

Abstract
Antimicrobial resistance (AMR) is a major global health threat that affects millions of people each year. Funding agencies worldwide and the global research community have expended considerable capital and effort tracking the evolution and spread of AMR by isolating and sequencing bacterial strains and performing antimicrobial susceptibility testing (AST). For the last several years, we have been capturing these efforts by curating data from the literature and data resources and building a set of assembled bacterial genome sequences that are paired with laboratory-derived AST data. This collection currently contains AST data for over 67 000 genomes encompassing approximately 40 genera and over 100 species. In this paper, we describe the characteristics of this collection, highlighting areas where sampling is comparatively deep or shallow, and showing areas where attention is needed from the research community to improve sampling and tracking efforts. In addition to using the data to track the evolution and spread of AMR, it also serves as a useful starting point for building machine learning models for predicting AMR phenotypes. We demonstrate this by describing two machine learning models that are built from the entire dataset to show where the predictive power is comparatively high or low. This AMR metadata collection is freely available and maintained on the Bacterial and Viral Bioinformatics Center (BV-BRC) FTP site ftp://ftp.bvbrc.org/RELEASE_NOTES/PATRIC_genomes_AMR.txt.
Keywords: artificial intellignence, machine learning, antimicrobial resistance, epidemiology, GWAS
Introduction
Antimicrobial resistance (AMR) occurs when a microorganism becomes resistant to a compound that is intended to kill or arrest its growth [1]. Resistance can spread rapidly through horizontal gene transfer mechanisms and the unintended selection of resistant strains [1–3]. This spread is typically fueled by the overuse or incorrect administration of antibiotics in both clinical and agricultural settings [1, 4–8], and the problem is worsened by a lack of uniform international policies governing antibiotic stewardship [8–11]. As a result, AMR causes considerable morbidity and mortality for patients worldwide, with correspondingly severe economic impacts [8, 12, 13]. Indeed, AMR is predicted to cause approximately 700 000 deaths worldwide each year, and costs 55 billion dollars annually in the USA due to health care related expenses and lost productivity [8, 12]. In the last few decades, the discovery and development of new antimicrobial compounds has not kept pace with the spread of resistance, so this global health concern that used to be managed by selecting from a variety of efficacious drugs has become more alarming as the arsenal of effective antibiotics has dwindled [1, 14, 15].
In the clinic and at the bench, AMR is typically assessed using antimicrobial susceptibility testing (AST) methods that detect the growth of an organism in the presence of a known concentration of an antibiotic [16, 17]. These are often measured as minimum inhibitory concentrations (MICs) or by measuring the size of a zone of inhibition in a disk diffusion or test-strip assay on a plate [16, 17]. Several diagnostic devices, including the BD Phoenix (Becton Dickinson) and VITEK 2 (bioMérieux) instruments, are commonly used to perform rapid AST testing in the clinic [16–18]. Establishing whether an organism is resistant or susceptible to an antibiotic is then performed by comparing the test results with clinical breakpoints, which are established by organizations such as the Clinical and Laboratory Standards Institute (CLSI) or the European Committee on Antimicrobial Susceptibility Testing (EUCAST) [19–21]. These AST data are frequently published in studies tracking AMR epidemiology and genetics [22–27].
Genome sequence data provide an alternative view of AMR, enabling researchers to assess the genetic mechanisms conferring AMR in each strain [28–30]. Many publicly available resources have been developed to help determine AMR phenotypes based on the presence of resistance-conferring SNPs and genes [31–39]. Coupling AST data with genome sequences also offers the potential to discover regions of the genome that are directly involved in resistance, have changed as a result of epistasis, or that correlate with the presence of AMR [28, 29]. This is typically done through genome wide association studies or with machine learning techniques [40–52]. The use of machine learning algorithms for predicting AMR phenotypes and identifying genomic regions associated with resistance has generated considerable interest in the literature over the last few years [53–55].
A major challenge in developing machine learning techniques for predicting phenotypes or identifying AMR-related genomic features is the difficulty in obtaining genome sequences paired with laboratory-derived AST data [41, 56–58]. Although it is customary for researchers to submit genomes to a public repository before publication, this is not a prerequisite for AST data [59, 60]. This makes it difficult to quickly collect datasets that are large enough for modeling. Once the data are obtained, other issues arise from ensuring that the dataset is balanced across phenotypes, phylogenetically and geographically diverse, and representative of the AMR mechanisms that exist in nature or the clinic [41, 42, 45, 61, 62]. In general, these data do not accumulate evenly. Sampling tends to reflect the prioritization of certain pathogens over others, along with the missions and needs of public health agencies and the populations that they serve [60, 63]. To help with this problem, several resources including NCBI, EMBL-EBI, the Relational Sequencing TB Data Platform, AR Isolate Bank and Pathogenwatch maintain sets of genomes that are paired with AST data for use in downstream analyses, including comparative genomics and modeling [64–69].
For several years, we have been collecting bacterial genomes and manually curating AST data from a variety of sources [70]. The purpose of this paper is to describe this AST data collection, use it to evaluate areas where sampling within the collection is comparatively shallow or deep, and demonstrate how it can be used for predicting AMR and guiding future research.
Characteristics of the AST data collection
The data collection consists of a set of assembled and uniformly annotated bacterial genomes with laboratory-derived AST data for each isolate [71]. The data are housed and maintained within the Bacterial and Viral Bioinformatics Resource Center (BV-BRC), which is the umbrella project operating the bacterial Pathosystems Resource Integration Center (PATRIC) [70]. The AST data for each genome have been curated primarily from the literature, data resources including NCBI, direct submissions to the PATRIC resource, and projects that have been supported by the United States National Institute of Allergy and Infectious Diseases (NIAID) (Table 1) [64]. In addition, at least 218 studies publishing AST data with sequenced genomes have been curated from the literature for use in this collection (Table S1). As of November 2020, the collection contains 67 817 bacterial genomes with some form of AST data covering 38 bacterial genera. These data are most frequently represented as either MICs or susceptible, intermediate and resistant (SIR) calls based on a standard set of breakpoints. In some cases, phenotypes have been recorded as ‘reduced susceptibility’, ‘non-susceptible’, etc. by the original authors, and these calls have been kept in their original form. In addition to MICs and SIR calls, the collection also contains diameter sizes for zones of inhibition. In cases where the genomes have been deposited in GenBank, the original assemblies were integrated into PATRIC [72]. However, many of the genomes corresponding to these isolates were deposited in either SRA or ENA as reads, and were subsequently assembled using the PATRIC assembly service prior to integration [40, 73, 74]. Approximately 36 000 genomes in this collection were assembled from reads.
