

Abstract
Delineating species boundaries in phylogenetic groups undergoing recent radiation is a daunting challenge akin to discretizing continuity. Here, we propose a general approach exemplified by American butterflies from the genus Junonia Hübner notorious for the variety of similar phenotypes, ease of hybridization, and the lack of consensus about their classification. We obtain whole-genome shotgun sequences of about 200 specimens. We reason that discreteness emerges from continuity by means of a small number of key players, and search for the proteins that diverged markedly between sympatric populations of different species, while keeping low polymorphism within these species. Being 0.25% of the total number, these three dozen ‘speciation’ proteins indeed partition pairs of Junonia populations into two clusters with a prominent break in between, while all proteins taken together fail to reveal this discontinuity. Populations with larger divergence from each other, comparable to that between two sympatric species, form the first cluster and correspond to different species. The other cluster is characterized by smaller divergence, similar to that between allopatric populations of the same species and comprise conspecific pairs. Using this method, we conclude that J. genoveva (Cramer), J. litoralis Brévignon, J. evarete (Cramer), and J. divaricata C. & R. Felder are restricted to South America. We find that six species of Junonia are present in the United States, one of which is new: Junonia stemosa Grishin, sp.n. (i), found in south Texas and phenotypically closest to J. nigrosuffusa W. Barnes & McDunnough (ii) in its dark appearance. In the pale nudum of the antennal club, these two species resemble J. zonalis C. & R. Felder (iii) from Florida and the Caribbean Islands. The pair of sister species, J. grisea Austin & J. Emmel (iv) and J. coenia Hübner (v), represent the classic west/east U.S.A. split. The mangrove feeder (as caterpillar), dark nudum J. neildi Brévignon (vi) enters south Texas as a new subspecies Junonia neildi varia Grishin ssp.n. characterized by more extensive hybridization with and introgression from J. coenia, and, as a consequence, more variable wing patterns compared with the nominal J. n. neildi in Florida. Furthermore, a new mangrove-feeding species from the Pacific Coast of Mexico is described as Junonia pacoma Grishin sp.n. Finally, genomic analysis suggests that J. nigrosuffusa may be a hybrid species formed by the ancestors of J. grisea and J. stemosa sp.n.
Introduction
A biologist interested in the early phases of animal speciation can hardly imagine a model system more promising than the butterflies from the genus Junonia Hübner (family Nymphalidae) (Kodandaramaiah & Wahlberg, 2007). Distributed around the globe and called buckeyes (America), pansies (Asia), commodores (Africa), or arguses (Oceania) in various parts of the world, they are abundant and accessible for studies. Junonia species are diverse in their eye-spotted wing patterns, variable geographically and seasonally, and differ in caterpillar foodplant preferences (Lalonde et al., 2018; Lalonde & Marcus, 2019b), adult behaviour and habitats. They develop from egg to adult in a few weeks (van der Burg et al., 2019), and are easy to rear and manipulate in the laboratory, including DNA injection into eggs for genetic engineering (Beaudette et al., 2014) and induced mating for hybridization, both within and between species (Hafernik, 1982; Paulsen, 1996).
Notoriously, the buckeyes (i.e. American Junonia) have been a source of taxonomic nightmare in the New World for over a century (Forbes, 1928; Turner & Parnell, 1985; Neild, 2008; Lalonde et al., 2018). Due to their recent speciation and incomplete reproductive isolation between species exacerbated by phenotypic plasticity within species (Vane-Wright & Tennent, 2011), it is challenging to assign Junonia phenotypic forms to taxa and to define species boundaries. Depending on a source, the number of Junonia species in North America varies between one and six (Forbes, 1928; Lalonde et al., 2018; Lalonde & Marcus, 2019b), and their relationship with South American buckeyes remain unclear (Brévignon & Brévignon, 2012; Lalonde & Marcus, 2019b). A definitive study that brings order to evolutionary and taxonomic treatment of U.S.A. Junonia is long overdue.
The most widely distributed U.S.A. species is the common buckeye, Junonia coenia Hübner (Fig. 1) (Scott, 1986). While this name was proposed for illustrated specimens without locality given (Hübner, 1822), phenotypic uniqueness of this species leaves no doubt about its identity. Per Hübner illustrations ([1822]: pl. [32], figs 1–4), warm-brown ground wing colour above, nearly white cream-coloured forewing bands extending almost to the inner wing margin on the body side of the eyespot, large fully ringed with a dark-brown circle eyespots by the forewing tornus and near the hindwing apex, a smaller eyespot at the hindwing tornus, well-developed hindwing orange band marginally from the eyespots, and intricately patterned contrasty hindwing underside with a row of prominent eyespots and a darker band interior to them imply that it is a summer brood of a butterfly from eastern U.S.A. found almost anywhere east of the 100th meridian (Scott, 1986). Resident in the more southern states, J. coenia disperses north during summer, reaching Canada (Layberry et al., 1998). Caterpillars are polyphagous and feed on diverse plants, with Agalinis Raf. and Castilleja Mutis ex L.f. being among their favourites (Hafernik, 1982; Scott, 1986; Lalonde et al., 2018; Lalonde & Marcus, 2019b).
Fig. 1.
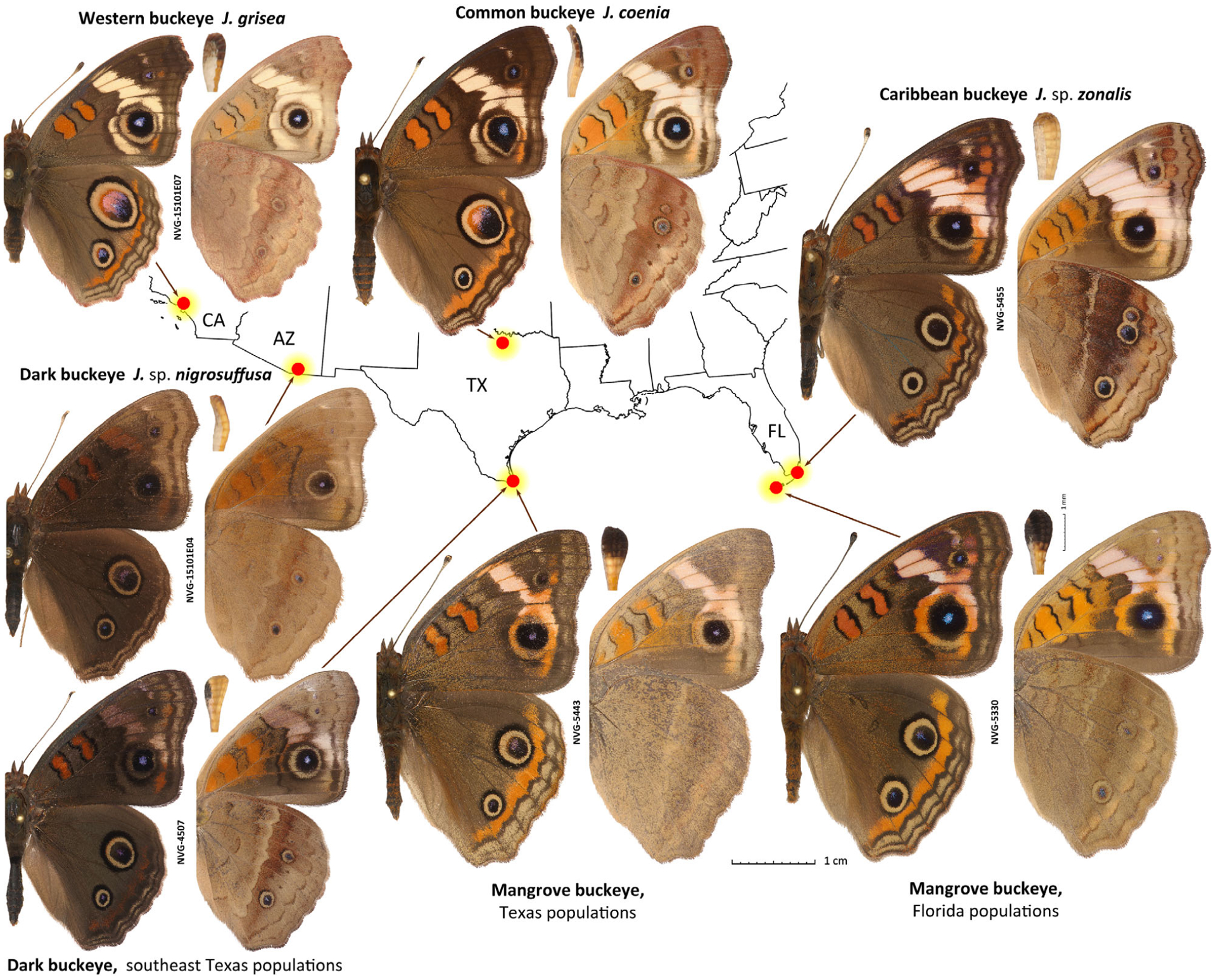
Seven major Junonia populations in the United States. Populations are denoted by their English names used in the text. Prior to this work, scientific names applied with confidence were: J. coenia (type locality not stated, judging from illustrated specimens should be eastern U.S.A.), J. grisea (type locality: CA, Los Angeles County), J. sp. nigrosuffusa in Arizona (type locality: AZ: Cochise and Pima Counties) and J. sp. zonalis (type locality: Cuba), but species versus subspecies status and species associations for nigrosuffusa and zonalis were unclear. The locality of each specimen is shown on the map outlining the states. States with specimens shown are indicated by two-letter codes. For each specimen, dorsal (left) and ventral (right) views and the antennal club in ventral view (between) are shown. All specimens are males, and data for specimens with voucher numbers are in Table S1. Junonia coenia is from the U.S.A.: Texas, Wise Co., LBJ National Grassland, ex larva, eclosed 11-Sep-1997. All images are to scale: 1 cm (specimens); 1 mm (antennal clubs). Specimen images were edited to remove imperfections such as loss of scales and small wing chips to emphasize the actual elements of wing patterns and shapes, and some images were flipped (left/right inverted). Images of specimens (except holotypes, which are unedited) shown in other figures were similarly enhanced.
Fig. 4.

Differences between males of the three Junonia species from south Texas. The three specimens are from the Gulf coast of the lower Rio Grande Valley in U.S.A.: Texas, Cameron County. Junonia coenia (NVG-5417, South Padre Island, north end of the city, east of SH100, GPS: 26.15477, −97.17263, 25-Dec-2015), dark buckeye from TX (NVG-5413, same data as for J. coenia) and mangrove buckeye from TX (NVG-5443, Port Isabel, east of Laguna Heights, north of SH100, GPS: 26.07962, −97.24556, 26-Dec-2015) are shown in left, middle and right columns, respectively (see Fig. 3 caption for explanations).
Western populations of this butterfly have greyer brown background wing colour above, with yellower (instead of with a touch of pink) postdiscal forewing band and a paler and less developed submarginal hindwing orange band, are typically less contrasty and weaker-spotted beneath, and were named Junonia coenia grisea Austin & J. Emmel (Fig. 1) (Austin & Emmel, 1998). Their caterpillars feed mostly on Plantago L., among many other plants (Shapiro & de Grassi, 2017) (Fig. 2). Originally described from California and said to be from ‘western United States and adjacent Mexico’, this subspecies was later characterized by a unique COI barcode haplotype in California present in some populations further west in Arizona (Gemmell & Marcus, 2015). These nearly 1% barcode differences imply a certain level of reproductive isolation from J. c. coenia, and it is not inconceivable that J. c. grisea could be a species-level taxon similar to Pterourus rutulus (Lucas) (western species) versus P. glaucus (Linnaeus) (eastern species) (Kunte et al., 2011). Arguments to treat J. grisea as a species have recently been put forward on the basis of extensive comparisons (Lalonde & Marcus, 2019b).
Fig. 2.

Caterpillars and pupae of Junonia grisea. All from U.S.A.: Arizona, Santa Cruz Co., Pajarito Mtns., California Gulch, 31.41856, −111.2380. (a–e) Fifth-instar caterpillar; (f–h) pupa. (a–c) and (f–k) show the same individual. Scale, 1 cm for all images but (d, e), which are magnified twice. (a–e) 4 April 2016; (f–h) 14 April 2016.
In addition to wing patterns, the colour of a scale-free portion of the antennal club beneath, termed the nudum (Evans, 1943), has been suggested to be a taxonomically important character (Neild, 2008; Calhoun, 2010). Nudum is a part of the antenna that senses smell (Yuvaraj et al., 2018), and thus the differences in nudum structure and colour may be linked to pheromone response and therefore to speciation. Easy to examine by looking through a magnifier at the tip of the antenna from below for a stripe without scales, this character can be observed in individuals of any wear, even if antennal scales are mostly rubbed off. However, the antennal club frequently shrivels when dry, and hence care needs to be taken to locate the nudum in specimens. Some Junonia species have a dark-brown nudum, almost black near the tip, while in other species the nudum is completely pale: cream, ivory or almost white. Subject to more significant colour variation in females, the colour of the nudum appears constant in males of the same species and has been used to reliably separate the two very similar species, Junonia evarete (Cramer) (pale nudum) and Junonia genoveva (Cramer) (dark nudum), in the Guianas (Neild, 2008). All J. coenia populations are characterized by the dark nudum of the antennal club, although the extent and intensity (from paler brown to black) of the dark area vary between specimens.
In southern Arizona, New Mexico, and Texas, a much darker phenotype occurs (Fig. 1) (Scott, 1986). The wings are dark-brown above, the cream band of J. coenia is replaced by a brown and poorly defined patch or not expressed at all, and the eyespots are typically smaller. In most specimens from western U.S.A., the hindwing eyespot near the apex is not much larger (1.5–2×) than the eyespot near the tornus. In contrast to the darker overall appearance, the nudum of the antennal club is pale without darkening at the tip (except in some females). The wing shape also differs from that in J. coenia: rounder and less produced at the apex. These dark-winged, pale-clubbed buckeyes were originally described from southeastern Arizona as J. coenia nigrosuffusa Barnes & McDunnough, 1916 (Barnes & McDunnough, 1916) (Fig. 3). However, the invariably pale antennal clubs of the dark buckeyes and the difference in wing shape suggest that they are not mere melanists of pale-winged, dark-clubbed coenia, but a distinct species, because melanic individuals tend to be darker in all their body parts and would not differ in wing shape. To complicate the picture further, both J. coenia and J. grisea can be dark-patterned to various degrees throughout their ranges, can have reduced eyespots, and may superficially appear very similar to nigrosuffusa. The dark, reduced spots phenotype in J. coenia may be induced by exposure of caterpillars to short days and cold temperatures (Nijhout, 1984; Dhungel & Otaki, 2013). Nevertheless, these dark J. coenia or grisea can be distinguished from true nigrosuffusa by the dark nudum near the tip of the antennal club, which is pale in nigrosuffusa. Therefore, instead of being assigned to J. coenia, in recent works, nigrosuffusa has been most frequently treated as a subspecies of J. evarete, a pale-clubbed, but not dark-winged South American buckeye (the neotype from Suriname) (Neild, 2008).
Fig. 3.
