

Summary
Provoked by sterile/nonsterile insults, prolonged monocyte mobilization and uncontrolled monocyte/macrophage activation can pose imminent or impending harm to the affected organs. Curiously, folate receptor beta (FRβ), with subnanomolar affinity for the vitamin folic acid (FA), is upregulated during immune activation in hematopoietic cells of the myeloid lineage. This phenomenon has inspired a strong interest in exploring FRβ-directed diagnostics/therapeutics. Previously, we have reported that FA-targeted aminopterin (AMT) therapy can modulate macrophage function and effectively treat animal models of inflammation. Our current investigation of a lead compound (EC2319) leads to discovery of a highly FR-specific mechanism of action independent of the root causes against inflammatory monocytes. We further show that EC2319 suppresses interleukin-6/interleukin-1β release by FRβ+ monocytes in a triple co-culture leukemic model of cytokine release syndrome with anti-CD19 chimeric antigen receptor T cells. Because of its chemical stability and metabolically activated linker, EC2319 demonstrates favorable pharmacokinetic characteristics and cross-species translatability to support future pre-clinical and clinical development.
Keywords: autoimmunity, inflammation, folate receptor beta, monocyte/macrophage activation, dihydrofolate reductase inhibitor, aminopterin, adjuvant arthritis, glomerulonephritis, autoimmune uveitis, cytokine release syndrome
Graphical abstract
Highlights
-
•
Functional folate receptor beta is transiently expressed on inflammatory monocytes
-
•
EC2319 is an enhancement of traditional dihydrofolate reductase inhibitors
-
•
EC2319 anti-monocyte activity correlates with local/systemic therapeutic benefit
-
•
EC2319 inhibition of cytokine release suggests emergency use for hyperinflammation
By repurposing the antifolate aminopterin, Lu et al. reveal a rationally designed single-agent therapy that effectively mitigates monocyte/macrophage dysfunction. The observed biological effect appears to be independent of root causes of acute inflammation, which is frequently associated with autoimmune and inflammatory diseases, viral or bacterial infections, and cytokine storm disorders.
Introduction
Whether acute or chronic, a common clinical characteristic associated with sterile and non-sterile host inflammatory response is monocyte activation and recruitment in the peripheral blood.1,2 Making up ∼10% of total leukocytes in humans, circulating monocytes comprise three subsets of ∼85% CCR2+ classical (CD14+ CD16−) and ∼15% CCR2− intermediate (CD14+ CD16+)/nonclassical (CD14lo CD16+) monocytes.3 Emerging first from the bone marrow, CCR2+ classical monocytes migrate rapidly to sites of inflammation after ∼1–2 days in the blood. CCR2− intermediate/nonclassical monocytes are differentiated sequentially from classical monocytes with extended lifespans (∼4 and ∼7 days, respectively) in favor of resolution of inflammation.3 Traditionally, human monocytes are considered non-proliferative cells, but recent studies have shown that classical monocytes are anti-apoptotic and capable of proliferating in vitro.4 In synovial tissues of inflammatory osteoarthritis, classical macrophages have been found to express a higher level of the proliferation marker Ki67.5 Importantly, rapidly recruited monocytes and monocyte-derived macrophages are major sources of pro-inflammatory cytokines (e.g., interleukin-6 [IL-6], IL-1, and tumor necrosis factor alpha [TNF-α]) that are important drivers of many acute-phase reactions.2 Consequently, uncontrolled or sustained monocyte mobilization contributes to endothelial dysfunction and subsequent tissue damage by infiltrating monocyte-derived macrophages. Among pathologic inflammatory conditions, altered monocyte phenotype and frequencies have been reported in rheumatoid arthritis (RA), juvenile idiopathic arthritis, pediatric uveitis, cardiovascular diseases, psoriasis, systemic lupus erythematosus, inflammatory bowel disease, multiple sclerosis, and diabetes complications.2,6, 7, 8, 9
Folate receptor beta (FRβ) is one of two glycosyl-phosphatidylinositol-anchored membranous folate-binding proteins (the other one is FRα), and the latter is frequently associated with cancer cells of epithelial origin.10 Both FR isoforms bind folic acid (FA) and FA-conjugated small-molecule drugs with (sub)nanomolar affinity and undergo endocytosis and rapid receptor recycling. For reasons not yet understood, a fully functional FRβ is upregulated on activated but not resting macrophages and on peripheral blood monocytes (albeit at a lower level and frequency).11,12 In exploratory clinical trials, 99 mTc-etarfolatide, a FR-specific radioimaging agent for cancer, has been found to accumulate preferentially in arthritic joints of individuals with RA13 and osteoarthritis.14 A [18F]fluoro-polyethylene glycol (PEG)-folate imaging agent has also been evaluated as a macrophage tracer for detecting clinical and subclinical arthritis.15 These first-in-human studies have provided proof of concept for FRβ-directed delivery of drugs with suitable mechanisms of action to control local and systemic inflammation.16
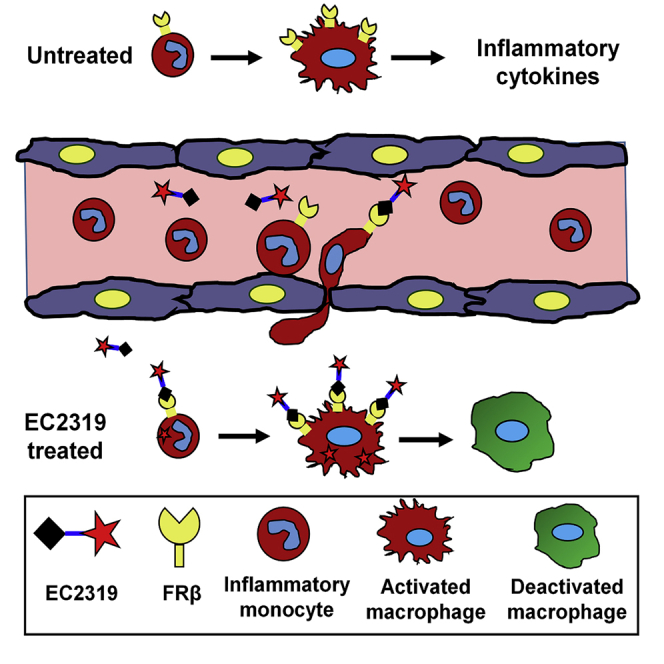
Previously in our investigation, aminopterin (AMT), a first-generation inhibitor of dihydrofolate reductase (DHFR) closely related to the RA anchor drug methotrexate (MTX), has emerged as a highly potent antifolate warhead for targeted delivery to activated macrophages.17,18 AMT and MTX enter cells primarily through the proton-coupled folate transporter in the upper gastrointestinal tract (at acidic pH optimum) and through the reduced folate carrier in normal tissues (at neutral pH).16,19 For MTX’s mechanism in RA, multiple hypotheses beyond folate antagonism exist, including adenosine signaling, cytokine modulation, generation of reactive oxygen species, and downregulation of some adhesion molecules.20 Compared with MTX, AMT is retained more efficiently in the cell because of its higher affinity for folylpolyglutamate synthase, but AMT itself has negligible binding affinity toward FRβ.17 In a FA-conjugated form with a saccharopeptidic spacer, EC0746 is a FR-targeted AMT therapy that exhibits a superior therapeutic index with ∼40× improvement in tolerability compared with unmodified AMT.17 At the cellular level, EC0746 demonstrates FR-specific DHFR inhibition and a cytostatic effect against RAW264.7 macrophages as well as cytokine-modulating activity in rat macrophages upon lipopolysaccharide/interferon-γ co-stimulation.17 Remarkably, EC0746 is highly effective in mitigating active inflammation in animal models of adjuvant-induced arthritis (AIA),17 experimental autoimmune uveitis (EAU),18 and experimental autoimmune encephalomyelitis.18 For clinical translation, an EC0746 synthetic analog, EC2319 (Figures 1 and 2), was chosen for further continued investigation because of its linker stability/metabolism and pharmacokinetic properties across species. To establish a potential pharmacokinetic-pharmacodynamic relationship, we began to investigate EC2319’s ability to shut down inflammatory monocytes as a way of preventing subsequent tissue damage brought on by monocyte-derived macrophages. In rodents, monocytes constitute between 1%–4% of total leukocytes, with CCR2+ monocytes identified in rats as the classical CD172+ CD43− inflammatory phenotype,21,22, and in mice, they are identified as classical Ly6Chigh CD43+ inflammatory and intermediate Ly6Chigh CD43high pro-inflammatory phenotypes.23,24 Regardless of the root causes of pathologic inflammatory conditions, EC2319’s anti-monocyte activity has been demonstrated unequivocally by three independent research institutes using various animal models. Considering this mechanism of action for potential immuno-oncology applications, we applied EC2319 in a co-culture human Nalm-6 leukemic model of severe cytokine release syndrome (CRS) with anti-CD19 chimeric antigen receptor (CAR) T cells and FRβ-transfected THP1 monocytes.
Figure 1.

Chemical structures and total synthesis
Shown is an abbreviated synthetic scheme of EC2319. The EC2319 construct comprises four functional components separated by the dashed lines: FA, an FR-targeting ligand; a Saccharo-peptidic spacer designed to reduce liver clearance; an L-cysteine methyl ester-derived releasable linker with a gem-dimethyl protected disulfide bond; and AMT, the active drug.
Figure 2.

EC2319 linker metabolism and species differences
(A) EC2319 has two predicted cleavage sites that allow formation of two active metabolites: the AMT adduct (via disulfide reduction) and the AMT parent drug (via enzymatic cleavage).
(B) Whole-blood stability of EC2319 (500 nM) spiked into EDTA-anticoagulated whole blood (rat/dog/human) and incubated for 30 and 60 min at 37°C. Plasma samples were extracted for LC-MS/MS quantitation of the intact molecule, AMT, and the AMT adduct.
(C) Formation of AMT after 1-h incubation at 37°C in 10% liver cytosols (rat/dog/human) that were pH adjusted to 4.5 or 7.4 and spiked with EC2319 (1 μM).
(D) GGH metabolism of EC2319 (L-cysteine methyl ester linker) compared against three synthetic analogs of alternative linkers (L-cysteine, cysteamine, or L-lysine). After 2-h incubation at 37°C, any AMT formed after enzymatic cleavage was quantified. All values represent the mean ± SD (n = 2).
Results
Compound design, synthesis, and characterization
Our lead compound, EC2319 (2,263 g/mol), was synthesized by a combination of solid- and solution-phase methods (Figure 1). Compared with EC0746,17 EC2319 shares FA as the targeting ligand, the same saccharo-(L)-amino acid peptidic spacer that helps reduce liver clearance,25,26 and the AMT warhead. However, instead of a hydrazide/disulfide-containing L-cysteine linker within EC0746, our current construct consists of an L-cysteine methyl ester-derived linker that forms a gem-dimethyl-protected disulfide bond on the spacer side (Figure S1). The attachment of a modified L-cysteine to the γ-carbonyl of AMT is designed to allow two potential cleavage sites for the intact EC2319 molecule (Figure 2A). The first, via reduction of the disulfide bond, results in release of γAMT-cysteine methyl ester, which is “trapped” ex vivo as γAMT-cysteine methyl ester-S-succinimidopropionic acid (referred to here as the AMT adduct) for liquid chromatography-tandem mass spectrometry (LC-MS/MS) quantification. The second site of cleavage is based on enzymatic cleavage of the amide bond at the γ position of AMT, likely by gamma-glutamyl hydrolase (GGH), leading to release of unmodified parent AMT drug.
