

Summary
G protein-coupled receptor (GPCR) kinases (GRKs) selectively phosphorylate activated GPCRs, priming them for desensitization1. How GRKs recognize these receptors is debated2–4, but a conserved region at their N-termini is essential for this process5–8. Herein we report a series of cryo-EM single particle reconstructions of light activated rhodopsin (Rho*) bound to rhodopsin kinase (GRK1), wherein the N-terminus of GRK1 forms a helix that docks into the open cytoplasmic cleft of Rho*. The helix also packs against the GRK1 kinase domain, stabilizing it in an active configuration. The complex is further stabilized by electrostatic interactions between basic residues conserved in most GPCRs and acidic residues conserved in GRKs. Density for the regulator of G protein signaling homology domain of GRK1 and the C-terminus of rhodopsin are not observed. Crosslinking with mass spectrometry analysis confirms these results and helps explain how the docking of GRKs with activated GPCRs allows for phosphorylation of multiple sites. We identify GRK1 residues whose mutation augments kinase activity and crosslinking with Rho*, as well as those involved in activation by acidic phospholipids. From these data, we present a general model for how a small family of protein kinases can recognize and be activated by hundreds of different GPCRs.
The activation of G protein-coupled receptors (GPCRs) instigates intracellular signaling cascades that modulate a vast array of cellular processes9. Desensitization of these GPCRs is triggered by a small family of GPCR kinases (GRKs), which selectively phosphorylate either the third cytoplasmic loop or the C-terminal tail of activated receptors, triggering the recruitment of arrestins and, typically, clathrin-mediated endocytosis1. Because maladaptive overexpression of GRKs exacerbates conditions such as heart failure and cancer, GRKs have emerged as potentially important therapeutic targets10,11.
The seven vertebrate GRKs (GRK1–7) belong to the AGC kinase family and as such contain a protein kinase domain followed by a C-terminal extension (C-tail) (Fig. 1a). Located centrally within the C-tail is the active site tether (AST), a loop that contributes to the ATP binding site12. The GRK kinase domain is inserted into a loop of a regulator of G protein signaling homology (RH) domain, a bi-lobed helical bundle that bridges the small and large lobes of the kinase domain13. GRKs also feature a highly conserved ~16 residue element at their extreme N-termini. Truncation, antibody blockade, or mutation of this element can result in complete loss of activity towards receptor substrates (Extended Data Table 1)5–8. However, the mechanism by which GRKs recognize and are in turn activated by GPCRs is unresolved2. Recent technical advances in cryo-EM have greatly facilitated structure determinations of GPCRs in complex with heterotrimeric G proteins and arrestins14–18, which also selectively recognize activated GPCRs. Unfortunately, analogous GPCR–GRK structures are currently lacking, likely because such complexes are transient, anionic lipid dependent, and of low to mid μM affinity19,20.
Figure 1. Trapping an activation-dependent complex between rhodopsin and GRK1.
