

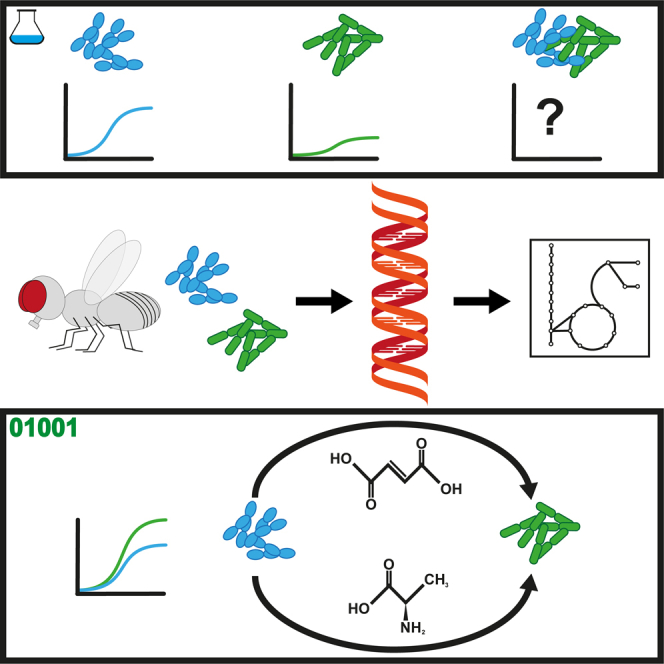
Summary
We know a lot about varying gut microbiome compositions. Yet, how the bacteria affect each other remains elusive. In mammals, this is largely based on the sheer complexity of the microbiome with at least hundreds of different species. Thus, model organisms such as Drosophila melanogaster are commonly used to investigate mechanistic questions as the microbiome consists of only about 10 leading bacterial species. Here, we isolated gut bacteria from laboratory-reared Drosophila, sequenced their respective genomes, and used this information to reconstruct genome-scale metabolic models. With these, we simulated growth in mono- and co-culture conditions and different media including a synthetic diet designed to grow Drosophila melanogaster. Our simulations reveal a synergistic growth of some but not all gut microbiome members, which stems on the exchange of distinct metabolites including tricarboxylic acid cycle intermediates. Culturing experiments confirmed our predictions. Our study thus demonstrates the possibility to predict microbiome-derived growth-promoting cross-feeding.
Subject areas: Microbial metabolism, Microbiome
Graphical abstract
Highlights
-
•
Reconstruction of six Drosophila gut bacteria genome-scale models
-
•
Gut bacteria consortium modeling reveals growth interdependencies
-
•
Certain Drosophila gut bacteria show growth-promoting cross-feeding
-
•
Experimental validation of the modeling-derived growth-promoting effect
Microbial metabolism; Microbiome
Introduction
Multicellular organisms are inhabited by a vast number of microorganisms, which is generally termed the microbiome. In humans, the number of associated bacteria is in the same range as the cells of the host (Sender et al., 2016). As an entity, the bacteria encode an overwhelming number of genes and thus expand the metabolic capabilities of the host enormously. We are still at the beginning of understanding this metabolic interplay. Yet, first reports demonstrated an importance of the microbes present in the gut, the so-called gut microbiome, in humans and model organisms for increasing nutrient availability and energy harvest (Krajmalnik-Brown et al., 2012), the production of important bioactive metabolites including branched-chain amino acids (Lin et al., 2017; Liu et al., 2020), the metabolism of pharmaceuticals applied to the host (Clayton et al., 2009; Haiser et al., 2013), or the release of metabolites which affect signaling pathways of the host (Martin et al., 2019; Shin et al., 2011). Thus, the microbiome affects the host far beyond nutrient access. The importance of the gut microbiome can be seen most prominently in times of a perturbation or altered microbiome composition, which has been linked to many human diseases such as diabetes (Hartstra et al., 2014; Komaroff, 2016), obesity (Tilg and Kaser, 2011; Turnbaugh and Gordon, 2009), autism (Vuong and Hsiao, 2016), or inflammatory bowel disease (Halfvarson et al., 2017). Based on the observation that a perturbed microbiome is linked to pathologies, microbiome-focused therapies appear possible. Indeed, microbiome transfer therapies proved effective for the treatment of infections with the pathogen Clostridium difficile (Weingarden et al., 2013) and many pro- and prebiotic dietary regimens are already used (Arora et al., 2013).
The microbiome of mammals with hundreds to thousands of different bacterial species is extremely complex. In addition, many of these species cannot be cultured ex vivo, which hinders detailed functional analyses. Simpler model organisms can help to overcome these limitations and thus provide access to targeted functional analyses. The microbiome of Drosophila melanogaster, for example, only consists of 5–20 different species (Douglas, 2019; Ludington and Ja, 2020), which makes it much easier to analyze. Still, the gut microbiome of Drosophila has a significant impact on many aspects of the hosts' life such as the survival under nutrient limiting conditions, the lifespan of the flies, or the locomotor behavior (Consuegra et al., 2020a; Keebaugh et al., 2018; Ridley et al., 2012; Schretter et al., 2018; Shin et al., 2011; Silva et al., 2020; Storelli et al., 2011, 2018). The most abundant Drosophila gut bacteria belong to the Lactobacilli, Acetobacter, and Enterococci genera. Key species of these bacteria are culturable under standard laboratory conditions (Adair et al., 2018; Broderick and Lemaitre, 2012; Erkosar et al., 2013).
The prominent Drosophila gut microbiome members Lactobacillus plantarum and Lactobacillus brevis are Gram-positive rod-shaped lactic acid-producing microaerophilic bacteria from the Firmicutes phylum, which promote the systemic growth of fly larvae under nutrient-limiting conditions (Storelli et al., 2011). In humans, several lactobacilli strains have been shown to confer host health benefits (Marco et al., 2010), and a decline in their abundance is commonly associated with diseases (Aron-Wisnewsky et al., 2020; Heeney et al., 2018; Lee et al., 2020; Schwarzer et al., 2016). Acetobacter in contrast are Gram-negative, acetic acid-producing bacteria within the class of alpha-proteobacteria. They can be isolated from a variety of sources such as fruits and flowers and are often used to generate fermented food, e.g., vinegar (Azuma et al., 2009). Acetobacter species are major constituents of the Drosophila gut microbiome. Like lactobacilli (Storelli et al., 2011), they contribute to a successful larval development under nutrient-limiting conditions (Shin et al., 2011). This growth-promoting effect was demonstrated to stem on the secretion of acetic acid, which interferes with the insulin signaling pathway of the fly (Shin et al., 2011). This observation underpins the importance of secreted metabolites in terms of an interaction not only with the host but also likely with other members of the gut microbiome. At this point, the beneficial as well as detrimental (e.g., in terms of competition for nutrients) interactions between the microbiome members are not clear. First analyses, however, detected a complex interplay between combinations of the bacterial species and the host, which shapes host fitness through life history trade-offs (Gould et al., 2018). Similarly, also studies with isolated bacteria using growth on agar-based solid media (Sommer and Newell, 2018) or chemically defined media (Aumiller et al., 2021) support growth-promoting effects among the bacterial species of the Drosophila gut microbiome.
In order to investigate such metabolic interactions, we isolated bacteria from laboratory-reared Drosophila and investigated their isolated growth in different media such as Lactobacillus-promoting MRS and Acetobacter-selective ACE media. Furthermore, we used a synthetic diet suitable to grow D. melanogaster (holidic Drosophila diet; HD) (Piper et al., 2014). Six bacterial strains were analyzed in total and we resequenced their respective genomes to reconstruct genome-scale metabolic networks. These were used in single and co-culture growth simulations using the BacArena software package (Bauer et al., 2017). Our results reveal co-operative growth of certain bacteria based on the exchange of distinct metabolites including tricarboxylic acid cycle (TCA) intermediates, certain sugars, as well as amino acids in the D- and L-form. In analogous growth experiments, we could confirm the growth-promoting effect of several identified metabolites. Thus, the simulations open the door to systematically investigate the metabolic interplay of gut microbiome constituents and to reveal beneficial metabolites, which can promote the growth of selected gut microbiome constituents.
Results
Bacterial isolation, species identification, and in vitro growth characteristics
We started our analysis with the isolation of bacteria from the intestine of white[-] and Oregon-R adult flies (see material and methods). First, we isolated in total six morphologically distinct colonies on either Lactobacillus growth-promoting MRS- or Acetobacter-enriching ACE-agar plates and subsequently extracted the respective genomic DNA of our pure cultures. The 16S rRNA gene region of all clones was amplified by PCR, subcloned, and sequenced to allow species identification by BLAST searches. In total, we isolated two L. plantarum, one L. brevis, two Acetobacter indonesiensis, and one Acetobacter pasteurianus strains (see Table 1).
Table 1.