In our standard tests, EC2319 displayed high FR-binding properties with relative affinity values of ∼0.493 and ∼0.313 for KB, a HeLa derivative (FRα) and Chinese hamster ovary (CHO)-β (FRβ) cells, respectively, compared with unmodified FA (Figure S2). EC2319 (2-h pulse exposure/70 h chase) also showed low relative 50% inhibitory concentration values against THP1-FRβ (human monocytes) and RAW264.7 (murine macrophages) at 8.7 nM and 2.9 nM, respectively (Figure S3). All EC2319 activities were cytostatic in nature, FR specific (independent of the α/β subtype), and fully reversible in the presence of excess FA. Cellular exposure to FA alone under the same experimental condition did not show an immediate biological effect against RAW264.7 macrophages or rat macrophages upon lipopolysaccharide and/or interferon-γ stimulation.17
Cross-species difference, linker metabolism, and pharmacokinetics
Prior to embarking on efficacy studies, cross-species differences of EC2319 metabolism and pharmacokinetics were studied. EC2319 (500 nM) was first spiked into rat, dog, and human whole blood for 30 and 60 min at 37°C (Figure 2B). Although EC2319 remained mostly intact during the initial 30 min of exposure for all species, a gradual increase in free AMT was observed over time in rat samples but not in dog or human samples. Very little of the AMT adduct was produced, and no changes in its production were observed over time for all species. Next, we spiked EC2319 (1 μM) in 10% rat, dog, and human liver cytosols and incubated at 37°C for 1 h at 2 different pH levels (4.5 and 7.4) (Figure 2C). Although the rat sample displayed a broader pH specificity in AMT release, the opposite was true at acidic pH (4.5) for dog and human samples, with a 4-fold or higher amount of AMT produced. Finally, we treated EC2319 with recombinant human GGH (STAR Methods; Figure 2D) and detected ∼2% free AMT (i.e., ∼20 nM) after 2 h of incubation at 37°C. To confirm that EC2319’s L-cysteine methyl ester linker was prone to GGH-mediated linker degradation, we included control conjugate compounds that had L-cysteine (no methyl ester), cysteamine, or L-lysine methyl ester within their linkers proximal to the γ-carbonyl of AMT (Figure S1). Importantly, as predicted based on enzymatic specificity, the latter two FA-targeted AMT analogs failed to respond to GGH treatment (Figure 2D).
Pharmacokinetically, in female Lewis rats (Figures 3A and 3C), subcutaneously administered EC2319 (500 nmol/kg) presented a plasma maximum drug concentration (Cmax) of 682 nM ∼0.5 hours after the dose and cleared rapidly from the blood with a terminal half-life of ∼0.463 h. For the two active metabolites, AMT and the AMT adduct, there was a 30-min delay (i.e., ∼1 h after the dose) in reaching Cmax values of 38.2 nM and 12.5 nM, respectively, and both cleared more slowly than EC2319 with terminal half-lives of ∼1.11 h and 1 h. The corresponding area under the curve (AUClast) values for EC2319, AMT, and the AMT adduct were 1,017, 106, and 33.2 nM/h, which amounted to 88%, 9.17%, and 2.87% of total AUClast systemic exposure (i.e., intact + metabolites), respectively. Further, we investigated EC2319 pharmacokinetics in male beagle dogs via intravenous and subcutaneous administrations at 1 mg/kg and 2.43 mg/kg, respectively (Figure 3B). For both dosing routes, EC2319 was the predominant species detected in the systemic circulation and averaged around 97%–98% of total AUClast (Figure 3C). Unmodified AMT and the AMT adduct were found to be present at an average around 1.37%–2.99% and 0.431%–0.489% of the total AUClast, respectively. Although a similar amount of the AMT adduct was released from EC2319 via both routes, ∼2.2× more unmodified AMT was released via the subcutaneous route. Within experimental variability, subcutaneous administration of EC2319 in dogs was readily bioavailable with an estimated bioavailability of ∼100%. Regardless of the dosing routes, the terminal half-lives of EC2319, AMT, and the AMT adduct in dogs were projected to be 1.01–1.21, 2.00–2.74, and 1.28–2.14 h, respectively. The nearly 3- to 7-fold differences in AUClast between EC2319-derived AMT and the AMT adduct in dogs confirmed that the amide bond cleavage adjacent to γ-carbonyl AMT of the intact molecule was the major metabolic pathway. Based on the AUClast ratio of active AMT-containing metabolites (AMT and AMT adduct) over total systemic exposure (i.e., intact EC2319, AMT, and AMT adduct), subcutaneously administered EC2319 in rats produced ∼3× and ∼6.7× more AMT and the AMT adduct compared with EC2319 in dogs, respectively.
Figure 3.

Pharmacokinetic studies in rats and dogs
(A) Lewis rats (n = 3) with jugular vein catheters (Envigo) were given a single subcutaneous injection of EC2319 at 500 nmol/kg at time zero.
(B) Beagle dogs (n = 2) were given a single intravenous or subcutaneous injection of EC2319 at 1 and 2.43 mg/kg, respectively. Rat or dog plasma samples at various time points were analyzed by LC-MS/MS to quantitate EC2319, AMT, and the AMT adduct levels. The plasma concentration (nM) as a function of time was plotted over 12–24 h after injection as indicated.
(C) The table summarizes the values of pharmacokinetic data analysis, percentage of total AUC0-last, and EC2319 bioavailability in dogs. All values represent the mean ± SD.
Correlating FR-specific monocyte response to EC2319 anti-arthritic activity
When AIA is induced, Lewis rats present rapid onset and disease progression mediated by monocyte accumulation in the joints.27 Monocyte influx starts around 7 days and maximizes around 14–21 days, when arthritis is fully developed. Previously, we have shown that EC074617 and a FA-based conjugate of everolimus (an inhibitor of mammalian target of rapamycin)28 were more efficacious when dosed twice a week (BIW) rather than once a week (SIW). Further, we elected to start EC2319 treatment 9 days after AIA induction at 500 nmol/kg for 2 weeks (BIW × 2) without and with a FA-based high-affinity competitor (STAR Methods; Figure 4). Similar to EC0746, EC2319 showed FR-specific anti-arthritic activity in (1) overall disease control (Figure 4A, left), (2) reducing body weight loss (Figure 4A, right), (3) alleviating paw swelling (Figure 4B; ∼97%), and (4) splenomegaly (Figure 4C; ∼55%). Histologically (Figures 4D–4F), EC2319-treated animals had ∼84% (peri-articular inflammation), 93% (bone loss/resorption), 65% (cartilage loss), and 87% (pannus formation) decreases in individually scored parameters, representing an overall decrease of ∼85% in the summed scores (Figure 4E). EC2319-treated animals that also received the FA competitor experienced severe joint deterioration that were indistinguishable from that observed in arthritic control animals (Figure 4F). This strongly indicates that the observed therapeutic effects in non-competed animals were indeed specific to FRβ-targeted cells (i.e., monocytes/macrophages).
Figure 4.

EC2319’s anti-arthritic activity in AIA rats
Starting 9 days after induction, AIA rats with progressing disease were left untreated (n = 7) or treated with a BIW dosing regimen of EC2319 at 500 nmol/kg without or with a 500-fold molar excess of FA competitor (n = 5).
(A) Visual arthritis score plotted over days since first treatment (left) and percent change in body weight (right).
(B) Total paw weight (open bars, right y axis) and percent increase in paw weight (closed bars, left y axis), which was reduced by ∼95% in the EC2319-treated group.
(C) Spleen weight (open bars, right y axis) and percent increase in spleen weight (closed bars, left y axis), which was reduced by ∼55% in the EC2319-treated group.
(D) Individual and sum histological scores of ankle joints (right and left hind paws combined) on a scale of 0–10 for inflammation, bone loss/resorption, cartilage loss, and pannus formation.
(E) Mean summed histological score with a maximum score of 40 per rat.
(F) Representative photomicrographs (20×; scale bars, 500 μm) of the ankle closest to the mean summed histological score.
All values are expressed as mean ± SEM. One-way ANOVA with Tukey’s post hoc analysis: ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05 compared with the arthritic control in (B), (C), and (E). Kruskal-Wallis test with Dunn’s post hoc analysis: ∗p < 0.05 in (D).
To help establish the pharmacodynamic effect of EC2319, we studied CD172+ CD43– inflammatory monocytes within peripheral blood mononuclear cells (PBMCs) isolated from healthy and AIA rats (Figure 5A). Compared with healthy controls, this monocyte subpopulation was elevated by 5- to 6-fold in AIA rats 16 days after induction (Figure 5B, top panel). Biweekly subcutaneous EC2319 treatment (1,000 nmol/kg, BIW × 1), initiated 9 days after induction, caused a dramatic reduction of CD172+ CD43− monocytes as opposed to a minor reduction of CD172+ CD43+ monocytes (Figure 5B, top panel). The same treatment had no effect on healthy rat monocytes, and it did not affect T or B lymphocytes from healthy or AIA rats (Figure 5B, bottom panel). Next, we determined that EC2319’s effect on CD172+ CD43− monocytes was dose dependent (30–750 nmol/kg, BIW × 1) (Figure 5C) and FR specific because its activity (500 nmol/kg, BIW × 1.5) was blocked by a 500-fold molar excess of the FA competitor (Figure 5D). Finally, an elusive population of CD11bhi CD45+ PBMCs was found to express functional FRβ detectable by anti-rat FRβ monoclonal antibody29 and FA-Alexa Fluor 647 (Figure 5E). However, this FRβ+ subset of inflammatory monocytes was almost invisible and rarely detectable in AIA rats (Figure 5F). Our data suggest that EC2319 possesses a mode of action that is directed specifically to the monocyte-macrophage lineage of cells under inflammatory conditions in vivo.
Figure 5.

EC2319’s anti-monocyte activity in AIA rats
(A) Flow cytometry gating strategy of CD45+ PBMCs isolated from healthy and AIA rats for identification of classic (R1, CD172+ CD43−) and non-classical (R2, CD172+ CD43+) monocytes. Further gating on CD172− leukocytes (R3) allowed identification of T cells (CD3ε+ major histocompatibility complex [MHC] class II−) and B cells (CD3ε− MHC class II+).
(B) Healthy (n = 3) and AIA (n = 5) rats were left untreated or treated with EC2319 for 1 week (1,000 nmol/kg, BIW × 1). Shown are analyses of blood monocytes (top panel) and T and B lymphocytes (bottom panel).
(C) EC2319 dose response (30–750 nmol/kg, BIW × 1) with respect to R1 monocytes in AIA rats (n = 5).
(D) EC2319 activity (500 nmol/kg, BIW × 1.5) on R1 monocytes blocked by the innocuous FA competitor in AIA rats (n = 5).
(E and F) Analysis of functional FRβ in CD45+ CD11bhi PBMCs in the same study animals as in (D). Shown are histograms from an arthritic control animal (E) and individual data plotted with the group mean lines (F).
Values in (B)–(D) are expressed as mean ± SEM. One-way ANOVA with Tukey’s post hoc analysis: ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05 for the comparing groups in (B) and (C). Kruskal-Wallis test with Dunn’s post hoc analysis: ∗p < 0.05 in (D).