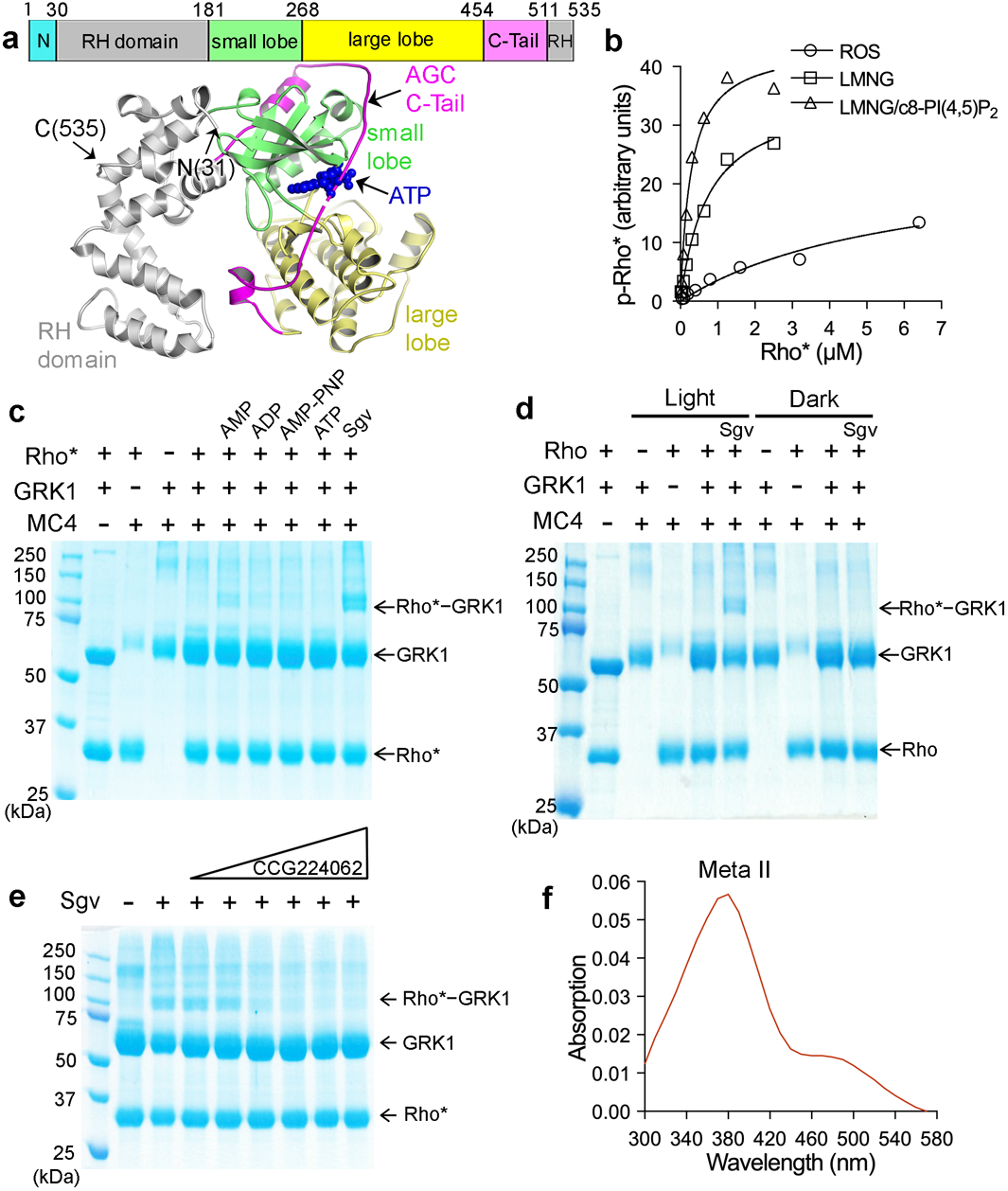
a) Primary structure of C-terminally truncated bovine GRK1 (residues 1–535) used in this study and its crystal structure in complex with ATP (PDB entry 3C4W45), wherein the N-terminus is disordered and the AST loop is partially ordered. b) Michaelis-Menten kinetics curves from a representative experiment out of three repeats with Rho* as substrate. Rhodopsin was solubilized with LMNG or LMNG/c8-PI(4,5)P2 and its kinetics was compared with rhodopsin in ROS. c) Crosslinking between Rho* and GRK1 in the presence of either 1 mM AMP, ADP, AMP-PNP, ATP, or 0.8 mM Sgv, an adenosine analog that helps stabilize the active conformation of GRKs. d) Light dependence of the crosslinking reaction. e) Crosslinking in the presence of Sgv was progressively inhibited by increasing amounts (10–630 μM) of the GRK inhibitor CCG224062 (IC50=0.1 nM)27, which stabilizes an inactive kinase domain conformation. For gel source data for c-e, see Supplementary Fig. 10a–c. f) The purified, crosslinked Rho*–GRK1 complex showed an absorption peak at 380 nm, consistent with Rho* stabilized in its Meta ll state.
