

Summary
Ligation of the inhibitory receptor PD‐1 on T cells results in the inhibition of numerous cellular functions. Despite the overtly inhibitory outcome of PD‐1 signalling, there are additionally a collection of functions that are activated. We have observed that CD4+ T cells stimulated through the T‐cell receptor and PD‐1 primarily do not proliferate; however, there is a population of cells that proliferates more than T‐cell receptor stimulation alone. These highly proliferating cells could potentially be associated with PD‐1‐blockade unresponsiveness in patients. In this study, we have performed RNA sequencing and found that following PD‐1 ligation proliferating and non‐proliferating T cells have distinct transcriptional signatures. Remarkably, the proliferating cells showed an enrichment of genes associated with an activated state despite PD‐1 signalling. Additionally, circulating follicular helper T cells were significantly more prevalent in the non‐proliferating population, demonstrated by enrichment of the associated genes CXCR5, CCR7, TCF7, BCL6 and PRDM1 and validated at the protein level. Translationally, we also show that there are more follicular helper T cells in patients that respond favourably to PD‐1 blockade. Overall, the presence of transcriptionally and functionally distinct T cell populations responsive to PD‐1 ligation may provide insights into the clinical differences observed following therapeutic PD‐1 blockade.
Keywords: PD‐1, proliferation, RNA sequencing, T cells
We have observed that CD4+ T cells stimulated through the T‐cell receptor and PD‐1 primarily do not proliferate; however, there is a population of cells that proliferates more than T‐cell receptor stimulation alone. In this study, we have performed RNA sequencing and found that following PD‐1 ligation the proliferating T cells showed an enrichment of genes associated with an activated state despite PD‐1 signaling. Additionally, circulating follicular helper T cells were significantly more prevalent in the non‐proliferating population, demonstrated by enrichment of the associated genes CXCR5, CCR7, TCF7, BCL6 and PRDM1 and validated at the protein level.

Abbreviations
- CFSE
carboxyfluorescein succinimidyl ester
- ICI
Immune checkpoint inhibitors
- scRNAseq
Single cell RNA sequencing
- TCR
T cell receptor
INTRODUCTION
Immune checkpoint therapy, specifically antibodies that target PD‐1 or its ligands, blocks the interaction between the inhibitory receptor PD‐1 expressed on T cells and its ligands PDL1 and PDL2 [1, 2]. It is well established that PDL2 binds to PD‐1 with a higher affinity than does PDL1 [1, 3, 4]. Expression differences between these two ligands are also known, with PDL1 expression being expressed more ubiquitously than PDL2 [3, 5, 6, 7]. Though the expression of both ligands is upregulated under inflammatory conditions, the differences in pattern of expression, with PDL2 restricted to antigen presenting cells, support differential functional scenarios or signalling for the two ligands.
High expression of PD‐1 ligands on tumour cells represents a hijacking of the immune checkpoint system that dampens T cell‐mediated tumour clearance. Despite the success of PD‐1 targeting checkpoint inhibitors in near 30% of patients with an array of tumour types, many patients do not respond to the treatment [8]. Within those patients that do not respond, clinical evidence is mounting, suggesting that a significant portion experiences an acceleration of disease following treatment. This observation of accelerated disease has been termed hyperprogressive disease and is estimated to occur in up to 10% of patients treated with PD‐1 targeting therapy [9, 10, 11, 12, 13, 14, 15, 16, 17].
The immune checkpoint field has progressed to identifying numerous factors predictive of responsiveness to PD‐1 blockade. The contribution of the T‐cell compartment can be better understood by identifying differences in PD‐1 signalling [18]. There is an increased understanding that PD‐1 is not universally inhibitory to all T‐cell function. Similar to CTLA‐4 [19, 20, 21, 22], PD‐1 may activate individual T‐cell functions or even distinct T‐cell subsets [23]. More recently, using a phosphoproteomic approach, we showed that not all the pathways downstream of PD‐1 are inhibitory, and some unique cellular functions are clearly stimulated by this ‘inhibitory’ receptor [2]. Moreover, the expression level of PD‐1 is differential among the different T‐cell subsets, and this may correlate with potency of PD‐1 signalling [24].
A majority of previous efforts have focused on CD8+ T cell‐mediated cytotoxicity as the key to understanding immune checkpoint responsiveness. While cytotoxic CD8+ T cells are of critical importance to mounting an effective anti‐tumour immune response [25, 26]. the contribution of other T cell subsets, including CD4+ subsets, cannot be overlooked. In fact, CD4+ T cells expressing cytolytic effector proteins were found to be clonally expanded in tumours and could kill autologous tumours in vitro [26, 27, 28, 29, 30]. The gene signature of these cytotoxic CD4+ T cells was associated with response to anti‐PD‐1 [26]. Of particular interest, these T cells were found in proliferating and non‐proliferating states within the tumour [26].
In order to better understand the complete effects of PD‐1 signalling on T‐cell function, and the possible heterogeneity among T cells, we sorted T cells based on function into proliferating and non‐proliferating populations. RNA sequencing on these two populations reveals a distinct transcriptional profile in T cells that are functionally stimulated, specifically enhanced proliferation, following PD‐1 ligation.
MATERIALS AND METHODS
Isolation of CD4+ human T cells and T‐cell proliferation assay
Mononuclear cells were isolated from whole blood from healthy donors by Lymphoprep density gradient (Stemcell Technologies). CD4+ cells were then purified by CD4 microbeads (Miltenyi) and allowed to rest in enriched media (RPMI 1640, 10% heat inactivated fetal bovine serum, 1% penicillin–streptomycin, 1X non‐essential amino acids, 1mM sodium pyruvate and IL‐2 40 IU/mL) for 72 hours. For plate bound stimulation, 48‐well plates (Corning) were coated with either 5 µg/mL anti‐CD3 (UCHT1, Biolegend) and 5 µg/mL human serum albumin, 5 µg/mL anti‐CD3 and 5 µg/mL PDL1 (Biolegend), or 5 µg/mL anti‐CD3 and 5 µg/mL PDL2 (Biolegend) for 16 hours at 4 degrees. Prior to stimulation, 5 x 105 cells per condition were stained with 1 µM carboxyfluorescein succinimidyl ester (CFSE; Biolegend) for 20 minutes at 37℃ protected from light. The CFSE was neutralized with warm media, and the stained cells were washed in PBS, and resuspended in enriched media without IL‐2 and containing 1 µg/mL anti‐CD28 (CD28·2, Biolegend) then added to the appropriate stimulation wells. Where indicated, 100 ng/mL rhIL‐15 (Biolegend) was added to the cells. Cells were incubated with stimulation for 5 days at 37℃ with 5% CO2. On day 5, cells were either sorted based on CFSE intensity, stained for protein expression and analysed by flow cytometry. Each of these end‐points is detailed below.