Anti-monocyte activity in experimental glomerulonephritis
In a rat model of anti-glomerular basement membrane (anti-GBM)-induced glomerulonephritis (Figure 6A), administration of polyclonal anti-GBM antibody causes rapid onset (∼4 h) and multiple surges of monocyte/macrophage infiltration on approximately days 6 and 11.30 Compared with healthy rats, a preliminary study showed that anti-GBM rats had a surge of CD172+ CD43− inflammatory monocytes in the blood that peaked on day 6 and quickly disappeared by day 12 (Figure 6B). To coincide with the timing of acute monocyte influx, the first EC2319 dose (575 nmol/kg) was initiated 8 h after induction on day 0, and the same dosage was repeated on day 3. For comparison with oral MTX, we employed a single high dose of MTX (2.07 mg/kg or 4.56 μmol/kg) administered 8 h after induction on day 0, or two “EC2319-matching” low doses of MTX (575 nmol/kg) administered similarly at 8 h on day 0 and again on day 3. Based on day 6 whole-blood analysis, EC2319 treatment in the acute phase of glomerulonephritis remarkably abridged CD172+ CD43− monocytes, whereas oral MTX treatments were ineffective (Figure 6C). More importantly, EC2319’s anti-monocyte activity was also FR-specific in anti-GBM rats and reversible in the presence of a 300-fold molar excess of the FA competitor. Using multiple parameters to assess kidney injury and M1/M2 macrophage ratios in anti-GBM rats, EC2319 activity was confirmed in organ levels.31
Figure 6.

EC2319’s anti-monocyte activity in anti-GBM rats and EAU mice
(A) Glomerulonephritis was induced in Wistar Kyoto (WKY) rats (n = 3) on day 0 with anti-GBM antibody, followed by EC2319 treatment (8 h and on day 3) and fluorescence-activated cell sorting (FACS) analyses, as indicated.
(B) CD172+ CD43- monocytes found in PBMCs isolated from anti-GBM rats on days 6 and 12 post induction and compared to healthy littermates.
(C) CD172+ CD43− monocytes as percentage of total blood leukocytes isolated from anti-GBM rats treated with EC2319 (575 nmol/kg) without or with a 300-fold molar excess of the FA competitor or oral MTX in two ways.
(D) Flowchart illustrating a daily dosing schedule of EC2319 (subcutaneous) and BLZ945 (oral gavage) in EAU mice (n = 10).
(E) Flow cytometry gating strategy on CD45+ CD11b+ NK1.1− mouse leukocytes for identification of classical (R1, Ly6Chigh CD43+) and intermediate (R2, Ly6Chigh CD43high) monocytes.
(F) Mean effect of EC2319 and BLZ945 treatment on R1 and R2 monocyte subpopulations per 100 μL mouse blood as bar graphs with healthy controls (no EAU) and individual data points.
Values represent mean ± SEM. One-way ANOVA with Tukey’s post hoc analysis: ∗∗∗p < 0.001 and ∗p < 0.05 for the comparing groups in (B) and (C). Kruskal-Wallis test with Dunn’s post hoc analysis: ∗∗∗p < 0.001 and ∗∗p < 0.01 in (F). ns, not significant.
Anti-monocyte activity in EAU
To demonstrate EC2319 activity in a third disease model, mice were induced with EAU by uveitogenic immunization against the human IRBP161–180 peptide plus concurrent administration of pertussis toxin (STAR Methods; Figure 6D).32 BLZ945, an orally active inhibitor of colony-stimulating factor 1 receptor (CSF-1R),33 was used as a positive control compound. Starting on the day of immunization, EC2319 (1000 nmol/kg, subcutaneous) and BLZ945 (200 mg/kg, oral gavage) were administered daily for 10 consecutive days (days 0–9). On day 10, 100–200 μL anticoagulated whole-blood samples from EAU mice and healthy control animals were collected for analysis of circulating monocytes that included classical (Ly6Chigh CD43+) and intermediate (Ly6Chigh CD43high) subsets in mice (Figure 6E, R1 and R2 gates).23 Elevated classical (R1) and intermediate monocytes (R2) observed in EAU mice were reduced significantly to background levels by EC2319 and BLZ945, whereas the R2 subpopulation was only reduced significantly by EC2319 (Figure 6F).
Downregulation of monocyte activation in experimental CRS
Before setting up a co-culture model of CRS aggravated by FR+ monocytes, we first profiled THP1-FRβ cell activation inducible by non-lethal combination of interferon (IFN)-γ and TNF-α, two major Th1 cytokines secreted by activated T cells. Forty-eight hours after IFN-γ/TNF-α co-stimulation (500 IU/mL each), THP1-FRβ cells (3 × 105 cells in 1 mL, starting density in 6-well plates) produced high levels of IL-6 and IL-1β in the culture medium (Figure 7A). In the absence of the FA competitor, both attributes of THP1-FRβ activation were inhibited by EC2319 pretreatment in a dose-dependent manner (0–10 μM, 2-h pulse/24-h chase) (Figure 7A). Next, in a triple co-culture fashion, we placed THP1-FRβ cells pretreated with EC2319 (0–10 μM, 2-h pulse/24-h chase) in the Transwell insert with CD19-CAR T cells premixed overnight with CD19+ Nalm-6 target cells at predetermined numbers at a 1:3 ratio in the main well (Figure 7B). In direct cell-to-cell contact, Nalm-6 cells are known to be highly sensitive to activated CD19-CAR T cells with massive production of IFN-γ and TNF-α.34 As necessary controls, THP1-FRβ cells placed in a Transwell insert were also exposed to CD19-CAR T cells or Nalm-6 cells alone in the main well. After 48 h of triple co-culture without direct cell-to-cell contact to Nalm-6-activated CD19-CAR T cells, THP1-FRβ cells were stimulated efficiently to produce IL-6 and IL-1β (Figure 7C). It should be noted that no IL-6 or IL-1β was produced by CD19-CAR T cells co-cultured with Nalm-6 in the absence of THP1-FRβ cells. EC2319-pretreated THP1-FRβ cells (same as above) were resistant to Nalm-6-stimulated CD19-CAR T cell activation with reduced IL-6 and IL-1β production (Figure 7C). Consistent with the aforementioned studies, EC2319’s CRS-modulating effect on THP1-FRβ cells was blocked by pre-incubation in the presence of the FA competitor. Our data suggest a possible second mechanism of action for EC2319 in attenuating acute monocyte responses provoked by excessive T cell activation.
Figure 7.

EC2319 suppression of IL-6/IL-1β release from THP1-FRβ monocytes activated by IFN-γ/TNF-α and Nalm-6-stimulated CD19-CAR T cells
(A) EC2319-pretreated THP1-FRβ cells were resistant to IFN-γ/TNF-α co-stimulation. THP1-FRβ cells (3 × 105/mL) were subjected to a 2-h pulse with EC2319 (0, 2 μM, and 10 μM) or 10 μM EC2319 plus a 100-fold excess of the FA competitor. After a 24-h “chase” in drug-free FFPRMI, recombinant human IFN-γ/TNF-α (500 IU/mL each) were added to the culture medium and incubated for 48 h. Shown are IL-6 and IL-1β levels detected in the culture medium (SEM, n = 4).
(B) Cartoon illustrating a 72-h co-culture assay: THP1-FRβ cells (3 × 105/mL) were pretreated with EC2319 as described in (A) and placed in the upper Transwell. CD19-CAR T cells were premixed with Nalm-6 cells at an E:T ratio of 1:3 overnight and placed in the bottom compartment. After 48 h of triple co-culture, the supernatants were collected for quantification of cytokine release.
(C) EC2319-pretreated THP1-FRβ cells were resistant to activation by Nalm-6-stimulated CD19-CAR T cells. Shown are IL-6 and IL-1β levels in the culture medium normalized per THP1-FRβ cell numbers (SEM, n = 4). Kruskal-Wallis test with Dunn’s post hoc analysis: ∗∗p < 0.01 and ∗p < 0.05 for the comparing groups.
Discussion
Inflammation is a double-edged sword with important benefits but collateral consequences that can lead to short-term and long-term damage to the body. For a broad range of pathologic conditions, there is a strong rationale in regulating acute/chronic monocyte and macrophage responses to curb systemic and local inflammation. For monocyte/macrophage-driven manifestations, subsets of peripheral blood monocytes may also serve as biomarkers of disease status and pharmacologic responses to therapeutic intervention.2 Although FRβ is a validated target for M1 and M2 macrophage phenotypes at the tissue level, little is known regarding the molecular basis of FRβ expression (gene versus functional protein) and how it may change during the lifespan of myeloid cells. In this respect, very low levels of functional FRβ were found on CD14highCD16− proinflammatory monocytes in healthy volunteers, and a reduction of FRβ+ blood monocytes was seen in individuals with renal cell carcinoma treated with folate-hapten-redirected immunotherapy.35 On the other hand, Samaniego et al.36,37 reported that FRβ expression is mostly associated with macrophage M2 polarization/differentiation involving activin A36 and the E26 transformation specific (ETS) domain transcription factor PU.1.37 In addition, Machacek et al.38 reported that FRβ is a negative CD11b/CD18 (Mac-1) regulator of trafficking and homing of a subset of human macrophages on collagen. Given the plasticity of monocyte-macrophage-lineage cells in response to inflammatory stimuli, exactly how FRβ plays a role in regulating monocyte trafficking, recruitment, and differentiation remains an open question.
Although a few FR-targeted therapeutic agents have advanced into clinical trials for treatment of solid tumors, no small-molecule FA-drug conjugate has yet entered into clinical testing for non-oncology applications. Compared with malignant cells in a solid tumor mass, inflammatory cells are more easily accessible in the blood and, because of capillary leakage, also in affected organ systems. To date, there has been no report of a FR-dependent, cell-targeted approach that effectively shuts down specific subsets of inflammatory/pro-inflammatory monocytes in acute or chronic pathologic immune disorders. Although animal models do not recapitulate all aspects of human diseases, they are valuable scientific tools for research and preclinical development. Except for the rare detection of FR functionality on rat CD11bhigh CD45+ PBMCs, our effort to identify an elusive FRβ+ monocyte population as a pharmacodynamic biomarker has not yet been successful. Although focusing on CD172+ CD43− classical monocytes in rat models of arthritis and glomerulonephritis, EC2319 given at progressive onset of both diseases clearly counteracted this population in a FR-dependent manner. In terms of downregulating classical and/or intermediate monocytes in EAU mice, EC2319 performed similarly as BLZ945 (if not better), a potent inhibitor of CSF-1R that depletes all myeloid cells because of complete blockade of the survival signal from CSF1. In a rat focal model of progressive chronic multiple sclerosis, EC2319 has been shown to significantly reduce lesion size and FRβ expression while improving inducible nitric oxide synthase/mannose receptor C type 1 ratios in macrophages and microglia.39 These data in various animal models strongly advocate for the reasonable concept that (1) transient FRβ expression occurs in a subpopulation of trafficking inflammatory/pro-inflammatory monocytes and (2) FRβ biology on monocyte-macrophage-lineage cells renders EC2319’s ability to specifically interfere with the process of monocyte recruitment and differentiation in vivo. Aside from its cytostatic and cytokine-modulating activity, there is still a missing link regarding the precise moment when EC2319 binds FRβ+ myeloid cells and executes its molecular mechanism in vivo. Our data suggest that FRβ is not likely to be involved directly in the initial process of monocyte egress and migration from the bone marrow. Because FRβ expression/functionality is increased abruptly in macrophages of M1 and M2 phenotypes,16 we hypothesize that FRβ is likely involved in monocyte perivascular and/or extravascular development. It would be challenging but interesting to track functional FRβ expression on bone marrow-derived monocytes as they traffic across the blood vessel wall into inflamed tissues. Because activated macrophages preferentially accumulate FA-targeted radiotracers, it would also be interesting to see whether EC2319’s pharmacologic effect on inflammatory monocytes in animal models translates to corresponding classical and/or intermediate monocytes in human diseases.