Table 1.
Characteristics of the AST data collection
| Genomes | 67 817 |
|---|---|
| Genera | 38 |
| Species* | 88 |
| Antibiotics | 128 |
| MICs | 324 134 |
| Phenotype calls (e.g. SIR) | 356 206 |
| Measurement methods | ~20 |
| Publication sources | ~218 |
*Count does not include unnamed species with a ‘sp.’ designation.
The data collection is mostly comprised of pathogens, with Mycobacterium tuberculosis, Salmonella enterica, Streptococcus pneumoniae and Neisseria gonorrhoeae currently having the largest number of genomes with AST data (Figure 1A). The data for many of the highly represented species are the result of sequencing efforts led by large consortia, such as CRyPTIC (Comprehensive Resistance Prediction for Tuberculosis: an International Consortium) and TB ARC (Tuberculosis Antibiotic Resistance Catalog Project), and genomic surveillance programs that are led by large public health agencies such as NARMS (the National Antimicrobial Monitoring System), which consolidates monitoring and outbreak tracking efforts from the United States Food and Drug Administration, Centers for Disease Control and Prevention, US Department of Agriculture, and other Public Health agencies [75–78]. However, a variety of smaller studies have published data covering over 88 species.
Figure 1.
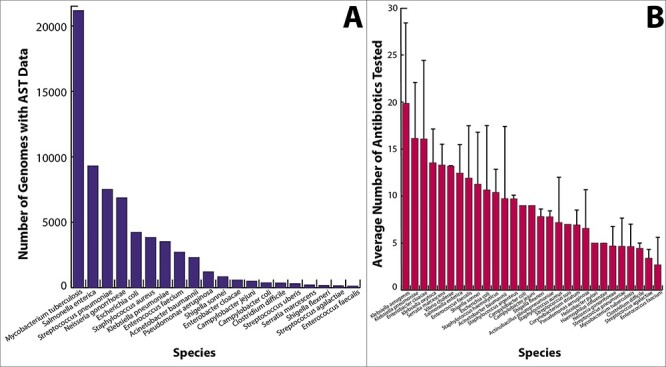
Histograms depicting (A) the number of genomes for the top organisms in the AST data collection, and (B) the average number of antibiotics tested per isolate. Whisker bars depict the standard deviation.
Across all organisms, data have been reported for approximately 128 antimicrobial compounds. Most of these compounds are well known antibiotics that are used extensively in the clinic. The compounds that have been tested on each isolate vary due to the organism’s physiology, risks to patients, and the original study design and scope (Figure 1B). For example, many studies have focused heavily on extended spectrum beta-lactamase producing Gram-negative pathogens, methicillin and vancomycin resistant Gram-positives, and resistance to antimycobacterial drugs in M. tuberculosis (Figure 2). Data on experimental compounds, topical antibiotics and antibiotics that are used in veterinary settings are present in the collection, but are less common. SIR determinations are more common than the MICs, and although many species have a handful of organisms with MIC data, the most deeply sampled MIC datasets correspond with A. baumannii, C. jejuni, E. cloacae, E. coli, K. pneumoniae, N. gonorrhoeae, P. aeruginosa, S. enterica and S. pneumoniae (Figure 3).
Figure 2.

Counts of susceptible and resistant isolates with sequenced genomes for each species and antibiotic in the AST data collection. The category (susceptible or resistant) with fewer genomes is depicted. The color scale is capped at 500 genomes. Antibiotic abbreviations are defined in Table S2. Unnamed species with a ‘sp.’ designation are not shown.
Figure 3.
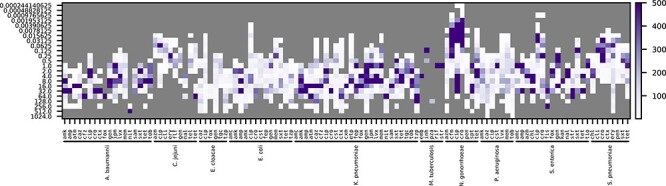
Counts of sequenced isolates with a given MIC for each species and antibiotic in the AST data collection. MICs are shown on the left. The color scale is capped at 500 genomes. Antibiotic abbreviations are defined in Table S2.
Data coverage and considerations
To our knowledge, this is one of the largest publicly available AST data collections that is paired with genome sequence data. Although it contains a large number of species, strains, genomes and antibiotic resistance phenotypes, there are sampling biases that exist due to the limited availability of data. For example, there are several important bacterial pathogens that are conspicuously underrepresented in the collection, including E. coli, which has only 4217 genomes with AST data. By comparison, there are just under 30 000 E. coli genomes in the PATRIC data resource. Considering that E. coli is one of the most intensely studied organisms and one of the most common Gram-negative bacterial pathogens in hospital systems worldwide, it is striking that such a small number of studies have published AST data and associated sequences for this pathogen [44, 79–84]. The related Shigella species are similarly underrepresented. In these cases, and that of other important surveillance organisms including Campylobacter, Enterobacter and Enterococcus, the efforts of genomic surveillance and source tracking projects are actively improving data coverage [85]. On the other hand, anaerobic organisms, including Clostridium difficile, are underrepresented due to the added difficulty in growing and testing these isolates. Many non-human pathogens commonly seen in veterinary and agricultural settings, and important environmental isolates, which may be acting as reservoirs of resistance, are also underrepresented. Despite these limitations, data continue to accumulate for more diverse organisms over time.
In addition to the variation in species sampling, the data collection also has biases that are related to the geographical sampling and the mission objectives of funding agencies. For instance, M. tuberculosis sampling skews heavily toward Asian, African and South American countries where tuberculosis is problematic and individual nations and the research community are focusing on surveillance (Figure 4). On the other hand, sampling of N. gonorrhoeae is heavily biased toward the USA, Canada and the UK. In some cases, the deepest sampling is the result of sequencing during outbreaks, which can also skew the data phylogenetically. For example, most of the S. enterica serovar Paratyphi and Typhi data are from outbreaks from the Indian Subcontinent and Southeast Asia [86–91]. We expect some of these sampling biases to become less dramatic as more studies publish AST data, however; local outbreaks, endemic disease burdens and biased geographic sampling will continue to remain important considerations when using these data for downstream studies.