Rhodopsin and rhodopsin kinase (GRK1)21,22 have co-evolved since the emergence of vertebrates in the rod outer segments (ROS) of photoreceptors to regulate scotopic vision, and as such represent a highly optimized system to study the GPCR–GRK interaction. Herein we report four cryo-EM structures of the bovine Rho*–GRK1 complex ranging from 7 – 4 Å resolution, crosslinking with mass spectroscopy (CLMS) analysis, and functional studies that reveal the molecular mechanism by which GRKs recognize activated GPCRs and a rare glimpse of a protein kinase being allosterically activated by its physiological substrate.
Isolation of the Rho*–GRK1 complex.
In initial attempts, we determined that the Km of GRK1 for Rho* in LMNG micelles decreased 6-fold relative to native ROS membranes (Km > 5.8 μM), and that addition of 5% c8-PI(4,5)P2 further decreased the Km by 2-fold, to 0.33 μM. In contrast, the Km for ATP remained constant (Fig. 1b and Extended Data Fig. 1a). We also documented a significant increase in the activity of GRK1 against Rho* when incorporated into nanodiscs containing negatively charged lipids (Extended Data Fig.1b–c), and then identified a cluster of basic residues in the GRK1 α0 helix, which bridges the small lobe and RH domain, that are involved in this activation (Supplementary Discussion and Extended Data Fig.1c–e). Despite these improvements in apparent affinity, an intact complex still could not be purified. Treating Rho* reconstituted in c8-PI(4,5)P2/LMNG micelles with the amine crosslinker DC423 or MC424 trapped a super-shifted species of ~100 kDa, consistent with the anticipated 1:1 Rho* (40 kDa):GRK1 (61 kDa) complex25 (Fig. 1c). Crosslinking was most efficient in presence of sangivamycin (Sgv), an adenosine analog with 180 nM Ki against GRK126 and that previously was used to trap GRK6 in what was proposed to be an active configuration6. Dark-adapted rhodopsin failed to crosslink (Fig. 1d) and an active site directed small molecule inhibitor (CCG224062) that stabilizes GRKs in an inactive state27 competed with crosslinking in a dose-dependent manner (Fig. 1e). The absorption spectrum of the purified complex also confirmed the presence of Meta II rhodopsin (Fig. 1f). Thus, the isolated Rho*–GRK1 complex was dependent on the activated state of both GPCR and GRK. Crosslinking did not strongly depend on the phosphorylation status of Rho* or whether its C-terminal tail was truncated (Supplementary Fig. 1).
Cryo-EM Analysis.
This purified complex (Supplementary Fig. 2) was vitrified for cryo-EM analysis, yielding a 7 Å severely anisotropic reconstruction (Fig. 2a, Extended Data Fig. 2, Extended Data Table 2). Interpretation of this map (Supplementary Discussion) suggested that affinity could be improved if GRK1 residues Ser5, Ser488, and Thr489, which are endogenous sites of autophosphorylation, were substituted with glutamic acids (GRK1S5E/EE). This variant exhibited a decreased Km for Rho*, enhanced crosslinking yield, and yielded an improved 5.8 Å reconstruction (Fig. 2b, Extended Data Fig. 3, Extended Data Table 2). Two Fabs (Fab1 and Fab6), selected against Sgv-bound GRK1, were then added to the Rho*–GRK1S5E/EE complex to increase particle size (by 50 kDa) and aid in particle alignment. These Fabs bind GRK1 with a low nanomolar affinity (KD ~ 2.5 nM) but have no effect on kinase activity towards Rho* (Supplementary Fig. 3). Imaging the Fab1 and Fab6 complexes yielded ~4 Å resolution cryo-EM reconstructions (Fig. 2c–d, Extended Data Figs. 4, 5, Extended Data Table 2, Supplementary Video 1, 2). A high degree of dynamic behavior and preferred orientation were however still evident in the data. When GRK1 in the Fab6 complex is superimposed with GRK1 in the other three structures, Rho* differs in orientation by ~5.5° relative to the small lobe (Supplementary Fig. 4). It is unclear how Fab6 stabilizes this unique state. Density for Rho* is superior in the Fab1 complex, whereas density for intracellular loop 1 (ICL1) and Helix 8 (H8) of Rho* are weak in the Fab6 complex, but the overall density for GRK1 is superior.