From a compound design perspective, placement of L-cysteine immediately before γ-carbonyl AMT in EC2319 drives the major AMT-releasing metabolic pathway in favor of enzymatic cleavage by GGH, a vital enzyme in folyl and antifolyl poly-γ-glutamate metabolism.40 However, we determined a species difference in EC2319 linker metabolism with higher AMT release in rodents than in dogs and in human samples (i.e., whole blood and liver cytosols). Regardless, EC2319’s FR-specific anti-arthritic and/or anti-monocyte activity in AIA and anti-GBM rats was observed consistently and fully reversible in the presence of the FA competitor. Thus, EC2319 appears to remain largely intact in the circulation and becomes metabolically activated when inside a cell. According to interspecies allometric scaling, an EC2319 dose of 500 nmol/kg (i.e., 1.13 mg/kg) in a 150-g rat is equivalent to ∼0.33 mg/kg in a 10-kg dog. However, based on total AUClast of EC2319 plus active metabolites, an effective dose of EC2319 at 500–1000 nmol/kg in a rat would have been equivalent to ∼0.535–1.07 mg/kg in a dog.
Engineered CAR T cell therapy is a rapidly emerging form of cancer treatment that has demonstrated considerable activity in hematological malignancies but not without significant challenges because of CRS.41 During CAR T cell activation, clinical manifestations of CRS can be worsened by monocyte/macrophage activation driven by T cell- and monocyte-derived cytokines, including IFN-γ, TNF-α, IL-6, and IL-1.42 Despite the commonly used corticosteroids or even tocilizumab (anti-IL-6R), there remain unmet medical needs in managing severe CRS and its associated neurotoxicity. Because of the difficulty of isolating FRβ+ primary human monocytes, we used FRβ-transfected THP1 monocytic leukemia cells, which are naturally “anti-inflammatory,” in production of pro-inflammatory cytokines (e.g., IL-6) upon lipopolysaccharide activation.43 Nevertheless, THP1-FRβ cells were confirmed to be activated by exogenous IFN-γ/TNF-α co-stimulation and in the presence of Transwell co-cultured CD19-CAR T cells and Nalm-6 cells (effector-to-target ratio [E:T] = 1:3). To demonstrate FR specificity, we elected to report IL-6 and IL-1β data because (1) both are key pleiotropic pro-inflammatory cytokines differentially required for CRS and neurotoxicity induced by CAR T cells,44 (2) both are released from activated THP1-FRβ but not from activated CD19-CAR T cells, and (3) both showed statistically significant changes upon THP1-FRβ activation and EC2319 pretreatment. Although the levels of THP1-FRβ-derived cytokines are likely different from the serum levels found in individuals receiving highly active CD19-CAR T cell therapy, our co-culture observation is relevant for the proposed mechanisms associated with CRS in the clinic.41,42 THP1-FRβ cells express a higher level of functional FRs compared with primary human monocytes.35 In this regard, our animal studies suggest that FRβ expression on host monocyte-macrophage-lineage cells may change dramatically under inflammatory conditions that lead to their hypersensitivity to EC2319 treatment at some point of that dynamic process. The aforementioned THP1-FRβ in vitro response provides a clue regarding EC2319’s general immune modulation capability against inflammatory monocytes/macrophages. Further studies are warranted to validate EC2319 anti-CRS activity in vivo.
As the coronavirus disease 2019 (COVID-19) pandemic persists across the globe, it appears that the complication of hyperinflammation and cytokine storms increase the risk of fatality in people with and without underlying medical conditions.45, 46, 47 The pathogenic environment has been shown to drive pulmonary accumulation of CD14+ CD16+ monocytes, trigger macrophage activation syndrome, and increase production of pro-inflammatory cytokines.45,48 Because elevated IL-6 and IL-1β levels are detected at local and systemic levels, tocilizumab, sarilumab (anti-IL-6R), siltuximab (anti-IL-6), and anakinra (IL-1Ra antagonist) have been used off label49 or fast tracked in clinical studies against COVID-19 (https://clinicaltrials.gov/ct2/show/NCT04330638). Because rodent classical CD172+ CD43− (rat) and Ly6Chigh CD43+ (mouse) monocytes correspond to classical CD14+ CD16− monocytes in humans that can develop into intermediate CD14+ CD16+ monocytes, we hypothesize that this COVID-19-provoked inflammatory cell population may also respond to EC2319 treatment, with the added advantage of inhibiting production of multiple pro-inflammatory cytokines with a single-agent therapy.
In summary, unresolved inflammation involving monocytes/macrophages can become a major challenge in acute and progressive chronic phases that requires special care to prevent tissue damage and organ dysfunction. Because of the FRβ biology apparently restricted to these specific immune cells, EC2319’s FR-directed “real-life” mode of action is an enhancement of traditional DHFR inhibitors. To boost a physician’s ability to manage pathologic inflammation regardless of the root causes, we also propose that EC2319’s utility goes beyond autoimmune/inflammatory diseases and may be explored for treating monocyte-macrophage activation syndrome as well as cytokine storms associated with adoptive cell therapies, therapeutic antibodies, and bacterial or viral infections.
Limitations of the study
To curtail hyperinflammation, EC2319 delivers a potent effect against bone marrow-derived monocytes that translates to its broad systemic and local activity. However, there remains a missing link regarding the precise moment when EC2319 binds FRβ+ myeloid cells and executes its molecular mechanism in vivo. The transient FRβ functionality on inflammatory subsets of circuiting blood monocytes makes the task difficult but may not be impossible with modern single-cell multi-omics analysis. Further, we demonstrated EC2319 anti-CRS activity by indirectly exposing monocytic THP1-FRβ cells to Nalm-6-activated CD19-CAR T cells. More “real-life” CRS model systems are needed to validate EC2319 activity in vivo. Research opportunities exist in different treatment modalities when monocyte-macrophage activation triggers cytokine storms in adoptive cell therapies, therapeutic antibodies, and bacterial or viral infection.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-rat CD45 | Thermo Fisher Scientific | clone OX1; RRID:AB_11217484 |
| Anti-rat CD3ε | Thermo Fisher Scientific | clone eBioG4.18; RRID:AB_11220081 |
| Anti-rat MHC class II | Thermo Fisher Scientific | clone OX17; RRID:AB_10736599 |
| Anti-rat CD11b | BD Bioscience | clone WT5 |
| Anti-rat CD172 | BD Bioscience | clone OX41; RRID:AB_2573173 |
| Anti-rat CD43 | BioRad | clone W3/13; RRID:AB_960292 |
| Anti-rat FRβ | Dr. Takami Matsuyama, Kagoshima University, Japan | IgM clone 4A67 |
| Anti-mouse IgM | Thermo Fisher Scientific | clone ll41; RRID:AB_467583 |
| Anti-mouse CD45 | Thermo Fisher Scientific | clone 30-F11; RRID:AB_10376146 |
| Anti-mouse CD11b | Thermo Fisher Scientific | clone M1/70; RRID:AB_11154207 |
| Anti-mouse NK1.1 | Thermo Fisher Scientific | clone PK136; RRID:AB_2539362 |
| Anti-mouse Ly6C | Thermo Fisher Scientific | clone HK1.4; RRID:AB_10804510 |
| Anti-mouse CD43 | Thermo Fisher Scientific | clone eBioR2/60; RRID:AB_465039 |
| Anti-glomerulonephritis (GBM) serum | Dr. Gabriela Garcia, University of Colorado Denver | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Aminopterin (AMT) | Millipore Sigma | Cat# A3411 |
| Aminopteroic acid | Millipore Sigma | Cat# A1784 |
| Methotrexate (MTX) | Millipore Sigma | Cat# A6770 |
| Folic acid (FA) | Millipore Sigma | Cat# F7876 |
| Folic acid, diammonium salt, [3′,5′,7,9-3H]-1% Potassium ascorbate solution,pH6 | Moravek | Cat# MT783K |
| BLZ945 | Novartis | N/A |
| Pteroyl-γGlu-D-Asp-D-Asp (folate competitor) | Synthesized in house | N/A |
| FA-Alexa Fluor® 647 (pteroyl-γGlu-L-Asp-L-Lys-L-Asp-L-Lys-L-Lys(AF-647)-OH) (fluorescent folate probe) | Synthesized in house | N/A |
| Light mineral oil | Millipore Sigma | Cat# M8410 |
| Human interphotoreceptor retinoid-binding protein (IRBP) peptide 161-180 | GenScript | Cat# RP20268 |
| Recombinant human gamma-glutamyl hydrolase (GGH) | Abnova | Cat# H00008836-P01 |
| Recombinant human IL2 | Miltenyi biotec | Cat# 130-097-744 |
| Recombinant human IFNγ | R&D Systems | Cat# 285-IF-100/CF |
| Recombinant human TNFα | R&D Systems | Cat# 210-TA-020/CF |
| RPMI1640 medium (complete) | ATCC | Cat# 30-2001 |
| RPMI1640 medium (folate free) | Mediatech | Cat# 98-303-CV |
| TexMACS™ medium | Miltenyi biotec | Cat# 130-097-196 |
| Human AB serum | Valley Biomedical | Cat# HP1022HI |
| Ficoll-Paque Premium gradient | GE Healthcare | Cat# 17-5442-02 |
| 24-well HTS transwell plate (Corning) | Millipore Sigma | Cat# CLS3396 |
| CountBright beads | Thermo Fisher Scientific | Cat# C36950 |
| Experimental models: Cell lines | ||
| KB subclone | ATCC (adapted to survive in folate deficient medium) | Cat# CCL-17 |
| RAW264.7 subclone | ATCC (adapted to survive in folate deficient medium) | Cat# TIB-71 |
| CHO-β | Dr. Manohar Ratnam, The University of Toledo | N/A |
| THP1-FRβ | Dr. Manohar Ratnam, The University of Toledo | N/A |
| Nalm-6 | ATCC | Cat# CRL-3273 |
| Experimental models: Organisms/strains | ||
| Lewis rat (female) | Envigo | Cat# LEW/SsNHsd RRID:RGD_737922 |
| Wistar Kyoto rat (male) | Charles River | Cat# strain code 008 RRID:RGD_1358112 |
| B10.RIII mouse (female) | The Jackson laboratory | Cat# 000457 RRID:IMSR_JAX:000457 |
| Beagle dog (male) | Provided by BASi | https://www.inotivco.com/ |
| Mycobacteria butyricum (heat-killed) | BD Diagnostic Systems | CAS 9007-81-2 |
| Mycobacterium tuberculosis (BD Difco™) | Fisher scientific | Cat# DF0640-33-7 |
| Pertussis toxin | List Biological Laboratories | Cat# 180 |
| Folate-deficient diet (rodent) | Envigo | Cat# TD.95247 |
| Software and algorithms | ||
| WinNonLin pharmacokinetics software | Certara | https://www.certara.com/software/phoenix-winnonlin/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Kaluza flow cytometry software | Beckman Coulter | https://www.beckman.com/flow-cytometry/software/kaluza/downloads |
| Biological samples | ||
| CD19-CAR T cells | Promab Biotechnology | Cat# PM-CAR1007-100M |
| Whole blood K2EDTA (human) | BioIVT | https://bioivt.com/human-whole-blood-pooled |
| Whole blood K2EDTA (dog) | BioIVT | https://bioivt.com/canine-whole-blood-pooled |
| Whole blood K2EDTA (rat) | BioIVT | https://bioivt.com/pooled-rat-whole-blood |
| Human liver cytosols | Xenotech | Cat# H0610.C |
| Dog liver cytosols | Xenotech | Cat# D1000.C |
| Rat liver cytosols | Xenotech | Cat# R1000.C |
| LEGENDplex™ human cytokine panels | BioLegend | Cat# 740721 |
| XTT cell viability assay kit | ATCC | Cat# 30-1011K |
| Histopathology | HistoTox Labs | https://histotoxlabs.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yingjuan J. Lu (yjlu001@gmail.com).