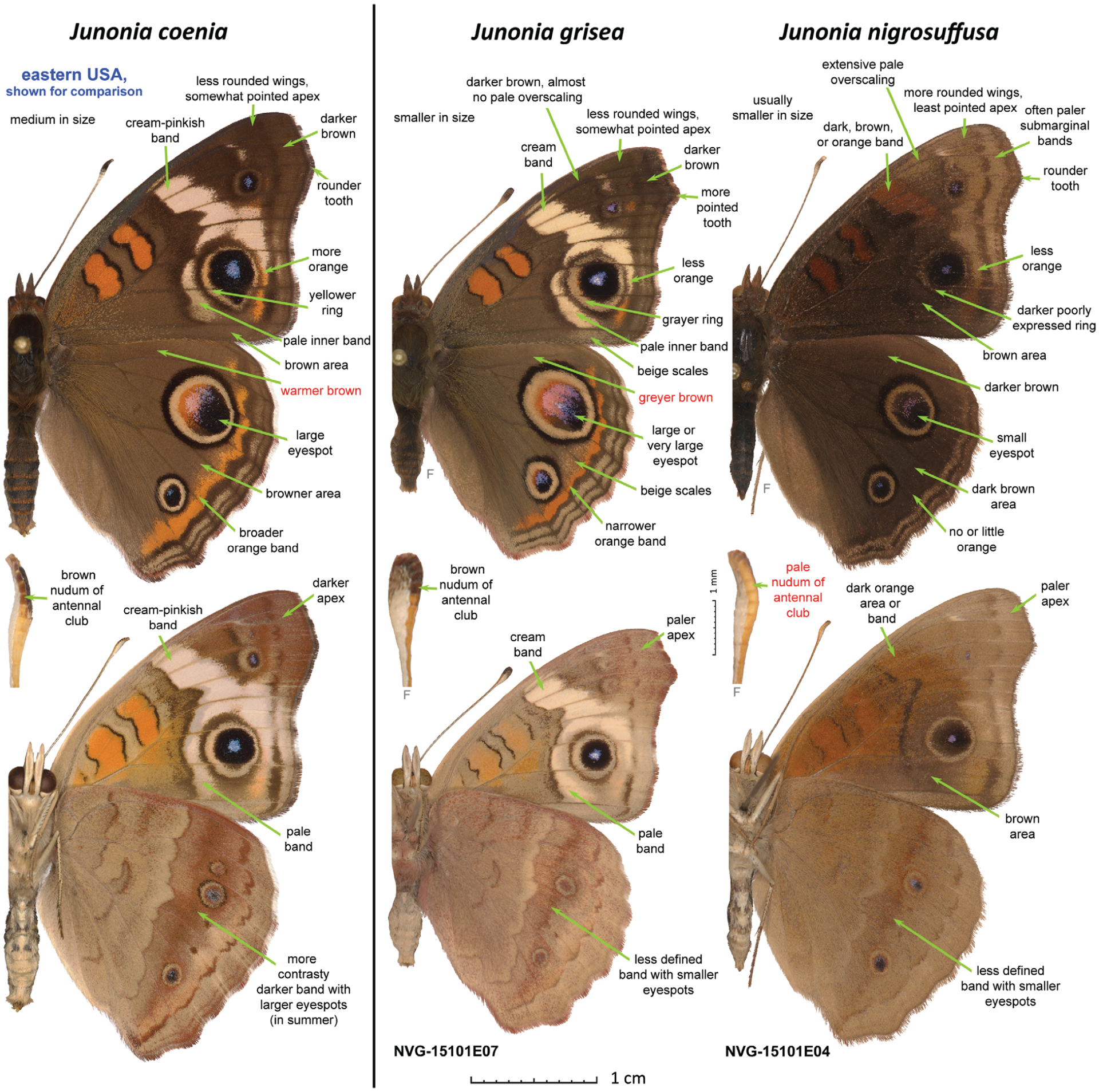
Differences between males of the two Junonia species from western U.S.A. and J. coenia. The three specimens are those shown in Fig. 1. Junonia coenia (U.S.A.: Texas, Wise Co., LBJ National Grassland, ex larva, eclosed 11-Sep-1997), J. grisea (NVG-15101E07; see Table S1 for the data), and J. nigrosuffusa (NVG-15101E04) are shown in the left, middle and right columns, respectively; dorsal and ventral views of the same specimen are shown above and below; and ventral view of the antennal club (shrivelled to various extents) is shown between them. All images are to scale: 1 cm scale (specimens); 1 mm (antennal clubs). Characters typical for males of each species are listed on the images with green arrows pointing to these features. A single most reliable character to identify each species is marked in red. ‘Nudum’ is a bare, scale-free portion of antenna beneath. ‘F’ indicates a flipped image (left/right inverted). Specimen images were edited to remove imperfections such as loss of scales and small wing chips to emphasize the actual elements of wing patterns and shapes.
In addition to southern Arizona, New Mexico and west Texas, dark-winged, pale-clubbed buckeyes phenotypically similar to nigrosuffusa occur in south Texas, generally near the Gulf Coast (Fig. 1) (Hafernik, 1982; Scott, 1986; Lalonde & Marcus, 2019b). While nigrosuffusa caterpillars mostly feed on Mimulus L. in Arizona, Texas populations are associated with Stemodia tomentosa (Mill.) Greenm. & Thomp. (Hafernik, 1982). As a result of a detailed and comprehensive study largely focused on the Gulf Coast in Texas, Hafernik (1982) concluded that dark-winged, pale-clubbed buckeyes (he used the name Junonia nigrosuffusa for them) are a species distinct from J. coenia, and their species integrity is maintained by a number of elaborate mechanisms of isolation and selection. Interestingly, Hafernik failed to find postzygotic isolation between any Junonia species he tried to cross in captivity. Both intraspecies and interspecies crosses and backcrosses of buckeyes from Texas, California and Guatemala were ‘highly fertile’ (Hafernik, 1982). Therefore, such hybridization studies may generally not be definitive to determine if certain populations are conspecific. However, the fieldwork and experiments to paint wings revealed mating preferences within each colour: white-banded males responded to white-banded females and dark males courted dark females. Additionally, dark buckeyes use S. tomentosa as the major caterpillar foodplant in Texas, but this tomentose plant is mechanically protected by dense pubescence from J. coenia: its caterpillars refuse S. tomentosa and die in the absence of other foodplants. Instead, J. coenia caterpillars feed on Agalinis, a plant that can be utilized by both species. Curiously, when the pubescence was rubbed off, J. coenia caterpillars accepted S. tomentosa as a food source. Finally, J. coenia populations shrink in numbers during winter, largely due to die-off of Agalinis plants that are annuals, while the populations of dark buckeyes expand on evergreen perennial S. tomentosa, possibly purging hybrids with reduced fitness. Hafernik (1982) concluded that despite interspecies hybridization due to the lack of postzygotic isolation, the two sympatric Junonia species maintain their integrity in south Texas at least in part due to these three factors: visually stimulated mating preference within each phenotype, separation by caterpillar foodplants, and differences in seasonal population dynamics (Hafernik, 1982).
Some Junonia populations along the southeastern coast and on the Caribbean islands have been associated with stands of black mangrove (Avicennia germinans L.), used as their caterpillar foodplant (Scott, 1986; Calhoun, 2010; Lalonde et al., 2018). These Mangrove buckeyes possess very dark antennal clubs and lack the dark ring around the forewing tornal eyespot (Fig. 1). Hence the eyespot is not separated from the pale band by a dark ring, which is particularly noticeable along the direction towards the apex. Their wings are more angular with a pointed forewing apex. In the U.S., the Mangrove buckeyes are found in southern Florida and near the Rio Grande Valley delta in Texas. They have been historically treated within the dark-clubbed South American species (J. genoveva) (Calhoun, 2010). However, two dark-clubbed mangrove-feeding Junonia taxa have recently been described – J. genoveva neildi Brévignon from Guadeloupe, later elevated to species (Brévignon, 2009), and J. litoralis Brévignon from French Guiana (Brévignon, 2009) – and it is not clear which name, if any, should refer to the U.S. populations. To complicate this picture further, recent studies suggested that both J. neildi (Florida mangrove-feeding populations) and J. litoralis (the name applied to Texas mangrove-feeding populations) occur in the U.S.A. (Lalonde & Marcus, 2019b). We found that, in south Texas, the mangrove buckeyes are fully sympatric with common buckeyes and dark buckeyes, and all three species can be found flying together in one meadow on the South Padre Island (Fig. 4).
In southern Florida (including the Florida Keys), J. coenia is sympatric with the dark-clubbed mangrove buckeyes, and with the third species characterized by a pale nudum and frequently referred to in the literature as J. evarete zonalis C. Felder & R. Felder, 1867 (Calhoun, 2010), recently elevated to J. zonalis zonalis by Brévignon & Brévignon (2012) without examination of specimens from Florida (Fig. 5). Hence, species assignment of U.S.A. specimens needs further study and it has been suggested recently that Florida populations are J. zonalis (Lalonde et al., 2018). The lectotype of zonalis is from Cuba (Neild, 2008), and we refer to these pale-clubbed butterflies as ‘Caribbean buckeyes’ (Fig. 1). In Florida, Caribbean buckeyes are associated with the blue porterweed (Stachytarpheta jamaicensis (L.) Vahl.) (Lalonde et al., 2018). Similar to dark-clubbed mangrove buckeyes, Caribbean buckeyes lack the dark ring around the forewing eyespot, but in addition to pale antennal clubs they differ in having a wider and whiter (less orange, more pinkish) forewing band with dark wedge on the inner edge extending less into it, and the vein M3 with fewer brown scales (Fig. 1) (Scott, 1986). Caribbean buckeyes became abundant in Florida in the early 1980s, but are now found only in localized populations (Lalonde et al., 2018). Early records exist from the beginning of the 20th century (Lalonde et al., 2018), but the lack of records between then and 1961 suggests that they may be periodically introduced from the Caribbean Islands, rather than being continuous residents in south Florida (Lalonde & Marcus, 2019a). Most literature sources assume that Caribbean buckeyes are conspecific with dark buckeyes from Texas and Arizona and with South American J. evarete (Calhoun, 2010); however, recent studies argue against such treatment (Lalonde et al., 2018; Lalonde & Marcus, 2019b).
Fig. 5.

Differences between males of the three Junonia species from Florida. The three specimens are from the U.S.A.: Florida, Monroe County. Junonia coenia (NVG-4879, see Table S1 for data), J. sp. zonalis (NVG-5455) and mangrove buckeye from FL (NVG-5330) are shown in left, middle and right columns, respectively (see Fig. 3 caption for explanations).
In summary, according to literature, Junonia in the United States is represented by at least five distinct phenotypes (major caterpillar foodplants are given in parentheses), two of which can be further divided into two geographic variants each (Fig. 1): (1) common buckeye J. coenia: eastern U.S.A., warmer-brown, pale-banded, dark-clubbed butterflies with ringed forewing eyespot (Agalinis); (2) western buckeye J. coenia? grisea: subspecies or species from the western U.S.A., greyer-brown, pale-banded, dark-clubbed, ringed forewing eyespot (Plantago); (3) dark buckeye J. (coenia? evarete? sp.?) nigrosuffusa: species attribution uncertain (may be a full species), southern U.S.A., dark-winged, pale-clubbed populations, ring around the forewing eyespot is present in paler individuals and is merging with the background in darker ones (Mimulus in AZ and Stemodia tomentosa in TX); (4) black mangrove buckeye J. (genoveva? neildi? litoralis?), status and species uncertain, southern Gulf Coast of Texas and Florida, larger, narrower orange bands, black nudum on the club, no ring around the forewing eyespot (Avicennia); (5) Caribbean buckeye J. (evarete? zonalis?) zonalis, species attribution unclear, south Florida, smaller, broader, white-pinkish bands, pale nudum, no ring around the forewing eyespot (Stachytarpheta).
As intermediates between these forms are frequently found, hybridization is assumed, and at one extreme all of them were thought to be overlapping populations of a single highly variable species, diverged in wing patterns and caterpillar foodplant preferences (Forbes, 1928). On the other extreme, all of these five phenotypes have been considered different species in publications (Lalonde et al., 2018; Lalonde & Marcus, 2019b). The most frequent treatment in the current literature is to divide U.S.A. Junonia into three species: common (phenotypes 1 and 2), mangrove (4), and tropical (3 and 5) buckeyes (Scott, 1986). A series of recent works on Junonia employed the analysis of a 654 bp segment of mitochondrial DNA encoding the N-terminal half of cytochrome c oxidase subunit 1, termed ‘barcode’ (due to its purported capacity to identify animal species; Hebert et al., 2003) on a large sample of specimens across the range (Brévignon & Brévignon, 2012; Pfeiler et al., 2012a, b; Borchers & Marcus, 2014; Gemmell et al., 2014; Gemmell & Marcus, 2015). Even combined with selected nuclear DNA (wingless gene and randomly amplified fingerprints) (Borchers & Marcus, 2014), these studies were not successful in pinpointing species boundaries, but revealed extensive introgression within Junonia instead. Largely basing their conclusions on phenotypic features with some contribution from DNA studies, recent works have suggested that six Junonia species are present in the U.S.A. (Lalonde et al., 2018, Lalonde & Marcus, 2019b). We reason that to address the questions of speciation and distinction between all these closely related Junonia populations, analysis of complete genomes offers the best chance for success.
Here, we rationalize available morphological and ecological data from the genomics perspective and clarify the taxonomy of Junonia from the United States by comparatively analysing their whole-genome shotgun sequences. First, we find proteins that differ the most between sympatric populations of different species. Second, we compute divergence in these proteins between different Junonia populations. Third, we treat as different species two populations that have divergences similar to that for sympatric populations of distinct species. If divergence is at the level of allopatric populations of the same species, we consider these populations to be conspecific. As a result, we conclude that the species J. genoveva, J. litoralis, J. evarete and J. divaricata C. & R. Felder are restricted to South America and delineate Junonia species present in the U.S.A.
Materials and methods
Specimens, DNA extraction, library preparation and sequencing
Specimens used in the analysis are listed in Table S1. These specimens were either collected in the field and preserved in RNAlater solution (minus wings, antennae and genitalia stored in glassine envelopes) specifically for this study, or older specimens pinned in collections were used. We used a shotgun approach to produce whole-genome sequence data. For fresh specimens stored in RNAlater, DNA was extracted from several pieces of tissue, including the head, thorax and abdomen. For pinned specimens, DNA was extracted from the abdomen (subsequently dissected to preserve genitalia and skin) or a leg using our published protocols (Li et al., 2019; Zhang et al., 2019). DNA libraries were prepared using NEBNext® Ultra™ II DNA Library Preparation Kit for Illumina® (San Diego, CA, U.S.A.), and details of the protocol were as described in our previous publications (Li et al., 2019, Zhang et al., 2019). The DNA library was sequenced for 150 bp using the Illumina X10 platform from both ends, targeting 5 Gb of data for each specimen (c. 10× genomic coverage). Sequence data generated in this project were deposited at NCBI under PRJNA596851, with Sequence Read Archive accessions SAMN13640881-SAMN13641088. Sequence alignments and trees were deposited at https://osf.io/xvume/.
Assembling protein and genomic sequences
We assembled protein-coding genes using protein sequences encoded by the published J. coenia genome (van der Burg et al., 2019) as a reference. Briefly, we split protein sequences of J. coenia into exons. We found the reads mapping to each exon from each specimen and assembled them into exons in a reference-guided way. Finally, the assembled exon sequences were concatenated to obtain protein sequence for each specimen. Details of this procedure were as previously described (Li et al., 2019, Zhang et al., 2019). These assembled sequences were used for the identification of putative speciation genes and species boundaries. For other analyses, we obtained the genomic sequence of each specimen by mapping the reads to the reference and performed single nucleotide polymorphism (SNP) calling as described in the following.
We removed the sequencing adapters and low-quality portions from sequencing reads using trimmomatic (Bolger et al., 2014), and using pear (Zhang et al., 2014) we merged read1 and read2 from the same fragment if their sequences overlapped. The resulting reads of each specimen were mapped to the reference genome using bwa (Li & Durbin, 2009). We kept only the reads that were mapped unambiguously in the correct orientation. Using the read mapping results, we computed the total sequencing depth for all recently collected specimens (after 2015) for each 100 bp window. We considered windows with too high or too low total depth to be less confident. For example, windows with high depth may cover repetitive regions and may not be suitable for phylogenetic studies, whereas windows with very low depth may be misassembled or highly variable. Therefore, we only used the windows with depth in between half of the median and twice of the median depth.
As many specimens we used were old and their DNA could have been contaminated, we developed and sequentially applied the following two protocols to clean up the alignments. Most of the contamination came from bacteria and fungi that was apparently living on the specimen, and some contamination was from other Lepidoptera species, probably from the relaxing chamber when many specimens were relaxed together. If < 50% of the reads from a sample could be mapped to the reference, we discarded this sample from analysis due to its high level of contamination (Table S2).
Protocol A. For each 30 bp sliding window applied to the alignment between reference and the reads, we clustered all the reads into groups of similar sequences using the following procedure. We ranked reads based on their sequence identity to the query from high to low. The first read initiates a cluster. Starting from the second read, a new read is compared to the first sequence of each cluster and assigned to the first cluster whose first sequence has no more than one mismatch from the current sequence. If a new read cannot be assigned to existing clusters, a new cluster is initiated with this read as the first member. For each cluster, we computed its size and the average number of mismatches to the query and we considered a cluster to be good if its size was at least half of the largest cluster size and the number of mismatches was no larger than minimal mismatches +2. If the number of good clusters was no more than 2, we marked the reads that were not in the good clusters as bad reads; otherwise, we marked all the reads as bad. All the bad reads were discarded.
Protocol B. For each read (a target), we obtained other reads that overlap with it and counted the number of other reads that were consistent and inconsistent with it. We considered a read to be consistent if it overlapped with the target read for at least 20 bp and the sequence identity in the overlapping region was > 95% (this allows one mismatch in every 20 bp from the overlapping region); otherwise, we considered a read to be inconsistent with the target read if it had at least two mismatches and the sequence identity was < 95%. If the number of consistent reads for a target was more than twice of the number of inconsistent reads, the target read was kept. Otherwise the target read was discarded.
We found the dominant nucleotide (frequency > 0.6) at each position for each sample from the sequence alignments after the cleaning procedures described and treated other positions (without mapped reads or without a dominant nucleotide) as gaps. The resulting sequences, consisting of these dominant nucleotides connected with gaps were used in the following analyses. For principal component analysis (PCA) analysis, heterozygous positions may be important, and we therefore identified up to two nucleotides per position using an in-house SNP calling script (https://github.com/QQcc/genomic_tools). Briefly, we only used positions with at least two different mapped reads and others were replaced with gaps. If a position had only one possible nucleotide, we considered this position as likely to be homozygous. Otherwise, we took the two most frequent nucleotides and decided whether it was homo- or heterozygous using Bayesian inference that uses the empirical error rates observed in the reads mapped to mitochondrial genomes (excluding D-loop region) and an expected heterozygosity level of 2%.
A similar pipeline was used to assemble the mitochondrial genomes using the mitogenome of Junonia coenia (GenBank accession: NC_028207) as reference (Teng et al., 2016). As the mitogenome is haploid, we modified protocol A to allow only one good cluster and we took the dominant nucleotide at each position for each sample to determine its sequence.
