


Abstract
DNA–protein interactions play essential roles in all living cells. Understanding of how features embedded in the DNA sequence affect specific interactions with proteins is both challenging and important, since it may contribute to finding the means to regulate metabolic pathways involving DNA–protein interactions. Using a massive experimental benchmark dataset of binding scores for DNA sequences and a machine learning workflow, we describe the binding to DNA of T7 primase, as a model system for specific DNA–protein interactions. Effective binding of T7 primase to its specific DNA recognition sequences triggers the formation of RNA primers that serve as Okazaki fragment start sites during DNA replication.
INTRODUCTION
Specific protein–DNA recognition is essential for a wide range of cellular processes, including DNA replication, repair and recombination (1). Determination of the specific binding preferences of proteins both in vivo (2–7) and in vitro (8–17) has been facilitated by recent technological advances in high-throughput testing. Computational analysis combined with high-throughput assays have identified protein–DNA binding preferences at the whole-genome level (8,11,16,18–25), and the information so obtained has been used to elucidate the mechanisms of gene expression regulation by transcription factors (TFs) and RNA polymerases in different organisms (20,24,26,27). While most studies on protein-DNA specificity rely on base readout through the major and the minor grooves and on shape readout through global and local shapes of double stranded DNA (28), little is known about the principles that govern the specific binding of a protein to single-stranded DNA. A clue may perhaps be drawn from a structural study of the binding of cold-shock proteins to single-stranded DNA, which revealed that sequence-specific DNA binding is mediated by base-stacking interactions of aromatic amino acids and that additional H-bonding is important for sequence recognition (29). Nonetheless, in-depth work to elucidate the specific binding of proteins to single-stranded DNA is still required.
In all cells, DNA replication serves as a metabolic pathway in which specific DNA–protein interactions take place (30). During DNA replication, double-stranded DNA is unwound to expose the two individual DNA strands; one is copied continuously (the leading strand) and the other is copied discontinuously (the lagging strand). On the lagging DNA strand, a DNA primase recognizes the DNA sequence used as the template for the synthesis of RNA primers and is thus responsible for elongating these RNA primers into the DNA segments known as Okazaki fragments. This process of RNA-primed DNA synthesis by a DNA polymerase is triggered exclusively by the recognition of a specific DNA sequence by the primase. This recognition is thus fundamental to the establishment of Okazaki fragments and consequently to the whole process of proper DNA replication. In prokaryotes, RNA primer formation occurs on pre-defined sequences on the genome that are specifically recognized by a DnaG-type primase (31) (Figure 1). Against this background, the focus of our study is the activity of the N-domain of gene 4 protein (helicase-primase, gp4) of bacteriophage T7 (known as T7 primase). The activity of this N-domain (comprising residues 1–271) is initiated by the sequence-specific binding of a DNA primase to 5′-GTC-3′ (32–34), which is then followed by the synthesis of a functional primer (35). Importantly, it is now known that even though a DNA primase recognizes a specific trinucleotide sequence, flexibility in the selection of initiation sites for Okazaki fragments is allowed (36), i.e. not every primase-DNA recognition sequence (PDRS) will become an Okazaki fragment start site. The reason for this flexibility is, however, not understood, and it is this enigma that we address in the current study: Although extensive research has been carried out on the interactions of DNA primase with DNA, it is still not clear why a DNA primase ignores the majority of trinucleotide recognition sites. In Escherichia coli, for example, DnaG primase ignores ∼97% of the trinucleotide recognition sites and initiates Okazaki fragments only every 1.5–2 kb (and not more frequently) (37). The literature offers two possible explanations for the effect of selective DNA sequence recognition by a primase: The first, well-explored possibility, is that other DNA replication proteins, such as DnaB (38–41), single-stranded DNA-binding protein (SSB) (42), or a clamp loader (43), affect the binding of the primase (DnaG) to DNA or even change the preferences for PDRSs on the genome. The second possibility is that a sequence larger than a trinucleotide is required for the specific binding of a primase. On the basis of the mismatch between the frequency of GTC sequences on the bacteriophage T7 genome and the actual size of the Okazaki fragments, it is reasonable to assume that only a sequence larger than a trinucleotide will lead to ‘effective binding’ of a DNA primase, i.e. binding yielding an RNA primer that marks the start site of an Okazaki fragment. It is known that a tetraribonucleotide (44), but not a di- or tri-ribonucleotide (45), can serve as a primer for the T7 DNA polymerase. However, it is still not known whether longer primers are required for more efficient DNA replication. The requirement for a primer of at least tetranucleotide size indicates that a primase recognition sequence must be larger than 5′-GTC-3′ [note that the 3′-cryptic C is essential for recognition but is not copied into the RNA primer (46)]. Many questions regarding such larger primase recognition sequences remained unanswered: In particular, what is the effect of sequences flanking the 5′-GTC-3′ on primase activity? Does primase-DNA binding affinity correlate with RNA primer synthesis? These questions cannot be addressed by using ‘classical’ techniques, and we have therefore developed a dedicated workflow to answer open questions of this type. In our first steps to investigate the above-described possibility of a larger DNA binding determinant, we applied high-throughput primase profiling (47), in which binding scores (median fluorescence intensities) for tens of thousands of DNA primase–DNA binding events on a protein-DNA binding microarray (PBM) were combined with biochemical analysis (47). This technology facilitated the analysis of the composition of sequences flanking the specific recognition site and their corresponding binding scores and confirmed that a GTC sequence is not sufficient for the recognition of DNA templates by T7 primase (47,48). Specifically, we showed that T7 primase has a high affinity for PDRSs composed of GTC with G/C/T-rich flanking sequences, leading to the formation of longer RNA primers (47). Although the development of high-throughput primase profiling facilitated the acquisition of massive amounts of data on DNA-primase-binding events, the means to systematically analyze this data in a way that will facilitate a comprehensive understanding of the recognition process are yet to be put in place. Specifically, analysis of this data will throw light on the principles for selection of PDRSs on a genome during DNA replication by enabling us to answer three key questions: (i) Is there information stored in the DNA sequence that is important for T7 primase binding? (ii) If we understand the principles of specific primase–DNA recognition, can we predict binding scores of T7 primase for a given DNA sequence? And (iii) Can we generate new DNA sequences with desired binding scores based on the sequence features embedded in the DNA? Answering the third question will enable us to ascertain which features embedded in specific DNA binding sequences govern the binding of DNA primase.
Figure 1.
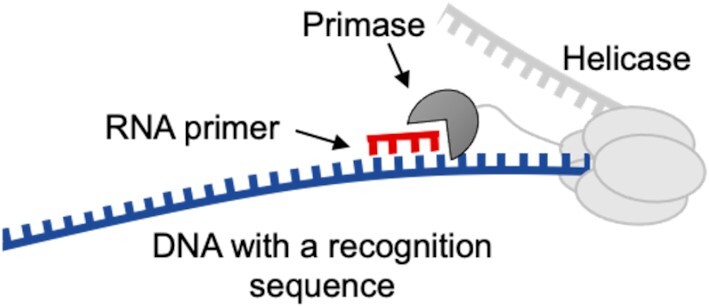
Schematic representation of primase binding to a single-stranded DNA template and synthesis of an RNA primer. Enzymes used in this study: a truncated primase domain (residues 1–271, 27 kDa) and a helicase-primase (residues 1–566, 63 kDa).
In concert with cutting-edge developments in biochemical technologies, current progress in computational science provides us with the opportunity to construct knowledge-based models that will help us to answer the above questions. Here, we describe an intelligent learning workflow that provides a comprehensive view of the principles that govern the design and activity of PDRSs with unprecedented flexibility and accuracy. We applied this workflow to elucidate the link between the larger context, i.e. the flanking nucleotides, of the primase recognition sequence and the synthesis of RNA primers that initiate Okazaki fragments. Whereas our initial data (47) showed that TA is better than GA, in the current study—using state-of-the-art machine learning analysis—we found a set of rules that allowed us to quantitatively predict primase binding and catalysis for any DNA sequence.