Sequencing results, genome reassembly, and generated genome-scale model summaries
| L. plantarum (A2) | L. plantarum (B2) | L. brevis (B6) | A. indonesiensis (A4) | A. indonesiensis (A5) | A. pasteurianus (B5) | |
|---|---|---|---|---|---|---|
| Genome assembly | ||||||
| Reads [#] | 3,587,296 | 3,638,786 | 3,277,616 | 3,319,800 | 3,387,326 | 3,239,340 |
| Used reads [#] | 2,902,970 | 3,125,105 | 1,917,270 | 2,060,518 | 2,036,898 | 2,077,587 |
| Used reads [%] | 86.2 | 90.3 | 59.6 | 62.2 | 60.3 | 66.9 |
| Unmapped [#] | 495,436 | 353,339 | 1,324,152 | 1,254,692 | 1,345,613 | 1,070,761 |
| Genes [#] | 3,676 | 3,559 | 2,595 | 3,352 | 3,364 | 3,091 |
| Ref. genome sequence length (bp) | 3,581,586 | 3,581,586 | 2,340,228 | 3,396,180 | 3,396,180 | 3,007,920 |
| Reference genome | BDGP2 | BDGP2 | ATCC 367 | NBRC 16471 | NBRC 16471 | BDGP5 |
| Metabolic models | ||||||
| Reactions [#] | 1,815 | 1,815 | 1,584 | 1,931 | 1,931 | 1,796 |
| Metabolites [#] | 1,567 | 1,567 | 1,411 | 1,763 | 1,763 | 1,673 |
| Genes [#] | 657 | 658 | 473 | 631 | 632 | 580 |
| Blocked reactions [%] | 40.1 | 40.1 | 41 | 43.3 | 43.3 | 43.7 |
| Unbalanced reactions [%] | 9.4 | 9.4 | 10.2 | 8.2 | 8.2 | 8.5 |
| Exchange reactions [%] | 9 | 9 | 9.9 | 7.5 | 7.5 | 7.7 |
| Bacterium | Isolate | Ref. genome | NCBI ID | Link |
|---|---|---|---|---|
| Acetobacter pasteurianus | B5 | BDGP5 | ASM245613v1 | https://www.ncbi.nlm.nih.gov/assembly/GCF_002456135.1/ |
| Acetobacter indonesiensis | A4 | NBRC 16471 | ASM799107v1 | https://www.ncbi.nlm.nih.gov/assembly/GCF_007991075.1/ |
| Acetobacter indonesiensis | A5 | NBRC 16471 | ASM799107v1 | https://www.ncbi.nlm.nih.gov/assembly/GCF_007991075.1/ |
| Lactobacillus plantarum | A2 | BDGP2 | ASM229018v1 | https://www.ncbi.nlm.nih.gov/assembly/GCF_002290185.1/ |
| Lactobacillus plantarum | B2 | BDGP2 | ASM229018v1 | https://www.ncbi.nlm.nih.gov/assembly/GCF_002290185.1/ |
| Lactobacillus brevis | B6 | ATCC 367 | ASM1446v1 | https://www.ncbi.nlm.nih.gov/assembly/GCF_000014465.1/ |
The upper part of the table summarizes the sequencing results in terms of the number of reads obtained for the six bacterial resequencing reactions. These sequencing results were mapped with the ASA³P software (Schwengers et al., 2020) to the respective reference genomes whose ID as well as NCBI accession is provided. The details of the mapping results in terms of the number and percent of used (as well as unmapped) reads, the number of detected genes, and the genome sequence length are provided. The resequenced genome sequences were subsequently used to build the genome-scale metabolic models (see materials and methods). The lower part of Table 1 provides the details of the six genome-scale models in terms of the number of reactions, metabolites, mapped genes, blocked and unbalanced, as well as exchange reactions. All sequencing, ASA3P, and model data are available at https://doi.org/10.17632/2tgjd6y4zb.1.
We tested next the growth of the different bacteria in three different growth media (Figures 1 and 2). On top of the commonly used semi-defined MRS (Lactobacillus enriching medium; see materials and methods) and ACE (promoting Acetobacter growth; see materials and methods) liquid culturing media, we also tested for growth in a chemically defined (holidic diet [HD]) growth medium sufficient to culture D. melanogaster (Piper et al., 2014). All isolated lactobacilli were able to grow on the MRS medium (Figure 1A). L. brevis, however, showed a lower total growth than the two L. plantarum isolates (Figure 1A). On the ACE medium, all lactobacilli only showed low growth (Figure 1B) demonstrating the selectivity of the growth medium. In line with previous results (Consuegra et al., 2020a), L. plantarum grew relatively well on the HD, whereas L. brevis again only showed a low growth (Figure 1C). To our surprise, growth of the Acetobacter isolates did not differ much on the MRS and ACE media (Figures 2A and 2B). A. indonesiensis isolates showed prominent growth on the HD (Figure 2C). A. pasteurianus, in contrast only showed a relatively poor growth on the HD (Figure 2C). An overview of the experimentally determined growth rates is provided as Figure S1.
Figure 1.

Wet-lab and in silico growth of Lactobacillus on different media
(A–C) Growth of the Lactobacillus isolates L. plantarum (A2, light green, dot), L. plantarum (B2, medium green, check), and L. brevis (B6, dark green, cross) on MRS (A), ACE (B), and HD (C) media. Growth of all bacteria was monitored for at least 45 h in a plate reader without shaking. All cultures were inoculated with a 1:1,000 dilution for MRS and ACE media and with a 1:100 dilution for the HD medium. All cultures had an optically dense pre-culture. Representative growth curves of at least three biologically independent experiments are shown. Growth curves show mean values of triplicate measurements.
(D–F) Simulated growth of the same bacteria in the same media as shown in (A–C). For the isolated bacteria, the genomes were resequenced and used to reconstruct genome-scale metabolic networks. These were used for growth simulations using the BacArena software package (Bauer et al., 2017) in combination with MRS (D), ACE (E), and HD (F) media. L. plantarum (A2, light green, dot), L. plantarum (B2, medium green, check), and L. brevis (B6, dark green, cross) on MRS (D), ACE (E), and HD (F) media. The simulations for each bacterium were run at least 12 times, and the computed growth curves represent the mean values. Detailed model data are available at https://doi.org/10.17632/2tgjd6y4zb.1. Wet-lab (A–C; small reaction tube) and in silico data (D–F; computer) are also indicated by the pictograms and labels on the right side of the figure.
Figure 2.

Wet-lab and in silico growth of Acetobacter on different media
(A–C) Growth of the Acetobacter isolates A. indonesiensis (A4, light orange, pentagon), A. indonesiensis (A5, medium orange, triangle), and A. pasteurianus (B5, dark orange, star) on MRS (A), ACE (B), and HD (C) media. Growth of all bacteria was monitored for at least 45 h in a plate reader with shaking. All cultures were inoculated with a 1:1,000 dilution for MRS and ACE media and with a 1:100 dilution for the HD medium. All cultures had an optically dense pre-culture. Representative growth curves of at least three biologically independent experiments are shown. Growth curves show mean values of triplicate measurements. For the isolated bacteria, the genomes were resequenced and used to reconstruct genome-scale metabolic networks. These were used for growth simulations using the BacArena software package (Bauer et al., 2017) in combination with MRS (D), ACE (E), and HD (F) media.
(D–F) A. indonesiensis (A4, light orange, pentagon), A. indonesiensis (A5, medium orange, triangle), and A. pasteurianus (B5, dark orange, star) on MRS (D), ACE (E), and HD (F) media. The simulations for each bacterium were run at least 12 times, and the computed growth curves represent the mean values. Detailed model data are available at https://doi.org/10.17632/2tgjd6y4zb.1. Wet-lab (A-C; small reaction tube) and in silico data (D-F; computer) are also indicated by the pictograms and labels on the right side of the figure.
The determination of growth of single species cultures is trivial, whereas the determination of the individual contribution of distinct species to the biomass production of a consortium is difficult. Yet, a better understanding of the mutual effect on the growth of bacterial consortia is an intriguing and important question. Modeling experiments are a possibility to overcome this obstacle. For the modeling, an exact knowledge of the nutritional content of the growth medium is very important. Thus, growth of the bacteria on HD was particularly important, as this diet allows the exact description of the input for the modeling experiments. In the past, we already benefitted from this for modeling the growth and metabolism of Drosophila larvae (Schönborn et al., 2019). In order to reconstruct the genome-scale metabolic networks of the isolated bacteria, our next step was to sequence their respective genomes using the Illumina MiSeq platform (see material and methods). In the following, the genomes were assembled using whole-genome information as a scaffold, which we obtained from the NCBI database.
Sequencing of the isolate genomes and model reconstruction
The sequencing runs resulted in 3.2–3.6 Mio reads per genome (see Table 1). The reads were mapped to the whole-genome sequences of L. plantarum BDGP2, L. brevis ATCC367, A. indonesiensis NBRC16471, and A. pasteurianus BDGP5, respectively, and further analyzed using the ASA³P software (Schwengers et al., 2020) (the complete dataset is available in the supplement). Between 60% and 90% of the total reads mapped to the respective reference strains (see Table 1).
We reconstructed the genome-scale metabolic models (for a summary cf. Table 1) of our isolated Drosophila gut bacteria using the gapseq pipeline (Zimmermann et al., 2021). As a last step in the model generation, we used gapseq's in-built gap filling algorithm to enable in silico growth of the models on the one hand for the ACE/MRS media and on the other hand for the HD medium (see material and methods and Data S1). This additional step takes composition differences of the varying media into consideration. The ACE and MRS media are semi-defined owing to chemically complex components, which makes the in silico representation of the growth environment more difficult. We could explain between 73% and 92% of the unknown complex ingredients (yeast extract, peptone, and meat extract) by the help of information from the literature or the respective manufacturer. For HD such problems do not exist, as this medium is chemically completely defined (Piper et al., 2014). The overview of the diet parametrization is provided in Figure S2 as well as Data S2. In the course of generating the models, we took great care to correct for stochiometric inconsistencies, mass and charge imbalances, as well as metabolite connectivity (see materials and methods section and Table 1). All models were tested for model quality using the MEMOTE tool (Lieven et al., 2020) and resulted in at least 77% model scores (see Data S3 and materials and methods).
In silico biomass and signature metabolite production by the different genome-scale metabolic network models
In order to model growth of the different isolated gut bacteria alone as well as in combination, we performed dynamic and agent-based simulations of bacterial population growth and metabolic fluxes using the BacArena software package (Bauer et al., 2017). In brief, BacArena allows growth simulation of single-species population and multi-species microbial communities in a spatially limited compartment, including the calculations of the changing medium composition due to the metabolite utilization and production by individual bacterial cells. Thus, the metabolism of the organisms is calculated in a time-resolved manner with the biomass production as the objective function (for information concerning the biomass production and objective function, please see material and methods as well as Data S1). BacArena provides the metabolic fluxes, growth pattern, and concentrations of the medium for each time point of each individual species present in the in silico experiment. This allows the determination of possible cross-feeding and/or physiological interactions in a multi-species in silico culture experiment.