RNA sequencing and bulk data analysis
For RNA sequencing following proliferation, CD4+ cells were isolated and stimulated as described above. On day 5 of the proliferation assay, cells were collected and sorted (BD Influx) by CFSE fluorescent intensity into proliferating and non‐proliferating populations (gating strategy in Figure S1). Sorted cells were captured directly into RNA isolation buffer and flash‐frozen. Total RNA was isolated from sorted cells using RNeasy Mini Prep (Qiagen). Library preparation (Illumina ultra‐low input RNA protocol) and sequencing were performed by Genewiz with mRNA enrichment, mRNA fragmentation, random priming, first‐ and second‐strand cDNA synthesis, end repair, 5’ phosphorylation, dA‐tailing, adaptor ligation, PCR enrichment and paired‐end sequencing using Illumina HiSeq 3000, PE 2x150. The RNA‐Seq data were analysed using Basepair software (https://www.basepairtech.com) with a pipeline that included the following steps: reads were aligned to the transcriptome derived from UCSC genome assembly (((hg19))) using STAR [31] with default parameters. Read counts for each transcript were measured using featureCounts [32]. Differentially expressed genes were determined using DESeq2 [33], and a cut‐off of 0·02 on adjusted P‐value (corrected for multiple hypotheses testing) was used for creating lists and heatmaps. Significant DEGs are included in heatmaps used to depict normalized read counts for each gene (row). GSEA was performed on normalized gene expression counts, using gene permutations for calculating P‐value. Molecular Signatures Database (MSigDB) was used to source gene sets for functional characterization of select DEG [34, 35, 36, 37, 38, 39, 40].
For subset analysis, the following gene set lists were used: Th1 cells CD3, CD4, CD94, CXCR3, CCR5, TBX21, STAT1, STAT4, IFNG, IL12R, IFNGR, IL2, LTA; Th2 cells CD3, CD4, CCR3, CCR4, CXCR4, GATA3, STAT6, IL4R, IL33R, IL17RB, CRTH2, DEC2, MAF, IL4, IL5, IL13, IL10, IRF4; Th17 cells CD3, CD4, CD161, CCR4, CCR6, RORC, STAT3, IL17A, IL23R, IL1R, RORA, IL17F, IL21, IL22, CCL20, BATF, NFKBIZ, IRF4, AHR, IL26; Th9 cells CD3, CD4, CCR6, TCRA/B, IRF4, GATA3, STAT6, SPI1, IL9, IL10; Th22 cells CD3, CD4, CCR4, CCR6, CCR10, AHR, BNC2, FOXO4, STAT3, IL22; iTreg cells CD3, CD4, CD25, IL7R, FOXP3, STAT5, SMAD2; Treg cells CD3, CD4, CD25, CTLA4, GITR, HELIOS, FOXP3; Tfh cells CD3, CD4, CD69, CXCR5, CCR7, STAT3, BCL6, IL6, IL7R, TCF7, LEF1, ICOS, IL12, STAT4, NSG2, ID3, P2RX7, CD83, PACSIN1, RGS10, POU6F1, SYNPO, TOX2, SH2B3, IL6RA, IL6ST, BCL2, NT5E, EGR2, CCND3, SLAMF6, CD200, CXCL10, PDCD1, CD40L, OX40, IL21, IL23, TGFB, IL21R, SH2D1A, IL2RA, IL2, PRDM1, HAVCR2, TMEM163, RAI14, CDKN1A, BACH2, CAPN3, TCF12.
Flow cytometry and sorting
For protein expression analysis following proliferation, CD4+ cells were isolated and stimulated as described above. On day 5 of the proliferation assay, cells were collected and stained with the following antibodies for surface protein expression: CD3 APC‐Cy7 (HIT3a, Biolegend), CD4 AlexaFluor 700 (RPA‐T4, Biolegend), CCR7 PE Dazzle 594 (G043H7, Biolegend) and CXCR5 Brilliant Violet 605 (J252D4, Biolegend). Following surface staining, cells were fixed and permeabilized with the True‐Nuclear Transcription Factor Buffer Set (Biolegend) then stained with the following antibodies for transcription factor protein expression: TCF1 PE (7F11A10, Biolegend) and Blimp1 AlexaFluor 647 (646702, R&D Systems). Alternatively, on day 5 of the proliferation assay, cells were collected and stained with the following antibodies for surface protein expression: CD3 APC‐Cy7 (HIT3a, Biolegend), CD4 AlexaFluor 700 (RPA‐T4, Biolegend), CD38 PECy7 (HB‐7, Biolegend), CD162/SELPG PE Dazzle 594 (KPL‐1, Biolegend), CD109 PE (W7C5, Biolegend), CD58/LFA‐3 APC (TS2/9, Biolegend), CCR2 BV510 (K036C2, Biolegend), CD86 BV758 (IT2·2, Biolegend), CD274/PDL1 BV650 (29E.2A3, Biolegend), PD‐1 BV711 (EH12.2H7, Biolegend) and CD49D/ITGA4 BV605 (9F10, Biolegend). Flow cytometry acquisition was done using the BD Fortessa and the BD LSRii with BD FACSDiva and data were analysed by FlowJo 10.1r7 and GraphPad Prism 8.
For restimulation experiments, CD4+ cells were isolated and stimulated as described above. On day 5 of the proliferation assay, cells were collected and sorted (BD Influx) by CFSE fluorescent intensity into proliferating and non‐proliferating populations as was described for RNA isolation. In this case, sorted cells were allowed to rest for 72 hours in enriched media. After 72 hours, cells were CFSE stained and stimulated with plate bound antibodies and ligands as described in the proliferation assay. On day 5 of the second proliferation assay, cells were collected and stained for surface protein and transcription factor expression as described.
Luminex
Primary human CD4+ T cells were isolated from peripheral blood and stimulated by anti‐CD3 and anti‐CD28 with or without PDL2. Supernatants were collected after 48 hours, and the levels of secreted cytokines were assayed by the Luminex 20 plex assay.
Single‐cell RNA data analysis
We analysed a publicly available data set GSE120575 [41] using Bio Turing Browser according to the author's pipeline. Tfh were defined based on the expression of the following markers: CD3, CD4, CD69, TCF7, ICOS, IL6ST, CCR7, LEF1, IL7R, CD200, TOX2, SH2D1A and CCND3 [42]. Responder (28 samples from 14 patients) and non‐responder (32 samples from 16 patients) groups were treated with anti‐PD‐1 antibody (Pembrolizumab), with matched pre‐ and post‐treatment samples. Each of the tumour samples was characterized based on radiologic assessments into progression/non‐responder or regression/responder. Tfh populations were cross validated with the other publicity available data sets (GSE144469 and GSE144735).
Ethics
The study was approved by the Institutional Review Board at Columbia University Medical Center, and all donors provided informed consent.
Statistical analysis
Graphs depict mean ±SEM. Statistical analyses for graphs were performed using non‐paired Student's t‐test using GraphPad Prism 8.
RESULTS
Proliferating T cells following T‐cell receptor (TCR), CD28 and PDL1/L2 stimulation have a distinct transcriptional profile
Peripherally circulating primary human CD4+ T cells proliferate in response to stimulation with anti‐CD3 and anti‐CD28 antibodies, as demonstrated by a reduction in CFSE fluorescence with each doubling (Figure 1a). PD‐1 ligation by either PDL1 or PDL2 results in an overall inhibition of proliferation, as demonstrated by an increase in cells that have undergone one or fewer doublings (Figure 1a,b; Figure S1). Focusing on those cells that proliferate following PD‐1 ligation reveals a population that underwent more than 5 doublings in response to PDL1 or PDL2, an additional doubling compared to anti‐CD3 and anti‐CD28 alone. This is demonstrated by the lower CFSE level in proliferating cells stimulated with PDL1 or PDL2 (Figure 1a (5+ doubling), c). With the goal of identifying differences between the proliferating cells with 5+ doublings and non‐proliferating (0–1 doubling) cells, we sorted the two populations following stimulation with anti‐CD3 and anti‐CD28 antibodies in combination with either PDL1 or PDL2 (gating strategy shown in Figure S2) and performed bulk RNA sequencing (Table S1–S3). These two populations are referred to as proliferating and non‐proliferating, and the combined stimulations with anti‐CD3, anti‐CD28 and either PDL1 or PDL2 are referred to as PDL1 or PDL2 stimulation for additional analysis.
