RTK that is activated by dimerization, leading to autophosphorylation of five key cytoplasmic tyrosine sites. These are (using the human sequence nomenclature) Tyr951 (involved in permeability regulation), Tyr1054/1059 (the active kinase site), Tyr1175 (ERK1/2 activation) multiple and Tyr1214 (multiple signaling pathways)(51–54).
A global Flk1 deletion in mice leads to a complete loss of vascular morphogenesis both in the yolk sac and in the embryo body as well as reduced numbers of CD34+ hematopoietic progenitors (50) (Table2). Flk1 null embryos die between E8.5–9.5 due to defective vasculogenesis and hematopoiesis (50). Mice embryos harboring a knock-in mutant Flk1 Y1173F allele (analogous to the human Flk1 Y1175F mutation) that disrupts activation of MAPK signaling (55) phenocopy almost exactly vasculogenic and hematopoietic defects seen in Flk1 null mice and die during the same time window, emphasizing the critical role of VEGFR2-MAPK signaling pathway for vascular development(51)(Table2). In contrast, mice carrying Flk1 Y1212F (51) or Y949F (54) alleles are viable and fertile, suggesting that signals from these two tyrosine phosphorylation sites are dispensable for embryonic development.
VEGFR2 Tyr1175 (Tyr1173 in mice) is a PLCγ1 docking site and is thought to be the principal means of VEGF-dependent ERK activation(55). Although de novo vasculogenesis is defective in both Flk1-null and Flk1 Y1173F mouse embryos that demonstrate compromised formation of all blood vessels including the dorsal aortae(50, 51), it is almost normal in PLCγ1-null embryos, which possess a pair of dorsal aortae with a well-developed lumen (32) (Table2). The dorsal aortae and the cardinal veins in PLCγ1-deficient zebrafish are also morphologically visible, indicating completion of vasculogenesis (32, 38) (Table 2). Such a crucial difference in de novo vasculogenesis suggests that PLCγ1 is likely not the exclusive effector for VEGFR2-Tyr1173-initiated signaling and that there is another non-PLCγ1-dependent mechanism that regulates vasculogenesis.
Embryos with Tie2-Cre-mediated endothelial deletion of ERK2 in the context of ERK1−/− (combined deletion of ERK1 and ERK2 in the endothelium) are alive at E9.5 but die by E10.5 due to defective angiogenesis (56). However, given that Tie2-Cre activation occurs in endothelial cells but not endothelial progenitors, this model does not provide any information about the role of ERK1/2 in vasculogenesis. Mice with global deletion of ERK1 are normal and fertile (57), whereas global deletion of ERK2 leads to embryonic lethality by E11.5 due to severe placental defects (58). Although no effect on vascular development has been reported, the survival of ERK2-null until E10.5 suggests that a primordial cardiovascular system between E7.5–9.5 can be established independently of ERK2. In light of the compensatory redundancy of ERK1 in ERK2-null embryo(59), a combined knockout model in mesodermal progenitors is necessary to answer the role of ERK1/2 in vasculogenesis.
It must be pointed out that observations in Flk1-null and Flk1 Y1173F embryos cannot be used to infer the presence of defective angiogenesis, because angiogenesis conceptually refers to the expansion of a capillary bed and thus cannot be tested by Flk1 deficiency or Flk1 Y1173F mutation which results in complete absence of vasculature. However, inducible endothelial-specific deletion of VEGFR2 results in a near complete compromise in angiogenesis in neonatal mouse retina (60, 61). In line with these observations, defective angiogenesis is a major phenotype in zebrafish and likely, mouse PLCγ1-deficient embryos and mouse embryos with endothelial deletion of ERK1/2. Collectively, these studies suggest that PLCγ1 is the primary signal transducer of VEGFR2-dependent activation of MAPK signaling and regulation of angiogenesis (Table 2).
The realization of the critical role of PLCγ in VEGFR2-dependent activation of MAPK signaling was a surprise because for RTKs this function is normally served by the Ras-Raf-MAPK axis. Indeed, virtually all other RTKs, including receptors for FGF(62), EGF(63), PDGF(64) and NGF(65), utilize the Ras-Raf1 cascade. For these receptors, RTK activation leads to Ras phosphorylation that then sets off the RAF-MEK-ERK phosphorylation cascade. However, in rat liver sinusoidal endothelial cells, VEGF does not induce substantial activation of Ras and a dominant-negative-Ras construct does not inhibit VEGF-induced MAPK signaling (66). This and similar results prompted a search for the alternative means of ERK activation by VEGFR2 that naturally focused on PLCγ1.
VEGF induces the recruitment and activation of PLCγ1 by VEGFR2 through binding of the cSH2 domain of PLCγ1 to the phosphorylated Tyr1175 residue of VEGFR2 (20, 55, 66–68). Tyr1175 is located in the C-terminal tail of VEGFR2, in the immediate proximity of the IVL consensus motif (Y1175IVL) which binds to SH2-containing proteins, such as PLCγ1 and Shb, both of which have been reported to bind to the same site, Tyr1175 (69, 70). Antibodies targeting phospho-Tyr1175 disrupt the VEGF-induced association of VEGFR2 and PLCγ1(55). The Y1175F mutant inhibits VEGFA-dependent MAPK signaling and activation of PLCγ1 (55). Phosphorylation of PLCγ1 is not detected in endothelial cells expressing the recruitment-deficient mutant VEGFR2 Y1173F or the kinase-dead mutant VEGFR2 K866R(20). Co-expression of both VEGFR2 mutants can restore the activation of PLCγ1 upon VEGF stimulation, possibly due to dimerization and functional compensation of both mutant VEGFR2 monomers(20). Together, these studies establish the existence of a VEGFR2 Tyr1175-PLCγ1-MAPK signaling axis that is critical to VEGF signaling and development of the vascular system. This model accounts for the phenotypic similarities in angiogenesis in VEGFR2- and PLCγ1-deficient animals and, consequently, has been broadly accepted in the field(52, 71, 72).
However, there are a number of studies that contradict the VEGFR2-PLCγ-MAPK consensus. Thus, several groups have reported that VEGF treatment in endothelial cells induces RAS activation as assessed by RAS-GTP pulldowns(73–75). As noted above, in some studies dominant-negative Ras has minimal effect on VEGF-induced activation of ERK1/2 in HUVECs(73, 74) and HAECs(76), but it abolishes VEGF-ERK signaling in HMVECs(77). Yet another study reported that dominant negative Ras completely blocks VEGF-ERK signaling HUVECs and inhibits HUVEC proliferation, branching and tubulogenesis (75). Finally, in the neonatal retina vasculature, p120RasGAP, an endogenous Ras inhibitor, is generally expressed in inactive endothelial cells in established vessels but is only present in a small number of endothelial cells in the angiogenic front(78). This spatial expression pattern is opposite to the distribution of phosphorylated ERK, which is detected in endothelial cells of the angiogenic front, where VEGF signaling is active, but not in fully formed vessels distal from the front (78). The association of Ras inhibition and downregulation of ERK activation suggests that Ras indeed plays a role in mediating VEGF-ERK signaling in the angiogenic front of the retinal vasculature.
