

Abstract
Genomic instability consists of a range of genetic alterations within the genome that contributes to tumor heterogeneity and drug resistance. It is a well-established characteristic of most cancer cells. Genome instability induction results from defects in DNA damage surveillance mechanisms, mitotic checkpoints and DNA repair machinery. Accumulation of genetic alterations ultimately sets cells towards malignant transformation. Recent studies suggest that miRNAs are key players in mediating genome instability. miRNAs are a class of small RNAs expressed in most somatic tissues and are part of the epigenome. Importantly, in many cancers, miRNA expression is dysregulated. Consequently, this review examines the role of miRNA dysregulation as a causal step for induction of genome instability and subsequent carcinogenesis. We focus specifically on mechanistic studies assessing miRNA(s) and specific subtypes of genome instability or known modes of genome instability. In addition, we provide insight on the existing knowledge gaps within the field and possible ways to address them.
Keywords: genomic instability, chromosomal instability, miRNA, DNA repair
1. Introduction
Cancer ranks as a leading cause of global deaths [1]. The GLOBOCAN 2020 database compiled by the International Agency for Research on Cancer (IARC) estimated 19.3 million new cases of cancer and almost 10 million cancer deaths across the globe for the year 2020 [1]. Cancer risk factors are best differentiated into genetic predispositions and external environmental and/or occupational exposures to carcinogens [2]. Inherited genetic mutations are believed to play a major role in 5 to 10% of all cancers, while the remaining 90–95% are caused by exposure to carcinogens such as sunlight, air pollution, smoke and heavy metals [2]. In most cases, cancer develops as a consequence of nuanced interplay of genetic susceptibilities and environmental, lifestyle and occupational exposures [3].
Cellular transformation, the key step in carcinogenesis, arises from different insults to cells resulting in alterations in epigenetics, chromosome structure and number and clonally selected heterotypic interactions, leading to attainment of malignancy, as evidenced by the six hallmark signatures [4]. The six hallmarks were first described in 2000: sustaining proliferative signaling, resisting cell death, evading growth suppressors, inducing angiogenesis, enabling replicative immortality, and activating invasion and metastasis [5]. Ten years later, two emerging hallmarks: deregulating cellular energetics and avoiding immune destruction, and two enabling characteristics: genomic instability (genome instability) and mutation, and tumor-promoting inflammation were added to the initial list of cancer hallmarks [6]. This organization of cancer traits enables the understanding of the complex biology of cancer and encourages progress in cancer research.
Research into mechanisms of carcinogenesis reveals that induction of genome instability is one of the earliest steps that launches the cells towards malignancy [7,8]. Genome instability, a feature of most cancer cells [9,10], refers to a range of genetic alterations within the genome that contributes to tumor heterogeneity and drug resistance [11]. Depending on the nature of the change involved, genome instability can be broadly classified into increased rates of base pair mutations, microsatellite instability, and chromosome instability (CIN). genome instability favors carcinogenesis as it increases the chances of acquiring oncogenic mutations enabling the onset of cancer hallmarks [5,6,12,13]. Consequently, under normal physiological conditions, a multitude of molecular machineries are involved in closely monitoring and maintaining genomic stability. These include maintenance of DNA replication fidelity in S phase (including telomere maintenance), smooth cell cycle progression and checkpoint control, accurate chromosome segregation in mitosis, and repair of DNA damage [12–14]. Dysregulation of one or more of these processes is well demonstrated to lead to genome instability which subsequently acts as harbinger of cancer.
While genome instability has been studied for a considerable time, new mechanisms are emerging in the last few years, enhancing our understanding of the contribution of genome instability to carcinogenesis [15]. A rapidly emerging paradigm in the field of genome instability and carcinogenesis is modulation of genes and pathways by microRNAs (miRNA). Recent studies have brought miRNA-mediated mitotic regulation and DNA damage response under the spotlight [16–19]. Despite the known association between dysregulated miRNA expression and genome instability, the precise mechanisms by which miRNAs can causally induce genome instability have remained elusive till recently.
miRNAs are endogenous short non-coding RNAs (containing ~22 nucleotides) that are part of the epigenome and play a significant role in the regulation of gene expression at the post-transcriptional level [20]. The biogenesis of the mature miRNA has been extensively described [21,22]. miRNAs are transcribed by RNA-polymerase II and processed by the enzymes drosha ribonuclease III (DROSHA) and dicer 1 ribonuclease III (DICER). Dicer and transactivation response element RNA-binding protein (TRBP) recruit Argonaute proteins (AGO2) to mediate the assembly of the RNA-induced silencing complex (RISC) [23]. RISC is a ribonucleoprotein complex that binds to a miRNA and guides it to the target mRNA to regulate translation [24]. miRNAs target cytosolic mRNAs by forming complementary base pairing, typically with the 3’ untranslated regions (3’-UTRs) of the target mRNAs within the RISC complex. This binding recruits (AGO2) to their cognate mRNA targets [25] leading to post-transcriptional gene silencing. miRNA targeting is reliant on base pairing of the seed region, nucleotides 2–7, of the miRNA to sites in mRNA 3′ UTRs, Such base pairing mediated mRNA targeting leads to decreased translation, deadenylation or degradation of the mRNA [26–28]. In cases of perfect base pairing of miRNAs with the seed regions of their target mRNA, the mRNA is degraded. However, imperfect base pairing leads to repression of mRNA translation and restoration of translation is possible when the repressor miRNA is degraded [29]. In addition, circular RNA, a novel class of endogenous non-coding RNA, may act as miRNA sponges protecting target mRNAs from miRNA-dependent degradation [30]. miRNAs play a complex and crucial role in many biological processes (cell proliferation, apoptosis, response to therapy, diseases, development) and can create complex feedback regulatory loops in a cell [31,32]. A single miRNA can target hundreds of mRNAs and each mRNA can be targeted by multiple miRNAs in different regions [29,32]. Occasionally, miRNAs also can bind to the 5’-UTR, promoter or the open reading frame (ORF) regions, creating a complex network of interactions [32]. In fact, miRNAs target more than 60% of protein-coding regions in the human genome which bears testimony to the widespread effects these molecules have on transcriptome and proteome diversity as well as function in a context dependent manner. miRNAs are frequently found dysregulated in human malignancies, acting as a novel class of oncogenes and tumor suppressors [24,31,33,34].
2. Review Focus and Literature Search
The present review focuses on the role of miRNA dysregulation as a causal step for induction of genome instability and subsequent carcinogenesis. We specifically highlight the studies that have established a mechanistic relationship between miRNA(s) and specific subtypes of genome instability or known modes of genome instability induction. With this objective, we carried out multiple literature searches employing the PubMed search engine between February and March 2021. The keywords used for this search included various combinations of the terms, “miRNA” AND “genomic instability”, “chromosomal instability”, “microsatellite instability”, “structural aberration”, “numerical aberration”, “aneuploidy”, “DNA damage”, “cell cycle”, “mitotic checkpoint complex”, “spindle assembly checkpoint” and “centrosome amplification” AND “cancer” or “carcinogenesis”. The search identified a list of 370 candidate articles. We further refined our search to exclude articles which were reviews, for which the full paper was not available, those which were retracted, had no data connecting the three search categories (miRNA, one form of genome instability and cancer) or were association studies with no mechanistic examination of the interaction of miRNA dysregulation and genome instability. A list of 27 research articles passed all the exclusion criteria and were thus included for this review. The details regarding selection of articles included in this review are depicted in Fig. 1.
Figure 1.
Summary of literature search strategy depicting screening procedures and exclusion criteria.
3. Chromosomal Instability
Chromosomal instability (CIN) is defined as an increased occurrence of chromosome segregation errors during mitosis. CIN can be classified as numerical or structural [35–38] (Fig. 2). Numerical CIN is characterized by gain or loss of whole chromosomes possibly due to errors in the distribution of DNA to daughter cells during cell division, while structural CIN is characterized by gains, losses or translocations of parts of one or multiple chromosomes without necessarily changing the number of chromosomes (Fig. 2) [35,39]. CIN has been recognized as a hallmark of cancer and a source of intratumor heterogeneity, increasing the chances of selection in favor of an advantageous karyotype [39]. Approximately 60–80% of human tumors exhibit chromosomal abnormalities suggestive of CIN, and most solid tumors are aneuploid as a consequence of increased rates of CIN [38,40,41]. Direct correlations between CIN and poor prognosis, metastasis, recurrence and drug resistance have been shown for multiple tumors [39,42]. Various mechanisms underlying CIN have been examined (Fig. 2), including defects in chromosome cohesion, centrosome copy number, kinetochore-microtubule attachment dynamics, cell cycle regulation and DNA damage repair components [43]. miRNAs are known to target and modulate the expression of important protein components of these biological processes [19,44,45]. In addition, numerous miRNAs are dysregulated during carcinogenesis as previously discussed [46,47]. Thus, it is clear that miRNAs promote genomic and chromosomal instability contributing to tumorigenesis. However, a deeper understanding of how miRNA changes can bring about genomic instability is lacking.
Figure 2.
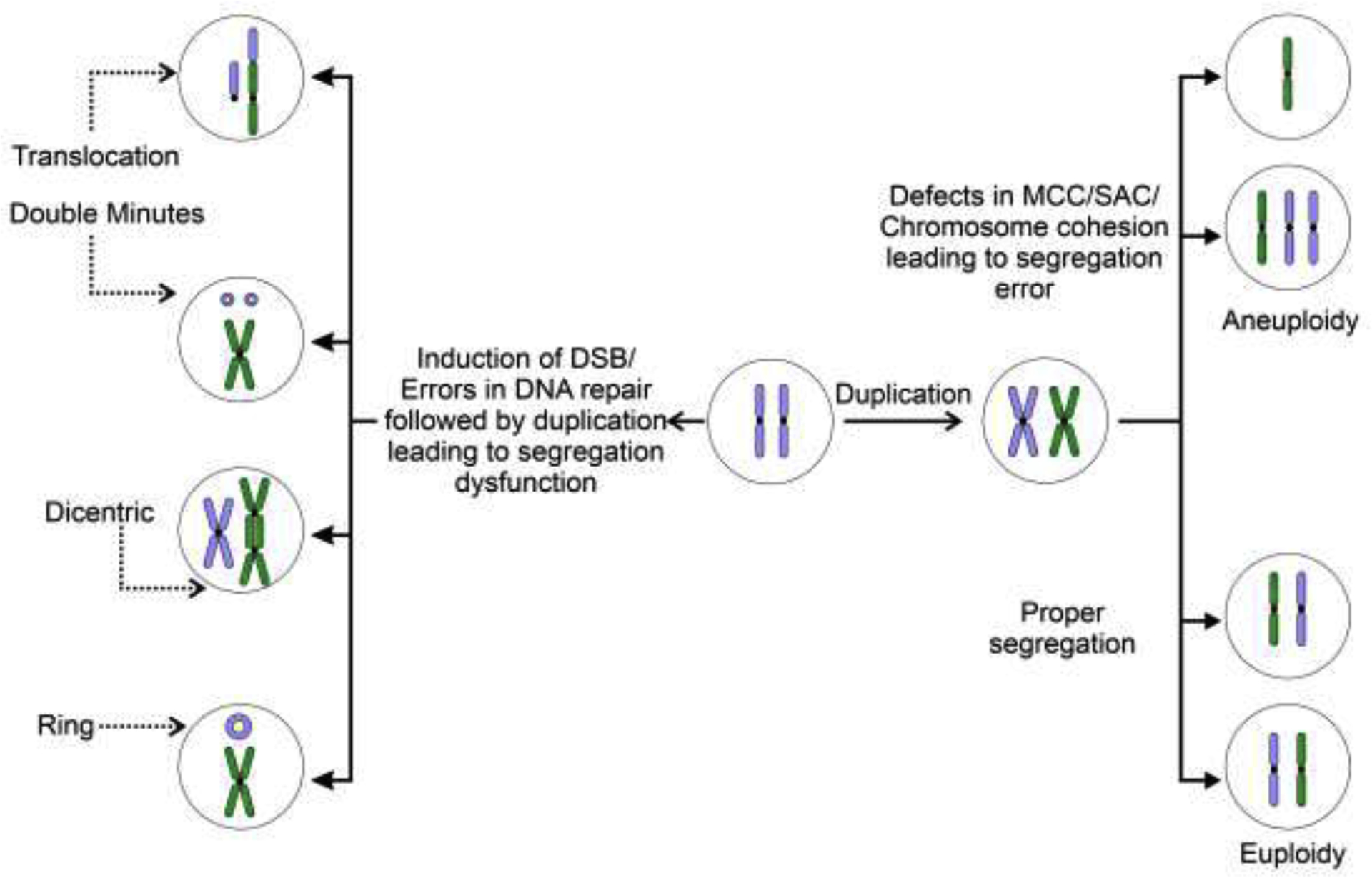
Simplified schematic representation of mechanisms leading to various forms of CIN (only the most common forms of CIN are shown). Numerical and structural chromosomal instability arise from improper segregation of chromosome(s) during mitosis. Numerical and structural alterations predispose chromosomes to subsequent chromosomal alterations, increasing instability.