As a starting point, we performed single bacteria growth simulations in the three different media MRS, ACE, and HD. An uncertain parameter was the amount of oxygen entering the system. Acetobacteraceae are aerophilic, whereas lactobacilli are microaerophilic and tolerate only a small amount of oxygen. Furthermore, it is still unknown how much oxygen is present in the larval and adult Drosophila gut. Given that our goal was to model the situation within the Drosophila gut where the two genera would meet each other, we performed all simulations in the presence of 0.1 mM oxygen, which represents a microaerobic situation (Ito et al., 2002).
Of the lactobacilli, the two L. plantarum models showed good growth on all media (Figures 1D–1F). L. brevis, in contrast, showed only limited biomass production in the MRS, ACE, and HD simulations (Figures 1D–1F). The A. indonesiensis and A. pasteurianus models all result in strong biomass production in simulations utilizing the ACE and MRS media (Figures 2D and 2E). On the HD, however, all Acetobacter strain model simulations only showed low biomass production (Figure 2F). When we compared our in silico growth simulation results to the actual wet-lab data (Figure S1), our lactobacilli simulations fitted the experimental data overall better. So far, the reasons for the discrepancies of the Acetobacter simulations are not clear. Yet, the appropriate simulation of growth magnitudes is inherently difficult using FBA (see discussion) and might depend on many parameters. For our experiments, however, we focused on the identification of growth dependencies and metabolite exchanges, which are only considering relative changes and are thus unaffected by these shortcomings.
Next, we investigated the production of certain signature metabolites by the different models. Several Lactobacillus species are able to use the phosphoketolase pathway and are thus heterolactic (Spector, 2009). On top of the lactobacilli signature metabolite lactate, heterolactic bacteria also produce acetate. Here, we thus tested for a possible heterolactic behavior of our L. plantarum and L. brevis models. For the Acetobacter models, we did not expect such a behavior and only a prominent production of acetate.
As flux-balance simulations can vary to some extent in terms of individual flux predictions due to stochastic effects, we performed the simulations 100 times to identify the most likely metabolite production behavior (Figure S3). Figures 3 and S4 show representative simulation results (Data S4 is an interactive version of Figure 3, which provides all predicted metabolite productions). Lactate production was mostly limited to L. plantarum (B2) on the MRS and ACE diets, L. plantarum (A2) on the ACE diet, and L. brevis (B6) on the HD (Figures 3 and S3). None of the Acetobacter models produced lactate (Figures S3 and S4).
Figure 3.

In silico production of signature metabolites by the different genome-scale metabolic network models
(A–C) Production of lactate by the L. plantarum (A2, light green, dot), L. plantarum (B2, medium green, check), and L. brevis (B6, dark green, cross) genome-scale models on MRS (A), ACE (B), and HD (C) media, respectively.
(D–F) Production of acetate by the A. indonesiensis (A4, light orange, pentagon), A. indonesiensis (A5, medium orange, triangle), and A. pasteurianus (B5, dark orange, star) genome-scale models on MRS (D), ACE (E), and HD (F) media, respectively. Please note that not all models produced the respective signature metabolite on the given medium. Metabolite production curves represent mean values of at least 12 simulation runs. An interactive version of the figure is available as Data S4 and detailed model data are available at https://doi.org/10.17632/2tgjd6y4zb.1.
All Acetobacter model simulations resulted in prominent acetate production on the ACE and MRS growth media (Figures 3D, 3E and S3). Yet, on the HD only A. pasteurianus (B5) was producing acetate (Figures 3F and S3). For the Lactobacilli, only the two L. plantarum models showed prominent acetate production on the MRS and ACE media (Figures S3 and S4). On the HD, all Lactobacilli showed acetate production (Figures S3 and S4). Altogether, our simulations thus reveal a heterolactic behavior of the isolated lactobacilli as well as demonstrate the expected metabolite production for the Acetobacter models. Next, we investigated the co-culturing behavior in silico.
Simulating the co-culturing of Lactobacillus and Acetobacter
Our key question was whether bacteria present in the gut could affect each other's growth. For other gut microbiome members of the fly such beneficial metabolite exchange behavior could be recently demonstrated (Consuegra et al., 2020a; Henriques et al., 2020). For the species isolated in this study, we detected prominent growth differences in the different growth media in vitro (Figures 1 and 2) as well as in silico (Figures 1 and 2). Our hypothesis was that the growth of co-cultures could be different from the growth of pure cultures based on the exchange of metabolites. If one is able to predict the impact of an exchange of metabolites between the different species of a gut microbiome as well as the impact of the metabolite exchange, one could design prebiotics, which means metabolites promoting the growth of a certain beneficial gut microbiome constituent. In order to test for such potential growth-promoting effects, we performed simulations comparing the mono-inoculations to all pair-wise combinations of Acetobacter and lactobacilli. In order to quantify potential growth effects, we first estimated the predicted biomass production after 45 h for the individual or co-cultured growth. Figures 4A–4C show the color-coded results for all individual and combined growth conditions on the MRS (A), ACE (B), and HD (C) media (all simulation data are available in the supplement). In Figures 4D–4F we highlight three detailed representative modeling outcomes from the overview representation in Figures 4A–4C (orange box in B relates to D, green box in B relates to E, and red box in C relates to F).
Figure 4.

In silico co-culturing of Lactobacillus and Acetobacter
(A–C) We simulated the growth of all individual as well as pair-wise combinations of the Lactobacilli and Acetobacter models on the MRS (A), ACE (B), and HD (C) media. The plots summarize the color-coded biomass produced after 45 h of simulated growth. Total amount of produced biomass from 0–250 pg: beige, equals no or weak growth; 250–500 pg of predicted biomass: light blue; equals intermediate growth, and 500–750 pg of predicted biomass: dark blue; equals strong growth.
(D–F) Detailed time-resolved data for three different examples of single organism growth simulations as well as the simulated growth of the combination of the bacteria. D (refers to orange box in B) shows an example of the most trivial growth behavior, where the combination of L. plantarum (A2, light green, dot) and A. pasteurianus (B5, dark orange, star) on the ACE medium limits the growth of each other based on the impact of space and resource competition. E (relates to green box in B) shows an example of a detrimental outcome of the combination of bacteria. L. plantarum (B2, medium green, check) and A. indonesiensis (A5, medium orange, triangle) grow individually well on the ACE medium. The combination, however, results in a prominent block of the Lactobacillus growth, perhaps due to resource competition effects. F (relates to red box in C) shows a probiotic activity of L. brevis (B6, dark green, cross) on the growth of A. indonesiensis (A4, light orange, pentagon) on the HD. Both bacteria individually only show minute biomass production on the HD, whereas the combination results in a prominent growth of A. indonesiensis (A4, light orange, pentagon).
First, we consider the predicted growth curves of singular (upper two panels) or combined (lowest panel) L. plantarum (A2) and A. pasteurianus (B5) on ACE medium (Figure 4D) as an example of a trivial growth behavior. Both bacteria individually grow very well on the ACE medium. When combined, however, the available space gets limiting and thus both bacteria just reach half of the arbitrarily set maximum possible biomass production of 750 pg. Thus, the two bacteria only affected their mutual growth in terms of a limitation of the available resources. The combination of bacteria, however, can also result in non-trivial growth effects. Simulations with the L. plantarum (B2) and A. indonesiensis (A5) models on the ACE medium, for example, result individually in very high biomass production (Figure 4E). Yet in combination, the Acetobacter model results in higher biomass production, whereas the Lactobacillus model results in much lower biomass production (Figure 4E). Thus, the presence of Acetobacter apparently limits the biomass production of the Lactobacillus model, perhaps by winning the competition about the available resources.
Most striking, however, the combination of L. brevis B6 and A. indonesiensis A4, which individually produce on the HD only very little biomass in simulations (Figure 4F), results in a surprisingly prominent biomass production of Acetobacter (Figure 4F). In fact, the combination of L. brevis (B6) and all Acetobacter models resulted in such a growth behavior (Figure 4C). Thus, only a small amount of Lactobacillus was necessary to allow prominent biomass production of the Acetobacter model and Lactobacillus serves as a probiotic for Acetobacter in our simulations.
Analysis for metabolites exchanged between Acetobacter and Lactobacillus
The results of our co-occurrence simulations suggest that growth interdependencies between the different gut bacteria exist. Ultimately, the simulations should result in predictions ready to test in in vivo experiments. Thus, we concentrated on the following on the growth simulations performed with the HD, as with this defined diet, we can control and fine-tune its constituents. In addition, this diet can also be used in the future to monitor the growth of the bacteria in combination with their natural host D. melanogaster. In terms of a probiotic activity of L. brevis for A. indonesiensis we envisioned that the Lactobacillus either removed a growth-inhibiting or secreted a growth-promoting factor thus enabling Acetobacter to produce biomass in our simulations. Thus, we monitored the excretion and uptake rates of both bacteria over time within the simulations. For an easier detection of a net efflux or uptake, we formed a quotient between the individual uptake rates and normalized the values (see materials and methods). This allowed us to plot the exchange reactions in a heatmap (Figure 5) where a positive value means that both bacteria take up or excrete the given metabolite and a negative value means that the bacteria show a reciprocal metabolite transport behavior. Thus, a negative value is consistent with the excretion of a given metabolite from one bacterium and the uptake of the same metabolite by the other species. Figure 5 shows the situation after 32 h of growth (see Data S5 for an interactive version of the figure providing the data for all time points). Many transport reactions had a positive sign, and thus the direction of the transport pointed in the same direction in both bacteria. Few reactions, however, consistently showed a negative sign, which is in line with an exchange of the given metabolite. Among those, D-Alanine, L-Arginine, D-Ribose, Acetaldehyde, Fumarate, and Butane-2,3-diol (BDOH) showed the most prominent exchange behavior.