FIGURE 1.
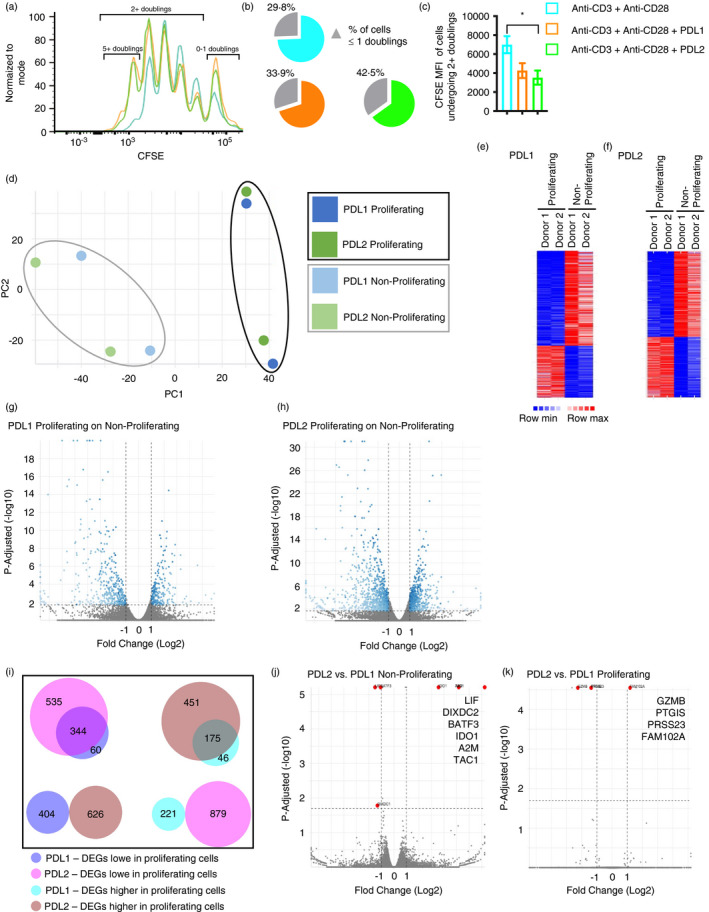
Proliferating T cells following anti‐CD3, anti‐CD28 and PDL1/L2 stimulation have a different transcriptional signature compared to anti‐CD3 and anti‐CD28. (ai). Flow cytometry of CFSE stained CD4+ human T cells stimulated with soluble anti‐CD28 and plate bound anti‐CD3 with or without PDL1 and PDL2. (b) The percentage of cells with 0–1 doublings is shown. (c) The median fluorescent intensity (MFI) of cells that underwent two or more doublings is shown. Data shown in A are representative of 15 independent donors and experiments. (d) Principle component analysis (PCA) of transcriptional profiles of proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28, and PDL1 or PDL2. (e) Heatmap of significant DEG based on the same threshold criteria; proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28 and PDL1. (f) Heatmap of significant DEG based on the same threshold criteria; proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28 and PDL2. (g) Volcano plot displaying differentially expressed genes (DEG) between proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28 and PDL1. Thresholds are set at adjusted P‐value 0·02 and log2 fold change ±1, and significant DEG are in blue. (h) Volcano plot displaying DEG between proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28 and PDL2. Thresholds are set at adjusted P‐value 0·02 and log2 fold change ±1, and significant DEG are in blue. (i) Comparisons of PDL1 and PDL2 significant DEG based on the same threshold criteria. DEG are either decreased (‘lower’) or increased (‘higher’) in proliferating cells. (j‐k) Volcano plot displaying DEG between non‐proliferating (j) or proliferating (k) cells stimulated with anti‐CD3, anti‐CD28 and either PDL1 or PDL2. Thresholds are set at adjusted P‐value 0·02 and log2 fold change ±1, and significant DEG are in red with the significant gene names indicated. Bars represent mean ±SEM; * P ≤ 0·05
We analysed differential gene expression between proliferating and non‐proliferating cells from two individuals. Principal component analysis (PCA) based on the differentially expressed genes (DEG) reveals that the transcriptional profile of proliferating cells clustered together, distinct from that of non‐proliferating cells for all donors analysed (Figure 1d). Through this analysis, we identified 625 (PDL1) and 1505 (PDL2) significant DEG with an adjusted P value ≤0·02 and log2 fold change ≤ −1 or ≥1 (Figure 1e,f,g,h). These analyses revealed that the proliferating and non‐proliferating cell populations exhibit unique transcriptional profiles.
We next sought to identify differences in DEG between PDL1 and PDL2 stimulation. While there was overlap between the two ligands in genes that were lower in proliferating cells (344 common DEG), there were also unique genes identified (60 (PDL1); 535 (PDL2)) (Figure 1i). Similar analysis of genes that were higher in proliferating cells again shows overlap (175 common DEG) and unique genes (46 (PDL1); 451 (PDL2)) (Figure 1i). No DEG was altered in opposite directions by PDL1 and PDL2 (Figure 1i). Differential gene expression analysis between non‐proliferating cells stimulated by PDL1, and those by PDL2 identified only 6 significant DEG (Figure 1j). The comparison between proliferating cells stimulated by PDL1, and those by PDL2 identified even fewer genes (Figure 1k). The expression levels of PD‐1 and PDL2 were not significantly different in proliferating and non‐proliferating cells, while the expression of PDL1 (CD274) was higher in non‐proliferating cells (Figure S3). Taken together, these initial comparisons suggest a distinct transcriptional profile in the proliferating population in response to PD‐1 signalling. Additionally, there is a high degree of transcriptional similarity in proliferating cells or in non‐proliferating cells stimulated with PDL1 as compared to PDL2.
Proliferating T cells exhibit an activated transcriptional signature
To understand the mechanism for the distinct gene expression profile of the proliferating population, we searched within the significant DEG list for enrichment of functional gene sets. The analysis showed conserved upregulation of pathways associated with T‐cell activation (cell activation, cell adhesion molecules (CAMs) and positive regulators of adhesion) and downregulation of pathways associated with T cell functional inhibition (negative regulators of activation, negative regulators of cell cycle) in the proliferating population (Figure 2a,b; Figure S4a,b). Therefore, CD4+ T cells that are highly proliferative following PDL1 or PDL2 stimulation exhibit a transcriptional profile that is not limited to proliferative genes, but also associated with genes related to other stimulatory functions such as cell activation and enhanced adhesion. Collectively, this clearly demonstrates that PD‐1 ligation has not induced an overall inhibitory state in these T cells.
FIGURE 2.