3.1. miRNA and Numerical CIN in Cancer
Alteration of chromosome number from the normal diploid state (or haploid in germ cells) is commonly referred to as numerical chromosomal aberration. Numerical aberration can be broadly classified into aneuploidy (different from diploid number, but not a perfect multiple of the haploid set) and polyploidy (perfect multiple of the haploid set greater than the diploid number). While numerical aberration is related to etiology of cancer and intellectual disability disorders [48–52], it is important to reiterate that aberrant chromosome number does not automatically lead to genomic instability [49]. Evidence regarding the role of polyploidy in cancer is an emerging field [51,53], with meager data available on role of miRNA in induction of polyploidy, if any. Thus, this review concentrates on the role of miRNA in aneuploidy and cancer.
The connection between aneuploidy and carcinogenesis is well established and has been the subject of multiple excellent reviews [49,50]. Aneuploidy is a hallmark of several cancers spanning across multiple tissue types [50] and can lead to carcinogenesis via induction of oncogene expression, epithelial-mesenchymal transition, chemoresistance and immune invasion [49,50,54,55]. A few studies have explored the effect of aneuploidy on miRNA expression [56,57]; however, the effect of miRNAs on aneuploidy remains poorly understood. The mechanistic relationship between miRNA regulation of aneuploidy have been examined in a few studies. Overexpression of miR-26a in breast cancer cell lines (MCF-7 and MDA-MB-231) and mouse embryonic fibroblasts (MEFs) induced mitotic and cytokinetic defects as well as aneuploidy and centrosome defects, enhancing tumorigenesis [58]. A higher proportion of cells containing >80 chromosomes was observed in MCF-7 and MDA-MB-231 overexpressing miR-26a. The same effect on chromosomes number was also seen in non-cancerous MEFs overexpressing miR-26a, indicating that this miRNA could be a key regulator in the acquisition of CIN [58]. It was further demonstrated that overexpression of miR-26a in MCF-7 cells, MDA-MD-231 cells and MEFs resulted in abnormal mitotic spindles, known as a major cause of incorrect attachment of kinetochores leading to mis-segregation of chromosomes [58,59]. In HEK293 cells and in renal cell carcinoma cell line 786-O, overexpression of miR-210 led to heightened induction of centrosome amplification resulting in multipolar spindle formation and aneuploidy in about 20% of cells [60]. The effects of miR-28-5p overexpression were investigated in HeLa, HCT-116, RPE-1, and IMCD-3 cell lines [61]. Mis-segregation errors such as multiple and single lagging chromosomes and increased aneuploidy were observed with miR-28-5p overexpression. Additionally, the authors demonstrated that inhibition of miR-28-5p restored mitotic checkpoint proficiency and chromosomal stability in 786-O renal cell carcinoma cells [61]. Together, these results suggest that aneuploidy observed in these cell lines could be a consequence of miR-28-5p modulation. Analysis of HeLa cells overexpressing miR-let-7b revealed increased monosomy and trisomy about two times as often as in control cells, as well as multipolar spindles [62]. In another study from the same group, HeLa and HCT-116 cells were transfected with miR-493-3p; these cells showed premature sister chromatid separation and aneuploidy [63]. More studies are necessary to unravel how dysregulated miRNA expression can impact aneuploidy and the mode of regulation involved in the process.
3.2. miRNA and Structural CIN in Cancer
Human cancers frequently harbor alterations in chromosome structure, ranging from chromosome arm-level deletions or amplifications to alterations connecting multiple chromosomes [64,65]. One or more forms of structural genomic rearrangements can be found in tumors. Chromosome amplification and deletion are the most common structural chromosome abnormalities, occurring in 88% of cancer samples, followed by unbalanced translocations [66]. The most frequently lost chromosomes are 8p and 17p, whereas the most frequently gained is the 8q; the two least changed are 2p and 2q [65]. As a consequence, structural CIN can alter DNA copy numbers, rearrange DNA sequences and perturb regulatory pathways, underlying disease pathogenesis [64]. Structural chromosomal abnormalities are believed to arise from induction of telomere erosion, double-strand DNA breaks (DSB), replication stress and chromothripsis [67–69]. A recent review discussed the involvement of miRNAs in regulatory networks such as Ataxia Telangiectasia Mutated (ATM) and Ataxia Telangiectasia And Rad3-Related Protein (ATR) pathways and the mismatch repair machinery, affecting the DNA damage/repair process in several types of solid tumors [70]. These observations add another layer of complexity on the regulation of chromosomal stability and tumor development. Although increasing evidence has suggested miRNAs as essential players in the regulation of the DNA damage/repair network, a few studies have explored the mechanisms by which miRNAs affect this network and ultimately lead to CIN. Ding et al., assessed the link between miR-497, the structure specific recognition protein 1 (SSRP1), and genomic instability in hepatocellular carcinoma (HCC) [71]. SSRP1 is involved in transcriptional regulation, DNA damage repair, and cell cycle regulation [72–74]. In HCC samples, SSRP1 expression was significantly upregulated compared with the paired non-neoplastic tissues. In these same samples, a greater fraction of copy number altered genome and higher p53 and RB1 mutation frequencies were detected [71]. Additionally, a negative correlation between miR-497 and SSRP1 expression levels was observed in HCC samples. To confirm these findings, the authors overexpressed miR-497 in HepG2 and LM3 cells, leading to decreased SSRP1 mRNA and protein levels. [71]. The authors speculated that HCC progression caused by accumulating extra copies of DNA and gene mutations in cells is due to the instability provoked by SSRP1 dysregulation via miR-497 modulation [71]. Overexpression of miR-223 in Jurkat cells decreased the percentage of cells showing chromosomal translocation phenotypes, including double minutes, cruciform structure, dicentrics, and rings [75]. Furthermore, mice with homozygous deletion of the miR-223 locus showed a two-fold increase in hematopoietic cell chromosomal translocations compared with the WT, validating the role for miR-223 in maintaining genomic stability [75].
Only two studies have evaluated the impact of miRNA dysregulation on chromosome number and structural abnormalities. In a recent study from our group, stable overexpression of miR-186 in immortalized but non-malignant HaCaT cell line resulted in 2.5-fold increase in the number of cells with supernumerary chromosomes after 4 weeks of passaging and a 4-fold increase in conjunction with chronic arsenic exposure (100 nM) at 8 weeks [76]. Additionally, miR-186 overexpression for 8 weeks (with or without chronic arsenic exposure) also gave rise to metaphases with >200 chromosomes, which roughly corresponds to 10N, indicating miR-186 overexpression is responsible for inducing endomitosis. Structural abnormalities such as dicentrics, ring chromosomes and double minutes were also seen in cells overexpressing miR-186 [76]. Pagotto et al examined the causal contribution of miR-155 overexpression to CIN induction [77], a common observation in multiple malignant tumors [78]. Transformed human adult dermal fibroblasts show high levels of miR-155-5p and have marked gain or loss of whole chromosomes, chromosomal amplifications, unbalanced translocations and deletions [77]. Using lentiviral transduction, the authors knocked down miR-155-5p in this cell line resulting in reversion to a near-diploid karyotype [77].
4. miRNA and Microsatellite Instability (MIN) in Cancer
Microsatellites are short motifs of 1–6 nucleotides that are tandemly iterated up to 50 times [79,80]. Such sequences are common in eukaryotes accounting for ~3% of the human genome [81]. Microsatellite repeats are characterized by somatic hypermutability arising out of a combination of replication slippage, indel slippage and point mutations [82,83]. Under normal physiological conditions, the repeat number of microsatellites is kept under strict control by the action of mismatch repair (MMR) [84]. However, MMR malfunction results in induction of hypermutation in microsatellite loci throughout the genome leading to microsatellite instability (MIN). MIN has been implicated in the etiology of diseases including neurodegenerative disorders and cancer [85]. First identified and most widely studied in colorectal cancer [86–88], the role of MIN in the pathogenesis of a multitude of cancers is currently well-established [88–92].
Since MMR system plays a crucial role in maintenance of microsatellite loci, miRNAs targeting and abrogating the normal expression pattern of the MMR pathway members are likely to induce MIN. Several studies have examined the effects of miRNA with respect to MMR modulation (reviewed in [70]). However, most of these studies are associative in nature and have not empirically examined if the differentially expressed miRNA is causal to the MIN or merely an outcome [93–95]. In a study on colorectal cancer derived data from the TCGA database, Ashizawa et al., demonstrated that miR-148a-3p was consistently downregulated in MMR deficient samples along with a concomitant induction of its putative target Programmed Death-Ligand 1 (PD-L1) expression [96]. They subsequently demonstrated that miR-148a-3p directly binds to 3’-UTR of PD-L1 suppressing its expression at the mRNA and protein level along with suppression of growth proliferation of colorectal cancer cell lines [96]. Unfortunately, the authors did not address the question if overexpression of miR-148a-3p mimic could lead to MIN directly. However, in another study on colorectal cancer, it was shown that miR-155 targets several members of the MMR pathway employing dual luciferase assay [97]. Using Colo-320 DM CRC cell line stably overexpressing miR-155, this group demonstrated that such overexpression led to the suppression of MutL Homolog 1 (MLH1), MutS Homolog 2 (MSH2) and MutS Homolog 6 (MSH6) expression along with a concomitant length change in reporter repeat sequences [97]. This provides unequivocal evidence that miR-155 overexpression is causal to MIN induction by modulation of MMR pathway. In another interesting study, Huang et al., demonstrated that miR-129-2 targets 3’UTR of SRY-Box Transcription Factor 4 (SOX4), which is induced in endometrial cancer tissues as well as cell lines [98]. They further showed that in the endometrial cancer tissue samples and cell lines, miR-129-2 expression is lost due to 5’ CpG hypermethylation leading to induction of its downstream target SOX4, which could be suppressed by pharmacological reactivation of miR-129-2 expression [98]. Such hypermethylation was also found to be positively correlated with MIN status in tumor tissues. However, the authors did not probe if the cell lines overexpressing miR-129-2 also showed high MIN, which would have demonstrated that hypermethylation mediated miRNA-silencing could play a causal role in bringing about MIN. Interestingly, in 2013, Koole et al., developed a fluorescence based MIN reporter system [99]. Employing this system, they screened a retroviral miRNA expression library and demonstrated that miR-21 overexpression leads to induction of MIN, possibly by targeting and downregulating the expression of the key MMR member MSH2 [99].
Ultimately, while existing literature demonstrates widespread association of dysregulated miRNA expression with MIN, mostly by targeting MMR family members, there are minimal direct mechanistic data causally linking the two. Studies are needed to examine if reversal of the candidate miRNA expression can rescue the MIN phenotype to provide clarity to the nature of the relationship between miRNA dysregulation and MIN. Advantage could be taken of the reporter systems such as the one developed by Koole et al., [99] for this purpose.
5. Checkpoints & Pathways Involved in Genomic Stability
5.1. DNA damage
DNA damage is continuously occurring in cells. This damage must be corrected in order to prevent the induction of genomic instability and subsequent neoplastic transformation [100]. Endogenous factors which damage DNA include reactive oxygen species (ROS) [101], replication errors (including base substitution, single base insertion, deletion errors, and strand slippage), topoisomerase action, base deamination, abasic site generation, and DNA methylation. Exogenous agents that damage DNA include ultraviolet (UV) and ionizing radiation (IR), alkylating agents, aromatic amines, polycyclic aromatic hydrocarbon, N-nitrosamines, estrogen, genotoxic and carcinogenic compounds, and environmental stresses [102]. Genomic instability, which occurs as a result of genome assault by endogenous and exogenous agents and is a characteristic of almost all human cancers, can be divided into two groups: small range aberrations such as driver point mutations, or large range aberrations which include chromosomal rearrangements [12,103–106]. The following section focuses on small range aberrations specifically.
