

Summary
The human gut microbiome harbors hundreds of bacterial species with diverse biochemical capabilities. Dozens of drugs have been shown to be metabolized by single isolates from the gut microbiome, but the extent of this phenomenon is rarely explored in the context of microbial communities. Here, we develop a quantitative experimental framework for mapping the ability of the human gut microbiome to metabolize small molecule drugs: Microbiome Derived Metabolism (MDM)-Screen. Included are a batch culturing system for sustained growth of subject-specific gut microbial communities, an ex vivo drug metabolism screen, and targeted and untargeted functional metagenomic screens to identify microbiome-encoded genes responsible for specific metabolic events. Our framework identifies novel drug-microbiome interactions that vary between individuals and demonstrates how the gut microbiome might be used in drug development and personalized medicine.
In brief
Each human has a diverse gut microbiome, which can metabolize drugs differently. In this resource, Javdan et al. present a way to capture and grow much of the unique diversity of human microbiomes in culture and also a way to detect many of our microbiome-derived metabolites. Together, they use these unique gut communities and the metabolomics pipeline to see how personalized microbiomes metabolize drugs in different ways.
Graphical Abstract
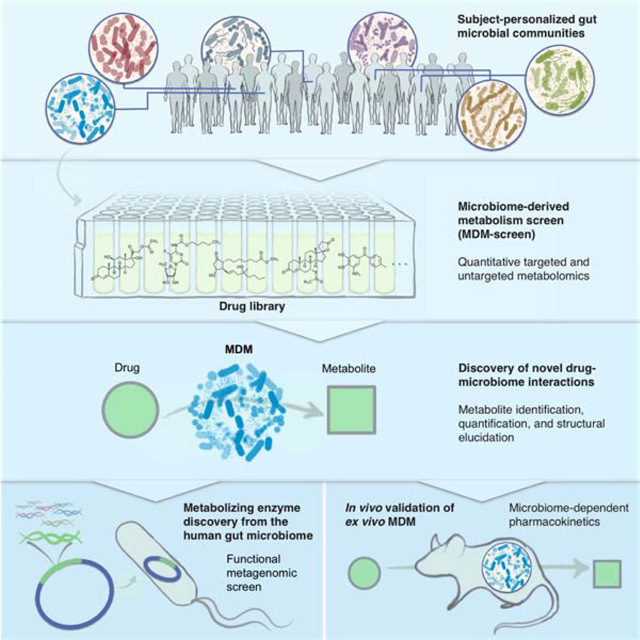
INTRODUCTION
The oral route is the most common route for drug administration. Upon exiting the stomach, drugs can be absorbed in the small and/or large intestine to reach systemic circulation and eventually the liver, or directly transported there via the portal vein. In the liver, drugs may be metabolized and secreted back to the intestines through bile, via enterohepatic circulation (Kimura et al., 1994; Li and Jia, 2013). Even parenterally administered drugs and their metabolites can reach the intestines through biliary secretion. Thus, whether prior to or after absorption, some administered drugs will spend a considerable amount of time in the small and large intestines, where our human gut microbiome resides. It is therefore important to study gut microbiome composition and function, specifically as it relates to drug interactions, while accounting for the significant variability between individuals (Falony et al., 2016).
Broadly speaking, the microbiome interacts with drugs both directly and indirectly. Indirect interactions include competition between microbiome-derived metabolites and administered drugs for the same host metabolizing enzymes (Clayton et al., 2009), microbiome effects on the immune system in anticancer immunotherapy (Iida et al., 2013; Sivan et al., 2015; Vetizou et al., 2015), microbiome reactivation of secreted inactive metabolites of the drug (Wallace et al., 2010), and overall microbiome effects on the levels of metabolizing enzymes in the liver and intestine (Meinl et al., 2009). Direct interactions include the partial or complete biochemical transformation of a drug into more or less active metabolites by microbiome-derived enzymes (termed herein: Microbiome-Derived Metabolism, or MDM).
The human gut microbiome harbors hundreds of bacterial species, encoding an estimated 100 times more genes than the human genome (Qin et al., 2010). This enormous diversity and richness represent a repertoire of yet-uncharacterized biochemical activities capable of metabolizing ingested chemicals (Backhed et al., 2005; Koppel et al., 2017). Although MDM has been observed in dozens of examples for the past 50 years, this process is still mostly overlooked in the drug development pipeline where little to no effort is spent on determining the specific role of MDM in pharmacokinetics (Ilett et al., 1990; Li and Jia, 2013; Scheline, 1973; Spanogiannopoulos et al., 2016). This is because of the vast complexity of the microbiome, and overwhelming technical challenge of testing hundreds of drugs against thousands of cultured isolates under multiple conditions. In contrast to liver-derived metabolism, we lack a systematic and standardized map of MDM, hindering our ability to reliably predict and eventually interfere with undesired microbiome effects on drug pharmacokinetics and pharmacodynamics.
To address this gap in knowledge, we developed a quantitative experimental workflow for mapping MDM of orally administered drugs using personalized gut microbiome-derived microbial communities (MDM-Screen). The methods and findings reported here provide a framework for discovering and characterizing novel cases of MDM, and for potentially incorporating an “MDM module” in the drug development pipeline.
RESULTS
Mapping the capacity of a single subject’s microbiome to metabolize hundreds of drugs
A major challenge in studying the capacity of the human gut microbiome to metabolize orally administered drugs is the diversity of bacterial species and strains involved (Almeida et al., 2019; Lloyd-Price et al., 2017; Nayfach et al., 2019; Pasolli et al., 2019; Qin et al., 2010). Because it is impractical to systematically screen thousands of isolated strains against hundreds of drugs, previous studies have relied mainly on monocultures of a selected set of representative species. However, gene expression and biochemical transformation profiles vary dramatically between a strain grown in monoculture versus in a mixed community. To address these challenges, we sought to develop an optimized ex vivo mixed culturing system that supports the growth of a large proportion of the species from a given microbiome sample and is amenable to high-throughput (HT) biochemical screens.
We began our screening efforts focused on a single microbiome donor (pilot donor, PD). To identify the medium and culturing period that can support the growth of a batch culture whose composition is maximally similar to the original PD microbiome, freshly collected and glycerol-stocked human feces from PD were cultured in 14 different media and sampled daily for four days. We then extracted DNA from all samples, amplified the V4 region of the bacterial 16S rRNA gene, and deeply sequenced the amplicons (Figure 1A). From the sequencing results, amplicon sequence variants (ASVs) were inferred and the taxonomic composition at different levels was determined for each sample (see STAR Methods) (Bokulich et al., 2018; Bolyen et al., 2018; Callahan et al., 2016; McDonald et al., 2012). We then quantified the differences between the various media and PD at the family level (using the Jensen-Shannon divergence, DJS), and the variant recovery from PD at the single ASV level.
Figure 1. Development of an ex vivo batch culturing system for the PD microbiome.

A) Schematic representation of the media selection procedure. B) Family level bacterial composition of the original fecal sample (far left), as well as that of PD ex vivo cultures grown anaerobically in 14 different media over two days (.01 and .02). See STAR Methods for full media names. 16S rRNA gene sequences that could not be classified at the family level, and families with less than 1% relative abundance in all samples are grouped into “Other”. Cultures are ordered according to their Jensen-Shannon (DJS) divergence from the original PD sample (upper axes, computed at the family level in base e). C) ASV level bacterial composition of the original PD fecal sample, and that of day two ex vivo cultures of PD, where each square represents one sample. Rainbow colored dots represent the relative abundance of individual ASVs that are above 1% in PD, while grey dots represent the combined relative abundance of all ASVs below 1% in PD. Samples are ordered by their Shannon diversity (H) at the ASV level, computed in base 2 and shown above each square. See also Figure S1, Table S2.
As expected, we observed a great level of variation in both the taxonomic composition and diversity between the different media and culturing periods. Some media led to highly diverse communities that captured portions of the original fecal diversity, while others became dominated almost exclusively by a single family. Among the 14 media commonly used in gut microbiome cultivation efforts (Rettedal et al., 2014), we identified one, modified Gifu Anaerobic Medium (mGAM), that supported the growth of a bacterial community most similar in composition and diversity to PD’s (Figures 1B, S1A). At the family level, mGAM cultures largely match the composition of PD, differing primarily in a commonly observed expansion of the facultative anaerobes, Enterobacteriaceae, at the expense of the obligate anaerobes, Ruminococcaceae (McDonald et al., 2018). Among all tested media, mGAM cultures showed the lowest DJS divergence from PD, becoming increasingly similar to the original sample as growth proceeds (Figure S1A).
Even at the single ASV level, mGAM cultures capture much of the diversity in PD (mGAM cultures have the highest Shannon diversity across all media, and the closest one to PD) (Figures 1C, S1A). In PD, there are 33 ASVs present above a relative abundance of 1%, 26 (79%) of which are present in mGAM day two culture. Overall, total shared ASVs between PD and mGAM day two account for 70% of the PD composition (by relative abundance), indicating that the mGAM culture recapitulates the bulk of the original community. Taken together, and consistent with previous reports showing that mGAM can support the growth of a wide variety of gut microorganisms in monoculture (Rettedal et al., 2014; Tramontano et al., 2018), our results support the use of mGAM day two cultures as a viable ex vivo batch culturing model for the PD microbiome.
With an optimized ex vivo culturing system for PD in hand, we next developed a combined biochemical/analytical chemistry approach to map the capacity of PD-derived microbial communities to metabolize clinically used, orally administered drugs (MDM-Screen) (Figure 2A). Three samples were prepared per drug of interest: 1) a 24-hour mGAM ex vivo culture of PD, incubated with the drug of interest (final concentration 33 μM in line with estimates of drug concentrations in the gastrointestinal tract) (Maier et al., 2018), 2) a similar culture incubated with a vehicle control (DMSO), and 3) an equal volume of sterile mGAM, incubated with the same drug concentration. Cultures and controls were then incubated for an additional 24 hours at 37°C in an anaerobic chamber, chemically extracted, and analyzed using High Performance Liquid Chromatography coupled with Mass Spectrometry (HPLC-MS). To verify the reproducibility, the entire procedure was repeated three consecutive times. We tested a diverse library of 575 orally administered drugs, and although the majority of the drugs in this library are currently being used in the clinic, less than 10% of them had been previously explored with respect to MDM (Table S1A). A drug was deemed MDM+ when: a) a new metabolite was observed when incubated with PD culture, or b) the drug was no longer detected when incubated with PD culture, indicating that it is either consumed entirely or metabolized into a molecule that fails our detection, and c) the drug was metabolized in the same manner during at least two of three independent experiments.
Figure 2. Screening of the PD microbiome against orally administered drugs identifies novel drug-microbiome interactions.

A) Schematic representation of MDM-Screen. A drug was considered MDM+ if a new metabolite is produced (e.g., drug 3) or if the drug is no longer detectable (e.g., drug 5) after incubation with the microbiome, as compared to abiotic media controls. B) A bar graph showing the pharmacological classes of MDM+ drugs discovered by MDM-Screen with the PD microbiome. “Others” include one drug each from 14 additional classes. C) Examples of MDM+ drugs where the drug is no longer detectable after incubation with the PD microbiome. D) Examples of MDM+ drugs where a new metabolite is discovered by MDM-Screen and fully characterized in this study. See also Table S1, Data S1,S2.
MDM-Screen identifies known and novel drug-microbiome interactions
Among the 575 drugs, we successfully analyzed 438 (76%); the remaining 137 failed MDM-Screen due to issues related to drug stability or incompatibilities with the extraction or chromatography methods employed (see Discussion). Of the successfully analyzed drugs, we identified 57 (13%) as MDM+. These spanned 28 pharmacological classes and even more based on their chemical structure (Figure 2B, Table S1B, Data S1). As expected, several previously reported MDM cases were identified. These include the nitroreduction of the muscle relaxant dantrolene (Kuroiwa et al., 1985), the antiepileptic clonazepam (Elmer and Remmel, 1984; Zimmermann et al., 2019b), and the antihypertensive drug nicardipine (Kuroiwa et al., 1986); hydrolysis of the isoxazole moiety in the antipsychotic risperidone (Mannens et al., 1993; Meuldermans et al., 1994); and azoreduction of the anti-inflammatory prodrug sulfasalazine (Azadkhan et al., 1982; Peppercorn and Goldman, 1972).
More importantly, we identified a suite of novel MDM cases (45 cases, ~80% of the MDM+ drugs): ten resulted from full depletion of the parent drug (or full conversion to a metabolite that evades our detection), while 35 resulted from the appearance of a new metabolite (Figures 2C,D, Table S1B, Data S1A). In most cases, the new metabolites showed a high-resolution mass spectrometry (HRMS) profile within a small difference from their parent drugs, and/or a similar tandem MS fragmentation (HRMS/MS) pattern, indicating that they are derivatives (Wang et al., 2016) (Table S1B, Data S1B). An aggregate statistical analysis of MDM+ and MDM- drugs revealed specific structural features that are significantly enriched in MDM+ drugs (e.g., a steroidal skeleton, nitro groups, ketones, among others) (see STAR Methods, Table S1C).
Although HRMS and HRMS/MS analyses can narrow down the number of possibilities for the molecular structure of a given metabolite, they are not sufficient for full structural determination. Thus, we selected seven MDM+ examples for detailed characterization of their resulting metabolites: spironolactone (anti-hypertensive), tolcapone (anti-Parkinson’s), misoprostol (anti-ulcer), mycophenolate mofetil (immunosuppressant), capecitabine (anticancer), and finally, hydrocortisone and hydrocortisone acetate (two steroidal anti-inflammatory drugs that produced an identical MDM metabolite). In all but one example, no direct drug-microbiome interactions had been previously reported. The exception was hydrocortisone where several metabolites had been previously reported from individual gut isolates (Ridlon et al., 2013; Winter et al., 1982), but the identity of the MDM metabolite we observed could not be accurately matched to any of them based on MS alone.
To unequivocally determine the structure of the resulting metabolites, we isolated them from scaled-up biochemical incubations with PD cultures, and elucidated their structures using Nuclear Magnetic Resonance (NMR) and/or comparison to an authentic standard (STAR Methods, Data S2). For hydrocortisone, we determined that MDM results in the reduction of the ketone group at C20, producing 20β-dihydrocortisone. For hydrocortisone acetate, the same modification occurs but is accompanied with deacetylation of the C21 hydroxyl group (Figure S2). While C20 ketone reduction was previously reported for hydrocortisone (to produce either 20β-dihydrocortisone or 20α-dihydrocortisone depending on the gut isolate incubated with it (Ridlon et al., 2013; Winter et al., 1982)), neither MDM deacetylation nor C20 ketone reduction were reported for hydrocortisone acetate. For capecitabine, we show that MDM results in complete deglycosylation, for misoprostol and mycophenolate mofetil, we observed an ester hydrolysis transformation, and in the case of spironolactone, a thioester hydrolysis one. None of these MDM transformations were previously reported for these drugs. Finally, for tolcapone, we observed two consecutive transformations, a typical nitroreduction followed by a relatively uncommon N-acetylation – neither of which had been previously linked to the microbiome for this drug (Figure 2D). Taken together, these results establish MDM-Screen as a viable method for identifying both known and novel biochemical modifications of structurally and pharmacologically diverse drugs by the gut microbiome.
Interestingly, based on already known pharmacokinetic studies in humans, some of these new MDM cases may have direct consequences on the activation or toxicity of the drugs involved. For example, in the case of spironolactone we observed the production of 7α-thiospironolactone, a postulated intermediate en route to the drug’s main active metabolite, 7α-thiomethylspironolactone (Gardiner et al., 1989; Sica, 2005). For the prodrugs misoprostol and mycophenolate mofetil, we observed the production of their active metabolites misoprostol acid (Schoenhard et al., 1985; Tsai et al., 1991) and mycophenolic acid (Bullingham et al., 1998), respectively. Interestingly, mycophenolic acid has been linked to the clinically observed gastrointestinal toxicity associated with mycophenolate mofetil use (Taylor et al., 2019), albeit generated via a different route – hydrolysis of a biliary secreted glucuronide conjugate by gut microbiome-derived β-glucuronidases. Finally, in the case of tolcapone, N-acetylamino-tolcapone has been detected systemically in humans post tolcapone administration, and was suggested to be involved in liver toxicity observed clinically following tolcapone use, yet the mechanism of its production remains unknown (Jorga et al., 1999; Smith et al., 2003). Our discovery that the same metabolite can be produced via MDM provides a possible explanation, and a potential link between the gut microbiome and tolcapone toxicity. In all four cases, additional experiments need to be performed to differentiate the contribution of human- and microbiome-derived metabolism to the observed drug pharmacokinetics and/or toxicity in humans.
