

Summary
Current therapies to treat coronavirus disease 2019 (COVID-19) involve vaccines against the spike protein S1 of SARS-CoV-2. Here, we outline an alternative approach involving chimeric antigen receptors (CARs) in T cells (CAR-Ts). CAR-T recognition of the SARS-CoV-2 receptor-binding domain (RBD) peptide induced ribosomal protein S6 phosphorylation, the increased expression of activation antigen, CD69 and effectors, interferon-γ, granzyme B, perforin, and Fas-ligand on overlapping subsets of CAR-Ts. CAR-Ts further showed potent in vitro killing of target cells loaded with RBD, S1 peptide, or expressing the S1 protein. The efficacy of killing varied with different sized hinge regions, whereas time-lapse microscopy showed CAR-T cluster formation around RBD-expressing targets. Cytolysis of targets was mediated primarily by the GZMB/perforin pathway. Lastly, we showed in vivo killing of S1-expressing cells by our SARS-CoV-2 CAR-Ts in mice. The successful generation of SARS-CoV-2 CAR-Ts represents a living vaccine approach for the treatment of COVID-19.
Subject areas: Cell biology, Immunology, Molecular microbiology
Graphical abstract
Highlights
-
•
Cytolytic CAR-Ts can be successfully developed against SARS-CoV-2
-
•
CAR-Ts binding to RBD peptide induced effectors IFN-γ, GZMB, Perforin and FasL
-
•
CAR-Ts with different hinge regions showed differences in target killing
-
•
SARS-CoV-2 CAR-Ts show successful in vivo killing of S1-expressing cells in mice
Cell biology; Immunology; Molecular microbiology
Introduction
SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) is a positive-sense single-stranded RNA virus and the cause of coronavirus disease 2019 (COVID-19). The severity of COVID-19 ranges from asymptomatic to mild self-limiting disease to severe and acute respiratory distress syndrome, neurological symptoms, and death (Amanat and Krammer, 2020; Tay et al., 2020; ter Meulen et al., 2006). SARS-CoV-2 has an incubation period of 4–7 days before symptom onset, followed in some cases by the progression to severe disease (Peng et al., 2020). Current therapies include the use of vaccines, convalescent plasma (Bloch et al., 2020), and re-purposed anti-viral treatments (Sarzi-Puttini et al., 2020) such as umifenovir (Xu et al., 2020) and remdesivir/ribavirin (Jean et al., 2020). Other approaches include the use of anti-interleukin-7 (IL-7), anti-IL-6 receptor antibodies, and corticosteroids to dampen the inflammatory immune response (Russell et al., 2020; Tharmarajah et al., 2021) and neutralizing monoclonal antibodies (mAbs) that bind to viral proteins that are critical for attachment and entry to mammalian cells (Altmann and Boyton, 2020).
SARS-CoV-2 contains glycosylated spike (S) protein that facilitates viral attachment and cell entry and plays a critical role in the elicitation of the host immune response (Grifoni et al., 2020; Meckiff et al., 2020; Premkumar et al., 2020; Varchetta et al., 2020; Wu et al., 2020). It binds to the human angiotensin-converting enzyme 2 (ACE2) (Hoffmann et al., 2020b; Letko et al., 2020), which is the target of neutralizing antibodies (Cao et al., 2020; Shi et al., 2020). The S protein forms a trimer where each monomer has two subunits (S1 and S2) separated by a cleavage site that is recognized by host cell proteases (Hoffmann et al., 2020a). The S1 subunit is composed of the signal peptide (SP), N terminal domain (NTD), and receptor-binding domain (RBD), whereas the S2 subunit mediates membrane fusion. The RBD region is comprised of a 5-stranded anti-parallel beta-sheet between the β4-7 strands and is an extended insertion of short β5-6 strands. The extension carries the receptor-binding motif (RBM) that binds to the ACE2 receptor for entry into respiratory and digestive epithelial cells (Lan et al., 2020). The three RBDs on the S protein head show conformational variability. In a closed conformation, the three RBDs are flat with the RBM occluded, whereas in an open conformation, one or more RBDs lift to expose the RBM.
Severe COVID-19 patients show marked lymphopenia (Braun et al., 2020; Cizmecioglu et al., 2020), increased pro-inflammatory cytokines and chemokines (Song et al., 2020), and high neutrophil levels in peripheral blood (Song et al., 2020; Varchetta et al., 2020). Several studies have delineated aspects of cell immunity that determines disease outcomes (Braun et al., 2020; Campbell et al., 2020; Cheng et al., 2020; Weiskopf et al., 2020). Mild cases are associated with antibody response, CD4 and CD8 T cell responses, whereas severe cases involve the loss of T cells and reduced antibody responses (Rydyznski Moderbacher et al., 2020; Weiskopf et al., 2020). To date, the vaccines have been approved, although the long-term efficacy of antibodies induced by these vaccines is still under evaluation (Amanat and Krammer, 2020). Other approaches include adoptive cell transfer of immune and stem cells (Shetty, 2020), serum from convalescent or recovered patients, and the use of hexapeptides corresponding to the ACE2-interacting domain of SARS-CoV-2 (AIDS) to inhibit S1 subunit binding to the ACE2 receptor (Paidi et al., 2021).
Because the long-term efficacy of antibodies induced by vaccines and the overall resistance to new variants is unclear, it will be important to consider alternate approaches. It is also unclear how well immunocompromised or aged individuals respond to immunization, especially for long-term immunity and immune memory. In influenza infection, CD8 T cells cannot prevent infection but are needed to resolve infections (Allan et al., 1990). In mouse models, T cells can promote recovery from lethal flu infections in the absence of B cells and antibodies (Epstein et al., 1998; Graham and Braciale, 1997). In humans, the number of influenza-specific cytolytic T cells (CTLs) correlates with the rate of viral clearance (McMichael et al., 1986). The presentation of viral peptides by major histocompatibility class 1 antigens activates CD8+ T cells for the development of effector functions against host cells presenting viral determinants. Effectors control viral expansion via the production of cytokines such as interferon-gamma (IFN-γ), the expression of Fas-ligand, and the release of granules containing perforin and granzymes such as granzyme B (GZMB) (Topham et al., 1997). In keeping with the importance of cellular-mediated immunity in COVID-19, a reduction in CD8 cells is correlated with a worse prognosis (Urra et al., 2020), whereas SARS-CoV-2-specific T cell responses promote disease resolution and reduce the severity of infection (Rydyznski Moderbacher et al., 2020). The fact that CD4 and CD8 T cell responses are induced during infection indicates that SARS-CoV-2 peptides can be processed and presented to T cells (Grifoni et al., 2020; Meckiff et al., 2020; Premkumar et al., 2020; Rydyznski Moderbacher et al., 2020; Weiskopf et al., 2020). Mass spectrometry has detected SARS-CoV-2 virus-derived peptides from the epithelial cells, or gargle, of patients with infections (Ihling et al., 2020; Nikolaev et al., 2020), whereas another study reported SARS-CoV-2 S protein expressed on the surface of infected cells, resulting in syncytia formation with ACE2-expressing cells (Buchrieser et al., 2021).
Although the development and use of chimeric antigen receptor (CAR) is an effective immunotherapy against hematologic cancers, the potential of CAR-T cells (CAR-Ts) in preventing or treating severe cases of COVID-19 has not yet been exploited (Kawalekar et al., 2016; Posey et al., 2016). CARs combine an antigen recognition domain of a specific antibody with an intracellular domain of the CD3ζ chain or FcγRI protein into a single chimeric protein (Gross et al., 1989; Irving and Weiss, 1991). The inclusion of intracellular motifs from the co-receptors CD28 and CD137 (4-1BB) extend the longevity of CAR-Ts (Jena et al., 2010; Maus and June, 2016). CAR-Ts against relapsed acute lymphoblastic leukemia (ALL) have been associated with durable and sustained remissions for up to 24 months (Maude et al., 2014). Severe cytokine-release syndrome in a minority of patients has been effectively treated with the anti-IL-6 receptor antibody, tocilizumab (Maude et al., 2014). Three CAR-T-based drugs (Breyanzi, Yescarta, and Kymriah) have been approved by Food and Drug Administration, whereas other trials are presently underway in many academic medical centers (Sadelain et al., 2009). In the case of SARS-CoV-2 (Grifoni et al., 2020; Meckiff et al., 2020; Premkumar et al., 2020; Rydyznski Moderbacher et al., 2020; Weiskopf et al., 2020), the induction of T cell responses with different clonotypes indicates that SARS-CoV-2-derived peptides are likely available for recognition by appropriately designed CAR-Ts.
Surprisingly, although some CAR-Ts have been used to target hepatitis B virus envelope proteins in mice (Krebs et al., 2013), the potential of CAR-Ts in the treatment of SARS-CoV-2 infection has not been explored (Seif et al., 2019). In this study, we aimed to evaluate the feasibility of anti-SARS-CoV-2 CAR immunotherapy. We report the design and generation of different CAR-Ts that recognize and kill target cells loaded with the RBD region of the SARS-CoV-2 spike protein. The CAR-Ts could be phenotypically segregated into subsets expressing effector molecules, granzyme B, perforin, IFN-γ, and FasL. Further, our work demonstrated that CAR-Ts against SARS-CoV-2 can be generated to elicit the in vitro and in vivo killing of RBD and S1-bearing target cells. This approach offers a new potential therapeutic approach for the treatment of COVID-19 for longer-term immunity. Future work will evaluate the efficacy of the different generated CAR-Ts for targeting the emerging threat caused by the different variants of SARS-CoV-2 Spike mutations.
Results
Anti-SARS-CoV-2 CAR design
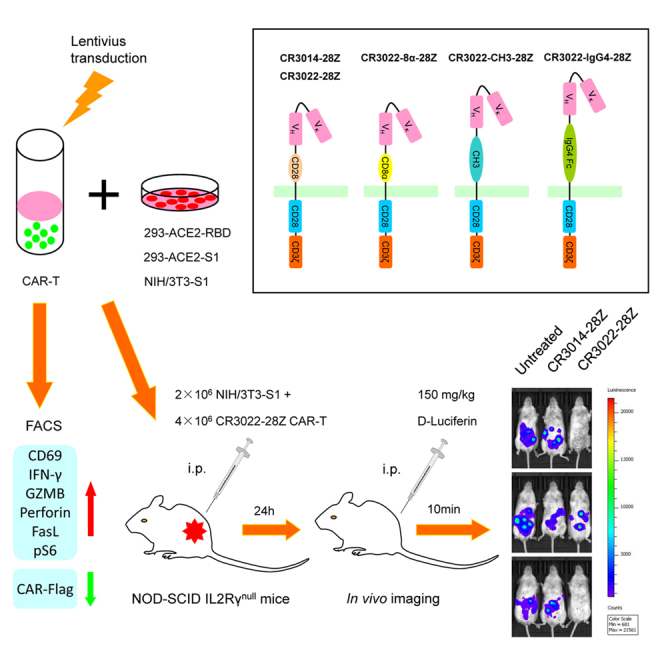
A neutralizing antibody from a convalescent SARS patient termed CR3022 binds to the RBD region of both SARS-CoV-1 and SARS-CoV-2 spike protein (Tian et al., 2020). Based on its sequence, the CR3022 single-chain variable fragment (scFv) was cloned into the bicistronic lentiviral vector (pHIV) followed by a Flag-tag sequence (DYKDDDDK), a hinge region, the CD28 transmembrane (TM), and intracellular domain and the CD3ζ intracellular domain. CARs are comprised of different hinge regions that included either a 39 amino acid (aa) CD28 hinge (CR3022-28Z) (residues 114–152), a 47 aa CD8α chain (residues 136–182) (CR3022-8a-28Z), a 119 aa CH3 region hinge (residues 221–327) (CR3022-CH3-28Z), or a 229 aa IgG4 region hinge (residues 99–327) (CR3022-IgG4-28Z) (Figure 1A). As a control, we generated a CAR lacking an scFv region (Flag-28Z) and another with the scFv region of another monoclonal antibody termed CR3014 that reacts only with the RBD region of SARS-CoV-1 (CR3014-28Z). The n represents the number of single experiments, each from a different, individual donor. This measure applied to all figures in the paper. Each pHIV bicistronic lentiviral construct contained IRES-EGFP, allowing for sorting and tracking the CAR-Ts.
Figure 1.