To maintain genomic integrity, all organisms have developed pathways and associated processes to respond to DNA damage and faithfully repair DNA [107]. The first step of the DNA damage response (DDR) is DNA lesion recognition followed by activation of the DNA damage signaling cascade which promotes a pause in cell-cycle progression and recruitment of appropriate downstream repair pathways (Fig. 3). Repair mechanisms specific for many types of DNA lesions have evolved including mismatch repair (MMR) for mispaired DNA bases; base excision repair (BER) for small, chemically altered DNA bases; nucleotide excision repair (NER) which repairs pyrimidine dimers and bulky chemical adducts; interstrand crosslink repair (ICL) which integrates distinct repair activities from several pathways to remove crosslinks [108,109]; single-strand break repair which repairs oxidative attack by ROS and altered or damaged bases as part of BER, or DNA topoisomerase 1 during transcription and DNA replication, and is involved in double-stranded DNA break repair [110]; and classical nonhomologous end joining (NHEJ), homologous recombination (HR), or microhomology-mediated end joining (MMEJ, or alternative NHEJ), for DNA double-strand breaks (DSBs) [111] (Fig. 3). Each of the aforementioned repair mechanisms are initiated by coordinated recruitment of precise, spatiotemporally regulated factors to sites of DNA damage [112]. The function and recruitment of DNA repair pathways have been thoroughly covered in other exceptional reviews [107,108,111,113]. In the next section, the repair pathways that are targeted by miRNAs simultaneously causing chromosomal instability are specifically discussed. Although both NER and BER protect against genomic instability and carcinogenesis [114–117], no study has experimentally demonstrated mechanistic relationships between miRNA and NER/BER function leading to genomic instability and cancer. Consequently, these DNA repair pathways have not been included in this review.
Figure 3.
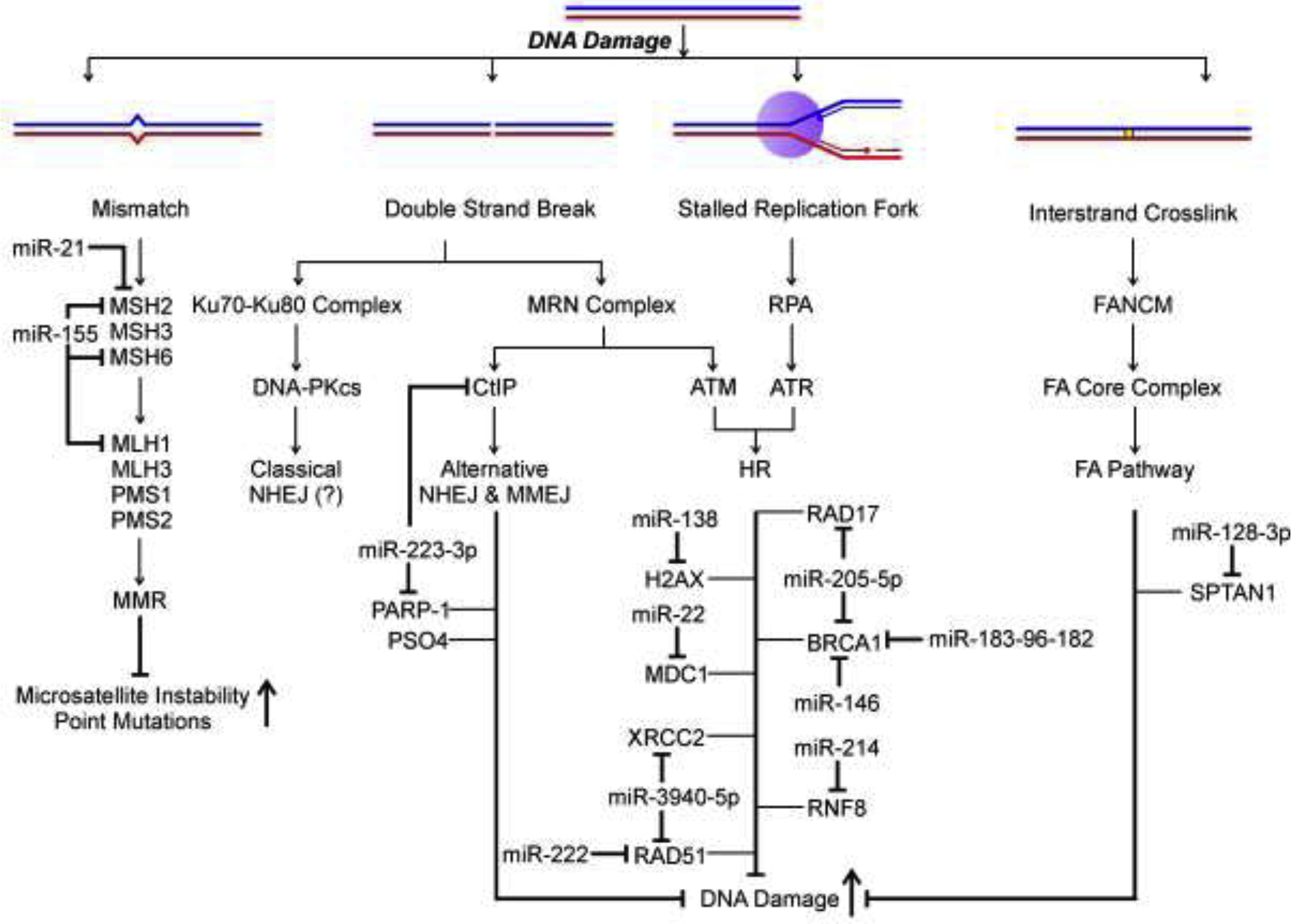
Simplified schematic representation summarizing the causal relationships by which miRNA dysregulation can lead to accumulation of heightened DNA damage. The figure includes only those forms of DNA damage, DDR and DNA repair pathways whose induction/perpetuation have been causally linked with at least one dysregulated miRNA expression (Mismatch Repair, Non-Homologous End Joining, Homologous Recombination and Interstrand Crosslink Repair). “(?)” denotes that so far no information is available for the miRNA modulation of the particular DNA damage pathway.
Numerous studies have demonstrated that inherited DNA repair gene mutations result in genomic instability and predispose individuals to the development of various cancers [12]. However, in sporadic cancers, mutations in known repair genes are limited, suggesting that genomic instability in many sporadic human cancers is due to other contributing factors. To date, few studies have investigated the miRNA regulation of DNA repair genes and direct induction of structural chromosomal instability by targeting miRNAs (Fig. 3). The majority of studies described in this section experimentally demonstrate repressed reporter activity due to miRNA targeting of specific mRNA sequences (detailed in Table 1), miRNA suppression of endogenous protein levels and chromosomal instability.
Table 1:
Experimental evidence for targeting of mRNA 3’UTRs by cognate miRNAs*
| miRNA | TargetGene | Binding Sequence on Target | Reference |
|---|---|---|---|
| miR-210 | E2F3 | 5' …UGUCUAAACGCACA… 3' | [60] |
| miR-148a-3p | CD274(PD-L1) | 5' …GCACUG… 3' | [96] |
| miR-129-5p | SOX4 | 5' …GCAAAA… 3' | [98] |
| miR-129-3p | SOX4 | 5' …AAGGGCU… 3' | [98] |
| miR-21 | MSH2 | 5' …AUAAGCUA… 3' | [99] |
| miR-155 | MLH1 | Not Described | [97] |
| miR-155 | MSH2 | Not Described | [97] |
| miR-155 | MSH6 | Not Described | [97] |
| miR-497 | SSRP1 | 5' …ACUCUGCUGCU… 3' | [71] |
| miR-497 | SSRP1 | 5' …AGGAUGUAG-CUGCUGCU… 3' | [71] |
| miR-146a-5p | MALAT1 | 5' …AGUUCUC… 3' | [128] |
| miR-216b-5p | MALAT1 | 5' …TAGAGAU… 3' | [128] |
| miR-22 | MDC1 | 5' …UGUCAUAAGUGGCAGCU… 3' | [140] |
| miR-222 | RAD51 | Not Described | [141] |
| miR-205 | RAD17 | 5' …GAUGUGAAGG… 3' | [132] |
| miR-205 | BRCA1 | 5' … GACUUCUG-GCUAUGCAAGG… 3' | [132] |
| miR-128-3p | SPTAN1 | 5'…AUCAUGUCACUGUGG…3' | [122] |
| miR-138 | H2AX | 5'… CAAGCACCUAGAUACCAGC… 3' | [126] |
| miR-214 | RNF8 | 5'…CAACCUGCUG…3' | [129] |
| miR-223-3p | PARP-1 | 5'…GGUAGAU..A..AAACUGAC.. | [75] |
| miR-223-3p | CTIP | 5'…CCG..A.AAGUAAAACGUGAGA..3' | [75] |
| miR-223-3p | PSO4 | 5'…AUGACCAGCUUAAAACUAGCU..3' | [75] |
| miR-26a | CHFR | 5' …UCUGGAAUAAUACUUGAA… 3' | [58] |
| miR-509-3-5p | PLK1 | 5' …GUGCCAUGUCUGCAGUG… 3' | [160] |
| miR-193a-3p | RASSF1 | 5' …GUGUGAGUGUGACAGGGCCAGUGG… 3' | [161] |
| let-7b | Aurora B | 5' … UCCCUUAUCUGUUUUCUACCUCC… 3' | [62] |
| miR-155 | BUB1 | 5' …CCCTTGTT.CCCTTAGCATTA… 3' | [77] |
| miR-125b | MAD 1 | 5' …UGCAUCAAUCGGACUCGGGGGC… 3' | [163] |
| miR-493-3p | MAD 2 | 5' …CCUUUUGACCUUCAU… 3' | [63] |
| miR-28-5p | MAD 2 | 5' …ACAAUGAAAUAUUGCUGUAUAGCUCCUU… 3' | [61] |
All listed studies assessed the interaction of miRNA and the specific target sequence by luciferase reporter assay. Sequences represented in bold and underline correspond to the binding nucleotides between mRNA and cognate miRNA.
Previous studies demonstrate that nonerythroid αII spectrin (SPTAN1) localizes to the nucleus, and plays important roles in repair of ICLs, chromosomal instability [118], and in cancer development and progression [119]. Following formation of ICLs, SPTAN1 acts as a scaffold and binds FA Complementation Group G (FANCG), FA Complementation Group A (FANCA), and Xeroderma Pigmentosum, Complementation Group F (XPF) [120,121]. In the lung cancer cell line A549, Zhang and colleagues showed that overexpression of miR-128-3p resulted in decreased SPTAN1 and increased chromosomal aberrations [122]. Furthermore, miR-128-3p targeting of the SPTAN1 3’-UTR was demonstrated via decreased luciferase reporter activity following co-transfection with miR-128-3p mimic and this outcome was reversed by addition of a miR-128-3p inhibitor [122] (Fig. 3).
DNA double-stranded breaks (DSBs) are highly toxic to cells because left unresolved, they lead to chromosomal rearrangements and structural variations. To repair DSBs, either homologous recombination (HR) or non-homologous end joining (NHEJ) can be used. The ATM (Ataxia telangiectasia mutated) signaling cascade within the HR pathway relies on initial recruitment of the MRN complex (MRE11 Homolog, Double Strand Break Repair Nuclease (MRE11)-RAD50 Double Strand Break Repair Protein (RAD50)-Nibrin (NBS1)} to DSBs followed by recruitment of ATM and subsequent phosphorylation of histone H2AX (H2A.X Variant Histone) at Serine 139 (γH2AX) by ATM [123,124]. As a result of γH2AX activation, repair proteins can be recruited, such as BRCA1 (Breast And Ovarian Cancer Susceptibility Protein 1), and further activation of ATM can occur, leading to DNA lesion resolution [125]. Targeting of H2AX by miR-138 has been demonstrated in U2OS cells [126] (Fig. 3). Overexpression of miR-138 resulted in chromosomal abnormalities, increased sensitivity to DNA-damaging agents, and reduced γH2AX foci formation following induced DNA damage, suggesting significantly diminished DNA repair activity. In another study, Weeraratne et al [127] demonstrated that overexpression of miR-183-96-182 cluster, a common occurrence in medulloblastoma, results in accumulation of DSB, suppression of BRCA1 protein expression along with a concomitant increase of cell migration and induction of epithelial mesenchymal transition, all of which contribute to carcinogenesis. Furthermore, it was recently shown that a long non-coding RNA, MALAT1 (Metastasis Associated Lung Adenocarcinoma Transcript 1), acts as a competing endogenous RNA and sequesters miR-146 by directly binding to it, thereby removing repression of BRCA1 expression in non-small cell lung carcinoma cell lines A549 and H1299, leading to activation of HR repair and protection of these cells from cisplatin-induced cytotoxicity [128]. Recruitment, but not expression of BRCA1 and Tumor Protein P53 Binding Protein 1(53BP1) to the sites of DNA damage was found to be regulated by miR-214 overexpression induced suppression of RNF8 expression (Ring Finger Protein 8) [129] in ovarian carcinoma cell lines (Fig. 3). Luciferase assay provided direct evidence that miR-218 directly binds to 3’UTR of RNF8 and downregulates its expression both at the mRNA and protein level and this suppression in turn leads to accumulation of DNA damage [129]. This heightened DNA damage could be rescued by either inhibiting the endogenous expression of miR-214, or by overexpressing RNF8 lacking 3’UTR sequence, providing strong causal link between miR-214 expression and DNA damage induction leading to carcinogenesis [129].