MATERIALS AND METHODS
Materials
All chemical reagents were of molecular biology grade and were obtained from Sigma. ATP and CTP were purchased from Roche Molecular Biochemicals.
Protein overexpression and purification
Full length gene 4 protein (gp4, residues 1–566, 63 kDa) was overexpressed and purified as previously described (49). The T7 primase domain (residues 1–271, 27 kDa) was overexpressed and purified as previously described (50).
Design of the DNA library
The analysis was based on previously collected data (47,48), specifically, on 25220 DNA sequences that include the T7 DNA-primase recognition sequence (5′-GTC-3′). The general pattern of each sequence was 5′-(N)17-GTC-(N)16-GTCTTGATTCGCTTGACGCTGCTG-3′, where (N)17 and (N)16 represent the variable regions flanking the GTC recognition site. The above data Ωset contained accurate binding scores for T7 primase to each DNA sequence, obtained by PBMs as described previously (47). Data acquisition was performed using a GenePix 4400A scanner (Molecular Devices), and data was analyzed using custom scripts to obtain fluorescence intensities for all sequences represented on the array.
Data preprocessing
Each PBM consisted of 5076 unique sequences and 25220 samples, 6 repetitions per sequence, and overall 151320 samples (instances). All scripts were written in Python (Python Software Foundation, version 3.7, http://www.python.org), Scikit learn (51), and the software PyCharm (community edition, https://www.jetbrains.com/pycharm/). The source code for the machine learning algorithms is available in the Github repository (https://github.com/csbarak/T7pdrs). This git repository also contains the data used for the analysis.
By extracting the coefficient of variation (52) for scores associated with each sequence (six repetitions), we observed that the stronger the score, the more stable the coefficient of variation (Supplementary Figure S1). Finally, each sequence's score was determined as its median score. For the stability evaluation, it was necessary to account for the different binding score ranges; thus, to eliminate the different scales of the standard deviation, we evaluated the binding score stability of each sequence by using the coefficient of determination (COD, Equation 1):
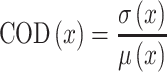 |
(1) |
where 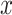 is a set of binding score repeats for a specific sequence; σ is the standard deviation of that sequence; and μ is the mean value of
is a set of binding score repeats for a specific sequence; σ is the standard deviation of that sequence; and μ is the mean value of 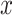 .
.
Method for sequence-based feature extraction
We tried out linear, quadratic and root weighting of the K-mers according to their distance from the GTC; e.g. while the 3-mer ‘ACA’ appears twice in the sequence ‘ACATGTCACAT’, the weighted linear count of ‘ACA’ would multiply its distance plus 1 from the kernel (GTC). However, this approach did not improve the performance of the model. While proper usage of the mer's location might lead to different results, using advanced algorithms to produce a more complex connection between features would limit our work's explainability and further exploration of the mer's effect. We therefore used simple K-mer counts and normalized by the length of the sequence to increase the generalization and prevent bias.
Principal component analysis
PCA is commonly used to reduce dimensions of datasets by de-correlating the features and extracting the linear combinations that hold the greatest variance. Thus, non-informative features are dropped, and the remaining features consist of highly variant linear combinations (principal components) of the original features. We used PCA on overlapping K-mer count instances so as to visualize the projected distribution of binding scores upon the three most significant principal components. Features were obtained by counting every combination of dimers, trimers, and tetramers in the DNA sequence (K-mer, Supplementary Figure S2). Different K values were used for the K-mer feature extraction, and all experiments resulted in a clear 5-cluster construct for the entire dataset. To compare data between clusters, we applied MinMax normalization (Equation 2) and colored each instance according to its relative strength.
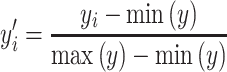 |
(2) |
where y = binding scores of the entire dataset, 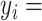 the ith binding score.
the ith binding score.
Conversion of the categorical DNA variables
The DNA data was converted to an array of integers by OHE, a process in which each nucleotide is represented by the following scheme: (A = [1000], C = [0100], G = [0010], T = [0001]). The N × 4 matrix represents every DNA oligonucleotide, and is used as input for both the Kmeans model and the WD-based hierarchical clustering model (53).
Kmeans
In the initial step of Kmeans, the distances of the sequence vectors in the training set from randomly located centroids are measured, with the number of centers (K) being considered as a hyperparameter. Then, the distance of every sequence from the centroid is computed using the Euclidean distance (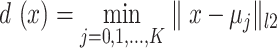 ). For the optimization step, each centroid's position (
). For the optimization step, each centroid's position (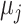 ) is moved to its own cluster's geometric mean. This process is repeated until a stop condition is met, which is usually determined by an improvement in the loss function. The loss function of the
) is moved to its own cluster's geometric mean. This process is repeated until a stop condition is met, which is usually determined by an improvement in the loss function. The loss function of the 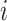 th iteration is the sum of the distances between all instances and their matching centroids (Equation 3):
th iteration is the sum of the distances between all instances and their matching centroids (Equation 3):
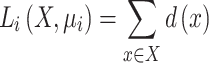 |
(3) |
where 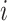 is the iteration number,
is the iteration number, 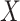 denotes the entire data matrix,
denotes the entire data matrix, 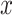 represents an OHE vector and
represents an OHE vector and 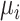 represents the set of centroids at the beginning of the
represents the set of centroids at the beginning of the 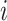 th iteration.
th iteration.
An optimized model is obtained when the difference in the value of the loss function between consecutive iterations is small enough (typically 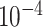 ) or the maximal number of iterations has been reached.
) or the maximal number of iterations has been reached.
Hierarchical clustering
Ward's criterion is used to determine which clusters should be merged by creating new data partitions in such a way that the sum of cluster variances of the newly offered partitions is kept low; in our case, it amounts to the smallest number of nucleotide changes between same-cluster sequences. Since the sum of the squared errors is minimized when each ‘word’ acts as its own cluster, the common way to choose the number of clusters 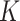 is to choose the
is to choose the 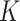 that maximizes the WD gap. Using this method, we can extract both
that maximizes the WD gap. Using this method, we can extract both 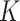 and the evolutionary stages of each cluster. WD calculates the similarity of two clusters (
and the evolutionary stages of each cluster. WD calculates the similarity of two clusters (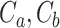 ) as the normalized distance of their corresponding cluster means (
) as the normalized distance of their corresponding cluster means (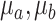 , Equation 4):
, Equation 4):
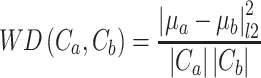 |
(4) |
The first step of the method initiates a cluster for each instance, and the second seeks the two most similar clusters in terms of WD. When found, these two clusters are united, and the second step is repeated until only one cluster remains.
Supervised learning: linear regression with L1 regularization (Lasso)
The Lasso algorithm performs linear regression under L1 regularization. Its output is a closed form equation that is generated under the constraint of having the smallest number of variables as possible. The algorithm complies with this constraint by applying a penalty for each variable taken into account in the closed form equation. Simple linear regression uses a weighted combination of features to generate a prediction based on (Equation 5):
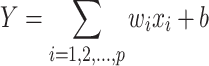 |
(5) |
where 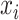 is the ith feature chosen from
is the ith feature chosen from 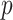 features, while
features, while 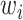 and
and 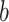 are the learned weights (usually found by minimizing the mean square error over the training set) and the learned bias, respectively.
are the learned weights (usually found by minimizing the mean square error over the training set) and the learned bias, respectively.
While a simple linear model uses the entire set of features, Lasso applies a loss function on the number of features. Moreover, compared to L2 regularization, L1 regularization facilitates the zeroing out of features rather than minimizing their weights, leading to the selection of a smart subset of features. Using Lasso on our data required two preprocessing stages; the first was extracting K-mer counts for obtaining a simple and general solution, and the second was applying a square root on the binding scores to better match their values for linear regression. The MinMax-wise normalized scores yielded a cross-validated result with a mean absolute error (MAE) value of 0.10, calculated using (Equation 6):
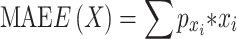 |
(6) |
where 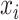 is the MAE of bin
is the MAE of bin 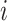 of the bins obtained by Kmeans, and
of the bins obtained by Kmeans, and 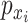 is the percentage of samples in that bin out of the entire data set.
is the percentage of samples in that bin out of the entire data set.