Design of anti-SARS-CoV-2 S1 CAR-Ts and effect on CD69 activation marker expression
(A) Design of anti-SARS-CoV-2 S1 CAR-Ts. The CR3022 scFv region was coupled to a Flag-Tag followed by different hinge regions and then the CD28 transmembrane (TM) domain and intracellular domain and a CD3ζ intracellular domain. The hinge regions included a 39 amino acid CD28 hinge (CR3022-28Z), a 47 aa CD8α chain (CR3022-8a-28Z), a 119 aa CH3 region hinge (CR3022-CH3-28Z), or a 229 aa IgG4 hinge (CR3022-IgG4-28Z). As a control, a CAR lacking a scFv region (Flag-28Z) and another with the scFv region of monoclonal antibody CR3014 (CR3014-28Z).
(B) Flow cytometric profiles of the CAR construct expression in T cells. CAR expression was detected by staining the Flag-tag, and the EGFP was used for cell sorting (n = 5) (also see Figures S1 and S2).
(C) Expression of surface activation antigen CD69 on CAR-Ts. (Left panel) Histogram showing CD69 expression on different CAR-Ts (Flag-28Z, CR3014-28Z, CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, and CR3022-IgG4-28Z). (Right panel) FACS profile showing CD69 expression on the different CARs after incubation with 293-ACE2 or 293-ACE2-RBD cells for 20 h. Data are shown as mean ± SD of triplicate samples. ∗∗∗∗p < 0.0001 (compared with Flag-28Z by one-way ANOVA, n = 5) (also see Figure S3).
(D) Increased CD69 expression on CR3022-28Z CAR-T in response to 293-ACE2 cells preincubated with different concentrations of the RBD peptide. CAR-Ts were co-cultured for 20 h with 293-ACE2 cells preincubated with various concentrations of the RBD peptide for 1 h prior to co-culture. Data are shown as mean ± SD of triplicate samples. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (two-tailed Student’s t test, n = 2).
(E) viSNE profiles of CAR and CD69 expression on CAR-Ts. (Left panel) Anti-Flag staining shows the surface expression of the Flag-tagged CARs in islands i, ii, and iii after 20 h of incubation with 293-ACE2 and 293-ACE2-RBD cells, whereas anti-CD69 staining shows the expression of CD69 in CAR-Ts (CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, or CR3022-IgG4-28Z CAR-Ts) (n = 3). (Right panels) Histograms showing changes in the percent representation of CAR-Ts in island i relative to total CAR-Ts (upper right panel) and in the relative mean fluorescent intensity (MFI) values in island i (lower right panel) (also see Figure S4).
(F) Induction of S6 phosphorylation in response to 293-ACE2-RBD cells. CR3022-8a-28Z CAR-Ts were incubated with 293-ACE2 or 293-ACE2-RBD cells for 20 min prior to the detection of S6 phosphorylation by flow cytometry. In parallel, CR3022-8a-28Z CAR-Ts were incubated with PMA and ionomycin for the same period. Data are shown as mean ± SD of triplicate samples. ∗p < 0.05, ∗∗p < 0.01 (compared with CAR-T only group by one-way ANOVA, n = 5).
(G) CR3022-28Z downregulation in response to 293-ACE2 cells preincubated with different concentrations of the RBD peptide. CAR-Ts were co-cultured for 20 h with 293-ACE2 cells preincubated with various concentrations of the RBD peptide for 1 h prior to the co-culture. Data are shown as mean ± SD of triplicate samples. ∗p < 0.05, ∗∗∗∗ p < 0.0001 (two-tailed Student’s t test, n = 2).
To generate CAR-Ts, peripheral blood mononuclear cells (PBMCs) from a given donor were stimulated in vitro with anti-CD3 and anti-CD28 antibodies for 72 h followed by spinoculation with lentiviral supernatant, further culturing for 4 days in X-VIVO15 serum-free medium followed by fluorescence-activated cell sorting (FACS) based on EGFP expression. Sorted CAR-Ts were then expanded in the same media supplemented with IL-2 for 2–3 weeks before their use in assays. Lentivirus-driven expression of the CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, or CR3022-IgG4-28Z constructs was confirmed by EGFP expression (Figure 1B). The CR3022-CH3-28Z construct was routinely observed to be expressed at slightly lower levels than the other constructs. CAR expression on the surface of cells was confirmed by anti-Flag staining (Figures 1B and S1). As a representative example, using CR3022-8a-28Z-transduced cells, anti-Flag-APC and EGFP showed a similar heterogeneous viSNE pattern for CAR receptor expression. It could roughly be divided into populations of cells with high (island i), intermediate (island ii), and low-nil expression (island iii). The islands showed some overlap but defined a general trend of heterogeneity within the CAR-T population. Lastly, flow cytometric staining with anti-CD4 and anti-CD8 antibodies showed similar proportions of CD8+ and CD4+ cells among the CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, or CR3022-IgG4-28Z-expressing CAR-Ts (Figure S2). These data showed that we were able to successfully transduce and maintain CAR-Ts.
The binding to the S1 RBD peptide activates SARS-CoV-2 CAR-Ts
We next assessed whether the RBD viral sequence presented on the surface of cells could activate the various CAR-Ts (Figure 1C). For this, 293-ACE2 cells were incubated with RBD peptide for 1 h (293-ACE2-RBD) prior to co-culturing with the CAR-Ts for 20 h. The expression of hACE2 on the 293-ACE2 cells was confirmed by flow cytometry (Figure S3). We initially examined changes in the expression of CD69 on CAR-Ts, a well-established activation marker on the surface of T cells (Cibrian and Sanchez-Madrid, 2017). Indeed, the co-culturing with 293-ACE2-RBD cells induced a marked increase in CD69 expression on the CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, or CR3022-IgG4-28Z CAR-Ts (left panel). As a negative control, neither Flag-28Z nor CR3014-28Z CAR-Ts showed an increase in CD69. Similarly, standard flow cytometry profiles of a representative donor showed that 293-ACE2-RBD cells induced an increase in CD69 expression on each of the CR3022 CAR-Ts (upper right panel), whereas no increase was seen with incubation with 293-ACE2 cells lacking RBD (lower right panel). Further, in a titration experiment, CD69 expression was seen to increase in response to increasing concentrations of the RBD peptide preincubated with the 293-ACE2 cells (Figure 1D). In this case, we observed a titratable increase in the response to peptide as low as 10 ng/mL. No response of the CR3014-28Z CAR-Ts was noted. The viability of cells in control and stimulation assays was similar. These data showed clearly that the presentation of the RBD peptide induced the expression of CD69 on the various CR3022 CAR-Ts.
We next assessed the heterogeneity of CD69 expression on CAR-Ts in more detail by viSNE and Cytobank analysis (Alfei et al., 2019; McLane et al., 2019) (Figure 1E). The CD69 population could be divided into three islands based on levels of expression (island i with the highest level, island ii with intermediate levels [yellow/turquoise], and island iii with low-nil levels of CD69 [dark blue]). Importantly, the CD69 expression patterns changed in response to 293-ACE2-RBD cells with an increase in the percentage of CAR-Ts in island i relative to the total CAR-T population (from 35%–40% to 65%–70%; upper right panel) and an increase in the relative mean fluorescent intensity (MFI) (from 1,000 to 1,800–2,000; lower right panel). No change in the expression of CD69 on Flag-28Z or CR3014-28Z was seen. By contrast, islands ii and iii showed much lower increase in expression and a decrease in their percent representation in the total CAR-T population (Figure S4, left and right panels, respectively).
We also transduced Jurkat T cells to express different CARs and monitored CD69 expression in response to RBD peptide presented by Vero cells (Figure S5). Vero cells were co-incubated with 200 or 1000 ng/mL RBD peptide. The CR3022 Jurkat cells, but not the CR3014 Jurkat cells, showed an increase in CD69 expression in response to peptide in the presence of Vero cells.
In addition, as a nearly intracellular signaling event, we assessed the phosphorylation of the ribosomal protein S6, a key signal transduction event that regulates cell division, protein translation, and metabolism (Roux et al., 2007) (Figure 1F). As an example, CR3022-8a-28Z CAR-Ts were incubated with 293-ACE2 or 293-ACE2-RBD cells for 20 min prior to the detection of S6 phosphorylation by flow cytometry. The incubation of CAR-Ts with 293-ACE2-RBD induced S6 phosphorylation (i.e. pS6) in the T cells, whereas no change was observed in response to incubation with the control, 293-ACE2 cells. As a positive control, the incubation of cells with PMA and ionomycin also increased S6 phosphorylation. These data indicate CAR-T engagement generated a key early intracellular signaling event needed for cell proliferation.
Lastly, the recognition of the RBD peptide led to the downregulation of the CARs from the surface of cells (Figure 1G). CAR downregulation has been previously reported (Li et al., 2020) and in the case of other receptors, due to receptor-mediated engagement (Monjas et al., 2004; San Jose et al., 2000; Schneider et al., 1999). The 20 h incubation period was sufficient to allow for both the induction of signaling and the subsequent downregulation of the receptors (Li et al., 2020). In agreement, the CR3022-28Z CAR receptors were downregulated with increasing RBD peptide concentrations in a manner that inversely mirrored the induction of CD69 expression. As a control, no titratable response of the CR3014-28Z CAR-T was observed. These data confirmed that the CR3022-based CARs became engaged in a specific manner when presented with the RBD peptide.
Anti-SARS-CoV-2 CAR-Ts kill RBD/S1-coated targets in vitro
We next assessed whether anti-SARS-CoV-2 CAR-Ts were cytolytic in killing SARS-CoV-2 RBD/S1-loaded cells (Figure 2). CAR-Ts were co-cultured with 293-ACE2-RBD cells at different effector to target ratios (20:1, 10:1, 5:1) for 5h for the assessment of killing. Each of the CR3022-based CAR-Ts was seen to kill the targets over various ratios (Figure 2A, left panel). The presence of the RBD peptide on the 293-ACE2-RBD cells was confirmed by staining with anti-S1 and the CR3022 antibody (right panel). By contrast, as a negative control, T cells expressing Flag-28Z or CR3014-28Z failed to kill targets. Interestingly, CAR-Ts with different hinge regions showed differences in the efficiency of killing. The CD8a (CR3022-8a-28Z) and IgG4 (CR3022-IgG4-28Z) CAR-T expressing cells were the most effective killers followed by CR3022-28Z and CR3022-CH3-28Z.
Figure 2.

Different CAR-Ts possess cytolytic activity that mediated the killing of RBD, S1-loaded, and S1-expressing target cells
(A) Different CAR-Ts kill targets loaded with the RBD peptide. (Left panel) 293-ACE2 cells were loaded with the RBD peptide followed by washing and incubation with CAR-Ts for 4–5 h prior to a measure of killing. (Right panel) The presence of RBD peptides on loaded 293-ACE2 cells was detected with anti-S1 and anti-RBD antibodies. Data are shown as mean ± SD of triplicate samples. ∗∗∗∗p < 0.0001 (compared with CR3014-28Z by two-way ANOVA, n = 4).
(B) CR3022 antibody blocks CR3022-8a-28Z and CR3022-IgG4-28Z kill RBD-coated targets. 293-ACE2-RBD cells were incubated with CR3022, or the isotype control, at 37°C for 30 min prior to the incubation with CAR-Ts. Data are shown as mean ± SD of triplicate samples. ∗∗∗∗p < 0.0001 (compared with isotype control groups by paired Student’s t test, two tailed, n = 2).
(C) Different CAR-Ts kill targets coated with S1 peptide. (Left panel) 293-ACE2 cells were loaded with the S1 peptide followed by washing and incubation with CAR-Ts for 4–5 h prior to a measure of killing. (Right panel) The presence of S1 peptides on loaded 293-ACE2 cells was detected with anti-S1 and anti-RBD antibodies. Data are shown as mean ± SD of triplicate samples. ∗∗∗∗p < 0.0001 (compared with CR3014-28Z by two-way ANOVA, n = 3).
(D) Different CAR-Ts kill NIH/3T3 cells expressing the S1 peptide. (Lower panel) The S1 overexpressing NIH/3T3 cells were sorted by FACS staining of S1. These cells showed high levels of anti-S1 staining, whereas the staining with the CR3022 antibody was lower. (Upper panel) S1-expressing NIH/3T3 cells were co-cultured with CAR-Ts for 4–5 h prior to a measure of killing. Data are shown as mean ± SD of triplicate samples. ∗∗∗∗p < 0.0001 (compared with CR3014-28Z by two-way ANOVA, n = 3) (also see Figure S6).