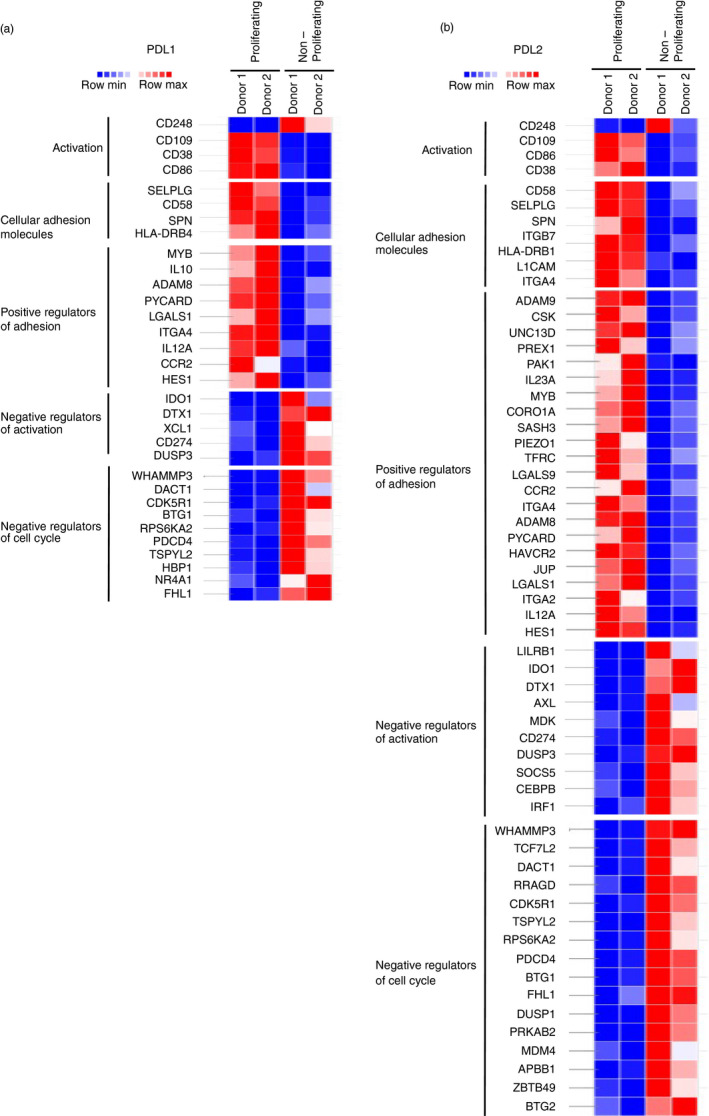
T cells that proliferate following PDL1/L2 stimulation have an activated transcriptional signature. (a‐b) Heatmap displaying significant DEG between proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28, and PDL1 or PDL2 that are within the indicated gene sets. Thresholds are set at adjusted P‐value 0·02 and log2 fold change ±1
Differences in cytokine inhibition by PD‐1 are associated with functional heterogeneity
To identify changes in cytokine secretion following stimulation, we focused on PDL2 because all of the genes identified to be significantly differentially regulated between the PDL1‐stimulated populations were also included in the PDL2 gene lists. An additional reason for the focus on PDL2 with respect to cytokine secretion is the more robust response to PDL2 stimulation that we and others have observed [1, 2]. relative to PDL1. Stimulation of primary human CD4+ T cells with anti‐CD3 and anti‐CD28 antibodies or in combination with PDL2 followed by cytokine quantification reveals that PD‐1 signalling has individualized effects on different cytokines (Table S4). While IL‐2 secretion is inhibited by PD‐1 signalling (Figure 3a), IL‐15, a cytokine that both structurally and functionally is similar to IL‐2, is unaffected (Figure 3a). Expression of IL‐2 was not significantly different between proliferating and non‐proliferating cells stimulated with PDL1 or PDL2, while others were differentially expressed in non‐proliferating cells (Figure 3b; Figure S5a). Specifically, IL‐15 was enriched in the non‐proliferating population. Population‐based assays to measure cytokine secretion cannot detect differences in response to stimulation on a cell‐by‐cell basis, but rather measure the net changes. For this reason, the secretion data taken together with the differential expression analysis suggest that proliferating and non‐proliferating cells may differentially regulate cytokine expression in response to PD‐1 ligation.
FIGURE 3.
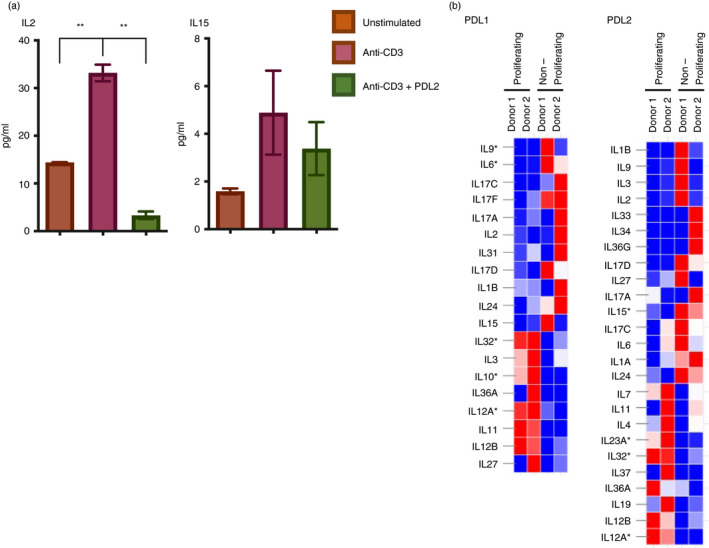
Differences in cytokine inhibition by PD‐1 are associated with functional heterogeneity. (a) Secreted levels of IL‐2 and IL‐15 from primary human CD4+ T cells stimulated with anti‐CD3 and anti‐CD28 with or without PDL2 quantified by Luminex. (b) Heatmaps displaying DEG between proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28, and PDL1 or PDL2. Thresholds are set at log2 fold change ±1, and significant DEG with adjusted P‐value 0·02 are indicated by *
To identify a role for IL‐15 in driving CD4+ T cell proliferation, we incorporated recombinant human IL‐15 into the stimulation. We found that the inclusion of IL‐15 did not change CD4+ proliferation in the context of anti‐CD3 and anti‐CD28 with or without PDL1/2; however, we observed a minor decrease in CFSE MFI within the total proliferating cells treated only with IL‐15 (Figure S5b).
Tfh cell‐associated genes are enriched in non‐proliferating compared to proliferating T cells
T cells, specifically CD4+ T cells, differentiate into different subsets including T helper (Th) 1, Th2, Th9, Th17, Th22, regulatory T cells (Treg) and follicular helper T cells (Tfh). To determine whether any of these subsets were enriched in the proliferating population, we looked for genes associated with these individual subsets within the DEG. We did not observe any association between proliferation status and genes associated with the Th1, Th2, Th9, Th17, Th22 or Treg subsets (Figure 4a–b). However, the genes associated with Tfh cells were significantly higher in the non‐proliferating population (Figure 4c–f; Figure S6a,b). Importantly, the transcription factors TCF7 and BCL6, which are important for Tfh differentiation [43], were strongly enriched in the non‐proliferating cells. Additionally, the transcription factor PRDM1, which is inhibitory to Tfh differentiation, was present at a decreased level in the non‐proliferating cells.