DNA damage can induce stalling of the replication machinery, resulting in failure to complete chromosomal duplication, chromosomal rearrangements, and cell death or neoplastic transformation. The most common pathway for stressed replication fork repair is the ATR pathway. Long stretches of Replication Protein A1 (RPA1) polymerized on single-stranded DNA (ssDNA) formed from uncoupling between Methylmalonyl-CoA Mutase (MCM), helicase, and DNA polymerase during replication recruits ATR and ATR Interacting Protein (ATRIP) to stalled forks [130]. RPA-coated ssDNA also recruits the RAD17 Checkpoint Clamp Loader Component- Replication Factor C Subunit 2–5 (RAD17-RFC2-5) clamp loader and RAD9 Checkpoint Clamp Component A-RAD1 Checkpoint DNA Exonuclease- HUS1 Checkpoint Clamp Component [RAD9-RAD1-HUS1 (9-1-1)] checkpoint clamps, resulting in DNA Topoisomerase II Binding Protein 1 (TOPBP1) and RAD9-HUS1-RAD1 Interacting Nuclear Orphan 1 (RHINO) recruitment. The ATR signaling cascade is then activated by engagement of ATR/ATRIP by TopBP1 and Rhino [131]. Both RAD17 and BRCA1 mRNA (important for ATR and ATM cascade signaling cascades, respectively) are specific targets of miR-205-5p in head and neck squamous cell carcinoma (HNSCC) as demonstrated by induced activity of luciferase reporters containing RAD17 and BRCA1 sequences in miR-205-5p overexpressing cells transfected with locked nucleic acid-205-5p oligos (Fig. 3). Additionally, overexpression of miR-205-5p resulted in increased DNA damage measured by comet assays and increased γH2AX foci [132].
The HR versus NHEJ decision in cells is dependent upon both the cell cycle stage and DNA end resection, where 5’ to 3’ nucleolytic processing of DNA ends commits repair to HR and prevents ligation by the NHEJ pathway [133]. Resection is initiated by the MRN complex and HR primarily occurs in the S and G2 phases of the cell cycle, whereas NHEJ can occur throughout the cell cycle and is dominant in G0/G1 and G2 [134]. Partial resection or binding of MRN to free DNA ends and recruitment of RB Binding Protein 8, Endonuclease (CTIP) to initiate resection prevents X-Ray Repair Cross Complementing (Ku) complex binding. Following resection, RPA binds to exposed ssDNA and is displaced by RAD51 Recombinase (Rad51) which catalyzes homologous pairing and strand invasion. Exposure to hexavalent chromium, a well characterized carcinogen, is known to induce DSBs followed by suppression of HR repair pathway [135,136] as well as differential miRNA expression [137,138]. A recent publication demonstrated that such HR activation in response to chromium exposure is causally mediated by suppression of miR-3940-5p [139]. Using Human bronchial epithelial cell line 16HBE and miR-3940-5p mimic, the authors demonstrated unequivocally that miR-3940-5p overexpression led to suppression of X-Ray Repair Cross Complementing 2 (XRCC2) protein as well as Rad51 foci blocking DSB repair, culminating in heightened DNA damage as measured by comet assay [69]. Induction of miR-22 expression as a consequence of AKT Serine/Threonine Kinase 1 (AKT1) activation was also demonstrated to lead to heightened accumulation of DSBs [140]. miR-22 was demonstrated to bring about DSB by directly targeting the 3’UTR of the key HR molecule Mediator of DNA Damage Checkpoint 1 (MDC1), suppressing its expression, which could be rescued by transfection with anti-miR-22 [140]. HR repair was also demonstrated to be abrogated by miR-222 through negative regulation of Rad51 expression in murine Balb/c 3T3 clone A31-1-1 cell line. miR-222 overexpression acted as an initiator for downstream cellular transformation through heightened DSB accumulation in response to γ-irradiation [141]. In classical NHEJ, X-Ray Repair Cross Complementing 6 (KU70) and X-Ray Repair Cross Complementing 5 (KU80) proteins heterodimerize (Ku complex) to broken DNA ends followed by subsequent recruitment of DNA ligase IV and ligase accessory proteins LIF Interleukin 6 Family Cytokine (LIF1) and Non-Homologous End-Joining Protein 1 (Nej1) to re-ligate broken ends [142]. Alternatively, the MMEJ pathway (alternative NHEJ pathway) is initiated by Poly(ADP-Ribose) Polymerase 1 (PARP1) and competes with the Ku heterodimer for DNA ends [143]. Within the MMEJ pathway, CTIP, Pre-MRNA Processing Factor 19 (PSO4), and Meiotic Recombination 11 Homolog 1 (MRE11) mediate 5’ end resection, DNA polymerase theta promotes microhomology annealing, and DNA ligase 1 or 3 with X-Ray Repair Cross Complementing 1 (XRCC1) ligate DNA. Targeting of PARP-1, CTIP, and Pso4 by miR-223-3p (Fig. 3) resulted in repressed chromosomal translocation in Jurkat cells [75]. Furthermore, deletion of miR-223-3p in mice resulted in increased chromosomal aberrations indicating that miR-223-3p acts to maintain chromosomal stability. The mechanistic relationship between miRNAs and the DNA damage response is summarized in Fig. 3.
5.2. Mitotic Checkpoint
Smooth progression through cell cycle is critical for maintenance of cellular and genomic integrity. Cells rely on precise mechanisms and checkpoints to ensure proper transition through different cell cycle stages culminating in proper cell division. Dysregulation of one or more of these mechanisms and/or checkpoints often results in faulty chromosomal segregation during mitosis [144]. The consequent chromosomal instability is known to significantly contribute to aneuploidy and has been recognized as a hallmark of cancer [6] Abnormal chromosome segregation and aneuploidy are caused by aberrant mitotic checkpoint activity and defects in mitotic spindle formation. In the following section, the role of miRNAs in regulating cell cycle, mitotic checkpoint and mitotic spindle formation and how their abrogation can lead to carcinogenesis is discussed.
The cell cycle consists of two stages, interphase, which consists of G1, S, and G2 phases, and the mitotic (M) phase. Progression through the four distinct phases of the cell cycle is regulated by cyclin dependent kinases (CDK’s) that complex with their cyclin partners (reviewed in [145–147]). During the G2 phase, cyclin A2-CDK1 and cyclin B-CDK1 complexes are formed. Entry into the M phase requires degradation of cyclin A2 and in late mitosis, cyclin B is degraded by the proteasome after polyubiquitinylation by the anaphasepromoting complex/cyclosome (APC/C) to allow for chromatid separation and mitosis. The mitotic checkpoint acts specifically in M phase to delay anaphase onset until sister chromatids are properly aligned and attached in a bipolar fashion to the mitotic spindle [148]. Failure of chromatids to properly align or attach correctly to the spindle prior to anaphase results in unequal segregation of chromosomes and results in aneuploidy (Fig. 2 and Fig. 4). Signaling of proper alignment is ultimately provided first by kinetochore complex formation on chromosomal centromeres, secondly through interaction of kinetochores and microtubules, thirdly by assembly of the KMN network complex, comprised of the Kinetochore Scaffold 1 (Knl1) complex, the MIS12 Kinetochore Complex Component (MIS12) complex and the NDC80 Kinetochore Complex Component (NDC80) complex, on kinetochores, and lastly, loading of the Mitotic Checkpoint Complex (MCC) complex onto the KMN network complex [149–152]. The MCC is a multiprotein complex comprised of numerous proteins, including BUB1 Mitotic Checkpoint Serine/Threonine Kinase B (BUBR1), BUB3 Mitotic Checkpoint Protein (BUB3), Mitotic Arrest Deficient 1 Like 1 (MAD1), Mitotic Arrest Deficient 2 Like 1 (MAD2), and Cell Division Cycle 20 (CDC20) that prevents anaphase until all chromosomes are stably attached to the spindle (Fig. 4). The downstream target of the MCC is the APC/C, an E3 ubiquitin ligase that targets many proteins for proteolytic degradation to regulate progression through mitosis [150]. The APC/C requires activation by one of two co-factors, CDC20 or Cadherin 1 (CDH1). Specific mechanisms which contribute to APC/C activation and inhibition have been extensively reviewed [150,153,154]. The APC/C catalyzes polyubiquitylation of securin, the inhibitory chaperone of separase, directing its degradation. The degradation of securin relieves separase inhibition, and separase then cleaves the cohesin complex that links sister chromatids allowing cells to proceed to anaphase.
Figure 4.
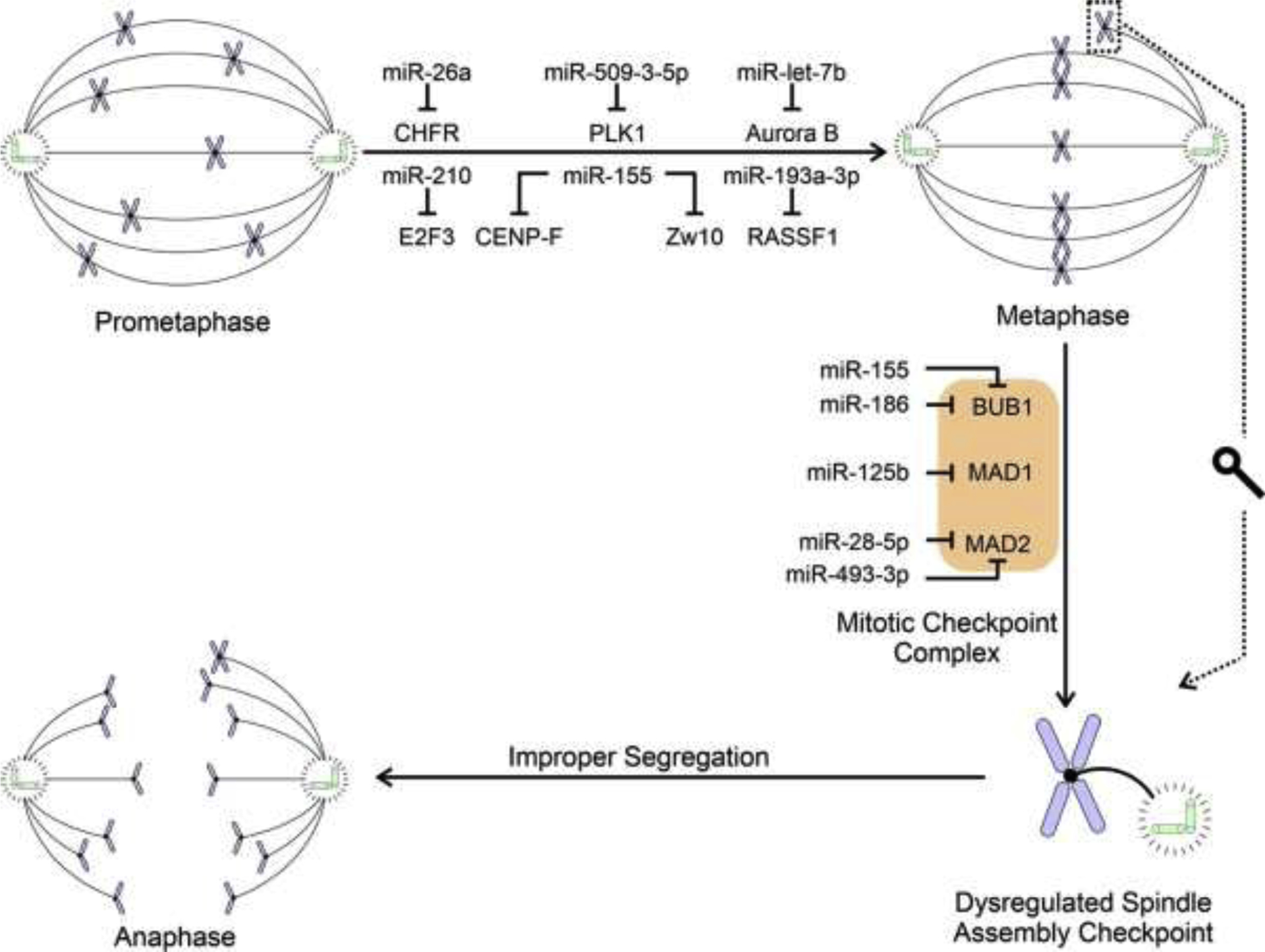
Simplified schematic representation summarizing the causal relationships by which miRNA dysregulation leads to mitotic disruption. The figure depicts how miRNA(s) disrupt prometaphase to metaphase transition and cause dysregulation of Spindle Assembly Checkpoint by targeting components of the Mitotic Checkpoint Complex leading to improper chromatid segregation and ultimately aneuploidy. Please note that no data are currently available causally linking miRNA dysregulation to disruption of other cell cycle stages.