Specificity was confirmed by CR3022 antibody blockade of killing (Figure 2B). The 293-ACE2-RBD cells were incubated with CR3022, or the isotype control, at 37°C for 30 min prior to the incubation with CAR-Ts. As shown in a representative experiment, the CR3022 antibody abrogated the killing of targets by the CR3022-8a-28Z and CR3022-IgG4-28Z CAR-Ts by 70%–80% at various effector to target ratios. By contrast, the isotype control antibody had no effect on killing.
We also assessed the killing of 293-ACE2 target cells that had been precoated with the full-length S1 peptide (Figure 2C). The presence of the S1 peptide on the cells was confirmed by staining with anti-S1 and the CR3022 antibody (right panel). CR3022-28Z, CR3022-8a-28Z, and CR3022-IgG4-28Z CAR-Ts killed S1 peptide-loaded targets, albeit at lower levels (Figure 2C, left panel). The CR3022-CH3-28Z CAR-T or the control CR3014-28Z CAR-T showed no killing. In this case, the CR3022-IgG4-28Z CAR-T was most effective. Overall, the CAR-Ts were less able to kill targets, with only 10%–30% for S1-peptide-coated cells compared with the 30%–60% for RBD-peptide-loaded target. This may be due to the mode of the specific RBD sequence presentation in the S1 protein as recognized by the CAR-Ts. Examples of the similar response of CAR-Ts from different donors to the RBD and S1 peptides are also shown in Figure S6.
We next assessed SARS-CoV-2 CAR-T killing of NIH/3T3 cells that had been transduced to express the S1 protein (Figure 2D). FACS profiles showed high levels of anti-S1 staining but a lower level of staining with the CR3022 antibody against the RBD peptide (lower panel). The reason for this difference is not clear but is likely related to the accessibility of the CR3022 epitope. The RBD is only accessible when at least two RBDs are open simultaneously (Yuan et al., 2020), and the frequency of the opening RBD is usually less than 30% (Wrobel et al., 2020). Despite this, the various CR3022 CAR-Ts were able to effectively kill targets (upper panel). Each of the CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, and CR3022-IgG4-28Z CAR-Ts killed 20%–50% of target cells, dependent on the CAR-T to target ratio. In this assay, the CR3022-28Z CAR-T and the CR3022-IgG4-28Z CAR-Ts were the most effective.
Overall, these data showed that the CAR-Ts could recognize and kill 293-ACE2 cells coated with the RBD and S1 peptides or the S1-expressing NIH/3T3 cells. The CAR-Ts with different-sized extracellular domains also varied in their killing efficacy of different target cells. These CAR-T variants will enable the eventual testing efficacy against S1 variants.
Anti-SARS-CoV-2 CAR-Ts form multicellular clusters with targets
The interaction of the CAR-Ts with the target cells was next examined using time-lapse live-cell microscopy (Figure 3). We analyzed the behavior of CR3022-8a-28Z CAR-Ts co-cultured with the 293-ACE2 and 293-ACE2-RBD cells over 20 h. The EGFP+ CAR-Ts were observed to be green due to EGFP expression, whereas the 293 cells were seen as red-orange due to the lentiviral transduction with mCherry. CR3022-8a-28Z formed clusters with 293-ACE2-RBD cells within 30–60 min and were present for 10–15 h followed by a slight reduction in clusters (Figure 3A, also see Video S2). No clusters were seen with 293-ACE2 negative control cells (see Video S1). On average, there were 3–5 CAR-Ts in each cluster, although this varied depending on the timing of the analysis (Figure 3B, also high-magnification examples from two separate experiments in right panel). No clusters were seen with the 293-ACE2 cells alone. Cluster diameter also increased from 60 min to 10–12 h with an average diameter of 30–40 μm as assessed by ZEN Lite software (Figure 3C). Similarly, the surface area of the clusters increased from 500 to 1500 μm2 over the same time period (Figure 3D). An example of tracking between CR3022-8a-28Z CAR-Ts co-cultured with the 293-ACE2 preloaded with S1 peptide is also shown (Video S3). Overall, these data confirmed that CR3022-8a-28Z CAR-Ts formed clusters with 293-ACE2 targets dependent on the presence of the RBD/S1 peptides.
Figure 3.

Time-lapse microscopy shows that CR3022-8a-28Z CAR-Ts form multicellular clusters in the killing of 293-ACE2-RBD target cells
CR3022-8a-28Z CAR-Ts were co-cultured with 293-ACE2 cells or 293-ACE2-RBD cells and subjected to time-lapse microscopy for 20 h (also see Videos S1, S2, and S3). Red-colored cells are 293-ACE2 cells or 293-ACE2-RBD transduced to express mcherry (see STAR Methods).
(A) Images of CAR-T cells with 293-ACE2 cells (left panels) or 293-ACE2-RBD cells (right panels) over 20 h. Examples of images of GFP+ CAR-Ts (green) in clusters with target cells (red-orange) (n = 2).
(B) (Left panel) Histogram showing the numbers of GFP+ cells per cluster over 20 h. Data are shown as mean ± SD of five individual clusters. ∗∗p < 0.01, ∗∗∗p < 0.001 (two-tailed Student’s t test, n = 2). (Right panel) Examples of 20× images of cell clusters between CR3022-8a-28Z CAR-Ts and 293-ACE2-RBD cells (n = 2).
(C) Histogram showing the diameter (μm) of clusters over a time course of 20 h. Data are shown as mean ± SD of five individual clusters. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (two-tailed Student’s t test, n = 2).
(D) Histogram showing the area (μm2) of clusters over a time course of 20 h. Data are shown as mean ± SD of five individual clusters. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (two-tailed Student’s t test, n = 2).
CR3022-8a-28Z CAR-Ts were co-cultured with the 293-ACE2 cells and tracked for 20 hours. EGFP+ CAR-Ts failed to form clusters with 293-ACE2.
CR3022-8a-28Z CAR-Ts were co-cultured with 293-ACE2 that had been pre-incubated with RBD peptide and tracked for 20 hours. EGFP+ CAR-Ts were observed to form clusters with 293-ACE2+RBD cells.
CR3022-8a-28Z CAR-Ts were co-cultured with 293-ACE2 that had been pre-incubated with S1 peptide and tracked for 20 hours. EGFP+ CAR-Ts were observed to form clusters with 293-ACE2+S1 cells.
IFN-γ is induced on the CD69 subset of SARS-CoV-2 CAR-Ts
We next examined the inducible expression of immune effector molecules on the CAR-Ts. The cytokine interferon-γ (IFN-γ) promotes differentiation into Th1 cells and the upregulation of MHC antigens on dendritic cells (Schoenborn and Wilson, 2007). Indeed, we observed an increase in IFN-γ expression in response to 293-ACE2-RBD cells on each of the CAR-Ts (Figure 4A). The highest increase in expression was seen on CR3022-28Z, CR3022-8a-28Z, and CR3022-IgG4-28Z CAR-Ts. In a separate experiment, viSNE analysis showed that IFN-γ was already widely expressed on the majority of CAR-Ts after their expansion in IL-2 supplemented media (Figure 4B, see CAR-T alone; left panel). A small increase in expression was observed after incubation with 293-ACE2 cells alone (middle panel). However, in response to 293-ACE2-RBD cells, the CAR-Ts caused a major change in the distribution of expressing cells and expression intensity of IFN-γ as seen by viSNE. There was a loss of cells in islands ii and iii concurrent with an increase in the density of cells in island i. Each of the CR3022-based CAR-Ts showed a similar trend. In keeping with this, there was an increase in the percentage of IFN-γ+ cells in island i relative to the total CAR-T population (Figure 4C) as well as an increase in the relative MFI for IFN-γ in island i (Figure 4D). Lastly, viSNE analysis showed that the same island of cells with increased IFN-γ expression in island i also showed an increase in CD69 expression (Figure 4B, right panels). Overall, these data indicated that the recognition of the RBD peptide by CAR-Ts caused a major increase in the expression of IFN-γ in the same subset of cells that showed an increase in CD69 expression.
Figure 4.

Expression of interferon γ (IFN-γ) in a CD69+ anti-SARS-CoV-2 CAR-Ts
(A) Mean fluorescent intensity (MFI) values of IFN-γ expression on Flag-28Z, CR3014-28Z, CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, and CR3022-IgG4-28Z CAR-Ts in response to 293-ACE2-RBD cells after 20 h of co-culture. Data are shown as mean ± SD of triplicate samples. ∗p < 0.05, ∗∗∗∗p < 0.0001 (compared with Flag-28Z by one-way ANOVA, n = 3).
(B) viSNE plots of the expression of IFN-γ on CAR-Ts following 3 weeks of expansion in IL-2 supplemented media (CAR-T alone) (left panels) or following an additional 20 h of incubation with 293-ACE2 cells (middle panel) or 293-ACE2-RBD cells (right panel). viSNE plots of CD69 expression on cells from the same donor (right panel) (n = 3).
(C) Histogram showing the changed distribution of cells expressing IFN-γ in viSNE islands. Percentage of IFN-γ+ cells relative to total population of CAR-Ts (n = 3).
(D) Histogram showing the changed expression of IFN-γ on individual cells in the viSNE islands. Relative MFI in islands i, ii, and iii (n = 3).
Granzyme B/perforin is induced in a subset of CD69+ anti-SARS-CoV-2 CAR-Ts
Given the ability of the various CAR-Ts to kill RBD or S1-bearing targets, we next assessed the expression of the cytolytic effectors, granzyme B (GZMB) and perforin (Figure 5). Both are key effector molecules that mediate the killing of targets (Kurschus and Jenne, 2010). Perforin is a pore-forming cytolytic protein that facilitates GZMB entry to kill target cells (Smyth et al., 1996). Indeed, each CR3022-based CAR-T showed an increase in the level of expression of GZMB (Figure 5A, upper panel) and perforin (Figure 5A, lower panel) when incubated with RBD loaded target cells. CR3022-28Z, CR3022-8a-28Z, and CR3022-IgG4-28Z CAR-Ts showed the highest level of GZMB and perforin expression. No increase was observed upon incubation with 293-ACE2 cells lacking RBD, or in the negative controls, Flag-28Z and CR3014-28Z.
Figure 5.

Expression of granzyme B (GZMB) and perforin on a subset of CD69+ anti-SARS-CoV-2 CAR-Ts
(A) Mean fluorescent intensity (MFI) values of GZMB and perforin expression on Flag-28Z, CR3014-28Z, CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, and CR3022-IgG4-28Z CAR-Ts in response to 293-ACE2-RBD cells after 18 h of co-culture. Data are shown as mean ± SD of triplicate samples. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (compared with Flag-28Z by one-way ANOVA, n = 4). (Upper panel) GZMB; (lower panel) perforin.
(B) viSNE plots for CD69, GZMB, and perforin expression in SARS-CoV-2 CAR-Ts following incubation with 293-ACE2 (upper panels) or 293-ACE2-RBD (lower panels) (n = 4).
(C) Histogram showing relative MFI values for GZMB (upper panel) and perforin (lower panel) in viSNE islands i, ii, and iii (n = 4).
(D) Percent representation of CD69hiGZMB+ perforin+ subset in response to 293-ACE2 and 293-ACE2-RBD cells (n = 4).
Unlike in the case of IFN-γ, GZMB and perforin were expressed in a highly localized subset of CD69+ CAR-Ts within island i and ii (termed this subset as sub-island i–iia) (Figure 5B). A basal level of GZMB and perforin expression was seen in CAR-Ts incubated with 293-ACE2 cells. Both molecules were expressed within the same subset of CAR-Ts. Incubation with 293-ACE2-RBD cells increased the expression of both proteins (lower panels). Each of the CAR-Ts showed a similar increase, except for the CR3022-CH3-28Z that showed a more modest increase. An increase in the MFI for GZMB (Figure 5C, upper panel) and for perforin (lower panel) was observed. No increase in GZMB in islands ii and iii was observed (upper panel), whereas a modest increase in perforin expression in the same islands was seen. Overall, there was a striking increase in the presence of CD69hiGZMB+perforin+ CAR-Ts in sub-island i–iia (Figure 5D). These data showed that the recognition of RBD peptides by CAR-Ts induced a detectable increase in the expression of GZMB and perforin within a subset of CD69 high expressing CAR-Ts.