Clustering with principal component analysis and t-sne
We performed PCA using smartpca from the eigensoft package on Junonia samples excluding the outgroups (Junonia vestina C. & R. Felder and Junonia villida [Fabricius]). eigensoft is a set of population genetic tools developed in the Reich laboratory focusing on population genetics of humans (Patterson et al., 2006), and among them, smartpca is used to perform PCA on the covariance of SNP frequencies at each position between samples. Principal component analysis can partition samples into populations. By default, smartpca reports the first ten components of PCA. When there are a small number of populations, e.g. three or four, the first two components are sufficient to separate them. However, when there are more populations, more than two components are frequently needed to separate all of them. We therefore applied the popular dimension reduction and clustering tool, t-sne (van der Maaten & Hinton, 2008), to reduce the first 10 principal components to two dimensions.
eigensoft analyses SNP data based on covariation of SNP frequencies between pairs of samples. As the covariance between each pair of samples is computed on the shared positions between them, having a lot of gaps in some specimens will cause the calculation for each pair to be done on very different positions (Fig. S1). In addition, we observe that in old museum specimens, variable regions in the genome tend to degrade faster than conserved ones (Fig. S2). Therefore, regions that are missing (gaps) in old museum specimens are, on average, more divergent than the regions that are present. If we include a lot of segments that are gaps in the old museum specimens, we will underestimate their divergence from other specimens because of the shortage of more divergent regions. These two factors imply that these analyses should preferably be done on sequence alignments with low and similar amounts of gaps in each specimen.
From the SNP-calling results, we quantified the fraction of gaps in each sample. Samples with at least 70% gaps were excluded. We grouped the remaining samples into three groups (groups 1, 2, and 3 in Table S2) based on the gap ratio. Starting from the group with the highest gap ratio, we removed positions with gaps in > 25% of samples from this group. This approach allowed us to keep positions with few gaps and yet ensured that each sample contained a similar number of gaps in the remaining positions (Table S2). We further filtered the alignment to retain only the biallelic loci: positions with only two possible nucleotides from all samples. As a rare nucleotide at a position might result from sequencing errors, we required each nucleotide to be present in at least three samples.
The processed alignments were converted to input files ([input].snp, [input].geno, and [input].ind) according to the instructions online (https://github.com/chrchang/eigensoft/tree/master/CONVERTF). In addition, we specified the following parameters in a file: genotypename: [input].geno, snpname: [input].snp, indivname: [input].ind, evecoutname: [output].evec, evaloutname: [output].eval, altnormstyle: NO, numoutevec: 10, numoutlieriter: 5, numoutlierevec: 10, out-liersigmathresh: 6 qtmode: 0. Finally, we ran Eigensoft by: smartpca -p [parameter file]. The eigensoft results were further analysed using t-sne (van der Maaten & Hinton, 2008). We used the tsne module from sklearn package <https://scikit-learn.org/stable/> of Python through the following commands: tsne = TSNE(n_components = 2, verbose = 1, perplexity = 25, n_iter = 20 000); output = tsne.fit_transform(input). The parameter ‘perplexity’ needs to be adjusted according to the average size (higher perplexity for larger size) of each population. The results from both eigensoft and t-sne were visualized using matplotlib <https://matplotlib.org/> from Python.
Test for species: determining species boundaries
Our test of whether a pair of populations should be treated as different species is based on the rationale that despite extensive hybridization and gene exchange between Junonia species, genes that are important for the different phenotypes that distinguish different species should not readily flow between species. We term these ‘putative speciation genes’. We identify such genes using sympatric species: these fly together but still maintain their integrity and are phenotypically distinct.
We partition all the Junonia samples into populations based on the phenotypes and geographical locations (Table S1, column ‘group’), and in this analysis, we only used populations with at least three sequenced specimens. We identify ‘speciation genes’ from the three sympatric populations in Texas and the three in Florida. For each population, we derive the consensus sequence for each protein by taking the dominant (≥ 60%) amino acid at each position. We compare the sequence of each specimen in the population with this consensus and calculate the divergence as the fraction of positions that are different. The average divergence over all specimens in a population is used as a measurement of divergence within a population. Divergence between populations is calculated as the fraction of different positions between the consensus sequences of both populations.
For each locality (FL or TX), we compared the lowest divergence between populations with the highest divergence within each population and the ratio between these two values is used to select putative speciation genes. The cutoffs were determined by observing the distribution of the ratios computed for different proteins (details in the Results and discussion section), and proteins higher than a cutoff were considered as putative speciation genes for populations in one locality. The overlap (36 proteins) between the putative speciation genes identified from TX populations and FL populations was used as our final set of putative speciation genes to test other populations. For each pair of populations, we computed the divergence between the consensus sequences of each population for each protein in this final set of 36 putative speciation genes, and the average divergence naturally partitioned the pairs into two groups: different species and the same species with a break in between.
Phylogenetic analysis with iq-tree
We performed whole-genome alignment between Junonia reference genome and Heliconius melpomene (Davey et al., 2016) using lastz (Harris, 2007). We extracted the scaffolds that were aligned to the Z chromosome of the Heliconius genome and considered them as likely to be Z-linked scaffolds. We validated them further by calculating the sequencing depth of each scaffold in all female specimens: Z-linked scaffolds are expected to show only half of the depth compared with other scaffolds.
We selected representative specimens for each species as those with fewer gaps its sequence. We quantified the gap fraction in each specimen sequence, and grouped the specimens (excluding outgroups) into two groups based on the gap ratio. Starting from the group with the highest gap ratio, we removed positions with gaps in > 40% of samples from this group. This approach allowed us to remove positions with large fraction of gaps and yet ensured that each sequence contained a similar number of gaps in the remaining positions. We partitioned the remaining positions to Z-linked and autosome-linked. The autosome-linked positions were split into 100 partitions (172 468 positions each) and phylogenetic analysis by iq-tree with the best substitution model inferred by the program, TVM + F + R4 (Nguyen et al., 2015), was done on each partition. A consensus was derived from these trees using sumtrees.py (Sukumaran & Holder, 2010). Because there are fewer Z-linked positions (658 684 in total), instead of splitting them to 100 partitions, we randomly sampled 100 000 positions from the alignment for 100 times and applied iq-tree to each sample (model: TVM + F + R3). Again, we used sumtree.py to obtain a consensus between the 100 trees on different samples. For mitochondrial genomes, we used bootstrap to generate 100 replicates and used the substitution model TIM2 + F + R3.
Resolving the incongruence between autosome and Z chromosome tree
As the phylogenetic tree based on Z chromosome and autosomes revealed different topologies regarding the placement of J. nigrosuffusa, we probed which topology should represent the species’ history and which is likely to be a result of introgression using a test similar to the one used in the analysis of mosquito genomes (Fontaine et al., 2015). The assumption of this analysis is that if the tree based on a genomic region dates to earlier time point compared with trees based on other regions, that tree is more likely to represent the true species history and other trees are more affected by introgression between species, which makes sequences more similar across species. We split the genome into 10 kb windows and derived consensus sequences for J. nigrosuffusa, J. coenia, J. grisea, and J. vestina as outgroup in each window. We built phylogenetic trees using the four consensus sequences and rooted the tree with J. vestina. We computed the distance from the root to each leaf except the outgroup, and kept only trees where the maximal distance is no more than two times the minimal distance. We excluded the trees with very different distances from the root to leaf because those regions may have elevated evolutionary rate in some species, and the branch length will be poorly correlated with time. We classified the remaining trees into three possible topologies: (J. coenia, J. grisea), J. nigrosuffusa; (J. coenia, J. nigrosuffusa), J. grisea; and (J. nigrosuffusa, J. grisea), J. coenia. For each topology, we computed the average leaf branch length (T1) of the sister species. We compared the distribution of T1 for different topologies, and the one with the largest T1 represents more ancient event, and thus is more likely to reflect the history of these species.
Inferring the hybrid origin of Junonia nigrosuffusa
The following analyses were used to infer the hybrid origin of J. nigrosuffusa. First, we counted the number of unique dominant nucleotides in each species. We considered a nucleotide to be dominant if it was present in at least three specimens and at least 75% of all specimens, and we considered a dominant nucleotide to be unique for a species if the frequency for it in every other species was < 20%: we did not require the frequency to be 0 to account for frequent gene flow between Junonia species. Second, we computed F3 statistics (Reich et al., 2009) for each species. F3 is a test of whether a species has allele frequency in between two other species and is thus a possible hybrid species. Starting from the input to eigensoft, we selected all the biallelic loci and summarized the frequency of each nucleotide in each species. This result was used as input of ‘threepop’ program from treemix (−k 100) (Pickrell & Pritchard, 2012). For each species A, we computed the F3 values against all possible pairs of another two species B and C, and we took the lowest Z-score among all possible combinations of B and C as an indicator of whether species A is a hybrid species.
The previous two tests reviewed J. nigrosuffusa as the species with the lowest number of unique dominant nucleotide and the only one with negative F3, suggesting its hybrid origin. Therefore, we continued to assign the origin for each 1000 bp genomic window along the Z chromosome. We constrained this analysis to the Z chromosome because recombination and introgression are expected to be more frequent in autosomes, making it more difficult to decompose them. For each 1000 bp window in the Z chromosome, we obtained the consensus sequence for each species and detected the species whose consensus sequence is the most similar (sequence identity at least 0.2% higher than others) to J. nigrosuffusa. The majority of the windows were assigned to J. coenia, J. grisea or J. stemosa, and therefore we used these three species to assign each window to its most closely related species.
Identification and functional analysis of putative speciation genes in Junonia
We focused on pairs of U.S.A. Junonia species: J. coenia, J. grisea, J. stemosa, J. nigrosuffusa, J. zonalis, and J. neildi. For each pair of these species (15 pairs in total), we identified the putative speciation genes as those that are divergent between but show low polymorphism within each species. We detected such proteins using the fixation index (Fst) and the enrichment of divergent positions in a protein, as described previously (Cong & Grishin, 2018). We selected the proteins that are detected as putative speciation genes in at least four species, and obtained a total of 550 proteins. We analysed the function of these proteins using gene ontology (GO) terms and visualized the enriched GO terms by revigo (Supek et al., 2011).
Results and Discussion
Clustering Junonia populations
The main goal of this study is to delineate species boundaries in Junonia populations from the United States using genomic data. We sequenced specimens from the seven known major groups of U.S.A. populations across their ranges (Fig. 6a): (i) J. coenia from the eastern U.S.A., including specimens from south Florida and south Texas; (ii) J. grisea from the western U.S.A., from coastal California to New Mexico; (iii) J. sp. nigrosuffusa from south-central U.S.A., notably from southeast Arizona and west Texas, and from central Mexico; (iv) dark buckeye populations in south Texas; (v) J. sp. zonalis from south Florida and the Caribbean Islands; (vi) mangrove buckeyes from Florida; and (vii) mangrove buckeyes from south Texas and eastern Mexico. Furthermore, we also sequenced specimens from three other populations with caterpillars feeding or expecting to feed on black mangrove: (viii) those from the northwestern Mexico; (ix) J. neildi from the Caribbean islands; and (x) J. litoralis from French Guiana, its type locality. Finally we added three species described from South America: (xi) J. genoveva, a non-mangrove-feeding black nudum species whose name was formerly applied to North American mangrove buckeyes; (xii) J. evarete, the name that was historically used for North American population with pale nudum of antennae such as nigrosuffusa; and (xiii) J. divaricata, a species widely distributed in South America, but traditionally considered a synonymous of J. evarete before its distinction has been noticed recently (Brévignon, 2009; Brévignon & Brévignon, 2012).
Fig. 6.
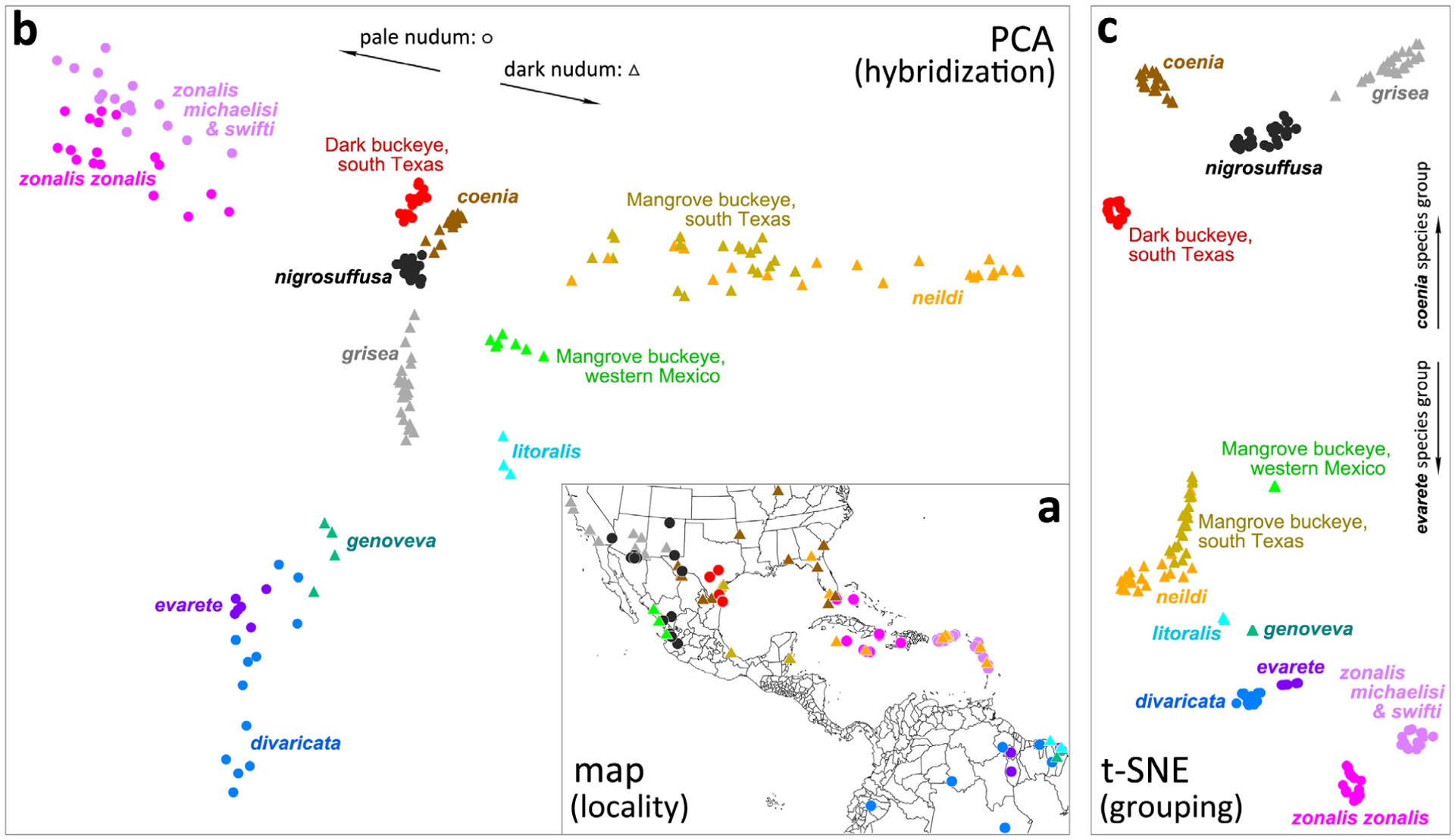
Clustering Junonia populations. (a) Map of localities of specimens used in the analysis; (b) principal component analysis (PCA) plot showing specimens as points in the plane of the second and fourth principal components; (c) t-sne clustering of specimens based on their PCA coordinates. Circles correspond to populations with pale nudum of the antennal club, and triangles denote populations with a dark nudum. Distinct taxa are coloured differently, and the colour scheme is the same for all three panels in the figure (as well as in Figs 16,17).
We obtained genomic data for 207 specimens, on average 5.1 Gb (± 2.2 Gb) of data per specimen, targeting a sequencing depth of about 10-fold. However, because we used dry specimens from museum collections not preserved for the purpose of genomic study, genomic DNA in them frequently degraded into short fragments of about 100 bp. Even if we sequenced 150 bp from both ends, the effective sequence we got from a 100 bp genomic DNA fragment would be 100 bp. Therefore, after removing the sequencing adapters with trimmomatic and merging the forward and reverse reads of the same DNA fragment, the average amount of data dropped to 2.7 Gb (± 1.5 Gb), corresponding to a sequencing depth of five-fold (Table S2). On average, c. 90% of the processed reads could be mapped to the reference genome of J. coenia. Five outlier specimens with < 50% sequences mapped were removed. We derived genomic sequences of these specimens from the results of SNP calling, and these genomes were c. 72% complete (28% gaps) on average. Seven samples with very incomplete genomes (> 70% gaps) were further removed from the analyses. Therefore, a total of 195 samples with decent data quality were used in the following analyses.