Figure 4.
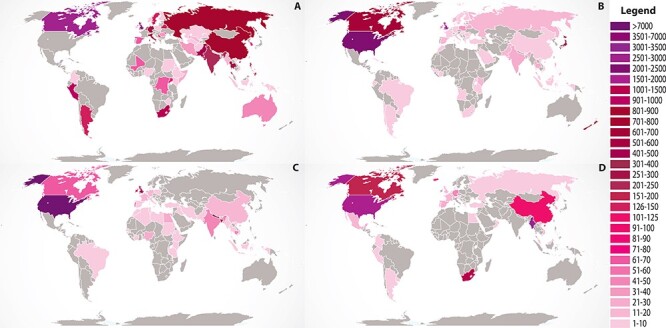
The country of isolation for genomes in the AST data collection. The top four species with the most genomes are shown: (A) Mycobacterium tuberculosis, (B) Neisseria gonorrhoeae, (C) Salmonella enterica and (D) Streptococcus pneumoniae. The legend depicts the number of genomes from each country. Global map courtesy of Free Vector Maps.com.
Because the sampling of genomes with AST data often reflects certain study goals, such as surveillance for resistance to certain classes of antibiotics, sequencing during outbreaks, and sequencing in the event of antimicrobial treatment failure, there can be differences in the underlying AMR gene content of the genomes in the AST data collection when compared with other publicly available genomes, or what might be expected in naturally occurring bacterial populations. As an illustration of this, Table S3 shows examples where there are large differences in the fraction of PATRIC genomes encoding various AMR genes versus the subset of these genomes represented in the AST data collection. For example, there are currently 13% more E. coli genomes encoding TEM-type beta lactamases in the AST data collection versus all E. coli genomes in PATRIC, and there are 18% fewer genomes encoding OXA-23-type beta-lactamases in the A. baumannii genomes in the AST data collection versus all of A. baumannii genomes in PATRIC. These sampling differences should be taken into consideration when conducting analyses and evaluating the generalizability of models that are built from the data.
Using machine learning to predict AMR phenotypes and genomic regions
Advancements in artificial intelligence (AI) methods, and the development of software packages that support their use, have led to an explosion of interest in using these methods to advance our understanding of biology [92, 93]. One of the reasons for gathering and curating this AST data collection is to help advance our ability to predict AMR phenotypes and identify genomic regions that may be associated with resistance.
Many machine learning studies building predictive models from AST data have been published in recent years. The way that these models are constructed can differ depending on the research objectives, but models are typically built either as classifiers, which predict discrete phenotypes such as SIR calls, or as regressors, which predict a numeric value in a rage, such as MICs. In either case, the features are often derived from the genome sequence data, and studies have used nucleotide and amino acid k-mers of various lengths, SNPs, gene content, phylogeny and combinations thereof to predict AMR phenotypes [41–46, 51, 52, 94, 95]. These features are given to the machine learning algorithm in the form of a matrix of frequencies or counts of each feature, and the machine learning identifies the features that best distinguish each category in order to make the prediction.
In previous work, we have built machine learning models using nucleotide k-mers of various sizes as features and several ensemble methods, including adaptive boosting (AdaBoost), random forest and extreme gradient boosting (XGBoost), to predict both SIR calls and MICs [41–43, 49]. The experimental design and computing requirements often dictate the choice of algorithm, parameters and k-mer lengths for these models. For example, we have used k-mer lengths ranging in size from 7 to 31 nucleotides. Shorter oligonucleotide k-mer lengths usually result in smaller matrix files, lower memory usage and faster compute times because there are fewer k-mer combinations. This can enable modeling on a larger number of genomes, but if the k-mer size is too small, the model accuracy begins to decay. Also, as the k-mer length decreases, they occur more frequently in the genome, making the interpretation of the important features more challenging. In our experience, k-mers that are 7–10 nucleotides in length are usually sufficient for good computing times and accuracies, and slightly longer k-mers of approximately 14–15 nucleotides usually provide the uniqueness needed for feature lookup.
Many of the machine learning algorithms allow the user to extract the top features that were used by the model to make the prediction. This offers the potential of using the algorithms as a research tool. When AMR models are based on sequence data, such as nucleotide k-mers or alignments, the top features often correspond with regions of the genome that are known to be involved in resistance [42, 43, 45, 46, 48, 51, 52, 61, 96]. Several previous studies building AMR prediction models have found top features in known AMR genes, resistance-conferring SNPs in housekeeping genes and integration sites for AMR conferring genetic elements, such as SCCmec [41–43, 45, 46, 48, 49, 51, 52, 61, 83, 96]. Other features, including SNPs in transporters and virulence factors that are correlated with resistance are also often found to contribute to the models [96]. In addition, these models frequently identify features in regions of the genome that are not known to be involved in resistance, such as SNPs in genes encoding metabolic functions [43, 46, 51, 52, 96]. In these cases, the direct relationship between the features found by the models and AMR phenotypes can be difficult to interpret. It is possible that some of these features may have a previously unrecognized role in AMR, or that they are compensatory epistatic changes that occurred as resistance became fixed in a given lineage [46, 51, 96]. In any case, the features found by a machine learning model can be thought of as a set of testable hypotheses, and they offer a useful starting point for downstream analyses.
The AST data collection as an exemplar data frame for modeling
To demonstrate the utility of the AST data collection for modeling, we built a classifier for predicting susceptible and resistant phenotypes across all of the species in the collection with at least 75 resistant and susceptible genomes (Figure 5). This represents a large number of genomes and compute resource consumption relative to models previously built by the group. To accommodate the large number of genomes, we used 7-mer oligonucleotides as features and trained the model using XGBoost, as described previously [41–43]. Overall, the F1 score for the entire model, averaged over each fold of a 5-fold cross-validation, is 0.925 [0.924–0.927, 95% confidence interval]. We also built a regression model to predict MICs using 7-mers and XGBoost using similar parameters (Figure 6). In this case, the average accuracy for the model, within ±1 two-fold dilution step (which is the limit of resolution for most AST methods), is similarly high when there is sufficient sampling of genomes corresponding to a given MIC value for a species. In both cases, the models show that most of the AMR phenotypes in the collection with good sampling depth are predictable.
Figure 5.