FasL (CD95L) is preferentially expressed on CAR-Ts with high CD69 expression
A second mechanism of cytolytic-mediated killing involves Fas ligand (FasL) (Figure 6). FasL is a tumor necrosis factor (TNF) superfamily member that binds to the Fas receptor on target cells, leading to their apoptosis and death (Russell and Ley, 2002). Each CR3022 CAR-T increased FasL expression upon incubation with 293-ACE2-RBD cells (Figure 6A). In this case, the CR3022-IgG4-28Z CAR-T showed the greatest increase. viSNE analysis showed that FasL was broadly expressed on the majority of CAR-Ts that are seen with the other markers (Figure 6B). As in the case of CD69, three islands of cells could be defined based on expression (islands i, ii, iii). Unlike for CD69, an increase in FasL was observed (albeit to a lesser extent) in both islands i and ii. The increase in the positive signal in island ii was accompanied by the loss of FasL negative cells (see dark blue), as most evident in the viSNE patterns for the CR3022-IgG4-28Z CAR-Ts. As negative controls, the incubation of CAR-Ts with soluble RBD peptide without 293-ACE2 cells or the incubation of Flag-28Z cells with 293-ACE2 or 293-ACE2-RBD showed background staining (upper panels). This indicated that SARS-CoV-2 recognition led to an increase in FasL on a broader population of cells than observed for CD69 (Figures 6B and 6C). As an alternate system, we also assessed changes in FasL expression in response to NIH/3T3 cells that had been stably transduced to express the S1 peptide (Figure 6D). In this case, CR3022-8a-28Z cells also showed a broad increase in FasL expression. These data showed that the recognition of the RBD peptide or the S1 protein on target cells induced the expression of FasL and that the pattern of induced expression was broader than seen with the expression of CD69, IFN-γ, or GZMB/perforin.
Figure 6.

FasL expression on anti-SARS-CoV-2 CAR-Ts
(A) Relative mean fluorescent intensity (MFI) values for FasL expression on Flag-28Z, CR3014-28Z, CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, and CR3022-IgG4-28Z CAR-Ts in response to 293-ACE2-RBD cells after 24 h of co-culture. Data are shown as mean ± SD of triplicate samples. ∗p < 0.05, ∗∗∗p < 0.001 (compared with Flag-28Z by one-way ANOVA, n = 3).
(B) viSNE plots defining subsets of CD69 and FasL expression on CAR-Ts. Shown are the profiles of Flag-28Z, CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, and CR3022-IgG4-28Z CAR-Ts under conditions where cells were incubated for 24 h with soluble RBD peptide in the absence of 293-ACE2 cells or with 293-ACE2 or 293-ACE2-RBD cells.
(C) Histogram showing the increase in FasL expression on islands i, ii, and iii on Flag-28Z, CR3014-28Z, CR3022-28Z, CR3022-8a-28Z, CR3022-CH3-28Z, and CR3022-IgG4-28Z CAR-Ts after incubation with 293-ACE2-RBD cells (n = 3).
(D) viSNE profiles of FasL expression in CR3022-8a-28Z CAR-T following incubation without (upper panel) or with NIH/3T3-S1 cells (lower panel) (n = 2).
(E) The blockade of FasL failed to interfere with the killing of anti-SARS-CoV-2 CAR-Ts. Two anti-FasL blocking antibodies were assessed for the ability to inhibit CR3022-28Z, CR3022-8a-28Z, and CR3022-IgG4-28Z CAR-T killing of target cells loaded with RBD peptide. Data are shown as mean ± SD of triplicate samples. ∗p < 0.05, ∗∗p < 0.01 (compared with mouse IgG1 isotype group by paired Student’s t test, two tailed, n = 2).
Given this, we next assessed the importance of FasL expression to the killing of RBD-coated cells by adding anti-FasL blocking antibodies to the killing assay (Figure 6E). CR3022-28Z, CR3022-8a-28Z, and CR3022-IgG4-28Z CAR-Ts were incubated with 293-ACE2-RBD cells in the presence or absence of anti-FasL blocking mAbs, #100419 or #NOK-1, for 5 h prior to and during the killing assay. NOK-1 has been previously reported to block cell-mediated killing (Kayagaki et al., 1995). A minor reduction in the killing was observed in some ratios of effector to target cells for CR3022-28Z and CR3022-8a-28Z CAR-Ts; however, the patterns were not consistent. No statistically significant effect was observed on the killing by CR3022-IgG4-28Z CAR-Ts. From these data, we observed that the FasL pathway does not play a major role in the killing of RBD-coated ACE2-expressing cells in our systems.
Anti-SARS-CoV-2 CAR-Ts kill S1-expressing targets in vivo
Lastly, it was important to assess whether CAR-Ts could elicit the killing of targets in vivo in mice (Figure 7). For this, we injected a mixture of 2 × 106 NIH/3T3-S1 cells and 4 × 106 CR3014-28Z/CR3022-28Z CAR-Ts to NOD-SCID IL2Rγnull mice via the intraperitoneal route. The NIH/3T3-S1 cells, in addition to expressing S1 from SARS-CoV-2, express luciferase, and the presence of NIH/3T3-S1 cells can be assessed by injecting D-luciferin. For analysis, bioluminescence images were acquired at 24 h after injection of the cells by administering D-luciferin intraperitoneally (i.p.). We found that the adoptive transfer of CR3022-28Z reduced the presence of target cells in mice (Figure 7A). Although injection with NIH/3T3-S1 cells alone resulted in nearly 4 × 109 luminescence (p/s), injection of NIH/3T3-S1 cells with CR3022-28Z CAR-Ts reduced the luminescence measurement to <1 × 109, and CR3014-28Z CAR-Ts had no effect on the presence of the target (Figure 7B). In comparison to the untreated samples, the CR3022-28Z reduced the presence of NIH/3T3-S1 cells (i.e. luminescence) by 80%. However, relative to the CR3014-28Z, the luminescence was reduced by 60%. There was a loss of signal in 5/8 mice (shown for 2/3 mice). Overall, these data confirm that CAR-Ts also are capable of killing S1 expressing cells in vivo in mice.
Figure 7.

The in vivo elimination of NIH/3T3 cells expressing SARS-CoV-2 S1
(A) Images of NIH/3T3-S1 cells in mice. NIH/3T3-S1 cells express luciferase and the presence of luminescence after D-luciferin injection indicates the presence of NIH/3T3-S1 cells. A mixture of 2 × 106 NIH/3T3-S1 cells and 4 × 106 CR3014-28Z or CR3022-28Z CAR-Ts were injected into NOD-SCID IL2Rγnullmice through the intraperitoneal route. Twenty-four hours later, D-luciferin was injected and bioluminescence images were acquired. 3 mice are shown out of 8 mice per group in total.
(B) Histogram showing the significant reduction of NIH/3T3-S1 cells in vivo by CR3022-8a-28Z but not by CR3014-28Z CAR-T cells. Data are shown as mean ± SD of 8 mice. ∗p < 0.05, ∗∗∗p < 0.001 (compared with untreated group by one-way ANOVA). This experiment was repeated twice.
We next assessed whether the SARS-CoV-2 CAR-Ts could also block viral entry (Figure S7). We expected that the efficiency of the blockade would be low, given the difficulty in having cells compete for smaller-scale protein-protein interactions. Recombinant pseudo-typed VSV particles containing SARS-CoV-2 spike protein were used to mimic SARS-CoV-2 cell infection and cell entry. The SARS-CoV-2 pseudo-virus particles encode DsRed in the virus genome. DsRed was strongly expressed after the SARS-CoV-2 pseudo-virus entry into ACE2-expressing cells (Millet et al., 2019). From this, using SARS-CoV-2 as a control with an infection rate of 60%, we observed that co-incubation with CAR-Ts expressing the various constructs reduced viral entry in response to different ratios of T-cells to target. As expected, the efficacy of blockade was low and observed for both CR3014- and CR3022-based CAR-Ts (Figure S7). Both Flag-28Z and CR3014-28Z reduced binding to the same degree as CAR-Ts expressing specific receptors. Overall, the data showed that CAR-Ts are unlikely to sterically block viral entry into cells, but rather limit viral infection, as seen in the case of most viral infections by mediating CTL killing responses against infected cells expressing viral antigens (Moskophidis and Kioussis, 1998).
Discussion
Given the ongoing threat of SARS-CoV-2 and the uncertainties related to the long-term efficacy of vaccines against new variants of the virus, new therapeutic approaches may be needed. The development of CAR therapy has been proven effective in the treatment of various types of cancers (Kawalekar et al., 2016; Posey et al., 2016). In this study, we showed that it is possible to generate CAR-Ts capable of responding to the SARS-CoV-2 and elicit the in vitro and in vivo killing of cells loaded with RBD/S1 peptides or that express the S1 protein. Our findings outline the potential use of anti-SARS-CoV-2 CAR-Ts as a therapeutic option against COVID-19.
CR3022-based CAR-Ts were stimulated by the recognition of the RBD peptide as shown by the increased expression of CD69 and the phosphorylation of ribosomal S6, a key regulator of metabolism, translation, and cell division (Roux et al., 2007). CD69 expression was induced in a peptide concentration-dependent manner, with responses to cells that had been loaded with a peptide-concentrations as low as 10 ng/mL. This denotes a remarkable sensitivity to the peptide. CD69 expression also promotes the retention of T cells in the lymphoid organs (Cibrian and Sanchez-Madrid, 2017) and serves as a marker for resident memory T cells (TRMs) (Osborn et al., 2019; Ziegler et al., 1994). In this way, the increased CD69 expression on CAR-Ts may alter their residency for enhanced responses against SARS-CoV-2-infected cells.
Exposure to 293-ACE2-RBD cells also potently downregulated CARs from the surface of CAR-Ts. This property has been previously reported for CD19-CARs (Li et al., 2020) and is another measure of receptor-mediated engagement (Monjas et al., 2004; San Jose et al., 2000; Schneider et al., 1999). In this manner, the level of CAR downregulation inversely mirrored the effect of different peptide concentrations on CD69 expression. While confirming that CAR-Ts were responsive to the RBD peptide, the loss of the surface CAR might ultimately limit the longevity of responses against targets as seen in tumor models (Li et al., 2020).
Importantly, SARS-CoV-2 CAR-Ts were cytolytic in the specific killing of target cells loaded with either the RBD peptide, S1 peptide, or that express the S1 protein. Specificity was seen by the fact that CR3014-based CAR-Ts failed to kill SARS-CoV-2-expressing target cells. In keeping with killing, time-lapse microscopy showed the formation of multicellular CAR-T clusters around single-target cells. Further, CAR-Ts with different-sized hinge regions varied in the efficacy of killing where the IgG4 hinge CAR-T gave the most consistent results in the killing of targets. However, these differences appeared to vary depending on the mode of RBD or S1 presentation. For example, the IgG4 hinge CAR-Ts predominated in response to RBD peptide loaded on 293-ACE cells, whereas CD28 and IgG4 hinge CAR-Ts were most effective in response to S1 peptide presented on the surface of NIH/3T3 cells. The different hinge regions may therefore affect the ability of CARs to bind and respond to peptides depending on their mode of presentation. By contrast, we did not observe blockade of viral entry into cells based on the use of recombinant pseudo-typed VSV particles containing SARS-CoV-2 spike protein. These were used to mimic SARS-CoV-2 cell infection and cell entry. CAR-Ts are therefore unlikely to sterically block viral entry into cells but rather limit viral infection in a classic fashion involving CTL-like killing of infected cells expressing viral antigens (Moskophidis and Kioussis, 1998; Rouse and Sehrawat, 2010).
The extent of SARS-CoV-2 viral peptide antigen expression during infection remains to be established. Various labs have shown that COVID-19 is accompanied by a robust induction of CD4 and CD8 responses against a variety of SARS-CoV-2 peptide antigens (Grifoni et al., 2020; Meckiff et al., 2020; Premkumar et al., 2020; Rydyznski Moderbacher et al., 2020; Weiskopf et al., 2020). Given that CD4 and CD8 T cells are generated by the presentation of peptide antigens, these findings clearly show that peptides derived from various regions of SARS-CoV-2 are presented during infection. The same peptides could be targeted by CAR-Ts. Further, mass spectrometry has detected SARS-CoV-2-virus-derived peptides in patients (Nikolaev et al., 2020), whereas SARS-CoV-2-infected cells undergo syncytia formation of ACE2-expressing cells, which can be facilitated by antigen recognition (Buchrieser et al., 2021).