Materials availability
EC2319 and its associated intellectual property are the property of Novartis Institute for Biomedical Research and will only be made available to 3rd parties under agreed upon terms and execution of appropriate contracts.
Experimental model and subject details
Cell lines
KB cells are a human cancer cell line known for its elevated expression of FRα.17 The CHO-FRβ cell line was produced by stable integration and amplification of a human FRβ cDNA expression construct in CHO-K1 cells and obtained from Manohar Ratnam, Department of Biochemistry and Cancer Biology, The University of Toledo (Toledo, Ohio). RAW264.7, a subclone of mouse macrophage-derived tumor cells expressing a murine FR.17 THP1-FRβ is a non-adherent human promonocytic leukemia cell line engineered to express human FRβ (also courtesy of Dr. Manohar Ratnam). All cells were cultured in a folate-free RPMI1640 medium (Mediatech, Manassas, VA) containing 10% heat-inactivated fetal calf serum and antibiotics, FFRPMI, and maintained under a 5% CO2 atmosphere using standard cell culture techniques at 37°C. For co-culture studies, cryopreserved CD19-CAR T cells were purchased from Promab Biotechnology (Richmond, CA). The Nalm-6 leukemia cell line50 (American Type Culture Collection, ATCC) was used as CD19+ target cells and maintained in complete RPMI1640 medium.
Experimental models
Lewis rats (female, ∼175-200 g or 12-14 weeks of age; RRID:RGD_737922) were purchased from Envigo (Indianapolis, IN) and housed at Purdue University Animal Facility for AIA and pharmacokinetic studies. Wistar Kyoto (WKY) rats (male, ∼172-219 g or 7-8 weeks of age; RRID:RGD_1358112) were purchased from Charles River (Wilmington, MA) for anti-GBM glomerulonephritis studies (University of Colorado Denver, Denver, CO). B10.RIII mice (female, 25 ± 1.5 g or 8-12 weeks of age; RRID:IMSR_JAX:000457) for EAU studies (Novartis Institutes for Biomedical Research, Cambridge, MA) were purchased from The Jackson laboratory (Bar Harbor, ME). All animals were allowed to acclimate for at least 1 week before use. All experiments were performed according to animal protection guidelines issued by Purdue Animal Use and Care Committee, the IACUC at the University of Colorado Denver, and the Novartis Animal Welfare Policy, respectively.
Method details
Compound syntheses
Materials
N10-trifluoroacetylpteroic acid (N10-TFA pteroic acid) was purchased from Irvine Chemistry Lab (Anaheim, CA). Fmoc-3,4;5,6-di-O-isopropylidene-1-amino-1-deoxy-D-glucitol-γ-glutamate (Fmoc-saccharo-Glu-OH) was prepared according to the literature procedure.26 Aminopteroic acid was purchased from Cambridge Major Laboratories (Germantown, WI). Peptide synthesis reagents L-glutamic acid γ-methyl ester-α-t-butyl ester (H-Glu(Ot-Bu)-OMe), Fmoc-L-glutamic acid γ-t-butyl ester (Fmoc-Glu(Ot-Bu)-OH), Fmoc-L-glutamic acid α-t-butyl ester (Fmoc-Glu-Ot-Bu), Boc-S-(3-nitro-2-pyridylthio)-L-cysteine (Boc-Cys(Npys)-OH), and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) were purchased from Chem-Impex International (Wood Dale, IL). S-trityl-L-penicillamine on 2-chloro-trityl-resin (H-Pen(Trt)-2-Cl−Trt-resin) was purchased from Bachem Americas (Torrance, CA). N, N-Dimethylformamide (DMF), isopropanol (IPA), triisopropylsilane (TIPS), ethanedithiol (EDT), N, N-diisopropoylethylamine (DIPEA), trifluoroacetic acid (TFA), methanol (MeOH), N-methyl-2-pyrollidone (NMP), lithium hydroxide hydrate (LiOH·H2O), toluene, acetonitrile, diethyl ether, trimethylsilyl diazomethane (TMS-diazomethane), dimethylsulfoxide (DMSO), and ammonium bicarbonate were purchased from Sigma-Aldrich (St. Louis, MO).
Chromatography/NMR
Ammonium bicarbonate buffers used for analytical and preparative reverse phase chromatography were prepared and adjusted to pH 7 upon addition of the appropriate amount of acetic acid. All compounds purified by reversed phase chromatography were lyophilized to remove buffer and acetonitrile. Compounds were lyophilized for no fewer than two days. Reactions were monitored with a Waters Acquity H Class UPLC equipped with PDA detector and SQ mass detector using a Waters UPLC BEH C18 1.7 μm, 2.1 X 50 mm column. Reverse phase preparative chromatography was performed on a Biotage Isolera One utilizing SNAP Ultra C18 columns. 1H NMR were obtained on an Agilent 500 MHz NMR. All experiments were conducted at 25°C.
Synthesis of folate spacer 3
H-Pen(Trt)-2-chlorotrityl-resin (1) was subjected to a 7-step Fmoc solid-phase peptide synthesis as described below to provide compound 3.
Table: Amino acids used in each coupling step for the synthesis of 3
| Coupling step | Reagent | mmol | Equivalent | MW (g/mol) | Amount (g) |
|---|---|---|---|---|---|
| H-Pen(Trt)-2-Cl−Trt-resin (loading 0.56 mmol/g) | 4.05 | 7.25 | |||
| 1 | Fmoc-saccharo-Glu-OH | 8.1 | 2 | 613 | 5.0 |
| 2 | Fmoc-Glu(Ot-Bu)-OH | 8.1 | 2 | 425 | 3.4 |
| 3 | Fmoc-saccharo-Glu-OH | 6.5 | 1.6 | 613 | 3.9 |
| 4 | Fmoc-Glu(Ot-Bu)-OH | 8.1 | 2 | 425 | 3.4 |
| 5 | Fmoc-saccharo-Glu-OH | 6.5 | 1.6 | 613 | 3.9 |
| 6 | Fmoc-Glu-Ot-Bu | 8.1 | 2 | 425 | 3.4 |
| 7 | N10-TFA-Pteroic Acid | 7.1 | 1.8 | 408 | 2.9 |
The resin was added to a peptide synthesis vessel and then swelled with DMF for 10 minutes. Before each amino acid (AA) coupling step, the resin was treated with 20% piperidine in DMF for Fmoc deprotection (3X 10 minutes) and subsequently washed with 3X DMF, IPA, and DMF again. The Fmoc deprotection via piperidine treatment was repeated a second time to ensure complete Fmoc deprotection. For each coupling step, the appropriate amino acid, DMF, DIPEA (2X eq. of AA), and PyBOP (1.0X eq. of AA) were added to the rector. The reaction mixture was agitated with argon bubbling (overnight for the first Fmoc-saccharo-Glu-OH coupling and 1 hour for all of the other coupling steps) and washed with 3X DMF, IPA, and DMF again. The sequence was continued until all 7 coupling steps were completed. The peptide was cleaved from the resin by treatment with a TFA/H2O/TIPS/EDT (92.5, 2.5, 2.5, 2.5) cleavage solution with argon bubbling for 1 hour. The cleavage solution was then poured into diethyl ether to affect precipitation of the crude peptide. After isolation of the solid by filtration, the crude peptide was treated with aqueous sodium carbonate (pH 10) under argon bubbling for 1 hour to cleave the TFA protecting group. The resulting crude material was purified by a Biotage C18 column (0.1% TFA solvent A, acetonitrile solvent B; gradient 0% A to 35% A). The pure fractions were concentrated to remove the acetonitrile and the resulting solutions were passed back through a Biotage C18 column (water and acetonitrile eluents) to desalt the sample. After freeze drying, pure 3 (2.7g, 40% yield, > 98% purity) was recovered as a yellow solid. ESI-MS m/z [M + 2H]2+ = 854.9. 1H-NMR (500 MHz, D2O): δ 8.62, (s, 1H), 7.51 (d, J = 7.5 Hz, 2H), 6.64 (d, J = 7.5 Hz, 2H), 4.51 (s, 2H), 4.35-4.33 (m, 1H), 4.31-4.29 (m, 2H), 4.26-4.23 (m, 1H), 4.15-4.07 (m, 3H), 3.77-3.71 (m, 3H), 3.71-3.68 (m, 1H), 3.66-3.60 (m, 6H), 3.56-3.49 (m, 6H), 3.33-3.24 (m, 3H), 3.16-3.09 (m, 3H), 2.46-2.36 (m, 3H), 2.36-2.14 (m, 11H), 2.04-1.72 (m, 12H), 1.35 (s, 3H), 1.27 (s, 3H).
Synthesis of α-t-butyl protected AMT 5
Aminopteroic acid (4, 12 g, 38.6 mmol), H-Glu(OMe)-Ot-Bu HCl salt (10.8g, 42.5 mmol, 1.15 eq.), and PyBOP (30 g, 57.6 mmol, 1.5 eq.) were suspended in DMF (200 mL). To the suspension was added TEA (19.5 mL, 140 mmol, 3.6 eq.). After one hour, the reaction was poured into H2O (900 mL) and the resulting mixture was filtered through a Buchner funnel. The filter cake was washed with H2O (900 mL). The damp crude solid was transferred into a bottle, frozen, and placed on a freeze dryer for several days to give ca. 20 g of crude adduct. A portion of this adduct (10 g, ca. 19.5 mmol) was suspended in MeOH (30 mL) and H2O (30 mL). A solution of LiOH·H2O (1.6 g, 38.1 mmol, 2 eq.) in a minimum amount of H2O was added and the reaction was allowed to stir for 30 minutes. The reaction mixture was decanted, and the solution was diluted with water and loaded onto a Biotage C18 column and purified (water and acetonitrile as eluents) to give 5 (3 g, 31% yield) was a yellow solid. ESI-MS m/z [M+H]+ = 497.5. 1H-NMR (500 MHz, DMSO-d6): δ 8.68 (s, 1H), 7.68 (d, J = 8.8 Hz, 2H), 6.71 (d, J = 8.8 Hz, 2H), 3.98 (t, J = 6.3 Hz, 1H), 2.05 (m, 2H), 1.84 (m, 2H), 1.35 (s, 9H).
Synthesis of H-Cys(Npys)-OMe
Boc-Cys(Npys)-OH (3.81 g, 10.2 mmol) was dissolved in toluene (45 mL) and MeOH (45 mL). To this solution was added a solution of TMS-diazomethane in diethyl ether (9 mL of a 2M solution, 1.8 eq.) dropwise. After 10 minutes, the solvent and excess reagent were removed under reduced pressure and dried under high vacuum for several hours to yield the desired methyl ester (4 g, quantitative yield). A portion of this material (1.3g, 3.4 mmol) was treated with the standard TFA/H2O/TIPS cleavage solution (12 mL; 95, 2.5, 2.5) for 45 minutes. The cleavage solution was removed under reduced pressure and the resulting residue was placed on the high vacuum for 2 hours. The resulting crude H-Cys(Npys)-OMe was used in the following reaction without further purification. ESI-MS m/z [M+H]+ = 290.2.