If Ras is indeed involved, it may utilize a VREGFR2 site different than Tyr1175. Mass spectrometry and proximal ligation assays have identified VEGFR2 Tyr1212 as the binding site for Grb2(53), a putative effector required for Ras activation by RTKs. The Y1212F mutation in VEGFR2 partially compromises VEGF-induced ERK1/2 phosphorylation and nuclear accumulation in primary mouse lung endothelial cells (53), but exerts only a minimal effect on embryonic(51) and postnatal vascular development(53). Gene Set Enrichment Analysis (GSEA) detects substantial downregulation of a set of genes in K-Ras signaling in endothelial cells expressing VEGFR2 Y1212F, suggesting a role of Ras in mediating signaling downstream of phosphorylated Tyr1212 (53). Together, this evidence suggests that Ras plays a role in mediating VEGFR2 Tyr1212-Grb2-MAPK signaling pathway, thus bringing a closure to PLCγ1 versus Ras debate in mediating VEGFR2-MAPK signaling (Fig.2A).
To summarize, utilization of PLCγ1 instead of Ras as the primary effector responsible for ERK activation is a unique feature of VEGFR2 signaling. The phenotypes of VEGFR2 and PLCγ1 deficient animal models fall into the same angiogenic and hematopoietic categories in general, underscoring PLCγ1 as the primary VEGFR2 effector in development. The formation of primordial vasculature in PLCγ1-deficient mouse and fish models suggest the existence of additional effector(s) mediating VEGFR2 signaling during vasculogenesis. Like other RTKs, VEGFR2 signals through Grb2-SOS-Ras to activate MAPK. However, given the lack of embryonic phenotype(51) and the mild defect in postnatal angiogenesis(53) in VEGFR2 Y1212F mice, the role of Grb2-SOS-Ras is likely additive and complementary to PLCγ1-PKC in mediating VEGFR2-MAPK signaling in vascular development (Fig.2A).
Uncertain role of PLCγ1 in VEGFR3 and VEGFR1signaling
The VEGF receptor VEGFR3 (encoded by Flt4) is indispensable for lymphatic development(79), but its distribution and function is not restricted to the lymphatic system. VEGFR3 is also detected in the endothelium of the dorsal aorta and the yolk sac blood islands as early as E9.0(80). VEGFR3-null mouse embryos do not survive beyond E12 possibly due to defective vascular remodeling(80) but unlike embryos lacking VEGFR2, vasculogenesis and angiogenesis in VEGFR3-null mice embryos is generally normal(22). In both postnatal and adult blood vasculature, VEGFR3 is upregulated in angiogenic endothelial cells under physiological and pathological circumstances and tends to be silenced in mature, quiescent, vessels(81), (82), (83, 84).
It is not clear whether PLCγ1 interacts with the kinase domain of activated VEGFR3. VEGFR3 can heterodimerize with VEGFR2 in lymphatic and blood Ecs(85–88). In lymphatic endothelial cells, VEGF-C but not VEGF-A induces formation of a VEGFR2/VEGFR3 complex (88). VEGF-C stimulates activation and phosphorylation of PLCγ1 in lymphatic endothelial cells, but at a lower level than VEGF-A(89). Because VEGF-C activates both VEGFR2 and VEGFR3, it is possible that the observed low level PLCγ1 activation in lymphatic endothelial cells due to partially activated VEGFR2. Lymphatic development in mice with Lyve1 Cre-mediated lymphatic deletion of VEGFR2 is generally intact and these mice show no structural or functional abnormalities, although the lymphatic plexus in the adult ear skin exhibits lower vascular density than that in wild-type mice (90). Silencing VEGFR2 in lymphatic endothelial cells does not substantially alter VEGF-C-induced ERK and AKT activation(88), suggesting that, in response to VEGF-C, VEGFR3 can signal independently of VEGFR2. VEGF-C can activate PLCγ1 in HEK 293T cells expressing VEGFR2, but not in cells expressing only VEGFR3(91). Collectively, it is possible that VEGFR3 activates ERK in a signaling pathway independently of PLCγ1, a hypothesis that requires further investigation.
VEGFR1 (encoded by Flt1) is commonly considered as a decoy receptor given its much higher affinity for VEGF-A than VEGFR2 and the lack of clear intracellular signaling response. It is thought to sequester VEGF-A, thereby controlling its access to VEGFR2 (92). Flt1-null embryos die at E8.5–9.0 due to vascular overgrowth and disorganization(93). Mice expressing Flt1 lacking the intracellular domain are viable and normal, suggesting that the major physiological function of VEGFR1 is conferred by its extracellular domain and that the intracellular domain is dispensable(94). In vitro studies indicate that several tyrosine phosphorylation sites in the cytoplasmic domain of VEGFR1 are associated with effector molecules, including PLCγ1, NCK, GRB2, and SHP2. PLCγ1 can bind to phosphorylated Tyr1169, Tyr1213, and Tyr1333 in VEGFR1 and activate PKC/MAPK signaling(92, 95), and mutation of Tyr1169 suppresses the binding of PLCγ1(92). The precise function of PLCγ1 as a signal transducer of VEGFR1 requires further study.
Regulation of other RTKs and non-RTK receptor signaling by PLCγ1
In addition to VEGFRs, endothelial cells rely on other RTKs that also signal through PLCγ1, including FGFR(96, 97), EGFR(98, 99), PDGFR(100, 101) and TrkB(102, 103). The common feature of these non-VEGFR RTKs is the use of Ras as the primary transducer to activate MAPK signaling. Due to the limited number of studies conducted in the endothelial system, this section will cover PLCγ1 biology from various cell types using fibroblast growth factor receptor (FGFR) signaling as an example.
PLCγ1 in FGFR signaling
FGFRs have many features in common with other RTKs. Ligand binding induces receptor dimerization and subsequent autophosphorylation of multiple tyrosine sites in the intracellular domain that induces kinase activity(96, 97). For FGFR1, intracellular signal transduction is mediated by direct interaction with several cytosolic effector proteins, including the scaffold proteins Frs2α, Crkl, Grb2, and Grb14; PLCγ1; and the Stat family of transcription factors (96, 104). Frs2a, a docking protein anchored to the plasma membrane and constitutively associated with the receptor, is thought to be the primary platform for effector recruitment to modulate FGFR1 signaling(105, 106). Upon ligand binding and activation, the kinase domain of FGFR1 phosphorylates tyrosine residues in Frs2α. Phosphorylated Frs2α in turn recruits Grb2, which then complexes with SOS, leading to activation of Ras-MAPK pathways(97, 107). In parallel with activation of the Frs2-Grb2-SOS-Ras cascade, FGFR1 also activates PLCγ1. When phosphorylated, Tyr766 in the FGFR1 C-terminal tail directly recruits PLCγ1 through its cSH2 domain(20, 108–110), in a manner similar to the interaction of PLCγ1 and phosphorylated Tyr1175 in VEGFR2 (20). FGFR1 phosphorylated at Tyr766 is also reported to bind to Grb14, which competes with PLCγ1 to inhibit its activation (111, 112).
FGFR1 null mutants die as early as E6.5 due to defective formation of the primitive endoderm beginning at E4.5 (104). Mice expressing a form of FGFR1 with point mutations at tyrosine phosphorylation sites that disrupt the interaction of the receptor with its 4 direct effectors display severely retarded development and early embryonic lethality by E11.5(104). A mutation that disrupts the binding of FGFR1 to Frs2α results in neonatal lethality and severe developmental defects in the skeleton(104), whereas mice bearing mutant FGFR1 with defective binding in all of the other three effectors (Crkl, PLCγ1 and Grb14) are viable with only mild skeletal defects(104) (Table 1). These observations indicate that FGFR1 uses multiple effectors to regulate development, during which Frs2α is the primary effector for FGFR1 signal transduction, with PLCγ1 and other effector proteins playing important but additive roles.