Expanding MDM-Screen to multiple subjects
Next, we sought to expand our framework to accommodate multiple subjects. To accomplish this goal, we needed to first design a generalizable quantitative metric for assessing the best culturing medium for microbiome samples (Figure 3A, 3B). In our analysis of PD’s ex vivo cultures, we applied a variety of metrics and found a medium that was the best trade-off between richness, evenness, and compositional similarity. However, this approach is not scalable to a large number of donors and also ignores the role of community biomass, which may lead to suboptimal media selection. Therefore we developed a metric called Expected Number of Detectable Strains (ENDS), a corrected richness metric where the contribution of each ASV is weighed by the probability that its metabolite can be detected while considering total biomass (STAR Methods, Figure 3B, Methods S1). The core idea is that we desire a medium that supports the highest number of different bacterial ASVs, while ensuring that the metabolic contributions of these ASVs are detectable by our experimental method. ENDS utilizes two data inputs related to the ex vivo culture composition: relative abundance at a given taxonomic level and total community biomass, and two inputs related to the instrument detection sensitivity: a model of instrument background noise and a model of instrument measurement noise. Using this information, a simple mechanistic model of MDM metabolite production, and estimations of statistical power, we compute the probabilities that metabolic reactions performed by each strain will be detected in the ex vivo culture (see STAR Methods for a detailed mathematical description of ENDS).
Figure 3. Identifying the optimal medium for multi-donor MDM-Screen.

A) Schematic representation of the media selection procedure for D1–20. B) Schematic representation of the ENDS metric. Using 16S rDNA sequencing and biomass measurements, absolute abundances of ASVs (orange and grey strains) in different ex vivo communities are measured and metabolite production from each member of the community is estimated using a simple mathematical model. Using instrument noise properties, distributions of metabolite measurements from each ASV (orange and grey distributions) are estimated and compared to instrument noise (white distribution). Statistical power estimation is then used to compute metabolism detection probabilities for each ASV and the condition maximizing ENDS (the sum of these probabilities) is selected. C) ASV abundance heat map of the original fecal samples and ex vivo microbial communities for each donor. Each box corresponds to samples from a single donor, with the original fecal sample shown on the far left followed by different ex vivo media in the order specified above the heat map. Only ASVs above 5% in at least one sample are shown, with all remaining ASVs aggregated into ‘Other’. The taxonomic classification of each ASV (on the order level) is indicated by the color bar on the left. D) Histogram of ex vivo community biomass for all donors in different media conditions. E) Comparison of shared ASVs within (self, i.e., the ASV richness) and between (non-self) donor fecal samples. ‘***’ indicates p < 0.001, permutation test. F) Comparison of shared ASVs between donor fecal samples and ex vivo cultures originating from the same donor (self) versus ones originating from other donors (non-self). ‘***’ indicates p < 0.001, permutation test. G) Average ENDS of different media conditions at varying metabolite production rates (quantified as AUC normalized to an internal standard). ENDS was computed for each ex vivo culture assuming a p-value significance cutoff of 0.01 and three replicates. For each media condition, ENDS was averaged across all donors. H) Average fractional recovery of different taxa in BG ex vivo communities as a function of relative abundance in the original donor fecal sample. The fractional recovery was calculated for all donors and then averaged. See also Figure S1, Table S2.
With this quantitative framework in hand, we collected additional fresh fecal samples from 20 healthy donors (D1–20) and processed them in the same manner as PD. We then cultured each sample in nine representative media and used 16S rDNA sequencing to determine the composition of the cultured communities as previously described (Figures 3A-C, S1, Table S2A). To measure community biomass, one ml of each culture was pelleted and weighed (STAR Methods, Table S2C,D). We observed a wide variation in culture characteristics, with the richness ranging from 20–135 ASVs and biomass density ranging from 2–27.9 g/L (Figure 3D). mGAM and BB media consistently performed well with all 20 donors. Interestingly, mGAM had moderate ASV richness and high biomass, while BB yielded a much lower biomass with high richness and did not suffer from the Enterobacteriaceae expansion observed in mGAM. We calculated that a 70/30 BB/mGAM mixture would yield an optimal medium with moderate biomass, high richness, and a reduced Enterobacteriaceae expansion (Methods S1B), thus we included this mixture (named BG) as a 10th medium in our culturing trials.
Next, we wondered whether the ex vivo cultured communities are truly personalized per subject, an important prerequisite if cultured communities are to be used for assessing inter-individual variability in MDM. Personalization between cultures was clearly observed at the ASV level, with clear specific patterns unique to individual donors and their cultures (Figure 3C). We found significantly more ASVs shared between donor feces and their self ex vivo cultures than non-self (47.1 vs. 27.5 ASVs, p < 0.001, permutation test), partially recapitulating the inherent personalization between the donor fecal samples (Figure 3E,F). Moreover, we identified 167 ASVs that were unique to one of the 20 donors in their fecal samples (8.4 ASVs per donor on average) and were concordantly unique to the same donor in their ex vivo cultures (Table S2F). Finally, we grew and sequenced multiple replicates of mGAM and BG ex vivo cultures from different donors (all 20 donors, three replicates each for BG, and eight donors, six replicates each for mGAM). The analysis of these replicates, whether cultured from the same or separate glycerol stock aliquots, revealed a high correlation between ASV abundances of replicates from the same donor and ensured that the community assembly process was replicable (Pearson correlation coefficient > 0.9) (Methods S1A, Figure S1). These analyses confirm that our approach results in personalized and replicable microbial communities.
To select a single medium that would be on average optimal for use in a 20-donor screen, we computed the average ENDS of all media across a range of reaction rates (Figure 3G). We found that BG is on average an optimal medium at reaction rates we estimated to be the most physiologically relevant (Methods S1D). In addition, BG recapitulates a large portion of the microbial community in the original fecal sample. On average, BG cultures recover 76.6% of the ASVs above 1% in the original fecal sample, which translates to 84.7%, 88.3%, 92.7% recovery rate on the species, genus, and family levels of taxa above 1% in the original sample, respectively (Figure 3H). In terms of recovery of all elements, BG recovers 43.3%, 57.3%, 60.6%, and 62.8% on the ASV, species, genus, and family levels, respectively. BG is also on average the closest in composition to the original sample (DJS = 0.16) and has the highest average diversity (H = 4.3). We therefore selected BG as the medium to use in our 20-donor screen.
We next sought to develop a HT, quantitative metabolomic approach to assess MDM inter-individual variability with a subset of drugs. Several improvements were made to the original drug metabolism screen. All experimental steps including incubation, chemical extraction, and HPLC-HRMS analysis were performed in microtiter 96-well plates instead of individual tubes, at a 400 μl volume instead of 3 ml. This lowered the amount of drug used per incubation, allowed us to perform triplicated reactions simultaneously, and streamlined our chemical extraction and analysis procedures. We also spiked a known concentration of an internal standard prior to the chemical extraction, which allowed us to precisely quantify partial, in addition to complete, drug depletion.
We chose a 23 drug subset to test the ability of our quantitative approach to reveal potential inter-individual variabilities in MDM under the MDM-Screen conditions. Thirteen drugs had at least one defined metabolite with a known chemical structure, allowing us to unambiguously compare their levels between samples (Figure S3, Table S3). For all 20 donors, ex vivo cultures (in BG medium) were incubated in triplicates in a 96-well microtiter plate with each of the 23 drugs at a final concentration of 33 μM, or with DMSO (Figure 4A). In addition, an abiotic medium-drug plate as well as a heat-killed-microbiome-drug (HKM-drug) plate were prepared in the same manner. After 24-hour incubation, culture and control plates were chemically extracted and analyzed using HPLC-HRMS (STAR Methods).
Figure 4. A HT, quantitative metabolomic approach to assess inter-individual variability in MDM using personalized microbial communities.

A) Schematic representation of quantitative MDM-Screen with 20 donors and 23 selected drugs. B) Heat map of drug depletion showing the mean fraction of drug remaining after 24 hours for each donor-drug combination. The fraction remaining is computed relative to the medium-drug control, and fractions above 1 are truncated to 1 for simplicity. C) Heat map of metabolite production showing the mean level of metabolite after 24 hours, normalized to the maximum level of that metabolite across all donors. Metabolites in red were discovered using the untargeted metabolomics approach, while ones in black were discovered previously or by MDM-Screen with the PD microbiome (Table S3B). In B and C, “*” indicates statistically significant metabolism in the donor condition as compared to controls. The upper inset axes represent inter-individual variability in MDM using the Shannon entropy (calculated in base 2) of the distribution of donors with significant and non-significant metabolism. D) Cumulative histogram of the number of significant donors for both metabolite production and parent drug depletion. For parent drugs, the y-axis is normalized to the total number of drugs tested (23), and for metabolite production, it is normalized to the total number of metabolites produced (32). Levels of metabolite production (measured by HPLC-HRMS in AUC normalized to an internal standard) for four drugs, with the variability entropy indicated above. Filled data points indicate that the replicates are significantly higher than control conditions, while hollow data points indicate that they are not. F) The upper three scatter plots show significant negative correlation between drug depletion and metabolite production, with the Pearson correlation coefficient indicated above. The line shown is a linear regression fit of the data. The lower bar plot indicates the Pearson correlation coefficient between remaining drug levels and total metabolite production for all computed cases. ‘*’ indicates an FDR corrected two-sided t-test p < 0.01. For drugs with multiple metabolites, we sum the normalized AUC of all metabolites. G) Correlation between drug depletion and metabolite production for nicardipine before and after inclusion of metabolites discovered by untargeted metabolomics. See also Table S3.
We calculated percent drug remaining and metabolite level, to assess drug depletion and metabolite production in the presence of microbiome cultures, respectively. Both metrics were calculated using Area Under the Curve (AUC) integration with normalization to the internal standard (see STAR Methods, Table S3J-M). We determined statistical significance for metabolite production using a one-sided Welch’s t-tests between the donor-drug condition and the donor-DMSO, medium-drug, and HKM-drug conditions and corrected the resulting p-values for multiple hypotheses using the Benjamini-Hochberg method, requiring that tests against all three control conditions be significant at a level of 0.01 (Benjamini and Hochberg, 1995). For drug depletion, we used the same method with the donor-drug and HKM-drug conditions as controls and included an additional fold-change cutoff of two (Figure 4B,C, Table S3N-P). We also performed untargeted metabolomics analyses for new metabolite discovery, by identifying unique molecular features from all samples, determining statistical significance using similar methods as for the targeted metabolomics, and verifying the metabolite’s relationship to the parent drug based on their HRMS/MS fragmentation pattern (Wang et al., 2016) (STAR Methods, Table S3C,D). All verified metabolites from the untargeted metabolomics approach were then quantified using the same targeted metabolomics workflow described above (Figure 4C, Table S3N-P).
We observed cases of consistently negative MDM across donors (ketoconazole, praziquantel, ropinirole, and torsemide), consistently positive MDM in either drug depletion (misoprostol, nicardipine, spironolactone), metabolite production (tolcapone, vorinostat), or both (clonazepam, risperidone, and sulfasalazine), and variable MDM (Figure 4B-D). This variability was in drug depletion (ketoprofen, levonorgestrel), metabolite production (misoprostol, nicardipine, spironolactone), or both (capecitabine, clofazimine, digoxin, hydrocortisone, lovastatin, mycophenolate mofetil, sulindac, vorinostat). We quantified this variability by computing the Shannon entropy (in base 2) of the distribution of metabolizers and non-metabolizers, denoted as HV. This metric is maximal (HV = 1) when half of donors metabolize the drug and is minimal when the drug is either always or never metabolized (HV = 0).
The observed variability ranged widely from 1/20 to 19/20 donors deemed MDM+ for a given type of drug depletion or metabolite production. In the case of digoxin, for example, 3/20 donors (HV = 0.61) produced the known metabolite dihydrodigoxin in statistically significant amounts (Figure 4C,E). Inter-individual variability in digoxin MDM has been clinically known for decades, where significant reduction of the drug into dihydrodigoxin and related metabolites occurs in only a subset of patients (Lindenbaum et al., 1981). These results demonstrate that our screen can quantitatively assess the inter-individual variability of MDM between personalized gut microbial communities cultured under identical ex vivo conditions. Follow-up studies will need to be performed to evaluate whether our screening results directly correlate with clinical outcomes.
Next, we sought to determine whether the depletion of drugs in our screen can be explained by the production of associated metabolites. If changes in drug levels are primarily due to conversion to a detected metabolite, there should exist a strong negative correlation between depletion and metabolite production, corresponding to a stoichiometric mass balance. The absence of such a correlation, on the other hand, would suggest additional events that are not accounted for (e.g., the production of additional unknown or undetectable metabolites, the conversion of the initial metabolite into a third one, or bacterial consumption of the parent drug). For drugs with variable MDM (HV > 0.5 for at least one metabolite or the parent drug), we computed the Pearson correlation coefficient of the drug signal and the sum of known metabolite signals in all donor-drug ex vivo samples. We then determined whether a drug has statistically significant correlation by performing t-tests, correcting p-values using the Benjamini-Hochberg method, and requiring FDR corrected p < 0.01. For vorinostat and digoxin, for example, we found significant negative correlation between metabolite production and drug depletion (Pearson correlation coefficient of −0.91 and −0.79, respectively), suggesting that the majority of drug depletion can be explained by the production of the quantified metabolite (Figure 4F). Nicardipine, on the other hand, exhibited a very poor correlation initially (Pearson correlation coefficient of −0.22), implying that additional unknown factors are at play. Interestingly, our untargeted metabolomics pipeline detected 11 additional metabolites of nicardipine, which upon inclusion in the analysis resulted in a stronger negative correlation (Pearson correlation coefficient of −0.6, FDR corrected p = 0.0102) (Figure 4G, Table S3E). Since our screen is based on microbial communities and not individual strains, it provides a powerful platform to discover interacting factors that influence drug and metabolite levels under realistic conditions – as exemplified by the varying number of nicardipine metabolites observed per personalized community.
Next, we assessed whether we could predict MDM using taxonomic data. We computed Spearman correlations between absolute abundances of taxonomic elements (at different levels) in the BG ex vivo cultures and measured drug and metabolite levels in matching donors, but found no significant correlations – even in specific cases of MDM where metabolism has been previously attributed to a single species (e.g., digoxin reduction by Eggerthella lenta) (Haiser et al., 2013). This is likely due to a combination of two factors. First, as has been previously observed (Haiser et al., 2013) (Maini Rekdal et al., 2019), taxonomic classifications may not reflect the presence or absence of gene variants that encode strain-specific drug-metabolizing enzymes, even at the ASV level. Second, the observed level of MDM may not be monotonically dependent on a single taxon’s abundance if confounding community effects are at play. Examples of such effects include the contribution of several community members to the production of the metabolite(s), the consumption of the drug or metabolite(s), or the inhibition of the metabolite producing or drug depleting bacterium or enzyme (for a mathematical analysis of the impact of these factors on the correlation, see Methods S1C). These results emphasize the importance of considering whole community effects in MDM. While our ex vivo communities may not fully recapitulate all possible community effects that occur in humans, they represent an important step towards identifying and quantifying them.
Linking MDM to specific genes in the human microbiome
Next, we sought to link the observed biochemical transformations to specific microbiome-derived enzymes. We picked two representative cases of MDM transformations: MDM deglycosylation of capecitabine into deglycocapecitabine and C20 ketone reduction of hydrocortisone into 20β-dihydrocortisone. Two main approaches had been previously employed to identify genes responsible for a specific MDM transformation: comparative transcriptomics, which assumes that the expression of metabolizing enzymes is induced in the presence of their substrates (e.g. digoxin) (Haiser et al., 2013; Koppel et al., 2018), and homology-based discovery, which assumes that related classes of enzymes metabolize similar substrates (e.g., levodopa) (Maini Rekdal et al., 2019; van Kessel et al., 2019). For capecitabine deglycosylation, we elected to use a homology-based approach.
Characterizing the genetic basis of MDM deglycosylation using a homology-based approach
To identify a specific microbiome-derived isolate where a homology-based approach can be employed, we explored the ability of a limited panel of bacterial isolates to deglycosylate capecitabine, including strains isolated originally from PD. Interestingly, capecitabine deglycosylation was mainly performed by Proteobacteria (including Escherichia coli), and one of two tested Bacteroidetes: Parabacteroides distasonis, providing genetically tractable organisms for functional studies (Figure S4). In humans, thymidine phosphorylase (TP) and uridine phosphorylase (UP), both part of the pyrimidine salvage pathway, catalyze the deglycosylation of 5’-deoxy-5-fluorouridine (a late metabolite of capecitabine) to yield 5-fluorouracil (5-FU) (Temmink et al., 2007). To test whether bacterial homologs of human TP and/or UP are responsible for the observed MDM deglycosylation of capecitabine, we generated strains of E. coli BW25113 that are knockouts for TP (ΔdeoA), UP (Δudp), or both, and compared their ability to metabolize capecitabine to that of wild type E. coli (WT) (Figure 5A). While WT E. coli efficiently deglycosylates capecitabine (~30% conversion rate), the deglycosylating activity of Δudp and the ΔdeoA/Δudp strains is significantly diminished (less than 4% conversion rate, p-value <0.001, two-tailed t-test) (Figure 5B). Surprisingly, the ΔdeoA strain showed a significant increase in its deglycosylating activity in comparison to WT (~ 50% conversion rate, p-value <0.01, two-tailed t-test), possibly due to a compensating mechanism (e.g., overexpression of udp) in the absence of deoA. These results indicate that microbiome-derived UP is, at least in part, responsible for the deglycosylation of capecitabine.