We evaluated the results with MAE, and obtained the expected error in terms of a weighted MAE, where the weights refer to the percentage of clustered sequences (Equation 7).
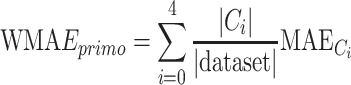 |
(7) |
where 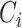 is the ith cluster,
is the ith cluster, 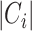 is the number of sequences belonging to the ith cluster,
is the number of sequences belonging to the ith cluster, 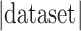 is the size of the entire dataset and
is the size of the entire dataset and 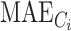 is the mean absolute error of the ith cluster.
is the mean absolute error of the ith cluster.
Our main goal was to develop a predictive model with as small an error as possible, while maintaining model explainability and simplicity. Examining the results of different regression models (Supplementary Table S1), we see that the smallest error was achieved using XGBoost, yet the difference between the errors of XGBoost and those of Lasso is about 0.5% MAE. In contrast to the decision-tree-based XGBoost, Lasso generates a closed predictive equation (i.e. 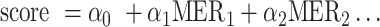 ), and combined with Lasso's L1 regularization, it constrains the number of features and the coefficients needed for the prediction. In addition, in contrast to support-vector-machine (SVM)-based models, Lasso enables limiting the coefficients to positive values, which could lead to a meaningful K-mer addition approach. Lastly, with Lasso the bias can be neutralized, meaning that the prediction is dependent solely on the K-mer count. Increasing the bias further enables a decrease in the variance and therefore a precise prediction.
), and combined with Lasso's L1 regularization, it constrains the number of features and the coefficients needed for the prediction. In addition, in contrast to support-vector-machine (SVM)-based models, Lasso enables limiting the coefficients to positive values, which could lead to a meaningful K-mer addition approach. Lastly, with Lasso the bias can be neutralized, meaning that the prediction is dependent solely on the K-mer count. Increasing the bias further enables a decrease in the variance and therefore a precise prediction.
In summary, in this study, we chose to use Lasso, since it provides good performance and a closed predictive expression that is short and (intentionally) consists of non-negative coefficients. Other regression models also generated an expected error that was less than 10% MAE (Supplementary Table S1), meaning that the data collection and preprocessing techniques were highly informative regarding the researched binding score.
Oligoribonucleotide synthesis assay
Synthesis of oligoribonucleotides by DNA primase was performed as described previously (47). The reaction mixture contained 5 μM DNA sequences generated by our machine-learning prediction algorithms described above, 1 mM ATP, 1 mM [α-32P]ATP, and T7 primase in a buffer containing 40 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 10 mM DTT and 50 mM potassium glutamate. After incubation at room temperature for 10 min, the reaction was terminated by adding an equal volume of sequencing buffer containing 98% formamide, 0.1% bromophenol blue, and 20 mM EDTA. The samples were loaded onto 25% polyacrylamide sequencing gel containing 7 M urea and visualized using autoradiography.
RESULTS
The overall structure of our study comprised the following stages of analysis of the data obtained using PBMs for quantitative measurements of T7 primase–DNA binding, after preprocessing of the data (Scheme 1): (step 1) preparation of a PBM-driven benchmark data; (step 2) clustering the PDRSs containing DNA sequences; (step 3) training a regression model and (step 4) predicting the score of new DNA sequences, and generating novel DNA sequences with the desired binding scores for T7 primase. These steps are elaborated below, as are the data preprocessing and step 5 (Scheme 1), which is biochemical validation.
Scheme 1.
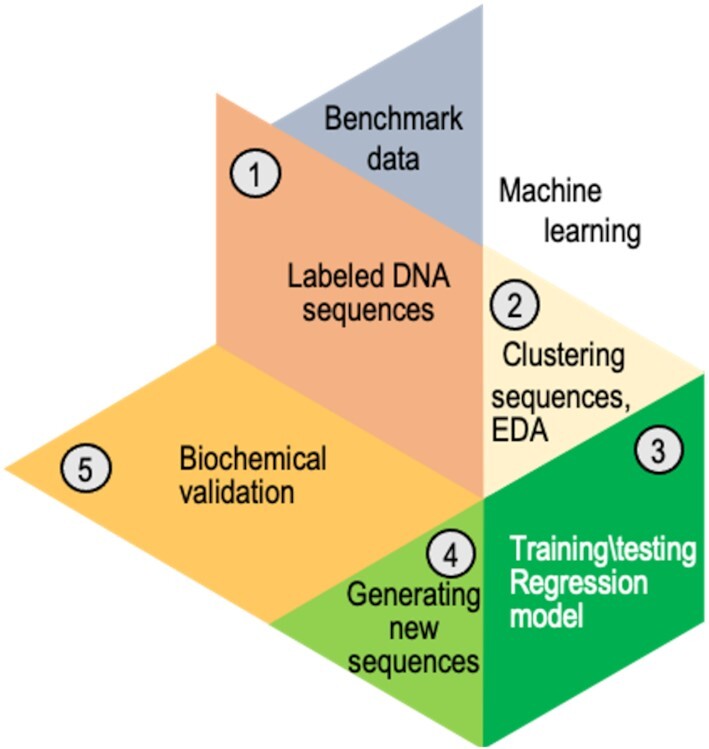
Analysis workflow after preprocessing the data from the primase–DNA binding microarray. The benchmark dataset containing DNA sequences for the training set was preprocessed (step 1). The DNA sequences were clustered into five bins using exploratory data analysis (EDA), i.e. unsupervised algorithms (step 2). A different regressor was trained for every cluster. Several regression algorithms were used; linear regression with L1 regularization provided the best results. To predict the binding scores of a new DNA sequence, the sequence was assigned to a specific bin, and its score for primase binding was predicted using that bin's regressor (step 3). Novel DNA sequences (PDRSs) with high binding score for primase were generated (step 4). It was then possible to examine the ability of those PDRSs to bind primase and induce the synthesis of RNA primers (step 5).
Data preprocessing and vectorization of DNA sequences
Data were acquired for His-tagged T7 primase produced and purified as described previously (47). All algorithms described for data preprocessing and analysis are written in Python and are publicly available (https://github.com/csbarak/T7pdrs). Before the data analysis, considerable attention was paid to data preprocessing, as the success of the subsequent application of machine-learning algorithms depended on the explicit presentation of the data in a way that facilitated the extraction of meaningful features and the removal of distracting outliers. The preprocessing of PBM-derived DNA-primase binding data comprised four steps: data cleansing, data filtration, embedding of the sequences into vectors, and data normalization, as follows. Using the PDRSs as meaningful ‘words’ on the basis of their sequence features, where each sequence was assigned to its PBM-driven binding score, we focused on the sequences that could potentially serve as Okazaki fragment start sites. We started with the preparation of a ‘lexicon’ of DNA ‘words’, each comprising a larger context of GTC-containing sequences that allow effective binding of T7 primase, i.e. the binding of T7 primase that yields RNA primers. The starting point for the preprocessing was the notion that while an average size of ∼64 nucleotides (Figure 2A) is the expected distance between two GTC sequences, the experimentally obtained size of an Okazaki fragment is 1000–6000 nucleotides (Figure 2B, marked in the red range box). We thus posited that GTC-containing PDRSs must be larger than a trinucleotide sequence to meet the frequency on the genome that would allow the creation of Okazaki fragments of sizes that were observed previously.
Figure 2.