To visualize the relationships between these populations (Fig. 6a) and to probe hybridization between them, we used PCA of their genomic data, excluding J. vestina and J. villida used as outgroups (Patterson et al., 2006). The first principal component mostly separates J. grisea from the rest (Fig. S1), and its eigenvalue is more than two times of other components (Fig. S2). This is probably due to the fact that we sampled a lot more of each north American species and that J. grisea probably experienced faster evolution (reflected by longer branch in the nuclear tree in Fig. 16a) compared with other species. Similarly, the third principal component also mostly reflects the separation between J. grisea and other species in the mainland U.S.A. However, the combination of the second and fourth components displays a picture that separates more species in a way that is consistent with the clustering we obtained from all first ten principal components. The plane of the second and fourth components revealed a T-shaped arrangement of populations and is used here in the illustration (Fig. 6b).
Fig. 16.
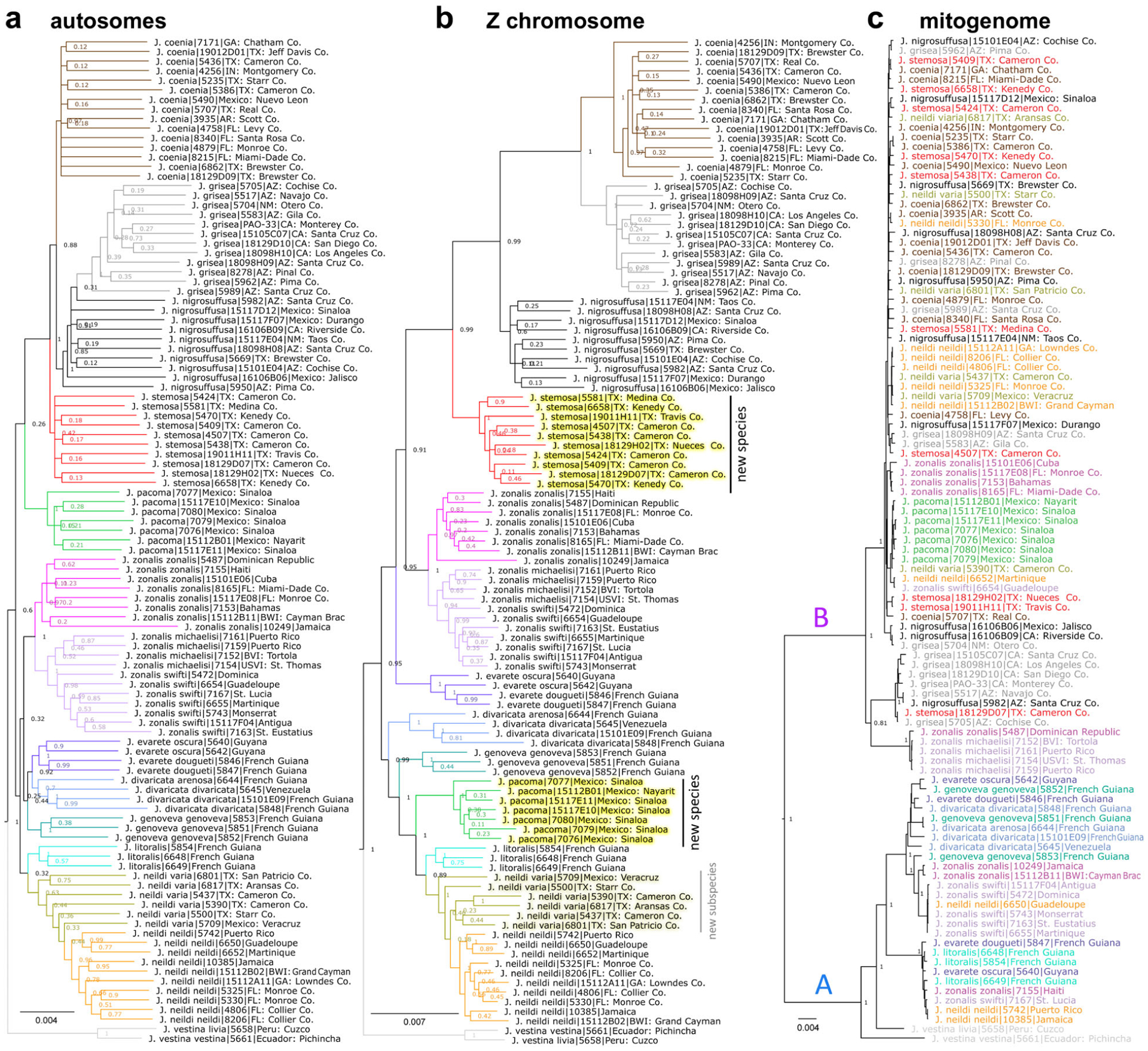
Genomic trees of Junonia. The trees are based on concatenated genomic regions from: (a) autosomes; (b) Z chromosome; (c) mitogenome. Colour scheme for the taxa is the same as in Fig. 6; the names are coloured instead of the tree branches in the mitogenome tree. New taxa are indicated in the Z chromosome tree.
The vertical dimension of this plot (fourth component) roughly separates North and South American populations (cf. Fig. 6a). The horizontal dimension (second component) shows the variation of the Caribbean populations, with white (left) and black (right) nudum species as ‘arms’ stretching far out. The sympatric populations of J. zonalis (pale nudum) and J. neildi (black nudum) from the islands are widely separated from each other on the plot, while the centre of the plot is occupied by the U.S.A. populations. The ‘streaks’ of specimens between the centres of populations on the PCA plot indicate hybridization between these populations. We see the grey triangles (J. grisea) line up towards the cloud of black dots (J. nigrosuffusa), indicating gene flow between them caused by hybridization. Similarly, J. coenia and J. nigrosuffusa clouds approach each other, and J. neildi with Texas mangrove buckeyes cluster around the trendline directed at J. coenia. The most unexpected result is the distinction of dark buckeye populations from south Texas (red circles). Despite being quite similar in appearance to J. nigrosuffusa, they form a cloud separated from the true J. nigrosuffusa in Arizona and Mexico and not suggesting ongoing hybridization. Then, mangrove buckeyes from the Pacific coast of Mexico form a distinct group, as well as South American J. litoralis. While all mangrove-feeding populations are on the right side of the plot, they form three distinct clouds of points: (i) J. neildi are together with the mangrove buckeyes from south Texas and eastern Mexico; (ii) mangrove buckeyes from western Mexico; and (iii) J. litoralis.
Although PCA analysis reveals clustering of points, it is largely aimed at detecting patterns of hybridization, so the clouds of points are not compact for hybridizing populations. We applied t-sne (van der Maaten & Hinton, 2008) analysis to the first 10 PCA components. The large number of populations make it impossible to decompose the variation between them in just two dimensions in PCA, while the machine learning-based dimension reduction tool, t-sne can cluster samples using information from more dimensions. Clustering with t-sne detects 12 distinct populations (Fig. 6c). The four clusters in the upper part of the plot form the coenia species group characterized by the dark ring around the larger eyespot on the forewing and their exclusively northern distribution in the American Junonia range (Mexico and the U.S.A.). All other populations, mostly from South America and the Caribbean Islands, lack the dark ring (Figs 1,3–5), except for the mangrove buckeyes from western Mexico, who share a number phenotypic similarities with J. nigrosuffusa, and some mangrove buckeyes from south Texas, who hybridize with J. coenia. Most notably, we see that mangrove buckeyes from the U.S.A. cluster with J. neildi, and not with J. litoralis. We see no evidence of the true J. litoralis presence in North America. Finally, J. zonalis forms two clusters: the nominotypical J. zonalis, which includes specimens from the U.S.A., and J. z. michaelisi Fruhstorfer together with J. z. swifti Brévignon, which are from Puerto Rico and the Lesser Antilles. Although the PCA and t-sne analyses reveal the groups of Junonia populations, they do not suggest which groups are biological species and which groups are better considered as subspecies.
Delineating Junonia species
Biological species are defined by a certain level of reproductive isolation. Because speciation is a continuous process, this isolation is not complete during its early stages and hybridizations abound. Nevertheless, we see the distinction of phenotypes (Figs 1,3–5) that persists over time (Hafernik, 1982) and correlates with the distinction of genotypes in our clustering analysis (Fig. 6). We want to investigate whether the continuous speciation process in Junonia populations leads to discrete species separated from each other by genomic hiatus, i.e. the discontinuity in sequence space that separates individuals in different clusters. We reason that discreteness originates from continuity through a small number of players. In this case, such players could be ‘speciation’ proteins, which show low polymorphism within each species, but diverged prominently between species. While the causality between these proteins and speciation needs special investigation, such proteins can be used as markers for species boundaries.
To find speciation proteins, we turn to sympatric populations of Junonia that form distinct genotypic clusters (and thus are somewhat isolated reproductively) and have been considered to be distinct species based on their morphological and biological distinctness (Hafernik, 1982; Scott, 1986; Lalonde et al., 2018; Lalonde & Marcus, 2019b). We take three populations in south Florida (common, Caribbean and mangrove buckeyes; Fig. 5) and three populations in south Texas (common, dark and mangrove buckeyes; Fig. 4). We assume that each of the two triplets is composed of three distinct biological species because they can be found flying in the same meadow and yet maintain their genomic integrity (Fig. 6). This genomic distinctness can only be explained by a certain level of reproductive isolation and manifests in phenotypic distinctness immediately apparent to an observer (Figs 1,3–5). Among many species concepts that have been proposed (Aldhebiani, 2018), here we have chosen to adopt what Claridge described as a ‘broadly based biological species concept’ (Claridge, 2017), which in our view is similar to the ‘genomic integrity species definition’ of Sperling (2003). That is, species are defined by a reproductive barrier, which is not absolute but porous (Mallet et al., 2016); hybrids are characterized by lower fitness and usually are eliminated from the population. These concepts do not rule out the possibility that at very low probability, some hybrids may become more adapted to certain environmental conditions and evolve into a hybrid species. Hybrid species in animals are rare, but they do not negate the biological species concept (Barrera-Guzman et al., 2018). Using this abstract concept, here we devise a concrete and objective method to delineate species from genomic data and apply it to Junonia populations.
For each triplet of sympatric Junonia species (three in Florida and three in Texas), we select specimens collected at the same or nearby locality, and measure divergence in proteins between species (three pairs of species in each triplet) and between specimens of the same species and analyse the distribution of their ratio among all proteins (Fig. 7a,b; see the Materials and methods section for details). Divergence is simply the fraction of different amino acids between two proteins. The interspecies divergence is measured as the fraction of different amino acids between the two consensus sequences of both species, while the intraspecies divergence is measured as the average fraction of different amino acids between the consensus sequence and sequences of individual specimens. Proteins that show large divergence between species but small divergence within species (and thus their large ratios) are more likely to be associated with speciation. To make this ratio criterion even more stringent, out of three species pairs in a triplet, we take the one with the smallest divergence between them, and out of three species we take the one with the largest divergence within it.
Fig. 7.
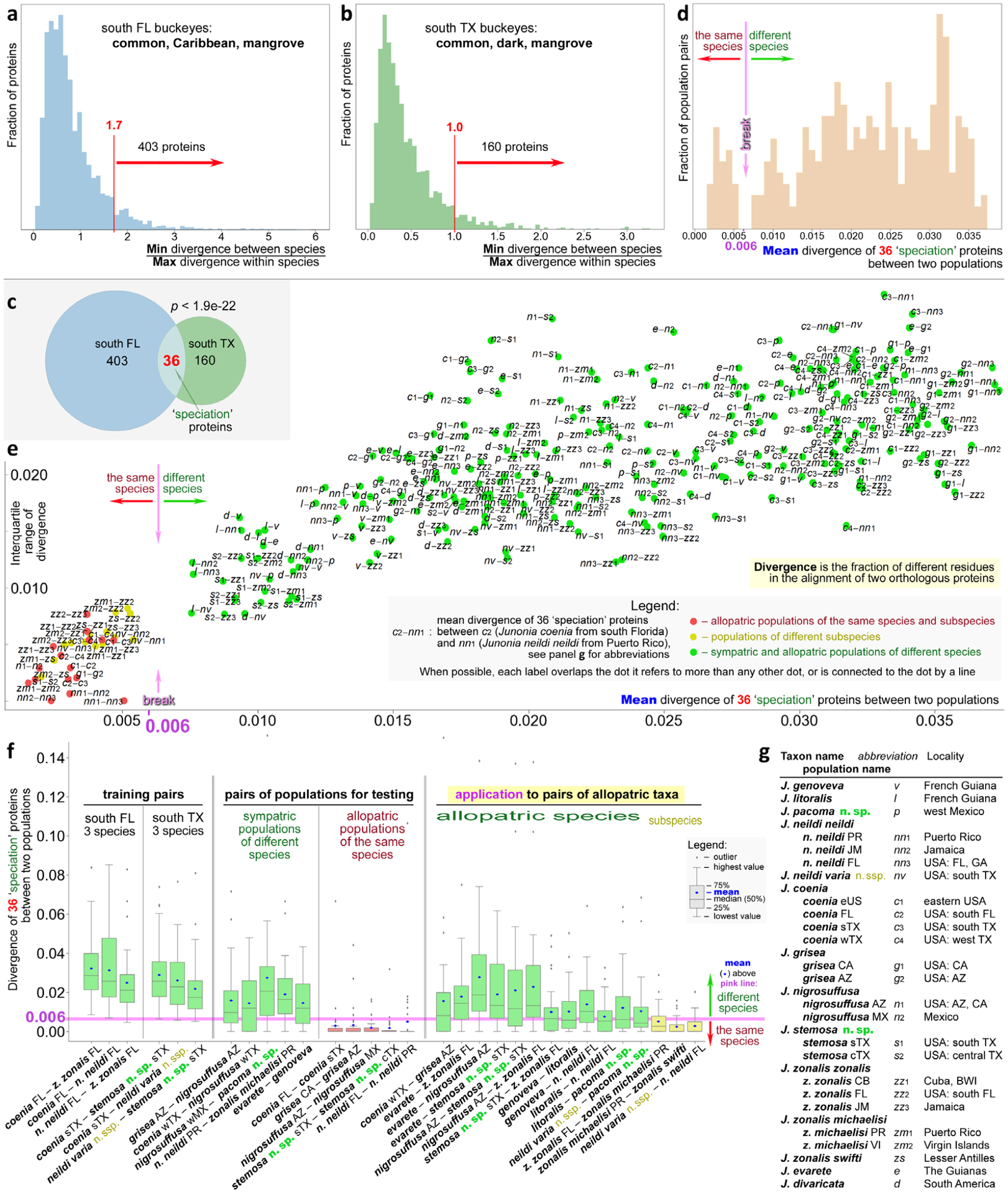
Delineating Junonia species and subspecies. Histogram of divergence in proteins among the sympatric populations of three species in south Florida (a) and south Texas (b). Cutoffs for selection of more diverging proteins are marked in red. (c) Venn diagram of the divergent proteins in three Florida and three Texas species showing a statistically significant overlap used to define 36 ‘speciation’ proteins. (d) Histogram of mean divergence of 36 ‘speciation’ proteins in all pairs of populations. The speciation cutoff around 0.006, revealed as a break in the distribution, is marked. (e) Standard deviation of divergence of 36 ‘speciation’ proteins versus the mean divergence shown for all pairs of populations listed and abbreviated in (g). (f) Boxplots for divergence of 36 ‘speciation’ proteins in selected pairs of populations. (g) Populations used in the analysis are shown in this figure with their abbreviations. In panels (d)–(f), green refers to a pair of populations of different species, yellow marks pairs of different subspecies and red indicates populations of the same species. Pink corresponds to the speciation cutoff.
The ratios of divergences show a wide distribution among proteins, both in Florida and in Texas, but tend to be larger in Florida, suggesting larger genomic divergence in Florida species than in Texas species (Fig. 7a,b). Most proteins show a low ratio. There are two possible reasons for this low ratio: (i) the lack of divergence between species, and the variation in proteins largely reflects the polymorphisms in these proteins; and (ii) there are multiple distinct alleles in these proteins, but they correlate poorly with species due to incomplete lineage sorting or introgression. However, both distributions reveal a fraction of proteins showing much higher values, and they are more likely to be relevant to speciation.
A more natural way to place a boundary is to find a more prominent minimum (probably representing a natural break between possible speciation drivers and those proteins that can more freely flow between species) along the right shoulder and take the proteins with the ratios larger than that around the minimum. We took the values 1.7 and 1 for the Florida and Texas populations, respectively (Fig. 7a,b). In all, 403 and 160 proteins passed the cutoff for the Florida and Texas populations, respectively.
To further maximize the chances of finding proteins that are more relevant to speciation rather than random outliers in the distributions, we take proteins that pass the ratio cutoff in both Florida and Texas populations, i.e. the intersection of 403 and 160 proteins, resulting in 36 proteins (Fig. 7c). Although it is possible that speciation mechanisms in Florida and Texas are not completely the same, taking the proteins in common will probably increase the proportion of true speciation genes. Notably, the number of proteins we get from the intersections of the 403 and 160 proteins, i.e. 36, is highly nonrandom, with a value of P < 1e–21. That means if we perform, for instance, 1 000 000 000 000 000 000 00 random comparisons like this, we still do not expect to find the overlap of more than 35 proteins.