Figure 5. Genetic basis and widespread nature of MDM deglycosylation among the FPs and in human gut metagenomes.

A) Genetic organization of the udp and deoA loci in the genome of E. coli BW25113. B) A bar graph indicating percent conversion of capecitabine to deglycocapecitabine by wild type E. coli BW25113 (WT), and Δudp, ΔdeoA, and ΔdeoA/Δudp mutants (each tested in triplicate). *** indicates p-value <0.001, while ** indicates p-value <0.01, two-tailed t-test. Error bars represent the standard deviation. C) Biochemical reaction catalyzed by thymidine and uridine phosphorylases on their natural substrates. D) MDM deglycosylation of the oral anticancer drug trifluridine leads to its premature inactivation, since trifluorothymine is no longer active. E) MDM deglycosylation of the anticancer prodrug doxifluridine leads to its premature activation, since 5-FU is the intended active metabolite. F) Heat maps indicating the prevalence and median abundance (in RPKM) of E. coli-derived deoA and udp across six gut metagenomic cohorts. G) Jitter plots of E. coli-derived deoA and udp abundances (in RPKM) in the same cohorts. See also Figures S4,S5, Table S4.
Capecitabine is one of several generations of antimetabolite chemotherapeutic agents, many of which are prodrugs for 5-FU, and are known collectively as the oral fluoropyrimidines (FPs) (Lamont and Schilsky, 1999; Longley et al., 2003). Importantly, oral FPs’ bioavailability and toxicity vary widely among patients (Cleary et al., 2017; Zampino et al., 1999), but the human gut microbiome’s contribution to this variability had not been explored. To determine whether deglycosylation occurs with other FPs, and whether the same enzymes are involved, we investigated the MDM of two additional oral FPs (doxifluridine and trifluridine) using WT and mutant E. coli. Unlike with capecitabine, almost complete deglycosylation was observed for both drugs with WT E. coli, and the activity was dependent on both TP and UP (Figures S4,S5). These results indicate a level of deglycosylation specificity for TP/UP amongst the FPs (Figure 5C). Remarkably, the consequences of the same modification differ depending on the tested drug. For trifluridine, the resulting metabolite (trifluorothymine) is inactive (Figures 5D, S4): trifluridine is typically incorporated intact into DNA to cause cytotoxicity (Lenz et al., 2015). Such a premature intestinal inactivation by the microbiome may thus be an unknown contributor to the established low bioavailability of trifluridine, in addition to the known contribution of human TP (Cleary et al., 2017). For doxifluridine, however, the resulting metabolite is the active 5-FU (Figures 5E, S5). This premature activation of the prodrug may therefore lead to gastrointestinal toxicity – again, a side effect commonly associated with oral doxifluridine (Kim et al., 2001; Min et al., 2000). Additional studies are necessary to directly correlate the level of MDM deglycosylation of different FPs in humans to their clinically observed pharmacokinetics and/or toxicity.
Since capecitabine was significantly and variably metabolized into deglycocapecitabine in 17/20 donors, we sought to examine the representation of FP deglycosylating enzymes in the gut microbiome of the human population at large. We specifically focused on enzymes that we experimentally verified to have a role in FP deglycosylation: E. coli-derived TP and UP. Overall, we analyzed six large and diverse human cohorts: the Human Microbiome Project (HMP-1-1 and HMP-1-2, 299 subjects from the USA) (Human Microbiome Project, 2012; Lloyd-Price et al., 2017), the Metagenomics of the Human Intestinal Tract Consortium (MetaHIT, 219 subjects from Spain and 176 subjects from Denmark) (Nielsen et al., 2014), a Chinese cohort (194 subjects) (Qin et al., 2012), and a Fijian cohort (Fijicomp, 192 subjects) (Brito et al., 2016). We mapped fecal metagenomic reads from each of the cohort samples to the DNA sequence of deoA and udp, and calculated two metrics: prevalence, i.e., the percent of subjects from each cohort that are positive for a given gene, and abundance of the gene amongst positive samples (calculated in Reads Per Kbp per Million of sequenced reads, or RPKM) (STAR Methods, Table S4B,C). Interestingly, we found that both genes were most prevalent in non-Western cohorts (Fijicomp, 74/76% positive subjects, and Chinese, 63/70% positive subjects for deoA/udp, respectively) in comparison to Western ones (24/26% on average, for deoA/udp, respectively), and that their abundance per positive samples varies widely within and between cohorts (from ~10−1 to ~102 RPKM) (Figures 5F,G, Table S4B,C). These results indicate that FP deglycosylating enzymes are both widespread and variable in the gut microbiome of diverse human cohorts (even when considering the contribution of a single bacterial species, E. coli), and further highlight the importance of considering MDM deglycosylation of FPs in clinical studies.
An untargeted functional metagenomic screening approach for identifying metabolizing enzymes
Although the homology-based approach was relatively straightforward in identifying responsible species and enzymes for the deglycosylation of FPs, it is not widely applicable. Unlike pyrimidine phosphorylases, oxidoreductases (the enzyme class likely responsible for hydrocortisone reduction to 20β-dihydrocortisone) are extremely diverse and typically substrate specific, with numerous homologs found per bacterial genome. Moreover, a homology-based approach usually requires the identification of an isolated strain that performs the modification of interest and its use as the basis for genetic manipulations and functional analyses. These two limitations motivated us to employ an orthogonal strategy that is not reliant on neither enzymatic homology nor isolated strains.
While no human gut microbiome-derived enzymes had previously been deemed responsible for converting hydrocortisone into 20β-dihydrocortisone, a cat microbiome-derived enzyme had: a 20β-hydroxysteroid dehydrogenase (20β-HSDH) from the cat fecal isolate Butyricicoccus desmolans ATCC 43058 (Devendran et al., 2017). Neither this enzyme, nor close homologs thereof (at 60% protein sequence identity or above) could be identified in a deep metagenomic sequencing dataset that we generated from the PD fecal DNA (STAR Methods). We therefore decided to use this example as a test case for developing an untargeted functional metagenomic screening strategy for metabolizing enzymes. In typical functional metagenomic screens, metagenomic DNA is cloned into a vector that replicates in E. coli and functional screens are performed in either a selective manner (e.g., for antibiotic resistance or an engineered circuit for survival) (Genee et al., 2016; Sommer et al., 2009; Uribe et al., 2019), or a visual readout (e.g., a colorimetric or antibacterial one) (Brady et al., 2002; Cohen et al., 2015; Gillespie et al., 2002; Rondon et al., 2000). Here, we use a functional metagenomic screen where the readout is a specific MDM transformation that is detected by MS. For metagenomic genes that are successfully expressed and produce functional gene products in E. coli, this approach would allow access to enzymes encoded by cultured and not-yet cultured members of the microbiome, and to ones that share no close homology with previously characterized enzymes. Two major technical challenges in this strategy, however, are to produce a large-enough metagenomic library that captures the majority of the genetic content in the complex microbiome, and to develop a HT analytical chemistry approach that permits the screening of such a library.
We isolated metagenomic DNA from PD and used it to construct a ~3 X 106-member clone library (PD-CL) in an E. coli expression vector (insert size 2–4 Kbp) (Figure 6A). To determine whether PD-CL is truly representative of the genetic content in PD, we deeply sequenced a representative pool that contains ~105 unique clones (PD-CL-100) and compared it to the deeply sequenced PD fecal metagenome. We then mapped metagenomic reads from either PD or PD-CL-100 to assembled scaffolds from the PD metagenome (25,529 scaffolds ≥ 2 Kbp). Satisfyingly, reads from PD-CL-100 (which represents only 3% of the full PD-CL) map to 21% of the PD scaffolds, including ones that originate from all major phyla and varying coverages in the PD microbiome (Figure 6B, Table S4A). These results indicate that PD-CL represents a large component of the genetic content in PD, and that it is adequate for use in functional metagenomics screens.
Figure 6. A functional metagenomic screening approach to identify a metabolizing enzyme.

A) Schematic representation of the functional metagenomic screening approach. B) A scatter plot comparing the coverage of assembled PD scaffolds (≥ 2 Kbp, in RPKM) in the two metagenomic datasets (PD and PD-CL-100). Dots representing PD metagenomic scaffolds are colored and sized on the basis of their phylum-level taxonomic assignments and lengths, respectively, and as indicated in the key on the right. For ease of visualization, only scaffolds with RPKM values ≤ 10 are shown in this plot (97% of all scaffolds ≥ 2 Kbp) (see also Table S4A for the entire dataset). C) Functional metagenomic screening of the PD-CL library. Beginning with pools containing 2–6 X 104 unique clones, pools were selected and further sub-pooled based on their functional ability to convert hydrocortisone to 20β-dihydrocortisone. Produced 20β-dihydrocortisone levels were quantified using HPLC-HRMS as AUC normalized to an internal standard. For each round, the pool producing the highest normalized signal of 20β-dihydrocortisone (signified by a red dot with a black outline) was selected for further sub-pooling, until a unique clone encoding a 20β-HSDH activity was identified. A single 20β-HSDH gene from the positive metagenomic clone was further verified by heterologous expression in E. coli, when cloned as the native sequence (cloned) or synthesized as codon-optimized for E. coli (synth.), in comparison to an empty-vector control (empty vector). D) Genetic organization of two unique clones identified using functional metagenomic screening for the 20β-HSDH activity (PD-CL-Hyd-red-1 and PD-CL-Hyd-red-2), in comparison to their corresponding scaffold assembled from the PD metagenome. E) A bar graph indicating the count of PD fecal metatranscriptomic reads that mapped to the discovered 20β-HSDH gene (red) and its flanking genes (grey). F) Heat maps indicating the prevalence and median abundance (in RPKM) of 20α-HSDH (from C. scindens) and 20β-HSDH (from the PD metagenome) across six gut metagenomic cohorts. G) Jitter plots of 20α-HSDH (from C. scindens) and 20β-HSDH (from the PD metagenome) abundances (in RPKM) in the same cohorts. See also Figure S6, Table S4.
During the construction of PD-CL, we split it into 80 pools of 2–6 X 104 unique clones (UCs) each, and preserved them in corresponding glycerol stocks (see STAR Methods). We tested each of these pools for the ability to convert hydrocortisone into 20β-dihydrocortisone, and identified six that showed significant metabolism. To reach a single functional clone, we performed 10-fold serial dilutions of a selected positive pool of 2 X 104 UCs, by following positive sub-pools at the 2 X 103, 2 X 102, and 2 X 101 UC levels. We then plated the 20-UCs positive sub-pool, and screened individual clones in a 96-well plate format to reach a single positive clone: Hyd-red-1 (Figures 6C,S6).
Sequencing of Hyd-red-1 revealed that it likely originated from a Bifidobacterium sp. Analysis of the genetic context of Hyd-red-1 in a PD scaffold revealed a single putative oxidoreductase in the cloned insert (Figure 6D). We then cloned and heterologously expressed this single gene, and showed that it is indeed a 20β-HSDH (Figure 6C,D). A second round of screening of PD-CL performed in a similar manner revealed a different clone, Hyd-red-2, harboring the same gene and confirming our findings (Figure 6D). These results indicate that combining MDM-Screen with a functional metagenomics approach is a valid strategy to link MDM transformations to metabolizing enzymes from diverse bacteria without the need for bacterial isolation.
We then sought to further probe the biological relevance of the discovered 20β-HSDH. Since it was discovered by heterologous expression of PD-derived DNA in E. coli, we wondered if it is actually expressed under host colonization conditions. To answer this question, we isolated RNA from PD, subjected it to deep metatranscriptomic sequencing, and mapped resulting reads to the PD scaffold harboring the 20β-HSDH gene (see STAR Methods). We observed robust expression of the 20β-HSDH gene in PD-derived metatranscriptomic data, but not of neighboring genes, suggesting that it is expressed individually and not as part of a gene cluster (Figure 6E). To determine whether the identified 20β-HSDH is unique to PD or widespread in the human population, we mapped fecal metagenomic reads from the same six human cohorts mentioned above to the DNA sequence of its gene, and to that of a previously identified 20α-HSDH gene from the gut microbiome isolate Clostridium scindens ATCC 35704 for comparison (which converts hydrocortisone to 20α-dihydrocortisone) (Ridlon et al., 2013). While the C. scindens-derived 20α-HSDH gene was rare (present in only 0.5% of subjects, on average), the PD-derived 20β-HSDH gene was widespread in all cohorts (present in 36% of subjects, on average) and its abundance varied widely between subjects and cohorts (Figures 6F,G, Table S4B,C).
Although Bifidobacterium adolescentis had been known to convert hydrocortisone into 20β-dihydrocortisone for almost 40 years (Winter et al., 1982), no responsible enzymes have been identified from it. Interestingly, while this manuscript was under revision, a different study published the crystal structure of a 20β-HSDH from B. adolescentis L2–32 (which is 98% identical to the 20β-HSDH we identified from the PD microbiome), further corroborating our findings (Doden et al., 2019). As mentioned above (Figure 2D), we also observed the production of 20β-dihydrocortisone from hydrocortisone acetate when incubated with PD. This transformation would require two steps: deacetylation at the C21 hydroxyl, by a yet-unidentified enzyme, and reduction of the ketone at C20 by a 20β-HSDH. Interestingly, when we incubated hydrocortisone acetate with either P. distasonis or C. bolteae, it was deacetylated to yield hydrocortisone but not further reduced, implying that the two metabolic steps at play here can be uncoupled and performed by different members of the microbiome in a sequential manner (Figure S2).
MDM deglycosylation occurs in vivo
Although MDM-Screen is able to uncover novel microbiome-drug interactions, it is unclear whether these results (observed ex vivo) can be recapitulated within the gastrointestinal tract of a live mammalian host (in vivo). To address this question, we sought to monitor one MDM transformation, MDM deglycosylation of FPs, in an in vivo pharmacokinetic study that is performed in a microbiome-dependent manner. Capecitabine was amongst the initial hits that resulted from MDM-Screen, and its modification yields a novel metabolite (deglycocapecitabine) that has not been previously reported in humans or animals, we selected its MDM deglycosylation as a test case for in vivo studies and a proxy for other FPs. We treated two groups of C57BL/6 mice with a cocktail of antibiotics for 14 days to eliminate their native microbiome, then colonized one group with PD while the control group remained non-colonized (see STAR Methods). The two groups were then treated with a single human-equivalent oral dose of capecitabine (755 mg/kg), and blood and feces were collected from each mouse at 0, 20, 40, 60, 120, and 240 minutes post drug administration (Figure 7A,B). We then quantified capecitabine and its metabolites in the serial fecal and blood samples using HPLC-HRMS. In blood samples, capecitabine and its major liver-derived metabolite (5’-deoxy-5-fluorocytidine), but not deglycocapecitabine, were readily detected and showed no significant differences between the two groups (Figure S7). In fecal samples, however, deglycocapecitabine was detected from animals colonized with PD as early as 20 min after dosing, and was almost completely absent in non-colonized ones (Figure 7C). These results indicate that – at least in the case of FP deglycosylation – MDM transformations observed ex vivo by MDM-Screen are recapitulated in vivo (i.e., in mice); establishing the same results in humans awaits further studies. They also suggest that MDM deglycosylation of certain FPs (e.g., doxifluridine, which is prematurely activated into 5-FU upon deglycosylation) should be investigated as a potential contributor to their undesired intestinal toxicity observed in the clinic, although future in vivo studies with different dosing regimens and a variety of FPs need to be performed.
Figure 7. MDM deglycosylation occurs in vivo.

A) Schematic representation of the microbiome-dependent pharmacokinetic experiment performed here. B) Design of the capecitabine pharmacokinetic experiment. Mice are treated with antibiotics for 14 days, then colonized with PD (N=6) or left uncolonized (N=6). On the pharmacokinetic experiment day, a single human-equivalent dose is administered to mice using oral gavage, and serial sampling of blood (B) and feces (F) is performed at 0, 20, 40, 60, 120, and 240 minutes post dosing. C) HPLC-HRMS based quantification of deglycocapecitabine in fecal samples from mice colonized with PD in comparison to uncolonized ones. Metabolite AUC per gram of feces is normalized by the AUC of the internal standard (see STAR Methods). Error bars represent the standard error of the mean. The difference between the two conditions is significant (p < 0.01, determined by testing the intersection null hypothesis with marginal two-tailed t-tests using the Bonferroni correction to control family-wise error rate). See also Figure S7.