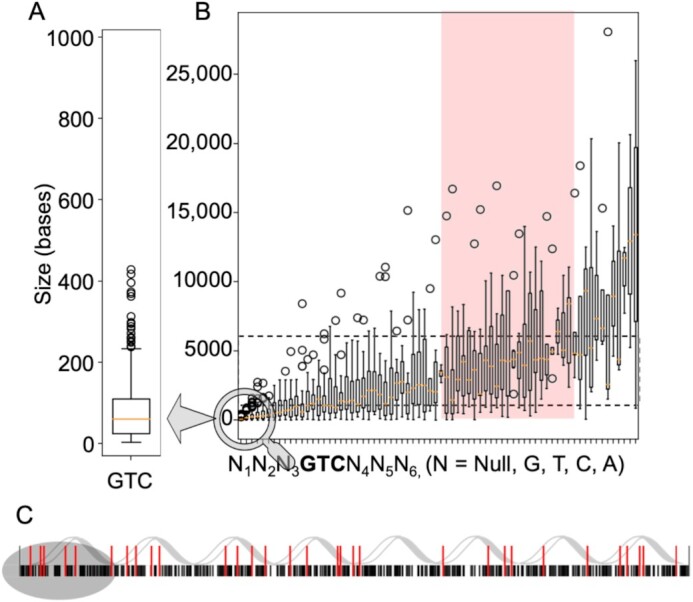
Frequency and distribution of GTC-containing primase–DNA recognition sequences (PDRSs) on the bacteriophage T7 genome. (A) The frequency of the occurrence of GTC in a random sequence is every 43 = 64 bases (approximately as indicated by the orange line). (B) Calculated size distribution of the DNA sequence between GTC-containing PDRSs on the T7 genome that matches the actual size of Okazaki fragments. These PDRSs consist of 0–3 nucleotides flanking the GTC sequence and are distributed at an inter-PDRS distance that ranges between 1000–6000 nucleotides, which yield Okazaki fragments of the same size. (C) All combinations of possible T7 PDRSs (5′-GTC-3′) on the genome are considered. Black lines represent the frequency of GTCs; red lines represent the frequency of large-context GTC-containing PDRSs that match experimental values for Okazaki fragment sizes (62); black lines: frequency of GTC every 64 bases.
Since DNA sequences constitute a form of categorical data represented by nucleotides, the preprocessing step was required to convert the plain representation of DNA sequences into a meaningful numeric representation. Such a representation of DNA sequences was obtained by using One Hot Encoding (OHE). In this way, a categorical sequence was converted into an array of integers in which each nucleotide was represented by four unit vectors: (A = [1000], C = [0100], G = [0010], T = [0001]), e.g. the sequence ACCG was encoded as 1000|0100|0100|0010. Every DNA sequence, represented by a 144-dimensional vector, was fed as an input into both a Kmeans model (using Euclidian distance) and a Ward-method-based (53) hierarchical clustering model (see below).
Defining the mathematical descriptors (features) of the PDRSs
The challenge in the selection of descriptors in the DNA sequences derives from the fact that only a limited number of features that have chemical/physical meaning are useful for model construction and from difficulties in converting DNA sequences into vectors of numbers. Importantly, nucleotides, being categorical variables, cannot be treated in terms of ordinal data. Since ‘hand-crafted’ features extracted from DNA sequences did not improve prediction of primase binding scores (Supplementary Figure S1), we utilized the K-mer method for feature extraction (54). In brief, the K-mer is a frequency vector that counts all possible combinations of short sequences (of size K) in larger DNA sequences. As the K parameter increases, the number of possible combinations increases, while the frequency of each mer decreases (Supplementary Figure S2), giving sparser, yet more detailed, data. Since the K-mer method can be implemented with different normalization and striding techniques, even with insufficient structural information, we used a 1-step stride to allow overlap between mers and we then normalized the extracted K-mer counts.
Extracting features from the DNA sequences of the microarray using the K-mer approach allowed us to find association rules for those DNA sequences (unsupervised algorithms). The sequential features obtained were also used to generate a prediction model of primase–DNA binding, based on primase-binding data collected from PBM experiments (supervised algorithms).
Exploratory data analysis
As is customary, we started with exploratory data analysis, which is unsupervised in nature (i.e. the primase binding scores were ignored). The goal here was to produce a meaningful visualization of the data with the aim to obtain new insights. To this end, we reduced the dimensionality of the data using principal component analysis (PCA), and applied various clustering algorithms, which revealed a meaningful cluster structure with respect to the binding score. After applying PCA to the data, i.e. to the 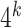 dimensions existing for each 36-mer oligonucleotide, we found that the top three principal components explained 64% of the total variance. Therefore, we concluded that this number of principal components enables the production of a meaningful 3D picture.
dimensions existing for each 36-mer oligonucleotide, we found that the top three principal components explained 64% of the total variance. Therefore, we concluded that this number of principal components enables the production of a meaningful 3D picture.
To interpret the clusters generated using PCA, the binding values obtained for the T7 primase of all the DNA sequences were normalized and used to color code the data points in the clusters (Figure 3A). The most striking result to emerge from the color-coded data was its arrangement into five clusters—one homogeneous cluster of DNA sequences that are strongly bound to T7 primase (colored red in Figure 3A), two homogeneous clusters of DNA sequences with weaker binding to T7 primase (colored blue), and two inhomogeneous clusters with uniformly distributed binding scores. This organization of the data points into meaningful clusters indicates that: (i) there are hidden descriptors within the DNA sequence that are essential for primase binding and (ii) the sequence descriptors obtained by the K-mer approach (Figure 3B) are more than adequate for describing primase–DNA binding.
Figure 3.
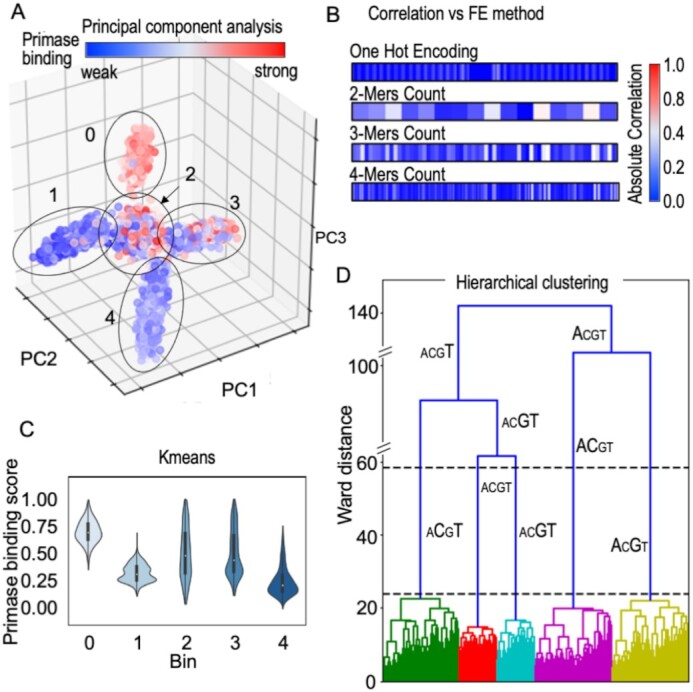
Inference from DNA sequences without labeled responses for T7 primase binding (unsupervised learning). (A) Dimensionality reduction algorithm PCA tri-plot used to visualize the data by selecting the top three principal components. Assignment of the binding scores (labels) to each data point shows an uneven distribution across two clusters (2, 3) and a homogeneous distribution in three clusters (0, for strong binding to T7 primase, and 1 and 4 for weak binding). (B) Correlation between the primase binding score and feature extraction (FE) using different methods: OHE, 2-mer, 3-mer and 4-mer counts. K-mers were used as descriptors for the PCA analysis. (C) Kmeans clustering on one-hot encoded DNA sequences. Clustering was performed by measuring pairwise distances of DNA sequences from the centroid of each cluster. Violin plots representing the distribution of the binding scores assigned to each data point in the clusters are shown. Three clusters show evenly distributed scores (0, 1, 4) and two show a less homogeneous score distribution (2, 3). Each cluster is represented by a ‘mean word’ (centroid): (0) GTTTTGTTTTGTTTTTGTCGTGTGGTTGTGGTGGTA; (1) CTTTTTTTCTTTTTTCGTCCTTTTTTTTTTTCCCCA; (2) GAAGAAAATCCATAGGGTCAACCGGGTTATGTTAAA; (3) CCACAAAAAAAAAAAAGTCCAACCCACAAAACCCC A; (4) GGAAAGGAGAGAAAAAGTCAAAAAAGAAAGAAAGAA. (D) Hierarchical clustering. The x-axis shows DNA sequences emerging into clusters, and the y-axis shows the induced Ward distance of each stage. Letter sizes indicate the letter's frequency in each cluster. The maximal Ward distance gap is indicated between the dashed black lines. The figures were created using the Python package Seaborn and Matplotlib.