We used sympatric populations to find these proteins: out of all J. coenia specimens we used only those from south Texas in the Texas comparisons and only those from south Florida to carry out Florida comparisons. Therefore, our routine for selecting ‘speciation’ proteins did not impose any restriction on the similarity (or the lack of it) between J. coenia from south Texas and J. coenia from south Florida. We would like to extrapolate the results of this analysis on other sympatric and, more importantly, allopatric populations.
We studied the divergence in these 36 ‘speciation’ proteins in various populations of Junonia. To develop more objective and general criteria for species delineation in Junonia, we computed divergence in these 36 proteins for all pairs of sympatric and allopatric populations and we sequenced and plotted a histogram of the mean (over these 36 proteins) divergence between these pairs (Fig. 7d). Again, divergence is measured as the fraction of different amino acids between the two consensus sequences of both populations. Interestingly, the resulting distribution shows a break: there are no pairs with the mean divergence from 0.005 to 0.008 (0.5–0.8% positions, on average, differ in these 36 proteins for a pair of populations). There are a number of pairs with low divergence, c. 0.3%, and a very large number of pairs with high divergence, as high as 3.5%. We expect that the high divergence corresponds to different species, and low divergence would be for the allopatric populations of the same species. And the natural cutoff at c. 0.006 may correspond to an objectively defined speciation boundary.
Other properties of the distribution of divergence of the 36 ‘speciation’ proteins may offer additional insights. They are shown in a boxplot (Fig. 7f). First, we checked the distribution of divergences for the species pairs that were used to find the 36 proteins: the triplets of sympatric species in Florida and Texas. We found that in all six distributions the mean (blue dot) was above the 0.006 cutoff and the distributions were broad: the maximal value (top of the ‘whisker’) and the interquartile range (green box) were large. Such a result is expected, because these species were used to find the proteins that show large divergence between species. Then we turned to sympatric populations of other species, not included in the selection of these 36 ‘speciation’ proteins, e.g. J. grisea and J. nigrosuffusa from southeast Arizona, J. neildi and J. zonalis from Puerto Rico, or even South American J. evarete and J. genoveva. They showed the same trends, although the means and the upper bounds of these distributions were smaller, but for these pairs, the high divergences were not biased by the selection of proteins (Fig. 7f). The mean values of divergence are above the 0.006 cutoff in all these pairs. These pairs are the positive control of the method. Next, we compared the divergence of allopatric populations of what is considered the same species, but from distant parts of the range. We took J. coenia from Florida and south Texas, J. grisea from California and Arizona, and J. nigrosuffusa from Arizona and central Mexico. The divergence of 36 ‘speciation’ proteins in these pairs is very low, with the mean value much lower than 0.006 and even the upper quartile being lower (red boxes in Fig. 7f). These pairs are the negative control of the method.
The positive and negative controls confirmed that the method was performing as expected. We applied it to allopatric population of uncertain relationships to decide whether they represent different species or would be better considered as subspecies. In these comparisons we selected the two populations that were closest to each other geographically. We reasoned that such populations may have been in contact at some point and may be more similar to each other than more geographically distant populations, making the comparisons more challenging. The logic behind our approach follows the tradition that if allopatric populations are similar in divergence to that of sympatric populations of the same species, they should be considered species (Baker & Bradley, 2006). We used the 0.006 cutoff to decide on the species status and assigned names to populations based on divergence in these 36 ‘speciation’ proteins. Box plots for several key pairs of populations are shown in Fig. 6(f) (rightmost panel). All pairs we tested passed the cutoff for speciation and these populations are better to be treated as species. We confirmed recent findings that J. coenia and J. grisea are species by comparing west Texas populations of J. coenia with Arizona populations of J. grisea. Next, we confirmed that Caribbean J. zonalis is a species distinct from South American J. evarete, and J. nigrosuffusa is a species distinct from them both. Furthermore, we found that J. genoveva, J. litoralis and J. neildi are three species, although J. litoralis and J. neildi are particularly close to each other. It should be noted that, to date, we have only sequenced specimens from near the type locality of J. litoralis. Additional specimens across the J. litoralis range would need to be analysed to learn more about variation in this species in order to better understand its relationship with J. neildi. On the other hand, we found that J. zonalis zonalis and J. zonalis michaelisi are indeed best considered as subspecies of the same species (yellow box in Fig. 7f). The same applies to J. zonalis swifti.
To investigate the divergence between Junonia populations further, we computed it for all-to-all pairs of populations and plotted the standard deviation of divergence of 36 ‘speciation’ proteins versus its mean (Fig. 7e). Abbreviations for populations are explained in Fig. 6(g). In this figure can be seen the speciation gap around the mean 0.006, the pairs that pass the cutoff for speciation (green points) and populations that probably belong to the same species (red and yellow points). Comparisons of different subspecies are shown as yellow points. Because subspecies are usually defined more arbitrarily, sometimes only by minor variation in phenotype, one would not expect a break between the points corresponding to the comparison of different subspecies (yellow) and different populations of the same subspecies (red). Nevertheless, comparisons of different subspecies are usually characterized by higher mean divergence, but some subspecies pairs, such as J. zonalis michaelisi and J zonalis swifti, show some of the lowest mean divergences, indicating closeness between these subspecies.
To study functions of the 36 ‘speciation’ proteins, we looked for their Drosophila orthologues (as the most similar proteins in their sequences) in FlyBase (Thurmond et al., 2019). The orthologues were found for 22 of them, and eight are functionally related to developmental processes, especially morphogenesis (Table S3). Therefore, divergence in these proteins may result in phenotypic differences between different species, contributing to the observed phenotypic separation in sympatric species. One of the proteins (JC_0012055-RA), orthologous to a Drosophila gene (CG9858), is involved in male mating behaviour and another protein (JC_0014643-RA) is a putative steroid hormone receptor. These proteins may be behind the differences in mating behaviour and selection of mates, contributing to prezygotic isolation.
Notably, 24 of the 36 ‘speciation’ proteins map to the Z chromosome in Heliconius (Martin et al., 2013). A reduced level of introgression in sex chromosomes has been reported for humans (Sankararaman et al., 2014; Wolf & Akey, 2018), monkeys (Cortes-Ortiz et al., 2019), Drosophila (Meiklejohn et al., 2018) and Heliconius (Martin et al., 2013). This lower introgression makes sex chromosomes better markers for speciation because more frequent autosomal introgression will mix the gene pool and reduce the distinction between species. In addition to the selected 36 ‘speciation’ proteins, we carried out the same analyses on all Z-linked proteins, and the results show a similar trend (Fig. S3): pairs of different species show larger divergence in Z-linked proteins than conspecific populations or subspecies, allowing us to delineate species based on the divergence in Z chromosome between populations. By contrast, this analysis repeated on autosomal proteins (Fig. S4) does not separate different species and conspecific populations. This result suggests that discontinuity between species may originate from the apparent genomic continuity caused by interspecific hybridization through a small number of key proteins, mostly encoded by the Z chromosome. This observation is consistent with the long-recognized importance of sex chromosomes in speciation (Coyne, 1992, 2018).
We further analysed the functions of Z-linked proteins using GO terms (Table S4). Z-linked gene products are enriched in both molecules that may be related to mate recognition and factors governing morphogenesis (GO:0035317, imaginal disc-derived wing hair organization; GO:0045314, regulation of compound eye photoreceptor development). For example, Z-linked genes are enriched in ionotropic glutamate receptors (GO:0008328), which are chemosensory molecules (Rytz et al., 2013), possibly involved in sensing of pheromones (Koh et al., 2014). In addition, the Z chromosome encodes disproportionally more proteins related to sperm (GO:0035045) and egg formation (GO:0048601), and fertilization (GO:0009566). Introgression of such genes may impair the fertility in the hybrids due to Dobzhansky–Muller hybrid incompatibilities between molecules involved in sperm/egg production from different species (Orr, 1996). Taken together, the Z chromosome’s resistance to introgression may be a crucial factor for maintaining the genetic integrity of different species when they meet and hybridize.
Description of new Junonia taxa
The most significant finding of our work that follows from this analysis is that the dark buckeyes from south Texas are a new species distinct from J. nigrosuffusa that is named here as:
Junonia stemosa Grishin sp.n.
http://zoobank.org/urn:lsid:zoobank.org:act:EC4E5167-C23E-4BD8-B23E-261282DDA317.
Fig. 8.

Junonia stemosa sp.n. compared with Junonia nigrosuffusa. The holotype is shown in full expanse with its labels at two-thirds of the scale. Dorsal and ventral views are on the left and right, respectively; species name and voucher number are between the views; general locality is by the antenna; ‘F’ indicates flipped image (left/right-inverted). Holotype (HT), syntype (ST) and paratypes (PT) are labelled. See Table S1 for specimen data by their voucher numbers; J. stemosa without the number is in line 165.
Fig. 11.

Caterpillars and pupae of Junonia stemosa. All from U.S.A.: Texas, Cameron Co., South Padre Island, N end of the city, E of SH100, GPS: 26.15501, −97.17254. (a) Fourth instar; (b–h) fifth-instar caterpillars; (i–k) pupa. (c), (e), and (f) show the same individual; (b) and (d) show the same individual; (g) and (h) are the same individual. Scale: 1 cm for all images but (f–h), which are magnified twice the scale. (a) 31-Dec-2015; (b, d, g, h) 5-Jan-2016; (c, e, f) 29-Dec-2016; (i–k) 18-Jan-2016.
Diagnosis.
This species is most similar to J. nigrosuffusa in having white nudum of antennal club, less angular wings, and darker appearance. Females are easier to distinguish from J. nigrosuffusa than males by their brighter, orange-coloured forewing discal bands and typically larger eyespots on hindwing. Most males differ from J. nigrosuffusa by the darker (rather than paler) area near the apex of forewing, more extensive orange scaling in postdiscal forewing band and submarginal hindwing band. Definitive determination is possible by means of DNA characters: JC_0012618-RA.e5:G142T, JC_0010165-RA.e3:T141A, JC_0010158-RA.e1:A162G, JC_0010158-RA.e1:A135G, JC_0011457-RA.e11:G459A, JC_0005817-RA.e3:T159A, JC_0011476-RA.e18:T525A, JC_0012618-RA.e5:A267T, JC_0012618-RA.e5:A295T, JC_0012095-RA.e1:A60T. See Supplementary file S1 for the DNA sequences of exon with these characters.
Type locality.
U.S.A.: Texas, Cameron County, South Padre Island, N end of the city, E of SH100, GPS: 26.15501, −97.17254.
Distribution and phenology.
The records exist from central and south Texas: from Austin to Houston areas and south to the Rio Grande Valley. The species is most abundant near the Gulf coast, with the westernmost record being from Medina County (west of San Antonio). It is replaced with J. nigrosuffusa in western Texas (Brewster & Jeff Davis Counties) and westwards. In south Texas, the species flies throughout the year, usually building up in numbers during late autumn and winter. The species becomes scarce towards northern limits of the range.
Etymology.
This species is named for its commonly used caterpillar foodplant, Stemodia tomentosa. The name is a fusion of the plan genus and species names: stem[odia toment]osa. The name is an adjective.
Type material.
Holotype male (Fig. 8, shown full expanse, with its labels), to be deposited at the National Museum of Natural History, Smithsonian Institution, Washington, DC, U.S.A. (USNM) upon publication, with the following labels: white printed || U.S.A.: TEXAS: Cameron Co., | South Padre Island, N end | of the city, E of SH100, elev. | 2 m, 26.15501°, −97.17254° | larva collected 26 Dec 2015 | on Stemodia tomentosa, adult | ecl. 14 Jan 2016 Grishin N.V. ||; white printed || DNA sample ID: | NVG-5578 | c/o Nick V. Grishin ||; red printed || HOLOTYPE | Junonia stemosa | Grishin ||. 100 paratypes: 63 males and 37 females, all from U.S.A.: Texas, the following counties: Travis, Comal, Bexar, Medina, Aransas, Neuces, Kleberg, Kenedy, Cameron, Hidalgo; see Table S1 for their data (‘PT’ in the column ‘Type status’).
Further notes on identification and phenotypic variation.
Due to extensive individual and seasonal variation, we failed to find a single very reliable wing pattern character to distinguish the two dark buckeyes, J. nigrosuffusa (western species) and J. stemosa sp.n. (eastern species). Wing pattern differences are statistical and exceptions exist. As stated in the diagnosis, the most noticeable difference in males (and many females) is paler apical third of forewing dorsal compared with basal third in J. nigrosuffusa. While J. stemosa frequently has a somewhat paler apex and paler submarginal bands, the area just distal of the postdiscal paler band (basad of the apical eyespots) is typically not much paler than the ground colour near the base of forewing. Thus in J. stemosa, discal darker-brown band stands out more prominently from the ground brown colour, and the postdiscal band is typically paler than the area distad. In J. nigrosuffusa, the discal darker-brown band is not prominently darker than the ground brown colour basad of the band, and the wing apex is usually heavily overscaled with beige scales, making it approximately the same colour (or paler) as the postdiscal pale band. Similarly, the paler area inward of the large forewing eyespot, between the eyespot and the dark-brown discal band, is prominently paler than the brown ground colour basad of the dark-brown band in J. nigrosuffusa, but is the same colour or only slightly paler in J. stemosa. The J. nigrosuffusa specimen shown in Fig. 8 (NVG-15101E08) is an exception as it lacks the paler inner area altogether; however, its forewing apex is still paler than the ground colour.
Junonia stemosa has more orange scales than J. nigrosuffusa. For instance, in most males, the pale postdiscal band is overscaled with orange, or may be completely orange (Fig. 8, NVG-5425). In J. nigrosuffusa, the band is usually overscaled in beige and purple, and only occasionally with orange. Males of J. stemosa usually have a better developed submarginal orange band on hindwing dorsal, typically completely absent in J. nigrosuffusa, and tornus of forewing has better developed submarginal orange than J. nigrosuffusa, above and beneath. Moreover, the orange cell bars are usually darker in J. nigrosuffusa than in J. stemosa. Specimens in Figs 8 and 9 mostly agree with the last three trends.
Fig. 9.

Wing pattern variation in Junonia stemosa sp.n. All specimens are paratypes and are reared from caterpillars collected on Stemodia tomentosa in U.S.A.: Texas, Cameron Co., South Padre Island, N end of the city, E of SH100, GPS: 26.15501, −97.17254 on 26-Dec-2015. Dorsal and ventral views are on the left and right, respectively; voucher number is between the views; ‘F’ indicates flipped image (left/right-inverted). See Table S1 for specimen data by their voucher numbers.
In females, the easiest to observe difference is the coloration of paler postdiscal bands. In J. nigrosuffusa, the bands are less coloured, are greyer or semi-white and tinted purple rather than orange, and the orange is darker where developed. In J. stemosa, the bands are usually wider and tinted with orange or are pure and brighter orange (see Fig. 9, NVG-5580, for an exception), and the hindwing submarginal orange band is wider, especially in the middle, J. coenia-like. This band is vestigial or interrupted in the middle, J. grisea-like, but darker orange in J. nigrosuffusa. Veins crossing the pale postdiscal forewing band are with heavier dark overscaling in J. nigrosuffusa, and therefore they stand out more from the pale-grey colour of the band. The apex distad of the band is overscaled with beige and appears paler than the ground colour by the wing bases in most J. nigrosuffusa females, but is darker than the base ground colour just distad of the pale postdiscal band in J. stemosa. In both sexes, but in females in particular, the apical eyespot on hindwing is frequently very large, J. coenia-like in J. stemosa, but is mostly small in J. nigrosuffusa. Finally, when making determinations, it is important to mind the exceptions from every wing pattern general trend.
Suggested English name.
Twintip buckeye, for the English name of the major foodplant of its caterpillars, grey-woolly twintip (Stemodia tomentosa).
Junonia stemosa sp.n. is distinct from others according to the divergence in 36 ‘speciation’ proteins (Fig. 7f). In particular, it shows mean divergence more than 0.006 from other species with white nudum of antennae: J. evarete from South America, J. nigrosuffusa from Arizona, and J. zonalis from Florida.
Next, divergence comparisons among mangrove-feeding allopatric populations reveal that Junonia from the Pacific coast of northern Mexico is a distinctive new species that is named here as:
Junonia pacoma Grishin sp.n.
http://zoobank.org/urn:lsid:zoobank.org:act:5C69F7E6-D083-44C3-8EDF-D2F1D91B5ABE.
(Fig. 12)
Fig. 12.

Junonia pacoma sp.n. The holotype is shown in full expanse with its labels at two-thirds of the scale; other specimens are paratypes. Dorsal and ventral views are on the left and right, respectively; species name and voucher number are between the views; general locality is by the antenna; ‘F’ indicates flipped image (left/right-inverted). Holotype (HT) and paratypes (PT) are labelled. See Table S1 for specimen data by their voucher numbers.
Diagnosis.
This species is similar to J. grisea in dark, but not black, nudum of antennae and typically cream-coloured forewing bands, but differs by these bands being narrower and more prominently crossed by dark veins. Differs from J. neildi by narrower and less orange forewing bands, more prominent dark rings around the large forewing eyespot, and paler (not as black) nudum. Definitive determination is possible by means of DNA characters: JC_0012597-RA.e3:C244A, JC_0012597-RA.e3:C245A, JC_0012597-RA.e3:C255T, JC_0006987-RA.e12:A437C, JC_0006987-RA.e12:A1035G, JC_0008910-RA.e3:G724A, JC_0011448-RA.e1:C714A, JC_0008910-RA.e3:C720T, JC_0011448-RA.e1:T717C, JC_0007955-RA.e4:A3000G. See Supplementary file S1 for the DNA sequences of exon with these characters.