FIGURE 4.
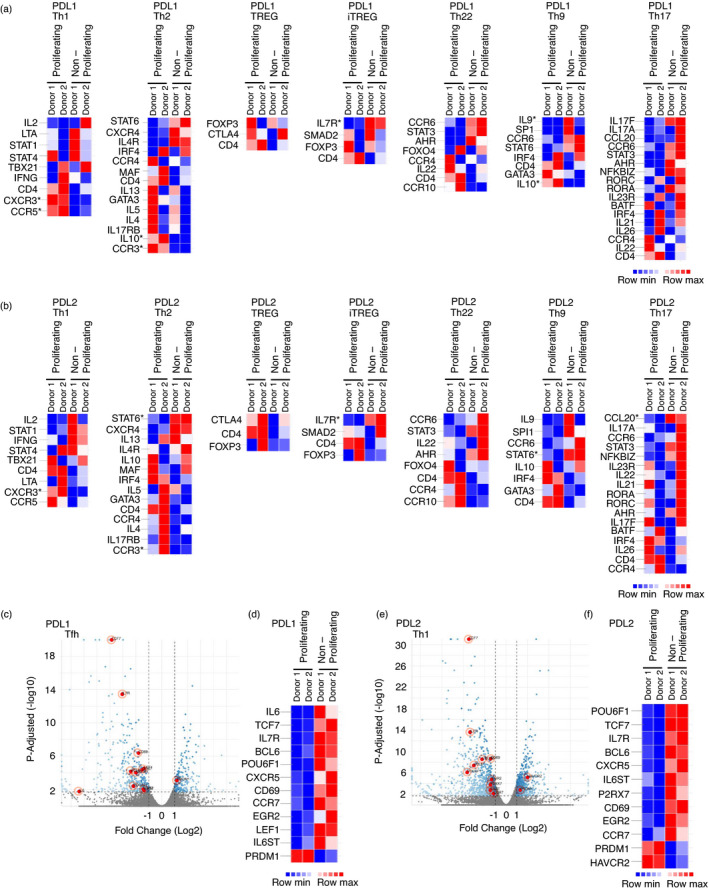
T cells that proliferate following PDL1/L2 stimulation have decreased expression of Tfh cell associated genes. (a‐b) Heatmaps displaying DEG between proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28, and PDL1 or PDL2. Thresholds are set at log2 fold change ±1, and significant DEG with adjusted P‐value 0·02 are indicated by *. (c–d) Volcano plot (c) displaying DEG between proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28 and PDL1. Thresholds are set at adjusted P‐value 0·02 and log2 fold change ±1, and significant DEG are in blue. DEG from the Tfh subset are indicated in red with those that are significant shown in the heatmap (d). (e–f). Volcano plot (e) displaying DEG between proliferating and non‐proliferating cells stimulated with anti‐CD3, anti‐CD28 and PDL2. Thresholds are set at adjusted P‐value 0·02 and log2 fold change ±1, and significant DEG are in blue. DEG from the Tfh subset are indicated in red with those that are significant shown in the heatmap (f).
To link these differences in RNA expression of TCF7, CCR7, CXCR5 and PRDM1 with protein expression and proliferation status, we performed another CFSE proliferation assay and compared the expression of these proteins with CFSE fluorescence. We found that non‐proliferating cells expressed more TCF1, CCR7 and CXCR5 and less BLIMP1 at the protein level (Figure 5a). Utilizing the surface expression of these proteins to identify the Tfh cell population, we show that Tfh cells are absent from the proliferating population (Figure 5b,c). Together, this supports the enriched presence of Tfh cells in the non‐proliferating population following stimulation with either PDL1 or PDL2.
FIGURE 5.
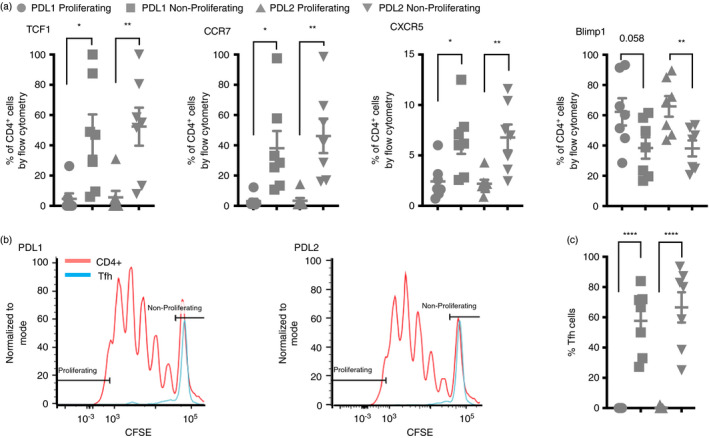
Tfh cell‐associated proteins are enriched within the proliferating population. (a) Flow cytometry data with events first gated on CD3+ events followed by CD4+ events followed by CFSE expression level. Proliferating cells were gated as the top 5% of proliferating cells based on CFSE fluorescent intensity. Shown is the per cent of CD4+ cells positive for each of the indicated proteins that were proliferating or non‐proliferating. Note TCF1 protein is encoded for by TCF7, and Blimp1 protein is encoded for by PRDM1. (b) Flow cytometry data with events again gated on CD3+ events followed by CD4+ events then CCR7+CXCR5+TCF1+Blimp1− to identify the Tfh cells. CFSE fluorescence is shown to compare proliferation of the total CD4+ population to the Tfh population. (c) The percent of cells from the identified Tfh population that were proliferating (top 5%) or non‐proliferating. *P ≤ 0·05, **P ≤ 0·01
Patients that respond favourably to PD‐1 blockade have more Tfh cells
Since Tfh cells were enriched among the non‐proliferating cells, we hypothesized that in these cells the PD‐1 pathway is, at least primarily, inhibitory, and accordingly that these cells will be susceptible to PD‐1 blockade in vivo. To test this, we analysed a publicly available single‐cell RNA sequencing data set (GSE120575) of melanoma patients pre‐ and post‐treatment with pembrolizumab, a PD‐1‐targeting antibody. Subset analysis suggested that Tfh were 6% of mononuclear cells (Figure 6a). Of the Tfh identified, 64% were isolated from patients that responded favourably to the treatment, while 36% of the cells were from patients that failed to respond to the same intervention (Figure 6b). Additionally, in comparing pre‐ and post‐treatment samples by patient, responders tended to have more Tfh cells pre‐treatment (Figure 6c). This suggests that the Tfh might predict response to PD‐1 blockade in vivo.
FIGURE 6.