Using the Kmeans algorithm, we were able to shed light on the distributions of the binding scores within the five clusters. Interestingly, preprocessing using OHE, converge into clusters containing similar score distributions to the five-cluster structure obtained by PCA. The Kmeans iterative algorithm partitions the data space into sub-spaces, thereby assigning a matching label (the cluster number) to each instance according to its location.
Clustering of the unlabeled DNA sequences in Kmeans relied on the sequence distances from the corresponding cluster centroids. As each sequence was represented using OHE, the distance between two sequences could be described as the number of changes needed in one sequence to convert it into the other sequence. In the Kmeans analysis, aligning the PBM-driven binding score for each DNA sequence revealed that each cluster exhibited a clear trend, as the group of binding scores to the primase was distributed unevenly with long tails (Figure 3C), which means that each cluster held exceptions.
While Kmeans computes distances of instances from the centroids, clustering of unlabeled DNA sequences using Ward's minimum variance method (53) allowed us to track of the evolution of clusters (Figure 3D). The maximal Ward distance (WD) gap was obtained using five clusters, as observed from PCA and Kmeans for the same dataset of DNA sequences. Furthermore, we can see in Figure 3D that each colored branch holds a sub-group with two highly repeating letters (CT, GT, AC, AG) or a uniform distribution of the letters (ACGT).
Predicting the binding score
After clustering the DNA sequences in the microarray into groups with common features, the next step was to predict the outcome of T7 primase–DNA binding for a given DNA sequence. To increase the accuracy, each cluster was fitted separately. We used GTC-containing DNA sequences and their corresponding PBM-driven binding scores as input and output pairs for the training set, respectively. The PBM-driven data comprises the continuous numerical binding values for DNA sequences, i.e. the type of data that regression models are aimed to solve.
We extracted sequence-based features (SBF) inspired by pseudo K-tuple nucleotide composition (55). We modified the ordinary method for SBF extraction by neglecting locality-based features, since distance was interpreted as the number of nucleotides between a mer and its closest 5′-GTC start site. Our modified method for feature extraction provided us with sequence-wise normalized K-mers (see Materials and Methods for normalization of the K-mers). We then tested several regression algorithms using criteria that can differentiate between uninformative and highly informative SBFs (Supplementary Table S1). Thereafter, we applied the L1 regularized linear regression [least absolute shrinkage and selection operator (Lasso) (56)] model on each bin separately with the aim to emphasize meaningful mers and to prevent overfitting of the model. (Lasso's output is a closed form expression that is generated under fewer, yet more meaningful, coefficients by applying a penalty for each variable.) In addition, we extracted an expected performance measure separately for each bin obtained by Kmeans, as the mean absolute errors (MAE, an error estimate for the regression) for our results were uneven across bins (Table 1). As we trained five Lasso models on five bins, each bin's prediction was obtained using a different set of coefficients. While the overall performance of the prediction model is obvious, examining the performance for each bin is more precise. Lasso's performance differed across bins, where bins 0 and 1 each generated an error of about 15%, and bins 2, 3 and 4 generated a relatively small error of 6% (Table 1). We found that pre-treating the data using Kmeans decreased the prediction error by approximately 10% (Table 2).
Table 1.
Results summary of 5-Fold-MCCV* in each clusters
| Bin | MAE* | STD | Mean MAE (%) | Mean STD (%) | Bin weight (% of instances from data) |
|---|---|---|---|---|---|
| 0 | 0.168 | 0.005 | 16.8 | 0.5 | 15.0 |
| 1 | 0.138 | 0.007 | 13.8 | 0.7 | 19.4 |
| 2 | 0.067 | 0.004 | 6.7 | 0.4 | 13.3 |
| 3 | 0.060 | 0.003 | 6.0 | 0.3 | 25.3 |
| 4 | 0.077 | 0.003 | 7.0 | 0.3 | 26.8 |
*MAE, mean absolute error; MCCV, Monte Carlo cross-validation.
Table 2.
Effect of K-mers on model prediction
| MAE* | K = 1 | K = 2 | K = 3 | K = 4 |
|---|---|---|---|---|
| BNS = 1 | 0.171 | 0.112 | 0.103 | 0.084 |
| BNS = 5 | 0.110 | 0.102 | 0.093 | 0.079 |
Ratio 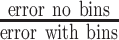
|
1.55 | 1.09 | 1.10 | 1.06 |
Decrease in error in % 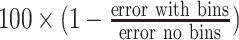
|
35% | 9% | 10% | 6% |
*MAE, mean absolute error.
To force all the coefficients of the Lasso model to be positive for every K-mer feature, we extracted the largest 10 coefficients of each bin and investigated the effect of the K-mer features on the model. The trained models were cross validated on 5-fold of the training dataset and tested on a small test-set taken from the PBM results; the set consisted of 16 sequences, divided into two groups with significantly different primase binding signals (Figure 4A), namely, eight sequences that showed weak binding to primase, and eight sequences with strong binding. Given the training data distribution, our models predicted binding of primase with any GTC-containing sequence, with an MAE of <12% (Figure 4).
Figure 4.
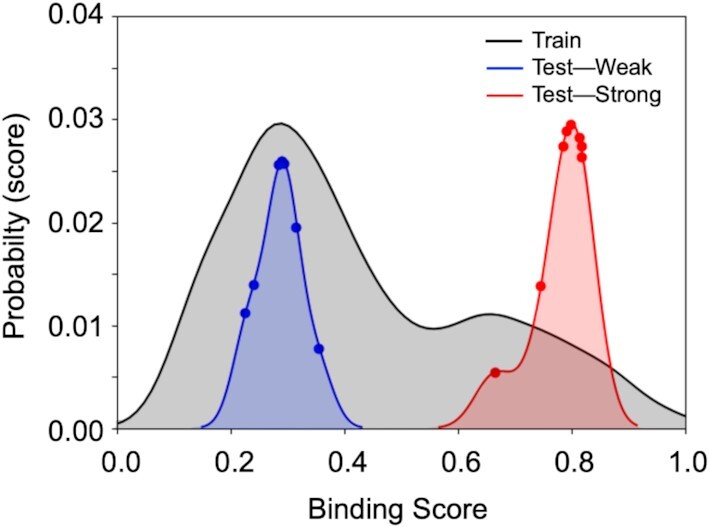
Results for linear regression with L1 regularization (Lasso). After cleaning, the training set contained 3150 instances (DNA sequences), whereas the test set contained 16 instances. Prediction of scores by using the regression model was performed on 16 DNA sequences with known scores, eight of which showed weak binding to T7 primase (blue graph) and eight showed strong binding to T7 primase (red graph). In accordance with the training-set double distribution (black graph), the predicted binding of the two test groups are distributed in weak and strong binding scores areas, respectively. Although the probability of finding DNA sequences with strong binding to primase is low, the model accurately predicted all DNA sequences that belong to the strong binding group. DNA sequences and their empirical and predicted scores are presented in Supplementary Table S2.
To identify the most influential nucleotides for accurate prediction of a binding score for a given PDRS, we used a different perspective on the data. Each nucleotide position before and after the GTC sequence was regarded as a feature and trained against gradient boosting machine (GBM) and random forest models. GBM and random forest models can handle categorical data, such as DNA sequences, and can easily explore the importance of each feature for the model. The results indicate that nucleotides adjacent to the GTC sequence are the most essential for primase binding, whereas distant nucleotides are much less influential (Supplementary Figure S3).
Biochemical validation
On the genome, the initial step of sequence specific (PDRS) binding is followed by synthesis of a dinucleotide (5′-AC-3′), which is then extended into a functional primer by DNA primase. It has previously been shown that A/G-containing sequences that flank the specific recognition site increase primase–DNA binding affinity in comparison to T/G-containing sequences (14). Since binding to DNA is a pre-requisite for primase activity, the strength of binding affects the catalytic activity of the primase and the yield of the RNA product. We used qualitative biochemical assays to experimentally validate the prediction model described above. The validation provided insights into the features embedded in the DNA sequences that are important for the binding and catalytic activity of DNA primase.