DISCUSSION
In the current study, we developed a quantitative experimental workflow for assessing the ability of the human gut microbiome to directly metabolize orally administered drugs, using a combination of microbial community cultivation, small molecule structural analysis, quantitative metabolomics, functional genomics and metagenomics, and mouse colonization assays. Several key differences set our approach apart from previous studies in this area. First, instead of relying on single isolates in performing the initial screen, we use well-characterized, subject-personalized microbial communities. Despite the technical challenges associated with characterizing and maintaining stable microbial communities in batch cultures, three main advantages make this strategy worth pursuing: i) the extent of a biochemical transformation performed by single isolates cultured individually may be different than that performed by the same isolates when cultured as part of a complex community; ii) the net result of several members of the microbiome acting on the same drug can only be identified in mixed communities and not in single-isolate experiments, unless all pairwise and higher order permutations are tested; and iii) our strategy is “personalized”. The results obtained here – including the extent and type of certain modifications – are specific to the strain-level composition of each donor’s microbiome. MDM-Screen thus has a good potential for assessing inter-individual variability in MDM.
Second, most previous studies have focused on certain drug / species combinations that have historically been deemed important (e.g., have been readily observed in humans), or that are manageable experimentally. By default, our microbial-community setup allows us to screen a wider range of combinations, which enabled us to expand in either the drug or subject spaces, and to discover drug-microbiome interactions never reported before. Notably, while this manuscript was under revision, an elegant study reported the screening of 271 orally administered drugs against 76 bacterial isolates of the human gut microbiome (Zimmermann et al., 2019a). Two thirds of the tested drugs were shown to be significantly depleted by at least one of the tested isolates, further emphasizing the great potential of gut microbes to metabolize orally administered small molecule drugs. We view these two approaches as complementary: while screening drugs against optimized, well-characterized, donor-derived microbial communities in MDM-Screen provides a personalized view of drug metabolism that takes into account strain-level and community-wide contributions, screening drugs against a set of well-studied gut isolates streamlines the identification and characterization of specific taxon-drug and gene-drug interactions. Combined together, the results from the two approaches serve as a valuable resource for the scientific community to further study the mechanistic details and pharmacological consequences of newly discovered drug-microbiome interactions.
Despite these advances, our approach is still subject to several limitations. First, 24% of the drugs tested failed to be analyzed using the general analytical chemistry workflow described in MDM-Screen. These drugs fell into one or more of three main categories: unstable after overnight incubation in no-microbiome controls, could not be extracted using ethyl acetate, or could not be analyzed using reverse phase chromatography. An alternative chemical analysis method will need to be developed for these molecules in order to assess their MDM. Second, we focused initially on oral drugs, yet several parenteral drugs and their liver-derived metabolites may be subject to important MDM transformations after biliary secretion. Third, even in our most diverse ex vivo cultures, we fail to support the growth of 100% of the community in the original sample. This limitation can potentially be overcome by utilizing multiple distinct media conditions that each capture unique portions of the community. ENDS provides the theoretical framework for selecting an optimal ensemble of media conditions and we show in STAR Methods how to compute a version of ENDS that estimates the number of detectable strains gained by testing additional media.
We developed our screen in two stages. We began with a single human sample, PD, and incubated its ex vivo culture with 575 drugs. We then transitioned into a HT format with more rigorous methods for media selection, drug and metabolite quantification, and metabolite discovery, and used these methods to screen ex vivo cultures from 20 human donors against 23 drugs. A simultaneous expansion into hundreds of drugs and hundreds of donor samples is necessary to reveal the complete biochemical potential of MDM: It is very likely that the types of MDM transformations observed here are an underestimation of all possible ones. With the HT experimental approach and automatic targeted and untargeted metabolomic analyses developed here, we have laid the groundwork for this expansion. Finally, and most relevant from a clinical stand point, a direct comparison between drug metabolism outcomes in humans and in MDM-Screen for the same cohort of donors is important to establish which MDM transformations can be observed in humans, and to quantify the magnitude by which inter-individual variability in MDM-Screen recapitulates that which occurs in humans. Our quantitative framework – on both the microbial community and metabolomic angles provides the necessary tools to perform such comparison.
STAR★METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mohamed S. Donia donia@princeton.edu)
MATERIALS AVAILABILITY
All unique/stable reagents generated in this study are available from the Lead Contact, but we may require a completed Materials Transfer Agreement if there is potential for commercial application.
DATA AND CODE AVAILABILITY
The sequencing datasets generated during this study are available in Table S2 and at NCBI (BioProject number PRJNA593062). The metabolomics datasets generated during this study are available in Tables S1, S3, and at MassIVE (Accession ID MSV000084641). The code generated during this study is available at GitHub (https://github.com/jaimegelopez/personalized_community_MDM_screen).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subject samples
Fecal samples were collected under Princeton University IRB#11606 at the Princeton University Department of Molecular Biology. Twenty healthy volunteers were recruited via e-mails sent to the departmental listserv as well as flyer advertisements. Volunteers gave informed consent prior to sample collection. Eligibility criteria included age (18 and above) and health status (feeling well at time of sample collection, no diabetes, gastrointestinal, oral, or skin infections, diseases, malignancies, or antibiotic use three months prior to or during sample collection).
Bacterial strains and conditions
The following media were pre-reduced by incubation in the anaerobic chamber for 24 hours before inoculation with the corresponding isolate’s glycerol stock: (PYG for Anaerostipes caccae and Clostridium bolteae; RCM for Prevotella bivia, Parabacteroides distasonis; mGAM for Serratia marcescens, Enterococcus faecalis TYG’11, Anaerococcus prevotii, Escherichia coli TYG’1 and Escherichia coli TYG’2; BHI for Lactobacillus gasseri; LB for Salmonella enterica and Escherichia coli BL21). See below the complete media names. Cultures were grown overnight at 37°C in the same anaerobic chamber (70% N2, 25% CO2, 5% H2).
MegaX DH10β E. coli (used for the metagenomic library construction) and BL21-DE3 E. coli (used for the heterologous expression of the discovered 20β-HSDH genes) were cultured aerobically in LB medium, at 37°C.
E. coli BW25113 wild type and mutants that harbor a replacement of deoA or udp with a kanamycin resistance gene were obtained from the Keio collection (Baba et al., 2006) and cultured in LB medium at 37°C. Clean TP knockout (ΔdeoA), UP knockout (Δudp), and TP/UP double knockout (ΔdeoA/Δudp) strains were obtained as explained below, and also cultured in LB medium at 37°C.
Mice
8–10-weeks old male and female mice (25–30 g) C57BL/6 mice were purchased from Jackson laboratories. All animals were housed and maintained in a certified animal facility and all experiments were conducted according to USA Public Health Service Policy of Humane Care and Use of Laboratory Animals. All protocols were approved by the Institutional Animal Care and Use Committee, protocol 2087-16 (Princeton University). The sex and number of animals are specified for the pharmacokinetic study.
METHOD DETAILS
Fecal sample processing for PD and D1–20
Freshly collected human fecal material from the healthy donors (~ 30 min from collection for PD, transported on ice; <15 min from collection for the rest of the donors, transported without ice) was brought into an anaerobic chamber (70% N2, 25% CO2, 5% H2). One gram of the sample was suspended in 15 ml of sterile phosphate buffer supplemented with 0.1% L-cysteine (PBSc) in a 50 ml sterile falcon tube. The suspension was left standing still for 5 min to let insoluble particles settle. The supernatant was mixed with an equal volume of 40% glycerol in PBSc. Aliquots (1 ml) of this suspension were placed in sterile cryogenic vials and frozen at −80 °C until use (Goodman et al., 2011). Samples were assigned de-identifying numbers (PD, and D1-D20), and stored in the Donia laboratory.
ex vivo culture of PD
A small aliquot (~20 μl) from a PD glycerol stock was used to inoculate 10 ml of 14 different media: Liver Broth (Liver), Brewer Thioglycolate Medium (BT), Bryant and Burkey Medium (BB), Cooked Meat Broth (Meat), Thioglycolate Broth (TB), Luria-Bertani Broth (LB) (obtained from Sigma Aldrich, USA), Brain Heart Infusion (BHI), MRS (MRS), Reinforced Clostridium Medium (RCM), M17 (M17) (obtained from Becton Dickinson, USA), modified Gifu Anaerobic Medium (mGAM) (obtained from HyServe, Germany), Gut Microbiota Medium (GMM (Goodman et al., 2011)), TYG, and a 1:1 mix of each (BestMix), and cultures were incubated at 37 °C in an anaerobic chamber. One ml was harvested from each culture each day for 4 consecutive days, and centrifuged to recover the resulting bacterial pellets.
ex vivo culture of D1–20
30 μl from each donor glycerol stock was used to inoculate 3 ml of 10 different pre-reduced media in replicates: Liver Broth (Liver), Bryant and Burkey Medium (BB), Thioglycolate Broth (TB), Luria-Bertani Broth (LB) (obtained from Sigma Aldrich, USA), Brain Heart Infusion (BHI), MRS (MRS), Reinforced Clostridium Medium (RCM), modified Gifu Anaerobic Medium (mGAM) (obtained from HyServe, Germany), Gut Microbiota Medium (GMM (Goodman et al., 2011)), and a 70:30 mix of BB:GAM (BG), and cultures were incubated at 37°C in an anaerobic chamber. One ml was harvested from each culture after 48 hours and centrifuged to recover the resulting bacterial pellets.
16S rRNA gene amplicon sequencing and analysis
DNA was extracted from all pellets using the Power Soil DNA Isolation kit (Mo Bio Laboratories, USA, now Qiagen), the 16S rRNA gene was amplified (~250 bp, V4 region), and Illumina sequencing libraries were prepared from the amplicons according to a previously published protocol and primers (Caporaso et al., 2012). Libraries were further pooled together at equal molar ratios and sequenced on an Illumina HiSeq 2500 Rapid Flowcell (PD samples) or or MiSeq (D1-D20 samples) as paired-end reads. For PD samples, these reads were 2X175 bp with an average depth of ~100,000 reads, while for D1-D20 samples the reads were 2X150bp with an average depth of ~30,000 reads. Also included were 8 bp Index reads, following the manufacturer’s protocol (Illumina, USA). Raw sequencing reads were filtered by Illumina HiSeq Control Software to generate Pass-Filter reads for further analysis. Different samples were de-multiplexed using the index reads. Amplicon sequencing variants (ASVs) were then inferred from the unmerged paired-end sequences using the DADA2 plugin within QIIME2 version 2018.6 (Bolyen et al., 2018; Callahan et al., 2016). For PD samples, the forward reads were trimmed at 165 bp and the reverse reads were trimmed at 140 bp. For D1–20 samples, the forward reads were trimmed at 150bp and the reverse reads trimmed at 140bp. All other settings within DADA2 were default. Taxonomy was assigned to the resulting ASVs with a naive Bayes classifier trained on the Greengenes database version 13.8 (Bokulich et al., 2018; McDonald et al., 2012). Only the target region of the 16S rRNA gene was used to train the classifier. Downstream analyses were performed in either MATLAB or Python (Hunter, 2007; McKinney, 2010; Oliphant, 2006). See Table S2.
Measurement of biomass for cultured D1–20
30 μl from each donor glycerol stock was cultured in 10 different pre-reduced media as previously described. One ml was harvested from each culture after 48 hours and centrifuged to recover the resulting bacterial pellets. The pellets were weighed in Eppendorf tubes and the mass was subtracted from that of the empty tube prior to pellet collection. See Data Table S2.
ex vivo screening of the drug library for PD
In an anaerobic chamber, a small volume (~100 μl) of a PD glycerol stock was diluted in 1 ml of mGAM, then 20 μl of this solution was used to inoculate 3 ml of mGAM in culture tubes. Cultures were grown for 24 hours at 37 °C in an anaerobic chamber. After 24 hours, 10 μl of each drug (575 total drugs, a subset of the SCREEN-WELL® FDA approved drug library, Enzo Life Sciences, Inc. with each molecule having a concentration of 10 mM in DMSO) or of a DMSO control were added to the growing microbial community. In addition, 10 μl of each drug was also incubated similarly in a no-microbiome, mGAM control. The no-drug control distinguishes microbiome-derived small molecules from ones that result from MDM, and the no-microbiome control distinguishes cases of passive drug degradation or faulty chemical extraction from those of active MDM. PD-DMSO control pellets from several batches of the screen were analyzed using high-throughput 16S rRNA gene sequencing as described above to ensure the maintenance of a similarly diverse microbial composition. Experiments and controls were allowed to incubate under the same conditions for a second 24-hour period. After incubation, cultures were extracted with double volume of ethyl acetate and the organic phase was dried under vacuum using a rotary evaporator (Speed Vac). This extraction method recovers organic molecules from both cells and broths of the cultures, and therefore is not affected by cases of bacterial sequestration of the parent drugs. The dried extracts were suspended in 250 μl MeOH, centrifuged at 15000 rpm for 5 min to remove any particulates, and analyzed using HPLC-MS (Agilent Single Quad, column: Poroshell 120 EC-C18 2.7μm 4.6 × 50mm, flow rate 0.8 ml/min, 0.1% formic acid in water (solvent A), 0.1% formic acid in acetonitrile (solvent B), gradient: 1 min, 0.5% B; 1–20 min, 0.5%−100% B; 20–25 min, 100% B). If drugs were deemed positive for MDM in one or both of the two runs, they were analyzed a third time using both HPLC-MS and HR-HPLC-MS/MS (Agilent QTOF, column: Poroshell 120 EC-C18 2.7μm 2.1×100 mm, flow rate 0.25 ml/min, 0.1% formic acid in water (solvent A), 0.1% formic acid in acetonitrile (solvent B), gradient: 1 min, 0.5% B; 1–20 min, 0.5%−100% B; 25–30 min, 100% B). We tested each drug twice, along with matching no-drug and no-microbiome controls. For final verification and consensus determination, a third trial was performed for drugs that showed a positive MDM on either or both of the first two trials. For selected molecules, cultures were scaled up and metabolites were purified and their structures were elucidated using NMR and/or comparison to an authentic standard obtained commercially using HPLC-HRMS/MS (see below). See Data S1 for the chromatograms of all MDM+ metabolites.
Structural elucidation of selected metabolites
One ml of PD glycerol stock was used to inoculate 100 ml mGAM medium and cultured for 24 hours at 37 °C in an anaerobic chamber. After 24 hours, 2 ml of 10 mM of either capecitabine, hydrocortisone, tolcapone, or misoprostol solutions were added to the PD culture and incubated for another 24 hours. After the second 24 hours, the cultures were extracted with double the volume of ethyl acetate and the organic solvent layer was dried under vacuum in a rotary evaporator. The dried extract was then suspended in MeOH and partitioned by reversed phase flash column chromatography (Mega Bond Elut-C18 10g, Agilent Technology, USA) using the following mobile phase conditions: solvent A, water with 0.01% formic acid; solvent B acetonitrile with 0.01% formic acid, gradient, 100% A to 100% B in 20% increments. Fractions containing the metabolites of interest were identified by HPLC-MS, and reverse phase HPLC was used to purify each metabolite using a fraction collector. The purified metabolites were subjected to NMR and HR-MS/MS analysis. For misoprostol, hydrocortisone, spironolactone, and mycophenolate mofetil, detailed HPLC-HRMS/MS comparisons with authentic standards were also performed. Structural elucidation details of capecitabine, hydrocortisone, tolcapone, spironolactone, misoprostol, and mycophenolate mofetil metabolites are detailed in Data S2.
Molecular networking analysis in PD screen
Raw data files were converted to the .mzXML format using ProteoWizard and uploaded to the Global Natural Products Social Molecular Networking (GNPS) online platform (http://gnps.uscd.edu) (Wang et al., 2016). The data was first filtered, removing MS/MS peaks within +/−17 Da of the precursor m/z. MS/MS spectra were window filtered by choosing only the top 6 peaks in the +/− 50 Da window throughout the spectrum. Before networking, the data was dereplicated using MS-cluster with a parent mass and MS/MS fragment mass tolerance of 0.1 Da and minimum fragment intensity of 1000. Following this consensus spectra were removed if they contained less than 2 spectra. Molecular ion networking was then performed, requiring that two ions have a cosine similarity of 0.5 and share at least 3 peaks in order to be linked. Connections were removed if the ions did not appear in each other’s top 10 most similar ions. Molecular networks were visualized and mined using Cytoscape (Shannon et al., 2003). We call the two compounds (parent drug and metabolite) related if they are in the same connected component of the graph. In the cases where either the metabolite or the parent drug or both were not picked up in the molecular ion networking analysis, we deem the linkage “undetermined”. There are several reasons why the metabolites or drugs are not picked up in the analysis, including the abundance of the ions and the number and abundance of fragment ions. See Data S1B for figures of all molecular ion networks of linked metabolites and parent drugs, and Table S1 for the GNPS web links of all molecular ion networking analyses.