Figure 2. Cryo-EM single particle reconstructions reveal the prominent role of the GRK1 αN helix and AST in forming the interface with Rho*.
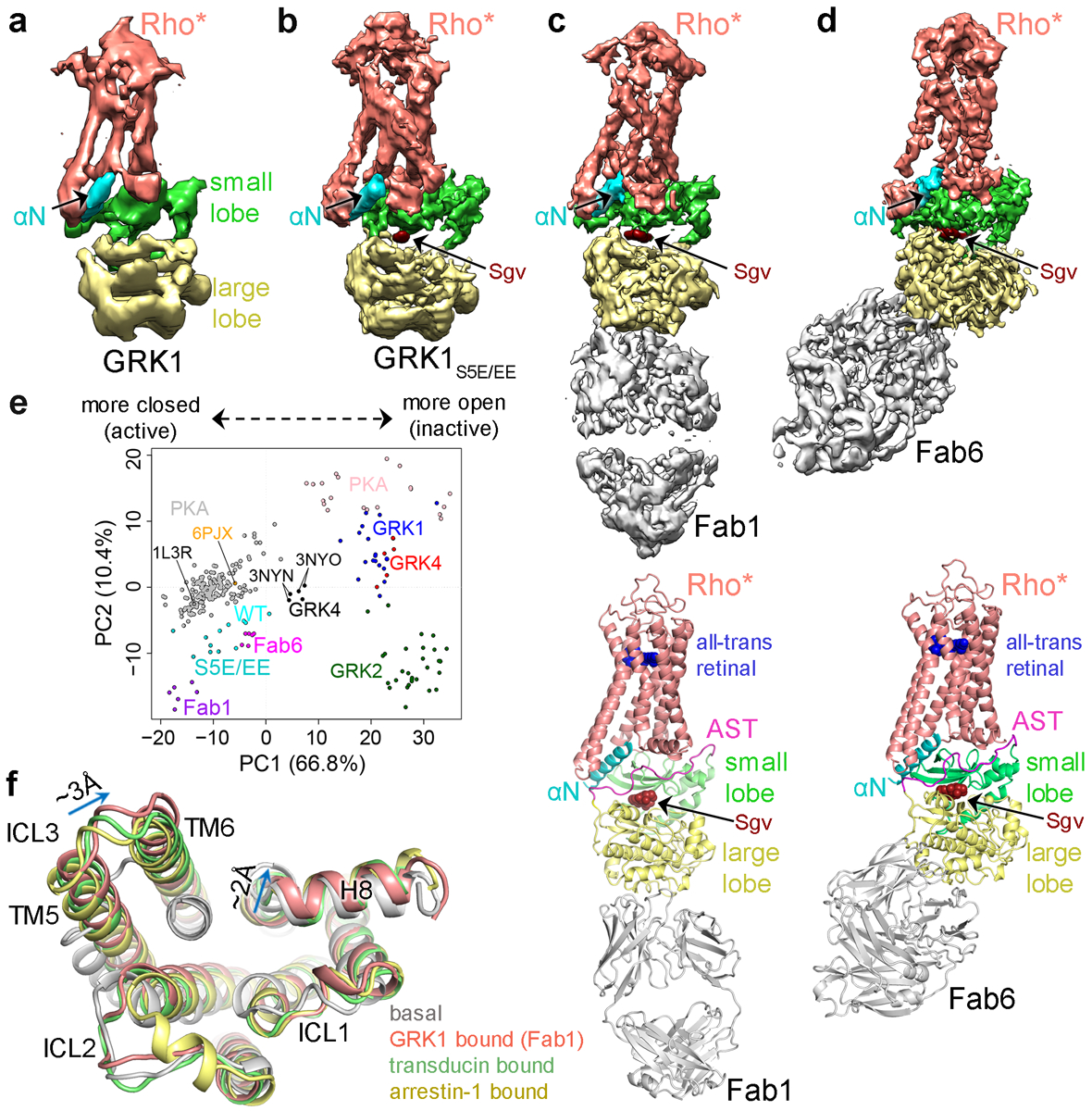
Sharpened maps of the 7 Å Rho*–GRK1 (a) and 5.8 Å Rho*–GRK1S5E/EE (b) complexes. Sharpened map and model of the Fab1–Rho*–GRK1S5E/EE complex (c) and the Fab6–Rho*–GRK1S5E/EE complex (d). e) Principal component (PC) analysis is consistent with a transition state-like conformation of the GRK1 kinase domain while in complex with Rho*. Each circle represents an individual experimental structure of PKA (grey active, pink inactive) or of the kinase domain from a member of one of the three GRK subfamilies: GRK1 (representing GRK1 and 7; various colors active, dark blue inactive), GRK2 (GRK2 and 3; green inactive), and GRK4 (GRK4, 5 and 6; black active, red inactive). Each structure was projected into the PC1-PC2 subspace, wherein PC1 and PC2 represent the directions with the largest conformational variance (67% and 10%, respectively). Molecular Dynamics Flexible Fitting (MDFF) models 1–6 (Supplementary Table 1) of Rho*–GRK1(WT) are colored cyan, those of Rho*–GRK1S5E/EE (S5E/EE) dark cyan, Rho*–GRK1S5E/EE–Fab1 (Fab1) magenta, and Rho*–GRK1S5E/EE−Fab6 (Fab6) purple. PDB entry 1L3R is the transition state-like structure of PKA, 3NYN/3NYO correspond to GRK6·Sgv/AMP complexes, and 6PJX is the Ca2+·CaM–GRK5 complex. PC1 and PC2 conformational changes are depicted in Supplementary Videos 3 and 4, respectively. f) Overlay of basal (PDB entry 1F8846), GRK1-bound (7MTA), transducin-bound (6OYA14), and arrestin-1 bound (5W0P32) Rho*. Key differences between GRK1-bound and transducin or arrestin-1-bound Rho* structures are indicated with arrows.
Structure of the Rho*–GRK1 complex.