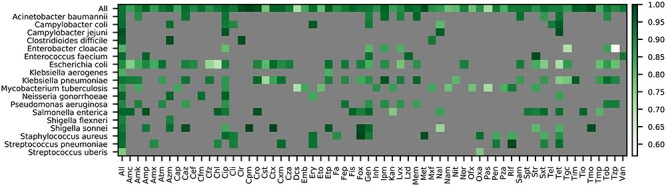
F1 scores for an XGBoost model built to classify susceptible and resistant phenotypes. One model was built using nucleotide 7-mers as input features for all species with at least 75 susceptible and resistant genomes. The coloring depicts the F1 score for each antibiotic-species combination based on a 5-fold cross-validation. The ‘All’ category depicts the average F1 score for each species and antibiotic, respectively. Antibiotic abbreviations are defined in Table S2.
Figure 6.
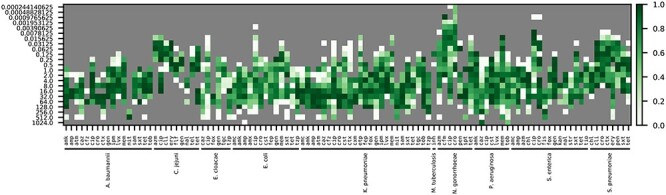
Accuracies for an XGBoost model built to predict MICs. One model was built for all species with at least two different MICs, and 450 genomes for each antibiotic using nucleotide 7-mers as input features. The coloring depicts accuracy for predicting the MIC for each antibiotic and species based on a 5-fold cross-validation. Accuracies are reported within ±1 two-fold dilution step, which is the limit of resolution for most automated MIC detection methods. Antibiotic abbreviations are defined in Table S2.
In general, the quality of these models often tracks with sampling, with the undersampled bins usually having lower accuracies. However, there are cases where the sampling is relatively deep, but the F1 scores and accuracies remain low. For example, in K. pneumoniae there are 860 and 528 genomes with AST data for tetracycline and tigecycline, but the F1 scores for these antibiotics are only 0.763 and 0.781, respectively. In these cases, more detailed examination of the data is required to understand the low F1 scores. For example, there could be issues with the underlying dataset relating to biases in sampling, phylogeny, genome quality, AST methods, etc. There could also be a genuine biological reason for the low accuracies such as phenotypes that are not hard-coded in the genome, including gene expression differences or persistence phenotypes. In either scenario, the model serves as a useful starting point for both curation and research. It is also important to recognize that in the reverse case, where accuracies are high but sampling is low, more data are needed to ensure that the models remain generalizable. In these cases, it is often best to retrain a model as new data become available.
As data coverage improves and new modeling strategies are applied to the data, we will likely see incremental improvements in AMR prediction accuracies and their ability to generalize. In particular, several studies describing deep learning-based approaches have been published, and could offer a framework for improvement [45, 48, 82, 97]. This may eventually lead to diagnostic quality models that could be applied to whole genome or metagenomic sequence data.
Similar modeling approaches have also been used in source tracking projects to predict host organisms for E. coli and S. enterica strains [98–102], and may eventually be used on other phenotypes as data become available. An incisive use of machine learning models for feature extraction is also beginning to help disentangle our understanding of the epistatic changes and other previously unrecognized genomic changes that are correlated with AMR [46, 51, 96].
Concluding remarks
The objective in collecting and curating this AST dataset is to provide a resource to the research community that can be reused and shared. Well-curated structured datasets are required for building high-quality machine learning models and other tools. This is especially important in the biological sciences. The AST data provide a quantifiable phenotype for bacteria, so they offer one of the best opportunities for advancing AI in this space. Indeed, several machine learning studies have made use of this data collection, and they illustrate the many different ways that the data can be used for modeling [41–43, 45, 46, 49, 51, 52, 61, 96, 103, 104]. In addition to modeling, the data offer a means of identifying genomic regions involved in resistance and could aid in the development novel antibiotics and countermeasures to help prevent the spread and burden of AMR.
This data collection would not exist without the many contributions to science by the research community. We have curated the data as a means to advance the field, but consideration should be given to the authors of the original studies. Collecting these data from the literature, which includes the curation of data from supplemental spread sheets, tables and figures can be challenging and labor intensive. We strongly encourage data generators to submit AST data to NCBI using their structured antibiogram template. This ensures that the data are publicly available and long-lasting. It also reduces the potential errors that can result from secondary curation efforts.
As the field advances toward having millions of bacterial genomes, we should take a moment to look back upon the value of this massive data generation effort. In many cases, genomes are sequenced for the purposes of observing a natural phenomenon, such as looking for resistance genes or monitoring an outbreak. However, AI methods can be used to enable the design of targeted sequencing experiments aimed at solving specific biological problems and filling gaps in our current knowledgebase. As the cost of sequencing has gone down, projects that aim to sequence large strain collections with diverse phenotypes have become achievable and could lead to a wide range AI-enabled discoveries. For example, in addition to AMR, there are many other phenotypes that could be measured and collected, such as virulence and growth requirements, but sequenced genomes with these data remain surprisingly scarce [105, 106]. It is also possible that the aggregation of different types of metadata could lead to improvements in the accuracy or interpretability of AMR and other phenotype prediction models. In our opinion, more attention should be given to generating and storing metadata to improve the overall value and reusability of genome sequences. To this end, we hope that this AST data collection will serve as a reference point for designing targeted studies that will more rapidly improve our knowledge and ability to fight AMR.
Key Points
Antimicrobial resistance (AMR) is a major global health threat.
A necessary tool in studying and predicting AMR are genomes paired with phenotypes from laboratory-derived antimicrobial susceptibility tests (AST) for a given isolate.
In this paper, we describe a curated collection of over 67 000 publicly available genomes with AST data and demonstrate how it can be used to predict resistance phenotypes.
Supplementary Material
Acknowledgments
We thank the members of the BV-BRC and iSENTRY teams for their helpful comments. We are extremely grateful to the members of the research community who are publishing AST data and genomes for use in the public domain.
Margo VanOeffelen is a research assistant at the Fellowship for Interpretation of Genomes.
Marcus Nguyen is a data scientist at the University of Chicago and Argonne National Laboratory.
Derya Aytan-Aktug is a PhD student at the Technical University of Denmark specializing in Data Science and Microbiology.