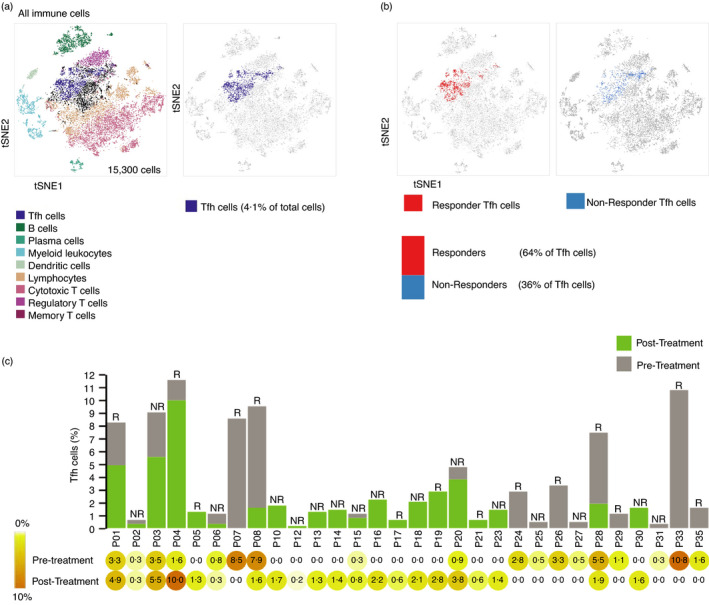
Patients that respond favourably to PD‐1 blockade have more Tfh cells. (a) tSNE (t‐distributed stochastic neighbour embedding) plot of CD45+ cells of melanoma tumours from patients treated with pembrolizumab retrieved from data set GSE120575. Different colours represent 9 clusters (cell types) defined by Bio Turing Browser. Position of Tfh cells from the tSNE plot is shown in purple. (b) tSNE plots of Tfh cells corresponding to responders (red; left) and non‐responders (blue; right), respectively. Bar representing the percentage of Tfh cells among responders vs. non‐responders is shown. (c) Bar graph representing Tfh cell counts as per cent of total cell counts, with pre‐ and post‐treatment values indicated as grey and green, respectively. R = responder, NR = non‐responder. % values are shown beneath the bar graph in the heatmap
DISCUSSION
We have observed distinct functional instances in which PD‐1 ligation leads to inconsistency in inhibition. We propose that the differences in these observations can be attributed to heterogeneity within the stimulated population wherein a subpopulation of cells may be inhibited while another is not. Whole population‐based methods of detection will not appreciate these differences. By sorting CD4+ T cells first by function before transcriptome analysis, we are able to associate PD‐1 function with genetic signatures, and potentially identify T‐cell subsets that respond differentially to PD‐1 ligation. We demonstrate that the functional phenotype of enhanced proliferation is present in cells co‐stimulated with PDL1 or PDL2. Furthermore, though there were few genes differentially regulated between PDL1 and PDL2 proliferating or non‐proliferating the list of DEGs following PDL2 co‐stimulation was larger than that following PDL1. In this way, the PDL1 gene signature can be considered a subset of the PDL2 gene signature. Of course, it must be noted that following this functional sort with single cell RNA sequencing would lend further power to this study and is a promising path forward.
We report here that PD‐1 ligation leads to activation of some T‐cell functions. Through the quantification of secreted cytokines following stimulation of the T‐cell receptor and PD‐1 ligation, we demonstrate that the levels of numerous cytokines are decreased, while others remain elevated. In the case of IL‐15, we show that IL‐15 transcripts are more abundant in the non‐proliferating cells than proliferating cells. At the protein level, through Luminex, we show that there is no overall difference in secreted levels of IL‐15 when PDL2 is added to anti‐CD3 stimulation. Combined, these data may suggest that the differential in expression from the proliferating and non‐proliferating cells in the anti‐CD3 and PDL2‐combined stimulation yields no net change in IL‐15 secretion. We have previously reported that stimulation of the T cell receptor and PD‐1 ligation leads to increased phosphorylation of a number of serine and threonine sites [2]. This finding gives evidence for signalling downstream of PD‐1 that may be SHP‐2 independent. Further, some of these phospho‐sites are associated with activation of downstream functions, while others are associated with inhibition of downstream functions.
The surprising observation of a population of CD4+ T cells with enhanced proliferation following PD‐1 ligation compared to T‐cell receptor stimulation alone has important implications because of the essential role proliferation plays in T‐cell function in the context of the immune response to tumours. Understanding the differences between the proliferating and non‐proliferating populations holds promise for identifying PD‐1 signalling mediators that may enhance current therapeutic targeting strategies. As a field, we now better appreciate the contribution of CD4+ T cells in mounting an effective anti‐tumour immune response. Tumour‐infiltrating CD4+ T cells are heterogeneous in subset distribution and function, and the gene signature of these subsets can be correlated with response to anti‐PD‐1 [26]. Differential signalling in subsets is exemplified by CTLA‐4, which is an inhibitory molecule in most T cell subsets though plays an essential activating role in Treg function [44, 45]. Additionally, understanding that populations of CD4+ T cells respond to PD‐1 ligation differently has implications beyond proliferation. Even the characterization of PD‐1 expression as a marker of T‐cell exhaustion is complex due to the fact that PD‐1 blockade could restore function in some ‘exhausted’ T cells [46]. It has been convenient to classify PD‐1 as an inhibitor of T‐cell function; however, the true signalling events underlying the functional consequences of PD‐1 ligation are proving to be far more nuanced. Better understanding the relationship between PD‐1 and IL‐15 regulation has important implications for tumour immunology. The intratumoural levels of IL‐15 have been previously shown to play an essential role in tumour regression [47], although it has also been shown that IL‐15 may cause upregulation of surface PD‐1 [48, 49]. These two findings appear to be at odds, but may suggest a potential feedback or T‐cell inhibition mechanism. It also provides a potential explanation for why PD‐1 ligation does not inhibit IL‐15 expression. IL‐15 has been previously shown to promote CD8+ T‐cell proliferation [50, 51], and in this study, we demonstrate that IL‐15 stimulation alone increased baseline proliferation of CD4+ T cells. A more in‐depth study is required to better understand the contribution of IL‐15 stimulation to TCR‐mediated CD4+ T‐cell proliferation. Additionally, we report that transcript levels of IL‐15 are enriched within the populations of T cells that were non‐proliferating following anti‐CD3, anti‐CD28, and either PDL1 or PDL2. Tfh cells were also enriched within the non‐proliferating population, leaving open the possibility of a link between the two.
Single‐cell RNA sequencing (scRNAseq) of tumours from patients with hyperprogressive disease following immune checkpoint inhibitors (ICI) uncovered a predictive gene expression signature [52]. That study focused primarily on the signature within the tumour cells. Our data presented here provide complementary insight into the T cell transcriptional signatures that may correlate with responsiveness to anti‐PD‐1. We show additional support for the importance of Tfh cells in immune checkpoint function and overall immune function. We have found an enrichment of the Tfh signature in the non‐proliferating population, and an enrichment of Tfh cells in tumours of patients that responded to PD‐1 blockade. Notably, Tfh cells do not proliferate following TCR stimulation, however do secrete many cytokines. Within the tumour, a new expression profile has been used to identify Tfh‐like cells that are CD4+FOXP3−PD‐1HI and accumulate in tumours as a function of tumour burden, and functionally, the presence of these cells correlates with tumour regression in response to combined anti‐PD‐1 and anti‐CTLA‐4 [53]. Of particular interest, response to CTLA‐4 blockade was found to be enhanced in Tfh‐deficient mice [53]. The presence of Tfh within breast and colorectal tumours was associated with positive prognosis, where Tfh gene signatures correlated with increased overall survival [54, 55]. The correlation of Tfh with improved disease‐free survival was influenced by the intratumoural localization of the Tfh cells along with the co‐localized cell types [55].