Figure 5.

Flux of exchange reactions during the co-culturing of Acetobacter and Lactobacillus on the HD
We simulated the combined growth of Acetobacter and Lactobacillus on the HD and monitored the respective fluxes of the exchange reactions (thus, the fluxes representing an uptake or excretion of a given metabolite) over time. Exchange reactions are defined as reactions (or passages) where metabolites can flow in and out of the metabolic network and therefore the organism or cell. They can be subjected to different constraints such as diffusion or Michaelis-Menten kinetics of metabolite transporters, but for most reactions, only boundary thresholds can be set as the real-world flux rates are unknown. Further information on exchange reaction is found in Cotten and Reed (2013); Orth et al. (2010). For the sake of simplicity, we combined the individual fluxes into a normalized quotient, where a positive sign represents the same directionality (e.g., both bacteria secrete a given metabolite) of the individual fluxes and a negative sign represents opposite directionalities (e.g., one bacterium secretes a given metabolite and the other consumes it). The heatmap represents the flux ratios at 32 h of growth (an interactive version of the plot for all time points is provided as Data S5). Gray color represents that the respective metabolite is either not present or only transported by one of the two bacteria (not shown in color scale on the right); green color opposite and lilac color same flux directionalities. Multiple metabolites consistently show opposite flux directionalities across bacterial species combinations and across the time line.
Growth-promoting effect of singular metabolites added to Acetobacter cultures
We tested next whether the addition of any of the metabolites shown in Figure 5 to the HD growth medium simulations is sufficient to improve the growth of A. indonesiensis, which alone showed only poor biomass production on the HD medium (Figure 6A). Of the 43 metabolites tested (Figure S5), only 10 metabolites showed a growth-promoting effect in silico. Those were indeed enriched for the metabolites, which showed a predicted exchange from one bacterial species to the other (negative sign in Figure 5). The in silico addition of the TCA intermediate fumarate, for example, resulted in prominently increased predicted biomass production (Figure 6B). The same growth-promoting effect is visible in the in silico prediction of D-Ribose added to the HD medium (Figure 6C). No growth-promoting effect was visible when D-Alanine was added to the HD medium in the in silico prediction of A. pasteurianus B5 (Figure 6D), whereas biomass production of A. indonesiensis A4 and A5 was promoted (Figure S5). Thus, the simulations suggested that already the exchange of a singular metabolite between the bacterial species could result in a growth-promoting effect.
Figure 6.

Growth-promoting effect of singular added metabolites
(A–D) In silico biomass production of A. pasteurianus (B5) on the standard HD. In silico biomass production of A. pasteurianus (B5) on HD with 10 mM (B) Fumarate, (C) Ribose, and (D) D-Alanine.
(E–G) Actual growth measurements of A. pasteurianus (B5) on HD (dark orange) with Fumarate (E), Ribose (F), or D-Alanine (G) (10 nM, 0.1 μM, 0.1 mM, 1 mM, and 100 mM; black color and different dashed lines). In silico experiments (A, B, C, and D) are represented by the computer, whereas the wet-lab experiments (E, F, and G) are represented by the small reaction tube pictograms.
Finally, we tested for the experimental validation of the predicted growth-promoting effects. For this purpose, we recorded growth curves of A. pasteurianus (B5) in HD containing varying concentrations of fumarate (Figure 6E), D-Ribose (Figure 6F), and D-Alanine (Figure 6G). With fumarate and D-Ribose, we selected metabolites that showed in silico a prominent growth-promoting effect on all Acetobacter species (Figure S5), whereas D-Alanine did not result in a full growth rescue of A. pasteurianus (B5), but only the other two Acetobacter species (Figure S5). D-Ribose alone was not sufficient to improve the growth of A. pasteurianus (B5) prominently (Figure 6F). Yet, the addition of fumarate and D-Alanine in different concentrations showed a prominent positive effect on the growth of the bacteria (Figures 6E and 6G).
Altogether, our results suggest that microbiome members are metabolically connected, thus affecting the growth of individual microbiome members. The strategy presented herein consisting of the isolation of distinct bacteria, their genome sequencing, and subsequent in silico modeling of growth and metabolism thus proved successful to identify metabolite exchange and growth-promoting metabolites. Future experiments targeted to investigate combinatorial effects of metabolite additions as well as the contribution of the hosts' metabolism will further extend our understanding of the complex interplay among the gut microbiome members.
Discussion
In this study, we analyzed multiple members of the Drosophila gut microbiome by a combination of in vitro and in silico experiments. In total, we isolated six bacterial strains from laboratory-reared Drosophila flies followed by in vitro growth experiments, resequencing, and genome assembly and in silico growth and metabolism modeling analyses.
First, we tested for a biomass production of the singular bacteria models on ACE, MRS, and HD. L. plantarum was able to generate high amounts of biomass on the ACE medium, whereas L. brevis was not (Figures 1D–1F). Similar growth was detected on the MRS medium and on HD. All Acetobacter models resulted in high biomass production on the ACE and MRS media and only very low biomass production on the HD (Figures 2D–2F). Our models mostly recapitulated the corresponding actual growth experiments (Figures 1, 2, and S1). Especially the poor growth of the L. brevis isolate was detected in vitro and in silico (Figures 1, 2, and S1). The reason for this growth deficit is to date not clear. For some of the organisms, such as A. indonesiensis on the ACE medium, the modeling results deviate from the actual measurements in terms of the magnitude of the effect (Figure S1). This is a problem seen in many modeling approaches, which might be based on a variety and most likely a combination of many parameters, including gaps in the model, confounding factors, and the lack of certain environmental conditions in the modeling procedure. Furthermore, the modeling procedure depends on the requirement to define “exchange reactions,” which are thresholds setting the boundaries for metabolic fluxes going into and out of the model. Although these thresholds can be controlled by different constraints such as diffusion or Michaelis-Menten kinetics of metabolite transporters (Cotten and Reed, 2013; Orth et al., 2010), for most of the reactions, these boundaries are not experimentally validated and thus the model itself is largely underdetermined. Furthermore, also the biology of the given bacterium might be a cause for deviations between the experimental and modeling data. A prerequisite of the FBA procedure is the assumption that an objective function is optimized in terms of a maximization. Often as well as in our study the optimized objective function is biomass production. Previous studies, however, demonstrated that several microorganisms operate at a sub-maximal growth rate (Fischer and Sauer, 2005; Schuetz et al., 2007, 2012). The reasons for this behavior are not yet always clear.
Altogether, these parameter variations and modeling uncertainties will result not only in deviations of the magnitude of, e.g., biomass production, but also in kinetic differences, e.g., in terms of the growth rate. With variations in the build-up of biomass, also the mass transfer will vary, thus potentially resulting in more prominent differences between the computed and wet-lab results. Important, these confounding characteristics of the modeling procedure apply to the single and the multiple species growth simulations. The latter, however, will of course be even more severely affected by differences in the growth rates of the individual species that make up the consortium as the mass ratios between the species will also affect the mass transfer of metabolites. Furthermore, also the details concerning the juxtaposition (directly neighbored versus located in, e.g., different compartments of the gut) as well as variations in the initial mass ratio, thus the relative abundance of each species, will prominently affect the individual growth rates and mass transfer. Further experimental data including, e.g., localization studies, measurements of the individual abundance of bacterial species, and metabolic labeling experiments to determine flux rates as well as refinements of the models will help to improve the modeling outcome in the future.
Future iterations and refinements of the models will also need to target the optimization of the growth condition parameterization. Our simulations using the HD medium is a first step in the direction of modeling the actual growth conditions within the fly gut, as all bacteria as well as the host can thrive on this medium. The standard diet most often used to rear Drosophila is complex and undefined, often containing live or dry yeast, molasses, or treacle, which makes the parameterization and modeling very complex. Also, the exact conditions within the gut are still not clear as, e.g., metabolite concentrations might vary along the anterior-posterior axis of the gut as well as across the diameter of the gut. Thus, further experimental and modeling work will be needed to decipher these details in the future.
On top of testing the biomass production, model validation also included the analysis of expected signature metabolite production. Acetobacter, for example, is known to oxidize sugars or ethanol to acetic acid (Raspor and Goranovič, 2008), whereas lactobacilli produce glucose-derived lactic acid as the main product (Hatti-Kaul et al., 2018). Both metabolic models were able to recapitulate this behavior (Figure 3). It is intriguing that the previously described heterolactic metabolism of lactobacilli (Spector, 2009) could also be recapitulated for our isolated bacteria (Figure S3) suggesting that our models result in realistic metabolic behavior predictions. Of note, however, some of the predictions need to be considered with care. Our simulations, for example, also revealed the production of H2O2 and also H2S. Both substances can act as inhibitors of bacterial growth, especially in higher concentrations (Alt et al., 1999; Reis et al., 1992). Nevertheless, some Acetobacter species were demonstrated to produce H2S under certain conditions (Ahmad et al., 2004). Thus, so far it is not clear whether the neutral or even positive effect of the presence of these substances on the growth (Figure S5) is real or based on the limitation of FBA to predict correctly growth-inhibiting and detrimental effects of certain metabolites.