Thomas Brettin is a strategic program manager for Computing and Life Sciences within the Computing, Environmental and Life Sciences Directorate at Argonne National Laboratory.
Emily M. Dietrich is the project manager of the NIH/NIAID-funded BV-BRC, as well as a technical science writer at the Computing, Environment and Life Sciences directorate at Argonne National Laboratory.
Ronald W. Kenyon is a senior scientist at the University of Virginia Biocomplexity Institute and Software Project Manager for the BV-BRC project.
Dustin Machi is a software engineer at the University of Virginia Biocomplexity Institute.
Chunhong Mao is a research associate professor at the University of Virginia Biocomplexity Institute.
Robert Olson is a senior software engineer in the Computing, Environment and Life Sciences Directorate of Argonne National Laboratory and University of Chicago, in Illinois, USA.
Gordon D. Pusch is a research scientist, bioinformatician and code developer for the Fellowship for Interpretation of Genomes.
Maulik Shukla is a project lead and computer scientist at Argonne National Laboratory, and data lead for the BV-BRC project.
Rick Stevens is a professor of Computer Science at the University of Chicago and the Associate Laboratory Director for Computing, Environment and Life Sciences at Argonne National Laboratory. He is the PI of the BV-BRC project.
Veronika Vonstein is a founding fellow and president of the Fellowship for Interpretation of Genomes.
Andrew Warren is a research assistant professor at the University of Virginia, senior bioinformatician for the BV-BRC project, and a coinvestigator for the Friend of Foe iSENTRY project.
Alice Rebecca Wattam is a research associate professor at the University of Virginia, bacterial outreach lead for the BV-BRC project, and a coinvestigator for the Friend or Foe iSENTRY project.
Hyunseung Yoo is a software engineer at Argonne National Laboratory, user-interface development lead for the BV-BRC project.
James J. Davis is a computational biologist at Argonne National Laboratory, bacterial services lead for the BV-BRC project, and a coinvestigator for the Friend or Foe iSENTRY project.
Contributor Information
Margo VanOeffelen, Fellowship for Interpretation of Genomes, Burr Ridge, IL, USA.
Marcus Nguyen, University of Chicago Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL, USA; Data Science and Learning Division, Argonne National Laboratory, Argonne, IL, USA.
Derya Aytan-Aktug, National Food Institute, Technical University of Denmark, Kgs. Lyngby, Denmark.
Thomas Brettin, University of Chicago Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL, USA; Computing Environment and Life Sciences, Argonne National Laboratory, Argonne, IL, USA.
Emily M Dietrich, University of Chicago Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL, USA; Computing Environment and Life Sciences, Argonne National Laboratory, Argonne, IL, USA.
Ronald W Kenyon, Biocomplexity Institute and Initiative, University of Virginia, Virginia, USA.
Dustin Machi, Biocomplexity Institute and Initiative, University of Virginia, Virginia, USA.
Chunhong Mao, Biocomplexity Institute and Initiative, University of Virginia, Virginia, USA.
Robert Olson, University of Chicago Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL, USA; Data Science and Learning Division, Argonne National Laboratory, Argonne, IL, USA.
Gordon D Pusch, Fellowship for Interpretation of Genomes, Burr Ridge, IL, USA.
Maulik Shukla, University of Chicago Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL, USA; Data Science and Learning Division, Argonne National Laboratory, Argonne, IL, USA.
Rick Stevens, Computing Environment and Life Sciences, Argonne National Laboratory, Argonne, IL, USA; Department of Computer Science, University of Chicago, Chicago, IL, USA.
Veronika Vonstein, Fellowship for Interpretation of Genomes, Burr Ridge, IL, USA.
Andrew S Warren, Biocomplexity Institute and Initiative, University of Virginia, Virginia, USA.
Alice R Wattam, Data Science and Learning Division, Argonne National Laboratory, Argonne, IL, USA; Biocomplexity Institute and Initiative, University of Virginia, Virginia, USA.
Hyunseung Yoo, University of Chicago Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL, USA; Data Science and Learning Division, Argonne National Laboratory, Argonne, IL, USA.
James J Davis, University of Chicago Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL, USA; Data Science and Learning Division, Argonne National Laboratory, Argonne, IL, USA; Northwestern Argonne Institute for Science and Engineering, Evanston, IL, USA.
Data availability
The AST data collection is currently maintained on the PATRIC FTP site (ftp://ftp.patricbrc.org/RELEASE_NOTES/PATRIC_genomes_AMR.txt), and is now also mirrored on the BV-BRC FTP site (ftp://ftp.bvbrc.org/RELEASE_NOTES/PATRIC_genomes_AMR.txt). Genomes and features can be downloaded by using the PATRIC and BV-BRC websites, FTP sites and the PATRIC command line interface tools (https://docs.patricbrc.org/cli_tutorial/#installing-the-cli-release). The modeling code and the AST data file used to build the models can also be found at https://github.com/BV-BRC/AMRMetadataReview_2021.
Funding
Federal funds from the United States Defense Advanced Research Projects Agency iSENTRY Friend or Foe program award [Contract No. HR0011150042 to J.J.D.], and by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, (under Contract No. 75N93019C00076 to R.S.).