Recent work established that miRNAs contribute to aneuploidy by modulation of genes important within the mitotic checkpoint, including mitotic spindle assembly. In parallel with the previous section, we heavily focused on studies which provide experimental validation of the specific nature of the miRNA-mRNA interactions and resulting modulation of expression (Table 1). Previous studies identified that Checkpoint With Forkhead And Ring Finger Domains (CHFR), an E3 ubiquitin-protein ligase, targets PLK1 (Polo-Like Kinase 1) and Aurora A [155,156]. PLK1 is important for the activation of the cyclin B/ Cyclin Dependent Kinase 1 (CDK1) complex and negatively regulates Aurora A, which localizes to the centrosome and is essential for spindle assembly [157–159]. Castellano and colleagues showed that miR-26a overexpression induced aneuploidy by targeting CHFR [58] (Fig. 4). However, partial rescue of the overexpressing miR-26a aneuploid phenotype was accomplished using overexpression of exogenous CHFR lacking its intact 3’ UTR. Experiments performed by Wang et al. also showed PLK1 repression by miR-509-3-5p and that miR-509-3-5p overexpression caused abnormal mitotic spindle assembly and lagging DNA [160].
Progression through mitotic division into anaphase is stimulated by phosphorylation of Ras Association Domain Family Member 1 (RASSF1) by Aurora A, leading to ubiquitylation and degradation of RASSF1. RASSF1 is a scaffolding protein that is frequently inactivated in many cancers and serves to control cell proliferation and cell cycle progression. Pruikkonen and Kallio showed that miR-193a-3p bound directly to the RASSF1 3’-UTR and that miR-193a-3p overexpression resulted in reduced RASSF1 expression and multipolar spindle formation [161]. Aurora B is a paralog of Aurora A that phosphorylates microtubule binding proteins at the outer kinetochore, weakening the affinity of the kinetochore for microtubules. Throughout mitosis, kinetochore and microtubule interactions are constantly created and destroyed until proper alignment of chromosomes occurs and kinetochore-microtubule attachments are stabilized [153]. Experiments performed by the Kallio lab established that the Aurora B 3’-UTR is a target of let-7b miRNA and that excess let-7b expression in cells results in polyploidy [62]. BUB1, MAD1, and MAD2 are key components of the MCC. BUB1 is required for efficient bipolar attachment of spindle microtubules to kinetochores [162]. Two studies demonstrated that both miR-186-5p (miR-186) and miR-155 overexpression results in diminished BUB1 protein and aneuploidy [76,77]. Formation of MAD1 and MAD2 complexes sequester CDC20 from APC/C to prevent premature activation until chromosomes are properly aligned. Previous studies identified that miR-125b, and miR-493-3p and miR-28-5p directly contribute to Mad1 and Mad2 regulation, respectively (Fig. 4) [61,63,163]. Overexpression of these miRNAs resulted in defective anaphase, including lagging chromosomes (miR-125b and miR-28-5p overexpression) and aneuploidy (miR-493-3p).
Overall, miRNAs target numerous proteins involved in the mitotic checkpoint. Furthermore, some of these miRNAs share the same mRNA targets (MAD2: miR-493-3p and miR-28-5p), whereas other miRNAs target multiple levels of protein pathways (CHFR:miR-26a > PLK1:miR-509-3-5p).
6. Knowledge Gaps, Future Directions and Conclusions
The data summarized in the previous sections provide evidence supporting the causal nature of a relationship between miRNA dysregulation and induction of several classes of genome instability. Nevertheless, considerable knowledge gaps exist that can be addressed in future experiments. Both genomic instability and dysregulated miRNA expression profiles are known to be operative in several cancer types [9,10,24,31,33,34]. However, in many cases, it has not been empirically established whether these two mechanisms are causally related in cancer. Ultimately, it is possible that miRNA dysregulation is playing a pivotal role in generating genomic instability in all or most of these cancer types but remains to be experimentally tested. Many studies that were not discussed within this review were omitted because they merely examine the association between differential miRNA expression and genome instability without providing experimental validation to demonstrate if such association is only correlative or is also causal in nature [93,94]. This is clear from Fig.1 which shows that the majority of the research articles that have probed the relationship between genome instability and miRNA only provided association data. To provide empirical evidence, methods such as overexpression and knockdown experiments can be used to increase our understanding of the relationship between miRNA(s) and genome instability.
Development of better assay systems is essential for exploring the mechanistic roles played by miRNA dysregulation in causing genome instability. Some advancement has been made in the field of high resolution and high throughput screening of MIN recently [99,164], but these methods have yet to be widely applied in the field. Application of genome-wide technologies will also be important in this regard. For example, structural and numerical CIN can be qualitatively and quantitatively examined employing techniques such as spectral karyotyping and array comparative genomic hybridization [165–168]. Yet, to our knowledge, no study has so far used any of these well-known techniques to explore the nature of miRNA-genome instability relationships. Furthermore, most studies have focused on the role of one miRNA on one or a few mRNA targets in genome instability induction. In the cellular context, it is likely that the dysregulated miRNA is acting as the harbinger of genome instability by acting on a complex targetome. In the future, it will be imperative to unravel the transcriptome-wide molecular targets of candidate miRNA(s) through next generation sequencing techniques such as high-throughput sequencing of RNAs isolated by crosslinking immunoprecipitation (HITS-CLIP) to truly understand how miRNA(s) can bring about genome instability and lead to carcinogenesis [169].
Much work also remains to be done regarding the role of miRNA dysregulation towards induction of genome instability via abrogation of cell cycle checkpoints, DDR and DNA repair systems. Currently, only a small subset of miRNAs targeting a few DDR pathway genes are demonstrated to affect genome instability (Fig. 3). It is quite probable that several other members involved in identification of damaged DNA lesions, factors which aid in recruitment of appropriate DNA repair systems, and specific DNA repair process (Fig. 3) and targeted by dysregulated miRNA expression leading to genome instability remain unidentified. Similar arguments can be made for molecules involved in cell cycle checkpoint maintenance (Fig. 4). Given the key role played by MCC in ensuring proper SAC and maintenance of genomic integrity [170], it is imperative that we develop a deep understanding of how miRNA dysregulation can affect each component of the MCC. However, as of now, data exists for only three of the six subunits (Fig. 4). Furthermore, ample experimental evidence demonstrates that genome instability can be induced as an outcome of inappropriate entry, progression or exit from G1 or S phases [171–173]. This represents a largely unexplored area of study that needs to be addressed with alacrity.
Acknowledgements
This work was supported by National Institute of Environmental Health Sciences Grants R21ES030334, R01ES027778 and P30ES030283 (JCS). ANN is supported by a National Institute of Environmental Health Sciences T32 post-doctoral training fellowship T32ES011564.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
- [1].Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F, Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA A Cancer J Clin. (2021) caac.21660. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- [2].Anand P, Kunnumakara AB, Sundaram C, Harikumar KB, Tharakan ST, Lai OS, Sung B, Aggarwal BB, Cancer is a Preventable Disease that Requires Major Lifestyle Changes, Pharm Res. 25 (2008) 2097–2116. 10.1007/s11095-008-9661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mbemi A, Khanna S, Njiki S, Yedjou CG, Tchounwou PB, Impact of Gene–Environment Interactions on Cancer Development, IJERPH. 17 (2020) 8089. 10.3390/ijerph17218089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fouad YA, Aanei C, Revisiting the hallmarks of cancer, Am J Cancer Res. 7 (2017) 1016–1036. [PMC free article] [PubMed] [Google Scholar]
- [5].Hanahan D, Weinberg RA, The hallmarks of cancer, Cell. 100 (2000) 57–70. 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- [6].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell. 144 (2011) 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [7].Fenech M, Chromosomal biomarkers of genomic instability relevant to cancer, Drug Discov Today. 7 (2002) 1128–1137. 10.1016/s1359-6446(02)02502-3. [DOI] [PubMed] [Google Scholar]
- [8].Willenbucher RF, Aust DE, Chang CG, Zelman SJ, Ferrell LD, Moore DH, Waldman FM, Genomic instability is an early event during the progression pathway of ulcerative-colitis-related neoplasia, Am J Pathol. 154 (1999) 1825–1830. 10.1016/S0002-9440(10)65438-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Saayman X, Esashi F, Breaking the paradigm: early insights from mammalian DNA breakomes, FEBS J. (2021). 10.1111/febs.15849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ferguson LR, Chen H, Collins AR, Connell M, Damia G, Dasgupta S, Malhotra M, Meeker AK, Amedei A, Amin A, Ashraf SS, Aquilano K, Azmi AS, Bhakta D, Bilsland A, Boosani CS, Chen S, Ciriolo MR, Fujii H, Guha G, Halicka D, Helferich WG, Keith WN, Mohammed SI, Niccolai E, Yang X, Honoki K, Parslow VR, Prakash S, Rezazadeh S, Shackelford RE, Sidransky D, Tran PT, Yang ES, Maxwell CA, Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition, Semin Cancer Biol. 35 Suppl (2015) S5–S24. 10.1016/j.semcancer.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Passerini V, Ozeri-Galai E, de Pagter MS, Donnelly N, Schmalbrock S, Kloosterman WP, Kerem B, Storchová Z, The presence of extra chromosomes leads to genomic instability, Nat Commun. 7 (2016) 10754. 10.1038/ncomms10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Negrini S, Gorgoulis VG, Halazonetis TD, Genomic instability — an evolving hallmark of cancer, Nat Rev Mol Cell Biol. 11 (2010) 220–228. 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- [13].Giam M, Rancati G, Aneuploidy and chromosomal instability in cancer: a jackpot to chaos, Cell Div. 10 (2015) 3. 10.1186/s13008-015-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wei Dai YY, Genomic Instability and Cancer, J Carcinog Mutagen. 05 (2014). 10.4172/2157-2518.1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cleal K, Baird DM, Catastrophic Endgames: Emerging Mechanisms of Telomere-Driven Genomic Instability, Trends Genet. 36 (2020) 347–359. 10.1016/j.tig.2020.02.001. [DOI] [PubMed] [Google Scholar]
- [16].Bhattacharjya S, Nath S, Ghose J, Maiti GP, Biswas N, Bandyopadhyay S, Panda CK, Bhattacharyya NP, Roychoudhury S, miR-125b promotes cell death by targeting spindle assembly checkpoint gene MAD1 and modulating mitotic progression, Cell Death Differ. 20 (2013) 430–42. 10.1038/cdd.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang T, Yang Z, Gao H, Advancements in the study of miRNA regulation during the cell cycle in human pituitary adenomas, J Neurooncol. 134 (2017) 253–258. 10.1007/s11060-017-2518-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wouters MD, van Gent DC, Hoeijmakers JHJ, Pothof J, MicroRNAs, the DNA damage response and cancer, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 717 (2011) 54–66. 10.1016/j.mrfmmm.2011.03.012. [DOI] [PubMed] [Google Scholar]