The main goal of our study was to test whether we can predict metabolic growth-promoting inter-species interactions. If possible, this could open up the door to design tailored prebiotics to promote or hinder the growth of certain gut microbiome members. For our simulations, we tested all pair-wise combinations of Acetobacter and Lactobacillus on the three different media ACE, MRS, and HD. Many combinations were neutral in a way that the growth of the singular bacteria was similar or identical in the singular and combination situation (Figures 4A–4C; the complete dataset is provided in the supplement). In case both bacteria showed high growth in single growth simulations, the combination resulted in a competitive situation, which caused both bacteria to grow less (e.g., Figure 4D). On top of these trivial situations, however, we also observed inhibitory and stimulatory interactions. The L. plantarum strain B2 and A. indonesiensis strain A5 result in comparable and high biomass production in the ACE medium when grown independently (Figure 4E). The combination, however, does not result in an equal reduction of the biomass to an intermediate level, but in contrast to a much stronger reduction of the Lactobacillus biomass production, whereas Acetobacter production got increased (Figure 4E). Likely, this effect is based on resource competition, which might also play a role within the gut of the Drosophila host. Even more astonishing was the stimulatory effect of combining the individually poor biomass producers L. brevis and either of the Acetobacter models, which we were able to track down to the exchange of selected metabolites (Figures 4F and S5). For fumarate and D-Alanine, we already were able to confirm the growth-promoting effect by simply adding these metabolites to the HD medium (Figure 6). Ribose, however, did not result in the expected growth rescue. At this point, the reasons for this discrepancy are unclear. Whether additional metabolites could also rescue the growth deficit to a similar extent is at this point unknown. Similarly, it is also not clear how the co-culturing of the organisms in the end affects each other as beneficial and competition effects most likely will play a role and thus a more complex growth effect will arise.
Fumarate and D-Alanine could affect the growth of the bacteria by different means. Thus, we considered different possibilities and cross-validated these using our modeling data. Formally, the metabolites could complement auxotrophies. Based on our modeling and experimental data, however, we exclude this possibility, as the bacteria also grow without the supplementation in the MRS or ACE media (Figures 1 and 2). Furthermore, the compounds could function as additional C- or N-source and enter the metabolism. Fumarate indeed is a central metabolite of the TCA. Thus, its uptake could enhance the overall capacity of the TCA. Various TCA intermediates further serve the biosynthesis of different amino acids, which potentially could also benefit biomass production. For Acetobacter pomorum a potential use of fumarate by the enzyme succinate dehydrogenase (EC1.3.5.1, present in TCA) was discussed where fumarate serves as an O-donor for the production of NAD+ and NADP+ from Aspartate (Consuegra et al., 2020b). D-Alanine, in contrast, could be converted first to L-Alanine and subsequently to pyruvate, which serves as a carbon and energy source. When we analyzed the corresponding flux differences of the modeling performed in the presence or absence of the metabolites in the HD (Figure S6 and Data S6 and S7), we indeed detected a number of corresponding flux changes. First, we consider the situation where D-Alanine was added to the HD. Here, we see an increase in the flux associated with the conversion of D-Alanine to Pyruvate, as expected (Figure S6A). Pyruvate production is further enhanced coming from oxaloglutarate (Figure S6B). Other prominent changes include the change of direction of the fluxes from fumarate to malate and oxaloacetate (Figures S6C and S6D), the production of isocitrate from citrate (Figure S6E), the production of S-Succinyl-dihydrolipoamide from oxaloglutarate (Figure S6F), or the enhanced production of glutamate-derived amino acids such as glutamine (Figure S6G). Many of these enzymatic reactions are also affected by adding fumarate to the HD. Overall, the fumarate-induced flux changes of the TCA reactions are, however, bigger as from D-Alanine. Fumarate also resulted in a third possibility to enhance the pyruvate production coming from oxaloacetate (Figure S6H). The fumarate addition induced higher flux changes, which might provide an explanation for the overall bigger growth rescue phenotype detected in the actual growth experiments (Figure 6E). A recent report also targeted the prediction of Drosophila gut microbiome metabolite interactions using in silico models (Ankrah et al., 2021). The authors independently also revealed that TCA intermediate metabolites appear to be prominently exchanged between gut microbiome members. In their simulations, the authors used different media than we did, but still found a similar range of exchanged metabolites. Reassuringly, many of the exchanged metabolites are shared by our and the published study. In our extended studies, however, we did not detect a prominent growth-promoting effect for some of these in our simulations (e.g., acetate, succinate, different individual amino acids). Yet, several metabolites detected in both studies (e.g., acetoin, acetaldehyde) clearly resulted in an individual growth-promoting activity (c.f. Figure S5 and Ankrah et al., 2021).
Our results support the possibility to use genome-scale models in combination with agent-based growth simulations to predict meaningful microbiome cooperativity. In the future, extending this approach to additional microbiome constituents and/or the metabolism of the host D. melanogaster will be exciting and perhaps pave the way to analyze also the much more complex microbiomes of higher organisms.
Limitations of the study
There are limitations in the modeling of growth-promoting bacterial metabolic interactions. On the one hand, this is true for the modeling side as outlined above. For example, FBA assumes optimization and maximization of a given parameter such as biomass production, yet organisms sometimes operate at a sub-optimal level. Furthermore, our knowledge of many parameters required for the modeling such as nutrient distribution along the gut, nutrient uptake rates, and transport reaction efficacies are unknown, which results in the necessity to make assumptions that are in the best case imprecise and in the worst case wrong. Further iterations and improvements on the modeling and experimental side might solve some of these shortcomings using, e.g., isotope labeling experiments. On the other hand, uncertainties concerning the biology exist. For example, we used laboratory-reared flies and detected the most prominent microbiome growth interactions on a minimal diet used for the growth of Drosophila. In the future, bacteria from wild-reared animals grown under natural conditions should be used, which, however, will be experimentally very challenging. Finally, our analyses were performed with simple consortia. Ultimately, complex mixtures with varying relative microbial species abundancies and consisting of more species will be required to estimate the true importance of metabolic cross-feeding phenomena among gut microbiota.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Acetobacter pasteurianus | This paper | B5 |
| Acetobacter indonesiensis | This paper | A4 |
| Acetobacter indonesiensis | This paper | A5 |
| Lactobacillus plantarum | This paper | A2 |
| Lactobacillus plantarum | This paper | B2 |
| Lactobacillus brevis | This paper | B6 |
| Chemicals, peptides, and recombinant proteins | ||
| L-arginine HCl | Sigma-Aldrich | Cat#A5131 |
| L-alanine | Sigma-Aldrich | Cat#A7627 |
| L-asparagine | Sigma-Aldrich | Cat#A0884 |
| L-aspartic acid | Sigma-Aldrich | Cat#A6683 |
| L-cysteine | Sigma-Aldrich | Cat#C1276 |
| L-glutamic acid monosodium salt monohydrate | Sigma-Aldrich | Cat#G5889 |
| L-glutamine | Sigma-Aldrich | Cat#G3126 |
| Glycine | Sigma-Aldrich | Cat#G7126 |
| L-histidine | Sigma-Aldrich | Cat#H8000 |
| L-isoleucine | Carbolution | Cat#CC10025 |
| L-leucine | Sigma-Aldrich | Cat#L8912 |
| L-lysine HCl | Sigma-Aldrich | Cat#L5626 |
| L-methionine | Sigma-Aldrich | Cat#M9625 |
| L-phenylalanine | Sigma-Aldrich | Cat#P2126 |
| L-proline | Sigma-Aldrich | Cat#P0380 |
| L-serine | Sigma-Aldrich | Cat#S4500 |
| L-threonine | Carl Roth | Cat#T206 |
| L-tryptophan | Sigma-Aldrich | Cat#T0254 |
| L-tyrosine | Sigma-Aldrich | Cat#T3754 |
| L-valine | Sigma-Aldrich | Cat#V0500 |
| Sucrose | Carl Roth | Cat#4661 |
| Cholesterol | Sigma-Aldrich | Cat#C8667 |
| choline chloride | Sigma-Aldrich | Cat#C1879 |
| myo-inositol | Sigma-Aldrich | Cat#I7508 |
| Inosine | Sigma-Aldrich | Cat#I4125 |
| Uridine | Sigma-Aldrich | Cat#U3750 |
| Tween20 | Sigma-Aldrich | Cat#P7949 |
| KH2PO4 | Grüssing Gmbh | Cat#120171000 |
| NaHCO3 | AppliChem | Cat#AP131638 |
| CaCl2.6H2O | Sigma-Aldrich | Cat#442909 |
| CuSO4.5H2O | AcrosOrganics | Cat#A0302205 |
| FeSO4.7H2O | Sigma-Aldrich | Cat#F7002 |
| MgSO4.7H2O | AppliChem | Cat#A6287 |
| MnCl2.4H2O | Sigma-Aldrich | Cat#M3634 |
| ZnSO4.7H2O | Sigma-Aldrich | Cat#Z0251 |
| thiamine (aneurin) | Sigma-Aldrich | Cat#T4625 |
| Riboflavin | Sigma-Aldrich | Cat#R4500 |
| nicotinic acid | Sigma-Aldrich | Cat#N4126 |
| Ca pantothenate | Sigma-Aldrich | Cat#P21210 |
| pyridoxine-HCL | Sigma-Aldrich | Cat#P9755 |
| Biotin | Sigma-Aldrich | Cat#B4501 |
| folic acid | Sigma-Aldrich | Cat#F7876 |
| HPLC | Fisher Scientific | Cat#231-791-2 |
| Fumarate | BLD Pharmatech Gmbh | Cat#BD131629 |
| D(-)-Ribose | AcrosOrganics | Cat#10320164 |
| D-Alanine | Carbolution | Cat#CC10041 |
| Acetic acid glacial | VWR Chemicals | Cat#KRAF20104 |
| Glucose | Fisher Scientific | Cat#10529190 |
| Sodium acetate | Grüssing Gmbh | Cat#121111000 |
| Cycloheximide | AppliChem | Cat#A0879 |
| Peptone | Carl Roth | Cat#8986.2 |
| Yeast Extract | BD Company | Cat#212750 |
| Beef Extract | Carl Roth | Cat#X975 |
| Triammonium citrate | Sigma-Aldrich | Cat#A1332 |
| Tween20 | Sigma-Aldrich | Cat#P7949 |
| Ethanol | Honeywell | Cat#32221 |
| MRS agar plates | Thermo Scientific | Cat#CM0361B |
| Proteinase K | Thermo Scientific | Cat#AM2546 |
| Lysozyme | Sigma-Aldrich | Cat#34046 |
| Phusion HF Polymerase | New England Biolabs | Cat#M0530 |
| Tris-HCL | Roche | Cat#10812846001 |
| EDTA | AppliChem | Cat#1.08452 |
| Triton™ X-100 | Sigma-Aldrich | Cat#X100 |
| Bleach | DanKlorix Hygiene Reiniger | N/A |
| Agar | Becton Dickinson | Cat# 10455513 |
| Polenta | Verival; Pronurel Bio | N/A |
| Soy flour | Bauck Hof | N/A |
| Yeast | Bruggeman | N/A |
| Treacle | Original Grafschafter Goldsaft | N/A |
| Malt extract | Demeter | N/A |
| Nipagin | Sigma-Aldrich | Cat# H3647 |
| Propionic acid | Acros Organics | Cat#AC149300010 |
| Tween80 | Sigma-Aldrich | Cat#P1754 |
| Critical commercial assays | ||
| TOPO TA Cloning Kit for Sequencing | Invitrogen | Cat#K4575J10 |
| QIAamp DNA Mini Kit | Qiagen | Cat#51304 |
| Deposited data | ||
| Raw and analyzed data | This paper | DOI: 10.17632/2tgjd6y4zb.1 |
| Experimental models: organisms/strains | ||
| D. melanogaster wildtype strain Oregon-R | Bloomington Drosophila Stock Center | BDSC: 5; FlyBase: FBsn0000276 |
| D. melanogaster white[1118] | Vienna Drosophila Resource Center | VDRC:60000 |
| Oligonucleotides | ||
| GM3F: AGAGTTTGATCMTGGC | Klindworth et al., 2013 | N/A |
| GM4R: TACCTTGTTACGACTT | Klindworth et al., 2013 | N/A |
| Software and algorithms | ||
| Python 3.8 | Python Software Foundation | https://www.python.org |
| R Studio 1.2.5042 | RStudio, Inc. | https://www.rstudio.com |
| R 3.6.1 | R Foundation for Statistical Computing | https://www.R-project.org |
| BacArena 1.8 | Bauer et al., 2017 | https://bacarena.github.io |
| gapseq 1.1 | Zimmermann et al., 2021 | https://github.com/jotech/gapseq |
| Plotly 4.14.3 | Plotly Technologies Inc. | https://plot.ly |
| Code to re-perform analyses and to recapitulate the plotting. | This paper | https://gitlab.com/Beller-Lab |
Resource availability
Lead contact
Further requests for resources should be directed to and will be fulfilled by the lead contact, Mathias Beller (mathias.beller@hhu.de).