In all reconstrutions, the conformation of Rho* closely resembles that in its complex with transducin and arrestin-1 (RMSD of 0.8 and 0.7 Å for 278 and 280 Cα atoms, respectively). The Rho* model spans residues 1–324, ending after H8, and the density was consistent with the presence of all-trans retinal, although strong density was only observed for the ionone ring (Extended Data Fig. 6a), consistent with the fact that excess 11-cis retinal consistently increased crosslinking yield (Extended Data Fig. 6b). The GRK1 models contain residues 6–24 (composing a single helix, αN), residues 182–508 (the kinase domain plus C-tail) and Sgv, for which there was strong density in all models (Extended Data Fig. 6a). Density for residues 491–508 is weak, implying a high degree of dynamics in this region. Density was entirely lacking for the RH domain (Extended Data Fig. 6c), and its positional heterogeneity in the complex is suggested by negative stain EM 2D class averages (Extended Data Fig. 6d).
In GRK1, αN packs against the AST and the small lobe, similar to its configuration in the GRK6·Sgv6 and Ca2+·CaM–GRK528 complexes (Extended Data Fig. 7a–d). GRK1-Asn12 is positioned to form hydrogen bonds with the side chain of GRK1-Arg191 on the small lobe, a position invariant in GRKs, but not other AGC kinases, essential for both GPCR and peptide phosphorylation (Extended Data Table1)5,29, and with GRK1-Asn480 in the AST (Fig. 3a–b). Consistent with this, the N12A mutation dramatically reduced crosslinking (Fig. 3d). GRK1-Phe15 and Phe22 are positioned to form hydrophobic contacts with hydrophobic residues in the small lobe and AST (Fig. 3c). Principal component (PC) analysis indicated that the GRK1 kinase domain in the Fab1 complex is in a more active state (based on the dominant component PC1) than in the other three reconstructions or in crystal structures of Sgv-bound GRK66 and GRK528 (Fig. 2e, Extended Data Fig. 7f–h)30,31. The differences in domain closure among the four reconstructions is likely a consequence of a high degree of dynamics in the complex, both in the hinge of the kinase domain as well as in the interface with Rho*, consistent with the heterogeneous local resolution of the Fab1 and Fab6 complex maps (Extended Data Figs. 4d, 5d).