Correlation between prediction of primase binding to PDRS and catalytic activity. The eight sequences with strong binding and the eight with weak binding to primase, which were used as the test set in the supervised learning part of this study (Figure 4), yielded RNA primers, as was expected from their PBM-driven binding values. Longer RNA primers were formed on DNA templates that were predicted to have higher binding affinity to the T7 primase (Supplementary Figure S4). This finding shows, for the first time, that the sequence descriptors embedded in the DNA sequence are sufficient to predict binding scores and that prediction of a binding sequence correlates well (96.9% Pearson correlation coefficient) with the formation of RNA primers (Supplementary Figure S4). The understanding of how sequential features embedded in the DNA are related to the binding of primase allows us not only to predict binding scores of a given PDRS but also to design novel PDRSs that yield high primase binding scores.
Exhaustive search for flanking sequences that yield novel PDRSs. Features important for DNA–primase binding were used in formulating design principles to generate novel GTC-containing DNA sequences with desired binding scores. Assuming that the DNA sequences originate from five different clusters (Figure 3) that require five different models, we generated two types of DNA sequence, as follows: (i) We selected DNA sequences from two homogeneous clusters of primase binding scores and exhaustively altered the non-‘GTC’ nucleotides to generate primase recognition DNA sequences (new PDRSs). Three altered sequences that did not exist in the training set and yielded the 10th, 50th and 90th percentile binding strengths were selected from the two clusters (clusters 0 and 4, Figure 3) for further biochemical evaluation. (ii) DNA sequences were generated in the same way, and two novel DNA sequences from each Kmean cluster, one that represented the strongest binding prediction and the other that represented weakest, were selected (overall 10 sequences). DNA sequences and their predicted binding scores are presented in Supplementary Table S3.
To characterize the effect of the DNA sequences (PDRSs) generated as described above, we quantified and compared RNA primer formation by T7 primase, where the generated PDRSs were used as templates for the synthesis (the complete list of the DNA sequences is presented in Supplementary Table S3). Specifically, we used [γ–32P]ATP to 5′ end-label the RNA primers, which ensured that each primer was labeled only once, and thus the intensity of the gel bands is proportional to the absolute amounts of the synthesized RNA primers. We found that the DNA sequences with higher binding scores for T7 primase showed improved RNA primer synthesis activity for each cluster (Figure 5). Moreover, the overall difference in the binding scores between the two clusters, 0 and 4, remained proportional to the amounts of RNA primers synthesized by the primase as evident from the intensities of the gel bands (Figure 5). Thus, larger amounts of RNA primers were synthesized against DNA templates of cluster 0 in comparison to the amounts of RNA primers synthesized by T7 primase if DNA templates from cluster 4 were used (Figure 5B). For the 10 DNA templates that represented weak/strong binding to primase from each of the five clusters (Figure 3), we found that the newly designed DNA sequence flanking the 5′-GTC-3′ sequence with higher binding scores for T7 primase showed improved RNA primer synthesis activity, as was to be expected (Supplementary Figure S5B).
Figure 5.
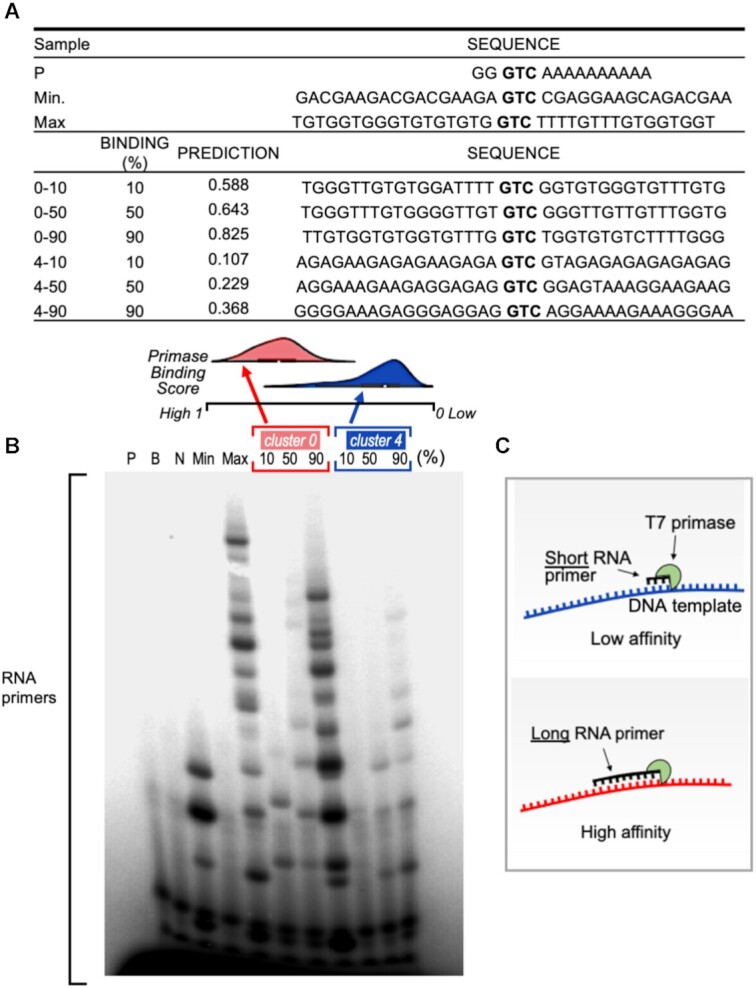
RNA primer synthesis catalyzed by the T7 primase on computer generated GTC-containing DNA templates. (A) Table summarizes the DNA template sequences used for the biochemical validation and their corresponded values. Three DNA sequences from each of the Kmeans clusters #0 and #4 that predicted the 10th, 50th and 90th percentile binding scores were selected in each cluster. (B) Top: Distribution of binding values for the two clusters. Note that cluster #0 shows stronger primase binding values, on average, than cluster #4. Bottom: Oligoribonucleotide synthesis by T7 primase. The standard reaction mixture contained oligonucleotides with the primase recognition sequence, a control oligonucleotide 5′-GGGTCA10-3′, and [γ-32P]ATP, CTP, GTP and UTP. After incubation, the radioactive products were analyzed by electrophoresis on a 25% polyacrylamide gel containing 7 M urea, and visualized using autoradiography. The pattern of primase activity remains identical when using the full-length helicase-primase (gene 4 protein, gp4) of bacteriophage T7 (Supplementary Figure S5). (C) illustration of the effect of primase-DNA binding affinity on the size of RNA primers.
The idea that a sequence impacts the shape of double-stranded DNA and, as a result, the specific binding of proteins has been studied extensively (57). However, it should be remembered that the DNA primase acts on single stranded DNA after unwinding of the double stranded by the DNA helicase. In our bacteriophage T7 system, both enzymes, the helicase and the primase, reside on the same polypeptide. Therefore, unwinding of the double-stranded DNA by the helicase is followed immediately by the action of the primase, which scans the newly formed single-stranded DNA for a recognition sequence (illustrated in Figure 1). Since the primase domain lags just slightly behind the helicase domain of gp4, we expect that folding of the single-stranded DNA will be negligible when the full length helicase-primase is used. In the microarray, due to limiting reaction conditions and the size of the tethered DNA oligonucleotides, local folding of the DNA may occur and affect primase–DNA binding (and activity). The binding values for all the DNA oligonucleotides used for the biochemical assays were compared to their corresponding propensity for secondary structure formation (Supplementary Table S3). Among the 19 DNA oligonucleotides, only two were predicted to form a stable secondary structure, and two were predicted to have weak folding propensity. The remaining 15 sequences were predicted to be unfolded. Neither of the two templates which showed likelihood to form secondary structures (Supplementary Table S3, cluster 2 a,b) showed RNA primer formation (Supplementary Figure S5), despite moderate predicted binding scores (Supplementary Table S2). Formation of secondary structures by the DNA templates may have an effect on primase binding and activity, however, no correlation was found between secondary structure formation and predicted binding scores.