Materials availability
This study did not generate new materials.
Experimental model and subject details
Fly strains and rearing
The fly lines that were used in this study are w1118 (white[-]) and Oregon-R. Flies were maintained at 25°C with 60–70% humidity and a 12 h light/dark cycle. Standard diet contains 0.5% agar (Becton Dickinson), 7.1% polenta (Verival, Pronurel Bio), 0.95% soy flour (Bauck Hof), 1.68% yeast (Bruggeman), 4% treacle (Original Grafschafter Goldsaft), 4.5% malt extract (Demeter). All diets contained 0.15% nipagin (Sigma-Aldrich) and 0.45% propionic acid (Acros Organics).
Isolation of bacterial species from Drosophila
In order to analyze different bacterial species from the gut microbiome of Drosophila, both white[-] and Oregon-R male flies (9 individuals) were surface sterilized by washing with 10% bleach, 70% ethanol and PBS before homogenization and plating on MRS and ACE agar plates. MRS agar plates (Oxoid, Thermo Scientific) contain (in 1000 mL dH2O): Agar (15 g), casein peptone, tryptic digest (10 g), meat extract (10 g), yeast extract (5 g), glucose (20 g), Tween 80 (1 g), K2HPO4 (2 g), Na-acetate (5 g), (NH4)2 citrate (2 g), MgSO4 x 7 H2O (0.2 g), MnSO4 x H2O (0.05 g), pH 6.2–6.5. ACE agar plates (Blum et al., 2013) contain: (in 1000 mL dH2O): Agar (15 g), yeast extract (8 g), casein peptone (15 g), glucose (10 g), after autoclaving: acetic acid (3 mL), ethanol (p.a.) (5 mL) and Cycloheximid (100 mg). The plates were incubated at 28°C for three to five days and single colonies were picked and isolated on new agar plates for three rounds to obtain pure cultures. These were then stored in glycerol stocks for later DNA extraction and analysis.
Method details
Single colony PCR and analysis of 16S rRNA genes
Of the different pure cultures single colonies were picked and transferred into PBS buffer containing 200 μg/ml Proteinase K and 10 mg/ml Lysozyme and incubated for 30 min at 37°C and 2 min at 95°C. The samples were centrifuged for 2 min at 13.000 rpm and the supernatant transferred to a new vial. The 16S rRNA Gen was amplified using the GM3F and GM4R primers (Klindworth et al., 2013) using the Phusion Polymerase (New England Biolabs) which produced a product of about 1500 bp. These PCR products were then ligated into the TOP TA Vector (TOPO TA Cloning Kit for Sequencing, Invitrogen) and transformed into chemocompetent E. coli DH5alpha according to the manufacturer's instructions. The vector including the insert was extracted from E. coli and the insert analyzed by Sanger sequencing (MWG Biotech). The DNA sequence was afterwards subjected to BLAST analysis to identify the isolated bacterial species.
DNA extraction from bacterial species for genome sequencing
The DNA extraction was performed using the Qiagen QiaAmp DNA Mini kit according to the manufacturer's recommendation, with the following modifications. Briefly, an inoculation loop was used to pick bacterial colonies from the pure cultures grown on ACE or MRS agar plates and the bacteria were resuspended in gram-positive lysis buffer (20 mg/ml lysozyme; 20 mM Tris·HCl, pH 8.0; 2 mM EDTA; 1.2% Triton®). The following lysis and purification steps were performed according to the kit's protocol for DNA extraction from gram-positive bacteria.
Liquid media bacteria growth experiments
For the bacterial growth experiment, we prepared pre-cultures in the respective semi-selective medium (MRS for Lactobacillus sp. and ACE for Acetobacter sp. (Blum et al., 2013)). Subsequently, we either directly used the optical dense overnight culture or adjusted it to an OD600 of 0.8. Next, we performed a 1:1000 (MRS and ACE) or 1:100 (HD) dilution and distributed the bacteria to transparent 96-well flat bottom plates (Sarstedt). The medium was covered with mineral oil and incubated in a BioTek Synergy Mx Plate Reader with (Acetobacter) or without (Lactobacillus) shaking for at least 48 hours. Optical density was measured every five minutes. Per experiment, all growth curves were measured in at least triplicate and the figures provide mean values.
Whole genome sequencing of isolated bacterial species
The isolated genomic DNA samples from the gut microbiota species were sequenced using the Illumina MiSeq platform following standard procedures. The library preparations and sequencing were performed by the Genomics and Transcriptomics Lab at the HHU.
Genome reassembly
For the genome reassembly the tool ASA³P (Schwengers et al., 2020) was used. ASA³P is an automatic, scalable assembly, annotation, and analysis pipeline for genomes of bacterial origin. The pipeline consists of four steps: Processing, characterization, comparative genomics, and reporting. Each step provides different analysis information about the used sequenced genome through different software tools and databases. While processing and reporting is mandatory, the steps of characterization and comparative genomics is optional and can be skipped by the user. The first step processing includes the task of quality control, genome assembly, scaffolding and annotation. The second step of characterization determines the taxonomy, performs a multi locus sequence typing (MLST) analysis, tries to detect antibiotic resistances (ABRs), a detection of virulence factors (VFs), performs a mapping by using quality clipped reads onto reference genomes provided by the user, and annotates single-nucleotide polymorphisms (SNPs). The third step of comparative genomics consists of the calculation of a phylogenetic tree and of a core, accessory and pan-genome while detecting isolate genes. The last step is a graphical presentation of the pipeline results. All ASA³P results are provided in the supplement.
Reconstruction of bacterial metabolic models
The sequenced genomes were used to reconstruct their genome-scale metabolic models using the gapseq analysis pipeline (Zimmermann et al., 2021). We used for the reconstruction and gap-filling step the MRS, ACE and HD as the growth medium. All metabolic models were created combining each genome sequence and every single medium. During the model generation process, we considered in particular stochiometric consistency, mass and charge balance as well as metabolite connectivity and introduced necessary changes following manual curation. In order to test for the quality of our models, we used the MEMOTE analysis pipeline (Lieven et al., 2020). All analysis results are provided as supplemental data. In brief, the models resulted in at least 77% model quality scores. Most importantly, the key requirements for the models all reached at least 99%. The score was only decreased by e.g. missing gene or metabolite annotation cross-references, which we do not focus on in the present manuscript and have no influence on flux predictions in constraint-based modeling. A central part of genome-scale metabolic models is the biomass reaction, which represents the metabolite consumption for the formation of all cell constituents. The biomass reaction is commonly, and also in this study, used as objective function for flux balance analysis (FBA) or FBA-derived simulation techniques. The gapseq software automatically adds a biomass reaction to the models based on the organism's Gram-staining phenotype in order to account for biomass composition differences due to differences in the structural characteristics of the cell wall. The exact biomass reaction stoichiometries in gapseq are directly derived from ModelSEED (Henry et al., 2010), which in turn derived the biomass reaction definitions from curated genome-scale metabolic models from Escherichia coli (Orth et al., 2011) as a proxy for Gram-negative bacteria and Bacillus subtilis (Oh et al., 2007) as a proxy for Gram-positive bacteria. The biomass compositions for all Lactobacilli models (Gram-positive) and Acetobacter models (Gram-negative) are provided in Data S1.
Constraint-based modeling
Flux balance analysis (FBA; (Orth et al., 2010)) was used to perform the growth and metabolic flux analysis. The mono- and co-culturing in silico experiments were performed using the BacArena tool (Bauer et al., 2017), which is also based on FBA.