Figure 3. Interactions within GRK1 and Rho* involve highly conserved elements in each protein family.
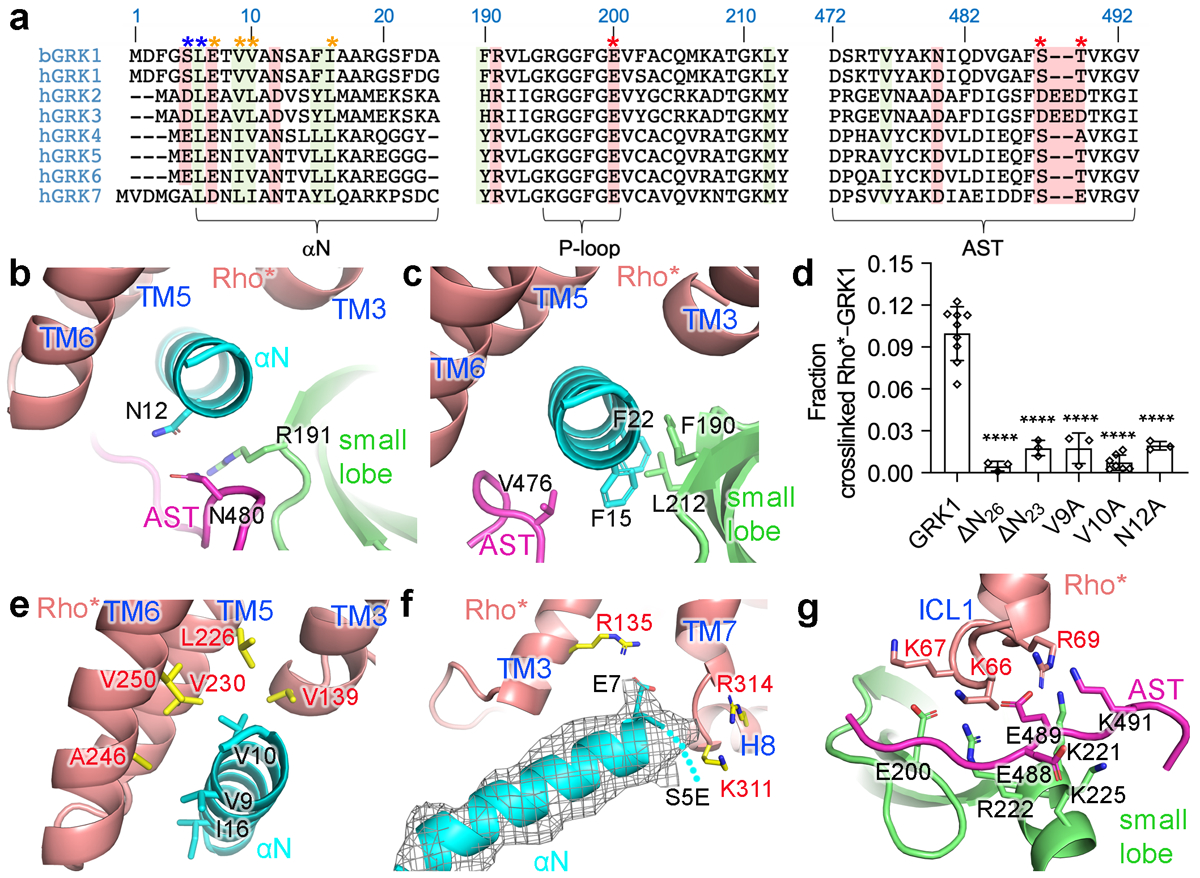
a) Sequence alignment of GRK N terminus, P-loop, and AST regions. The N-terminal Met residues in GRKs are expected to be cleaved, and in GRK2/3 the N-terminal residue is then acetylated47. Conserved hydrophobic residues involved in Rho* binding and kinase activation in GRK1 are highlighted in green, and conserved hydrophilic residues in red. Blue, orange, and red asterisks indicate GRK1 conserved residues interacting with Rho* H8, TM core, and ICL1, respectively. b, bovine; h, human. Sequence numbering above the alignment corresponds to bovine GRK1. b,c) Interactions of αN with the GRK1 small lobe and AST in the Fab1 complex. d) Crosslinking of GRK1 αN variants were compared to that of GRK1 using one-way ANOVA followed by a Dunnett’s multiple comparison test (****, P<0.0001). Error bars represent standard deviation from three (V9A, N12A, ΔN23, ΔN26) or seven (V10A) experiments. For gel source data, see Supplementary Fig. 10d. Hydrophobic (e) and electrostatic (f-g) interactions of GRK1 αN with the cytoplasmic cleft of Rho* in the Fab1 complex. The approximate position of GRK1-S5E backbone is indicated by a dotted line. Electron density of GRK1 αN is shown as a wire cage contoured at 16 σ. g) GRK1S5E/EE interaction with ICL1 of Rho*. Ser488 and Thr489 are autophosphorylation sites in GRK1 that were replaced by phosphomimetic glutamic acid residues in GRK1S5E/EE.