Previous studies proposed that the helicase may affect the specific binding of the DNA primase (58–60). To examine the effect of the helicase on primase binding and activity, we overexpressed and purified the 63-kDa full-length gp4 that contains both the helicase and the primase domains. Comparison between the full-length gp4 and the T7 primase domain showed no significant difference in activities and identical patterns of RNA primer formation (Supplementary Figures S5 and S6).
These results confirm our machine learning prediction model and indicate that higher binding affinity for PDRS recognition sequences is dictated by features embedded in the DNA sequence. Having completed the exhaustive search for flanking sequences that yield novel primase-DNA binding sequences described here, we are now in a position to design DNA templates that yield: (i) larger amounts of RNA primers and (ii) longer RNA primers that can serve as functional primers for T7 DNA polymerase (61). Both the length and the quantity of the RNA primers are likely to be essential for the ‘decision’ to start Okazaki fragments by DNA polymerase on the lagging DNA strand.
DISCUSSION
On the basis of PBM data for primase binding previously obtained for >150000 DNA sequences, this study set out to: (i) develop the means to predict the binding score of T7 primase for a given DNA sequence, (ii) describe the DNA sequence features essential for binding of the enzyme and (iii) generate novel sequences with a high propensity for T7 primase binding. The K-mer approach for feature selection in the DNA sequential data that was used here appears to cover all possible combinatorial pieces of information hidden in the DNA sequences and serves as an efficient strategy for feature extraction. The K-mer method, which simply counts explicit combinations of nucleotides in a DNA sequence, was superior to other accepted methods of ‘hand-crafted’ feature extraction from DNA sequences. Features obtained by the K-mer method clearly bear the DNA properties that are important for primase binding, as demonstrated by the unsupervised analysis in which clustered groups coincided with experimental binding scores. These DNA properties enable the formation of intermolecular forces (van der Waals, hydrogen, electrostatic and steric interactions) with the zinc-binding-motif of the primase important for specific DNA sequence recognition.
Although this study focused on DNA sequence recognition by T7 primase, the findings may well have bearing on rules hidden in DNA sequences that are crucial for other specific DNA-protein interactions. These findings thus contribute to our understanding of how DNA primase selects Okazaki fragments start sites on the genome and why only some of the possible priming sites initiate Okazaki fragments during DNA replication, while others do not, resulting in Okazaki fragments with a larger-than-expected average length. The implications of this study are that design principles for any DNA sequence with a desired binding affinity to T7 primase can indeed be generated computationally. Furthermore, PDRSs can be designed to yield an RNA primer with a particular content. In conclusion, state-of-the-art carefully selected learning methods, such as those used here, have enormous analytical potential for predicting specific protein-DNA interactions, but require large amounts of data, a requirement than can indeed be met by using PBMs. Our results for T7 DNA primase as a model system can be generalized to other primases, with improved sensitivity and specificity to their DNA recognition sequence.
DATA AVAILABILITY
All datasets and python codes for algorithms used for preprocessing and analysis are available in the GitHub repository (https://github.com/csbarak/T7pdrs).
Supplementary Material
Contributor Information
Adam Soffer, Department of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel; Data Science Research Center, Ben-Gurion University of the Negev, Beer-Sheva, Israel; School of Computer and Electrical Engineering, Ben-Gurion University of the Negev, Beer-Sheva, Israel.
Sarah A Eisdorfer, Department of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel.
Morya Ifrach, Department of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel.
Stefan Ilic, Department of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel.
Ariel Afek, Department of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel.
Hallel Schussheim, Department of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel.
Dan Vilenchik, Data Science Research Center, Ben-Gurion University of the Negev, Beer-Sheva, Israel; School of Computer and Electrical Engineering, Ben-Gurion University of the Negev, Beer-Sheva, Israel.
Barak Akabayov, Department of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel; Data Science Research Center, Ben-Gurion University of the Negev, Beer-Sheva, Israel.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Israel Science Foundation (ISF) [1023/18]. Funding for open access charge: Israel Science Foundation [1023/18].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lodish H.F., Berk A., Zipursky S.L., Matsudaira P., Baltimore D., Darnell J.. 2000; 4th ednNY: W. H. Freeman; Molecular Cell Biology. [Google Scholar]
- 2. Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E.. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008; 132:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giresi P.G., Kim J., McDaniell R.M., Iyer V.R., Lieb J.D.. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007; 17:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hesselberth J.R., Chen X., Zhang Z., Sabo P.J., Sandstrom R., Reynolds A.P., Thurman R.E., Neph S., Kuehn M.S., Noble W.S.. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods. 2009; 6:283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson D.S., Mortazavi A., Myers R.M., Wold B.. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007; 316:1497–1502. [DOI] [PubMed] [Google Scholar]
- 6. Ren B., Robert F., Wyrick J.J., Aparicio O., Jennings E.G., Simon I., Zeitlinger J., Schreiber J., Hannett N., Kanin E.. Genome-wide location and function of DNA binding proteins. Science. 2000; 290:2306–2309. [DOI] [PubMed] [Google Scholar]
- 7. Rhee H.S., Pugh B.F.. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011; 147:1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Badis G., Berger M.F., Philippakis A.A., Talukder S., Gehrke A.R., Jaeger S.A., Chan E.T., Metzler G., Vedenko A., Chen X.. Diversity and complexity in DNA recognition by transcription factors. Science. 2009; 324:1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berger M.F., Philippakis A.A., Qureshi A.M., He F.S., Estep P.W., Bulyk M.L.. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 2006; 24:1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fordyce P.M., Gerber D., Tran D., Zheng J., Li H., DeRisi J.L., Quake S.R.. De novo identification and biophysical characterization of transcription-factor binding sites with microfluidic affinity analysis. Nat. Biotechnol. 2010; 28:970–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G.. DNA-binding specificities of human transcription factors. Cell. 2013; 152:327–339. [DOI] [PubMed] [Google Scholar]
- 12. Maerkl S.J., Quake S.R.. A systems approach to measuring the binding energy landscapes of transcription factors. Science. 2007; 315:233–237. [DOI] [PubMed] [Google Scholar]
- 13. Noyes M.B., Christensen R.G., Wakabayashi A., Stormo G.D., Brodsky M.H., Wolfe S.A.. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell. 2008; 133:1277–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Riley T.R., Slattery M., Abe N., Rastogi C., Liu D., Mann R.S., Bussemaker H.J.. SELEX-seq: a method for characterizing the complete repertoire of binding site preferences for transcription factor complexes. Methods Mol. Biol. 2014; 1196:255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Warren C.L., Kratochvil N.C., Hauschild K.E., Foister S., Brezinski M.L., Dervan P.B., Phillips G.N., Ansari A.Z.. Defining the sequence-recognition profile of DNA-binding molecules. PNAS. 2006; 103:867–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K.. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014; 158:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zykovich A., Korf I., Segal D.J.. Bind-n-Seq: high-throughput analysis of in vitro protein–DNA interactions using massively parallel sequencing. Nucleic Acids Res. 2009; gkp802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Birney E., Stamatoyannopoulos J.A., Dutta A., Guigó R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E.. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007; 447:799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carlson C.D., Warren C.L., Hauschild K.E., Ozers M.S., Qadir N., Bhimsaria D., Lee Y., Cerrina F., Ansari A.Z.. Specificity landscapes of DNA binding molecules elucidate biological function. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:4544–4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. ENCODE Project Consortium The ENCODE (ENCyclopedia of DNA elements) project. Science. 2004; 306:636–640. [DOI] [PubMed] [Google Scholar]