References
- 1. Michael CA, Dominey-Howes D, Labbate M. The antimicrobial resistance crisis: causes, consequences, and management. Front Public Health 2014;2:145–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wintersdorff CJH, Penders J, Niekerk JM, et al. . Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front Microbiol 2016;7:173–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baker S, Thomson N, Weill F-X, et al. . Genomic insights into the emergence and spread of antimicrobial-resistant bacterial pathogens. Science 2018;360:733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hollis A, Ahmed Z. Preserving antibiotics, rationally. N Engl J Med 2013;369:2474–6. [DOI] [PubMed] [Google Scholar]
- 5. Goossens H, Ferech M, Vander Stichele R, et al. . Outpatient antibiotic use in Europe and association with resistance: a cross-national database study. The Lancet 2005;365:579–87. [DOI] [PubMed] [Google Scholar]
- 6. Riedel S, Beekmann SE, Heilmann KP, et al. . Antimicrobial use in Europe and antimicrobial resistance in Streptococcus pneumoniae. Eur J Clin Microbiol Infect Dis 2007;26:485. [DOI] [PubMed] [Google Scholar]
- 7. Shallcross LJ, Davies DSC. Antibiotic overuse: a key driver of antimicrobial resistance. Br J Gen Pract 2014;64:604–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ventola CL. The antibiotic resistance crisis: part 1: causes and threats. P T 2015;40:277–83. [PMC free article] [PubMed] [Google Scholar]
- 9. Nouwen JL. Controlling antibiotic use and resistance. Clin Infect Dis 2006;42:776–7. [DOI] [PubMed] [Google Scholar]
- 10. Sartelli M, Labricciosa FM, Barbadoro P, et al. . The global alliance for infections in surgery: defining a model for antimicrobial stewardship—results from an international cross-sectional survey. World Journal of Emergency Surgery 2017;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klein EY, Van Boeckel TP, Martinez EM, et al. . Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc Natl Acad Sci 2018;115:E3463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dadgostar P. Antimicrobial resistance: implications and costs. Infection and drug resistance 2019;12:3903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Naylor NR, Atun R, Zhu N, et al. . Estimating the burden of antimicrobial resistance: a systematic literature review. Antimicrob Resist Infect Control 2018;7:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jackson N, Czaplewski L, Piddock LJV. Discovery and development of new antibacterial drugs: learning from experience? J Antimicrob Chemother 2018;73:1452–9. [DOI] [PubMed] [Google Scholar]
- 15. Spellberg B, Gilbert DN. The future of antibiotics and resistance: a tribute to a career of leadership by John Bartlett. Clin Infec Dis 2014;59(Suppl 2):S71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vasala A, Hytönen VP, Laitinen OH. Modern tools for rapid diagnostics of antimicrobial resistance. Front Cell Infect Microbiol 2020;10:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reller LB, Weinstein M, Jorgensen JH, et al. . Antimicrobial susceptibility testing: a review of general principles and contemporary practices. Clin Infect Dis 2009;49:1749–55. [DOI] [PubMed] [Google Scholar]
- 18. Eigner U, Schmid A, Wild U, et al. . Analysis of the comparative workflow and performance characteristics of the VITEK 2 and phoenix systems. J Clin Microbiol 2005;43:3829–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kahlmeter G, Giske CG, Kirn TJ, et al. . Point-counterpoint: differences between the European committee on antimicrobial susceptibility testing and clinical and laboratory standards institute recommendations for reporting antimicrobial susceptibility results. J Clin Microbiol 2019;57:e01129–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. CLSI . Performance Standards for Antimicrobial Susceptibility Testing. CLSI supplement M100. Wayne, PA: Clinical and Laboratory Standards Institute, 2018. [Google Scholar]
- 21. The European Committee on Antimicrobial Susceptibility Testing . Breakpoint tables for interpretation of MICs and zone diameters, version 11.0, 2021. http://www.eucast.org. (4 August 2021, date last accessed).
- 22. Hatakeyama S, Ohama Y, Okazaki M, et al. . Antimicrobial susceptibility testing of rapidly growing mycobacteria isolated in Japan. BMC Infect Dis 2017;17:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karlowsky JA, Adam HJ, Golden AR, et al. . Antimicrobial susceptibility testing of invasive isolates of Streptococcus pneumoniae from Canadian patients: the SAVE study, 2011–15. J Antimicrob Chemother 2018;73:vii5–vii11. [DOI] [PubMed] [Google Scholar]
- 24. Salminen MK, Rautelin H, Tynkkynen S, et al. . Lactobacillus bacteremia, species identification, and antimicrobial susceptibility of 85 blood isolates. Clin Infect Dis 2006;42:e35–44. [DOI] [PubMed] [Google Scholar]
- 25. Davidson KE, Byrne BA, Pires AFA, et al. . Antimicrobial resistance trends in fecal salmonella isolates from northern California dairy cattle admitted to a veterinary teaching hospital, 2002-2016. PLoS One 2018;13:e0199928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Esteban-Cuesta I, Dorn-In S, Drees N, et al. . Antimicrobial resistance of Enterobacter cloacae complex isolates from the surface of muskmelons. Int J Food Microbiol 2019;301:19–26. [DOI] [PubMed] [Google Scholar]
- 27. Marr I, Sarmento N, O’Brien M, et al. . Antimicrobial resistance in urine and skin isolates in Timor-Leste. Journal of Global Antimicrobial Resistance 2018;13:135–8. [DOI] [PubMed] [Google Scholar]
- 28. Hendriksen RS, Bortolaia V, Tate H, et al. . Using genomics to track global antimicrobial resistance. Front Public Health 2019;7:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McDermott PF, Davis JJ. Predicting antimicrobial susceptibility from the bacterial genome: a new paradigm for one health resistance monitoring. J Vet Pharmacol Ther 2020;44:223–237. doi: 10.1111/jvp.12913. Epub 2020 Oct 3. PMID: 33010049. [DOI] [PubMed] [Google Scholar]
- 30. Su M, Satola SW, Read TD. Genome-based prediction of bacterial antibiotic resistance. J Clin Microbiol 2019;57:e01405–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bortolaia V, Kaas RS, Ruppe E, et al. . ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother 2020;75:3491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gupta SK, Padmanabhan BR, Diene SM, et al. . ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother 2014;58:212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alcock BP, Raphenya AR, Lau TTY, et al. . CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res 2019;48:D517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lakin SM, Dean C, Noyes NR, et al. . MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res 2016;45:D574–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hunt M, Mather AE, Sánchez-Busó L, et al. . ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microbial genomics 2017;3:e000131–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clausen PTLC, Zankari E, Aarestrup FM, et al. . Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J Antimicrob Chemother 2016;71:2484–8. [DOI] [PubMed] [Google Scholar]