Enrichment analysis for drugs in PD screen
The results of our screen against 438 drugs allow for an aggregate analysis of MDM by the PD microbiome. We hypothesized that members of the microbiome would be more likely to metabolize natural compounds or derivatives thereof due to a higher probability of prior exposure. To test this hypothesis, we first annotated each of the MDM+ or MDM− drugs to one of three categories: naturally occurring molecules (i.e., molecules directly derived from humans, plants, or microbes; e.g., hydrocortisone; N=30), derivatives of naturally occurring molecules (i.e., a semisynthetic derivative or a close structural mimic of a natural product, e.g., hydrocortisone acetate; N=90), and synthetic molecules (e.g., nicardipine; N=318). By comparing the fraction of MDM+ drugs in the first two categories (natural + derivative, 26 out of 120, 21.6%) to that of the third category (synthetic, 31 out of 318, 10%), we revealed a significant difference (p < 0.001, two-tailed proportions z-test, n based on the number of molecules with and without the classification). Intrigued, we decided to examine differences in MDM at lower levels of drug classification. We observed a significantly higher hit rate among steroids (steroids: 16 out of 28, 57.1%; non-steroid: 41 out of 410, 10%, p < 0.001, two-tailed proportions z-test), including hormonal steroids, corticosteroids, bile acids, and derivatives thereof. In fact, the high hit rate of the steroid class is the major contributor to the observed difference between the hit rates of natural/derivative and synthetic groups, which is abolished upon exclusion of the steroids (non-steroid natural/derivative: 10 out of 94, 10.6%; non-steroid synthetic: 31 out of 316, 9.8%) (Table S1). The high hit rate among steroids is in-line with the idea that the microbiome is more likely to metabolize compounds it frequently encounters, as steroids (e.g., bile acids) are normally present in the gut at high concentrations (Northfield and McColl, 1973). The fact that ~10% of fully synthetic molecules tested in our screen are metabolized by PD indicates the presence of a yet-unexplored range of biochemical activities encoded by the gut microbiome that are capable of recognizing foreign substrates.
Other than natural and synthetic classifications, we also looked more closely at functional groups that are enriched or depleted in MDM+ drugs. We generated a list of 94 common functional groups and structural features and searched for them within all of the drugs tested in our screen. To determine whether certain functional groups are enriched in MDM positive drugs, we aggregated the SMARTS of common functional groups and the SMILES of all drugs within our screen. We then searched for these functional groups within the drugs using the obgrep function within Open Babel (O’Boyle et al., 2011). We then tested for enrichment or depletion of these groups within MDM+ drugs using two-sided proportion z-tests, correcting the resulting p-values using the Benjamini-Hochberg method and requiring that the false discovery rate (FDR) corrected p value is less than 0.01. The n for these tests is based on the number of molecules with and without the functional group. Not surprisingly, we observed an enrichment of the following functional groups in MDM+ drugs: nitro groups (FDR corrected p = 3e-16), ketones (FDR corrected p = 3e-8), carbonyl groups with one carbon attachment (FDR corrected p = 8e-4), azo groups (FDR corrected p = 0.001), imines (FDR corrected p = 0.002), and alkenes (FDR corrected p = 0.001). These results are consistent with common reduction and hydrolysis reactions often performed by gut bacteria. On the other hand, we observed a general depletion of arenes and nitrogen atoms in MDM+ drugs (FDR corrected p = 7e-5 and FDR corrected p = 1e-7, respectively). However, when we excluded steroids and repeated the analysis we found that the depletions in arenes and nitrogen atoms were no longer statistically significant (corrected p = 0.7 and corrected p = 0.7, respectively). This indicates that the original statistically significant depletions were the result of steroids being a highly modified class that generally does not contain these functional groups, rather than the functional groups themselves being important predictors for the lack of metabolism (Figure 2D and Table S1). The exclusion of steroids, on the other hand, did not affect the observed enrichments we found for nitro groups, imines, azo groups and ketones (FDR corrected p < 0.01). It is important to note that the results of our analysis of MDM enrichment are based on a single subject’s microbiome, and should be repeated in the future with data from a much larger set of donors.
Gene abundance analysis in metagenomic cohorts
The following datasets were used for the metagenomic analysis of the genes of interest in this study: HMP-1-1 (Human Microbiome Project, 2012), HMP-1-2 (Lloyd-Price et al., 2017), MetaHIT (Nielsen et al., 2014), Chinese (Qin et al., 2012), and Fijicomp (Brito et al., 2016). Raw sequencing data were obtained using the accession numbers of the associated manuscripts, and pre-processed as previously described (Sugimoto et al., 2019). Quality-filtered reads were mapped to each gene using Bowtie2 (--end-to-end, --fast, --score-min L,−0.6,−0.3) (Langmead and Salzberg, 2012), and gene abundance (in Reads per Kbps per Million reads, RPKM) as well as gene breadth coverage (in percent of gene length) were calculated.
We only considered the gene as “present” if the reads cover greater than 50% of the gene, otherwise the quantification RPKM is considered zero. For datasets with multiple samples per subject we aggregated the quantifications. If one or more samples corresponding to a single subject met the coverage threshold, we present the average RPKM of these samples as the RPKM of the subject. If no samples met the threshold, we consider the overall RPKM of the subject to be zero. See Table S4 for tabulated results of this analysis.
ENDS (Expected Number of Detectable Strains)
A metric (“ENDS”) was developed to estimate the number of strains for which MDM reactions can be experimentally detected in ex vivo cultures. ENDS answers the following question: if all of the ASVs in the ex vivo culture performed an MDM reaction at a given rate r (in units of normalized metabolite signal per unit biomass per time), how many ASVs’ reactions will be detected in our screen given the culture composition and measurement instrument? This framework is needed to incorporate the potentially confounding impact of community biomass in the media selection process. For example, if biomass is not considered, a high-diversity low-density community where bacterial load is too low to produce detectable metabolite levels would be favored over a lower diversity community with high enough bacterial load to produce detectable metabolite levels. Formally, we are computing the expected value of the number of the reactions detected:
Where E[Ns] is the expected number of detectable microbes, and B(xi) is the probability of microbe i’s reaction being detected with an absolute population of size xi. How can we construct B(xi)? In statistical terms, B(xi) is equivalent to the power (the probability of deciding there is a reaction when the reaction is actually present) of the hypothesis testing method used to analyze the data. In this case, we are using a one-sided unequal variances t-test with cutoff α. Now that we have a framework for calculating B(xi), we must relate a given xi to a null and alternative distribution of measurements.
We assume that the metabolite measurements are composed of two types of signal: background noise, X and compound signal, Y. Both signals are assumed to be normally distributed (we show in Methods S1F that an empirically estimated power function provides similar results). The background noise is the signal present when no actual metabolite is present. The compound signal is the portion of the signal due to measurement of an actual metabolite. The measurements in the control condition are modeled by X while the measurements in the experimental conditions are modeled as
We will now relate the population abundance xi to the mean of Y, μ1. In real terms, μ1 is the average level of a metabolite produced by xi. We assume the production metabolite is governed by the dynamics where [M] is the concentration of the metabolite and r is the rate of metabolite production per cell. If we assume the drug is added at stationary phase such that for all t after drug addition, the total amount of metabolite produced is where τ is the incubation time. The r can vary widely, and to account for this it can be set using the rate of a known MDM reaction (see Methods S1D).
We must now estimate the distribution of X. We estimate this by computing the mean and standard deviation of spurious peaks detected when samples not containing the compound being measured is quantified.
Now we must define the standard deviation of Y, f(μ2). This is clearly dependent on the instrument being used. By plotting the standard deviations of triplicate measurements from our machine against their mean, we can estimate f(μ2). To ensure we are capturing only measurement signal, we based our model only on measurements that largely composed of measurement signal (more than three standard deviations above the mean of the null distribution). We have found that a power law fits the data well. With the distributions of X and Z, we then estimate the B(xi) using existing methods (Harrison and Brady, 2004).
What if we want to create an ensemble of multiple culture conditions to detect even more microbial reactions? To do this we can define the expected number of new detectable microbes gained if we include another media . For each ASV in all media, we take the product of the probability that the reaction is not detected in the existing media and the probability that the reaction is detected in the new media.
Where m is the number of media in the existing ensemble, and is the abundance of ASV i in existing media j.
In our actual computation of ENDS, we exclude samples with less than 10,000 reads and take the optimal medium as the one with the highest ENDS averaged across all twenty donors.
High-throughput screen with D1–20
A new medium, BG, was formulated (see Methods S1-B). BG was made as follows: BB powder and mGAM powder were reconstituted in water and autoclaved as per manufacturer instructions (Sigma and Hyserv, respectively). Liquid BB medium was mixed with liquid mGAM in a 70:30 ratio, respectively. For each donor (D1-D20), 500 μl of a donor’s glycerol stock was used to inoculate 50 ml of pre-reduced BG medium and incubated for 24 hours in an anaerobic chamber at 37°C. The culture was transferred to a sterile Nunc 96-well plate (Fisher Scientific) with each well containing 400 μl of culture. 13.2 μl of each drug stock solution (1mM in DMSO), or 13.2 μl of DMSO as a vehicle-only control, were pipetted and resuspended in 4 adjacent wells of the 96-well plate (quadruplicates) (see Table S3). Selected drugs included MDM+ drugs from our original screen (N=15), MDM- drugs randomly (N=4) or rationally (N=2) selected, and drugs reported in the literature to be MDM+ but were deemed negative in our screen (N=2). The plate was incubated for 24 hours anaerobically at 37°C. For chemical extraction, 10 μl of the internal standard (voriconazole, 1mM) was pipetted into the wells. For chemical extraction, 800 μl of ethyl acetate was pipetted into the wells and resuspended three times. 400 μl was pipetted into an Agilent 96-well plate and dried under Nitrogen with a 96-well blow-down evaporator (Fisher Analytical). For HPLC-HRMS analysis, the plate was resuspended in 300 μl methanol and left for 10 min at room temperature prior to centrifugation at 3900 RPM for 10min at 4°C. 60 μl were carefully decanted from the top into a new plate with 60 μl methanol and run on an Agilent 6545 LC/QTOF machine (0.5 μl injection, only three of the four replicates for each drug were run on the machine). The remaining 240 μl were dried and stored at −20°C for future runs. An abiotic BG-drug plate and a heat-killed-microbiome-drug plate were prepared and analyzed using the same method, serving as controls to estimate non-enzymatic drug degradation or metabolite production. To prepare the heat-killed-microbiome plate, 500 μl of D20 glycerol stock was used to inoculate 50 ml of pre-reduced BG media and incubated for 24 hours in an anaerobic chamber at 37°C. The culture was heat-killed at 100°C for 30 minutes while keeping the flask containing it sealed (and therefore maintaining the anaerobic conditions). Drug incubation, chemical extraction, and HPLC-HRMS analysis for the control plates were performed as previously described for the donor-drug plates.
Targeted quantitative metabolomics analysis
HPLC-HR-MS analysis was performed on an Agilent 6545 LC/QTOF machine. The Autosampler compartment was kept at 7°C, and the column was kept at 25°C. Reverse phase chromatography was performed using an Agilent Eclipse Plus C18 RRHD column 1.8 μM (2.1 × 50 mm) column (Agilent, USA) with the gradient 95%A, 5%B to 5%A, 95%B in 12 minutes, then 95%B for 2 minutes, followed by initial conditions (95%A, 5%B) for 3 min to re-equilibrate the column (A = 0.1% formic acid in water and B = 0.1% formic acid in Acetonitrile). The flow rate was 0.4 ml/min. The samples in this study were run in one of two modes: a high resolution 4GHz mode and a high dynamic range 2GHz mode. MS acquisition parameters for the 4GHz mode were set as follows: positive ion polarity, 0.5min delay before MS measurement, 325°C gas temperature, 10 L/min drying gas flow rate, 20 psi nebulizer pressure, 325°C sheath gas temperature, 12 L/min sheath gas flow rate, 4000 V capillary voltage, 500 V nozzle voltage, 135 V fragmentor voltage, 45 V skimmer voltage, MS and MS/MS mass range of 100 – 1700 m/z, acquisition of 5 MS1 spectra/s, acquisition of 3 MS2 spectra/s, 20 eV collision energy, a maximum of 2 precursors per cycle, and a precursor selection threshold of 200 counts absolute or 0.01% relative. The system was run in auto MS/MS mode. For the 2GHz mode, the parameters were the same as the 4GHz mode with the following changes: acquisition of 8 MS1 spectra/s, acquisition of 6 MS2 spectra/s, maximum of 5 precursors per cycle, precursor selection threshold of 2000 counts.
To verify that the concentration of internal standard (voriconazole) used in the screen was below the saturation limit of the machine, we constructed a standard curve of voriconazole. 12μL of 1mM voriconazole was added to 228μL of methanol and serial dilutions were performed by a factor of three to cover the concentration ranges of 40 μM to 0.165 μM. These samples were run on the 2GHz setting described above to match the setting used for drug quantification in the screen.
Drugs and their detected metabolites were quantified in the MS1 of all samples using MassHunter Quantitative Analysis with the Agile2 integrator. The metabolites quantified here were either ones that we previously discovered during the PD screen, ones that were previously reported in the literature, or novel metabolites from the multi-donor screen identified using untargeted metabolomics and verified by molecular networking (See below, Table S3, Figure S3). For quantification of dihydrodigoxin, we required a highly sensitive integration method to distinguish between dihydrodigoxin and the isotopes of digoxin, since parent and metabolite eluted at similar retention times. To do this, we performed integrations within MassHunter Qualitative, specifying a mass range of 805.43 – 805.44 m/z for dihydrodigoxin. We verified this method could differentiate the two compounds using authentic standards of dihydrodigoxin and digoxin, showing that it could accurately quantify dihydrodigoxin and that it did not detect dihydrodigoxin when only digoxin was present.
Following quantification, all further data processing was performed in MATLAB. For each plate, we remove any samples whose internal standard AUC was greater than three interquartile ranges above the third quartile or below the first quartile. In order to correct for differences in extraction efficiency, all peak areas in a given sample were divided by the corresponding internal standard area. This ratio was then used for hypothesis testing and all other downstream analyses. For drug depletion, unadjusted p-values were obtained by one-sided Welch’s t-tests testing whether drug levels are significantly lower in the donor-drug conditions than in controls; p-values were computed for tests against controls where the drug was incubated with BG medium (medium-drug) and incubated with the heat-killed-microbiome (HKM-drug) controls. For all metabolite quantification statistical tests, n is the number of replicates passing quality control (maximum 3). For metabolite production, unadjusted p-values were obtained for one-sided Welch’s t-tests testing whether metabolite levels are significantly higher in donor-drug conditions than in control conditions; p-values were computed for tests against medium-drug, HKM-drug, as well as donor-DMSO (where the cultures are incubated with only the vehicle, DMSO) controls. Correction for multiple hypotheses was performed using the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995). Metabolite production and drug depletion p-values were adjusted separately. For depletion to be considered significant, we required FDR corrected p value < 0.01 for both the medium-drug and HKM-drug adjusted p-values and also depletion was required to be greater than 50% relative to both controls. For metabolite production to be considered significant, we required that FDR corrected p value < 0.01 for the medium-drug, HKM-drug and donor-DMSO p-values.
We then looked for correlation between the results of the targeted metabolomics and the compositions of the BG communities, We restricted the drugs and metabolites tested by requiring that they must have at least one associated compound (parent drug or metabolite) with an inter-individual variability entropy > 0.5, and that there exists at least one sample with more than 20% of the drug remaining relative to medium-drug controls. We tested only taxonomic elements present in at least three samples with a biomass of at least 1 mg/L in at least one sample, and corrected the resulting p- values for multiple hypotheses at each taxonomic level using the Benjamini-Hochberg method. The n for these tests is based on the number of observations used to compute the correlations. Spearman correlation was computed for this analysis. The mean BG community composition for each donor was used for the correlation analysis. We tested taxonomic elements at the ASV, species, genus, and family levels.
Untargeted metabolomics analysis
In order to extract all features in a given sample, we used the batch recursive feature extraction method for small molecules and peptides within Profinder 8 (Agilent). We changed the following settings in the method from default: compound ion count threshold set to two ions, alignment retention time tolerance set to 0.2 min, minimum MFE score set to 90, minimum file prevalence set to 2, expected retention time set to ±1 min, retention time contribution to matching score set to 90, expected MS1 mass variation set to 10 ppm, expected retention time tolerance set to 0.2 min, absolute height threshold for EIC integration set to 2500 counts, and final absolute height threshold set to 5000. We then analyzed the resulting feature abundances in MATLAB. We remove any sample whose internal standard AUC is less than 106. We then perform hypothesis testing using similar statistical methods as for metabolite production in the targeted metabolomics, except that we utilize the multiple hypothesis correction of Storey (Storey, 2002) in place of the Benjamini-Hochberg method and require a fold-change cut-off of two relative to all controls. We combine features if their retention times and estimated molecular weights differ by less than 0.2 min and 0.01 Da, respectively. We then remove all ions already quantified in our screen and all features that are statistically significant for more than one drug.