Synthesis of pyridyl-disulfanyl activated cysteine methyl ester-AMT adduct 6
Compound 5 (1.53 g, 3.08 mmol) was suspended in NMP (30 mL). To this suspension was added TEA (2.4 mL, 5.5 eq.), PyBOP (3.5 g, 2.2 eq.), and H-Cys(Npys)-OMe (crude residue from the above reaction, re-constituted in 5 mL NMP, 1.1 eq.). The reaction was stirred for 45 minutes. Diethyl ether was added, and the resulting precipitate was recovered by centrifugation. The solid pellet was washed with water (2X) and separated by centrifugation. The resulting residue was dissolved in TFA/H2O/TIPS (12 mL; 95, 2.5, 2.5) and stirred for one hour. Diethyl ether was added, and the resulting precipitate was recovered by centrifugation. The resulting solid was placed on the high vacuum for 2 hours. The resulting residue was dissolved in DMSO (6 mL) and loaded onto a Biotage C18 column (ammonium bicarbonate (pH 7) and acetonitrile eluents) to give 6 (1.6 g, 68% yield) as a yellow solid after freeze drying. ESI-MS m/z [M+H]+ = 712.5. 1H-NMR (500 MHz, CD3OD): δ 8.80 (dd, J = 4.8, 1.8, 1H), 8.68 (s, 1H), 8.60 (dd, J = 8.2, 1.5 Hz, 1H), 7.65 (d, J = 8.8 Hz, 2H), 7.55 (dd, J = 8.4, 4.8 Hz, 1H), 6.71 (d, J = 8.8 Hz, 2H), 4.56 (dd, J = 8.6, 5.2 Hz, 1H), 3.98 (t, J = 6.3 Hz, 1H), 3.55 (s, 3H), 3.20 (dd, J = 13.9, 5.2 Hz, 1H), 3.07 (dd, J = 13.7, 8.8 Hz, 1H), 2.22 (t, J = 2.0 Hz, 2H), 2.02 (m, 1H), 1.88 (m, 1H).
Synthesis of EC2319
Compound 3 (1.67 g, 0.98 mmol) was dissolved in DMSO (15 mL) and purged with argon for 10 minutes. To this solution was added TEA (1.4 mL, 10 eq.) followed by adduct 6 (700 mg, 1 eq.) in DMSO (5 mL). The reaction was allowed to stir for 20 minutes with continued argon bubbling. The reaction mixture was poured into cold H2O (200 mL) with stirring and then purified with a Biotage C18 column (ammonium bicarbonate (pH 7) and acetonitrile eluents). After freeze drying, EC2319 (1.4g, 64% yield, > 98% purity) was recovered as a yellow solid. ESI-MS m/z [M+2H]2+ = 1133.5. 1H-NMR (500 MHz, D2O): δ 8.63 (s, 1H), 8.57 (s, 1H), 7.53 (dd, 4H), 6.65 (d, J = 8.8 Hz, 2H), 6.59 (d, J = 8.8 Hz, 2H), 4.45 (br, 4H), 4.35 (s, 2H), 4.19 (m, 2H), 4.16-4.07 (m, 7H), 4.07-4.01 (m, 2H), 3.65-3.46 (m, 15H), 3.42-3.35 (m, 6H), 3.26-3.14 (m, 3H), 3.08-2.92 (m, 4H), 2.96-2.88 (m, 1H), 2.20-2.00 (m, 14H), 2.00-1.70 (m, 14H), 1.22 (s, 3H), 1.14 (s, 3H).
Cell proliferation assay
The FR-specific activity of EC2319 in-vitro was assessed without and with FA at 100-fold molar excess against THP1-FRβ and RAW264.7 cells. In a 96-well microtiter plate assay, THP1-FRβ at 3 × 104 cells/well (round-bottom) and RAW264.7 at 2 × 104 cells/well (flat) were treated with 10-fold serial dilutions of EC2319 (0-10 μM) in FFRPMI medium without and with 100-fold molar excess of FA. After a 2 h pulse-exposure, the drug-containing media were replaced with drug free media and the cells were allowed to incubate further for 70 h (referred in the text as a 70 h “chase”). EC2319’s EC50 values (half-maximal effective concentration) were obtained by a standard XTT cell viability assay (ATCC, Manassas, VA) by adding XTT (2,3-bis(2-methoxy-4-nitro-5-sulfo-phenyl)-2H-tetrazolium-5-carboxanilide) to the remaining media for an additional 4 h following the manufacturer’s instructions.
Relative affinity assay
EC2319’s FR-specific binding affinity was determined according to established methods.17 Briefly, either 100,000 FR-positive KB (FRα) or CHO-β (FRβ) cells were seeded into each well of 24-well Falcon plates and incubated overnight (> 75% confluent) in folate free RPMI media containing 10% heat-inactivated fetal calf serum (FFRPMI/HIFCS). Typically, no more than five plates (120 test wells) were used for any given experiment. Spent incubation medium was replaced with FFRPMI containing 100 nM of 3H-FA in the absence and presence of increasing concentrations of unlabeled FA or EC2319. Cells were incubated for 1 h (in triplicate wells) in an incubator at 37°C and then rinsed 3 times with 0.5 mL of PBS. Five hundred microliters of 1% sodium dodecylsulfate in PBS were added to each well; after 5 min, cell lysates were collected, transferred to individual vials containing 5 mL of scintillation cocktail, and then counted for radioactivity. The relative FR-binding affinity (r.a.) value was defined as the inverse molar ratio of compound required to displace 50% of 3H-FA bound to KB and CHO-β with FA itself set at 1; that is, values < 1 reflect weaker affinity than FA, and values > 1 reflect stronger affinity.
Whole blood stability and linker metabolism studies
To study cross-species differences, EC2319 (500 nM) was first spiked into EDTA-anticoagulated whole blood (rat/dog/human) and incubated for 30 and 60 minutes at 37°C. Plasma samples were extracted for quantitation of EC2319 and its major metabolites (AMT, AMT adduct) by LC-MS/MS. For linker metabolism studies, liver cytosols (10 mg/mL protein) from various species (rat/dog/human) were diluted 20X in 0.5 M sodium acetate buffer (pH 4.5) in a final volume of 500 μL (i.e., 10% cytosol). Reactions in the liver cytosols were initiated by the addition of 1 μL of EC2319 stock to yield 1 μM final concentration and the reaction mixtures were incubated at 37°C for 1 hour. To test non-disulfide mediated linker cleavage, a final mixture of 100 μL 0.1 M sodium acetate buffer (pH 4.5) containing 1 μM EC2319, 0.09 ng human GGH, and 20 mM dithiothreitol were incubated for 2 hours at 37°C. At the termination of all reactions, samples were processed prior to LC-MS/MS analysis.
Pharmacokinetic studies
Female Lewis rats with a rounded tip jugular vein catheter (Harlan) were given a single subcutaneous dose of EC2319 at 500 nmol/kg. Whole blood samples collected at 1, 10, and 30 minutes, 1, 2, 3, 4, 8, 12, and 19 hours post dose, were directly placed into tubes containing 1.7 mg/mL K3EDTA, 0.425 mg/mL N-Maleoyl-β-alanine, 1 mg/mL mannitol, and 0.00375% acetic acid (for immediate quenching of free thiols). Rat plasma samples were obtained by centrifugation for 3 minutes at ∼2000xg and stored at −80°C until LC-MS/MS quantitation. The analytes included were the intact EC2319 molecule, unmodified AMT, and the AMT adduct. To study EC2319 pharmacokinetics in non-rodents, male beagle dogs (∼7-12.4 kg) housed at BASi (Mt. Vernon, IN) were given a single intravenous (1 mg/kg) or subcutaneous (2.43 mg/kg) dose of EC2319. In the group receiving the intravenous dose, blood samples were collected from a peripheral vein at predose, 2, 5, 15, and 30 minutes, 1, 2, 4, 8, and 12 hours after dosing. In the group receiving the subcutaneous dose, samples were collected from a peripheral vein at pre-dose, 15, 30, and 45 minutes, 1, 2, 3, 4, 8, and 24 hours after dosing. In both cases, blood samples were collected into tubes containing K3EDTA fortified with N-Maleoyl-β-alanine, mannitol, and acetic acid as described above. The dog plasma samples were obtained by centrifugation under refrigeration for at least 10 minutes at ∼2000xg and analyzed by LC-MS/MS. All pharmacokinetic parameters were calculated using the WinNonLin pharmacokinetics software including the maximum drug concentration in plasma (Cmax), time to Cmax (Tmax), areas under the curve from time zero to the time of last measurable concentration (AUClast), and terminal half-life (t1/2) were obtained. The absolute bioavailability (F) was calculated by using the following formula: AUC (subcutaneously)/AUC (intravenously).
Treatment of adjuvant-induced arthritis
To induce AIA, Lewis rats pre-fed with a folate-deficient diet (Envigo) for 9 days were intradermally inoculated (at the base of tail) with 0.5 mg of heat-killed Mycobacteria butyricum (BD Diagnostic Systems, Sparks, MD) in 100 μL light mineral oil (Sigma). During the course of disease development, AIA rats were scored 3 times a week according to the previously established criteria.17 Each paw was graded with a score of 0–4, with a maximum possible score of 16 for each animal. For efficacy assessment, AIA rats with desired arthritis scores were distributed evenly on day 0 across the control and EC2319 treatment groups without and with a FR-specific competitor (n = 5-7). Subcutaneous administration of EC2319 (500 nmol/kg) was carried out biweekly for two consecutive weeks (i.e., BIW x 2). The therapeutically irrelevant FA competitor, Pteroyl-γGlu-D-Asp-D-Asp, was used in 500-fold molar excess to block EC2319 activity in-vivo. Changes in arthritis score and body weight of each animal were monitored 3 times a week. Upon study completion (∼4 days after the last dose), rats were euthanized and processed for paw weight (all four limbs cut at the hairline) and spleen weight. The percent increases in paw weight (paw swelling) and spleen weight (splenomegaly) were calculated by the formula: 100 x (Wc – Wo) / (Wt - Wo), where Wc = Mean paw or spleen weight in the AIA control group of animals, Wo = Mean paw or spleen weight of healthy control animals, Wt = Mean paw or spleen weight in the treated group of animals. The removed hind paws were immersion-fixed in 10% buffered formalin and submitted for histopathology. All tissue preparation and staining were performed at HistoTox Labs, Inc. (Boulder, CO, U.S.A.). The processed animal tissues were examined microscopically by Laura Hoon-Hanks, a board-certified veterinary pathologist blinded to treatment conditions at the time of evaluation. To obtain histological scores, a numerical grading system for joint damage was used for each hind foot including periarticular inflammation (0-5), bone loss/resorption (0-5), and cartilage damage/loss (0-5) and pannus formation (0-5) at tibiotalar joint.51,52 The maximal individual and sum histology scores per rat were therefore at 10 and 40, respectively. In three separate studies for monocyte assessment by flow cytometry (Figure 5), whole blood samples were taken via cardiac puncture from healthy or AIA rats treated biweekly with EC2319 (starting on day 0, 9 days post induction). Initially, whole blood samples were collected four days after the second EC2319 dose (30-1000 nmol/kg, BIW x 1, on days 0 and 3) (Figures 5B and 5C). Subsequently to demonstrate FR specificity (Figures 5D–5F), AIA rat blood samples were taken 4 days after the third EC2319 dose (500 nmol/kg, BIW x 1.5, on days 0, 3, and 7) without and with the FA competitor at 500-fold molar excess.
Treatment of anti-GBM Induced glomerulonephritis
Anti-GBM autoantibody targeting the alpha-3 chain of type IV collagen is a rare form of glomerulonephritis that can lead to glomerular crescent formation and rapid loss of kidney function. To induce rat glomerulonephritis, anti-GBM serum was prepared in Gabriela Garcia’s laboratory at University of Colorado Denver according to previously established methods.53 Since the maximum glomerular deposition of IgG occurs one hour after anti-GBM antibody injection,54 all treatments were initiated 8 hours after antibody injection (i.e., day 0 = 8 hours). Thus, male WKY rats pre-fed with folate-deficient diet for 10 days were intravenously injected on day 0 with anti-GBM serum (25 μL per 100 g of body weight) and maintained on the diet for a total of 16-20 days. Initially, whole blood samples were taken from healthy or anti-GBM rats on days 6 and 12 post induction for monocyte analysis in isolated PBMCs. To demonstrate FR-specific activity, anti-GBM rats were given two doses of EC2319 (575 nmol/kg) on days 0 and 3 without and with 300-fold molar excess of the FA competitor. For comparison to EC2319, oral MTX was given in two ways, either at 575 nmol/kg on days 0 and 3 or at 4.56 μmol/kg (i.e., 2.07 mg/kg) on day 0 only. The latter 8-fold higher MTX dose was equivalent to that of 20 mg/week in RA patients (∼60 kg body weight). In this case, whole blood samples (100 μL) were collected from the tail vein on day 6 (3 days after the last EC2319 dose) and shipped overnight on ice for the next-day flow cytometry analysis.