FGFR1 activates PLCγ1, which, in addition to the signal from Frs2α, contributes to ERK activation in various cell types(20, 104, 113). However, the magnitude of such regulation could be context-dependent and/or cell type-specific. Endothelial cells do not show Ca2+ mobilization in response to FGF stimulation, unlike that which occurs in response to VEGF (114). Whether this is because FGF-induced PLCγ1 activation is modest compared to that induced by VEGF or because of other yet unknown factors remain unknown. It is not clear if FGF signaling is dispensable for angiogenesis: Although developing mouse embryos carrying a combined endothelial knockout of FGFR1 and FGFR2 do not show a vascular phenotype (115), both lymphatic and blood vascular development are impaired in mice carrying a double FGFR1/FGFR3 endothelial-specific deficiency (116). Endothelial Frs2α deletion, which results in nearly complete cessation of FGF signaling, leads to severe developmental abnormalities(117). The interpretation of these data, however, are complicated by the role of Frs2α in VEGFR signaling(117).
Together, canonical Ras-MAPK is the primary signaling pathway for FGFR (and other non-VEGF RTKs), whereas the role of PLCγ1 is additive (Fig.2B). In endothelial cells, although PLCγ1-dependent Ca2+ and PKC signaling is a unique feature for VEGFR2 signaling, the primary RTK that drives angiogenesis, the role of PLCγ1 for FGF signaling is likely minimal in vascular development.
Non-canonical activation of PLCγ1
Several studies have suggested that PLCγ1 can be activated by a non-canonical pathway different from the well-established direct activation through binding of the SH2 domain of PLCγ1 to the kinase domain of an RTK. PIP3 is a PI3K-catalytic lipid product in the plasma membrane that binds to PH domain-containing proteins, such as PDK1 and PLCγ1(12, 118). In response to growth factor stimulation, PDK1 and PLCγ1 can be simultaneously recruited by PIP3 and form a complex at the juxtamembranous region, where PLCγ1 can be activated by PDK1-dependent phosphorylation of Tyr783 (11–13, 118). Although this mechanism suggests a role for PLCγ1 in PI3K signaling, the precise details and biological relevance of such a mechanism requires further examination.
PLCγ1 in integrin-mediated signaling
PLCγ1 has been proposed to be involved in fibronectin/integrin-dependent cell adhesion, spreading and migration. Fibronectin induces the phosphorylation of Tyr783 in MEFs(119) and endothelial cells (120), likely through interaction with the Src family kinase and integrin adapter GIT1(120). PLCγ1 deficiency impairs cell adhesion and migration on fibronectin by decreasing the activation of Cdc42 and Rac1(121). PLCγ1 has been reported to be present in focal adhesion complexes (122–124), where its association with GIT1 and the RhoGTPase GEF β-Pix is essential for integrin-dependent activity(121). Further study to test the role of extracellular matrix signaling in activating PLCγ1 is required.
PLCγ1-dependent Ca2+ and PKC signaling in endothelial cells
RTK-dependent activation of PLCγ1 results in the hydrolysis of the membrane lipid PIP2 to generate IP3 and DAG. These lipid metabolites subsequently activate two critical signaling pathways, Ca2+-NFAT and cPKC (Fig.2A). IP3 triggers Ca2+ mobilization into the cytosol from two Ca2+ reservoirs, the ER and the extracellular medium(6). Binding of IP3 to IP3 receptors (IP3Rs) on the ER results in a fast but short-lived Ca2+ release from the ER(6). In contrast, Ca2+ influx from the extracellular medium through Ca2+ channels in the plasma membrane is long-lasting and can be activated by the initial Ca2+ release from the ER (store-operated Ca2+ entry; SOCE) or IP3 (non-SOCE-dependent)(6). The surge of cytosolic Ca2+ leads to activation of Ca2+-dependent enzymes, including kinase calmodulin (CaM) and phosphatase calcineurin(CaN) (6, 125). CaM interacts with a regulatory domain of CaN, leading to a conformational change that exposes the active site of CaN(126). Activated CaN subsequently dephosphorylates and activates NFAT(127), which induces a conformational change that exposes the nuclear localization domain of NFAT(128, 129). In the nucleus, NFAT functions as a transcription factor to regulate the expression of target genes(130).
VEGF/PLCγ1-mediated Ca2+-NFAT signaling is involved in various biological effects in endothelial cells, including proliferation, migration and angiogenic sprouting(131–133). VEGF triggers a distinct, dose-dependent pattern of Ca2+ waveforms in endothelial cells that impacts cell motility mediated by myosin light chain kinase (MLCK) and proliferation partially due to NFAT nuclear translocation(133). Live imaging of zebrafish with genetically engineered fluorescent Ca2+ indicator revealed that Ca2+ oscillations occur both in tip and stalk cells during sprouting angiogenesis from the dorsal aorta in response to VEGF signaling, and that DLL4/Notch signaling controls the tip cell-stalk cell selection by regulating Ca2+ oscillations in these cells(132). In addition to angiogenesis, Ca2+ signaling has also been implicated in regulating endothelial permeability(134).
An increase in intracellular Ca2+ also activates PKC signaling, which underlies the ability of VEGFR2 to activate the Raf1/MEK/ERK pathway. PKCs are a family serine-threonine kinases that are essential regulators of numerous biological activities and are therapeutic targets in various cardiovascular, diabetes, and inflammatory diseases(135, 136). They are divided into 3 subcategories, including the conventional, Ca2+-dependent PKCs (PKCα, PKCβ and PKCγ, which are activated by a combination of DAG and Ca2+), novel PKCs (PKCδ, PKCε, PKCη and PKCθ, which are activated by DAG alone but not by Ca2+) and atypical PKCs (PKCζ and PKCλ/ι, which are activated independently of either DAG or Ca2+)(135, 136). cPKCs have been reported to phosphorylate and activate Raf1, and therefore, induce the entire Raf1/MEk/ERK cascade(137–139).
PKCα and PKCß are thought to be the principal PKC isoforms in endothelial cells and therefore the key PKCs involved in VEGFR2-dependent activation of ERK signaling(140). Unlike phorbol esters which activate DAG-sensitive PKCs and induce a translocation of PKCα, β and γ from the cytosol to the juxtamembrane region(129, 140), VEGF stimulation induces translocation of only PKCβ, suggesting that this PKC isoform specifically responds to VEGF signaling(66). An inhibitor targeting PKCβ partially blocks VEGF-induced proliferation in endothelial cells (140) and tumor angiogenesis in vivo(76). Furthermore, overexpression of PKCβ2 (but not of PKCα, PKCδ or PKCζ) induces proliferation and migration of bovine retinal endothelial cells whereas overexpression of a dominant-negative form of PKCβ2 decreases both processes (141). Dominant-negative PKCβ2 also blocks VEGF-induced phosphorylation of the tight junction protein occludin and reduces endothelial permeability(142). Despite this seemingly convincing in vitro data, knockout studies have produced different results (Table 2). Mice with a global deletion of PKCβ (both the β1 and β2 isoforms, which are encoded by the same gene) or PKCα are viable and histologically normal without a cardiovascular phenotype (143, 144), suggesting compensation by other PKC isoforms or by stimulation of VEGF-induced, PKC-independent ERK1/2 activation.