In terms of mechanism, there are two well-established modes of killing by cytolytic T cells, one involving Fas-FasL and another perforin/granzymes (Williams and Bevan, 2007). The loss of perforin and Fas/Fas ligand can cause severe autoimmune and inflammation in mice (Peng et al., 1998; Spielman et al., 1998). In our model, the expression of FasL and GZMB/perforin increased in response to RBD-presenting cells. However, the blockade of FasL with antibodies had only a weak effect on the killing of targets by the anti-SARS-CoV-2 CAR-Ts, and there was low to negligible level of Fas expression on the target cells. The observation points to the GZMB/perforin pathway as a major mechanism by which our anti-SARS-CoV-2 CAR-Ts kill targets. Whether the Fas-FasL pathway may also participate in the killing of other target cells remains for future studies.
In this context, the effectors GZMB and perforin were expressed within a relatively small subset of CD69 high expressing anti-SARS-CoV-2 CAR-Ts. Perforin allows for GZMB into the target cell for cell death, although GZMB may also enter via other pathways (Smyth et al., 1996). Although GZMB and perforin were expressed in the same subset, not all cells expressing high levels of CD69 expressed GZMB and perforin. This indicates that factors other than the mere activation of cells influenced whether these effectors were expressed.
By contrast, FasL and IFN-γ were more widely expressed on populations of CAR-Ts. viSNE analysis showed that FasL expression occurred in both CD69 high and intermediate populations, whereas the increase in IFN-γ tended to be more restricted to the CD69 high population. These differences underscore the heterogeneity in the responses of individual SARS-CoV-2 CAR-Ts. IFN-γ has both anti-viral and immunoregulatory properties (Schroder et al., 2004) and can alter the transcription of multiple genes including major histocompatibility antigens on antigen-presenting cells (Schroder et al., 2004). Higher levels of type I interferons have also been associated with a lower risk of SARS-CoV-2 infection and amelioration of disease severity (Sui et al., 2021). IFN-γ also primes alveolar macrophages against secondary bacterial infections, which may have relevance to COVID-19-associated pulmonary disease (Yao et al., 2018). It could also assist by inducing STAT3 activation, which can activate GZMB expression for enhanced CTL effector function (Lu et al., 2019). The secretion of IFN-γ by CAR-Ts would, therefore, be expected to promote these beneficial events during the progression of SARS-CoV-2 infection.
CAR-Ts could be most effective in patients who are immunocompromised or who respond weakly to the virus or vaccines (Grifoni et al., 2020; Meckiff et al., 2020; Premkumar et al., 2020; Rydyznski Moderbacher et al., 2020; Weiskopf et al., 2020). Further, they may provide longer term memory responses in patients who fail to develop sufficient memory against vaccines. CAR-Ts against relapsed ALL cancers have shown sustained remissions for 24 months (Maude et al., 2014). The therapeutic approach could involve the retroviral transduction of CARs into T cells extracted from patient blood followed by the reinfusion of CAR-Ts into the patient, a method presently followed in CD19-CAR-T therapy against cancer (Pfeiffer et al., 2018). The use of anti-SARS-CoV-2 CAR-Ts may be more costly than vaccination but may provide longer-term protective immunity (Thomas et al., 2006). One complication may involve the induction of inflammation associated with CAR-T therapy. In this context, CAR-Ts would need to be used carefully in the clinic, possibly before infection by the virus or following the resolution of inflammatory disease associated with severe COVID-19. In this context, anti-IL-6 receptor antibodies have been successful in treating inflammation associated with the use of CAR-Ts in cancer therapy (Guedan et al., 2019). Its applicability will need to be assessed in a carefully controlled clinical trial.
During the final preparation of this paper, two groups reported the successful generation of natural killer and macrophage CARs in in vitro assays (Fu et al., 2021; Ma et al., 2021). Unlike T cells of the adaptive immune system, these cells are part of the innate immune system with distinct co-receptors that regulate function. NK cells possess activating receptors and inhibitory receptors that include killer-cell immunoglobulin-like receptors (Vivier et al., 2011), whereas macrophages carry other innate receptors such as Mac1 that help mediate phagocytosis (Galdiero et al., 2013). The work on these papers was confined to in vitro studies, but, if shown to operate in vivo, as in the case of our CAR-Ts, these reagents could complement our SARS-CoV-2 reactive CAR-T approach. Future studies will be needed to determine the application of CAR therapy to the treatment of COVID-19.
Limitations of the study
By employing CARs against the RBD or S1, we have documented the successful generation of reactive CAR-Ts with a capacity to kill peptide expressing targets using in vitro and in vivo models. As mentioned, the lab of Sette and others have shown the robust induction of CD4 and CD8 responses during SARS-CoV-2 infection, indicating that viral peptide antigens are expressed and presented by cells. The precise nature of the peptides and cells involved during infections is an active area and will be of great utility in designing new cytolytic CAR-Ts against infected cells. In terms of killing, the relative contributions of the perforin-granzyme versus the Fas-FasL effector pathways in different contexts need to be determined. As mentioned, although there was a lack of involvement of FasL in our system, future studies will be needed to explore the participation of FasL and IFN-γ in the different systems involving other cell types and peptides. Further, the behavior of different CARs in vivo and the relative contribution of CD4 and CD8 CAR-Ts need to be defined as does the longevity of CAR-T immune response involving aspects such as CAR-T memory. Lastly, the degree to which CAR-Ts will control SARS-CoV-2 infections in a patient or viral spread in populations remains to be assessed.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BUV395 conjugated Mouse Anti-Human CD4 Antibody | BD Biosciences | Cat. #: 564724; RRID: AB_2738917 |
| Brilliant Violet 785™ conjugated anti-human CD8a Antibody | Biolegend | Cat. #: 301046; RRID: AB_2563264 |
| Brilliant Violet 421™ conjugated anti-DYKDDDDK Tag Antibody | Biolegend | Cat. #: 637321: RRID: AB_2750051 |
| PE-CF594 conjugated Mouse Anti-Human Granzyme B Antibody | BD Biosciences | Cat. #: 562462: RRID: AB_2737618 |
| Alexa Fluor® 647 conjugated Mouse Anti-Human Perforin Antibody | BD Biosciences | Cat. #: 563576: RRID: AB_2738287 |
| Brilliant Violet 421™ conjugated anti-human CD178 (Fas-L) Antibody | Biolegend | Cat. #: 306411: RRID: AB_2716104 |
| APC conjugated anti-DYKDDDDK Tag Antibody | Biolegend | Cat. #: 637307; RRID: AB_2561496 |
| APC-Cy™7 conjugated Mouse Anti-Human CD69 Antibody | BD Biosciences | Cat. #: 560912; RRID: AB_396862 |
| PE-Cyanine7 conjugated IFN gamma Monoclonal Antibody (XMG1.2) | Invitrogen | Cat. #: 25-7311-82; RRID: AB_469680 |
| Recombinant Anti-SARS-CoV-2 Spike Glycoprotein S1 antibody [CR3022] | Abcam | Cat. #: ab273074; RRID: AB_2847846 |
| SARS/SARS-CoV-2 Coronavirus Spike Protein (subunit 1) Polyclonal Antibody | Invitrogen | Cat. #: PA5-81795; RRID: AB_2788969 |
| CD178 (Fas Ligand) Monoclonal Antibody (NOK-1) | Invitrogen | Cat. #: 16-9919-81; RRID: AB_469281 |
| Human Fas Ligand/TNFSF6 Antibody | R&D Systems | Cat. #: MAB126-SP; RRID: AB_2246667 |
| Human ACE-2 Antibody | R&D Systems | Cat. #: AF933-SP; RRID: AB_355722 |
| anti-human CD3 Antibody (OKT-3) | Bio X Cell | Cat. #: BE0001-2; RRID: AB_1107632 |
| anti-human CD28 Antibody (9.3) | Bio X Cell | Cat. #: BE0248; RRID: AB_2687729 |
| FITC conjugated Rabbit anti-Goat IgG (H+L) Secondary Antibody | Invitrogen | Cat. #: 31509; RRID: AB_228394 |
| Alexa Fluor® 647 conjugated Donkey anti-rabbit IgG (minimal x-reactivity) Antibody | Biolegend | Cat. #: 406414; RRID: AB_2563202 |
| eFluor® 450 conjugated Phospho-S6 (Ser235, Ser236) Monoclonal Antibody (cupk43k) | Invitrogen | Cat. #: 48-9007-42; RRID: AB_2574117 |
| Normal Rabbit IgG Control | R&D Systems | Cat. #: MAB1050-500; RRID: AB_2313773 |
| Ultra-LEAF™ Purified Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | Cat. #: 400166; RRID: AB_2313773 |
| Ultra-LEAF™ Purified Mouse IgG2b, κ Isotype Ctrl Antibody | Biolegend | Cat. #: 400348; RRID: AB_2313773 |
| Bacterial and virus strains | ||
| Pseudotyped ΔG-DsRed (G∗ΔG-DsRed) rVSV | Kerafast | Cat. #: EH1018-PM |
| NEB® Stable Competent E. coli | New England Biolabs | Cat. #: C3040I |
| Biological samples | ||
| Human Peripheral blood mononuclear cells | HÉMA-QUÉBEC | https://www.hema-quebec.qc.ca/index.fr.html |
| Chemicals, peptides, and recombinant proteins | ||
| Fixable Viability Stain 575V | BD Biosciences | Cat. #: 565694; RRID: AB_2869702 |
| Fixable Viability Dye eFluor™ 780 | Invitrogen | Cat. #: 65-0865-14; |
| X-tremeGENE™ HP DNA Transfection Reagent | Roche | Cat. #: 6366236001; |
| Tag-it Violet™ Proliferation and Cell Tracking Dye | Biolegend | Cat. #: 425101; |
| X-VIVOTM 15 Serum-free Hematopoietic Cell Medium | Lonza | Cat. #: 04-418Q |
| Protamine sulfate salt from salmon | Sigma-Aldrich | Cat. #: P4020 |
| Human Recombinant IL-2, ACF | STEMCELL | Cat. #: 78145 |
| Protein Transport Inhibitor (Containing Brefeldin A) | BD Biosciences | Cat. #: 555029; RRID: AB_2869014 |
| SARS-CoV-2 (COVID-19) S protein RBD, His Tag | Acrobiosystem | Cat. #: SPD-C52H3 |
| Biotinylated SARS-CoV-2 (COVID-19) S1 protein, His,Avitag™ | Acrobiosystem | Cat. #: S1N-C82E8 |
| Phorbol 12-myristate 13-acetate | Sigma-Aldrich | Cat. #: P8139 |
| Ionomycin from Streptomyces conglobatus | Sigma-Aldrich | Cat. #: I9657 |
| IVISbrite D-Luciferin Ultra Bioluminescent Substrate in RediJect Solution (XenoLight) | PerkinElmer | Cat. #: 770505 |
| Hygromycin B | Gibco | Cat. #: 10687010 |
| Critical commercial assays | ||
| Intracellular Fixation & Permeabilization Buffer Set | Invitrogen | Cat. #: 88-8824-00; |
| Foxp3 / Transcription Factor Staining Buffer Set | Invitrogen | Cat. #: 00-5523-00 |
| CytoTox 96® Non-Radioactive Cytotoxicity Assay | Promega | Cat. #: G1780 |
| Platinum™ SuperFi II Green PCR Master Mix | Invitrogen | Cat. #: 12369010 |
| Wizard® SV Gel and PCR Clean-Up System | Promega | Cat. #: A9281 |
| Wizard® Plus SV Minipreps DNA Purification Systems | Promega | Cat. #: A1330 |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | Cat. #: E0554S |
| NucleoBond® Xtra Maxi EF Kit | TAKARA | Cat. #: 740424.10 |
| Quick Ligation™ Kit | New England Biolabs | Cat. #: M2200S |
| Deposited data | ||
| raw data in this study | This paper | https://doi.org/10.17632/3y3m8btcnk.2 |
| Experimental models: Cell lines | ||
| 293T | ATCC | Cat. #: CRL-3216 |
| Vero | ATCC | Cat. #: CCL-81 |
| Jurkat E6-1 | ATCC | Cat. #: TIB-152 |
| 293-hACE2 | Laboratory of H. Uri Saragovi | N/A |
| NIH/3T3-SARS-CoV-2 S1 | Laboratory of H. Uri Saragovi | N/A |
| NIH/3T3-SARS-CoV-2 S1-mCherry-luciferase | This paper | N/A |
| Experimental models: Organisms/strains | ||
| NOD-SCID IL2Rγnull mice | The Jackson Laboratory | Cat. #: 005557 |
| Oligonucleotides | ||
| Flag-F for mutagenesis: GATTACAAGGACGATGACGAC | This paper | N/A |
| Flag-R for mutagenesis: gtccttgtaatcGCAGATGGCGTCGGTGATC | This paper | N/A |
| IgG4CH3-F for mutagenesis: GGCCAGCCAAGAGAACC | This paper | N/A |
| IgG4CH3-R for mutagenesis: AGGACATGGAGGACAAGGAG | This paper | N/A |
| HIV-F for sequencing: TCAAGCCTCAGACAGTGGTTC | This paper | N/A |
| HIV-R for sequencing: AAGCGGCTTCGGCCAGTAAC | This paper | N/A |
| CR3022-F for sequencing: AATACCGCCTACCTGCAGTG | This paper | N/A |
| Recombinant DNA | ||
| pHIV-EGFP | Addgene | Cat. #: 18121; RRID: Addgene_18121 |
| pMD2.G | Addgene | Cat. #: 12259; RRID: Addgene_12259 |
| psPAX2 | Addgene | Cat. #: 12260; RRID: Addgene_12260 |
| pCDH-EF1a-eFFly-mCherry | Addgene | Cat. #: 104833; RRID: Addgene_104833 |
| pGBW-m4137382 | Addgene | Cat. #: 149539; RRID: Addgene_149539 |
| pHIV-Flag-28Z-EGFP | This paper | N/A |
| pHIV-CR3014-28Z-EGFP | This paper | N/A |
| pHIV-CR3022-28Z-EGFP | This paper | N/A |
| pHIV-CR3022-8a-28Z-EGFP | This paper | N/A |
| pHIV-CR3022-CH3-28Z-EGFP | This paper | N/A |
| pHIV-CR3022-IgG4-28Z-EGFP | This paper | N/A |
| Software and algorithms | ||
| FlowJo V10.0 | Tree Star | https://www.flowjo.com/ |
| Premium Cytobank | Cytobank | https://www.cytobank.org/ |
| ZEN Lite | Zeiss | https://www.zeiss.com/microscopy/int/home.html |
| Aura Imaging Software V3.2 | Spectral Instruments Imaging | https://spectralinvivo.com/ |
| Living Image Software | PerkinElmer | https://www.perkinelmer.com/ |
| GraphPad Prism® software v8.0 | GraphPad | https://www.graphpad.com |
| Other | ||
| cover-glass bottom dish | SPL life sciences | Cat. #: 100350 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Christopher E Rudd (christopher.e.rudd@umontreal.ca).