Type locality.
Mexico: Sonora, Isla de la Piedra.
Distribution and phenology.
The records are known from the coastal areas of Mexican states of Sonora and Nayarit from August, September, November, December and January.
Etymology.
This species is named for its distribution along the Pa[cific] co[ast of ] M[exico] + a. The name is an adjective.
Type material.
Holotype male (Fig. 12, shown full expanse, with its labels), deposited at the C.P. Gillette Museum of Arthropod Diversity, Department of Bioagricultural Sciences, Colorado State University, Fort Collins, CO, U.S.A. (CSUC), with the following labels: white printed || MEX: Sinaloa | Isla de Piedra | XI-24-2003 | P. A. Opler ||; white printed and handwritten || MEXC11 | Junonia litoralis × grisea | Brévignon, 2009 | det. J.M.Marcus 2015 ||; white printed || DNA sample ID: | NVG-15117E10 | c/o Nick V. Grishin ||; red printed || HOLOTYPE ♂ | Junonia pacoma | Grishin ||. 10 paratypes: seven males and three females, all from Mexico: Sinaloa and Nayarit; see Table S1 for their data (‘PT’ in the column ‘Type status’).
Suggested English name.
Pacific mangrove buckeye, to reflect its distribution and the major caterpillar foodplant, black mangrove.
Junonia pacoma sp.n. is distinct from others according to the divergence in 36 ‘speciation’ proteins (Fig. 7f). In particular, it shows mean divergence more than 0.006 from other mangrove feeding populations, J. litoralis from South America and J. neildi from Florida.
Finally, we find that mangrove-feeding populations in south Texas and eastern Mexico are closely related to Caribbean species J. neildi and display mean divergence < 0.006 from it. Nevertheless, divergence between these populations and the nominal J. neildi is larger than that between different populations of geographically uniform species such as J. coenia, J. grisea and J. nigrosuffusa and is more similar to that between different subspecies. Thus, these western Gulf Coast populations of J. neildi get a new name:
Junonia neildi varia Grishin ssp.n.
http://zoobank.org/urn:lsid:zoobank.org:act:77CB6888-C500-440C-B7A2-59C8E383926A.
Fig. 10.

Habitat and caterpillar foodplants of Junonia. U.S.A.: Texas: (a–c) Grey-woolly twintip (Stemodia tomentosa), foodplant of J. stemosa sp.n., Cameron Co., South Padre Island, N end of the city, E of SH100, 26.15501, −97.17254, 26-Dec-2015. (d–m) Black mangrove (Avicennia germinans), foodplant of J. neildi varia sp.n.; (d–j) Cameron Co., Port Isabel, E of Laguna Heights, N of SH100, 26.07962, −97.24556, 25-Dec-2015; (k–m) San Patricio Co., Portland, Indian Point Park, 27.85351, −97.35338, 21-Aug-2016.
Fig. 13.

Junonia neildi varia ssp.n. The holotype is shown in full expanse with its labels at two-thirds of the scale; other specimens are paratypes. Dorsal and ventral views are on the left and right, respectively; species name and voucher number are between the views; general locality is by the antenna; ‘F’ indicates flipped image (left/right-inverted). Holotype (HT) and paratypes (PT) are labelled. See Table S1 for specimen data by their voucher numbers.
Fig. 14.

Wing pattern variation in Junonia neildi varia ssp.n. All specimens are paratypes and are reared from caterpillars collected on Avicennia germinans in U.S.A.: Texas, San Patricio Co., Portland, Indian Point Park, 27.85351, −97.35338 on 21-Aug-2016, except NVG-6805, which was collected as an adult at the same locality. Dorsal and ventral views are on the left and right, respectively; voucher number is between the views; ‘F’ indicates flipped image (left/right-inverted).. See Table S1 for specimen data by their voucher numbers.
Diagnosis.
This subspecies is similar to the nominal J. neildi and differs in rounder wings, more developed traces of the dark ring around the larger forewing eyespot, paler bands on the forewing, and a more contrasty and variegated pattern of hindwing below. It is characterized by larger variation in size, colours and patterns compared with the nominotypical subspecies. These phenotypic features, including higher variability, are likely a result of ongoing hybridization with J. coenia, hybridization more common in Texas than in Florida. More definitive determination is possible by means of DNA characters. Individuals of this subspecies do not possess most of the following characters defining J. coenia: JC_0012077-RA.e1:T983C, JC_0010137-RA.e4:C615T, JC_0012055-RA.e1:T63C, JC_0010158-RA.e1:T117G, JC_0010146-RA.e11:T126A, JC_0012466-RA.e1:A1374T, JC_0012095-RA.e1:C387A, JC_0010167-RA.e1:T1069C, JC_0010133-RA.e7:G996C, JC_0012968-RA.e3:A1296C, and do not have most of the following characters defining J. neildi neildi: JC_0012957-RA.e10:C54T, JC_0010023-RA.e3:G139C, JC_0015968-RA.e13:T78A, JC_0017531-RA.e6:G174A, JC_0002947-RA.e1:C78T, JC_0005568-RA.e1:G111A, JC_0012776-RA.e9:C67T, JC_0018108-RA.e3:G741A, JC_0014627-RA.e2:T100A, JC_0002853-RA.e132:A135T. See Supplementary file S1 for the DNA sequences of exon with these characters.
Type locality.
U.S.A.: Texas, Aransas County, Rockport, Goose Island State Park, Goose Island, GPS: 28.12535, −96.98178.
Distribution and phenology.
This subspecies inhabits the western Gulf coast in south Texas (Aransas County and southwards) and Mexico.
Etymology.
This subspecies is named for it is variable appearance due to extensive introgression from and ongoing hybridization with J. coenia, more variegated ventral wing pattern compared with the nominal subspecies, and more variegated phenotype of the caterpillars with extensive yellow dorsal patterns. The name is an adjective.
Type material.
Holotype male (Fig. 13, shown full expanse, with its labels), to be deposited at the National Museum of Natural History, Smithsonian Institution, Washington, DC, U.S.A. (USNM) upon publication, with the following labels: white printed || U.S.A.: TEXAS: Aransas Co., | Rockport, Goose Island State | Park, Goose Island, elev. 2 m | 28.12535°, −96.98178° | 21 Aug 2016 Grishin N.V. ||; white printed || DNA sample ID: | NVG-6810 | c/o Nick V. Grishin ||; red printed || HOLOTYPE | Junonia neildi | varia Grishin ||. 30 paratypes: 21 males and nine females, all from U.S.A.: Texas, the following counties: Aransas, San Patricio, Cameron, Starr; see Table S1 for their data (‘PT’ in the column ‘Type status’). Specimens NVG-5709 (Mexico: Veracruz) and NVG-15117F02 (Belize) were excluded from the type series because their classification needs further study.
In summary, we selected 36 ‘speciation’ proteins that we use to objectively define Junonia species according to a natural cutoff corresponding to a prominent and the only break in the distribution of divergences among all pairs of Junonia populations we analyse. According to this cutoff, there are six Junonia species in the United states: J. coenia, grisea, nigrosuffusa, stemosa sp.n. (dark buckeyes from south Texas), zonalis (Caribbean buckeyes) and neildi (mangrove buckeyes). We see that J. litoralis is a species restricted to South America. Additionally, mangrove-feeding populations from northwestern Mexico are described as J. pacoma sp.n., and western Gulf coast populations of J. neildi are described as J. n. varia ssp.n.
Phylogenomics and the origins of Junonia species
We constructed phylogenetic trees from three genomic regions: autosomes, Z chromosome and mitochondrial genome (Fig. 16a–c). Mitogenomes that include COI barcodes have been used extensively in the studies of Junonia populations (Pfeiler et al., 2012a, 2012b; Borchers & Marcus, 2014; Gemmell et al., 2014; Gemmell & Marcus, 2015; Lalonde et al., 2018; Lalonde & Marcus, 2019b). Here, we introduce phylogenetic analysis based on the Z chromosome. Males of butterflies have two copies of Z, while females have Z and W. In Z, recombination is reduced to half of that in autosomes. Recombination following interspecies hybridization (which are frequent in Junonia species) generates a plethora of hybrid alleles that challenge the traditional phylogenetic reconstruction method. Therefore, we expect that the lower recombination rate may make Z chromosomes more suitable for phylogeny inference. A similar conclusion has been derived from other heavily introgressed species complex (Fontaine et al., 2015).
The three trees reveal different topologies. The mitogenome tree (Fig. 16c) recapitulates previous findings based on COI barcodes. Similar to the barcodes, it reveals two major haplotypes, A and B. The haplotype A is present in specimens from South America and the Caribbean and also includes the two J. vestina specimens used as the outgroups. The haplotype B is largely continental North American with some Caribbean specimens mixed in. While the COI barcodes have been used in Junonia classification, even nearly complete mitogenomes do not discriminate species. Names of different species are shown in different colours (Fig. 16c) and they are extensively intermixed. Interestingly, most J. grisea specimens possess a somewhat distinct mitochondrial haplotype; however, some grisea have typical coenia haplotype. Nevertheless, due to extensive hybridization and shallow mitochondrial DNA divergence, mitogenomes offer little help in Junonia classification, confirming previous observations based on a larger set of specimens (Peters & Marcus, 2017).
The tree constructed from autosomes in nuclear genome reveals populations and species (Fig. 16a). The coenia groups species (J. coenia, grisea, nigrosuffusa and stemosa sp.n.) form a monophyletic group. Notably, J. stemosa sp.n. is sister to the other three species and is not grouped with J. nigrosuffusa, despite extensive wing pattern similarities. Likewise, phenotypically more similar J. coenia and J. grisea are not sisters. Instead, J. grisea forms a clade with sympatric J. nigrosuffusa, but J. nigrosuffusa is paraphyletic. The four species are monophyletic in the Z chromosome tree, and the Z tree is incongruent with the autosome tree (Fig. 16a,b). While J. stemosa sp.n. is still sister to the other three species, the major difference between the autosome tree and the Z tree is in the position of J. nigrosuffusa. Junonia coenia and J. grisea are close sisters in the Z tree, which exhibits topology (cg)n, where c, g, and n stand for J. coenia, grisea and nigrosuffusa, respectively. The autosomal topology is (gn)c.
To determine which topology represents the species tree and which is confounded by incomplete lineage sorting, introgression and hybridization, we resort to the method based on the level of divergence in 10 kb genomic regions grouped by tree topologies they produce (Fontaine et al., 2015) (Fig. 17a). The rationale is that extensive introgression in a certain genomic region will make two species similar to each other in that region, and thus the consensus sequences for different species will be very similar to each other. By contrast, the regions resistant to introgression will show larger interspecific divergence, and reveal the true history of species.
Fig. 17.
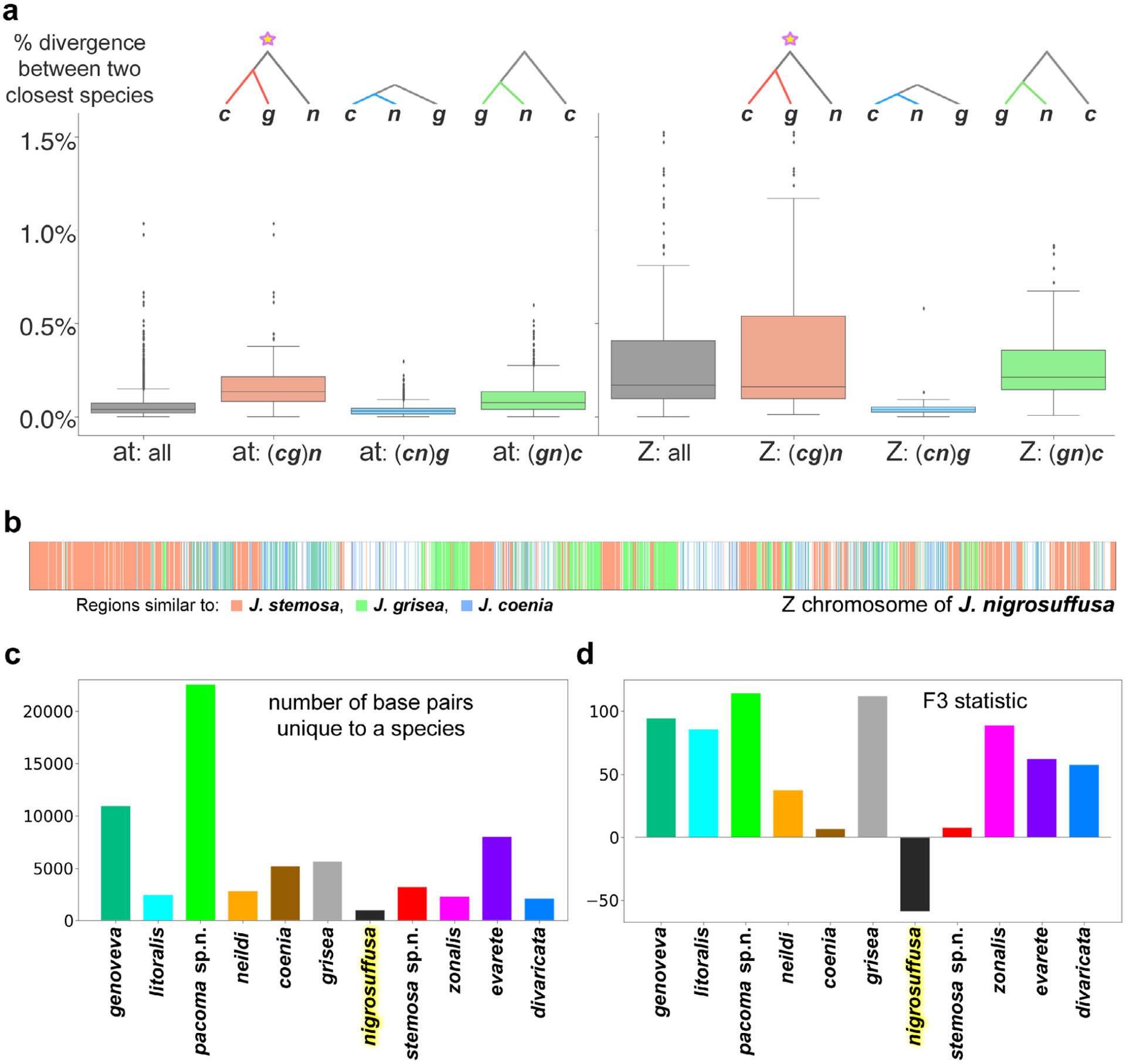
Species tree and the hybrid origin of Junonia nirgosuffusa. (a) Boxplots of sequence divergence for DNA segments by the tree topology they produce: ‘at’, autosomes; ‘Z’, Z chromosome. The species abbreviations are: c for Junonia coenia, g for Junonia grisea, and n for J nigrosuffusa. (b) Z chromosome of J. nigrosuffusa is coloured by the origin of its regions: red from J. stemosa, green from J. grisea and blue from J. coenia. (c) Number of positions occupied by a base pair unique to each species (not present in any other species). (d) F3 statistic computed for all species analysed in this work. Colours of species in (c) and (d) correspond to Fig. 6.
We perform the analysis separately for autosomes and the Z chromosome. We divide the genomic alignment into 10 kb windows and obtain the consensus sequences for each of the species, J. coenia (c), J. grisea (g), J. nigrosuffusa (n), and J. vestina (outgroup), and build phylogenetic tree for each window. We classify the trees into the three possible topologies (grouping c with g, c with n and g with n) and compute divergences between the two closest species for each topology. The boxplots of these distances for each topology are shown in Fig. 17(a). Both in autosomes and Z chromosome, trees with the topology (cg)n show the largest divergence, and much larger than the (cn)g topology. As previously suggested (Fontaine et al., 2015), the largest average divergence indicates the most ancient split between the sister taxa in this topology, implying that it corresponds to the species tree. Other topologies, with smaller average divergence and thus more recent split between the sister taxa, indicate evolutionary events after speciation and are expected to correspond to introgression and hybridization. Interestingly, while the concatenated autosome tree results in the topology (gn)c, the divergence analysis of autosome segments suggests that (cg)n had the most ancient split and thus corresponds to the species tree, the same result as in Z chromosome regions. This (cg)n topology is the one revealed by the concatenated Z tree. Superiority of the Z chromosome to reveal the species tree is probably due to its lower rate of recombination and the presence of many genes that control phenotype and mating, making these genes more resistant to introgression. However, the topology of the autosome tree that groups the sympatric J. grisea and J. nigrosuffusa together ((gn)c) is likely to be a result of hybridization and introgression between these two species, as suggested by smaller average divergence (and thus more recent age) of autosome (and Z chromosome) segments that follow this topology.