PLCγ1 in the nucleus
Phosphoinositide metabolism takes place not only in the plasma membrane and cytoplasm, but also in the nucleus(24, 145). Multiple PLC isozymes (PLCβ1, γ1, δ1, and ζ) are reported to be physically present in the nucleus and involved in various nuclear biological activities as phosphoinositol-specific lipases or non-enzymatic regulators(77), although not in endothelial cells. In the brain, nerve growth factor can induce nuclear trans-localization of PLCγ1, where it functions as a GEF through its SH3 domain to activate the nuclear GTPase PIKE(24, 145). PLCγ1 is also implicated in NaCl-induced activation of the osmoprotective transcription factor tonicity-responsive enhancer/osmotic response element-binding protein (TonEBP/OREBP; also called NFAT5) in the nuclei of fibroblasts and HEK293 cells(146). Because these observations are likely context-dependent and cell type-specific, the potential nuclear function of PLCγ1 in endothelial cells requires further examination.
Conclusions and perspectives
The past two decades have brought us a better understanding of PLCγ1-dependent biology and signaling in the endothelium. PLCγ1 plays a critical role in embryonic vascular development and postnatal angiogenesis and serves as the primary means for VEGFR2-dependent activation of MAPK signaling. It is also critical to the development of multiple endothelial-derived malignancies. Yet there are critical gaps in our understanding of its signaling and biological role, including as its function as the key transducer of VEGFR2-dependent activation of ERK1/2 pathway. In particular, it is not clear what determines the preferential use of PLCγ1 (rather than Ras) by VEGFR2 as the primary means of MAPK activation during vascular morphogenesis. Furthermore, the biological reason for this preference for VEGFR2 versus other RTKs is also unclear. Another unresolved issue is the role of PLCγ1 versus Ras in mediating VEGFR2-ERK signaling during postnatal angiogenesis. PLCγ1 is required for specification of arterial identity during vasculogenesis of the dorsal aorta in zebrafish embryo. However, its role in regulation of artery-vein differentiation during angiogenesis has not been elucidated.
It is also unknown whether PLCγ1 is involved in maintenance of the quiescent endothelium in adult vasculature or if it is involved in mediating VEGF-induced increase in endothelial permeability. Although the role of PLCγ1 in mediating VEGFR2-induced ERK1/2 activation is well established, it is not clear if the same pathway operates for VEGFR3 in lymphatic and blood endothelial cells or whether PLCγ1 is required for lymphatic development and normal lymphatic function in the adult? Finally, although there are substantial phenotypic differences between endothelial PLCγ1 compared to cPKC knockouts, it is not clear which cPKC isoform mediates PLCγ1-dependent ERK activation or whether another kinase is also involved. Resolution of these issues will help improve our understanding of VEGF signaling and endothelial biology.
Funding:
This work was funded by NIH grants HL149343 and HL 107205 to MS.
Footnotes
Competing interests: The authors declare that they have no competing interests.
References and Notes
- 1.Berridge MJ, Irvine RF, Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature 312, 315–321 (1984). [DOI] [PubMed] [Google Scholar]
- 2.Majerus PW, Connolly TM, Deckmyn H, Ross TS, Bross TE, Ishii H, Bansal VS, Wilson DB, The metabolism of phosphoinositide-derived messenger molecules. Science 234, 1519–1526 (1986). [DOI] [PubMed] [Google Scholar]
- 3.Suh PG, Park JI, Manzoli L, Cocco L, Peak JC, Katan M, Fukami K, Kataoka T, Yun S, Ryu SH, Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep 41, 415–434 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Nakamura Y, Fukami K, Regulation and physiological functions of mammalian phospholipase C. J Biochem 161, 315–321 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Balla T, Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93, 1019–1137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munaron L, Intracellular calcium, endothelial cells and angiogenesis. Recent Pat Anticancer Drug Discov 1, 105–119 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Yang YR, Choi JH, Chang JS, Kwon HM, Jang HJ, Ryu SH, Suh PG, Diverse cellular and physiological roles of phospholipase C-gamma1. Adv Biol Regul 52, 138–151 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Katan M, Families of phosphoinositide-specific phospholipase C: structure and function. Biochim Biophys Acta 1436, 5–17 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Koss H, Bunney TD, Behjati S, Katan M, Dysfunction of phospholipase Cgamma in immune disorders and cancer. Trends Biochem Sci 39, 603–611 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Essen LO, Perisic O, Cheung R, Katan M, Williams RL, Crystal structure of a mammalian phosphoinositide-specific phospholipase C delta. Nature 380, 595–602 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Maffucci T, Falasca M, Phosphoinositide 3-kinase-dependent regulation of phospholipase Cgamma. Biochem Soc Trans 35, 229–230 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Raimondi C, Calleja V, Ferro R, Fantin A, Riley AM, Potter BV, Brennan CH, Maffucci T, Larijani B, Falasca M, A Small Molecule Inhibitor of PDK1/PLCgamma1 Interaction Blocks Breast and Melanoma Cancer Cell Invasion. Sci Rep 6, 26142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, Schlessinger J, Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J 17, 414–422 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji QS, Chattopadhyay A, Vecchi M, Carpenter G, Physiological requirement for both SH2 domains for phospholipase C-gamma1 function and interaction with platelet-derived growth factor receptors. Mol Cell Biol 19, 4961–4970 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katan M, Williams RL, Phosphoinositide-specific phospholipase C: structural basis for catalysis and regulatory interactions. Semin Cell Dev Biol 8, 287–296 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Hajicek N, Charpentier TH, Rush JR, Harden TK, Sondek J, Autoinhibition and phosphorylation-induced activation of phospholipase C-gamma isozymes. Biochemistry 52, 4810–4819 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hajicek N, Keith NC, Siraliev-Perez E, Temple BR, Huang W, Zhang Q, Harden TK, Sondek J, Structural basis for the activation of PLC-gamma isozymes by phosphorylation and cancer-associated mutations. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poulin B, Sekiya F, Rhee SG, Intramolecular interaction between phosphorylated tyrosine-783 and the C-terminal Src homology 2 domain activates phospholipase C-gamma1. Proc Natl Acad Sci U S A 102, 4276–4281 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulin B, Sekiya F, Rhee SG, Differential roles of the Src homology 2 domains of phospholipase C-gamma1 (PLC-gamma1) in platelet-derived growth factor-induced activation of PLC-gamma1 in intact cells. J Biol Chem 275, 6411–6416 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Huang Z, Marsiglia WM, Basu Roy U, Rahimi N, Ilghari D, Wang H, Chen H, Gai W, Blais S, Neubert TA, Mansukhani A, Traaseth NJ, Li X, Mohammadi M, Two FGF Receptor Kinase Molecules Act in Concert to Recruit and Transphosphorylate Phospholipase Cgamma. Mol Cell 61, 98–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Bunney TD, Khosa S, Mace K, Beckenbauer K, Askwith T, Maslen S, Stubbs C, de Oliveira TM, Sader K, Skehel M, Gavin AC, Phillips C, Katan M, Structural insights and activating mutations in diverse pathologies define mechanisms of deregulation for phospholipase C gamma enzymes. EBioMedicine 51, 102607 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeBell K, Graham L, Reischl I, Serrano C, Bonvini E, Rellahan B, Intramolecular regulation of phospholipase C-gamma1 by its C-terminal Src homology 2 domain. Mol Cell Biol 27, 854–863 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi JH, Park JB, Bae SS, Yun S, Kim HS, Hong WP, Kim IS, Kim JH, Han MY, Ryu SH, Patterson RL, Snyder SH, Suh PG, Phospholipase C-gamma1 is a guanine nucleotide exchange factor for dynamin-1 and enhances dynamin-1-dependent epidermal growth factor receptor endocytosis. J Cell Sci 117, 3785–3795 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Ye K, Aghdasi B, Luo HR, Moriarity JL, Wu FY, Hong JJ, Hurt KJ, Bae SS, Suh PG, Snyder SH, Phospholipase C gamma 1 is a physiological guanine nucleotide exchange factor for the nuclear GTPase PIKE. Nature 415, 541–544 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Bunney TD, Esposito D, Mas-Droux C, Lamber E, Baxendale RW, Martins M, Cole A, Svergun D, Driscoll PC, Katan M, Structural and functional integration of the PLCgamma interaction domains critical for regulatory mechanisms and signaling deregulation. Structure 20, 2062–2075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harden TK, Hicks SN, Sondek J, Phospholipase C isozymes as effectors of Ras superfamily GTPases. J Lipid Res 50 Suppl, S243–248 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen W, Yan J, Zhang M, Structural characterization of the split pleckstrin homology domain in phospholipase C-gamma1 and its interaction with TRPC3. J Biol Chem 281, 12060–12068 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Behjati S, Tarpey PS, Sheldon H, Martincorena I, Van Loo P, Gundem G, Wedge DC, Ramakrishna M, Cooke SL, Pillay N, Vollan HKM, Papaemmanuil E, Koss H, Bunney TD, Hardy C, Joseph OR, Martin S, Mudie L, Butler A, Teague JW, Patil M, Steers G, Cao Y, Gumbs C, Ingram D, Lazar AJ, Little L, Mahadeshwar H, Protopopov A, Al Sannaa GA, Seth S, Song X, Tang J, Zhang J, Ravi V, Torres KE, Khatri B, Halai D, Roxanis I, Baumhoer D, Tirabosco R, Amary MF, Boshoff C, McDermott U, Katan M, Stratton MR, Futreal PA, Flanagan AM, Harris A, Campbell PJ, Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet 46, 376–379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner MJ, Ravi V, Menter DG, Sood AK, Endothelial cell malignancies: new insights from the laboratory and clinic. npj Precision Oncology 1, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaque JP, Gomez-Lopez G, Monsalvez V, Varela I, Martinez N, Perez C, Dominguez O, Grana O, Rodriguez-Peralto JL, Rodriguez-Pinilla SM, Gonzalez-Vela C, Rubio-Camarillo M, Martin-Sanchez E, Pisano DG, Papadavid E, Papadaki T, Requena L, Garcia-Marco JA, Mendez M, Provencio M, Hospital M, Suarez-Massa D, Postigo C, San Segundo D, Lopez-Hoyos M, Ortiz-Romero PL, Piris MA, Sanchez-Beato M, PLCG1 mutations in cutaneous T-cell lymphomas. Blood 123, 2034–2043 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Argeson AC, Druck T, Veronese ML, Knopf JL, Buchberg AM, Huebner K, Siracusa LD, Phospholipase C gamma-2 (Plcg2) and phospholipase C gamma-1 (Plcg1) map to distinct regions in the human and mouse genomes. Genomics 25, 29–35 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Ji QS, Winnier GE, Niswender KD, Horstman D, Wisdom R, Magnuson MA, Carpenter G, Essential role of the tyrosine kinase substrate phospholipase C-gamma1 in mammalian growth and development. Proc Natl Acad Sci U S A 94, 2999–3003 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao HJ, Kume T, McKay C, Xu MJ, Ihle JN, Carpenter G, Absence of erythrogenesis and vasculogenesis in Plcg1-deficient mice. J Biol Chem 277, 9335–9341 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Homma Y, Takenawa T, Emori Y, Sorimachi H, Suzuki K, Tissue- and cell type-specific expression of mRNAs for four types of inositol phospholipid-specific phospholipase C. Biochem Biophys Res Commun 164, 406–412 (1989). [DOI] [PubMed] [Google Scholar]
- 35.Tanaka O, Kondo H, Localization of mRNAs for three novel members (beta 3, beta 4 and gamma 2) of phospholipase C family in mature rat brain. Neurosci Lett 182, 17–20 (1994). [DOI] [PubMed] [Google Scholar]
- 36.Mizuguchi M, Yamada M, Kim SU, Rhee SG, Phospholipase C isozymes in neurons and glial cells in culture: an immunocytochemical and immunochemical study. Brain Res 548, 35–40 (1991). [DOI] [PubMed] [Google Scholar]
- 37.Shirane M, Sawa H, Kobayashi Y, Nakano T, Kitajima K, Shinkai Y, Nagashima K, Negishi I, Deficiency of phospholipase C-gamma1 impairs renal development and hematopoiesis. Development 128, 5173–5180 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Lawson ND, Mugford JW, Diamond BA, Weinstein BM, phospholipase C gamma-1 is required downstream of vascular endothelial growth factor during arterial development. Genes Dev 17, 1346–1351 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Covassin LD, Siekmann AF, Kacergis MC, Laver E, Moore JC, Villefranc JA, Weinstein BM, Lawson ND, A genetic screen for vascular mutants in zebrafish reveals dynamic roles for Vegf/Plcg1 signaling during artery development. Dev Biol 329, 212–226 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gore AV, Monzo K, Cha YR, Pan W, Weinstein BM, Vascular development in the zebrafish. Cold Spring Harb Perspect Med 2, a006684 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Udan RS, Culver JC, Dickinson ME, Understanding vascular development. Wiley Interdiscip Rev Dev Biol 2, 327–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato Y, Dorsal aorta formation: separate origins, lateral-to-medial migration, and remodeling. Dev Growth Differ 55, 113–129 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Cleaver O, Krieg PA, VEGF mediates angioblast migration during development of the dorsal aorta in Xenopus. Development 125, 3905–3914 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Fu G, Chen Y, Yu M, Podd A, Schuman J, He Y, Di L, Yassai M, Haribhai D, North PE, Gorski J, Williams CB, Wang D, Wen R, Phospholipase C{gamma}1 is essential for T cell development, activation, and tolerance. J Exp Med 207, 309–318 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu M, Chen Y, Zeng H, Zheng Y, Fu G, Zhu W, Broeckel U, Aggarwal P, Turner A, Neale G, Guy C, Zhu N, Chi H, Wen R, Wang D, PLCgamma-dependent mTOR signalling controls IL-7-mediated early B cell development. Nat Commun 8, 1457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kunze K, Spieker T, Gamerdinger U, Nau K, Berger J, Dreyer T, Sindermann JR, Hoffmeier A, Gattenlohner S, Brauninger A, A recurrent activating PLCG1 mutation in cardiac angiosarcomas increases apoptosis resistance and invasiveness of endothelial cells. Cancer Res 74, 6173–6183 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Prenen H, Smeets D, Mazzone M, Lambrechts D, Sagaert X, Sciot R, Debiec-Rychter M, Phospholipase C gamma 1 (PLCG1) R707Q mutation is counterselected under targeted therapy in a patient with hepatic angiosarcoma. Oncotarget 6, 36418–36425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sala G, Dituri F, Raimondi C, Previdi S, Maffucci T, Mazzoletti M, Rossi C, Iezzi M, Lattanzio R, Piantelli M, Iacobelli S, Broggini M, Falasca M, Phospholipase Cgamma1 is required for metastasis development and progression. Cancer Res 68, 10187–10196 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Manso R, Rodriguez-Pinilla SM, Gonzalez-Rincon J, Gomez S, Monsalvo S, Llamas P, Rojo F, Perez-Callejo D, Cereceda L, Limeres MA, Maeso C, Ferrando L, Perez-Seoane C, Rodriguez G, Arrinda JM, Garcia-Bragado F, Franco R, Rodriguez-Peralto JL, Gonzalez-Carrero J, Martin-Davila F, Piris MA, Sanchez-Beato M, Recurrent presence of the PLCG1 S345F mutation in nodal peripheral T-cell lymphomas. Haematologica 100, e25–27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC, Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66 (1995). [DOI] [PubMed] [Google Scholar]