Materials availability
These unique CAR reagents will be made available upon request.
Experimental model and subject details
Cell lines
Cell lines included human embryonic kidney 293 and 293T cells, Vero (naturally express monkey ACE2) cells, NIH/3T3 fibroblast cells and the human T lymphoblastic leukemia Jurkat cells (American Type Culture Collection (ATCC). 293, 293T and Vero cells were cultured in DMEM supplemented with 10% heat-inactivated FBS (Gibco), 2 mM L-Glutamine (Corning), 10 mM HEPES (Corning), 100 U/ml Penicillin and Streptomycin (Sigma). By contrast, Jurkat cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, 2 mM L-Glutamine, 10 mM HEPES, 100 U/ml Penicillin and Streptomycin. Transduced 293 cells stably over-expressing human ACE2 and transduced NIH/3T3 cells stably expressing S1 domain of SARS-CoV-2 spike glycoprotein were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 10 mM HEPES and drug selection by 50 μg/ml hygromycin B. The 293-hACE2 cells and NIH/3T3-S1 cells were also transduced with firefly luciferase and mCherry by using the lentiviral vector pCDH-EF1a-eFFly-mCherry (Addgene), followed by sorting based on mCherry expression. All cell lines were confirmed for gene transduction before experiments.
Mouse study
Experimental procedures were approved by the Animal Care Committee of the CR-HMR (#2021-2509). The NOD-SCID IL2Rγnull mice were purchased from The Jackson Laboratory (Cat# 005557) and bred in the animal facility of the Research Center of Maisonneuve Rosemont Hospital, Montreal (#2017-1346, 2017-JA-001). Both female and male mice, the age of which range from 9 to 15 weeks old, were included and randomized to treatment groups based on sex, age and weight.
Human subjects
Human blood from healthy donors was provided by HÉMA-QUÉBEC with donors’ written informed consent and experiments using human PBMC were approved by the Research Ethics Committee of CIUSSS de l'Est-de-l'Île-de-Montréal (protocol # 2019-1184).
Method details
CAR constructs
The sequence of anti-SARS & SARS-CoV-2 spike glycoprotein antibody CR3022 and anti-SARS spike glycoprotein antibody CR3014 were obtained from a patent. To construct CAR expression vector, the VH and Vκ of CR3022 were fused with a (Gly4Ser)3 linker. The resulting single chain variable fragment (scFv) was linked to a Flag-tag (DYKDDDDK), a spacer, the CD28 trans-membrane and intracellular domain (68AA, 153-220, GenBank: NP_006130.1) and the CD3ζ intracellular domain (112AA, 52-163, GenBank: NP_000725.1) in tandem. The spacer is either a truncated CD28 hinge (39AA, 114-152, GenBank: NP_006130.1), a CD8a hinge (47AA, 136-182, GenBank: NP_001759.3), the CH3 domain of human IgG4 (119AA, UniProt ID: P01861) or a mutated heavy chain of human IgG4 (CH2-CH3, 229AA, 99-327, UniProt ID: P01861) as described elsewhere (Hudecek et al., 2015). The whole CAR construct was codon-optimized and synthesized by GeneArt (Thermo Scientific) and sub-cloned into pHIV-EGFP vector between EcoRI and XbaI cloning site.
Plasmids
The lentiviral vector pHIV-EGFP was a kind gift from Bryan Welm& Zena Werb (Addgene # 18121; http://n2t.net/addgene:18121; RRID: Addgene_18121) (Welm et al., 2008). The packaging plasmids pMD2.G (Addgene # 12259; http://n2t.net/addgene:12259; RRID: Addgene_12259) and psPAX2 (Addgene # 12260; http://n2t.net/addgene:12260; RRID: Addgene_12260) were kind gifts from Didier Trono. The lentiviral vector pCDH-EF1a-eFFly-mCherry was a kind gift from Irmela Jeremias (Addgene # 104833; http://n2t.net/addgene:104833; RRID: Addgene_104833) (Ebinger et al., 2016). The plasmid pGBW-m4137382, encoding the SARS-CoV-2 surface glycoprotein (Spike) for VSV pseudotyping, was a kind gift from Ginkgo Bioworks (Addgene # 149539; http://n2t.net/addgene:149539; RRID: Addgene_149539).
Preparation, titration and infection of pseudo-typed SARS-CoV-2 virus
293T cells were pre-plated in 6 well plates in 2 ml complete DMEM per well 24h prior to transfection. After being cultured overnight, 3 μg pGBW-m4137382 was mixed with 8 μl X-tremeGENE HP DNA transfection reagent and incubated for 30 min at room temperature before being added to the plate. After 24 h of transfection, the supernatant was discarded, and the transfected cells were infected with pseudotyped ΔG-DsRed rVSV at multiplicity of infection (MOI) of 5 at 37°C. After 1 h of infection, 1 ml fresh complete RPMI-1640 medium was added to the plate. The next day, the virus supernatant was collected, and the titre determined by serial dilution.
For infection assay, 293-ACE2 cells were pre-labeled with Tag-it Violet™ Tracking Dye (Biolegend) according to the manufacturer’s instruction. Then, 2×105 per tube of labeled 293-ACE2 cells were co-cultured with 5×105 per tube of effector cells at 37°C in the presence of pseudo-typed SARS-CoV-2 virus (MOI = 0.1) in a final volume of 500 μl complete RPMI-1640 medium. After 20 h of incubation, cells were washed and stained for viability, Flag and CD69 and analyzed by FACS. All the experiments were carried out in the laboratory of biosafety level 2.
Preparation of lentivirus and generation of CAR-Jurkat cells and CAR-T cells
293T cells were co-transfected with CAR-encoding vectors, pMD2.G and psPAX2 by using X-treme GENE HP DNA transfection reagent (Roche) according to the manufacturer’s instruction. After overnight transfection, the supernatant was replaced with fresh RPMI-1640 complete medium. The supernatant containing lentivirus was harvested at 48 h and 72 h post-transfection and stored at −80°C.
To generate CAR-T cells, 3 × 106 PBMC were resuspended in 1ml X-VIVO15 serum-free medium (Lonza) with soluble 1 μg/ml anti-hCD28 antibody (Clone # 9.3) and seeded into a 24 well plate pre-coated with 2 μg/ml anti-hCD3 antibody (Clone # OKT-3). After 72 h of stimulation, cells were washed 2 times, and the medium was exchanged with 2 ml virus supernatant containing 10 μg/ml protamine sulfate and spinoculated at 1200 g for 60 min at 32°C. After 1 h of incubation at 37°C, the supernatant was replaced with 2 ml fresh X-VIVO15 serum-free medium and cultured for 4 days before FACS sorting based on EGFP expression. The sorted CAR-T cells were expanded in X-VIVO15 medium with 1000 IU/ml recombinant human IL-2 (Stemcell) and sub-cultured every 2–3 days with fresh media.
To generate CAR-Jurkat, 2 × 105 Jurkat cells were resuspended with 1 ml virus supernatant containing 10 μg/ml protamine sulfate and spinoculated at 1200 g for 60 min at 32°C. After 1 h of incubation at 37°C, supernatant was replaced with 2 ml fresh RPMI-1640 complete medium and the transduced cells were sub-cultured for 3 passages before sorting with cell sorter (SONY SH800) based on EGFP expression.
Flow cytometry
For flow cytometric surface staining (anti-Flag, CD69 and FasL), CAR-Ts and other cells were incubated with fluorochrome-conjugated antibodies at 4°C for 30min followed by washing and fixation with 4 percent PFA followed by additional washing and analysis by either FACS Celesta (BD) or Fortessa X-20 (BD) as previously described (Raab et al., 2010). In certain instances, indirect two step staining was conducted with an unconjugated primary antibody for 30min on ice (anti-S1, anti-RBD) followed by washing and incubation with a conjugated secondary antibody for another 30 min at 4°C and analyzed by FACS. For intracellular staining (IFN-γ, GZMB, perforin, pS6), cells were permeabilized by using the intracellular staining kit (Invitrogen), according to the manufacturer’s protocol. Data analysis was performed by using FlowJo V10.0 (Tree Star) or t-Distributed Stochastic Neighbor Embedding (t-SNE) and Cytobank analysis (Alfei et al., 2019; McLane et al., 2019).
In vitro functional assays
Three types of in vitro functional assays were conducted. In one case, freshly expanded CAR-Ts (106/cell/ml) were incubated with 293-ACE2-RBD or 293-ACE2 cells (106cells CAR-Ts and 5.0 × 105/ml 293-ACE2 cells) for 20 min prior to permeabilization kit (Invitrogen) and staining with phospho-S6 (1:1000 dilution). As a control, phorbol ester (PMA) (Sigma) and ionomycin (Sigma) was also used to stimulate the CAR-T for 20 min. In another instance, the same proportions of cells were incubated for 18 hrs followed by cell permeabilization and staining with anti-GZMB, anti-perforin and anti-IFN-g, or for 24 h and stained for CD69 and FasL. In the third in vitro assay, we measured the cytolytic killing of target cells. 293-ACE2 target cells were either left untreated or were coated with the RBD-His (#SPD-C52H3, Acro Biosystems) or S1-His peptides (#S1N-C82E8, Acro Biosystems). For coating the 293-ACE2 cells, 1 μg/ml RBD or S1 protein was added to 1 × 106/ml cells at 37°C for 1 hr followed by a gentle wash in media before used in experiments with CAR-Ts. In other experiments, NIH/3T3 cells expressing S1 was also used as described above. The effector cells were isolated from the same donor for each individual experiment and harvested after 2–3 weeks of cellular expansion in culture in vitro with IL-2 supplement media.
For the cytotoxicity assay, 1 × 104 per well of target cells were co-cultured with different effector cells at the ratio of 1/20, 1/10 and 1/5 in U-bottom 96-well plates (Corning) in a final volume of 100 μl RPMI-1640 medium (5% FBS, phenol red free) per well. All samples were set in triplicate. After 4 h of incubation at 37°C, 50 μl supernatant of each well was collected to measure the LDH release by using CytoTox 96® Non-Radioactive Cytotoxicity Assay kit (Promega) according to the manufacturer’s protocol. The cytotoxicity was calculated as follows: Cytotoxicity (%) = (Experimental Release – Effector Spontaneous Release – Target Spontaneous Release)/(Target Maximum Release– Target Spontaneous Release) × 100.