The Rho* and GRK1 interfaces in the Fab1 or Fab6 complexes bury 1,600 and 1,300 Å2 of accessible surface area, respectively, comparable to and overlapping with that buried in transducin14 and arrestin-132 complexes with Rho* (1,100 and 1,400 Å2, respectively). Deviations in the backbone structure of GRK1-bound Rho* from transducin-bound or arrestin-1-bound Rho* (Fig. 2f, Supplementary Fig. 5) are well supported in the reconstructions. The cytoplasmic ends of Rho* transmembrane (TM) 5 and 6 helices in GRK1-bound Rho* adopt the expected outwards conformation relative to the TM core of dark state rhodopsin33 but TM6 is rotated away from its position in rhodopsin by ~20° in the GRK1 complex, ~4° further than in the other two complexes, resulting in a shift of up to ~3 Å at the end of ICL3. This difference is mandated by contacts formed by αN and residues 474 and 475 in the AST loop of GRK1 with TM5, TM6, and ICL3. Rho* H8 is also shifted away from the receptor core by ~2 Å relative to the arrestin and transducin complexes. The end of TM1 and first half of ICL1 shift with H8, more so in Fab6 than Fab1. Key drivers for this difference may be the electrostatic interactions formed between the AST and ICL1, and the contacts between the N-terminus of αN in GRK1 and H8. The end result is a wider, more open cytoplasmic cleft in Rho* when bound to GRK1.
Conservation of the Rho*–GRK1 Interface.