- 51.Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M, Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci U S A 102, 1076–1081 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simons M, Gordon E, Claesson-Welsh L, Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17, 611–625 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Testini C, Smith RO, Jin Y, Martinsson P, Sun Y, Hedlund M, Sainz-Jaspeado M, Shibuya M, Hellstrom M, Claesson-Welsh L, Myc-dependent endothelial proliferation is controlled by phosphotyrosine 1212 in VEGF receptor-2. EMBO Rep 20, e47845 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X, Padhan N, Sjostrom EO, Roche FP, Testini C, Honkura N, Sainz-Jaspeado M, Gordon E, Bentley K, Philippides A, Tolmachev V, Dejana E, Stan RV, Vestweber D, Ballmer-Hofer K, Betsholtz C, Pietras K, Jansson L, Claesson-Welsh L, VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat Commun 7, 11017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takahashi T, Yamaguchi S, Chida K, Shibuya M, A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J 20, 2768–2778 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srinivasan R, Zabuawala T, Huang H, Zhang J, Gulati P, Fernandez S, Karlo JC, Landreth GE, Leone G, Ostrowski MC, Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS One 4, e8283 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J, Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 286, 1374–1377 (1999). [DOI] [PubMed] [Google Scholar]
- 58.Hatano N, Mori Y, Oh-hora M, Kosugi A, Fujikawa T, Nakai N, Niwa H, Miyazaki J, Hamaoka T, Ogata M, Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 8, 847–856 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Fremin C, Saba-El-Leil MK, Levesque K, Ang SL, Meloche S, Functional Redundancy of ERK1 and ERK2 MAP Kinases during Development. Cell Rep 12, 913–921 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K, VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci U S A 112, 761–766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D, Rocha SF, Langen UH, Stehling M, Nagasawa T, Ferrara N, Borggrefe T, Adams RH, Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol 19, 915–927 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Klint P, Kanda S, Kloog Y, Claesson-Welsh L, Contribution of Src and Ras pathways in FGF-2 induced endothelial cell differentiation. Oncogene 18, 3354–3364 (1999). [DOI] [PubMed] [Google Scholar]
- 63.Satoh T, Endo M, Nakafuku M, Akiyama T, Yamamoto T, Kaziro Y, Accumulation of p21ras.GTP in response to stimulation with epidermal growth factor and oncogene products with tyrosine kinase activity. Proc Natl Acad Sci U S A 87, 7926–7929 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Satoh T, Endo M, Nakafuku M, Nakamura S, Kaziro Y, Platelet-derived growth factor stimulates formation of active p21ras.GTP complex in Swiss mouse 3T3 cells. Proc Natl Acad Sci U S A 87, 5993–5997 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muroya K, Hattori S, Nakamura S, Nerve growth factor induces rapid accumulation of the GTP-bound form of p21ras in rat pheochromocytoma PC12 cells. Oncogene 7, 277–281 (1992). [PubMed] [Google Scholar]
- 66.Takahashi T, Ueno H, Shibuya M, VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 18, 2221–2230 (1999). [DOI] [PubMed] [Google Scholar]
- 67.Guo D, Jia Q, Song HY, Warren RS, Donner DB, Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J Biol Chem 270, 6729–6733 (1995). [DOI] [PubMed] [Google Scholar]
- 68.Wu LW, Mayo LD, Dunbar JD, Kessler KM, Baerwald MR, Jaffe EA, Wang D, Warren RS, Donner DB, Utilization of distinct signaling pathways by receptors for vascular endothelial cell growth factor and other mitogens in the induction of endothelial cell proliferation. J Biol Chem 275, 5096–5103 (2000). [DOI] [PubMed] [Google Scholar]
- 69.Holmqvist K, Cross MJ, Rolny C, Hagerkvist R, Rahimi N, Matsumoto T, Claesson-Welsh L, Welsh M, The adaptor protein shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J Biol Chem 279, 22267–22275 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Cunningham SA, Arrate MP, Brock TA, Waxham MN, Interactions of FLT-1 and KDR with phospholipase C gamma: identification of the phosphotyrosine binding sites. Biochem Biophys Res Commun 240, 635–639 (1997). [DOI] [PubMed] [Google Scholar]
- 71.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L, Signal transduction by vascular endothelial growth factor receptors. Biochem J 437, 169–183 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Shibuya M, VEGFR and type-V RTK activation and signaling. Cold Spring Harb Perspect Biol 5, a009092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doanes AM, Hegland DD, Sethi R, Kovesdi I, Bruder JT, Finkel T, VEGF stimulates MAPK through a pathway that is unique for receptor tyrosine kinases. Biochem Biophys Res Commun 255, 545–548 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Yashima R, Abe M, Tanaka K, Ueno H, Shitara K, Takenoshita S, Sato Y, Heterogeneity of the signal transduction pathways for VEGF-induced MAPKs activation in human vascular endothelial cells. J Cell Physiol 188, 201–210 (2001). [DOI] [PubMed] [Google Scholar]
- 75.Meadows KN, Bryant P, Pumiglia K, Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J Biol Chem 276, 49289–49298 (2001). [DOI] [PubMed] [Google Scholar]
- 76.Yoshiji H, Kuriyama S, Ways DK, Yoshii J, Miyamoto Y, Kawata M, Ikenaka Y, Tsujinoue H, Nakatani T, Shibuya M, Fukui H, Protein kinase C lies on the signaling pathway for vascular endothelial growth factor-mediated tumor development and angiogenesis. Cancer Res 59, 4413–4418 (1999). [PubMed] [Google Scholar]
- 77.Faenza I, Fiume R, Piazzi M, Colantoni A, Cocco L, Nuclear inositide specific phospholipase C signalling - interactions and activity. FEBS J 280, 6311–6321 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Westenskow PD, Kurihara T, Aguilar E, Scheppke EL, Moreno SK, Wittgrove C, Marchetti V, Michael IP, Anand S, Nagy A, Cheresh D, Friedlander M, Ras pathway inhibition prevents neovascularization by repressing endothelial cell sprouting. J Clin Invest 123, 4900–4908 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Secker GA, Harvey NL, VEGFR signaling during lymphatic vascular development: From progenitor cells to functional vessels. Dev Dyn 244, 323–331 (2015). [DOI] [PubMed] [Google Scholar]
- 80.Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K, Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 282, 946–949 (1998). [DOI] [PubMed] [Google Scholar]