In certain instances, antibodies to the RBD epitope or FasL were used to block the response. For blocking the RBD epitope, 1 μg/ml RBD protein was added to 1 × 106/ml 293-ACE2 cells and incubated at 37°C for 1 h. After that, the cells were washed twice, re-suspended with 400 μl RPMI-1640 medium (5% FBS, phenol red free) and split into two EP tubes equally. 1 μg/ml anti-RBD antibody (clone # CR3022, Abcam) or 1 μg/ml rabbit IgG isotype control antibody (Clone # 60024B, R&D) was added to each tube (5 × 105 293-ACE2-RBD in 200 μl medium), respectively, and incubated at 37°C for 30 min. Then cells were directly diluted with RPMI-1640 medium to a final volume of 2 ml/tube without wash, and co-cultured with different CAR-T cells at the ratio of 1/24, 1/12, 1/6 and 1/3 in a U-bottom 96-well plate (Corning).
Similarly, for the blockade of FasL, 2 μg/ml anti-hFasL antibody (NOK-1 or 100419) was added to 1×106/ml CAR-T cells, incubated at 37°C for 1 h and co-cultured with target cells without wash.
Time-lapse imaging
5 × 104 293-ACE2-mCherry cells were resuspended with 500 μl RPMI-1640 medium (5% FBS, phenol red free) and pre-plated in a cover-glass bottom dish (SPL life sciences) at 37°C. After being cultured overnight, 1 μg/ml RBD protein was added to cells and incubated at 37°C for 1 h. Then 2×106 CR3022-8a-28Z-EGFP CAR-T cells were resuspended with 1.5 ml RPMI-1640 medium (5% FBS, phenol red free) and added to the dish for co-culture in an inverted fluorescence microscope with climate control (Zeiss Observer Z1). Images were captured from the beginning of the co-culture with a 2 min interval for 24 h. Videos were exported and analysed by using ZEN Lite software (Zeiss).
In vivo cytolytic assay
Mice were injected i.p. with either 2 × 106 NIH/3T3-S1-Luc cells (NT group) or a combination of 2 × 106 NIH/3T3-S1-Luc cells and CAR-T cells in the ratio of 1/2 premixed in 300 ul of RPMI-1640 medium (CR3014-28z and CR3022-28z groups). At 24 h post-implantation, mice were injected i.p. with 150 mg/kg RediJect D-Luciferin (PerkinElmer; #770505). Mice were anaesthetized with 2% isoflurane and transferred to the IVIS Lumina III (PerkinElmer) for in vivo imaging at 10 min post-injection. Mice from three different groups were imaged simultaneously with 20 sec of exposure. Bioluminescence images were overlapped with bright field images of mice and displayed in the same intensity range. Images were acquired and analyzed by using Aura Imaging Software (Spectral Instruments Imaging). The total radiance (Photon/s and counts) was measured and calculated for each region of interest (ROI).
Quantification and statistical analysis
The n represents individual donors from a random pool of donors provided by the blood service Hema-Quebec (Quebec). The response of CAR-Ts was compared from one donor to another. This measure applied to all figures in the paper. Although CAR-T cell numbers were expanded in vitro from a given individual donor, all cells were used at a single time from the individual donor for cytolytic killing assays and flow cytometry. No repeat experiments from individual donors was done due to the lack of repeat access to individual donors. Statistical analysis was performed by using GraphPad Prism® software v8.0 (GraphPad). Two-tailed Student’s t-test was used to compare two normally distributed independent groups with continuous endpoints. The one-way ANOVA was applied when more than two groups were compared and the two-way ANOVA was applied when there are two independent variables. Dunnett’s post hoc test was used for multiple comparisons. All experiments were repeated at least two times, and all experimental data in figures and texts are shown as the mean ± standard deviation. p values less than 0.05 was considered statistically significant.
Acknowledgments
C.E. Rudd was supported by Canadian Institutes of Health Foundation grant (159912), Canada.
Author contributions
C.E. Rudd and X.Y. Guo conceptualized and coordinated study, whereas C.E. Rudd was responsible for writing and coordinating the study. X.Y. Guo and C.E. Rudd designed the CAR constructs, whereas X.Y. Guo conducted the research. A. Kazanova helped with the overall planning of in vitro and in vivo experiments, flow cytometry, and viSNE analysis and cooperated with X.Y. Guo in the running of the in vivo experiment. H.U. Saragovi helped to provide conceptual expertise on hACE2-expressing 293 cells and the S1-expressing NIH/3T3 cells used in some of the cytotoxicity assays. S. Thurmond provided expertise on viral expansion and in vitro studies.
Declaration of interests
The authors declare no competing financial interests.
Inclusion and diversity
One or more of the authors of this paper received support from a program designed to increase minority representation in science.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103295.
Supplemental information
Data and code availability
All datasets in this study have been deposited at Mendeley Data https://data.mendeley.com/datasets/3y3m8btcnk/2 and will be shared further by the lead contact upon request. DOI is listed in the key resources table. Any additional information required to re-analyze the data reported in this paper is available upon request.
References
- Alfei F., Kanev K., Hofmann M., Wu M., Ghoneim H.E., Roelli P., Utzschneider D.T., von Hoesslin M., Cullen J.G., Fan Y., et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571:265–269. doi: 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- Allan W., Tabi Z., Cleary A., Doherty P.C. Cellular events in the lymph node and lung of mice with influenza. Consequences of depleting CD4+ T cells. J. Immunol. 1990;144:3980–3986. [PubMed] [Google Scholar]
- Altmann D.M., Boyton R.J. SARS-CoV-2 T cell immunity: specificity, function, durability, and role in protection. Sci. Immunol. 2020;5:eabd6160. doi: 10.1126/sciimmunol.abd6160. [DOI] [PubMed] [Google Scholar]
- Amanat F., Krammer F. SARS-CoV-2 vaccines: status report. Immunity. 2020;52:583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch E.M., Shoham S., Casadevall A., Sachais B.S., Shaz B., Winters J.L., van Buskirk C., Grossman B.J., Joyner M., Henderson J.P., et al. Deployment of convalescent plasma for the prevention and treatment of COVID-19. J. Clin. Invest. 2020;130:2757–2765. doi: 10.1172/JCI138745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun J., Loyal L., Frentsch M., Wendisch D., Georg P., Kurth F., Hippenstiel S., Dingeldey M., Kruse B., Fauchere F., et al. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature. 2020;587:270–274. doi: 10.1038/s41586-020-2598-9. [DOI] [PubMed] [Google Scholar]
- Buchrieser J., Dufloo J., Hubert M., Monel B., Planas D., Rajah M.M., Planchais C., Porrot F., Guivel-Benhassine F., Van der Werf S., et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2021;40:e107405. doi: 10.15252/embj.2020107405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K.M., Steiner G., Wells D.K., Ribas A., Kalbasi A. Prediction of SARS-CoV-2 epitopes across 9360 HLA class I alleles. bioRxiv. 2020 doi: 10.1101/2020.1103.1130.016931. [DOI] [Google Scholar]
- Cao Y., Su B., Guo X., Sun W., Deng Y., Bao L., Zhu Q., Zhang X., Zheng Y., Geng C., et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients' B cells. Cell. 2020;182:73–84.e16. doi: 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M.H., Zhang S., Porritt R.A., Noval Rivas M., Paschold L., Willscher E., Binder M., Arditi M., Bahar I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc. Natl. Acad. Sci. U S A. 2020;117:25254–25262. doi: 10.1073/pnas.2010722117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibrian D., Sanchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017;47:946–953. doi: 10.1002/eji.201646837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizmecioglu A., Cizmecioglu H.A., Goktepe M.H., Emsen A., Korkmaz C., Tasbent F.E., Colkesen F., Artac H. Apoptosis-induced T cell lymphopenia is related to COVID-19 severity. J. Med. Virol. 2020;93:2867–2874. doi: 10.1002/jmv.26742. [DOI] [PubMed] [Google Scholar]
- Ebinger S., Ozdemir E.Z., Ziegenhain C., Tiedt S., Castro Alves C., Grunert M., Dworzak M., Lutz C., Turati V.A., Enver T., et al. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell. 2016;30:849–862. doi: 10.1016/j.ccell.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein S.L., Lo C.Y., Misplon J.A., Bennink J.R. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J. Immunol. 1998;160:322–327. [PubMed] [Google Scholar]
- Fu W., Lei C., Ma Z., Qian K., Li T., Zhao J., Hu S. CAR macrophages for SARS-CoV-2 immunotherapy. Front. Immunol. 2021;12:669103. doi: 10.3389/fimmu.2021.669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdiero M.R., Garlanda C., Jaillon S., Marone G., Mantovani A. Tumor associated macrophages and neutrophils in tumor progression. J. Cell. Physiol. 2013;228:1404–1412. doi: 10.1002/jcp.24260. [DOI] [PubMed] [Google Scholar]
- Graham M.B., Braciale T.J. Resistance to and recovery from lethal influenza virus infection in B lymphocyte-deficient mice. J. Exp. Med. 1997;186:2063–2068. doi: 10.1084/jem.186.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S., et al. Targets of T Cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020;181:1489–1501 e1415. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross G., Gorochov G., Waks T., Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transpl. Proc. 1989;21:127–130. [PubMed] [Google Scholar]
- Guedan S., Ruella M., June C.H. Emerging cellular therapies for cancer. Annu. Rev. Immunol. 2019;37:145–171. doi: 10.1146/annurev-immunol-042718-041407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Pohlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell. 2020;78:779–784 e775. doi: 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280 e278. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudecek M., Sommermeyer D., Kosasih P.L., Silva-Benedict A., Liu L., Rader C., Jensen M.C., Riddell S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015;3:125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihling C., Tanzler D., Hagemann S., Kehlen A., Huttelmaier S., Arlt C., Sinz A. Mass spectrometric identification of SARS-CoV-2 proteins from gargle solution samples of COVID-19 patients. J. Proteome Res. 2020;19:4389–4392. doi: 10.1021/acs.jproteome.0c00280. [DOI] [PubMed] [Google Scholar]
- Irving B.A., Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64:891–901. doi: 10.1016/0092-8674(91)90314-o. [DOI] [PubMed] [Google Scholar]
- Jean S.S., Lee P.I., Hsueh P.R. Treatment options for COVID-19: the reality and challenges. J. Microbiol. Immunol. Infect. 2020;53:436–443. doi: 10.1016/j.jmii.2020.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena B., Dotti G., Cooper L.J. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 2010;116:1035–1044. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawalekar O.U., O'Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Jr., Patel P.R., Guedan S., Scholler J., Keith B., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Kawasaki A., Ebata T., Ohmoto H., Ikeda S., Inoue S., Yoshino K., Okumura K., Yagita H. Metalloproteinase-mediated release of human Fas ligand. J. Exp. Med. 1995;182:1777–1783. doi: 10.1084/jem.182.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs K., Bottinger N., Huang L.R., Chmielewski M., Arzberger S., Gasteiger G., Jager C., Schmitt E., Bohne F., Aichler M., et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145:456–465. doi: 10.1053/j.gastro.2013.04.047. [DOI] [PubMed] [Google Scholar]
- Kurschus F.C., Jenne D.E. Delivery and therapeutic potential of human granzyme B. Immunol. Rev. 2010;235:159–171. doi: 10.1111/j.0105-2896.2010.00894.x. [DOI] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Letko M., Marzi A., Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020;5:562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Qiu S., Chen J., Jiang S., Chen W., Jiang J., Wang F., Si W., Shu Y., Wei P., et al. Chimeric antigen receptor designed to prevent ubiquitination and downregulation showed durable antitumor efficacy. Immunity. 2020;53:456–470.e456. doi: 10.1016/j.immuni.2020.07.011. [DOI] [PubMed] [Google Scholar]