In silico growth media
In silico experiments used parametrized versions of the experimentally used MRS, ACE and HD media (Supp. Table 1). MRS and ACE medium are semi-defined as the contain complex ingredients such as yeast extract. Therefore, we obtained compositional information from the suppliers of the respective media ingredients (see Data S2). For some media components, which are required to run the simulations, no quantitative information could be obtained. Those compounds were manually curated and added. We limited the number of such manually added compounds to the absolute minimum and provide all media information as supplemental data. The parametrized HD medium is based on the protocol of (Piper et al., 2014), which is completely synthetic and thus did not require any modifications.
Calculation of predicted relative flux ratios
To identify reactions with a higher flux and reactions corresponding to a crosstalk between Lactobacillus brevis B6 and the Acetobacter sp. we calculated a predicted relative flux ratio for each reaction and time point.
We calculated the predicted relative flux ratio as followed:
| (Equation 1) |
where is the flux of the of Lactobacillus B6 at time point t in , is the flux of the of Acetobacter sp. at time point t in .
If the predicted relative flux ratio value is between 1 and -1 we calculated the values as followed:
| (Equation 2) |
where is the unitless predicted relative flux ratio. We choose this representation of the value range between 1 and -1 to highlight the higher flux value between Lactobacillus B6 and the Acetobacter sp.
Calculation of cumulative flux values
In order to analyse the metabolic impact of an additional metabolite in the holidic diet towards the bacteria grown on the media we calculated the cumulative flux for each time point.
First, we calculated the sum of flux values:
| (Equation 3) |
where is the sum of flux values over the time t with medium in , is the flux value at a time point with medium M in .
Next, we calculated the difference of the sum flux values between the standard holidic diet HD and the medium M:
| (Equation 4) |
where is the difference between the summed flux values of HD and medium M over the time t.
Finally, we calculated the cumulative flux as followed:
| (Equation 5) |
where is the cumulative flux value between HD and the medium M for a reaction in . The cumulative flux value can also be calculated for a group of reactions.
Quantification and statistical analysis
Figures represent averaged or representative results of multiple independent experiments or simulations. The figure legends provide details concerning the N of experiments or simulations. Analyses and Plots were performed with custom Python scripts.
Additional resources
All data is available at data.mendeley.com under the URL https://doi.org/10.17632/2tgjd6y4zb.1.
Acknowledgments
We would like to thank all past and present members of the laboratory for helpful comments and support and the Genomics and Transcriptomics Lab (GTL) of the Heinrich Heine University Düsseldorf for the resequencing of the isolated bacterial species. Funding: The project was financed through the German “Bundesministerium für Bildung und Forschung (BMBF)” grant 031A 306 (to M.B.) and in part by a scholarship of the Jürgen Manchot Foundation (to J.W.S.). S.W. acknowledges support by the Cluster of Excellence 2167 - “Precision medicine in chronic inflammation” - Deutsche Forschungsgemeinschaft. The funders had no role in the study design, analysis, interpretation of the results or the writing of the manuscript.
Author contributions
J.W.S., F.A.S., and M.B. designed experiments. F.A.S., K.M.E., I.A., and A.D. conducted experiments. J.W.S., F.A.S., K.M.E., and M.B. analyzed the data. J.W.S. and K.M.E. performed the resequencing analysis and assembled the bacterial genomes. S.W. reconstructed the bacterial genome-scale metabolic networks, performed the gap filling, and identified the biomass functions. J.W.S. performed the flux-balance simulations and plotted the figures. J.W.S., F.A.S., I.A., S.W. and M.B. wrote the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103216.
Supplemental information
Data and code availability
-
•
Genome resequencing data, the genome-scale metabolic networks and bacterial growth data, as well as all data required to reproduce the figures are deposited at Mendeley Data and is available as of the date of publication at https://doi.org/10.17632/2tgjd6y4zb.1.
-
•
All original code was additionally deposited at our GitLab account and can be accessed via https://gitlab.com/Beller-Lab.
-
•
For any additional questions or information please contact the lead contact.
References
- Adair K.L., Wilson M., Bost A., Douglas A.E. Microbial community assembly in wild populations of the fruit fly Drosophila melanogaster. Isme J. 2018;12:959–972. doi: 10.1038/s41396-017-0020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad I., Sharma J., Ahmad F. Isolation and characterization of resistance traits of indigenous strains of Acetobacter diazotrophicus associated with sugarcane. Sugar Tech. 2004;6:41–46. doi: 10.1007/bf02942616. [DOI] [Google Scholar]
- Alt E., Leipold F., Milatovic D., Lehmann G., Heinz S., Schömig A. Hydrogen peroxide for prevention of bacterial growth on polymer biomaterials. Ann. Thorac. Surg. 1999;68:2123–2128. doi: 10.1016/s0003-4975(99)00832-2. [DOI] [PubMed] [Google Scholar]
- Ankrah N.Y.D., Barker B.E., Song J., Wu C., McMullen J.G., Douglas A.E. Predicted metabolic function of the gut microbiota of Drosophila melanogaster. Msystems. 2021;6 doi: 10.1128/msystems.01369-20. e01369–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron-Wisnewsky J., Vigliotti C., Witjes J., Le P., Holleboom A.G., Verheij J., Nieuwdorp M., Clément K. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroentero. 2020;17:279–297. doi: 10.1038/s41575-020-0269-9. [DOI] [PubMed] [Google Scholar]
- Arora T., Singh S., Sharma R.K. Probiotics: interaction with gut microbiome and antiobesity potential. Nutr. Burbank Los Angeles Cty Calif. 2013;29:591–596. doi: 10.1016/j.nut.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Aumiller K., Stevens E., Scheffler R., Güvener Z.T., Tung E., Grimaldo A.B., Carlson H.K., Deutschbauer A.M., Taga M.E., Marco M.L., Ludington W.B. A chemically-defined growth medium to support Lactobacillus – Acetobacter community analysis. Biorxiv. 2021;2021 doi: 10.1101/2021.05.12.443930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma Y., Hosoyama A., Matsutani M., Furuya N., Horikawa H., Harada T., Hirakawa H., Kuhara S., Matsushita K., Fujita N., Shirai M. Whole-genome analyses reveal genetic instability of Acetobacter pasteurianus. Nucl. Acids Res. 2009;37:5768–5783. doi: 10.1093/nar/gkp612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer E., Zimmermann J., Baldini F., Thiele I., Kaleta C. BacArena: individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput. Biol. 2017;13:e1005544. doi: 10.1371/journal.pcbi.1005544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum J.E., Fischer C.N., Miles J., Handelsman J. Frequent replenishment sustains the beneficial microbiome of Drosophila melanogaster. mBio. 2013;4 doi: 10.1128/mbio.00860-13. e00860–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick N.A., Lemaitre B. Gut-associated microbes of Drosophila melanogaster. Gut Microbes. 2012;3:307–321. doi: 10.4161/gmic.19896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton T.A., Baker D., Lindon J.C., Everett J.R., Nicholson J.K. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consuegra J., Grenier T., Akherraz H., Rahioui I., Gervais H., da Silva P., Leulier F. Metabolic cooperation among commensal bacteria supports Drosophila juvenile growth under nutritional stress. Iscience. 2020;23:101232. doi: 10.1016/j.isci.2020.101232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consuegra J., Grenier T., Baa-Puyoulet P., Rahioui I., Akherraz H., Gervais H., Parisot N., da Silva P., Charles H., Calevro F., Leulier F. Drosophila-associated bacteria differentially shape the nutritional requirements of their host during juvenile growth. PLoS Biol. 2020;18:e3000681. doi: 10.1371/journal.pbio.3000681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten C., Reed J.L. Mechanistic analysis of multi-omics datasets to generate kinetic parameters for constraint-based metabolic models. Bmc Bioinform. 2013;14:32. doi: 10.1186/1471-2105-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas A.E. Simple animal models for microbiome research. Nat. Rev. Microbiol. 2019 doi: 10.1038/s41579-019-0242-1. [DOI] [PubMed] [Google Scholar]
- Erkosar B., Storelli G., Defaye A., Leulier F. Host-intestinal microbiota mutualism: “learning on the fly. Cell Host Microbe. 2013;13:8–14. doi: 10.1016/j.chom.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Fischer E., Sauer U. Large-scale in vivo flux analysis shows rigidity and suboptimal performance of Bacillus subtilis metabolism. Nat. Genet. 2005;37:636–640. doi: 10.1038/ng1555. [DOI] [PubMed] [Google Scholar]
- Gould A.L., Zhang V., Lamberti L., Jones E.W., Obadia B., Korasidis N., Gavryushkin A., Carlson J.M., Beerenwinkel N., Ludington W.B. Microbiome interactions shape host fitness. Proc. Natl. Acad. Sci. 2018;115:201809349. doi: 10.1073/pnas.1809349115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser H.J., Gootenberg D.B., Chatman K., Sirasani G., Balskus E.P., Turnbaugh P.J. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science (New York, N.Y.) 2013;341:295–298. doi: 10.1126/science.1235872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfvarson J., Brislawn C.J., Lamendella R., Vázquez-Baeza Y., Walters W.A., Bramer L.M., D’Amato M., Bonfiglio F., McDonald D., Gonzalez A. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017;2:17004. doi: 10.1038/nmicrobiol.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartstra A.V., Bouter K.E.C., Bäckhed F., Nieuwdorp M. Insights into the role of the microbiome in obesity and type 2 diabetes. Diabetes Care. 2014;38:159–165. doi: 10.2337/dc14-0769. [DOI] [PubMed] [Google Scholar]