For the remaining statistically significant features, we use molecular ion networking to verify the metabolite’s relationship to the parent drug based on their HR-MS/MS fragmentation pattern. In order to gather data with a large enough number of MS2 spectra per parent ion we reran samples of interest on our QTOF HPLC-HRMS/MS instrument (Agilent) using the same column and conditions, and the 4GHz settings listed above (instead of the 2GHz one used in the multi-donor screen). In order to minimize the number of samples run a second time, we identified the minimal set of 81 samples that would allow us to detect and perform molecular ion networking on all novel metabolites found in the original 1380 donor-drug samples. For molecular ion networking, we used the Global Natural Product Social Molecular Networking (GNPS) server using the same settings used for the PD molecular networking. In order to determine whether a metabolite is linked to its parent by the molecular ion networking, we first identify whether the drug and metabolite are present in the network. For this, we require that the mass and retention time found in the molecular ion networking differ by less than 0.2 min and 0.02 Da, respectively, from the properties reported by the initial donor-drug stage of the pipeline. We call the two compounds related if they are in the same connected component of the graph. In the cases where either the metabolite or the parent drug or both were not picked up in the molecular ion networking analysis, we deem the linkage “undetermined”. There are several reasons why the metabolites or drugs are not picked up in the analysis, including the abundance of the ions and the number and abundance of fragment ions.
Isolate screen for capecitabine
Three-ml overnight seed cultures of the following bacteria in their respective media were obtained as described above: Anaerostipes caccae, Clostridium bolteae, Prevotella bivia, Parabacteroides distasonis, Serratia marcescens, Enterococcus faecalis TYG’11, Anaerococcus prevotii, Escherichia coli TYG’1, Escherichia coli TYG’2, Lactobacillus gasseri, Salmonella enterica, and Escherichia coli BL21. This panel was selected from three of the most abundant Phyla that normally inhabit the gut microbiome (Firmicutes, Bacteroidetes, and Proteobacteria), spans 10 bacterial genera (12 strains in total), and includes three strains isolated originally from PD using standard techniques (Enterococcus faecalis TYG11, Escherichia coli TYG1, and Escherichia coli TYG2). 20 μl of these seed cultures were inoculated into 3 ml of the same selected pre-reduced medium, and incubated at 37°C under the same anaerobic conditions for an additional 24 hours. After 24 hours, 10 μl of the 10 mM drug solution in DMSO, or of a DMSO control were added to the growing microbial culture and incubated for another 24 hours. In addition, 10 μl of each drug were incubated for 24 hours under the same conditions in a no-bacterium, medium-only control. After incubation, cultures were extracted with ethyl acetate and the organic phase was dried under vacuum in a rotary evaporator. Extracts were suspended in 250 μl of MeOH and analyzed using HPLC-MS as described above in the PD ex vivo screen. See Figure S4.
Metagenomic library construction
Metagenomic DNA was extracted from approximately 0.25 g of PD stool (stored at −80°C in RNAlater (Thermo Fisher Scientific, Massachusetts, USA)) using the PowerSoil DNA Isolation kit (MO BIO Laboratories California, now Qiagen, USA) according to manufacturer’s instructions. DNA was quantified using a NanoDrop 2000 (Thermo Fisher Scientific, Massachusetts, USA).
The vector pGFPuv (Clonetch Laboratories) was used as the parent vector for the metagenomic library. The pGFPuv plasmid was linearized by using PCR and Phusion High-Fidelity DNA Polymerase (New England Biolabs, Massachusetts, USA) and the 2.5kb product was excised and extracted from the gel after gel electrophoresis and using the QIAquick Gel Extraction kit according to manufacturer’s instructions. PCR primers for linearization were: forward, pGFP-IF-F = TAATGAATTCCAACTGA GCGCCGG and reverse, pGFP-IF-R = CATAGCTGTTTCCTGT GTGAAATTG. DNA was quantified using a NanoDrop 2000 and incubated with Dpn1 (New England BioLabs, Massachusetts, USA) at 37°C for one hour followed by incubation at 80°C for 20 minutes to heat inactivate the Dpn1 enzyme. To prevent re-ligation of the vector, Antarctic Phosphatase (New England Biolabs, Massachusetts, USA) was added to the Dpn1 treated mixture and incubated at 37°C for 15 min and 70°C for five minutes to heat inactivate the phosphatase. The product was then purified using the QIAquick PCR Purification kit (Qiagen, Maryland, USA) according to manufacturer’s instructions and quantified with a NanoDrop 2000.
Extracted DNA samples were brought to 150 μl in Milli-Q water. DNA was sheared via a g-TUBE (Covaris, Massachusetts, USA) according to manufacturer’s instructions using an accuSpin Micro 17R Microcentrifuge (Thermo Fisher Scientific, Massachusetts, USA). DNA fragment size was validated using the Agilent 2100 Bioanalyzer (Agilent Technologies, California, USA). Sheared DNA was subjected to gel electrophoresis, and the 2–4 Kbp product was excised from the gel. Gel extraction was performed using Zymoclean Large Fragment DNA Recovery Kit (Zymo Research, California, USA) according to the manufacturer’s instructions.
Sheared DNA was end-repaired using the End-It DNA End-Repair Kit (Epicentre (Illumina), California, USA) according to manufacturer’s instructions. The end-repaired DNA was then purified by isopropanol precipitation. Blunt-end ligation of DNA with linearised pGFPuv was performed with T4 DNA ligase (New England Biolabs, Massachusetts, USA) according to manufacturer’s instructions at 16°C overnight using 100 ng vector and 360 ng insert to achieve a 1:3 molar ratio. Ligated plasmids were purified by precipitation with Pellet Paint NF Co-Precipitant (EMD Millipore, Massachusetts, USA) and brought to 4 μl in Milli-Q water. 2 μl ligated plasmid were transformed into MegaX DH10b Electrocompetent Cells (Thermo Fisher Scientific, Massachusetts, USA) according to manufacturer’s instructions, and recovered in 1 ml of LB medium.
The 2 × 1 ml transformation products for each ligation were combined and brought up to 20.2 ml in LB/ampicillin and divided into 20 × 1 ml pools. Each 1 ml pool was cultured in a shaking incubator at 30°C overnight and mixed with 1 ml 50% glycerol the following day, gently vortexed, and stored at −80°C for future screening. Serial dilutions were made from the remaining 200 μl of transformation product and spread on LB/ampicillin plates which were incubated overnight at 37°C, to calculate the library titer of unique clones the following day. Random clones were picked, checked for the presence of insert, and used to estimate insert size by PCR (most clones harbored inserts in the 2–4 Kbps range). This procedure was repeated 4 times, with an average yield of 2–6 X 104 unique clones per pool (80 total pools) and a total of ~3 X 106 unique clones.
Functional screening of the metagenomic library
2 μl from each of the initial 80 pools containing 2–6 X 104 unique clones were added to 3 ml of LB-carbenicillin (LB-carb, 100 μg/ml) in glass culture tubes and grown at 37°C for 1 hour. After 1 hour, 10 μl of 10 mM hydrocortisone (in DMSO) was added to each culture and incubated at 37°C for 20 hours.
After a total of 20 hours of growth post hydrocortisone addition, cultures were chemically extracted as follows: 5 μl of 10 mM voriconazole was added to the cultures, followed by the addition of 7 ml of ethyl acetate solvent using a glass pipette. The resulting solution was vortexed on a medium-high setting twice, allowing for the mixture to separate between vortexing steps. After the last vortexing step, the mixture was left undisturbed for 5 minutes. 5 ml of the top organic layer of solvent was transferred to a 4 ml glass tube using a chemically resistant 1 ml tip and pipette. The samples were then dried using a Labconco CentriVap Concentrator for 1.5 hours. Dried samples are then resuspended in 250 μl of methanol and sonicated for 5 minutes. Samples are then transferred to a 1.5 ml Eppendorf tube and centrifuged. 180 μl of the clear solution was then deposited in glass LCMS vial and analyzed using HPLC-HRMS on an Agilent 6545 LC/QTOF instrument using the same column and gradient as described for the D1-D20 plates, and the 4GHz MS settings. An identical quantitative workflow was employed to identify the pools with the highest signal for the hydrocortisone metabolite/internal standard ratio.
Selected pools were then plated for colony forming units (CFU) counting. CFU counting was done as follows: 5 μl of each glycerol stock was taken and diluted into 95 μl of LB-carb by pipetting up and down. 5 μl was taken out from the first tube and moved to the second, with pipetting to ensure proper mixing. Serial dilutions were done four more times for a total of five dilutions. 50 μl of each serial dilution was plated onto LB-carb agar plates and spread evenly across the entire surface. Plates were incubated at 37°C C overnight and checked for colonies the next day. The number of colonies on a plate were counted and used to calculate CFUs for the original positive sample glycerol stock.
The positive glycerol stock sample was then diluted to obtain sub-pools of 2,000 clones (based on the CFU counts) per sub-pool with a total of 20 sub-pools to be screened for each positive sample. These 20 sub-pools were then tested for hydrocortisone metabolism and glycerol stocked following the same protocol outlined above for the 20,000 level sub-pools, except that hydrocortisone was added after 4 hours of culture instead of 1 hour and that a glycerol stock was made from each sub-pool 12 hours after the culture was initiated. Glycerol stock from the top 2,000-clone sub-pool that produced the hydrocortisone metabolite was then subjected to CFU counting, further sub-pooled and tested at the 200-clone level. The top 200-clone sub-pool was treated the same way and tested at the 20-clone level.
Glycerol stock from the sub-pool positive for hydrocortisone metabolite at the 20-clone level was then serially diluted and plated onto five LB-carb plates and incubated overnight at 37°C. The next day, the plates are retrieved and single colonies were picked and grown in 4 X 96 deep-well plates with 500 uL LB-carb per well. The deep-well plates were then grown at 37°C at 220 RPM shaking overnight. The next day, 100 uL of culture were saved from each well into another 96-well plate for glycerol stock using a multichannel pipette.
10 μl from each well of each row were pooled before inoculating a new glass culture tube containing 3 ml LB-carb. The resulting 32 culture tubes corresponding to the 32 pooled rows were grown at 37°C for 4 hours before the addition of 10 μl of 10 mM hydrocortisone, after which samples were grown at 37°C for 20 hours.
After 20 hours, 5 μl of 10 mM voriconazole and chemical extraction with ethyl acetate were performed as described previously and analyzed using HPLC-HRMS. The positive hit indicated which row the hydrocortisone metabolite originated from. 5 μl of each well in the positive row were used to inoculate a new glass culture tube with 3 ml LB-carb, which was then grown for 4 hours at 37°C, after which hydrocortisone was added and the metabolism screen proceeded as previously described. A single well from the positive row was then identified as a positive hit. See Figure S6.
The glycerol stock of the positive well was then used to inoculate a 5 ml LB-carb culture that was grown overnight at 37°C and shaken at 220 RPM. The next day, the plasmid containing metagenomic DNA was isolated from the culture using a QIAprep ® Spin Miniprep Kit (Qiagen, USA) according to manufacturer’s instructions and subjected to DNA sequencing (Sanger) to identify the metagenomic insert on the plasmid. End-sequences were compared to the PD metagenome using BLASTn.
Heterologous expression of PD-derived 20-HSDH
The Bifidobacterium sp. reductase identified from the functional metagenomic screening of PD metagenomic DNA was codon optimized for E. coli and ordered as a gBlock from IDT (USA). Primers annealing to the gBlock insert were designed to contain overhang restriction enzyme cut sites that would recognize restriction digested pet28a vector. The forward primer contains the cut site for NdeI, and the reverse primer contains the cut site for NotI. Primer sequences are as follows (5’ to 3’):
Forward: CTGGTGCCGCGCGGCAGCCATATGGCAGATGAATCATCGAAGATTCC Reverse: GGTGGTGCTCGAGTGCGGCCTTAGAAAACTGAATACCCACCGTCC PCR product was gel extracted and cloned into a double-digested pet28a vector using the InFusion cloning kit (Takara). InFusion product was transformed into chemically competent BL21-DE3 E. coli cells and grown in LB-carb media. Plasmid was recovered using a QIAprep ® Spin Miniprep Kit (Qiagen, USA) and sequenced (Sanger) using the T7 promoter to confirm presence of the codon optimized gBlock Bifidobacterium sp. reductase.
To clone the native-sequence of the Bifidobacterium sp. reductase identified from the functional metagenomic screening of PD metagenomic DNA, primers designed with restriction cut sites (as described above) were used to clone it from the metagenomic clone into a double-digested pet28a vector. Primer sequences are as follows (5’ to 3’):
Forward: CTGGTGCCGCGCGGCAGCCATATGGCAGACGAATCATCGAAGATTC
Reverse: GGTGGTGCTCGAGTGCGGCCCAAATGGGGTACGGTGATTAGAAGAC
Plasmid was recovered using a QIAprep ® Spin Miniprep Kit (Qiagen, USA) and sequenced (Sanger) using the T7 promoter to confirm presence the correct insert.
Cultures of BL21-DE3 E. coli harboring the codon optimized and native reductase were grown from glycerol stock in LB-carb overnight at 37°C, 220 RPM. The next day, cultures were back diluted to an OD600 of 0.05, grown at 37°C, 220RPM until OD600 was 0.4 and induced with a final concentration of 1mM IPTG. After four hours of growth (from the time of back dilution), 10 uL of 10 mM hydrocortisone were added. All cultures were then grown for 20 hours at 37°C, 220 RPM, chemical extraction and HPLC-HRMS analysis were performed as described above.
Metagenomic and metatranscriptomic analysis
PD metagenomic sequencing: metagenomic DNA of PD, prepared as described above, was sheared to a mean size of ~500 bps using a Covaris E220 sonicator (Covaris, USA). An Illumina sequencing library was prepared from the sheared DNA using the automated Apollo 324™ NGS Library Prep System and the PrepX DNA library kit (WaferGen, USA) according to the manufacturer’s protocol. This step included DNA end repairing, A-tailing, adapter ligation, and limited PCR amplification. After examination on Bioanalyzer (Agilent, CA) DNA HS chips for size distribution, and quantification by Qubit fluorometer (Invitrogen, CA), the library was sequenced on Illumina HiSeq 2500 Rapid Flowcell as paired-end reads, along with 8 bps Index reads, following the manufacturer’s protocol (Illumina, USA). Raw sequencing reads were filtered by Illumina HiSeq Control Software to generate Pass-Filter reads for further analysis.
PD metatranscriptomic sequencing: Total nucleic acid (DNA and RNA) was prepared from the RNAlater-preserved PD stool sample using the AllPrep PowerViral DNA/RNA Kit (Qiagen, USA). DNA was digested and removed using Turbo DNase (ThermoFisher Scientific, USA) following the manufacturer’s instructions, and remaining DNA-free RNA concentration and quality were measured using a Nano Drop 2000 and a Bioanalyzer (Agilent, USA). This RNA was further subjected to ribosomal RNA depletion using the Ribo-Zero Gold rRNA Removal Kit (Epidemiology) (Illumina, USA), and a strand-specific Illumina RNA-seq library was prepared from it as described for the PD metagenomic DNA sample.
PD-CL-100 metagenomic library sequencing: plasmids were isolated using the QIAprep Spin Miniprep Kit (Qiagen, USA) from two sub-pools containing a total 100,000 Unique Clones. An Illumina sequencing library was prepared and sequenced from the resulting DNA as described above, except that it was sequenced on a NovaSeq 6000 instead of the HiSeq 2500.
PD metagenomic sequencing yielded 35,527,955 reads (2 X 175 bps), PD metatranscriptomic sequencing yielded 30,796,174 reads (2 X 150 bps), and PD-CL-100 yielded 69,173,413 reads (2 X151 bps).
Raw Illumina reads were filtered using PRINSEQ, according to the following parameters: minimum average quality score of 30, maximum percentage of undetermined reads of 2%. Trimming on each end was implemented until a minimum average quality score of 30 is reached. Trimmed reads that are shorter than ½ the original read length were discarded (Schmieder and Edwards, 2011). SPAdes was used to assemble the resulting pairs and singletons of filtered reads, using default parameters (Bankevich et al., 2012).
For PD-CL-100, Bowtie2 was used to identify and remove reads mapping to the pGFP-UV plasmid backbone using the following settings: --end-to-end --sensitive. Reads that do not map to the backbone were then aligned to the SPAdes scaffolds produced from the PD metagenomic dataset, also using Bowtie2 (--very-sensitive-local) (Langmead and Salzberg, 2012). Reads from the PD metagenomic dataset itself were also aligned to their corresponding SPAdes-produced scaffolds using the same settings. RPKM values and coverage breadths for each PD scaffold equal or longer than 2 Kbps were calculated on the basis of the Bowtie2 alignment results for each the two read datasets (PD metagenome and PD-CL-100). A PD scaffold was considered “present” in PD-CL-100 if metagenomic reads from PD-CL-100 covered at least 50% of the scaffold’s length, otherwise the quantification RPKM is considered zero. To infer the taxonomy of the PD scaffolds, we ran kraken-1.1.1 with standard settings and the “MiniKraken DB_8GB” pre-built database constructed from complete bacterial, archaeal, and viral genomes in RefSeq (as of Oct. 18, 2017) (Wood and Salzberg, 2014). See Data Table S4.