- Lu C., Klement J.D., Ibrahim M.L., Xiao W., Redd P.S., Nayak-Kapoor A., Zhou G., Liu K. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J. Immunother. Cancer. 2019;7:157. doi: 10.1186/s40425-019-0635-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M.T., Badeti S., Chen C.H., Kim J., Choudhary A., Honnen B., Reichman C., Calianese D., Pinter A., Jiang Q., et al. CAR-NK cells effectively target SARS-CoV-2-spike-expressing cell lines in vitro. Front. Immunol. 2021;12:652223. doi: 10.3389/fimmu.2021.652223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus M.V., June C.H. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin. Cancer Res. 2016;22:1875–1884. doi: 10.1158/1078-0432.CCR-15-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLane L.M., Abdel-Hakeem M.S., Wherry E.J. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 2019;37:457–495. doi: 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- McMichael A.J., Michie C.A., Gotch F.M., Smith G.L., Moss B. Recognition of influenza A virus nucleoprotein by human cytotoxic T lymphocytes. J. Gen. Virol. 1986;67:719–726. doi: 10.1099/0022-1317-67-4-719. [DOI] [PubMed] [Google Scholar]
- Meckiff B.J., Ramirez-Suastegui C., Fajardo V., Chee S.J., Kusnadi A., Simon H., Eschweiler S., Grifoni A., Pelosi E., Weiskopf D., et al. Imbalance of regulatory and cytotoxic SARS-CoV-2-reactive CD4(+) T cells in COVID-19. Cell. 2020;183:1340–1353.e1316. doi: 10.1016/j.cell.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet J.K., Tang T., Nathan L., Jaimes J.A., Hsu H.L., Daniel S., Whittaker G.R. Production of pseudotyped particles to study highly pathogenic coronaviruses in a biosafety level 2 setting. J. Vis. Exp. 2019 doi: 10.3791/59010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monjas A., Alcover A., Alarcon B. Engaged and bystander T cell receptors are down-modulated by different endocytotic pathways. J. Biol. Chem. 2004;279:55376–55384. doi: 10.1074/jbc.M409342200. [DOI] [PubMed] [Google Scholar]
- Moskophidis D., Kioussis D. Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J. Exp. Med. 1998;188:223–232. doi: 10.1084/jem.188.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev E.N., Indeykina M.I., Brzhozovskiy A.G., Bugrova A.E., Kononikhin A.S., Starodubtseva N.L., Petrotchenko E.V., Kovalev G.I., Borchers C.H., Sukhikh G.T. Mass-spectrometric detection of SARS-CoV-2 virus in scrapings of the epithelium of the nasopharynx of infected patients via nucleocapsid N protein. J. Proteome Res. 2020;19:4393–4397. doi: 10.1021/acs.jproteome.0c00412. [DOI] [PubMed] [Google Scholar]
- Osborn J.F., Hobbs S.J., Mooster J.L., Khan T.N., Kilgore A.M., Harbour J.C., Nolz J.C. Central memory CD8+ T cells become CD69+ tissue-residents during viral skin infection independent of CD62L-mediated lymph node surveillance. PLoS Pathog. 2019;15:e1007633. doi: 10.1371/journal.ppat.1007633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paidi R.K., Jana M., Mishra R.K., Dutta D., Raha S., Pahan K. ACE-2-interacting domain of SARS-CoV-2 (AIDS) peptide suppresses inflammation to reduce fever and protect lungs and heart in mice: implications for COVID-19 therapy. J. Neuroimmune Pharmacol. 2021;16:59–70. doi: 10.1007/s11481-020-09979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S.L., Moslehi J., Robert M.E., Craft J. Perforin protects against autoimmunity in lupus-prone mice. J. Immunol. 1998;160:652–660. [PubMed] [Google Scholar]
- Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Broad and strong memory CD4(+) and CD8(+) T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A., Thalheimer F.B., Hartmann S., Frank A.M., Bender R.R., Danisch S., Costa C., Wels W.S., Modlich U., Stripecke R., et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol. Med. 2018;10:e9158. doi: 10.15252/emmm.201809158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey A.D., Jr., Schwab R.D., Boesteanu A.C., Steentoft C., Mandel U., Engels B., Stone J.D., Madsen T.D., Schreiber K., Haines K.M., et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 2016;44:1444–1454. doi: 10.1016/j.immuni.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premkumar L., Segovia-Chumbez B., Jadi R., Martinez D.R., Raut R., Markmann A., Cornaby C., Bartelt L., Weiss S., Park Y., et al. The receptor binding domain of the viral spike protein is an immunodominant and highly specific target of antibodies in SARS-CoV-2 patients. Sci. Immunol. 2020;5:eabc8413. doi: 10.1126/sciimmunol.abc8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M., Wang H., Lu Y., Smith X., Wu Z., Strebhardt K., Ladbury J.E., Rudd C.E. T cell receptor "inside-out" pathway via signaling module SKAP1-RapL regulates T cell motility and interactions in lymph nodes. Immunity. 2010;32:541–556. doi: 10.1016/j.immuni.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse B.T., Sehrawat S. Immunity and immunopathology to viruses: what decides the outcome? Nat. Rev. Immunol. 2010;10:514–526. doi: 10.1038/nri2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux P.P., Shahbazian D., Vu H., Holz M.K., Cohen M.S., Taunton J., Sonenberg N., Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell B., Moss C., George G., Santaolalla A., Cope A., Papa S., Van Hemelrijck M. Associations between immune-suppressive and stimulating drugs and novel COVID-19-a systematic review of current evidence. Ecancermedicalscience. 2020;14:1022. doi: 10.3332/ecancer.2020.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell J.H., Ley T.J. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 2002;20:323–370. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J., et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell. 2020;183:996–1012 e1019. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M., Brentjens R., Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 2009;21:215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Jose E., Borroto A., Niedergang F., Alcover A., Alarcon B. Triggering the TCR complex causes the downregulation of nonengaged receptors by a signal transduction-dependent mechanism. Immunity. 2000;12:161–170. doi: 10.1016/s1074-7613(00)80169-7. [DOI] [PubMed] [Google Scholar]
- Sarzi-Puttini P., Giorgi V., Sirotti S., Marotto D., Ardizzone S., Rizzardini G., Antinori S., Galli M. COVID-19, cytokines and immunosuppression: what can we learn from severe acute respiratory syndrome? Clin. Exp. Rheumatol. 2020;38:337–342. [PubMed] [Google Scholar]
- Schneider H., Martin M., Agarraberes F.A., Yin L., Rapoport I., Kirchhausen T., Rudd C.E. Cytolytic T lymphocyte-associated antigen-4 and the TCR zeta/CD3 complex, but not CD28, interact with clathrin adaptor complexes AP-1 and AP-2. J. Immunol. 1999;163:1868–1879. [PubMed] [Google Scholar]
- Schoenborn J.R., Wilson C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- Schroder K., Hertzog P.J., Ravasi T., Hume D.A. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Seif M., Einsele H., Loffler J. CAR T cells beyond cancer: Hope for Immunomodulatory therapy of infectious diseases. Front. Immunol. 2019;10:2711. doi: 10.3389/fimmu.2019.02711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty A.K. Mesenchymal stem cell infusion shows promise for combating coronavirus (COVID-19)- induced pneumonia. Aging Dis. 2020;11:462–464. doi: 10.14336/AD.2020.0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R., Shan C., Duan X., Chen Z., Liu P., Song J., Song T., Bi X., Han C., Wu L., et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020;584:120–124. doi: 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- Smyth M.J., Sutton V.R., Kershaw M.H., Trapani J.A. Xenospecific cytotoxic T lymphocytes use perforin- and Fas-mediated lytic pathways. Transplantation. 1996;62:1529–1532. doi: 10.1097/00007890-199611270-00030. [DOI] [PubMed] [Google Scholar]
- Song J.W., Zhang C., Fan X., Meng F.P., Xu Z., Xia P., Cao W.J., Yang T., Dai X.P., Wang S.Y., et al. Immunological and inflammatory profiles in mild and severe cases of COVID-19. Nat. Commun. 2020;11:3410. doi: 10.1038/s41467-020-17240-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman J., Lee R.K., Podack E.R. Perforin/Fas-ligand double deficiency is associated with macrophage expansion and severe pancreatitis. J. Immunol. 1998;161:7063–7070. [PubMed] [Google Scholar]
- Sui Y., Bekele Y., Berzofsky J.A. Potential SARS-CoV-2 immune correlates of protection in infection and vaccine immunization. Pathogens. 2021;10:138. doi: 10.3390/pathogens10020138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay M.Z., Poh C.M., Renia L., MacAry P.A., Ng L.F.P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 2020;20:363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Meulen J., van den Brink E.N., Poon L.L., Marissen W.E., Leung C.S., Cox F., Cheung C.Y., Bakker A.Q., Bogaards J.A., van Deventer E., et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. Plos Med. 2006;3:e237. doi: 10.1371/journal.pmed.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharmarajah E., Buazon A., Patel V., Hannah J.R., Adas M., Allen V.B., Bechman K., Clarke B.D., Nagra D., Norton S., et al. IL-6 inhibition in the treatment of COVID-19: a meta-analysis and meta-regression. J. Infect. 2021;82:178–185. doi: 10.1016/j.jinf.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P.G., Keating R., Hulse-Post D.J., Doherty P.C. Cell-mediated protection in influenza infection. Emerg. Infect. Dis. 2006;12:48–54. doi: 10.3201/eid1201.051237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X., Li C., Huang A., Xia S., Lu S., Shi Z., Lu L., Jiang S., Yang Z., Wu Y., Ying T. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020;9:382–385. doi: 10.1080/22221751.2020.1729069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham D.J., Tripp R.A., Doherty P.C. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 1997;159:5197–5200. [PubMed] [Google Scholar]
- Urra J.M., Cabrera C.M., Porras L., Rodenas I. Selective CD8 cell reduction by SARS-CoV-2 is associated with a worse prognosis and systemic inflammation in COVID-19 patients. Clin. Immunol. 2020;217:108486. doi: 10.1016/j.clim.2020.108486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varchetta S., Mele D., Oliviero B., Mantovani S., Ludovisi S., Cerino A., Bruno R., Castelli A., Mosconi M., Vecchia M., et al. Unique immunological profile in patients with COVID-19. Cell Mol. Immunol. 2020;18:604–612. doi: 10.1038/s41423-020-00557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E., Raulet D.H., Moretta A., Caligiuri M.A., Zitvogel L., Lanier L.L., Yokoyama W.M., Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf D., Schmitz K.S., Raadsen M.P., Grifoni A., Okba N.M.A., Endeman H., van den Akker J.P.C., Molenkamp R., Koopmans M.P.G., van Gorp E.C.M., et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020;5:eabd2071. doi: 10.1126/sciimmunol.abd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welm B.E., Dijkgraaf G.J., Bledau A.S., Welm A.L., Werb Z. Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell. 2008;2:90–102. doi: 10.1016/j.stem.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.A., Bevan M.J. Effector and memory CTL differentiation. Annu. Rev. Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- Wrobel A.G., Benton D.J., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020;27:763–767. doi: 10.1038/s41594-020-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Ong Y.K., Wang Y. Role of adjunctive treatment strategies in COVID-19 and a review of international and national clinical guidelines. Mil. Med. Res. 2020;7:22. doi: 10.1186/s40779-020-00251-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y., Jeyanathan M., Haddadi S., Barra N.G., Vaseghi-Shanjani M., Damjanovic D., Lai R., Afkhami S., Chen Y., Dvorkin-Gheva A., et al. Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell. 2018;175:1634–1650.e1617. doi: 10.1016/j.cell.2018.09.042. [DOI] [PubMed] [Google Scholar]
- Yuan M., Wu N.C., Zhu X., Lee C.-C.D., So R.T.Y., Lv H., Mok C.K.P., Wilson I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science. 2020;368:630–633. doi: 10.1126/science.abb7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler S.F., Ramsdell F., Alderson M.R. The activation antigen CD69. Stem Cells. 1994;12:456–465. doi: 10.1002/stem.5530120502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CR3022-8a-28Z CAR-Ts were co-cultured with the 293-ACE2 cells and tracked for 20 hours. EGFP+ CAR-Ts failed to form clusters with 293-ACE2.
CR3022-8a-28Z CAR-Ts were co-cultured with 293-ACE2 that had been pre-incubated with RBD peptide and tracked for 20 hours. EGFP+ CAR-Ts were observed to form clusters with 293-ACE2+RBD cells.
CR3022-8a-28Z CAR-Ts were co-cultured with 293-ACE2 that had been pre-incubated with S1 peptide and tracked for 20 hours. EGFP+ CAR-Ts were observed to form clusters with 293-ACE2+S1 cells.
Data Availability Statement
All datasets in this study have been deposited at Mendeley Data https://data.mendeley.com/datasets/3y3m8btcnk/2 and will be shared further by the lead contact upon request. DOI is listed in the key resources table. Any additional information required to re-analyze the data reported in this paper is available upon request.