- 37. Feldgarden M, Brover V, Haft DH, et al. . Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob Agents Chemother 2019;63:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zankari E, Allesøe R, Joensen KG, et al. . PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J Antimicrob Chemother 2017;72:2764–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bradley P, Gordon NC, Walker TM, et al. . Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and mycobacterium tuberculosis. Nat Commun 2015;6:10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davis JJ, Wattam AR, Aziz RK, et al. . The PATRIC bioinformatics resource center: expanding data and analysis capabilities. Nucleic Acids Res 2019;48:D606–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nguyen M, Brettin T, Long SW, et al. . Developing an in silico minimum inhibitory concentration panel test for Klebsiella pneumoniae. Sci Rep 2018;8:421–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nguyen M, Long SW, McDermott PF, et al. . Using machine learning to predict antimicrobial MICs and associated genomic features for nontyphoidal salmonella. J Clin Microbiol 2019;57:e01260–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nguyen M, Olson R, Shukla M, et al. . Predicting antimicrobial resistance using conserved genes. PLoS Comput Biol 2020;16:e1008319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pataki BÁ, Matamoros S, Putten BCL, et al. . Understanding and predicting ciprofloxacin minimum inhibitory concentration in Escherichia coli with machine learning. Sci Rep 2020;10:15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aytan-Aktug D, Clausen PTLC, Bortolaia V, et al. . Prediction of acquired antimicrobial resistance for multiple bacterial species using neural networks. mSystems 2020;5:e00774–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hyun JC, Kavvas ES, Monk JM, et al. . Machine learning with random subspace ensembles identifies antimicrobial resistance determinants from pan-genomes of three pathogens. PLoS Comput Biol 2020;16:e1007608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Khaledi A, Weimann A, Schniederjans M, et al. . Predicting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. EMBO Mol Med 2020;12:e10264–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shi J, Yan Y, Links MG, et al. . Antimicrobial resistance genetic factor identification from whole-genome sequence data using deep feature selection. BMC Bioinformatics 2019;20:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Davis JJ, Boisvert S, Brettin T, et al. . Antimicrobial resistance prediction in PATRIC and RAST. Sci Rep 2016;6:27930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jaillard M, Lima L, Tournoud M, et al. . A fast and agnostic method for bacterial genome-wide association studies: bridging the gap between k-mers and genetic events. PLoS Genet 2018;14:e1007758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kavvas ES, Catoiu E, Mih N, et al. . Machine learning and structural analysis of Mycobacterium tuberculosis pan-genome identifies genetic signatures of antibiotic resistance. Nat Commun 2018;9:4306–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kavvas ES, Yang L, Monk JM, et al. . A biochemically-interpretable machine learning classifier for microbial GWAS. Nat Commun 2020;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Camacho DM, Collins KM, Powers RK, et al. . Next-generation machine learning for biological networks. Cell 2018;173:1581–92. [DOI] [PubMed] [Google Scholar]
- 54. Lv J, Deng S, Zhang L. A review of artificial intelligence applications for antimicrobial resistance. Biosafety and Health 2021;3:22–31. [Google Scholar]
- 55. Anahtar MN, Yang JH, Kanjilal S. Applications of machine learning to the problem of antimicrobial resistance: an emerging model for translational research. J Clin Microbiol 2021;59:e0126020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lüftinger L, Májek P, Beisken S, et al. . Learning from limited data: towards best practice techniques for antimicrobial resistance prediction from whole genome sequencing data. Front Cell Infect Microbiol 2021;11:–610348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu Z, Deng D, Lu H, et al. . Evaluation of machine learning models for predicting antimicrobial resistance of Actinobacillus pleuropneumoniae from whole genome sequences. Front Microbiol 2020;11:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hicks AL, Wheeler N, Sánchez-Busó L, et al. . Evaluation of parameters affecting performance and reliability of machine learning-based antibiotic susceptibility testing from whole genome sequencing data. PLoS Comput Biol 2019;15:e1007349–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Aarestrup FM, Koopmans MG. Sharing data for global infectious disease surveillance and outbreak detection. Trends Microbiol 2016;24:241–5. [DOI] [PubMed] [Google Scholar]
- 60. Babu Rajendran N, Mutters NT, Marasca G, et al. . Mandatory surveillance and outbreaks reporting of the WHO priority pathogens for research & discovery of new antibiotics in European countries. Clin Microbiol Infect 2020;26:943.e941–6. [DOI] [PubMed] [Google Scholar]
- 61. Drouin A, Letarte G, Raymond F, et al. . Interpretable genotype-to-phenotype classifiers with performance guarantees. Sci Rep 2019;9:4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mahfouz N, Ferreira I, Beisken S, et al. . Large-scale assessment of antimicrobial resistance marker databases for genetic phenotype prediction: a systematic review. J Antimicrob Chemother 2020;75:3099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. World Health Organization . GLASS whole-genome sequencing for surveillance of antimicrobial resistance. Geneva, 2020;1–42.
- 64. Sayers EW, Beck J, Brister JR, et al. . Database resources of the National Center for biotechnology information. Nucleic Acids Res 2019;48:D9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Matamoros S, Hendriksen RS, Pataki BÁ, et al. . Accelerating surveillance and research of antimicrobial resistance – an online repository for sharing of antimicrobial susceptibility data associated with whole-genome sequences. Microb Genom 2020;6:e000342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Starks AM, Avilés E, Cirillo DM, et al. . Collaborative effort for a centralized worldwide tuberculosis relational sequencing data platform. Clin Infect Dis 2015;61:S141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. CDC & FDA . Antibiotic Resistance Isolate Bank. Atlanta (GA): CDC, April 2021. [Google Scholar]
- 68. Sánchez-Busó L, Yeats CA, Taylor B, et al. . A community-driven resource for genomic epidemiology and antimicrobial resistance prediction of Neisseria gonorrhoeae at Pathogenwatch. Genome medicine 2021;1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pathogenwatch : A Global Platform for Genomic Surveillance. https://pathogen.watch (4 August 2021, date last accessed).
- 70. Antonopoulos DA, Assaf R, Aziz RK, et al. . PATRIC as a unique resource for studying antimicrobial resistance. Brief Bioinform 2017;20:1094–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brettin T, Davis JJ, Disz T, et al. . RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep 2015;5:8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Clark K, Karsch-Mizrachi I, Lipman DJ, et al. . GenBank. Nucleic Acids Res 2016;44:D67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Leinonen R, Sugawara H, Shumway M, et al. . The sequence read archive. Nucleic Acids Res 2011;39:D19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Harrison PW, Ahamed A, Aslam R, et al. . The European Nucleotide Archive in 2020. Nucleic Acids Res 2020;49:D82–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Manson AL, Cohen KA, Abeel T, et al. . Genomic analysis of globally diverse Mycobacterium tuberculosis strains provides insights into the emergence and spread of multidrug resistance. Nat Genet 2017;49:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.CRyPTIC Consortium and the 100,000 Genomes Project. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N Engl J Med 2018;379:1403–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Food and Drug Administration (FDA) . NARMS Now. Rockville, MD: U.S. Department of Health and Human Services. https://www.fda.gov/animal-veterinary/national-antimicrobial-resistance- monitoring-system/narms-now-integrated-data (4 August 2021, date last accessed). [Google Scholar]
- 78. CRyPTIC . Comprehensive Resistance Prediction for Tuberculosis: an International Consortium. http://www.crypticproject.org (4 August 2021, date last accessed).