- Hatti-Kaul R., Chen L., Dishisha T., Enshasy H.E. Lactic acid bacteria: from starter cultures to producers of chemicals. Fems Microbiol. Lett. 2018;365 doi: 10.1093/femsle/fny213. [DOI] [PubMed] [Google Scholar]
- Heeney D.D., Gareau M.G., Marco M.L. Intestinal Lactobacillus in health and disease, a driver or just along for the ride? Curr. Opin. Biotech. 2018;49:140–147. doi: 10.1016/j.copbio.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques S.F., Dhakan D.B., Serra L., Francisco A.P., Carvalho-Santos Z., Baltazar C., Elias A.P., Anjos M., Zhang T., Maddocks O.D.K., Ribeiro C. Metabolic cross-feeding in imbalanced diets allows gut microbes to improve reproduction and alter host behaviour. Nat. Commun. 2020;11:4236. doi: 10.1038/s41467-020-18049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry C.S., DeJongh M., Best A.A., Frybarger P.M., Linsay B., Stevens R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 2010;28:977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- Ito T., Okabe S., Satoh H., Watanabe Y. Successional development of sulfate-reducing bacterial populations and their activities in a wastewater biofilm growing under microaerophilic conditions. Appl. Environ. Microb. 2002;68:1392–1402. doi: 10.1128/aem.68.3.1392-1402.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keebaugh E.S., Yamada R., Obadia B., Ludington W.B., Ja W.W. Microbial quantity impacts Drosophila nutrition, development, and lifespan. Iscience. 2018;4:247–259. doi: 10.1016/j.isci.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., Glöckner F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl. Acids Res. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaroff A.L. The microbiome and risk for obesity and diabetes. Jama. 2016;317:355–356. doi: 10.1001/jama.2016.20099. [DOI] [PubMed] [Google Scholar]
- Krajmalnik-Brown R., Ilhan Z.-E., Kang D.-W., DiBaise J.K. Effects of gut microbes on nutrient absorption and energy regulation. Nutr. Clin. Pract. Off. Publ. Am. Soc. Parenter Enter Nutr. 2012;27:201–214. doi: 10.1177/0884533611436116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N.Y., Shin M.J., Youn G.S., Yoon S.J., Choi Y.R., Kim H.S., Gupta H., Han S.H., Kim B.K., Lee D.Y. Lactobacillus attenuates progression of non-alcoholic fatty liver disease by lowering cholesterol and steatosis. Clin. Mol. Hepatol. 2020 doi: 10.3350/cmh.2020.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieven C., Beber M.E., Olivier B.G., Bergmann F.T., Ataman M., Babaei P., Bartell J.A., Blank L.M., Chauhan S., Correia K. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020;38:272–276. doi: 10.1038/s41587-020-0446-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R., Liu W., Piao M., Zhu H. A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids. 2017;49:2083–2090. doi: 10.1007/s00726-017-2493-3. [DOI] [PubMed] [Google Scholar]
- Liu Y., Wang Y., Ni Y., Cheung C.K.Y., Lam K.S.L., Wang Y., Xia Z., Ye D., Guo J., Tse M.A. Gut microbiome fermentation determines the efficacy of exercise for diabetes prevention. Cell Metab. 2020;31:77–91.e5. doi: 10.1016/j.cmet.2019.11.001. [DOI] [PubMed] [Google Scholar]
- Ludington W.B., Ja W.W. Drosophila as a model for the gut microbiome. PLoS Pathog. 2020;16:e1008398. doi: 10.1371/journal.ppat.1008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco M.L., de Vries M.C., Wels M., Molenaar D., Mangell P., Ahrne S., de Vos W.M., Vaughan E.E., Kleerebezem M. Convergence in probiotic Lactobacillus gut-adaptive responses in humans and mice. Isme J. 2010;4:1481–1484. doi: 10.1038/ismej.2010.61. [DOI] [PubMed] [Google Scholar]
- Martin A.M., Sun E.W., Rogers G.B., Keating D.J. The influence of the gut microbiome on host metabolism through the regulation of gut hormone release. Front Physiol. 2019;10:428. doi: 10.3389/fphys.2019.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y.-K., Palsson B.O., Park S.M., Schilling C.H., Mahadevan R. Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data∗. J. Biol. Chem. 2007;282:28791–28799. doi: 10.1074/jbc.m703759200. [DOI] [PubMed] [Google Scholar]
- Orth J.D., Conrad T.M., Na J., Lerman J.A., Nam H., Feist A.M., Palsson B.Ø. A comprehensive genome-scale reconstruction of Escherichia coli metabolism—2011. Mol. Syst. Biol. 2011;7:535. doi: 10.1038/msb.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth J.D., Thiele I., Palsson B. What is flux balance analysis? Nat. Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M.D., Blanc E., Leitão-Gonçalves R., Yang M., He X., Linford N.J., Hoddinott M.P., Hopfen C., Soultoukis G.A., Niemeyer C. A holidic medium for Drosophila melanogaster. Nat. Methods. 2014;11:100–105. doi: 10.1038/nmeth.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raspor P., Goranovič D. Biotechnological applications of acetic acid bacteria. Crit. Rev. Biotechnol. 2008;28:101–124. doi: 10.1080/07388550802046749. [DOI] [PubMed] [Google Scholar]
- Reis M.A.M., Almeida J.S., Lemos P.C., Carrondo M.J.T. Effect of hydrogen sulfide on growth of sulfate reducing bacteria. Biotechnol. Bioeng. 1992;40:593–600. doi: 10.1002/bit.260400506. [DOI] [PubMed] [Google Scholar]
- Ridley E.V., Wong A.C.-N., Westmiller S., Douglas A.E. Impact of the resident microbiota on the nutritional phenotype of Drosophila melanogaster. PLoS ONE. 2012;7:e36765. doi: 10.1371/journal.pone.0036765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönborn J.W., Jehrke L., Mettler-Altmann T., Beller M. FlySilico: flux balance modeling of Drosophila larval growth and resource allocation. Sci. Rep-uk. 2019;9:17156. doi: 10.1038/s41598-019-53532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schretter C.E., Vielmetter J., Bartos I., Marka Z., Marka S., Argade S., Mazmanian S.K. A gut microbial factor modulates locomotor behaviour in Drosophila. Nature. 2018;563:402–406. doi: 10.1038/s41586-018-0634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz R., Kuepfer L., Sauer U. Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 2007;3 doi: 10.1038/msb4100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz R., Zamboni N., Zampieri M., Heinemann M., Sauer U. Multidimensional optimality of microbial metabolism. Science. 2012;336:601–604. doi: 10.1126/science.1216882. [DOI] [PubMed] [Google Scholar]
- Schwarzer M., Makki K., Storelli G., Machuca-Gayet I., Srutkova D., Hermanova P., Martino M., Balmand S., Hudcovic T., Heddi A. Lactobacillus plantarum strain maintains growth of infant mice during chronic undernutrition. Science. 2016;351:854–857. doi: 10.1126/science.aad8588. [DOI] [PubMed] [Google Scholar]
- Schwengers O., Hoek A., Fritzenwanker M., Falgenhauer L., Hain T., Chakraborty T., Goesmann A. ASA3P: an automatic and scalable pipeline for the assembly, annotation and higher level analysis of closely related bacterial isolates. Plos Comput. Biol. 2020;16:e1007134. doi: 10.1371/journal.pcbi.1007134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R., Fuchs S., Milo R. Revised estimates for the number of human and bacteria cells in the body. Plos Biol. 2016;14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S.C., Kim S.-H.H., You H., Kim B., Kim A.C., Lee K.-A.A., Yoon J.-H.H., Ryu J.-H.H., Lee W.-J.J. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science (New York, N.Y.) 2011;334:670–674. doi: 10.1126/science.1212782. [DOI] [PubMed] [Google Scholar]
- Silva V., Palacios-Muñoz A., Okray Z., Adair K.L., Waddell S., Douglas A.E., Ewer J. The impact of the gut microbiome on memory and sleep in Drosophila. J. Exp. Biol. Jeb. 2020:233619. doi: 10.1242/jeb.233619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer A.J., Newell P.D. Metabolic basis for mutualism between gut bacteria and its impact on their host Drosophila melanogaster. Appl. Environ. Microb. 2018;85 doi: 10.1128/aem.01882-18. AEM.01882–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector M.P. Academic Press; 2009. Encyclopedia of Microbiology. Physiology Article Titles M 242–264. [DOI] [Google Scholar]
- Storelli G., Defaye A., Erkosar B., Hols P., Royet J., Leulier F. Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metab. 2011;14:403–414. doi: 10.1016/j.cmet.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Storelli G., Strigini M., Grenier T., Bozonnet L., Schwarzer M., Daniel C., Matos R., Leulier F. Drosophila perpetuates nutritional mutualism by promoting the fitness of its intestinal symbiont Lactobacillus plantarum. Cell Metab. 2018;27:362–377.e8. doi: 10.1016/j.cmet.2017.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H., Kaser A. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Invest. 2011;121:2126–2132. doi: 10.1172/jci58109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P.J., Gordon J.I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009;587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong H.E., Hsiao E.Y. Emerging roles for the gut microbiome in autism spectrum disorder. Biol. Psychiat. 2016;81:411–423. doi: 10.1016/j.biopsych.2016.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarden A.R., Chen C., Bobr A., Yao D., Lu Y., Nelson V.M., Sadowsky M.J., Khoruts A. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Gastrointest Liver Physiol. 2013;306:G310–G319. doi: 10.1152/ajpgi.00282.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann J., Kaleta C., Waschina S. gapseq: informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. Genome Biol. 2021;22:81. doi: 10.1186/s13059-021-02295-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Genome resequencing data, the genome-scale metabolic networks and bacterial growth data, as well as all data required to reproduce the figures are deposited at Mendeley Data and is available as of the date of publication at https://doi.org/10.17632/2tgjd6y4zb.1.
-
•
All original code was additionally deposited at our GitLab account and can be accessed via https://gitlab.com/Beller-Lab.
-
•
For any additional questions or information please contact the lead contact.