- 81.Ehling M, Adams S, Benedito R, Adams RH, Notch controls retinal blood vessel maturation and quiescence. Development 140, 3051–3061 (2013). [DOI] [PubMed] [Google Scholar]
- 82.Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, Breitman M, Alitalo K, Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A 92, 3566–3570 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R, Freitas C, Duarte A, Isoniemi H, Laakkonen P, Christofori G, Yla-Herttuala S, Shibuya M, Pytowski B, Eichmann A, Betsholtz C, Alitalo K, Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454, 656–660 (2008). [DOI] [PubMed] [Google Scholar]
- 84.Valtola R, Salven P, Heikkila P, Taipale J, Joensuu H, Rehn M, Pihlajaniemi T, Weich H, deWaal R, Alitalo K, VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in breast cancer. Am J Pathol 154, 1381–1390 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dixelius J, Makinen T, Wirzenius M, Karkkainen MJ, Wernstedt C, Alitalo K, Claesson-Welsh L, Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J Biol Chem 278, 40973–40979 (2003). [DOI] [PubMed] [Google Scholar]
- 86.Alam A, Herault JP, Barron P, Favier B, Fons P, Delesque-Touchard N, Senegas I, Laboudie P, Bonnin J, Cassan C, Savi P, Ruggeri B, Carmeliet P, Bono F, Herbert JM, Heterodimerization with vascular endothelial growth factor receptor-2 (VEGFR-2) is necessary for VEGFR-3 activity. Biochem Biophys Res Commun 324, 909–915 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Nilsson I, Bahram F, Li X, Gualandi L, Koch S, Jarvius M, Soderberg O, Anisimov A, Kholova I, Pytowski B, Baldwin M, Yla-Herttuala S, Alitalo K, Kreuger J, Claesson-Welsh L, VEGF receptor 2/−3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J 29, 1377–1388 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Deng Y, Zhang X, Simons M, Molecular controls of lymphatic VEGFR3 signaling. Arterioscler Thromb Vasc Biol 35, 421–429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jones D, Xu Z, Zhang H, He Y, Kluger MS, Chen H, Min W, Functional analyses of the bone marrow kinase in the X chromosome in vascular endothelial growth factor-induced lymphangiogenesis. Arterioscler Thromb Vasc Biol 30, 2553–2561 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dellinger MT, Meadows SM, Wynne K, Cleaver O, Brekken RA, Vascular endothelial growth factor receptor-2 promotes the development of the lymphatic vasculature. PLoS One 8, e74686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sase H, Watabe T, Kawasaki K, Miyazono K, Miyazawa K, VEGFR2-PLCgamma1 axis is essential for endothelial specification of VEGFR2+ vascular progenitor cells. J Cell Sci 122, 3303–3311 (2009). [DOI] [PubMed] [Google Scholar]
- 92.Sawano A, Takahashi T, Yamaguchi S, Shibuya M, The phosphorylated 1169-tyrosine containing region of flt-1 kinase (VEGFR-1) is a major binding site for PLCgamma. Biochem Biophys Res Commun 238, 487–491 (1997). [DOI] [PubMed] [Google Scholar]
- 93.Fong GH, Rossant J, Gertsenstein M, Breitman ML, Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376, 66–70 (1995). [DOI] [PubMed] [Google Scholar]
- 94.Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M, Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A 95, 9349–9354 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ito N, Wernstedt C, Engstrom U, Claesson-Welsh L, Identification of vascular endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of SH2 domain-containing molecules. J Biol Chem 273, 23410–23418 (1998). [DOI] [PubMed] [Google Scholar]
- 96.Brewer JR, Mazot P, Soriano P, Genetic insights into the mechanisms of Fgf signaling. Genes Dev 30, 751–771 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ornitz DM, Itoh N, The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4, 215–266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rotin D, Honegger AM, Margolis BL, Ullrich A, Schlessinger J, Presence of SH2 domains of phospholipase C gamma 1 enhances substrate phosphorylation by increasing the affinity toward the epidermal growth factor receptor. J Biol Chem 267, 9678–9683 (1992). [PubMed] [Google Scholar]
- 99.Rotin D, Margolis B, Mohammadi M, Daly RJ, Daum G, Li N, Fischer EH, Burgess WH, Ullrich A, Schlessinger J, SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: identification of Tyr992 as the high-affinity binding site for SH2 domains of phospholipase C gamma. EMBO J 11, 559–567 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liao HJ, Ji QS, Carpenter G, Phospholipase C-gamma1 is required for the induction of immediate early genes by platelet-derived growth factor. J Biol Chem 276, 8627–8630 (2001). [DOI] [PubMed] [Google Scholar]
- 101.Seedorf K, Millauer B, Kostka G, Schlessinger J, Ullrich A, Differential effects of carboxy-terminal sequence deletions on platelet-derived growth factor receptor signaling activities and interactions with cellular substrates. Mol Cell Biol 12, 4347–4356 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gu B, Huang YZ, He XP, Joshi RB, Jang W, McNamara JO, A Peptide Uncoupling BDNF Receptor TrkB from Phospholipase Cgamma1 Prevents Epilepsy Induced by Status Epilepticus. Neuron 88, 484–491 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McCarty JH, Feinstein SC, The TrkB receptor tyrosine kinase regulates cellular proliferation via signal transduction pathways involving SHC, PLCgamma, and CBL. J Recept Signal Transduct Res 19, 953–974 (1999). [DOI] [PubMed] [Google Scholar]
- 104.Brewer JR, Molotkov A, Mazot P, Hoch RV, Soriano P, Fgfr1 regulates development through the combinatorial use of signaling proteins. Genes Dev 29, 1863–1874 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Klint P, Kanda S, Claesson-Welsh L, Shc and a novel 89-kDa component couple to the Grb2-Sos complex in fibroblast growth factor-2-stimulated cells. J Biol Chem 270, 23337–23344 (1995). [DOI] [PubMed] [Google Scholar]
- 106.Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J, A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89, 693–702 (1997). [DOI] [PubMed] [Google Scholar]
- 107.Vasudevan HN, Mazot P, He F, Soriano P, Receptor tyrosine kinases modulate distinct transcriptional programs by differential usage of intracellular pathways. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mohammadi M, Honegger AM, Rotin D, Fischer R, Bellot F, Li W, Dionne CA, Jaye M, Rubinstein M, Schlessinger J, A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol Cell Biol 11, 5068–5078 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peters KG, Marie J, Wilson E, Ives HE, Escobedo J, Del Rosario M, Mirda D, Williams LT, Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature 358, 678–681 (1992). [DOI] [PubMed] [Google Scholar]
- 110.Bae JH, Lew ED, Yuzawa S, Tome F, Lax I, Schlessinger J, The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell 138, 514–524 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Browaeys-Poly E, Blanquart C, Perdereau D, Antoine AF, Goenaga D, Luzy JP, Chen H, Garbay C, Issad T, Cailliau K, Burnol AF, Grb14 inhibits FGF receptor signaling through the regulation of PLCgamma recruitment and activation. FEBS Lett 584, 4383–4388 (2010). [DOI] [PubMed] [Google Scholar]
- 112.Cailliau K, Perdereau D, Lescuyer A, Chen H, Garbay C, Vilain JP, Burnol AF, Browaeys-Poly E, FGF receptor phosphotyrosine 766 is a target for Grb14 to inhibit MDA-MB-231 human breast cancer cell signaling. Anticancer Res 25, 3877–3882 (2005). [PubMed] [Google Scholar]