- 79. Peleg AY, Hooper DC. Hospital-acquired infections due to gram-negative bacteria. N Engl J Med 2010;362:1804–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Toval F, Köhler C-D, Vogel U, et al. . Characterization of Escherichia coli isolates from hospital inpatients or outpatients with urinary tract infection. J Clin Microbiol 2014;52:407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kallonen T, Brodrick HJ, Harris SR, et al. . Systematic longitudinal survey of invasive Escherichia coli in England demonstrates a stable population structure only transiently disturbed by the emergence of ST131. Genome Res 2017;27:1437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Moradigaravand D, Palm M, Farewell A, et al. . Prediction of antibiotic resistance in Escherichia coli from large-scale pan-genome data. PLoS Comput Biol 2018;14:e1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tsang KK, Maguire F, Zubyk HL, et al. . Identifying novel β-lactamase substrate activity through in silico prediction of antimicrobial resistance. Microbial genomics 2021;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Van Camp P-J, Haslam DB, Porollo A. Prediction of antimicrobial resistance in gram-negative bacteria from whole-genome sequencing data. Front Microbiol 2020;11:1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gladstone RA, McNally A, Pöntinen AK, et al. . Emergence and dissemination of antimicrobial resistance in Escherichia coli causing bloodstream infections in Norway in 2002–17: a nationwide, longitudinal, microbial population genomic study. The Lancet Microbe 2021;2:e331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kuijpers LMF, Le Hello S, Fawal N, et al. . Genomic analysis of salmonella enterica serotype Paratyphi a during an outbreak in Cambodia, 2013–2015. Microbial genomics 2016;2:e000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Klemm EJ, Shakoor S, Page AJ, et al. . Emergence of an extensively drug-resistant Salmonella enterica Serovar Typhi clone Harboring a promiscuous plasmid encoding resistance to fluoroquinolones and third-generation Cephalosporins. MBio 2018;9:e00105–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tanmoy AM, Westeel E, De Bruyne K, et al. . Salmonella enterica Serovar Typhi in Bangladesh: exploration of genomic diversity and antimicrobial resistance. MBio 2018;9:e02112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ingle DJ, Nair S, Hartman H, et al. . Informal genomic surveillance of regional distribution of Salmonella Typhi genotypes and antimicrobial resistance via returning travellers. PLoS Negl Trop Dis 2019;13:e0007620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Britto CD, Dyson ZA, Mathias S, et al. . Persistent circulation of a fluoroquinolone-resistant Salmonella enterica Typhi clone in the Indian subcontinent. J Antimicrob Chemother 2019;75:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Thanh DP, Karkey A, Dongol S, et al. . A novel ciprofloxacin-resistant subclade of H58 Salmonella Typhi is associated with fluoroquinolone treatment failure. Elife 2016;5:e14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chicco D. Ten quick tips for machine learning in computational biology. BioData Mining 2017;10:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nguyen G, Dlugolinsky S, Bobák M, et al. . Machine learning and deep learning frameworks and libraries for large-scale data mining: a survey. Artif Intell Rev 2019;52:77–124. [Google Scholar]
- 94. ValizadehAslani T, Zhao Z, Sokhansanj BA, et al. . Amino acid k-mer feature extraction for quantitative antimicrobial resistance (AMR) prediction by machine learning and model interpretation for biological insights. Biology (Basel) 2020;9:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Břinda K, Callendrello A, Ma KC, et al. . Rapid inference of antibiotic resistance and susceptibility by genomic neighbour typing. Nat Microbiol 2020;5:455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Aytan-Aktug D, Nguyen M, Clausen PTLC, et al. . Predicting antimicrobial resistance using partial genome alignments. mSystems 2021;6:e00185–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Arango-Argoty G, Garner E, Pruden A, et al. . DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 2018;6:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wheeler NE, Gardner PP, Barquist L. Machine learning identifies signatures of host adaptation in the bacterial pathogen Salmonella enterica. PLoS Genet 2018;14:e1007333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lupolova N, Dallman TJ, Holden NJ, et al. . Patchy promiscuity: machine learning applied to predict the host specificity of Salmonella enterica and Escherichia coli. Microbial genomics 2017;3:e000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhang S, Li S, Gu W, et al. . Zoonotic source attribution of Salmonella enterica serotype Typhimurium using genomic surveillance data, United States. Emerg Infect Dis 2019;25:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wheeler NE. Tracing outbreaks with machine learning. Nat Rev Microbiol 2019;17:269–9. [DOI] [PubMed] [Google Scholar]
- 102. Munck N, Njage PMK, Leekitcharoenphon P, et al. . Application of whole-genome sequences and machine learning in source attribution of Salmonella Typhimurium. Risk Anal 2020;40:1694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Her HL, Wu YW. A pan-genome-based machine learning approach for predicting antimicrobial resistance activities of the Escherichia coli strains. Bioinformatics 2018;34:i89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Colbaugh R, Glass K. Predicting Antimicrobial Resistance via Lightly-Supervised Learning. In: 2019IEEE international conference on systems, Man and Cybernetics (SMC) IEEE, 2019. [Google Scholar]
- 105. Price MN, Wetmore KM, Waters RJ, et al. . Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 2018;557:503–9. [DOI] [PubMed] [Google Scholar]
- 106. Pincus NB, Ozer EA, Allen JP, et al. . A genome-based model to predict the virulence of Pseudomonas aeruginosa isolates. MBio 2020;11:e01527–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The AST data collection is currently maintained on the PATRIC FTP site (ftp://ftp.patricbrc.org/RELEASE_NOTES/PATRIC_genomes_AMR.txt), and is now also mirrored on the BV-BRC FTP site (ftp://ftp.bvbrc.org/RELEASE_NOTES/PATRIC_genomes_AMR.txt). Genomes and features can be downloaded by using the PATRIC and BV-BRC websites, FTP sites and the PATRIC command line interface tools (https://docs.patricbrc.org/cli_tutorial/#installing-the-cli-release). The modeling code and the AST data file used to build the models can also be found at https://github.com/BV-BRC/AMRMetadataReview_2021.