The intratumoural Tfh population may also be a highly proliferative population following ICI, and this may correlate with positive outcome. A study that performed scRNAseq on mouse models of triple negative breast cancer with or without ICI noted that the dominant CD4+ effector memory subset within responsive tumours is Tfh and that B‐cell activation in this context is dependent on this T‐cell population [56]. Additionally, this study reported that CD4+ cells from responsive tumours have high expression of proliferation genes [56]. This proliferating population of T cells is not directly analogous to the proliferating population studied here due to the fact that the cells in their study were exposed to PD‐1 and CTLA‐4 inhibitors. In fact, the T cells proliferating after treatment with ICI functionally are more closely modelled by the non‐proliferating population in this study, where instead of ICI, PD‐1 ligands were used. This raises the question of how the proliferating and non‐proliferating populations identified in this study would respond to treatment with ICI.
The Tfh subset plays an important role in ICI response in disease states other than cancer as well. Effective treatment with CTLA‐4‐Ig to block CD28 costimulation has been shown to decrease circulating Tfh cell frequency in patients with multiple sclerosis [57], Sjogren's syndrome [58], rheumatoid arthritis [59] and type 1 diabetes [45, 60]. Responsiveness to CTLA‐4‐Ig in type 1 diabetes patients was correlated with a lower baseline frequency of a population of Tfh cells [45]. An increase in Tfh cells following anti‐PD‐1 therapy has been correlated with an increased presence of thyroid autoantibodies and the development of Hashimoto's disease [61]. A proposed mechanism for the increase in Tfh cells following therapy is increased proliferation of this population with PD‐1 inhibition [61].
One remaining question coming out of this study is whether the proliferating population remains committed to this fate or if the cells are in a dynamic state capable of becoming inhibited by PD‐1 ligation in a subsequent challenge. To aim to address this, we stimulated cells from three individuals, sorted based on CFSE fluorescence for proliferating and non‐proliferating cells and then re‐stimulated the cells to again observe proliferation. While we did observe differences of proliferative capacity in the two populations of cells upon re‐stimulation, the individual variability was far too great to draw conclusions (Figure S7).
CONFLICT OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
M.S. and A.M. performed the conceptualization; M.S., A.S.T. and A.M. performed methodology; M.S. and S.M.B. involved in formal analysis and investigated the document; M.S. wrote original draft; M.S. and A.M. reviewed and edited the manuscript; A.M. contributed funding acquisition.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGMENT
Research reported in this publication was performed in the CCTI Flow Cytometry Core and supported in part by the Office of the Director, National Institutes of Health under awards S10RR027050 and S10OD020056.
Strazza M, Bukhari S, Tocheva AS, Mor A. PD‐1‐induced proliferating T cells exhibit a distinct transcriptional signature. Immunology. 2021;164:555–568. 10.1111/imm.13388
Funding information
This work was supported by grants from the NIH (AI125640, CA231277, AI150597), Cancer Research Institute (A.M.)
Contributor Information
Marianne Strazza, Email: ms5800@cumc.columbia.edu.
Adam Mor, Email: am5121@cumc.columbia.edu.
DATA AVAILABILITY STATEMENT
The data set generated during this study (GSE159774) is available at www.ncbi.nlm.nih.gov.geo (GEO).
REFERENCES
- 1. Philips EA, Garcia‐Espana A, Tocheva AS, Ahearn IM, Adam KR, Pan R, et al. The structural features that distinguish PD‐L2 from PD‐L1 emerged in placental mammals. J Biol Chem. 2020;295:4372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tocheva AS, Peled M, Strazza M, Adam K, Lerrer S, Nayak S, et al. Quantitative phosphoproteomic analysis reveals involvement of PD‐1 in multiple T cell functions. J Biol Chem. 2020;295:18036–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. [DOI] [PubMed] [Google Scholar]
- 4. Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, et al. Structural and functional analysis of the costimulatory receptor programmed death‐1. Immunity. 2004;20:337–47. [DOI] [PubMed] [Google Scholar]
- 5. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dong H, Zhu G, Tamada K, Chen L. B7–H1, a third member of the B7 family, co‐stimulates T‐cell proliferation and interleukin‐10 secretion. Nat Med. 1999;5:1365–9. [DOI] [PubMed] [Google Scholar]
- 7. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD‐L2 is a second ligand for PD‐1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. [DOI] [PubMed] [Google Scholar]
- 8. Wu X, Gu Z, Chen Y, Chen B, Chen W, Weng L, et al. Application of PD‐1 Blockade in Cancer Immunotherapy. Comput Struct Biotechnol J. 2019;17:661–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han XJ, Alu A, Xiao YN, Wei YQ, Wei XW. Hyperprogression: A novel response pattern under immunotherapy. Clin Transl Med. 2020;10:e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020;38:326–33. [DOI] [PubMed] [Google Scholar]
- 11. Ferrara R, Matos I. Atypical patterns of response and progression in the era of immunotherapy combinations. Future Oncol. 2020;16:1707–13. [DOI] [PubMed] [Google Scholar]
- 12. Kas B, Talbot H, Ferrara R, Richard C, Lamarque JP, Pitre‐Champagnat S, et al. Clarification of definitions of hyperprogressive disease during immunotherapy for non‐small cell lung cancer. JAMA Oncol. 2020;6:1039–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zang H, Peng J, Zheng H, Fan S. Hyperprogression After Immune‐Checkpoint Inhibitor Treatment: Characteristics and Hypotheses. Front Oncol. 2020;10:515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X, Wang F, Zhong M, Yarden Y, Fu L. The biomarkers of hyperprogressive disease in PD‐1/PD‐L1 blockage therapy. Mol Cancer. 2020;19:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Refae S, Gal J, Brest P, Giacchero D, Borchiellini D, Ebran N, et al. Hyperprogression under Immune Checkpoint Inhibitor: a potential role for germinal immunogenetics. Sci Rep. 2020;10:3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rauch DA, Conlon KC, Janakiram M, Brammer JE, Harding JC, Ye BH, et al. Rapid progression of adult T‐cell leukemia/lymphoma as tumor‐infiltrating Tregs after PD‐1 blockade. Blood. 2019;134:1406–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD‐1(+) regulatory T cells amplified by PD‐1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019;116:9999–10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD‐1 pathway. Sci Adv. 2020;6:eabd2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walker LS, Sansom DM. Confusing signals: recent progress in CTLA‐4 biology. Trends Immunol. 2015;36:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Melliere D, Veit R, Becquemin JP, Etienne G. Should all spontaneous popliteal aneurysms be operated on? J Cardiovasc Surg (Torino). 1986;27:273–7. [PubMed] [Google Scholar]
- 21. Pauken KE, Dougan M, Rose NR, Lichtman AH, Sharpe AH. Adverse events following cancer immunotherapy: obstacles and opportunities. Trends Immunol. 2019;40:511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chikuma S. CTLA‐4, an essential immune‐checkpoint for T‐cell activation. Curr Top Microbiol Immunol. 2017;410:99–126. [DOI] [PubMed] [Google Scholar]
- 23. Riley JL. PD‐1 signaling in primary T cells. Immunol Rev. 2009;229:114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peled M, Tocheva AS, Sandigursky S, Nayak S, Philips EA, Nichols KE, et al. Affinity purification mass spectrometry analysis of PD‐1 uncovers SAP as a new checkpoint inhibitor. Proc Natl Acad Sci U S A. 2018;115:E468–E77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E, et al. Intratumoral CD4(+) T cells mediate anti‐tumor cytotoxicity in human bladder cancer. Cell. 2020;181:1612–1625.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sledzinska A, Vila de Mucha M, Bergerhoff K, Hotblack A, Demane DF, Ghorani E, et al. Regulatory T cells restrain interleukin‐2‐ and blimp‐1‐dependent acquisition of cytotoxic function by CD4(+) T cells. Immunity. 2020;52:151–166.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sacher AG, St Paul M, Paige CJ, Ohashi PS. Cytotoxic CD4(+) T cells in bladder cancer‐A new license to kill. Cancer Cell. 2020;38:28–30. [DOI] [PubMed] [Google Scholar]
- 29. Nagasaki J, Togashi Y, Sugawara T, Itami M, Yamauchi N, Yuda J, et al. The critical role of CD4+ T cells in PD‐1 blockade against MHC‐II‐expressing tumors such as classic Hodgkin lymphoma. Blood Adv. 2020;4:4069–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maehara T, Kaneko N, Perugino CA, Mattoo H, Kers J, Allard‐Chamard H, et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J Clin Invest. 2020;130:2451–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 33. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gene Set: GO_CELL_ACTIVATION . 2020. Available from https://www.gsea‐msigdb.org/gsea/msigdb/cards/GO_CELL_ACTIVATION.html. [Accessed 2020 October 21 2020.]