Despite the meaningful results from the analysis of divergence, it is still puzzling why very similar species J. nigrosuffusa and J. stemosa sp.n. are not sisters in the trees we obtained. We observe that in both autosome and Z trees, J. nigrosuffusa occupies a position intermediate between J. grisea and J. stemosa sp.n. To offer further insights, we assign each 1 kb region in the Z chromosome (Fig. 17b) of J. nigrosuffusa to the most similar species in that region. The majority (> 95%) of such 1 kb windows can be assigned to three species: J. stemosa sp.n. (red), J. grisea (green) and J. coenia (blue). Those regions that cannot be unambiguously assigned to either of these species, mostly because the regions are similar in all these three species, are left white in Fig. 17(b). We see that the Z chromosome of J. nigrosuffusa is a mosaic of J. stemosa sp.n. and J. grisea, suggesting the possibility that J. nigrosuffusa is a hybrid species formed from the ancestors of J. stemosa sp.n. and J. grisea.
To further test this hypothesis, we computed the number of uniquely dominant (frequency > 0.75) SNPs in each species (Fig. 17c). Most of these positions originate from fixed mutations that occurred in each species individually. To account for some introgression or incomplete lineage sorting, we allowed such signature SNPs for each species to be present in other species, but at low frequency (< 0.2 for each other species). We found that J. nigrosuffusa has the smallest number of uniquely dominant SNPs, suggesting that its genome is largely a combination of chunks in some other genomes, rather than a result of independent evolution by mutations in the J. nigrosuffusa lineage. A standard method to detect hybrid individual or species is to compute F3 statistic (Reich et al., 2009). Negative values of F3 suggest hybrid origin. Out of 11 species we analysed, only J. nigrosuffusa displays a negative F3 value with an extremely significant Z-score of −58 (P < 1e–300), which further supports the hypothesis about its hybrid origin.
This hybrid origin obstructs phylogeny reconstruction, because a single binary tree cannot depict the hybridization scenario, and in trees constructed from concatenated genomic regions, J. nigrosuffusa (a hybrid species) is placed in between the closest relatives of its parent species, J. stemosa sp.n. and J. grisea. We note that this speciation occurred by hybridization of ancestors of J. stemosa sp.n. and J. grisea, not the present-day J. stemosa sp.n. and J. grisea. This conceptual distinction is important, because we also observe ongoing hybridization and possible introgression between J. nigrosuffusa and J. grisea, which is particularly common in the autosomes and results in paraphyly of J. nigrosuffusa in the autosome tree. Finally, the third topology (cn)g observed in the divergence analysis is likely to be most recent because regions supporting this topology show the smallest average divergence between J. nigrosuffusa and J. coenia (Fig. 17a). This result suggests some recent hybridization between J. nigrosuffusa and J. coenia, the two species that meet in west Texas and perhaps expanded their ranges to become sympatric only more recently.
Investigating how evarete group species are placed in the trees, we observe both similarities and incongruence between the autosome and Z chromosome trees. In both trees, J. zonalis subspecies group together, with the first split into the nominal subspecies and J. z. michaelisi together with J. z. swifti. Likewise, mangrove-feeding J. litoralis and J. neildi form a monophyletic group. However, the position of the third mangrove feeder, J. pacoma sp.n., differs in the two trees. In the Z tree, it is sister to other mangrove-feeding species, but in the autosome tree it is placed as a sister of the coenia species group, although with low support. By analogy with the analysis of the coenia group, we hypothesize that Z chromosome topology reveals the species tree, but the autosome tree is influenced by the hybridization between J. pacoma sp.n. and J. nigrosuffusa, the two sympatric but strongly distinct (according to the divergence analysis; Fig. 7e,f) species. Notably, J. pacoma sp.n. has the largest number of unique positions among all Junonia species we analysed (Fig. 17c), and thus is also most distinct from others according to this criterion.
In summary, phylogenetic trees constructed from various genomic regions are incongruent, and the mitogenome tree reveals very little about Junonia speciation in North America. Incongruence between the trees constructed from autosomes and Z chromosome can be explained by the ancient hybrid origin of the species J. nigrosuffusa and more recent ongoing hybridization between North American Junonia species. The Z chromosome regions are more resistant to hybridization and its topology is more likely to represent species tree, as evidenced by the divergence analysis of DNA segments. Both new species described here (J. stemosa and J. pacoma) form monophyletic clusters in both autosome and Z trees, but the new subspecies, J. neildi varia, does not, due to its hybridization with J. coenia. This hybridization is likely to be responsible for the wing pattern differences between J. n. varia and the nominal Caribbean neildi subspecies.
Functions of putative speciation genes in Junonia
Only a small number of proteins diverged between Junonia species, and an even smaller number are more resistant to introgression. We identified such putative speciation genes using methods similar to those described previously (Cong et al., 2015, 2016; Cong & Grishin, 2018). Briefly, for each species pair, we identify the proteins that are enriched (P < 0.01) in divergent positions. Of these proteins, we further select those with relatively high fixation indices (Fst > 0.5). The 11 species in this study comprise 55 pairs, and we identified speciation genes for each pair and considered the genes that repeatedly show up in at least 20% species pairs to be the putative speciation genes for the Junonia species complex, resulting in 706 such proteins. We annotated these proteins by finding the closest Drosophila protein from FlyBase (Thurmond et al., 2019) and transferred the GO terms from the Drosophila protein. We identified the GO terms that are more frequently associated with these ‘speciation genes’ using a binomial test (P < 0.01), and these GO terms in the categories of ‘biological process’ and ‘molecular function’ are summarized in Fig. 18(a,b).
Fig. 18.
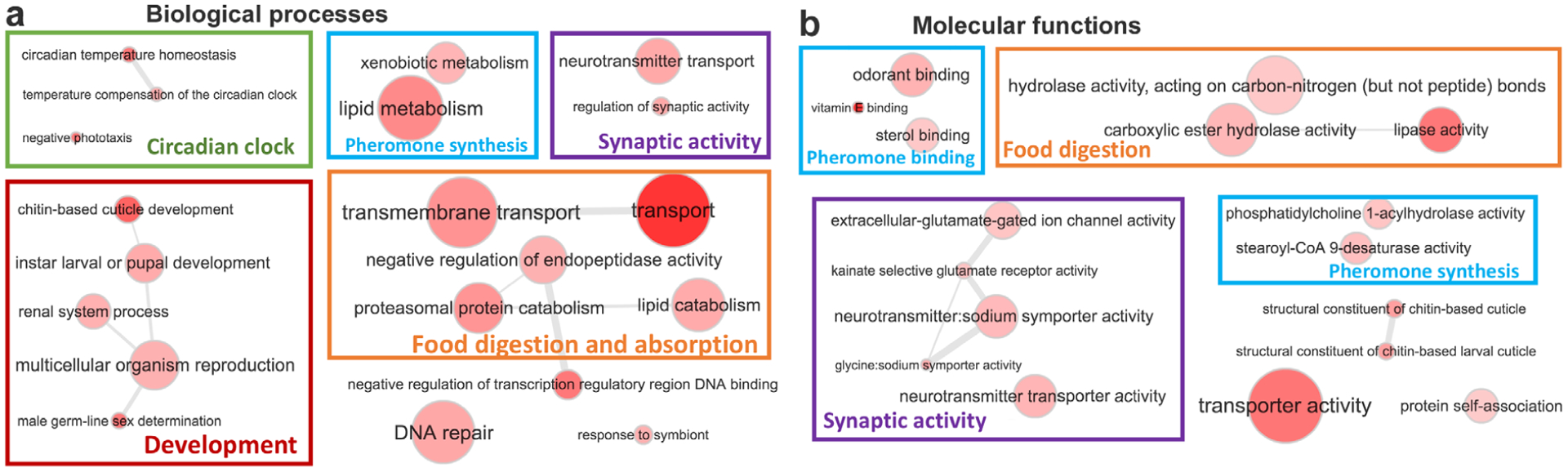
Functions of ‘speciation’ genes. Gene ontology (GO) database terms referring to biological processes (a) and molecular functions (b), clustered by major functional categories for proteins that show high divergence between Junonia species but have low polymorphism with each species.
This analysis reveals that the speciation genes tend to participate in development process, food digestion and absorption, circadian rhythm, pheromone synthesis, pheromone binding, and synaptic activity. Differences in developmental factors may account for the observed morphological differences; food digestion and absorption probably play a role in speciation of Junonia as these species frequently feed on different food plants; the involvement of circadian clock genes should be related to the fact that these species live in different latitudes and need to adapt to different photoperiod and temperature. The involvement of pheromone synthesis and binding proteins in speciation is more obvious, as such processes directly mediate the recognition of mates and facilitate mating.
The absence of developmental regulators, such as WntA, Wingless (Otaki, 2007, 2012; Martin & Reed, 2014; Zhang & Reed, 2016; Iwasaki et al., 2017; van der Burg et al., 2019), Splat, and Distal-less (Otaki, 2007, 2012, Martin & Reed, 2014, Zhang & Reed, 2016, Iwasaki et al., 2017, van der Burg et al., 2019), and components of biosynthetic pathways linked to wing colour pattern evolution in butterflies (Marcus, 2019) among speciation genes is intriguing but not surprising. It suggests that while colour patterns are evolving and diverging between species, proteins that directly regulate them do not change more than others, and speciation-linked changes are apparently taking place in regulators of these key proteins. Gene regulation (Wray et al., 2003) is carried out through interactions between cis- and trans-regulatory elements (Suryamohan & Halfon, 2015), where cis-regulatory regions are noncoding, relatively short specific DNA sequences that include promoters, enhancers and silencers (Riethoven, 2010). Trans-regulatory elements encode transcriptional factors, which are proteins that are being analysed here. For instance, clock is a general regulator of transcription (Trott & Menet, 2018). Trans-regulatory elements are longer in their DNA sequences and they are detected more confidently in draft genomes. Both their larger size and better data quality contribute to their better utility in the analyses presented here. Notably, cis- and trans-regulatory elements are expected to coevolve to maintain a functional gene regulation network. Therefore, our analyses of protein-coding sequences, including trans-regulatory elements, reflect the changes in gene regulation and provide extensive datasets to be analysed in the context of recent advances in our understanding of Junonia wing colour patterns and development (Otaki, 2007, 2012, Martin & Reed, 2014, Zhang & Reed, 2016, Iwasaki et al., 2017, van der Burg et al., 2019).
Dark and pale Junonia
Some confusion may arise from the English name ‘dark buckeye’ given to J. nigrosuffusa. While indeed all U.S.A. individuals of this species are dark in appearance (Fig. 19, top row), typically white-banded J. grisea (Fig. 19, bottom row) and J. coenia (Fig. 19, third row) also have a dark form, which may be induced by environmental conditions (Fig. 19, second row). These dark individuals of J. grisea and J. coenia may easily be mistaken for J. nigrosuffusa, J. stemosa, or hybrids between species (Fig. 19: compare top right with the right specimen in the second row). The unmistakable difference is in the colour of the nudum of the antennal club. The nudum is pale (whitish, yellowish or pale-brown) in J. stemosa and J. nigrosuffusa, and dark (dark-brown to nearly black) in J. coenia and J. grisea. If inspection of the nudum is not possible, the single most reliable character to distinguish between J. grisea/coenia and J. nigrosuffusa is the coloration of the area by the dorsal forewing costa near apex: between the postdiscal paler band (or its remnants in J. nigrosuffusa) and apical paler spots. This area is covered by extensive pale overscaling in J. nigrosuffusa and is mostly brown in J. grisea/coenia. Identification of J. stemosa may be more challenging, because many individuals, especially females, lack the pale overscaling, and their more rounded shape of wings may be the best character besides from the pale colour of the nudum. Then, some J. stemosa males (e.g. Fig.8, right specimen in the second row) and most females (Fig. 9, specimens on the right) may not be very dark, and the otherwise indistinct postdiscal forewing band is orange or even whitish, resembling J. grisea/coenia. Finally, to add to these complexities, a number of J. coenia individuals may have similar orange band. This form even received an infrasubspecific (and thus ICZN-unavailable) name, ‘tr. f. rubrosuffusa’ (W. D. Field). Not knowing the nudum colour, it may be impossible to identify such specimens with confidence, and the only other general character (which needs some practice to recognize) is the wing shape: more rounded in J. stemosa and J. nigrosuffusa, and more angular in J. coenia and J. grisea.
Fig. 19.

Dark and pale Junonia. See Table S1 for specimen data by their voucher numbers. Those without voucher numbers: J. coenia U.S.A.: Texas, Denton Co., Murrell Park, ex larva, eclosed 29-May-1998 (darker specimen, row 2), 4-Jun-1998 (paler, third row), J. grisea, same as NVG-5583. Dorsal and ventral views are on the left and right, respectively; species name and voucher number are between the views; general locality is by the antenna; ‘F’ indicates flipped image (left/right-inverted). Paratypes (PT) are labelled.
Conclusions
We devised a method to delineate species boundaries in Junonia using genomic data. As a result, we reached the following conclusions. First, we confirm that the mitochondrial DNA COI barcodes (and the entire mitochondrial DNA) are insufficient to mark out the differences between Junonia species, which apparently largely share the mitochondrial pool. We also confirmed the existence of two major barcode haplotypes: ‘A’, being largely South American and Caribbean, and ‘B’, being mostly North American. Second, in agreement with the recent studies (Gemmell et al., 2014; Lalonde et al., 2018; Lalonde & Marcus, 2019b), nuclear genomes reveal that J. evarete, J. genoveva and (as we additionally find here) J. litoralis are exclusively South American continental species, and application of these names to North American taxa is incorrect. Third, the Caribbean fauna does not seem to have these two species either, and is largely populated by J. zonalis and recently described J. neildi instead. Fourth, all available data agree best with the hypothesis that Junonia is represented by six species (one partitioned into two subspecies) in the United States. The mangrove buckeyes are J. neildi with its nominal subspecies in Florida and J. neildi varia Grishin, ssp.n. in southeast Texas. Similar to J. neildi in lacking the dark ring around the large forewing eyespot, J. zonalis zonalis populates Florida. Interestingly, while J. nigrosuffusa was formerly placed with J. zonalis (considered in turn to be a synonym of J. evarete), genomic data unambiguously show that J. nigrosuffusa belongs to the coenia group, along with J. coenia, J. grisea and a new species, the dark buckeye from southeast Texas: J. stemosa Grishin. Phenotypically, these four species are unified by the dark ring around the large forewing eyespot. Two Junonia species are sympatric in southeastern Arizona (J. grisea and J. nigrosuffusa) and in the Big Bend region of Texas (J. coenia and J. nigrosuffusa), and three are sympatric in the lower Rio Grande Valley of Texas (J. coenia, J. stemosa sp.n. and J. neildi varia ssp.n.) and Florida (J. coenia, J. neildi neildi and J. zonalis zonalis). Finally, we hypothesize about the origin of these species and suggest that J. nigrosuffusa is a hybrid species that originated from the ancestors of J. grisea and J. stemosa sp.n. In summary, we find that six Junonia species are present in the U.S.A. (one of which is described as new): J. neildi, J. coenia, J. grisea, J. nigrosuffusa, J. stemosa sp.n., and J. zonalis. South Texas populations of J. neildi are described as J. n. varia ssp.n. and west Mexican mangrove-feeding buckeyes as J. pacoma sp.n.
Supplementary Material
Appendix S1. DNA sequences with diagnostic nucleotide characters for the new taxa mapped to the reference genome of Junonia coenia.
Figure S1. PCA plot showing specimens as points in the plane of the first and third principal components. The shape and colour of the points are the same as in Fig. 6.
Figure S2. Eigenvalues for PCA of Junonia genomic SNPs with projections shown in Figs 6 and S1.
Figure S3. Interquartile range (y-axis) and mean (x-axis) of the divergence of all proteins encoded by the Z-chromosome for all pairs of populations listed in Fig. 7. Each dot represents one population pair, and pairs of different species, different subspecies, and the same subspecies are coloured in green, yellow, and red, respectively.
Figure S4. Interquartile range (y-axis) and mean (x-axis) of the divergence of a random sample of 600 autosomal proteins for all pairs of populations listed in Fig. 7. Each dot represents one population pair, and pairs of different species, different subspecies, and same subspecies are coloured in green, yellow, and red, respectively.
Table S1. Data for Junonia specimens used in this study.
Table S2. Quality of sequence data for specimens.
Table S3. Functional analysis of 36 ‘speciation’ proteins.
Table S4. Enriched GO terms associated with Z-linked proteins in Junonia.
Fig. 15.

Caterpillars and pupae of Junonia neildi varia ssp.n. All from U.S.A.: Texas, San Patricio Co., Portland, Indian Point Park, 27.85351, −97.35338. (a–k) Fifth-instar, caterpillars; (l) prepupa; (m–u) pupae. (a–c) and (f–k) show the same individual (NVG-6897). Scale: 0.5 cm (i–k); 1 cm (other panels). (a–c, p–r) 25-Aug-2016; (d, e, m–o) 23-Aug-2016; (l) 24-Aug-2016; (f–h, s–u) 28-Aug-2016. (m-o) NVG-6852; (l, p, q) NVG-6856; (s–u) NVG-6858.