Treatment of experimental autoimmune uveitis
Starting two weeks before immunization, B10.RIII mice were fed folate deficient diet and continued for a total of 24 days until euthanasia. To induce EAU, the animals were actively immunized on day 0 with a single subcutaneous injection of 50 μg of human interphotoreceptor retinoid-binding protein (IRBP) peptide 161-180 (IRBP161-180, SGIPYIISYLHPGNTILHVD; GenScript, cat.# RP20268), emulsified in complete Freund’s adjuvant (CFA) containing 2.5 mg/mL of Mycobacterium tuberculosis (BD Difco™). Pertussis toxin (List Biological Laboratories, Campbell, CA), a Th1-biased adjuvant, was given as 2 × 200 ng intraperitoneal doses on days 0 and 2 to enhance organ-specific autoimmunity. Starting on day 0, EAU mice were randomly distributed in three groups (n = 10) and treated once daily for 10 days with vehicle only, EC2319 (1000 nmol/kg, subcutaneous), and the positive control compound BLZ945 (200 mg/kg, oral gavage). On day 10, the animals were euthanized and whole blood samples (100 μL) were collected via cardiac puncture. For comparison, whole blood samples (200 μL) were also collected from naive mice. All samples were shipped overnight on ice for next-day flow cytometry analysis.
Flow cytometric analyses
All flow cytometry data were collected on a Beckman Coulter Gallios flow cytometer equipped with blue, red and violet lasers. For sample acquisition, whole blood samples in tubes containing 5 mM K2EDTA were analyzed on the same day for AIA rats or upon arrival on the following day for anti-GBM rats and EAU mice. For AIA rats, anticoagulated whole blood samples were diluted 1:1 with FFRPMI, carefully layered onto a Ficoll-Paque Premium gradient (GE Healthcare) and pelleted to separate PBMCs from granulocytes and red blood cells (RBC) according to the manufacturer’s instructions. After a RBC lysis step, PBMCs were washed 2x with PBS, filtered through a 40 μm nylon filter, and stained with fluorophore-conjugated antibodies to rat leukocyte surface markers. According to the literature, inflammatory monocytes in rats were identified as the classical CD172+ CD43- cell population.21,22 For comparison, rat T and B lymphocytes were identified as CD3ε + MHCII- and MHCII+ CD3ε - cell populations, respectively. For anti-GBM rats, CD172+ CD43- monocyte analysis was carried out in the same fashion on isolated PBMCs (Figure 6B) or on total leukocytes obtained from 100 μL of whole blood after RBC lysis (Figure 6C). For EAU mice and heathy controls, 100-200 μL of anticoagulated blood were pretreated with RBC lysis buffer and stained with fluorophore-conjugated antibodies to mouse leukocyte surface markers. Inflammatory and pro-inflammatory monocytes were identified as the classical Ly6Chigh CD43+ and intermediate Ly6Chigh CD43high phenotypes, respectively.23,24 Total leukocyte cells per 100 μL of whole blood was calculated with the addition of 52,500 CountBright beads (Thermo Fisher Scientific, Waltham, MA) just prior to flow cytometry data acquisition according to the manufacturer’s protocol.
Transwell triple co-culture assay
Prior to co-culture experiments, we elected to use a cocktail of IFNγ and TNFα to represent the products of activated CAR T cells to trigger monocyte activation. Thus, THP1-FRβ cells in FFRPMI (3 × 105 cells/mL, 6-well plates) were pulsed with 0, 2 and 10 μM of EC2319 for 2 hours without and with 100-fold excess of the FA competitor (at 10 μM EC2319). After a washing step, the cells were “chased” in drug-free FFRPMI for 24 hours before a cocktail of IFNγ/TNFα (500 IU/mL each, R&D Systems) was added. After 48 hours of IFNγ/TNFα co-stimulation, the cell culture supernatants were collected for multiplex cytokine analysis using LEGENDplex bead-based immunoassay. To prepare for triple co-culture experiments, cryopreserved CD19-CAR T cells were allowed to recover for 3 days in T cell culture medium [TexMACS™ medium (Miltenyi biotec, Auburn, CA)] supplemented with 2% heat-inactivated human AB serum and 50 IU/mL IL-2 (Miltenyi biotec, Auburn, CA). Afterward, viable CD19-CAR T cells (6.7 × 105) were transferred to the lower compartment of 24-well HTS transwell plates (Corning, Corning, NY) and incubated overnight with Nalm-6 cells in 600 μL FFRPMI at an effector-to-target (E:T) ratio of 1 to 3. Notably, all effector and target cells were prewashed with FFRPMI to remove any exogenous FA that could compete against EC2319 in the subsequent step. Meanwhile, THP1-FRβ cells (3 × 105 cells/mL, 6-well plates) were subjected to a 2-hour pulse of EC2319 plus 24-hour “chase” in drug-free FFRPMI as described above. On the day of triple co-culture, EC2319 pretreated THP1-FRβ cells were counted and adjusted to a desired density (2.5 × 105 in 200 μL) and seeded to the upper compartment of the transwell inserts without direct contact with the Nalm-6 and CD19-CAR T cells in the lower compartment. After 48 hours of triple co-culture, exhausted media (supernatants) were collected for quantification of cytokine levels.
Quantification and statistical analysis
Statistical analyses were performed using the computer program GraphPad Prism (GraphPad Software Inc., San Diego, CA). The data were tested for normality using the Shapiro-Wilk or D’Agostino & Pearson tests. If Gaussian distributions of the data were predicted from these tests, one-way ANOVA analysis was performed and differences between individual groups were assessed using Tukey’s post hoc testing. If evidence of non-normality in the data were indicated from the normality testing, Kruskal-Wallis tests were performed with a Dunn’s post hoc test to compare individual group means. If applicable, data was further analyzed across treatment groups using appropriate multiple comparison post-test. p < 0.05 was considered statistically significant in all tests.
Acknowledgments
Author contributions
Y.J.L. contributed to activity-based compound design, designed or co-designed in vitro/in vivo studies, and conceptualized/wrote the manuscript. L.W.W. designed/performed flow cytometry and statistical analyses. H.C. designed/performed co-culture experiments. I.R.V., F.Y., and P.J.K. designed/performed chemical synthesis. P.J.K., S.R., and M.P. designed/performed whole-blood stability, linker metabolism, and pharmacokinetic studies. G.G. designed/performed anti-GBM rat experiments. H.Y.W. and A.P.d.C. designed/performed EAU mouse experiments. E.W., V.C., and A.L. performed all in-house animal work. A.L. and C.D. performed cell viability and binding assays. P.S.L. provided general scientific consultation. C.P.L. supervised discovery research and edited/approved the manuscript.
Declaration of interests
I.R.V., C.P.L., F.Y., P.J.K., and Y.J.L. hold a patent in Japan (JP 6772186) for the design and synthesis of EC2319. There is a patent pending in the United States.
Published: October 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100422.
Contributor Information
Patrick J. Klein, Email: patrick.klein@novartis.com.
Christopher P. Leamon, Email: cpleamon@yahoo.com.
Supplemental information
Data and code availability
The published article includes all datasets generated or analyzed during this study.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Shi C., Pamer E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma W.-T., Gao F., Gu K., Chen D.-K. The Role of Monocytes and Macrophages in Autoimmune Diseases: A Comprehensive Review. Front. Immunol. 2019;10:1140. doi: 10.3389/fimmu.2019.01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel A.A., Zhang Y., Fullerton J.N., Boelen L., Rongvaux A., Maini A.A., Bigley V., Flavell R.A., Gilroy D.W., Asquith B. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017;214:1913–1923. doi: 10.1084/jem.20170355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ong S.M., Hadadi E., Dang T.M., Yeap W.H., Tan C.T., Ng T.P., Larbi A., Wong S.C. The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence. Cell Death Dis. 2018;9:266. doi: 10.1038/s41419-018-0327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wood M.J., Leckenby A., Reynolds G., Spiering R., Pratt A.G., Rankin K.S., Isaacs J.D., Haniffa M.A., Milling S., Hilkens C.M. Macrophage proliferation distinguishes 2 subgroups of knee osteoarthritis patients. JCI Insight. 2019;4:e125325. doi: 10.1172/jci.insight.125325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanter J.E., Hsu C.C., Bornfeldt K.E. Monocytes and Macrophages as Protagonists in Vascular Complications of Diabetes. Front. Cardiovasc. Med. 2020;7:10. doi: 10.3389/fcvm.2020.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walscheid K., Neekamp L., Heiligenhaus A., Weinhage T., Holzinger D., Heinz C., Kasper M., Foell D. Peripheral blood monocytes reveal an activated phenotype in pediatric uveitis. Clin. Immunol. 2018;190:84–88. doi: 10.1016/j.clim.2017.09.014. [DOI] [PubMed] [Google Scholar]
- 8.Golden J.B., Groft S.G., Squeri M.V., Debanne S.M., Ward N.L., McCormick T.S., Cooper K.D. Chronic Psoriatic Skin Inflammation Leads to Increased Monocyte Adhesion and Aggregation. J. Immunol. 2015;195:2006–2018. doi: 10.4049/jimmunol.1402307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Min D., Brooks B., Wong J., Salomon R., Bao W., Harrisberg B., Twigg S.M., Yue D.K., McLennan S.V. Alterations in monocyte CD16 in association with diabetes complications. Mediators Inflamm. 2012;2012:649083. doi: 10.1155/2012/649083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C., Ke J., Zhou X.E., Yi W., Brunzelle J.S., Li J., Yong E.L., Xu H.E., Melcher K. Structural basis for molecular recognition of folic acid by folate receptors. Nature. 2013;500:486–489. doi: 10.1038/nature12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Heijden J.W., Oerlemans R., Dijkmans B.A., Qi H., van der Laken C.J., Lems W.F., Jackman A.L., Kraan M.C., Tak P.P., Ratnam M., Jansen G. Folate receptor beta as a potential delivery route for novel folate antagonists to macrophages in the synovial tissue of rheumatoid arthritis patients. Arthritis Rheum. 2009;60:12–21. doi: 10.1002/art.24219. [DOI] [PubMed] [Google Scholar]
- 12.Xia W., Hilgenbrink A.R., Matteson E.L., Lockwood M.B., Cheng J.X., Low P.S. A functional folate receptor is induced during macrophage activation and can be used to target drugs to activated macrophages. Blood. 2009;113:438–446. doi: 10.1182/blood-2008-04-150789. [DOI] [PubMed] [Google Scholar]
- 13.Matteson E.L., Lowe V.J., Prendergast F.G., Crowson C.S., Moder K.G., Morgenstern D.E., Messmann R.A., Low P.S. Assessment of disease activity in rheumatoid arthritis using a novel folate targeted radiopharmaceutical Folatescan. Clin. Exp. Rheumatol. 2009;27:253–259. [PMC free article] [PubMed] [Google Scholar]