- [19].Wang Y, Taniguchi T, MicroRNAs and DNA damage response: Implications for cancer therapy, Cell Cycle. 12 (2013) 32–42. 10.4161/cc.23051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu X, Chen X, Yu X, Tao Y, Bode AM, Dong Z, Cao Y, Regulation of microRNAs by epigenetics and their interplay involved in cancer, J Exp Clin Cancer Res. 32 (2013) 96. 10.1186/1756-9966-32-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ha M, Kim VN, Regulation of microRNA biogenesis, Nat Rev Mol Cell Biol. 15 (2014) 509–524. 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- [22].Michlewski G, Cáceres JF, Post-transcriptional control of miRNA biogenesis, RNA. 25 (2019) 1–16. 10.1261/rna.068692.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Filipowicz W, Bhattacharyya SN, Sonenberg N, Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight?, Nat Rev Genet. 9 (2008) 102–114. 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- [24].Peng Y, Croce CM, The role of MicroRNAs in human cancer, Sig Transduct Target Ther. 1 (2016) 15004. 10.1038/sigtrans.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Catalanotto C, Cogoni C, Zardo G, MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions, IJMS. 17 (2016) 1712. 10.3390/ijms17101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bushati N, Cohen SM, microRNA Functions, Annu. Rev. Cell Dev. Biol 23 (2007) 175–205. 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- [27].Hayes J, Peruzzi PP, Lawler S, MicroRNAs in cancer: biomarkers, functions and therapy, Trends in Molecular Medicine. 20 (2014) 460–469. 10.1016/j.molmed.2014.06.005. [DOI] [PubMed] [Google Scholar]
- [28].Chipman LB, Pasquinelli AE, miRNA Targeting: Growing beyond the Seed, Trends in Genetics. 35 (2019) 215–222. 10.1016/j.tig.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].MacFarlane L-A, Murphy PR, MicroRNA: Biogenesis, Function and Role in Cancer, CG. 11 (2010) 537–561. 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J, The biogenesis, biology and characterization of circular RNAs, Nat Rev Genet. 20 (2019) 675–691. 10.1038/s41576-019-0158-7. [DOI] [PubMed] [Google Scholar]
- [31].Iorio MV, Croce CM, microRNA involvement in human cancer, Carcinogenesis. 33 (2012) 1126–1133. 10.1093/carcin/bgs140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lagos-Quintana M, Identification of Novel Genes Coding for Small Expressed RNAs, Science. 294 (2001) 853–858. 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- [33].Rupaimoole R, Slack FJ, MicroRNA therapeutics: towards a new era for the management of cancer and other diseases, Nat Rev Drug Discov. 16 (2017) 203–222. 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- [34].Tan W, Liu B, Qu S, Liang G, Luo W, Gong C, MicroRNAs and cancer: Key paradigms in molecular therapy (Review), Oncol Lett. (2017). 10.3892/ol.2017.7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Vargas-Rondón N, Villegas V, Rondón-Lagos M, The Role of Chromosomal Instability in Cancer and Therapeutic Responses, Cancers. 10 (2017) 4. 10.3390/cancers10010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bolhaqueiro ACF, Ponsioen B, Bakker B, Klaasen SJ, Kucukkose E, van Jaarsveld RH, Vivié J, Verlaan-Klink I, Hami N, Spierings DCJ, Sasaki N, Dutta D, Boj SF, Vries RGJ, Lansdorp PM, van de Wetering M, van Oudenaarden A, Clevers H, Kranenburg O, Foijer F, Snippert HJG, Kops GJPL, Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids, Nat Genet. 51 (2019) 824–834. 10.1038/s41588-019-0399-6. [DOI] [PubMed] [Google Scholar]
- [37].McClelland SE, Role of chromosomal instability in cancer progression, Endocrine-Related Cancer. 24 (2017) T23–T31. 10.1530/ERC-17-0187. [DOI] [PubMed] [Google Scholar]
- [38].Bakhoum SF, Cantley LC, The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment, Cell. 174 (2018) 1347–1360. 10.1016/j.cell.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tijhuis AE, Johnson SC, McClelland SE, The emerging links between chromosomal instability (CIN), metastasis, inflammation and tumour immunity, Mol Cytogenet. 12 (2019) 17. 10.1186/s13039-019-0429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, Lander ES, Meyerson M, Getz G, Absolute quantification of somatic DNA alterations in human cancer, Nat Biotechnol. 30 (2012) 413–421. 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cimini D, Merotelic kinetochore orientation, aneuploidy, and cancer, Biochim Biophys Acta. 1786 (2008) 32–40. 10.1016/j.bbcan.2008.05.003. [DOI] [PubMed] [Google Scholar]
- [42].Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z, A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers, Nat Genet. 38 (2006) 1043–1048. 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- [43].Thompson SL, Bakhoum SF, Compton DA, Mechanisms of chromosomal instability, Curr Biol. 20 (2010) R285–95. 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bueno MJ, Malumbres M, MicroRNAs and the cell cycle, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1812 (2011) 592–601. 10.1016/j.bbadis.2011.02.002. [DOI] [PubMed] [Google Scholar]
- [45].Wan G, Mathur R, Hu X, Zhang X, Lu X, miRNA response to DNA damage, Trends in Biochemical Sciences. 36 (2011) 478–484. 10.1016/j.tibs.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Caldas C, Brenton JD, Sizing up miRNAs as cancer genes, Nat Med. 11 (2005) 712–714. 10.1038/nm0705-712. [DOI] [PubMed] [Google Scholar]
- [47].Baer C, Claus R, Plass C, Genome-Wide Epigenetic Regulation of miRNAs in Cancer, Cancer Res. 73 (2013) 473–477. 10.1158/0008-5472.CAN-12-3731. [DOI] [PubMed] [Google Scholar]
- [48].Weaver BA, Cleveland DW, Does aneuploidy cause cancer?, Current Opinion in Cell Biology. 18 (2006) 658–667. 10.1016/j.ceb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- [49].Vishwakarma R, McManus KJ, Chromosome Instability; Implications in Cancer Development, Progression, and Clinical Outcomes, Cancers (Basel). 12 (2020). 10.3390/cancers12040824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Vasudevan A, Schukken KM, Sausville EL, Girish V, Adebambo OA, Sheltzer JM, Aneuploidy as a promoter and suppressor of malignant growth, Nat Rev Cancer. 21 (2021) 89–103. 10.1038/s41568-020-00321-1. [DOI] [PubMed] [Google Scholar]
- [51].Tanaka K, Goto H, Nishimura Y, Kasahara K, Mizoguchi A, Inagaki M, Tetraploidy in cancer and its possible link to aging, Cancer Sci. 109 (2018) 2632–2640. 10.1111/cas.13717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Molina O, Abad MA, Solé F, Menéndez P, Aneuploidy in Cancer: Lessons from Acute Lymphoblastic Leukemia, Trends in Cancer. 7 (2021) 37–47. 10.1016/j.trecan.2020.08.008. [DOI] [PubMed] [Google Scholar]
- [53].Schoenfelder KP, Fox DT, The expanding implications of polyploidy, Journal of Cell Biology. 209 (2015) 485–491. 10.1083/jcb.201502016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Vasudevan A, Baruah PS, Smith JC, Wang Z, Sayles NM, Andrews P, Kendall J, Leu J, Chunduri NK, Levy D, Wigler M, Storchová Z, Sheltzer JM, Single-Chromosomal Gains Can Function as Metastasis Suppressors and Promoters in Colon Cancer, Dev Cell. 52 (2020) 413–428.e6. 10.1016/j.devcel.2020.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z, Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells, Mol Syst Biol. 8 (2012) 608. 10.1038/msb.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Dürrbaum M, Kruse C, Nieken KJ, Habermann B, Storchová Z, The deregulated microRNAome contributes to the cellular response to aneuploidy, BMC Genomics. 19 (2018) 197. 10.1186/s12864-018-4556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Högnäs G, Hämälistö S, Rilla K, Laine JO, Vilkki V, Murumägi A, Edgren H, Kallioniemi O, Ivaska J, Aneuploidy facilitates oncogenic transformation via specific genetic alterations, including Twist2 upregulation, Carcinogenesis. 34 (2013) 2000–2009. 10.1093/carcin/bgt171. [DOI] [PubMed] [Google Scholar]
- [58].Castellano L, Dabrowska A, Pellegrino L, Ottaviani S, Cathcart P, Frampton AE, Krell J, Stebbing J, Sustained expression of miR-26a promotes chromosomal instability and tumorigenesis through regulation of CHFR, Nucleic Acids Res. (2017) gkx022. 10.1093/nar/gkx022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Silkworth WT, Nardi IK, Scholl LM, Cimini D, Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells, PLoS One. 4 (2009) e6564. 10.1371/journal.pone.0006564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Nakada C, Tsukamoto Y, Matsuura K, Nguyen TL, Hijiya N, Uchida T, Sato F, Mimata H, Seto M, Moriyama M, Overexpression of miR-210, a downstream target of HIF1α, causes centrosome amplification in renal carcinoma cells: miR-210 overexpression induces centrosome amplification, J. Pathol 224 (2011) 280–288. 10.1002/path.2860. [DOI] [PubMed] [Google Scholar]
- [61].Hell MP, Thoma CR, Fankhauser N, Christinat Y, Weber TC, Krek W, miR-28–5p Promotes Chromosomal Instability in VHL -Associated Cancers by Inhibiting Mad2 Translation, Cancer Res. 74 (2014) 2432–2443. 10.1158/0008-5472.CAN-13-2041. [DOI] [PubMed] [Google Scholar]
- [62].Mäki-Jouppila JHE, Pruikkonen S, Tambe MB, Aure MR, Halonen T, Salmela A-L, Laine L, Børresen-Dale A-L, Kallio MJ, MicroRNA let-7b regulates genomic balance by targeting Aurora B kinase, Mol Oncol. 9 (2015) 1056–1070. 10.1016/j.molonc.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tambe M, Pruikkonen S, Mäki-Jouppila J, Chen P, Elgaaen BV, Straume AH, Huhtinen K, Cárpen O, Lønning PE, Davidson B, Hautaniemi S, Kallio MJ, Novel Mad2-targeting miR-493–3p controls mitotic fidelity and cancer cells’ sensitivity to paclitaxel, Oncotarget. 7 (2016) 12267–12285. 10.18632/oncotarget.7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ly P, Brunner SF, Shoshani O, Kim DH, Lan W, Pyntikova T, Flanagan AM, Behjati S, Page DC, Campbell PJ, Cleveland DW, Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements, Nat Genet. 51 (2019) 705–715. 10.1038/s41588-019-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kou F, Wu L, Ren X, Yang L, Chromosome Abnormalities: New Insights into Their Clinical Significance in Cancer, Molecular Therapy - Oncolytics. 17 (2020) 562–570. 10.1016/j.omto.2020.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, Lazar AJ, Cherniack AD, Beroukhim R, Meyerson M, Genomic and Functional Approaches to Understanding Cancer Aneuploidy, Cancer Cell. 33 (2018) 676–689.e3. 10.1016/j.ccell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Russo A, Pacchierotti F, Cimini D, Ganem NJ, Genescà A, Natarajan AT, Pavanello S, Valle G, Degrassi F, Genomic instability: Crossing pathways at the origin of structural and numerical chromosome changes: Crossing Pathways at the Origin of Chromosome Changes, Environ. Mol. Mutagen 56 (2015) 563–580. 10.1002/em.21945. [DOI] [PubMed] [Google Scholar]