Acknowledgements
We acknowledge Texas Parks and Wildlife Department (Natural Resources Program Director David H. Riskind) for the permit no. 08-02Rev that made research based on material collected in Texas State Parks possible. We thank Robert K. Robbins, John M. Burns and Brian Harris (National Museum of Natural History, Smithsonian Institution, Washington, DC), Edward G. Riley, Karen Wright and John D. Oswald (Texas A & M University, College Station, TX), Rebekah Shuman Baquiran and Crystal Maier (The Field Museum of Natural History, Chicago, IL), David Grimaldi and Courtney Richenbacher (American Museum of Natural History, New York, NY), Vince F. Lee and the late Norm Penny (California Academy of Sciences, San Francisco, CA), Paul A. Opler and Boris Kondratieff (C.P. Gillette Museum of Arthropod Diversity, Department of Bioagricultural Sciences, Colorado State University, Fort Collins, CO), Weiping Xie (Los Angeles County Museum of Natural History, Los Angeles, CA), and Alex Wild (Biodiversity Center, University of Texas at Austin, Austin, TX) for facilitating access to the collections under their care and stimulating discussions, and Brian Banker, Jack S. Carter, Bill R. Dempwolf, Chris J. Durden and Jeff R. Slotten for specimens sampled for DNA used in this project. We are grateful to two anonymous reviewers for their suggestions which helped us to improve the manuscript. This work was supported in part by the grants from the National Institutes of Health (GM094575 and GM127390 to NVG) and the Welch Foundation (I-1505 to NVG). The authors declare that they have no conflicts of interest.
Footnotes
This published work has been registered on Zoobank, http://zoobank.org/urn:lsid:zoobank.org:act:C6E70C96-463D-4E6A-95CC-B0384B0EEEBA.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- Aldhebiani AY (2018) Species concept and speciation. Saudi Journal of Biological Sciences, 25, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin GT & Emmel JF (1998) New subspecies of butterflies (Lepidoptera) from Nevada and California. Systematics of Western North American Butterflies (ed. by Emmel TC), pp. 501–522. Marioposa Press, Gainesville, FL. [Google Scholar]
- Baker RJ & Bradley RD (2006) Speciation in mammals and the genetic species concept. Journal of Mammalogy, 87, 643–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes W & McDunnough JH (1916) Some new races and species of North American Lepidoptera. The Canadian Entomologist, 48, 221–226. [Google Scholar]
- Barrera-Guzman AO, Aleixo A, Shawkey MD & Weir JT (2018) Hybrid speciation leads to novel male secondary sexual ornamentation of an Amazonian bird. Proceedings of the National Academy of Sciences of the United States of America, 115, E218–E225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudette K, Hughes TM & Marcus JM (2014) Improved injection needles facilitate germline transformation of the buckeye butterfly Junonia coenia. BioTechniques, 56, 142–144. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M & Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchers TE & Marcus JM (2014) Genetic population structure of buckeye butterflies (Junonia) from Argentina. Systematic Entomology, 39, 242–255. [Google Scholar]
- Brévignon C (2009) Nouvelles observations sur le genre Junonia en Guyane Française. (Lepidopterar: Nymphalidae) Première Partie. Labillionea, CIX, 3–7. [Google Scholar]
- Brévignon L & Brévignon C (2012) Nouvelles observations sur le genre Junonia en Guyane Française. (Lepidoptera: Nymphalidae) (Seconde partie). Lépidoptères de Guyane, 7. 8–35. [Google Scholar]
- van der Burg KRL, Lewis JJ, Martin A, Nijhout HF, Danko CG & Reed RD (2019) Contrasting roles of transcription factors spineless and EcR in the highly dynamic chromatin landscape of butterfly wing metamorphosis. Cell Reports, 27, e1023. [DOI] [PubMed] [Google Scholar]
- Calhoun JV (2010) The identities of Papilio evarete Cramer and Papilio genoveva Cramer (Nymphalidae), with notes on the occurrence of Junonia evarete in Florida. News of the Lepidopterists’ Society, 52, 47–51. [Google Scholar]
- Claridge MF (2017) Insect species – concepts and practice. Insect Biodiversity: Science and Society (ed. by Foottit RG and Adler PH), pp. 527–546. John Wiley & Sons, Chichester, UK. [Google Scholar]
- Cong Q, Borek D, Otwinowski Z & Grishin NV (2015) Tiger swallowtail genome reveals mechanisms for speciation and caterpillar chemical defense. Cell Reports, 10, 910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Q & Grishin N (2018) Comparative analysis of Swallowtail transcriptomes suggests molecular determinants for speciation and adaptation. Genome, 61, 843–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Q, Shen J, Warren AD, Borek D, Otwinowski Z & Grishin NV (2016) Speciation in cloudless sulphurs gleaned from complete genomes. Genome Biology and Evolution, 8, 915–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Ortiz L, Nidiffer MD, Hermida-Lagunes J et al. (2019) Reduced introgression of sex chromosome markers in the Mexican howler monkey (Alouatta palliata x A. pigra) hybrid zone. International Journal of Primatology, 40, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA (1992) Genetics and speciation. Nature, 355, 511–515. [DOI] [PubMed] [Google Scholar]
- Coyne JA (2018) “Two rules of speciation” revisited. Molecular Ecology, 27, 3749–3752. [DOI] [PubMed] [Google Scholar]
- Davey JW, Chouteau M, Barker SL et al. (2016) Major improvements to the Heliconius melpomene genome assembly used to confirm 10 chromosome fusion events in 6 million years of butterfly evolution. G3 (Bethesda), 6, 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhungel B & Otaki JM (2013) Larval temperature experience determines sensitivity to cold-shock-induced wing color pattern changes in the blue pansy butterfly Junonia orithya. Journal of Thermal Biology, 38, 427–433. [Google Scholar]
- Evans WH (1943) A revision of the genus Aeromachus de N. (Lepidoptera: Hesperiidae). Proceedings of the Royal Entomological Society of London (B), 12, 97–101. [Google Scholar]
- Fontaine MC, Pease JB, Steele A et al. (2015) Mosquito genomics. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science, 347, 1258524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes WTM (1928) Variation in Junonia lavinia (Lepidoptera, Nymphalidae). Journal of the New York entomological Society, 36, 306–321. [Google Scholar]
- Gemmell AP, Borchers TE & Marcus JM (2014) Molecular population structure of Junonia butterflies from French Guiana, Guadeloupe, and Martinique. Psyche: A Journal of Entomology, 2014, 1–21. [Google Scholar]
- Gemmell AP & Marcus JM (2015) A tale of two haplotype groups: evaluating the New World Junonia ring species hypothesis using the distribution of divergent COI haplotypes. Systematic Entomology, 40, 532–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafernik JE (1982) Phenetics and ecology of hybridization in buckeye butterflies (Lepidoptera: Nymphalidae). University of California Publications in Entomology, 96, 1–109. [Google Scholar]
- Harris RS (2007) Improved Pairwise Alignment of Genomic DNA. PhD thesis,The Pennsylvania State University.
- Hebert PD, Cywinska A, Ball SL & deWaard JR (2003) Biological identifications through DNA barcodes. Proceedings of the Biological Sciences, 270, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner J (1822) pl. [32]. Sammlung Exotischer Schmetterlinge. Jacob Hübner, Augsburg. [Google Scholar]
- Iwasaki M, Ohno Y & Otaki JM (2017) Butterfly eyespot organiser: in vivo imaging of the prospective focal cells in pupal wing tissues. Scientific Reports, 7, 40705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodandaramaiah U & Wahlberg N (2007) Out-of-Africa origin and dispersal-mediated diversification of the butterfly genus Junonia (Nymphalidae: Nymphalinae). Journal of Evolutionary Biology, 20, 2181–2191. [DOI] [PubMed] [Google Scholar]
- Koh TW, He Z, Gorur-Shandilya S, Menuz K, Larter NK, Stewart S & Carlson JR (2014) The Drosophila IR20a clade of ionotropic receptors are candidate taste and pheromone receptors. Neuron, 83, 850–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunte K, Shea C, Aardema ML, Scriber JM, Juenger TE, Gilbert LE & Kronforst MR (2011) Sex chromosome mosaicism and hybrid speciation among tiger swallowtail butterflies. PLoS Genetics, 7, e1002274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde MML & Marcus JM (2019a) Entomological time travel: reconstructing the invasion history of the buckeye butterflies (genus Junonia) from Florida, USA. Biological Invasions, 21, 1947–1972. [Google Scholar]
- Lalonde MML & Marcus JM (2019b) Getting western: biogeographical analysis of morphological variation, mitochondrial haplotypes and nuclear markers reveals cryptic species and hybrid zones in the Junonia butterflies of the American southwest and Mexico. Systematic Entomology, 44, 465–489. [Google Scholar]
- Lalonde MLM, McCullagh BS & Marcus JM (2018) The taxonomy and population structure of the buckeye butterflies (genus Junonia, Nymphalidae: Nymphalini) of Florida, USA. Journal of the Lepidopterists’ Society, 72, 97–115. [Google Scholar]
- Layberry RA, Hall PW & Lafontaine JD (1998) The Butterflies of Canada. University of Toronto Press, Toronto, Canada. [Google Scholar]
- Li W, Cong Q, Shen J, Zhang J, Hallwachs W, Janzen DH & Grishin NV (2019) Genomes of skipper butterflies reveal extensive convergence of wing patterns. Proceedings of the National Academy of Sciences of the United States of America, 116, 6232–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H & Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L & Hinton G (2008) Visualizing Data using t-SNE. Journal of Machine Learning Research, 9, 2579–2605. [Google Scholar]
- Mallet J, Besansky N & Hahn MW (2016) How reticulated are species? BioEssays, 38, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JM (2019) Evo-Devo of butterfly wing patterns. Evolutionary Developmental Biology-A Reference Guide (ed. by Nuño de la Rosa L and Müller GB), pp. 1–13. Springer, Cham, Switzerland. [Google Scholar]
- Martin SH, Dasmahapatra KK, Nadeau NJ et al. (2013) Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Research, 23, 1817–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A & Reed RD (2014) Wnt signaling underlies evolution and development of the butterfly wing pattern symmetry systems. Developmental Biology, 395, 367–378. [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Landeen EL, Gordon KE et al. (2018) Gene flow mediates the role of sex chromosome meiotic drive during complex speciation. eLife, 7 e35468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neild AFE (2008) The Butterflies of Venezuela, Part 2: Nymphalidae II (Acraeinae, Libytheinae, Nymphalinae, Ithomiinae, Morphinae). Meridian, London. [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A & Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhout HF (1984) Colour pattern modification by coldshock in Lepidoptera. Journal of Embryology and Experimental Morphology, 81, 287–305. [PubMed] [Google Scholar]
- Orr HA (1996) Dobzhansky, Bateson, and the genetics of speciation. Genetics, 144, 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otaki JM (2007) Reversed type of color-pattern modifications of butterfly wings: a physiological mechanism of wing-wide color-pattern determination. Journal of Insect Physiology, 53, 526–537. [DOI] [PubMed] [Google Scholar]
- Otaki JM (2012) Structural analysis of eyespots: dynamics of morphogenic signals that govern elemental positions in butterfly wings. BMC Systems Biology, 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL & Reich D (2006) Population structure and eigenanalysis. PLoS Genetics, 2, e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen SM (1996) Quantitative genetics of the wing color pattern in the buckeye butterfly (Precis Coenia and Precis Evarete): evidence against the constancy of G. Evolution, 50, 1585–1597. [DOI] [PubMed] [Google Scholar]
- Peters MJ & Marcus JM (2017) Taxonomy as a hypothesis: testing the status of the Bermuda buckeye butterfly Junonia coenia bergi (Lepidoptera: Nymphalidae). Systematic Entomology, 42, 288–300. [Google Scholar]
- Pfeiler E, Johnson S & Markow TA (2012a) DNA barcodes and insights into the relationships and systematics of buckeye butterflies (Nymphalidae: Nymphalinae: Junonia) from the Americas. Journal of the Lepidopterists’ Society, 66, 185–198. [Google Scholar]
- Pfeiler E, Johnson S & Markow TA (2012b, 2012) Insights into Population Origins of neotropical Junonia (Lepidoptera: Nymphalidae: Nymphalinae) based on mitochondrial DNA. A Journal of Entomology, Psyche. [Google Scholar]
- Pickrell JK & Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genetics, 8, e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Thangaraj K, Patterson N, Price AL & Singh L (2009) Reconstructing Indian population history. Nature, 461, 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethoven JJ (2010) Regulatory regions in DNA: promoters, enhancers, silencers, and insulators. Methods in Molecular Biology, 674, 33–42. [DOI] [PubMed] [Google Scholar]
- Rytz R, Croset V & Benton R (2013) Ionotropic receptors (IRs): chemosensory ionotropic glutamate receptors in drosophila and beyond. Insect Biochemistry and Molecular Biology, 43, 888–897. [DOI] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Dannemann M et al. (2014) The genomic landscape of Neanderthal ancestry in present-day humans. Nature, 507, 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JA (1986) The Butterflies of North America: A Natural History and Field Guide. Standford University Press, Stanford, CA. [Google Scholar]
- Shapiro AM & de Grassi R (2017) The buckeye, Junonia coenia, uses the garden ornamental Russelia equisetiformis (Plantaginaceae) (“firecracker plant”) as a larval host in California. News of the Lepidopterists’ Society, 59, 201. [Google Scholar]
- Sperling FAH (2003) Butterfly molecular systematics: from species definitions to higher level phylogenies. Ecology and Evolution Taking Flight: Butterflies as Model Study Systems (ed. by Boggs C, Erlich P and Watt W), pp. 431–458. University of Chicago Press, Chicago, IL. [Google Scholar]
- Sukumaran J & Holder MT (2010) DendroPy: a Python library for phylogenetic computing. Bioinformatics, 26, 1569–1571. [DOI] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N & Smuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One, 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryamohan K & Halfon MS (2015) Identifying transcriptional cis-regulatory modules in animal genomes. Wiley Interdisciplinary Reviews: Developmental Biology, 4, 59–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng XX, Xie ZH & Zhang Y (2016) The complete mitochondrial genome of the common buckeye Junonia coenia (Insecta: Lepidoptera: Nymphalidae). Mitochondrial DNA A DNA Mapping, Sequencing, and Analysis, 27, 4351–4352. [DOI] [PubMed] [Google Scholar]
- Thurmond J, Goodman JL, Strelets VB et al. (2019) FlyBase 2.0: the next generation. Nucleic Acids Research, 47, D759–D765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott AJ & Menet JS (2018) Regulation of circadian clock transcriptional output by CLOCK:BMAL1. PLoS Genetics, 14, e1007156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TW & Parnell JR (1985) The identification of two species of Junonia Hübner (Lepidoptera: Nymphalidae): J. evarete and J. genoveva in Jamaica. Journal of Research on the Lepidoptera, 24, 142–153. [Google Scholar]
- Vane-Wright RI & Tennent WJ (2011) Colour and size variation in Junonia villida (Lepidoptera, Nymphalidae): subspecies or phenotypic plasticity? Systematics and Biodiversity, 9, 289–305. [Google Scholar]
- Wolf AB & Akey JM (2018) Outstanding questions in the study of archaic hominin admixture. PLoS Genetics, 14, e1007349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV & Romano LA (2003) The evolution of transcriptional regulation in eukaryotes. Molecular Biology and Evolution, 20, 1377–1419. [DOI] [PubMed] [Google Scholar]
- Yuvaraj JK, Andersson MN, Anderbrant O & Lofstedt C (2018) Diversity of olfactory structures: a comparative study of antennal sensilla in Trichoptera and Lepidoptera. Micron, 111, 9–18. [DOI] [PubMed] [Google Scholar]
- Zhang J, Cong Q, Shen J, Brockmann E & Grishin NV (2019) Genomes reveal drastic and recurrent phenotypic divergence in firetip skipper butterflies (Hesperiidae: Pyrrhopyginae). Proceedings of the Royal Society B: Biological Sciences, 286, 20190609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kobert K, Flouri T & Stamatakis A (2014) PEAR: a fast and accurate Illumina paired-end reAd mergeR. Bioinformatics, 30, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L & Reed RD (2016) Genome editing in butterflies reveals that Spalt promotes and distal-less represses eyespot colour patterns. Nature Communications, 7, 11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. DNA sequences with diagnostic nucleotide characters for the new taxa mapped to the reference genome of Junonia coenia.
Figure S1. PCA plot showing specimens as points in the plane of the first and third principal components. The shape and colour of the points are the same as in Fig. 6.
Figure S2. Eigenvalues for PCA of Junonia genomic SNPs with projections shown in Figs 6 and S1.
Figure S3. Interquartile range (y-axis) and mean (x-axis) of the divergence of all proteins encoded by the Z-chromosome for all pairs of populations listed in Fig. 7. Each dot represents one population pair, and pairs of different species, different subspecies, and the same subspecies are coloured in green, yellow, and red, respectively.
Figure S4. Interquartile range (y-axis) and mean (x-axis) of the divergence of a random sample of 600 autosomal proteins for all pairs of populations listed in Fig. 7. Each dot represents one population pair, and pairs of different species, different subspecies, and same subspecies are coloured in green, yellow, and red, respectively.
Table S1. Data for Junonia specimens used in this study.
Table S2. Quality of sequence data for specimens.
Table S3. Functional analysis of 36 ‘speciation’ proteins.
Table S4. Enriched GO terms associated with Z-linked proteins in Junonia.