Metratranscriptomic analysis: we first used BLASTn to map a database of quality filtered PD metatranscriptomic reads to a query of the PD scaffold harboring the 20β-HSDH gene, while specifying an e-value cutoff of 1e-30. Next, we matched these BLASTn-identified reads to either the 20β-HSDH gene or neighboring genes that we annotated in ~5 Kbps windows upstream and downstream of it. For this step, we used the Geneious assembler in Geneious (Kearse et al., 2012) with the following parameters: minimum overlap: 50 bps, minimum percent identity at overlap: 90%, and maximum percentage of mismatch per read: 20%. Matched reads per gene were counted and used to construct the bar graph in Figure 6E.
TP and UP gene deletions in E. coli BW25113
E. coli BW25113 mutants that harbor a replacement of deoA or udp with a kanamycin resistance gene were obtained from the Keio collection (Baba et al., 2006). Since the kanamycin resistance gene is flanked by FLP recognition target sites, we decided to excise it and obtain in-frame deletion mutants. Plasmid pCP20, encoding the FLP recombinase, was transformed to each of the mutants by electroporation, and transformants were selected on Ampicillin at 30 °C for 16 hours. 10 transformants from each mutant were then picked in 10 μl LB medium with no selection, and incubated at 42 °C for 8 hours to cure them from the temperature-sensitive pCP20 plasmid. Each growing colony was then streaked on three plates (LB-ampicillin, LB-kanamycin, and LB with no selection). Mutants that could only grow on LB, but not on LB-ampicillin (confirming the loss of the pCP20 plasmid), nor on LB-kanamycin (confirming the excision of the kanamycin resistance gene) were confirmed to harbor the correct deletion using PCR and DNA sequencing. Primers deoA-Check-F: 5’-CGCATCCGGCAAAAGCCGCCTCATACTCTTTTCCTCGGGAGGTTACCTTG-3’, deoA-Check-R: 5’-CAAATTTAAATGATCAGATCAGTATACCGTTATTCGCTGATACGGCGATA-3’, udp-Check-F: 5’-CGCGTCGGCCTTCAGACAGGAGAAGAGAATTACAGCAGACGACGCGCCGC-3’, and udp-Check-R: 5’-TGTCTTTTTGCTTCTTCTGACTAAACCGATTCACAGAGGAGTTGTATATG-3’ were used in PCR experiments to confirm the deletion of the deoA or upd genes and the kanamycin resistance gene replacing them (Baba et al., 2006). To construct the ΔdeoA/Δudp double knockout, the in-frame Δudp knockout obtained above was used as a starting point. Plasmid pKD46 expressing the Lambda Red recombinase was transformed to it using electroporation (Datsenko and Wanner, 2000) and transformants were selected on LB-Ampicillin at 30 °C for 16 hours. One Ampicillin-resistant transformant was then cultured at 30 °C in 50 ml of LB-Ampicillin, with an added 50 μl of 1 M L-arabinose to induce the expression of the recombinase. At an optical density of 0.4–0.6, electrocompetent cells were prepared from the growing culture by serial washes in ice cold 10% glycerol, and ~300 ng of a linear PCR product were transformed to it by electroporation. This PCR product was prepared by using the deoA-Check-F and deoA-Check-R primers on a template DNA prepared from the deoA mutant of the Keio library, in which a kanamycin resistance gene replaces deoA. After electroporation, transformants were selected on LB-kanamycin at 37 °C to induce the loss of the temperature sensitive pKD46 plasmid, cultured in LB-kanamycin overnight at 37 °C, and checked by PCR to confirm the correct recombination position. Finally, the kanamycin resistance gene was excised from the deoA locus by the FLP recombinase using the same strategy explained above, resulting in the final ΔdeoA/Δudp mutant
MDM-Screen of capecitabine using E. coli mutants
Wild type E. coli BW25113, and corresponding TP knockout (ΔdeoA), UP knockout (Δudp), and TP/UP double knockout (ΔdeoA/Δudp) strains were cultured overnight in LB medium (aerobically, shaking at 37 °C, 50 ml each). Triplicates of 3 ml for each strain were incubated with 10 μl of 10 mM capecitabine (in DMSO) for an additional 24 hours in an anaerobic chamber along with bacteria-only and media-only controls. Cultures were then extracted and analyzed as previously described, except for the addition of 20 μl of 0.25 mg/ml of an internal standard (voriconazole) prior to the extraction.
MDM-Screen of other FPs using E. coli mutants
Wild type E. coli BW25113, and corresponding TP knockout (ΔdeoA), UP knockout (Δudp), and TP/UP double knockout (ΔdeoA/Δudp) strains were cultured overnight in LB medium (aerobically, shaking at 37 °C, 50 ml each). Aliquots (100 μl) of each strain were used to inoculate 3 ml of M9 medium, which were grown again overnight (aerobically, shaking at 37 °C). 10 μl of 10 mM doxifluridine (in DMSO) or trifluridine (in methanol) were incubated with each culture for an additional 24 hours in an anaerobic chamber, along with bacteria-only and medium-only controls. Cultures were spun down and collected supernatants were lyophilized. The dried residues were then resuspended in 500 μl methanol and analyzed by HPLC-MS (Agilent Single Quad; column: Poroshell 120 EC-C18 2.7μm 4.6 × 100mm; flow rate: 0.6 ml/min; solvent A: 0.1% formic acid in water: solvent B: 0.1% formic acid in acetonitrile) and the following gradient: 1 min, 0.5% B; 1–20 min, 0.5%−35% B; 25–30 min, 35%−100% B; 30–35 min, 100% B. The structures of all resulting metabolites were confirmed by comparison to authentic standards. See Figure S4 and S5.
Microbiome-dependent pharmacokinetic experiment
Twelve C57BL/6 mice were treated with a commonly used cocktail of antibiotics (1 g/l of ampicillin, neomycin, metronidazole and 0.5 g/l vancomycin) in drinking water for 14 days (Planer et al., 2016). The antibiotic solution was supplemented with 5 g/l aspartame to make it more palatable (Karmarkar and Rock, 2013). During these two weeks, the gut microbiome composition was monitored by collecting feces from each mouse and performing molecular and microbiological analyses to make sure the microbiome is being cleared by the antibiotic treatment. On day 15, no antibiotics are administered for 24 hours (a washout period). On day 16, mice were separated into the two groups, 6 per group (3 males and 3 females). In group 1, mice remained non-colonized. In group 2, mice were administered 200 μl of freshly thawed PD glycerol stock using oral gavage. On day 17, the oral gavage was repeated the same way to ensure the colonization of the administered bacteria (fecal samples were collected on days 16 and 17 and cultured anaerobically to ensure colonization). On day 18, the pharmacokinetic experiment was performed by monitoring the fate of capecitabine in mouse blood and feces over time. A capecitabine dose equivalent to a single human dose and adjusted to the weight of the mice was administered by oral gavage (755 mg / kg, as a solution in 50 μl DMSO), then serial sampling of tail vein blood (by tail snipping), as well as fecal collection were performed at these time points (zero, 20 min, 40 min, 60 min, 2 hours, and 4 hours). Blood for each time point (30 μl) was collected using a 30 μl capillary tube and bulb dispenser (Drummond Microcaps, Drummond Scientific), quickly dispensed in 60 μl EDTA to prevent blood coagulation, and stored on ice for up to 4 hours and then frozen at −80 °C until further analysis. Feces were also collected at the same time points (even though defecation was left at will, we succeeded in collecting feces for most time points), stored on ice for up to 4 hours and then frozen in −80 °C until further analysis. After the 4-hour pharmacokinetic time point, mice were euthanized.
For chemical extraction, 2 μl of an internal standard solution (0.5 mg / ml of voriconazole) were added to the blood / EDTA solution mentioned above, and the sample was mixed using a vortex mixer. Next, 500 μl of ethyl acetate was added and mixed. The sample was then centrifuged briefly at 15000 rpm, and the organic layer was transferred to a glass tube and evaporated under vacuum using rotary evaporation (Speed Vac). The dried residue was dissolved in 100 μl of MeOH, and the solution was centrifuged at 15000 rpm and transferred to an autosampler vial for HR-HPLC-MS analysis. For fecal samples, pellets were weighed (for later normalizations), and suspended in 500 μl sterile Milli-Q water (Millipore Corporation, USA). 2 μl of an internal standard solution (0.5 mg / ml of voriconazole) were added to the sample, and the mixtures were extracted with 500 μl 1:1 ethyl acetate : MeOH. Fecal debris were then spun down and collected supernatants were dried under vacuum using a rotary evaporator (Speed Vac). The dried residues were suspended in 100 μl MeOH. The final solutions were centrifuged at 15000 rpm and transferred to autosampler vials. The prepared samples were analyzed by HR-HPLC-MS (Agilent QTOF). Chromatography separation was carried out on a Poroshell 120 EC-C18 2.7 μm 2.1 × 100 mm column (Agilent, USA) with the gradient: 99.5% A, 0.5% B to 100% B in 20 minutes and a flow rate of 0.25 ml/min, where A= 0.1% formic acid in water and B= 0.1% formic acid in acetonitrile. A 10 μl aliquot of the reconstituted extract was injected into the HR-HPLC-MS system, and the Area Under the Curve (AUC) was integrated for each metabolite and normalized by the internal standard’s AUC. Peak identities were confirmed by accurate mass, and by comparison of chromatographic retention time and MS/MS spectra to those of authentic standards.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed in MATLAB. p-values less than 0.01 (after correction for multiple hypotheses, if applicable) were considered significant. For comparisons of the means of two populations, Welch’s t-test was generally used. In cases where the independence assumption of this test was not met (as determined by the form of the null hypothesis), permutation tests were used instead. Comparison of multiple means was done via ANOVA. Comparisons of two proportions was done via a proportions z-test. For all analyses, the meaning and value of n and the measures of center, dispersion, and precision used can be found in the relevant main text or in Method Details.
Supplementary Material
Data S1. Chromatograms and molecular ion networking analyses of MDM+ Drugs with the PD microbiome, Related to Figure 2 and STAR Methods.
Data S2. Structural elucidation of selected MDM metabolites, Related to Figure 2.
Methods S1. Expanded methods for ex vivo culture characterization, Related to STAR Methods.
A) Four-day time course of PD cultured ex vivo in various media. Family level bacterial composition of the original PD fecal sample (far left), as well as that of PD ex vivo cultures grown anaerobically in 14 different media over four days (.01, .02, .03, .04). 16S rRNA gene amplicon sequences that could not be classified at the family level, and families with less than 1% relative abundance in all samples are grouped into “Other”. Cultures are ordered according to their Jensen-Shannon divergence (DJS) from the original PD sample (upper axes, computed at the family level), where lower values indicate higher similarity to PD. Note that cultures grown in mGAM are the most similar to PD. B) Family level bacterial composition of the original D1–20 fecal samples and corresponding BG cultures. “Other” includes 16S rRNA gene amplicon sequences not classified at the family level and taxa that are below 5% abundance in all sequenced samples. Replicate BG cultures for each donor labeled as “BG_1”, “BG_2”, and “BG_3”. See also STAR Methods, Table S2.
HPLC-MS analysis of hydrocortisone acetate (1) and hydrocortisone (2) incubated with PD mGAM.02 culture, mGAM.02 broth, P. distasonis culture, or C. bolteae culture. An HPLC chromatogram at an absorbance of 254 nm is shown for all samples, indicating the conversion of hydrocortisone acetate to hydrocortisone by both P. distasonis and C. bolteae, and the conversion of both hydrocortisone acetate (in two steps) and hydrocortisone (in one step) to 20β-dihydrocortisone (3) in the presence of the PD microbiome.
A) A heat map indicating the ability of each of 12 tested bacterial strains to perform capecitabine deglycosylation (d-G) in order to identify candidate species for the homology-based gene finding approach. E. faecalis TYG11, E. coli TYG1, and E. coli TYG2 were originally isolated from PD. B. HPLC-MS analysis of trifluridine (1) incubated with wild type E. coli BW25113 (WT), and Δudp, ΔdeoA, and ΔdeoA/Δudp mutants in M9 medium. An HPLC chromatogram at an absorbance of 250 nm is shown for all samples, indicating the conversion of trifluridine (1) to trifluorothymine (2) by wild type E. coli BW25113 (WT), Δudp, and ΔdeoA, but not the ΔdeoA/Δudp mutant.
HPLC-MS analysis of doxifluridine (1) incubated with wild type E. coli BW25113 (WT), and Δudp, ΔdeoA, and ΔdeoA/Δudp mutants in M9 medium. Extracted Ion Chromatograms for both doxifluridine (1) and its resulting MDM metabolite 5-fluorouracil (2) is shown for all samples, indicating the complete conversion of doxifluridine (1) to 5-fluorouracil (2) by wild type E. coli BW25113 (WT), Δudp, and ΔdeoA, but not the ΔdeoA/Δudp mutant.
Schematic representation of the screening scheme followed to identify the Bifidobacterium-derived 20β-HSDH from the PD microbiome metagenomic clone library.
HR-HPLC-MS based quantification of capecitabine and its human metabolite (5’-deoxy-5-fluorocytidine) in fecal and blood samples from mice colonized with PD in comparison to non-colonized ones. No significant difference in the levels of capecitabine were observed between the two groups. (p > 0.05, significance was determined by testing the intersection null hypothesis with marginal two-tailed t-tests using the Bonferroni correction to control family-wise error rate). Error bars represent the standard error of the mean.
Table S1. Results of MDM-Screen with the PD microbiome, Related to Figure 2 and STAR Methods.
Table S2. Tabular results of the 16S rRNA gene amplicon sequencing analyses of PD and D1–20, Related to Figure 1, Figure 3, and STAR Methods.
Table S3. Tabular results of the targeted and untargeted quantitative metabolomics analyses with the D1–20 microbiomes, Related to Figure 4 and STAR Methods.
Table S4. Results of the metagenomic analyses performed in this study, Related to Figure 5, Figure 6.
Highlights.
Development of subject-personalized ex vivo batch cultures of the gut microbiome
Discovery of diverse drug-microbiome interactions using MDM-Screen
MDM-Screen quantifies drug metabolism by personalized gut microbial communities
Functional genomic and metagenomic screens identify drug-metabolizing enzymes
Acknowledgments
We would like to thank Wei Wang and the Lewis Sigler Institute sequencing core facility for assistance with HT 16S rRNA gene amplicon sequencing, Matthew Cahn and Abhishek Biswas for assistance with sequencing data analysis, Shuo Wang for assistance with the functional group analysis, Joseph Koos, A. James Link, and Yuki Sugimoto for assistance with Mass Spectrometry, Riley Skeen-Gaar for assistance with statistical analysis, Joseph Sheehan and Zemer Gitai for assistance with obtaining the Keio library mutants, Laboratory Animal Resources at Princeton University for assistance with mouse studies, Janie Kim for illustrating the graphical abstract, and members of the Donia lab for useful discussions. We are grateful to the 21 anonymous donors who provided the fecal samples that made this project possible. Funding for this project has been provided by an Innovation Award from the Department of Molecular Biology, Princeton University, and an NIH Director’s New Innovator Award (ID: 1DP2AI124441), both to M.S.D. B.J. is funded by a New Jersey Commission on Cancer Research Pre-doctoral award (ID: DFHS18PPC056), Y-C.J.L. is funded by a training grant from the National Institute of General Medicine Sciences (ID: T32GM007388), and J.G.L. is funded by a National Science Foundation Graduate Research Fellowship (ID: 2017249408).