- 35. Gene Set: GO_NEGATIVE_REGULATION_OF_CELL_CYCLE . 2020. Available from https://www.gsea‐msigdb.org/gsea/msigdb/cards/GO_NEGATIVE_REGULATION_OF_CELL_CYCLE.html. [Accessed 2020 October 21 2020.]
- 36. Gene Set: GO_NEGATIVE_REGULATION_OF_CELL_ACTIVATION . 2020. Available from https://www.gsea‐msigdb.org/gsea/msigdb/cards/GO_NEGATIVE_REGULATION_OF_CELL_ACTIVATION.html. [Accessed 2020 October 21 2020.]
- 37. Gene Set: GO_POSITIVE_REGULATION_OF_CELL_ADHESION . 2020. Available from https://www.gsea‐msigdb.org/gsea/msigdb/cards/GO_POSITIVE_REGULATION_OF_CELL_ADHESION.html. [Accessed 2020 October 21 2020.]
- 38. Gene Set: KEGG_CELL_ADHESION_MOLECULES_CAMS . 2020. Available from https://www.gsea‐msigdb.org/gsea/msigdb/cards/KEGG_CELL_ADHESION_MOLECULES_CAMS.html. [Accessed 2020 October 21 2020.]
- 39. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sade‐Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175:998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weinstein JS, Lezon‐Geyda K, Maksimova Y, Craft S, Zhang Y, Su M, et al. Global transcriptome analysis and enhancer landscape of human primary T follicular helper and T effector lymphocytes. Blood. 2014;124:3719–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rao DA. T cells that help B cells in chronically inflamed tissues. Front Immunol. 2018;9:1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wing K, Onishi Y, Prieto‐Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA‐4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–5. [DOI] [PubMed] [Google Scholar]
- 45. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting co‐stimulatory molecules in autoimmune disease. Nat Rev Drug Discov. 2020;19:860–83. [DOI] [PubMed] [Google Scholar]
- 46. Verdon DJ, Mulazzani M, Jenkins MR. Cellular and molecular mechanisms of CD8(+) T Cell differentiation, dysfunction and exhaustion. Int J Mol Sci. 2020;21:7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Santana Carrero RM, Beceren‐Braun F, Rivas SC, Hegde SM, Gangadharan A, Plote D, et al. IL‐15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc Natl Acad Sci U S A. 2019;116:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hakim MS, Jariah ROA, Spaan M, Boonstra A. Interleukin 15 upregulates the expression of PD‐1 and TIM‐3 on CD4(+) and CD8(+) T cells. Am J Clin Exp Immunol. 2020;9:10–21. [PMC free article] [PubMed] [Google Scholar]
- 49. Berce PC, Bernstein RS, MacKinnon GE, Sorum S, Martin E, MacKinnon KJ, et al. Immunizations at Wisconsin Pharmacies: Results of a statewide vaccine registry analysis and pharmacist survey. Vaccine. 2020;38:4448–56. [DOI] [PubMed] [Google Scholar]
- 50. Liu K, Catalfamo M, Li Y, Henkart PA, Weng NP. IL‐15 mimics T cell receptor crosslinking in the induction of cellular proliferation, gene expression, and cytotoxicity in CD8+ memory T cells. Proc Natl Acad Sci U S A. 2002;99:6192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Van Acker HH, Anguille S, Willemen Y, Van den Bergh JM, Berneman ZN, Lion E, et al. Interleukin‐15 enhances the proliferation, stimulatory phenotype, and antitumor effector functions of human gamma delta T cells. J Hematol Oncol. 2016;9:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xiong D, Wang Y, Singavi AK, Mackinnon AC, George B, You M. Immunogenomic landscape contributes to hyperprogressive disease after anti‐PD‐1 immunotherapy for cancer. iScience. 2018;9:258–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zappasodi R, Budhu S, Hellmann MD, Postow MA, Senbabaoglu Y, Manne S, et al. Non‐conventional inhibitory CD4(+)Foxp3(‐)PD‐1(hi) T cells as a biomarker of immune checkpoint blockade activity. Cancer Cell. 2018;33:1017–1032.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gu‐Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. 2013;123:2873–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–95. [DOI] [PubMed] [Google Scholar]
- 56. Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia‐Recio S, Mott KR, et al. B cells and T follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell. 2019;179:1191–1206.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Glatigny S, Hollbacher B, Motley SJ, Tan C, Hundhausen C, Buckner JH, et al. Abatacept targets T follicular helper and regulatory T cells, disrupting molecular pathways that regulate their proliferation and maintenance. J Immunol. 2019;202:1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Verstappen GM, Meiners PM, Corneth OBJ, Visser A, Arends S, Abdulahad WH, et al. Attenuation of follicular helper T cell‐dependent B cell hyperactivity by abatacept treatment in primary sjogren's syndrome. Arthritis Rheumatol. 2017;69:1850–61. [DOI] [PubMed] [Google Scholar]
- 59. Fukuyo S, Nakayamada S, Iwata S, Kubo S, Saito K, Tanaka Y. Abatacept therapy reduces CD28+CXCR5+ follicular helper‐like T cells in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2017;35:562–70. [PubMed] [Google Scholar]
- 60. Bettelli E, Campbell DJ. Circulating TFH cells as a marker for early therapeutic intervention in T1D. Nat Immunol. 2020;21:1141–2. [DOI] [PubMed] [Google Scholar]
- 61. Torimoto K, Okada Y, Nakayamada S, Kubo S, Tanaka Y. Anti‐PD‐1 antibody therapy induces hashimoto's disease with an increase in peripheral blood follicular helper T cells. Thyroid. 2017;27:1335–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Table S1
Table S2
Table S3
Table S4
Data Availability Statement
The data set generated during this study (GSE159774) is available at www.ncbi.nlm.nih.gov.geo (GEO).