- 113.Huang J, Mohammadi M, Rodrigues GA, Schlessinger J, Reduced activation of RAF-1 and MAP kinase by a fibroblast growth factor receptor mutant deficient in stimulation of phosphatidylinositol hydrolysis. J Biol Chem 270, 5065–5072 (1995). [DOI] [PubMed] [Google Scholar]
- 114.McLaughlin AP, De Vries GW, Role of PLCgamma and Ca(2+) in VEGF- and FGF-induced choroidal endothelial cell proliferation. Am J Physiol Cell Physiol 281, C1448–1456 (2001). [DOI] [PubMed] [Google Scholar]
- 115.Oladipupo SS, Smith C, Santeford A, Park C, Sene A, Wiley LA, Osei-Owusu P, Hsu J, Zapata N, Liu F, Nakamura R, Lavine KJ, Blumer KJ, Choi K, Apte RS, Ornitz DM, Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc Natl Acad Sci U S A 111, 13379–13384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yu P, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS, Xie Y, Zhu J, Chen Z, De Smet F, Zhang J, Jin SW, Sun L, Sun H, Kibbey RG, Hirschi KK, Hay N, Carmeliet P, Chittenden TW, Eichmann A, Potente M, Simons M, FGF-dependent metabolic control of vascular development. Nature 545, 224–228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen PY, Qin L, Zhuang ZW, Tellides G, Lax I, Schlessinger J, Simons M, The docking protein FRS2alpha is a critical regulator of VEGF receptors signaling. Proc Natl Acad Sci U S A 111, 5514–5519 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Raimondi C, Chikh A, Wheeler AP, Maffucci T, Falasca M, A novel regulatory mechanism links PLCgamma1 to PDK1. J Cell Sci 125, 3153–3163 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tvorogov D, Wang XJ, Zent R, Carpenter G, Integrin-dependent PLC-gamma1 phosphorylation mediates fibronectin-dependent adhesion. J Cell Sci 118, 601–610 (2005). [DOI] [PubMed] [Google Scholar]
- 120.Jones NP, Peak J, Brader S, Eccles SA, Katan M, PLCgamma1 is essential for early events in integrin signalling required for cell motility. J Cell Sci 118, 2695–2706 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Jones NP, Katan M, Role of phospholipase Cgamma1 in cell spreading requires association with a beta-Pix/GIT1-containing complex, leading to activation of Cdc42 and Rac1. Mol Cell Biol 27, 5790–5805 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Plopper GE, McNamee HP, Dike LE, Bojanowski K, Ingber DE, Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol Biol Cell 6, 1349–1365 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Miyamoto S, Teramoto H, Coso OA, Gutkind JS, Burbelo PD, Akiyama SK, Yamada KM, Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol 131, 791–805 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cybulsky AV, McTavish AJ, Papillon J, Extracellular matrix stimulates production and breakdown of inositol phospholipids. Am J Physiol 271, F579–587 (1996). [DOI] [PubMed] [Google Scholar]
- 125.Hogan PG, Chen L, Nardone J, Rao A, Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17, 2205–2232 (2003). [DOI] [PubMed] [Google Scholar]
- 126.Rumi-Masante J, Rusinga FI, Lester TE, Dunlap TB, Williams TD, Dunker AK, Weis DD, Creamer TP, Structural basis for activation of calcineurin by calmodulin. J Mol Biol 415, 307–317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Armesilla AL, Lorenzo E, Gomez del Arco P, Martinez-Martinez S, Alfranca A, Redondo JM, Vascular endothelial growth factor activates nuclear factor of activated T cells in human endothelial cells: a role for tissue factor gene expression. Mol Cell Biol 19, 2032–2043 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rao A, Luo C, Hogan PG, Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15, 707–747 (1997). [DOI] [PubMed] [Google Scholar]
- 129.Masuda ES, Imamura R, Amasaki Y, Arai K, Arai N, Signalling into the T-cell nucleus: NFAT regulation. Cell Signal 10, 599–611 (1998). [DOI] [PubMed] [Google Scholar]
- 130.Crabtree GR, Calcium, calcineurin, and the control of transcription. J Biol Chem 276, 2313–2316 (2001). [DOI] [PubMed] [Google Scholar]
- 131.Hamdollah Zadeh MA, Glass CA, Magnussen A, Hancox JC, Bates DO, VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 15, 605–614 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yokota Y, Nakajima H, Wakayama Y, Muto A, Kawakami K, Fukuhara S, Mochizuki N, Endothelial Ca 2+ oscillations reflect VEGFR signaling-regulated angiogenic capacity in vivo. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Noren DP, Chou WH, Lee SH, Qutub AA, Warmflash A, Wagner DS, Popel AS, Levchenko A, Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci Signal 9, ra20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jho D, Mehta D, Ahmmed G, Gao XP, Tiruppathi C, Broman M, Malik AB, Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2 influx. Circ Res 96, 1282–1290 (2005). [DOI] [PubMed] [Google Scholar]
- 135.Mochly-Rosen D, Das K, Grimes KV, Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov 11, 937–957 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ, PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol 11, 103–112 (2010). [DOI] [PubMed] [Google Scholar]
- 137.Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marme D, Rapp UR, Protein kinase C alpha activates RAF-1 by direct phosphorylation. Nature 364, 249–252 (1993). [DOI] [PubMed] [Google Scholar]
- 138.Carroll MP, May WS, Protein kinase C-mediated serine phosphorylation directly activates Raf-1 in murine hematopoietic cells. J Biol Chem 269, 1249–1256 (1994). [PubMed] [Google Scholar]
- 139.Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR, Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem 278, 13061–13068 (2003). [DOI] [PubMed] [Google Scholar]
- 140.Xia P, Aiello LP, Ishii H, Jiang ZY, Park DJ, Robinson GS, Takagi H, Newsome WP, Jirousek MR, King GL, Characterization of vascular endothelial growth factor’s effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J Clin Invest 98, 2018–2026 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Suzuma K, Takahara N, Suzuma I, Isshiki K, Ueki K, Leitges M, Aiello LP, King GL, Characterization of protein kinase C beta isoform’s action on retinoblastoma protein phosphorylation, vascular endothelial growth factor-induced endothelial cell proliferation, and retinal neovascularization. Proc Natl Acad Sci U S A 99, 721–726 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Harhaj NS, Felinski EA, Wolpert EB, Sundstrom JM, Gardner TW, Antonetti DA, VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci 47, 5106–5115 (2006). [DOI] [PubMed] [Google Scholar]
- 143.Leitges M, Plomann M, Standaert ML, Bandyopadhyay G, Sajan MP, Kanoh Y, Farese RV, Knockout of PKC alpha enhances insulin signaling through PI3K. Mol Endocrinol 16, 847–858 (2002). [DOI] [PubMed] [Google Scholar]
- 144.Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, Tarakhovsky A, Immunodeficiency in protein kinase cbeta-deficient mice. Science 273, 788–791 (1996). [DOI] [PubMed] [Google Scholar]
- 145.Ye K, Snyder SH, PIKE GTPase: a novel mediator of phosphoinositide signaling. J Cell Sci 117, 155–161 (2004). [DOI] [PubMed] [Google Scholar]
- 146.Irarrazabal CE, Gallazzini M, Schnetz MP, Kunin M, Simons BL, Williams CK, Burg MB, Ferraris JD, Phospholipase C-gamma1 is involved in signaling the activation by high NaCl of the osmoprotective transcription factor TonEBP/OREBP. Proc Natl Acad Sci U S A 107, 906–911 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]