- 14.Kraus V.B., McDaniel G., Huebner J.L., Stabler T.V., Pieper C.F., Shipes S.W., Petry N.A., Low P.S., Shen J., McNearney T.A., Mitchell P. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthritis Cartilage. 2016;24:1613–1621. doi: 10.1016/j.joca.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verweij N.J.F., Yaqub M., Bruijnen S.T.G., Pieplenbosch S., Ter Wee M.M., Jansen G., Chen Q., Low P.S., Windhorst A.D., Lammertsma A.A. First in man study of [18F]fluoro-PEG-folate PET: a novel macrophage imaging technique to visualize rheumatoid arthritis. Sci. Rep. 2020;10:1047. doi: 10.1038/s41598-020-57841-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandrupatla D.M.S.H., Molthoff C.F.M., Lammertsma A.A., van der Laken C.J., Jansen G. The folate receptor β as a macrophage-mediated imaging and therapeutic target in rheumatoid arthritis. Drug Deliv. Transl. Res. 2019;9:366–378. doi: 10.1007/s13346-018-0589-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu Y., Stinnette T.W., Westrick E., Klein P.J., Gehrke M.A., Cross V.A., Vlahov I.R., Low P.S., Leamon C.P. Treatment of experimental adjuvant arthritis with a novel folate receptor-targeted folic acid-aminopterin conjugate. Arthritis Res. Ther. 2011;13:R56. doi: 10.1186/ar3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu Y., Wollak K.N., Cross V.A., Westrick E., Wheeler L.W., Stinnette T.W., Vaughn J.F., Hahn S.J., Xu L.C., Vlahov I.R., Leamon C.P. Folate receptor-targeted aminopterin therapy is highly effective and specific in experimental models of autoimmune uveitis and autoimmune encephalomyelitis. Clin. Immunol. 2014;150:64–77. doi: 10.1016/j.clim.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 19.O’Connor C., Wallace-Povirk A., Ning C., Frühauf J., Tong N., Gangjee A., Matherly L.H., Hou Z. Folate transporter dynamics and therapy with classic and tumor-targeted antifolates. Sci. Rep. 2021;11:6389. doi: 10.1038/s41598-021-85818-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman B., Cronstein B. Methotrexate mechanism in treatment of rheumatoid arthritis. Joint Bone Spine. 2019;86:301–307. doi: 10.1016/j.jbspin.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon S., Taylor P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 22.Yrlid U., Jenkins C.D., MacPherson G.G. Relationships between distinct blood monocyte subsets and migrating intestinal lymph dendritic cells in vivo under steady-state conditions. J. Immunol. 2006;176:4155–4162. doi: 10.4049/jimmunol.176.7.4155. [DOI] [PubMed] [Google Scholar]
- 23.Ziegler-Heitbrock L., Ancuta P., Crowe S., Dalod M., Grau V., Hart D.N., Leenen P.J., Liu Y.J., MacPherson G., Randolph G.J. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–e80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 24.Yang J., Zhang L., Yu C., Yang X.F., Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014;2:1. doi: 10.1186/2050-7771-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leamon C.P., Reddy J.A., Klein P.J., Vlahov I.R., Dorton R., Bloomfield A., Nelson M., Westrick E., Parker N., Bruna K. Reducing undesirable hepatic clearance of a tumor-targeted vinca alkaloid via novel saccharopeptidic modifications. J. Pharmacol. Exp. Ther. 2011;336:336–343. doi: 10.1124/jpet.110.175109. [DOI] [PubMed] [Google Scholar]
- 26.Vlahov I.R., Santhapuram H.K., You F., Wang Y., Kleindl P.J., Hahn S.J., Vaughn J.F., Reno D.S., Leamon C.P. Carbohydrate-based synthetic approach to control toxicity profiles of folate-drug conjugates. J. Org. Chem. 2010;75:3685–3691. doi: 10.1021/jo100448q. [DOI] [PubMed] [Google Scholar]
- 27.Issekutz A.C., Issekutz T.B. Monocyte migration to arthritis in the rat utilizes both CD11/CD18 and very late activation antigen 4 integrin mechanisms. J. Exp. Med. 1995;181:1197–1203. doi: 10.1084/jem.181.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Y., Parker N., Kleindl P.J., Cross V.A., Wollak K., Westrick E., Stinnette T.W., Gehrke M.A., Wang K., Santhapuram H.K. Antiinflammatory Activity of a Novel Folic Acid Targeted Conjugate of the mTOR Inhibitor Everolimus. Mol. Med. 2015;21:584–596. doi: 10.2119/molmed.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagai T., Kyo A., Hasui K., Takao S., Matsuyama T. Efficacy of an immunotoxin to folate receptor beta in the intra-articular treatment of antigen-induced arthritis. Arthritis Res. Ther. 2012;14:R106. doi: 10.1186/ar3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawasaki K., Yaoita E., Yamamoto T., Kihara I. Depletion of CD8 positive cells in nephrotoxic serum nephritis of WKY rats. Kidney Int. 1992;41:1517–1526. doi: 10.1038/ki.1992.221. [DOI] [PubMed] [Google Scholar]
- 31.Garcia G.E., Lu Y.J., Truong L.D., Roncal-Jiménez C.A., Miyazaki M., Miyazaki-Anzai S. A Novel Treatment for Glomerular Disease: Targeting the Activated Macrophage Folate Receptor with a Trojan Horse Therapy in Rats. Cells. 2021;10:2113. doi: 10.3390/cells10082113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agarwal R.K., Silver P.B., Caspi R.R. Rodent models of experimental autoimmune uveitis. Methods Mol. Biol. 2012;900:443–469. doi: 10.1007/978-1-60761-720-4_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giorgetti E., Panesar M., Zhang Y., Joller S., Ronco M., Obrecht M., Lambert C., Accart N., Beckmann N., Doelemeyer A. Modulation of Microglia by Voluntary Exercise or CSF1R Inhibition Prevents Age-Related Loss of Functional Motor Units. Cell Rep. 2019;29:1539–1554.e7. doi: 10.1016/j.celrep.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Singh N., Hofmann T.J., Gershenson Z., Levine B.L., Grupp S.A., Teachey D.T., Barrett D.M. Monocyte lineage-derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy. 2017;19:867–880. doi: 10.1016/j.jcyt.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen J., Hilgenbrink A.R., Xia W., Feng Y., Dimitrov D.S., Lockwood M.B., Amato R.J., Low P.S. Folate receptor-β constitutes a marker for human proinflammatory monocytes. J. Leukoc. Biol. 2014;96:563–570. doi: 10.1189/jlb.2AB0713-372R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samaniego R., Palacios B.S., Domiguez-Soto Á., Vidal C., Salas A., Matsuyama T., Sánchez-Torres C., de la Torre I., Miranda-Carús M.E., Sánchez-Mateos P., Puig-Kröger A. Macrophage uptake and accumulation of folates are polarization-dependent in vitro and in vivo and are regulated by activin A. J. Leukoc. Biol. 2014;95:797–808. doi: 10.1189/jlb.0613345. [DOI] [PubMed] [Google Scholar]
- 37.Samaniego R., Domínguez-Soto Á., Ratnam M., Matsuyama T., Sánchez-Mateos P., Corbí A.L., Puig-Kröger A. Folate Receptor β (FRβ) Expression in Tissue-Resident and Tumor-Associated Macrophages Associates with and Depends on the Expression of PU.1. Cells. 2020;9:1445. doi: 10.3390/cells9061445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Machacek C., Supper V., Leksa V., Mitulovic G., Spittler A., Drbal K., Suchanek M., Ohradanova-Repic A., Stockinger H. Folate Receptor β Regulates Integrin CD11b/CD18 Adhesion of a Macrophage Subset to Collagen. J. Immunol. 2016;197:2229–2238. doi: 10.4049/jimmunol.1501878. [DOI] [PubMed] [Google Scholar]
- 39.Elo P., Li X.G., Liljenbäck H., Gardberg M., Moisio O., Miner M., Virta J., Saraste A., Srinivasarao M., Pugh M. Efficacy and tolerability of folate-aminopterin therapy in a rat focal model of multiple sclerosis. J. Neuroinflammation. 2021;18:30. doi: 10.1186/s12974-021-02073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galivan J., Ryan T.J., Chave K., Rhee M., Yao R., Yin D. Glutamyl hydrolase. pharmacological role and enzymatic characterization. Pharmacol. Ther. 2000;85:207–215. doi: 10.1016/s0163-7258(99)00063-7. [DOI] [PubMed] [Google Scholar]
- 41.Murthy H., Iqbal M., Chavez J.C., Kharfan-Dabaja M.A. Cytokine Release Syndrome: Current Perspectives. ImmunoTargets Ther. 2019;8:43–52. doi: 10.2147/ITT.S202015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chou C.K., Turtle C.J. Insight into mechanisms associated with cytokine release syndrome and neurotoxicity after CD19 CAR-T cell immunotherapy. Bone Marrow Transplant. 2019;54(Suppl 2):780–784. doi: 10.1038/s41409-019-0602-5. [DOI] [PubMed] [Google Scholar]
- 43.Chanput W., Mes J.J., Wichers H.J. THP-1 cell line: an in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014;23:37–45. doi: 10.1016/j.intimp.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Norelli M., Camisa B., Barbiera G., Falcone L., Purevdorj A., Genua M., Sanvito F., Ponzoni M., Doglioni C., Cristofori P. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018;24:739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 45.Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., HLH Across Speciality Collaboration, UK COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore J.B., June C.H. Cytokine release syndrome in severe COVID-19. Science. 2020;368:473–474. doi: 10.1126/science.abb8925. [DOI] [PubMed] [Google Scholar]
- 47.Wang Q., Fang P., He R., Li M., Yu H., Zhou L., Yi Y., Wang F., Rong Y., Zhang Y. O-GlcNAc transferase promotes influenza A virus-induced cytokine storm by targeting interferon regulatory factor-5. Sci. Adv. 2020;6:eaaz7086. doi: 10.1126/sciadv.aaz7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gustine J.N., Jones D. Immunopathology of Hyperinflammation in COVID-19. Am. J. Pathol. 2021;191:4–17. doi: 10.1016/j.ajpath.2020.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan F.A., Stewart I., Fabbri L., Moss S., Robinson K., Smyth A.R., Jenkins G. Systematic review and meta-analysis of anakinra, sarilumab, siltuximab and tocilizumab for COVID-19. Thorax. 2021;76:907–919. doi: 10.1136/thoraxjnl-2020-215266. [DOI] [PubMed] [Google Scholar]
- 50.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bendele A. Animal models of rheumatoid arthritis. J. Musculoskelet. Neuronal Interact. 2001;1:377–385. [PubMed] [Google Scholar]
- 52.Bendele A.M., Chlipala E.S., Scherrer J., Frazier J., Sennello G., Rich W.J., Edwards C.K., 3rd Combination benefit of treatment with the cytokine inhibitors interleukin-1 receptor antagonist and PEGylated soluble tumor necrosis factor receptor type I in animal models of rheumatoid arthritis. Arthritis Rheum. 2000;43:2648–2659. doi: 10.1002/1529-0131(200012)43:12<2648::AID-ANR4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 53.Garcia G.E., Truong L.D., Chen J.F., Johnson R.J., Feng L. Adenosine A(2A) receptor activation prevents progressive kidney fibrosis in a model of immune-associated chronic inflammation. Kidney Int. 2011;80:378–388. doi: 10.1038/ki.2011.101. [DOI] [PubMed] [Google Scholar]
- 54.Unanue E.R., Dixon F.J. Experimental Glomerulonephritis. V. Studies on the Interaction of Nephrotoxic Antibodies with Tissue of the Rat. J. Exp. Med. 1965;121:697–714. doi: 10.1084/jem.121.5.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.