Footnotes
Declaration of Interests
M.S.D. is a member of the scientific advisory board of DeepBiome Therapeutics. A patent is being filed by Princeton University for the use of quantitative MDM-Screen to measure inter-individual variability in drug metabolism.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Almeida A, Mitchell AL, Boland M, Forster SC, Gloor GB, Tarkowska A, Lawley TD, and Finn RD (2019). A new genomic blueprint of the human gut microbiota. Nature 568, 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azadkhan AK, Truelove SC, and Aronson JK (1982). The disposition and metabolism of sulphasalazine (salicylazosulphapyridine) in man. Br J Clin Pharmacol 13, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2, 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, and Gordon JI (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920. [DOI] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y. (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 57, 289–300. [Google Scholar]
- Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, and Gregory Caporaso J. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2018). QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints 6, e27295v27292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SF, Chao CJ, and Clardy J. (2002). New natural product families from an environmental DNA (eDNA) gene cluster. J Am Chem Soc 124, 9968–9969. [DOI] [PubMed] [Google Scholar]
- Brito IL, Yilmaz S, Huang K, Xu L, Jupiter SD, Jenkins AP, Naisilisili W, Tamminen M, Smillie CS, Wortman JR, et al. (2016). Mobile genes in the human microbiome are structured from global to individual scales. Nature 535, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullingham RE, Nicholls AJ, and Kamm BR (1998). Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet 34, 429–455. [DOI] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton TA, Baker D, Lindon JC, Everett JR, and Nicholson JK (2009). Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci U S A 106, 14728–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JM, Rosen LS, Yoshida K, Rasco D, Shapiro GI, and Sun W. (2017). A phase 1 study of the pharmacokinetics of nucleoside analog trifluridine and thymidine phosphorylase inhibitor tipiracil (components of TAS-102) vs trifluridine alone. Invest New Drugs 35, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LJ, Kang HS, Chu J, Huang YH, Gordon EA, Reddy BV, Ternei MA, Craig JW, and Brady SF (2015). Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proceedings of the National Academy of Sciences of the United States of America 112, E4825–4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devendran S, Mendez-Garcia C, and Ridlon JM (2017). Identification and characterization of a 20beta-HSDH from the anaerobic gut bacterium Butyricicoccus desmolans ATCC 43058. J Lipid Res 58, 916–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doden HL, Pollet RM, Mythen SM, Wawrzak Z, Devendran S, Cann I, Koropatkin NM, and Ridlon JM (2019). Structural and biochemical characterization of 20beta-hydroxysteroid dehydrogenase from Bifidobacterium adolescentis strain L2–32. J Biol Chem 294, 12040–12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer GW, and Remmel RP (1984). Role of the intestinal microflora in clonazepam metabolism in the rat. Xenobiotica 14, 829–840. [DOI] [PubMed] [Google Scholar]
- Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, Kurilshikov A, Bonder MJ, Valles-Colomer M, Vandeputte D, et al. (2016). Population-level analysis of gut microbiome variation. Science 352, 560–564. [DOI] [PubMed] [Google Scholar]
- Gardiner P, Schrode K, Quinlan D, Martin BK, Boreham DR, Rogers MS, Stubbs K, Smith M, and Karim A. (1989). Spironolactone metabolism: steady-state serum levels of the sulfur-containing metabolites. J Clin Pharmacol 29, 342–347. [DOI] [PubMed] [Google Scholar]
- Genee HJ, Bali AP, Petersen SD, Siedler S, Bonde MT, Gronenberg LS, Kristensen M, Harrison SJ, and Sommer MO (2016). Functional mining of transporters using synthetic selections. Nature chemical biology 12, 1015–1022. [DOI] [PubMed] [Google Scholar]
- Gillespie DE, Brady SF, Bettermann AD, Cianciotto NP, Liles MR, Rondon MR, Clardy J, Goodman RM, and Handelsman J. (2002). Isolation of antibiotics turbomycin a and B from a metagenomic library of soil microbial DNA. Applied and environmental microbiology 68, 4301–4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, and Gordon JI (2011). Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 108, 6252–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, and Turnbaugh PJ (2013). Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 341, 295–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DA, and Brady AR (2004). Sample Size and Power Calculations using the Noncentral t-distribution. The Stata Journal 4, 142–153. [Google Scholar]
- Human Microbiome Project C. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD (2007). Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering 9, 90–95. [Google Scholar]
- Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al. (2013). Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilett KF, Tee LB, Reeves PT, and Minchin RF (1990). Metabolism of drugs and other xenobiotics in the gut lumen and wall. Pharmacol Ther 46, 67–93. [DOI] [PubMed] [Google Scholar]
- Jorga K, Fotteler B, Heizmann P, and Gasser R. (1999). Metabolism and excretion of tolcapone, a novel inhibitor of catechol-O-methyltransferase. Br J Clin Pharmacol 48, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmarkar D, and Rock KL (2013). Microbiota signalling through MyD88 is necessary for a systemic neutrophilic inflammatory response. Immunology 140, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NK, Min JS, Park JK, Yun SH, Sung JS, Jung HC, and Roh JK (2001). Intravenous 5-Fluorouracil Versus Oral Doxifluridine as Preoperative Concurrent Chemoradiation for Locally Advanced Rectal Cancer: Prospective Randomized Trials. Jpn J Clin Oncol 31, 25–29. [DOI] [PubMed] [Google Scholar]
- Kimura T, Sudo K, Kanzaki Y, Miki K, Takeichi Y, Kurosaki Y, and Nakayama T. (1994). Drug absorption from large intestine: physicochemical factors governing drug absorption. Biol Pharm Bull 17, 327–333. [DOI] [PubMed] [Google Scholar]
- Koppel N, Bisanz JE, Pandelia ME, Turnbaugh PJ, and Balskus EP (2018). Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel N, Maini Rekdal V, and Balskus EP (2017). Chemical transformation of xenobiotics by the human gut microbiota. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroiwa M, Inotsume N, Iwaoku R, and Nakano M. (1985). Reduction of Dantrolene by Enteric Bacteria. YAKUGAKU ZASSHI 105, 770–774. [DOI] [PubMed] [Google Scholar]
- Kuroiwa M, Inotsume N, and Nakano M. (1986). Reduction of nicardipine, calcium antagonist, with enteric bacteria. Yakugaku Zasshi 106, 698–702. [DOI] [PubMed] [Google Scholar]
- Lamont EB, and Schilsky RL (1999). The oral fluoropyrimidines in cancer chemotherapy. Clin Cancer Res 5, 2289–2296. [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz HJ, Stintzing S, and Loupakis F. (2015). TAS-102, a novel antitumor agent: a review of the mechanism of action. Cancer Treat Rev 41, 777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Jia W. (2013). Cometabolism of microbes and host: implications for drug metabolism and drug-induced toxicity. Clin Pharmacol Ther 94, 574–581. [DOI] [PubMed] [Google Scholar]
- Lindenbaum J, Tse-Eng D, Butler VP Jr., and Rund DG (1981). Urinary excretion of reduced metabolites of digoxin. Am J Med 71, 67–74. [DOI] [PubMed] [Google Scholar]
- Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, Creasy HH, McCracken C, Giglio MG, et al. (2017). Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, and Johnston PG (2003). 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3, 330–338. [DOI] [PubMed] [Google Scholar]
- Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, et al. (2018). Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini Rekdal V, Bess EN, Bisanz JE, Turnbaugh PJ, and Balskus EP (2019). Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannens G, Huang ML, Meuldermans W, Hendrickx J, Woestenborghs R, and Heykants J. (1993). Absorption, metabolism, and excretion of risperidone in humans. Drug Metab Dispos 21, 1134–1141. [PubMed] [Google Scholar]
- McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, Aksenov AA, Behsaz B, Brennan C, Chen Y, et al. (2018). American Gut: an Open Platform for Citizen Science Microbiome Research. mSystems 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, and Hugenholtz P. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney WG (2010). Data structures for statistical computing in python. Paper presented at: Proceedings of the 9th Python in Science Conference. [Google Scholar]
- Meinl W, Sczesny S, Brigelius-Flohe R, Blaut M, and Glatt H. (2009). Impact of gut microbiota on intestinal and hepatic levels of phase 2 xenobiotic-metabolizing enzymes in the rat. Drug Metab Dispos 37, 1179–1186. [DOI] [PubMed] [Google Scholar]
- Meuldermans W, Hendrickx J, Mannens G, Lavrijsen K, Janssen C, Bracke J, Le Jeune L, Lauwers W, and Heykants J. (1994). The metabolism and excretion of risperidone after oral administration in rats and dogs. Drug Metab Dispos 22, 129. [PubMed] [Google Scholar]
- Min JS, Kim NK, Park JK, Yun SH, and Noh JK (2000). A prospective randomized trial comparing intravenous 5-fluorouracil and oral doxifluridine as postoperative adjuvant treatment for advanced rectal cancer. Ann Surg Oncol 7, 674–679. [DOI] [PubMed] [Google Scholar]
- Nayfach S, Shi ZJ, Seshadri R, Pollard KS, and Kyrpides NC (2019). New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, Plichta DR, Gautier L, Pedersen AG, Le Chatelier E, et al. (2014). Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol 32, 822–828. [DOI] [PubMed] [Google Scholar]
- Northfield TC, and McColl I. (1973). Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 14, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, and Hutchison GR (2011). Open Babel: An open chemical toolbox. J Cheminform 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant TE (2006). A guide to NumPy (USA: Trelgol Publishing; ). [Google Scholar]
- Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, et al. (2019). Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 176, 649–662 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppercorn MA, and Goldman P. (1972). The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J Pharmacol Exp Ther 181, 555–562. [PubMed] [Google Scholar]
- Planer JD, Peng Y, Kau AL, Blanton LV, Ndao IM, Tarr PI, Warner BB, and Gordon JI (2016). Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature 534, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. [DOI] [PubMed] [Google Scholar]
- Rettedal EA, Gumpert H, and Sommer MO (2014). Cultivation-based multiplex phenotyping of human gut microbiota allows targeted recovery of previously uncultured bacteria. Nat Commun 5, 4714. [DOI] [PubMed] [Google Scholar]
- Ridlon JM, Ikegawa S, Alves JM, Zhou B, Kobayashi A, Iida T, Mitamura K, Tanabe G, Serrano M, De Guzman A, et al. (2013). Clostridium scindens: a human gut microbe with a high potential to convert glucocorticoids into androgens. J Lipid Res 54, 2437–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon MR, August PR, Bettermann AD, Brady SF, Grossman TH, Liles MR, Loiacono KA, Lynch BA, MacNeil IA, Minor C, et al. (2000). Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Applied and environmental microbiology 66, 2541–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheline RR (1973). Metabolism of foreign compounds by gastrointestinal microorganisms. Pharmacol Rev 25, 451–523. [PubMed] [Google Scholar]
- Schmieder R, and Edwards R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenhard G, Oppermann J, and Kohn FE (1985). Metabolism and pharmacokinetic studies of misoprostol. Dig Dis Sci 30, 126S–128S. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica DA (2005). Pharmacokinetics and Pharmacodynamics of Mineralocorticoid Blocking Agents and their Effects on Potassium Homeostasis. Heart Failure Reviews 10, 23–29. [DOI] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KS, Smith PL, Heady TN, Trugman JM, Harman WD, and Macdonald TL (2003). In vitro metabolism of tolcapone to reactive intermediates: relevance to tolcapone liver toxicity. Chem Res Toxicol 16, 123–128. [DOI] [PubMed] [Google Scholar]
- Sommer MOA, Dantas G, and Church GM (2009). Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325, 1128–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanogiannopoulos P, Bess EN, Carmody RN, and Turnbaugh PJ (2016). The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol 14, 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD (2002). A Direct Approach to False Discovery Rates. Journal of the Royal Statistical Society Series B (Statistical Methodology) 64, 479–498. [Google Scholar]
- Sugimoto Y, Camacho FR, Wang S, Chankhamjon P, Odabas A, Biswas A, Jeffrey PD, and Donia MS (2019). A metagenomic strategy for harnessing the chemical repertoire of the human microbiome. Science. [DOI] [PubMed] [Google Scholar]
- Taylor MR, Flannigan KL, Rahim H, Mohamud A, Lewis IA, Hirota SA, and Greenway SC (2019). Vancomycin relieves mycophenolate mofetil-induced gastrointestinal toxicity by eliminating gut bacterial beta-glucuronidase activity. Sci Adv 5, eaax2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temmink OH, de Bruin M, Turksma AW, Cricca S, Laan AC, and Peters GJ (2007). Activity and substrate specificity of pyrimidine phosphorylases and their role in fluoropyrimidine sensitivity in colon cancer cell lines. Int J Biochem Cell Biol 39, 565–575. [DOI] [PubMed] [Google Scholar]
- Tramontano M, Andrejev S, Pruteanu M, Klunemann M, Kuhn M, Galardini M, Jouhten P, Zelezniak A, Zeller G, Bork P, et al. (2018). Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat Microbiol 3, 514–522. [DOI] [PubMed] [Google Scholar]
- Tsai BS, Kessler LK, Stolzenbach J, Schoenhard G, and Bauer RF (1991). Expression of gastric antisecretory and prostaglandin E receptor binding activity of misoprostol by misoprostol free acid. Dig Dis Sci 36, 588–593. [DOI] [PubMed] [Google Scholar]
- Uribe RV, van der Helm E, Misiakou MA, Lee SW, Kol S, and Sommer MOA (2019). Discovery and Characterization of Cas9 Inhibitors Disseminated across Seven Bacterial Phyla. Cell host & microbe 25, 233–241 e235. [DOI] [PubMed] [Google Scholar]
- van Kessel SP, Frye AK, El-Gendy AO, Castejon M, Keshavarzian A, van Dijk G, and El Aidy S. (2019). Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat Commun 10, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, et al. (2015). Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, et al. (2010). Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330, 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, et al. (2016). Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J, Cerone-McLernon A, O’Rourke S, Ponticorvo L, and Bokkenheuser VD (1982). Formation of 20β-dihydrosteroids by anaerobic bacteria. J of Steroid Biochemistry 17, 661–667. [DOI] [PubMed] [Google Scholar]
- Wood DE, and Salzberg SL (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15, R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampino MG, Colleoni M, Bajetta E, Stampino CG, Guenzi A, and de Braud F. (1999). Pharmacokinetics of oral doxifluridine in patients with colorectal cancer. Tumori 85, 47–50. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, and Goodman AL (2019a). Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, and Goodman AL (2019b). Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Chromatograms and molecular ion networking analyses of MDM+ Drugs with the PD microbiome, Related to Figure 2 and STAR Methods.
Data S2. Structural elucidation of selected MDM metabolites, Related to Figure 2.
Methods S1. Expanded methods for ex vivo culture characterization, Related to STAR Methods.
A) Four-day time course of PD cultured ex vivo in various media. Family level bacterial composition of the original PD fecal sample (far left), as well as that of PD ex vivo cultures grown anaerobically in 14 different media over four days (.01, .02, .03, .04). 16S rRNA gene amplicon sequences that could not be classified at the family level, and families with less than 1% relative abundance in all samples are grouped into “Other”. Cultures are ordered according to their Jensen-Shannon divergence (DJS) from the original PD sample (upper axes, computed at the family level), where lower values indicate higher similarity to PD. Note that cultures grown in mGAM are the most similar to PD. B) Family level bacterial composition of the original D1–20 fecal samples and corresponding BG cultures. “Other” includes 16S rRNA gene amplicon sequences not classified at the family level and taxa that are below 5% abundance in all sequenced samples. Replicate BG cultures for each donor labeled as “BG_1”, “BG_2”, and “BG_3”. See also STAR Methods, Table S2.
HPLC-MS analysis of hydrocortisone acetate (1) and hydrocortisone (2) incubated with PD mGAM.02 culture, mGAM.02 broth, P. distasonis culture, or C. bolteae culture. An HPLC chromatogram at an absorbance of 254 nm is shown for all samples, indicating the conversion of hydrocortisone acetate to hydrocortisone by both P. distasonis and C. bolteae, and the conversion of both hydrocortisone acetate (in two steps) and hydrocortisone (in one step) to 20β-dihydrocortisone (3) in the presence of the PD microbiome.
A) A heat map indicating the ability of each of 12 tested bacterial strains to perform capecitabine deglycosylation (d-G) in order to identify candidate species for the homology-based gene finding approach. E. faecalis TYG11, E. coli TYG1, and E. coli TYG2 were originally isolated from PD. B. HPLC-MS analysis of trifluridine (1) incubated with wild type E. coli BW25113 (WT), and Δudp, ΔdeoA, and ΔdeoA/Δudp mutants in M9 medium. An HPLC chromatogram at an absorbance of 250 nm is shown for all samples, indicating the conversion of trifluridine (1) to trifluorothymine (2) by wild type E. coli BW25113 (WT), Δudp, and ΔdeoA, but not the ΔdeoA/Δudp mutant.
HPLC-MS analysis of doxifluridine (1) incubated with wild type E. coli BW25113 (WT), and Δudp, ΔdeoA, and ΔdeoA/Δudp mutants in M9 medium. Extracted Ion Chromatograms for both doxifluridine (1) and its resulting MDM metabolite 5-fluorouracil (2) is shown for all samples, indicating the complete conversion of doxifluridine (1) to 5-fluorouracil (2) by wild type E. coli BW25113 (WT), Δudp, and ΔdeoA, but not the ΔdeoA/Δudp mutant.
Schematic representation of the screening scheme followed to identify the Bifidobacterium-derived 20β-HSDH from the PD microbiome metagenomic clone library.
HR-HPLC-MS based quantification of capecitabine and its human metabolite (5’-deoxy-5-fluorocytidine) in fecal and blood samples from mice colonized with PD in comparison to non-colonized ones. No significant difference in the levels of capecitabine were observed between the two groups. (p > 0.05, significance was determined by testing the intersection null hypothesis with marginal two-tailed t-tests using the Bonferroni correction to control family-wise error rate). Error bars represent the standard error of the mean.
Table S1. Results of MDM-Screen with the PD microbiome, Related to Figure 2 and STAR Methods.
Table S2. Tabular results of the 16S rRNA gene amplicon sequencing analyses of PD and D1–20, Related to Figure 1, Figure 3, and STAR Methods.
Table S3. Tabular results of the targeted and untargeted quantitative metabolomics analyses with the D1–20 microbiomes, Related to Figure 4 and STAR Methods.
Table S4. Results of the metagenomic analyses performed in this study, Related to Figure 5, Figure 6.
Data Availability Statement
The sequencing datasets generated during this study are available in Table S2 and at NCBI (BioProject number PRJNA593062). The metabolomics datasets generated during this study are available in Tables S1, S3, and at MassIVE (Accession ID MSV000084641). The code generated during this study is available at GitHub (https://github.com/jaimegelopez/personalized_community_MDM_screen).