- 21. Hume M.A., Barrera L.A., Gisselbrecht S.S., Bulyk M.L.. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein–DNA interactions. Nucleic Acids Res. 2014; 43:D117–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K., Hannett N.M., Harbison C.T., Thompson C.M., Simon I.. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002; 298:799–804. [DOI] [PubMed] [Google Scholar]
- 23. Rohs R., West S.M., Sosinsky A., Liu P., Mann R.S., Honig B.. The role of DNA shape in protein–DNA recognition. Nature. 2009; 461:1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roy S., Ernst J., Kharchenko P.V., Kheradpour P., Negre N., Eaton M.L., Landolin J.M., Bristow C.A., Ma L., Lin M.F.. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010; 330:1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao Y., Stormo G.D.. Quantitative analysis demonstrates most transcription factors require only simple models of specificity. Nat. Biotechnol. 2011; 29:480–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mooney R.A., Davis S.E., Peters J.M., Rowland J.L., Ansari A.Z., Landick R.. Regulator trafficking on bacterial transcription units in vivo. Mol. Cell. 2009; 33:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Venters B.J., Pugh B.F.. Genomic organization of human transcription initiation complexes. Nature. 2013; 502:53–58. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28. Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Max K.E., Zeeb M., Bienert R., Balbach J., Heinemann U.. Common mode of DNA binding to cold shock domains. Crystal structure of hexathymidine bound to the domain-swapped form of a major cold shock protein from Bacillus caldolyticus. FEBS J. 2007; 274:1265–1279. [DOI] [PubMed] [Google Scholar]
- 30. Kornberg A., Baker T.A.. DNA Replication. 2005; 2nd ednSausalito, Calif: University Science Books. [Google Scholar]
- 31. Frick D.N., Richardson C.C.. DNA primases. Annu. Rev. Biochem. 2001; 70:39–80. [DOI] [PubMed] [Google Scholar]
- 32. Stratling W., Knippers R.. Function and purification of gene 4 protein of phage T7. Nature. 1973; 245:195–197. [DOI] [PubMed] [Google Scholar]
- 33. Wolfson J., Dressler D. Regions of single-stranded DNA in the growing points of replicating bacteriophage T7 chromosomes. PNAS. 1972; 69:2682–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tabor S., Richardson C.C.. Template recognition sequence for RNA primer synthesis by gene 4 protein of bacteriophage T7. PNAS. 1981; 78:205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Richardson C.C., Romano L.J., Kolodner R., LeClerc J.E., Tamanoi F., Engler M.J., Dean F.B., Richardson D.S.. Replication of bacteriophage T7 DNA by purified proteins. Cold Spring Harb. Symp. Quant. Biol. 1979; 43:427–440. [DOI] [PubMed] [Google Scholar]
- 36. Lee S.J., Zhu B., Hamdan S.M., Richardson C.C.. Mechanism of sequence-specific template binding by the DNA primase of bacteriophage T7. Nucleic Acids Res. 2010; 38:4372–4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Corn J.E., Pease P.J., Hura G.L., Berger J.M.. Crosstalk between primase subunits can act to regulate primer synthesis in trans. Mol. Cell. 2005; 20:391–401. [DOI] [PubMed] [Google Scholar]
- 38. Corn J.E., Pelton J.G., Berger J.M.. Identification of a DNA primase template tracking site redefines the geometry of primer synthesis. Nat. Struct. Mol. Biol. 2008; 15:163–169. [DOI] [PubMed] [Google Scholar]
- 39. Andrilenas K.K., Penvose A., Siggers T.. Using protein-binding microarrays to study transcription factor specificity: homologs, isoforms and complexes. Brief. Funct. Genomics. 2015; 14:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soultanas P. The bacterial helicase-primase interaction: a common structural/functional module. Structure. 2005; 13:839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thirlway J., Soultanas P.. In the Bacillus stearothermophilus DnaB-DnaG complex, the activities of the two proteins are modulated by distinct but overlapping networks of residues. J. Bacteriol. 2006; 188:1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Naue N., Beerbaum M., Bogutzki A., Schmieder P., Curth U.. The helicase-binding domain of Escherichia coli DnaG primase interacts with the highly conserved C-terminal region of single-stranded DNA-binding protein. Nucleic Acids Res. 2013; 41:4507–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chintakayala K., Larson M.A., Grainger W.H., Scott D.J., Griep M.A., Hinrichs S.H., Soultanas P.. Domain swapping reveals that the C- and N-terminal domains of DnaG and DnaB, respectively, are functional homologues. Mol. Microbiol. 2007; 63:1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhu B., Lee S.J., Richardson C.C.. Direct role for the RNA polymerase domain of T7 primase in primer delivery. PNAS. 2010; 107:9099–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kusakabe T., Richardson C.C.. Template recognition and ribonucleotide specificity of the DNA primase of bacteriophage T7. J. Biol. Chem. 1997; 272:5943–5951. [DOI] [PubMed] [Google Scholar]
- 46. Mendelman L.V., Richardson C.C.. Requirements for primer synthesis by bacteriophage T7 63-kDa gene 4 protein. Roles of template sequence and T7 56-kDa gene 4 protein. J. Biol. Chem. 1991; 266:23240–23250. [PubMed] [Google Scholar]
- 47. Afek A., Ilic S., Horton J., Lukatsky D.B., Gordan R., Akabayov B.. DNA sequence context controls the binding and processivity of the T7 DNA primase. iScience. 2018; 2:141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ilic S., Cohen S., Afek A., Gordan R., Lukatsky D.B., Akabayov B.. DNA sequence recognition by DNA primase using high-throughput primase profiling. J. Vis. Exp. 2019; 152:e59737. [DOI] [PubMed] [Google Scholar]
- 49. Lee S.J., Richardson C.C.. Essential lysine residues in the RNA polymerase domain of the gene 4 primase-helicase of bacteriophage T7. J. Biol. Chem. 2001; 276:49419–49426. [DOI] [PubMed] [Google Scholar]
- 50. Frick D.N., Baradaran K., Richardson C.C.. An N-terminal fragment of the gene 4 helicase/primase of bacteriophage T7 retains primase activity in the absence of helicase activity. PNAS. 1998; 95:7957–7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss O., Dubourg V.et al.. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 52. Brown C.E. Coefficient of Variation. 1998; Berlin, Heidelberg: Springer. [Google Scholar]
- 53. Ward J.H.J. Hierarchical grouping to optimize an objective function. J Am Stat Assoc. 1963; 58:236–244. [Google Scholar]
- 54. Chen W., Lei T.Y., Jin D.C., Lin H., Chou K.C.. PseKNC: a flexible web server for generating pseudo K-tuple nucleotide composition. Anal Biochem. 2014; 456:53–60. [DOI] [PubMed] [Google Scholar]
- 55. Dao F.Y., Lv H., Wang F., Ding H.. Recent advances on the machine learning methods in identifying DNA replication origins in eukaryotic genomics. Front. Genet. 2018; 9:613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tibshirani R. Regression shrinkage and selection via the lasso. J. Roy. Statist. Soc. Ser. A. 1996; 58:267–288. [Google Scholar]
- 57. Abe N., Dror I., Yang L., Slattery M., Zhou T., Bussemaker H.J., Rohs R., Mann R.S.. Deconvolving the recognition of DNA shape from sequence. Cell. 2015; 161:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van Eijk E., Paschalis V., Green M., Friggen A.H., Larson M.A., Spriggs K., Briggs G.S., Soultanas P., Smits W.K.. Primase is required for helicase activity and helicase alters the specificity of primase in the enteropathogen Clostridium difficile. Open Biol. 2016; 6:160272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Johnson S.K., Bhattacharyya S., Griep M.A.. DnaB helicase stimulates primer synthesis activity on short oligonucleotide templates. Biochemistry. 2000; 39:736–744. [DOI] [PubMed] [Google Scholar]
- 60. Tougu K., Marians K.J.. The interaction between helicase and primase sets the replication fork clock. J. Biol. Chem. 1996; 271:21398–21405. [DOI] [PubMed] [Google Scholar]
- 61. Romano L.J., Richardson C.C.. Characterization of the ribonucleic acid primers and the deoxyribonucleic acid product synthesized by the DNA polymerase and gene 4 protein of bacteriophage T7. J. Biol. Chem. 1979; 254:10483–10489. [PubMed] [Google Scholar]
- 62. Balakrishnan L., Bambara R.A.. Okazaki fragment metabolism. Cold Spring Harb. Perspect. Biol. 2013; 5:a010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets and python codes for algorithms used for preprocessing and analysis are available in the GitHub repository (https://github.com/csbarak/T7pdrs).