- [68].Zeman MK, Cimprich KA, Causes and consequences of replication stress, Nat Cell Biol. 16 (2014) 2–9. 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Quinlan AR, Hall IM, Characterizing complex structural variation in germline and somatic genomes, Trends Genet. 28 (2012) 43–53. 10.1016/j.tig.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tessitore A, Cicciarelli G, Del Vecchio F, Gaggiano A, Verzella D, Fischietti M, Vecchiotti D, Capece D, Zazzeroni F, Alesse E, MicroRNAs in the DNA Damage/Repair Network and Cancer, Int J Genomics. 2014 (2014) 820248. 10.1155/2014/820248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ding Q, He K, Luo T, Deng Y, Wang H, Liu H, Zhang J, Chen K, Xiao J, Duan X, Huang R, Xia Z, Zhou W, He J, Yu H, Jiao X, Xiang G, SSRP1 Contributes to the Malignancy of Hepatocellular Carcinoma and Is Negatively Regulated by miR-497, Molecular Therapy. 24 (2016) 903–914. 10.1038/mt.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kumari A, Mazina OM, Shinde U, Mazin AV, Lu H, A role for SSRP1 in recombination-mediated DNA damage response, J. Cell. Biochem 108 (2009) 508–518. 10.1002/jcb.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kundu LR, Seki M, Watanabe N, Murofushi H, Furukohri A, Waga S, Score AJ, Blow JJ, Horikoshi M, Enomoto T, Tada S, Biphasic chromatin binding of histone chaperone FACT during eukaryotic chromatin DNA replication, Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1813 (2011) 1129–1136. 10.1016/j.bbamcr.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Birch JL, Tan BC-M, Panov KI, Panova TB, Andersen JS, Owen-Hughes TA, Russell J, Lee S-C, Zomerdijk JCBM, FACT facilitates chromatin transcription by RNA polymerases I and III, EMBO J. 28 (2009) 854–865. 10.1038/emboj.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Srinivasan G, Williamson EA, Kong K, Jaiswal AS, Huang G, Kim H-S, Schärer O, Zhao W, Burma S, Sung P, Hromas R, MiR223–3p promotes synthetic lethality in BRCA1-deficient cancers, Proc Natl Acad Sci USA. 116 (2019) 17438–17443. 10.1073/pnas.1903150116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wu J, Ferragut Cardoso AP, States VAR, Al-Eryani L, Doll M, Wise SS, Rai SN, States JC, Overexpression of hsa-miR-186 induces chromosomal instability in arsenic-exposed human keratinocytes, Toxicology and Applied Pharmacology. 378 (2019) 114614. 10.1016/j.taap.2019.114614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Pagotto S, Veronese A, Soranno A, Lanuti P, Marco MD, Russo MV, Ramassone A, Marchisio M, Simeone P, Franchi PEG, Palka G, Costantini RM, Croce CM, Visone R, Hsa-miR-155-5p drives aneuploidy at early stages of cellular transformation, Oncotarget. 9 (2018) 13036–13047. 10.18632/oncotarget.24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bayraktar R, Van Roosbroeck K, miR-155 in cancer drug resistance and as target for miRNA-based therapeutics, Cancer Metastasis Rev. 37 (2018) 33–44. 10.1007/s10555-017-9724-7. [DOI] [PubMed] [Google Scholar]
- [79].Bagshaw ATM, Functional Mechanisms of Microsatellite DNA in Eukaryotic Genomes, Genome Biol Evol. 9 (2017) 2428–2443. 10.1093/gbe/evx164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gadgil R, Barthelemy J, Lewis T, Leffak M, Replication stalling and DNA microsatellite instability, Biophys Chem. 225 (2017) 38–48. 10.1016/j.bpc.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Subramanian S, Mishra RK, Singh L, Genome-wide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions, Genome Biol. 4 (2003) R13. 10.1186/gb-2003-4-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Leclercq S, Rivals E, Jarne P, DNA Slippage Occurs at Microsatellite Loci without Minimal Threshold Length in Humans: A Comparative Genomic Approach, Genome Biology and Evolution. 2 (2010) 325–335. 10.1093/gbe/evq023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Dieringer D, Two Distinct Modes of Microsatellite Mutation Processes: Evidence From the Complete Genomic Sequences of Nine Species, Genome Research. 13 (2003) 2242–2251. 10.1101/gr.1416703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Strand M, Prolla TA, Liskay RM, Petes TD, Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair, Nature. 365 (1993) 274–276. 10.1038/365274a0. [DOI] [PubMed] [Google Scholar]
- [85].Kovtun IV, McMurray CT, Features of trinucleotide repeat instability in vivo, Cell Res. 18 (2008) 198–213. 10.1038/cr.2008.5. [DOI] [PubMed] [Google Scholar]
- [86].Diao Z, Han Y, Chen Y, Zhang R, Li J, The clinical utility of microsatellite instability in colorectal cancer, Crit Rev Oncol Hematol. 157 (2021) 103171. 10.1016/j.critrevonc.2020.103171. [DOI] [PubMed] [Google Scholar]
- [87].Aaltonen LA, Peltomäki P, Leach FS, Sistonen P, Pylkkänen L, Mecklin JP, Järvinen H, Powell SM, Jen J, Hamilton SR, Clues to the pathogenesis of familial colorectal cancer, Science. 260 (1993) 812–816. 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- [88].Li K, Luo H, Huang L, Luo H, Zhu X, Microsatellite instability: a review of what the oncologist should know, Cancer Cell Int. 20 (2020) 16. 10.1186/s12935-019-1091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lorenzi M, Amonkar M, Zhang J, Mehta S, Liaw K-L, Epidemiology of Microsatellite Instability High (MSI-H) and Deficient Mismatch Repair (dMMR) in Solid Tumors: A Structured Literature Review, Journal of Oncology. 2020 (2020) 1–17. 10.1155/2020/1807929. [DOI] [Google Scholar]
- [90].Imai K, Yamamoto H, Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics, Carcinogenesis. 29 (2008) 673–680. 10.1093/carcin/bgm228. [DOI] [PubMed] [Google Scholar]
- [91].Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen H-Z, Reeser JW, Yu L, Roychowdhury S, Landscape of Microsatellite Instability Across 39 Cancer Types, JCO Precision Oncology. (2017) 1–15. 10.1200/PO.17.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yamamoto H, Watanabe Y, Maehata T, Imai K, Itoh F, Microsatellite instability in cancer: a novel landscape for diagnostic and therapeutic approach, Arch Toxicol. 94 (2020) 3349–3357. 10.1007/s00204-020-02833-z. [DOI] [PubMed] [Google Scholar]
- [93].Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T, A Polycistronic MicroRNA Cluster, miR-17-92, Is Overexpressed in Human Lung Cancers and Enhances Cell Proliferation, Cancer Res. 65 (2005) 9628–9632. 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- [94].Sarver AL, French AJ, Borralho PM, Thayanithy V, Oberg AL, Silverstein KA, Morlan BW, Riska SM, Boardman LA, Cunningham JM, Subramanian S, Wang L, Smyrk TC, Rodrigues CM, Thibodeau SN, Steer CJ, Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states, BMC Cancer. 9 (2009) 401. 10.1186/1471-2407-9-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lanza G, Ferracin M, Gafà R, Veronese A, Spizzo R, Pichiorri F, Liu C, Calin GA, Croce CM, Negrini M, mRNA/microRNA gene expression profile in microsatellite unstable colorectal cancer, Mol Cancer. 6 (2007) 54. 10.1186/1476-4598-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ashizawa M, Okayama H, Ishigame T, Thar Min AK, Saito K, Ujiie D, Murakami Y, Kikuchi T, Nakayama Y, Noda M, Tada T, Endo H, Fujita S, Sakamoto W, Saito M, Saze Z, Momma T, Ohki S, Mimura K, Kono K, microRNA-148a-3p regulates immunosuppression in DNA mismatch repair-deficient colorectal cancer by targeting PD-L1, Mol Cancer Res. (2019) molcanres.0831.2018. 10.1158/1541-7786.MCR-18-0831. [DOI] [PubMed] [Google Scholar]
- [97].Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, Adair B, Vannini I, Fanini F, Bottoni A, Costinean S, Sandhu SK, Nuovo GJ, Alder H, Gafa R, Calore F, Ferracin M, Lanza G, Volinia S, Negrini M, McIlhatton MA, Amadori D, Fishel R, Croce CM, Modulation of mismatch repair and genomic stability by miR-155, Proc Natl Acad Sci U S A. 107 (2010) 6982–6987. 10.1073/pnas.1002472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Huang Y-W, Liu JC, Deatherage DE, Luo J, Mutch DG, Goodfellow PJ, Miller DS, Huang TH-M, Epigenetic Repression of microRNA-129–2 Leads to Overexpression of SOX4 Oncogene in Endometrial Cancer, Cancer Res. 69 (2009) 9038–9046. 10.1158/0008-5472.CAN-09-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Koole W, Schäfer HS, Agami R, van Haaften G, Tijsterman M, A versatile microsatellite instability reporter system in human cells, Nucleic Acids Research. 41 (2013) e158–e158. 10.1093/nar/gkt615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ferguson DO, Alt FW, DNA double strand break repair and chromosomal translocation: lessons from animal models, Oncogene. 20 (2001) 5572–5579. 10.1038/sj.onc.1204767. [DOI] [PubMed] [Google Scholar]
- [101].Sies H, Jones DP, Reactive oxygen species (ROS) as pleiotropic physiological signalling agents, Nat Rev Mol Cell Biol. 21 (2020) 363–383. 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- [102].Chatterjee N, Walker GC, Mechanisms of DNA damage, repair, and mutagenesis, Environ Mol Mutagen. 58 (2017) 235–263. 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew Y-E, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan M-H, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR, Patterns of somatic mutation in human cancer genomes, Nature. 446 (2007) 153–158. 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mitelman F, Johansson B, Mertens F, The impact of translocations and gene fusions on cancer causation, Nat Rev Cancer. 7 (2007) 233–245. 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- [105].Mills KD, Ferguson DO, Alt FW, The role of DNA breaks in genomic instability and tumorigenesis, Immunol Rev. 194 (2003) 77–95. 10.1034/j.1600-065x.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- [106].Zhou J, Zhou XA, Zhang N, Wang J, Evolving insights: how DNA repair pathways impact cancer evolution, Cancer Biol Med. 17 (2020) 805–827. 10.20892/j.issn.2095-3941.2020.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Blackford AN, Jackson SP, ATM ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response, Mol Cell. 66 (2017) 801–817. 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- [108].Niraj J, Färkkilä A, D’Andrea AD, The Fanconi Anemia Pathway in Cancer, Annu Rev Cancer Biol. 3 (2019) 457–478. 10.1146/annurev-cancerbio-030617-050422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Moldovan G-L, D’Andrea AD, How the fanconi anemia pathway guards the genome, Annu Rev Genet. 43 (2009) 223–249. 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Caldecott KW, Single-strand break repair and genetic disease, Nat Rev Genet. 9 (2008) 619–631. 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- [111].Ciccia A, Elledge SJ, The DNA damage response: making it safe to play with knives, Mol Cell. 40 (2010) 179–204. 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Mortusewicz O, Leonhardt H, Cardoso MC, Spatiotemporal dynamics of regulatory protein recruitment at DNA damage sites, J Cell Biochem. 104 (2008) 1562–1569. 10.1002/jcb.21751. [DOI] [PubMed] [Google Scholar]
- [113].Deans AJ, West SC, DNA interstrand crosslink repair and cancer, Nat Rev Cancer. 11 (2011) 467–480. 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Rüthemann P, Balbo Pogliano C, Naegeli H, Global-genome Nucleotide Excision Repair Controlled by Ubiquitin/Sumo Modifiers, Front Genet. 7 (2016) 68. 10.3389/fgene.2016.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Garfinkel DJ, Bailis AM, Nucleotide Excision Repair, Genome Stability, and Human Disease: New Insight from Model Systems, J Biomed Biotechnol. 2 (2002) 55–60. 10.1155/S1110724302201023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Nemec AA, Wallace SS, Sweasy JB, Variant base excision repair proteins: contributors to genomic instability, Semin Cancer Biol. 20 (2010) 320–328. 10.1016/j.semcancer.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wallace SS, Murphy DL, Sweasy JB, Base excision repair and cancer, Cancer Lett. 327 (2012) 73–89. 10.1016/j.canlet.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].McMahon LW, Zhang P, Sridharan DM, Lefferts JA, Lambert MW, Knockdown of alphaII spectrin in normal human cells by siRNA leads to chromosomal instability and decreased DNA interstrand cross-link repair, Biochem Biophys Res Commun. 381 (2009) 288–293. 10.1016/j.bbrc.2009.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Ackermann A, Brieger A, The Role of Nonerythroid Spectrin αII in Cancer, J Oncol. 2019 (2019) 7079604. 10.1155/2019/7079604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Sridharan D, Brown M, Lambert WC, McMahon LW, Lambert MW, Nonerythroid alphaII spectrin is required for recruitment of FANCA and XPF to nuclear foci induced by DNA interstrand cross-links, J Cell Sci. 116 (2003) 823–835. 10.1242/jcs.00294. [DOI] [PubMed] [Google Scholar]
- [121].Lefferts JA, Wang C, Sridharan D, Baralt M, Lambert MW, The SH3 domain of alphaII spectrin is a target for the Fanconi anemia protein, FANCG, Biochemistry. 48 (2009) 254–263. 10.1021/bi801483u. [DOI] [PubMed] [Google Scholar]
- [122].Zhang R, Liu C, Niu Y, Jing Y, Zhang H, Wang J, Yang J, Zen K, Zhang J, Zhang C-Y, Li D, MicroRNA-128–3p regulates mitomycin C-induced DNA damage response in lung cancer cells through repressing SPTAN1, Oncotarget. 8 (2017) 58098–58107. 10.18632/oncotarget.12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Lanz MC, Dibitetto D, Smolka MB, DNA damage kinase signaling: checkpoint and repair at 30 years, EMBO J. 38 (2019) e101801. 10.15252/embj.2019101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Di Domenico EG, Romano E, Del Porto P, Ascenzioni F, Multifunctional role of ATM/Tel1 kinase in genome stability: from the DNA damage response to telomere maintenance, Biomed Res Int. 2014 (2014) 787404. 10.1155/2014/787404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Huen MSY, Sy SMH, Chen J, BRCA1 and its toolbox for the maintenance of genome integrity, Nat Rev Mol Cell Biol. 11 (2010) 138–148. 10.1038/nrm2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Wang Y, Huang J-W, Li M, Cavenee WK, Mitchell PS, Zhou X, Tewari M, Furnari FB, Taniguchi T, MicroRNA-138 modulates DNA damage response by repressing histone H2AX expression, Mol Cancer Res. 9 (2011) 1100–1111. 10.1158/1541-7786.MCR-11-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Weeraratne SD, Amani V, Teider N, Pierre-Francois J, Winter D, Kye MJ, Sengupta S, Archer T, Remke M, Bai AHC, Warren P, Pfister SM, Steen JAJ, Pomeroy SL, Cho Y-J, Pleiotropic effects of miR-183~96~182 converge to regulate cell survival, proliferation and migration in medulloblastoma, Acta Neuropathol. 123 (2012) 539–552. 10.1007/s00401-012-0969-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Huang J, Lin C, Dong H, Piao Z, Jin C, Han H, Jin D, Targeting MALAT1 induces DNA damage and sensitize non-small cell lung cancer cells to cisplatin by repressing BRCA1, Cancer Chemother Pharmacol. 86 (2020) 663–672. 10.1007/s00280-020-04152-7. [DOI] [PubMed] [Google Scholar]
- [129].Wang Z, Yin H, Zhang Y, Feng Y, Yan Z, Jiang X, Bukhari I, Iqbal F, Cooke HJ, Shi Q, miR-214-mediated downregulation of RNF8 induces chromosomal instability in ovarian cancer cells, Cell Cycle. 13 (2014) 3519–3528. 10.4161/15384101.2014.958413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Cortez D, Guntuku S, Qin J, Elledge SJ, ATR and ATRIP: partners in checkpoint signaling, Science. 294 (2001) 1713–1716. 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- [131].Maréchal A, Zou L, DNA damage sensing by the ATM and ATR kinases, Cold Spring Harb Perspect Biol. 5 (2013). 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Valenti F, Sacconi A, Ganci F, Grasso G, Strano S, Blandino G, Di Agostino S, The miR-205-5p/BRCA1/RAD17 Axis Promotes Genomic Instability in Head and Neck Squamous Cell Carcinomas, Cancers (Basel). 11 (2019). 10.3390/cancers11091347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Cejka P, DNA End Resection: Nucleases Team Up with the Right Partners to Initiate Homologous Recombination, J Biol Chem. 290 (2015) 22931–22938. 10.1074/jbc.R115.675942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ceccaldi R, Rondinelli B, D’Andrea AD, Repair Pathway Choices and Consequences at the Double-Strand Break, Trends Cell Biol. 26 (2016) 52–64. 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Browning CL, Wise JP, Prolonged exposure to particulate chromate inhibits RAD51 nuclear import mediator proteins, Toxicol Appl Pharmacol. 331 (2017) 101–107. 10.1016/j.taap.2017.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Browning CL, Qin Q, Kelly DF, Prakash R, Vanoli F, Jasin M, Wise JP, Prolonged Particulate Hexavalent Chromium Exposure Suppresses Homologous Recombination Repair in Human Lung Cells, Toxicol Sci. 153 (2016) 70–78. 10.1093/toxsci/kfw103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Jia J, Li T, Yao C, Chen J, Feng L, Jiang Z, Shi L, Liu J, Chen J, Lou J, Circulating differential miRNAs profiling and expression in hexavalent chromium exposed electroplating workers, Chemosphere. 260 (2020) 127546. 10.1016/j.chemosphere.2020.127546. [DOI] [PubMed] [Google Scholar]
- [138].Chen QY, Murphy A, Sun H, Costa M, Molecular and epigenetic mechanisms of Cr(VI)-induced carcinogenesis, Toxicol Appl Pharmacol. 377 (2019) 114636. 10.1016/j.taap.2019.114636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Li Y, Hu G, Li P, Tang S, Zhang J, Jia G, miR-3940-5p enhances homologous recombination after DSB in Cr(VI) exposed 16HBE cell, Toxicology. 344–346 (2016) 1–6. 10.1016/j.tox.2016.02.003. [DOI] [PubMed] [Google Scholar]
- [140].Lee J-H, Park S-J, Jeong S-Y, Kim M-J, Jun S, Lee H-S, Chang I-Y, Lim S-C, Yoon SP, Yong J, You HJ, MicroRNA-22 Suppresses DNA Repair and Promotes Genomic Instability through Targeting of MDC1, Cancer Res. 75 (2015) 1298–1310. 10.1158/0008-5472.CAN-14-2783. [DOI] [PubMed] [Google Scholar]
- [141].Rojas E, Martinez-Pacheco M, Rodriguez-Sastre MA, Ramos-Espinosa P, Valverde M, Post-transcriptional regulation of Rad51c by miR-222 contributes cellular transformation, PLoS ONE. 15 (2020) e0221681. 10.1371/journal.pone.0221681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Symington LS, Gautier J, Double-strand break end resection and repair pathway choice, Annu Rev Genet. 45 (2011) 247–271. 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- [143].Deriano L, Roth DB, Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage, Annu Rev Genet. 47 (2013) 433–455. 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- [144].Orr B, Godek KM, Compton D, Aneuploidy, Curr Biol. 25 (2015) R538–542. 10.1016/j.cub.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Vermeulen K, Van Bockstaele DR, Berneman ZN, The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer, Cell Prolif. 36 (2003) 131–149. 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Malumbres M, Barbacid M, Cell cycle, CDKs and cancer: a changing paradigm, Nat Rev Cancer. 9 (2009) 153–166. 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- [147].Otto T, Sicinski P, Cell cycle proteins as promising targets in cancer therapy, Nat Rev Cancer. 17 (2017) 93–115. 10.1038/nrc.2016.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].May KM, Hardwick KG, The spindle checkpoint, J Cell Sci. 119 (2006) 4139–4142. 10.1242/jcs.03165. [DOI] [PubMed] [Google Scholar]
- [149].Kops GJ, The kinetochore and spindle checkpoint in mammals, Front Biosci. Volume (2008) 3606. 10.2741/2953. [DOI] [PubMed] [Google Scholar]
- [150].Lara-Gonzalez P, Westhorpe FG, Taylor SS, The Spindle Assembly Checkpoint, Current Biology. 22 (2012) R966–R980. 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- [151].Sacristan C, Kops GJPL, Joined at the hip: kinetochores, microtubules, and spindle assembly checkpoint signaling, Trends in Cell Biology. 25 (2015) 21–28. 10.1016/j.tcb.2014.08.006. [DOI] [PubMed] [Google Scholar]
- [152].Barnum KJ, O’Connell MJ, Cell Cycle Regulation by Checkpoints, in: Noguchi E, Gadaleta MC (Eds.), Cell Cycle Control, Springer New York, New York, NY, 2014: pp. 29–40. 10.1007/978-1-4939-0888-2_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Kops GJPL, Snel B, Tromer EC, Evolutionary Dynamics of the Spindle Assembly Checkpoint in Eukaryotes, Current Biology. 30 (2020) R589–R602. 10.1016/j.cub.2020.02.021. [DOI] [PubMed] [Google Scholar]
- [154].Bates M, Furlong F, Gallagher MF, Spillane CD, McCann A, O’Toole S, O’Leary JJ, Too MAD or not MAD enough: The duplicitous role of the spindle assembly checkpoint protein MAD2 in cancer, Cancer Lett. 469 (2020) 11–21. 10.1016/j.canlet.2019.10.005. [DOI] [PubMed] [Google Scholar]
- [155].Derks S, Cleven AHG, Melotte V, Smits KM, Brandes JC, Azad N, van Criekinge W, de Bruïne AP, Herman JG, van Engeland M, Emerging evidence for CHFR as a cancer biomarker: from tumor biology to precision medicine, Cancer Metastasis Rev. 33 (2014) 161–171. 10.1007/s10555-013-9462-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Yu X, Minter-Dykhouse K, Malureanu L, Zhao W-M, Zhang D, Merkle CJ, Ward IM, Saya H, Fang G, van Deursen J, Chen J, Chfr is required for tumor suppression and Aurora A regulation, Nat Genet. 37 (2005) 401–406. 10.1038/ng1538. [DOI] [PubMed] [Google Scholar]
- [157].Lioutas A, Vernos I, Aurora A: working from dawn to dusk in mitosis, Cell Cycle. 13 (2014) 499–500. 10.4161/cc.27781. [DOI] [PubMed] [Google Scholar]
- [158].van de Weerdt BCM, Medema RH, Polo-Like Kinases: A Team in Control of the Division, Cell Cycle. 5 (2006) 853–864. 10.4161/cc.5.8.2692. [DOI] [PubMed] [Google Scholar]
- [159].Carvalhal S, Ribeiro SA, Arocena M, Kasciukovic T, Temme A, Koehler K, Huebner A, Griffis ER, The nucleoporin ALADIN regulates Aurora A localization to ensure robust mitotic spindle formation, MBoC. 26 (2015) 3424–3438. 10.1091/mbc.E15-02-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Wang X-H, Lu Y, Liang J-J, Cao J-X, Jin Y-Q, An G-S, Ni J-H, Jia H-T, Li S-Y, MiR-509-3-5p causes aberrant mitosis and anti-proliferative effect by suppression of PLK1 in human lung cancer A549 cells, Biochem Biophys Res Commun. 478 (2016) 676–682. 10.1016/j.bbrc.2016.08.006. [DOI] [PubMed] [Google Scholar]
- [161].Pruikkonen S, Kallio MJ, Excess of a Rassf1-targeting microRNA, miR-193a-3p, perturbs cell division fidelity, Br J Cancer. 116 (2017) 1451–1461. 10.1038/bjc.2017.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Bolanos-Garcia VM, Blundell TL, BUB1 and BUBR1: multifaceted kinases of the cell cycle, Trends Biochem Sci. 36 (2011) 141–150. 10.1016/j.tibs.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Bhattacharjya S, Nath S, Ghose J, Maiti GP, Biswas N, Bandyopadhyay S, Panda CK, Bhattacharyya NP, Roychoudhury S, miR-125b promotes cell death by targeting spindle assembly checkpoint gene MAD1 and modulating mitotic progression, Cell Death Differ. 20 (2013) 430–442. 10.1038/cdd.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Bacher JW, Sievers CK, Albrecht DM, Grimes IC, Weiss JM, Matkowskyj KA, Agni RM, Vyazunova I, Clipson L, Storts DR, Thliveris AT, Halberg RB, Improved Detection of Microsatellite Instability in Early Colorectal Lesions, PLoS One. 10 (2015) e0132727. 10.1371/journal.pone.0132727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Abdel-Rahman WM, Katsura K, Rens W, Gorman PA, Sheer D, Bicknell D, Bodmer WF, Arends MJ, Wyllie AH, Edwards PA, Spectral karyotyping suggests additional subsets of colorectal cancers characterized by pattern of chromosome rearrangement, Proc Natl Acad Sci U S A. 98 (2001) 2538–2543. 10.1073/pnas.041603298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Shaffer LG, Bejjani BA, Medical applications of array CGH and the transformation of clinical cytogenetics, Cytogenet Genome Res. 115 (2006) 303–309. 10.1159/000095928. [DOI] [PubMed] [Google Scholar]
- [167].Robledo C, García JL, Hernández JM, Clinical applications of BAC array-CGH to the study of diffuse large B-cell lymphomas, Methods Mol Biol. 973 (2013) 121–145. 10.1007/978-1-62703-281-0_8. [DOI] [PubMed] [Google Scholar]
- [168].Shinawi M, Cheung SW, The array CGH and its clinical applications, Drug Discov Today. 13 (2008) 760–770. 10.1016/j.drudis.2008.06.007. [DOI] [PubMed] [Google Scholar]
- [169].Zhang X, Shen B, Cui Y, Ago HITS-CLIP expands microRNA-mRNA interactions in nucleus and cytoplasm of gastric cancer cells, BMC Cancer. 19 (2019) 29. 10.1186/s12885-018-5246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Liu S-T, Zhang H, The mitotic checkpoint complex (MCC): looking back and forth after 15 years, AIMS Mol Sci. 3 (2016) 597–634. 10.3934/molsci.2016.4.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Almasan A, Linke SP, Paulson TG, Huang LC, Wahl GM, Genetic instability as a consequence of inappropriate entry into and progression through S-phase, Cancer Metastasis Rev. 14 (1995) 59–73. 10.1007/BF00690212. [DOI] [PubMed] [Google Scholar]
- [172].van Harn T, Foijer F, van Vugt M, Banerjee R, Yang F, Oostra A, Joenje H, te Riele H, Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling, Genes Dev. 24 (2010) 1377–1388. 10.1101/gad.580710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Ishikawa K, Ishii H, Saito T, DNA damage-dependent cell cycle checkpoints and genomic stability, DNA Cell Biol. 25 (2006) 406–411. 10.1089/dna.2006.25.406. [DOI] [PubMed] [Google Scholar]