Modeling indicates that the mode of interaction predicted by the Rho*–GRK1 complex will be compatible with other activated class A receptors such as the β2AR (Extended Data Fig. 7e, Supplementary Discussion). The most prominent feature in our maps is the αN of GRK1, which docks in the cytoplasmic cleft of Rho* and in roughly the same way as the C-terminal helix of transducin, although with opposite polarity and ~25° different trajectory14, and in a manner that overlaps with the finger loop of arrestin-1 (Extended Data Fig. 7i–p). This is in contrast to several recently proposed receptor docking models for GRKs wherein αN plays no direct role in the receptor interface2 (Supplementary Discussion). Consistent with the importance of αN, we showed that the crosslinking yield of N-terminal truncations GRK1ΔN23 and ΔN26 was dramatically reduced (Fig. 3d). Residues Val9, Val10, and Ile16, known to be important for agonist-dependent GPCR phosphorylation (Extended Data Table 1)5,6,34,35, pack against the hydrophobic inner surfaces of TM3, 5 and 6 (Fig. 3e). The V9A and V10A mutations also dramatically reduced crosslinking yield (Fig. 3d). Conversely, mutation of Rho*-Leu2265.61 or Val2305.65 (superscripts indicate Ballesteros–Weinstein nomenclature) to Ala in TM5 were observed to reduce Rho* phosphorylation by GRK136. Although density for GRK1-S5E was not observed, its side chain would complement those of Rho*-Lys3118.48 and -Arg3148.51 (Fig. 3f), which are highly conserved in Class A GPCRs (Supplementary Fig. 6). An acidic residue is in fact conserved at the position of GRK1-Ser5 in most GRKs (Fig. 3a), and mutation of the homologous residue in GRK2 (D3A/K) substantially reduced its ability to phosphorylate the β2 adrenergic receptor (β2AR)34. GRK1-Glu7, highly conserved among GRKs but less important for catalytic activity (Extended Data Table1)6,34,35, is positioned so that it could form a salt bridge with Rho*-Arg1353.50, one of the most highly conserved residues in Class A receptors (Fig. 3f).
The Rho* ICL1, which is highly conserved in both sequence and structure among Class A and B1 GPCRs (Supplementary Fig. 6), forms more extensive interactions with GRK1 than with either transducin or arrestin-1. ICL1 contacts the AST loop near GRK1-Ser488 and Thr489 (Fig. 3g). The rapid autophosphorylation of these residues in vitro or in cells enhances Rho* phosphorylation37,38. In our models, phospho-Ser488 could be coordinated by Rho*-Lys66 and GRK1-Lys225 and Lys491, and phospho-Thr489 by Rho*-Arg69 and GRK1-Lys221 and Arg222 (Fig. 3f). We validated this hypothesis via the GRK1EE and GRK1S5E/EE mutants, which exhibited lower Km (S5E/EE) and enhanced crosslinking (EE and S5E/EE) (Extended Data Fig. 8). Autophosphorylation of the homologous residues in GRK5 promotes its activity against activated receptors39,40 and GRK2/3 features a cluster of phosphomimetic residues (DEED) predicted to be at this ICL1 contact site (Fig. 3a). Rho*-Lys67 in ICL1 is also positioned such that it could form a salt bridge with GRK1-Glu200 (Fig. 3f), an invariant P-loop residue in GRKs that is conserved as Arg/Lys in most other AGC kinases. Thus, there is a striking number of complementary interactions formed between conserved basic residues in Rho* and acidic residues in GRK1, which may explain the well-known sensitivity of GRK-mediated GPCR phosphorylation to ionic strength. Interestingly, Class B2, C, and F GPCRs do not retain the signatures in H8 or ICL1 (Supplementary Discussion and Supplementary Fig. 7).
Rho*-ICL2 could not be easily modeled in part because of its continuity with the micelle boundary, but it would be in position to interact with the β1 strand region of the small lobe of GRK1. It would also sterically collide with the GRK1 α0 helix if this element were situated as observed in prior GRK1 crystal structures (Supplementary Fig. 8a). ICL3 contacts the AST at residues Arg474 and Thr475 (Supplementary Fig. 8b), but mutations at these positions in GRK1 did not measurably affect activity when tested previously (Extended Data Table1)5. However, by virtue of being located at the periphery of the interface, GPCRs with longer ICL3 loops would not only have adequate room to dock, but also have easy access to the active site of the bound GRK.
Validation by CLMS.
The lysine residues on GRK1 that crosslinked with Rho* identified by crosslinking with mass spectroscopy (CLMS) were all located on the kinase domain (Fig. 4a, Supplementary Fig. 9, Supplementary Table 2–3). Dead-end analysis, which identifies lysines modified by crosslinker and then quenched with either water or Tris buffer, was consistent with the RH domain not directly contributing to receptor binding, because all of its lysines were detected in this analysis (Fig. 4a, cyan spheres, Supplementary Table 4). Lysines in the kinase domain that did not react are not accessible to solvent in the observed complex (Fig. 4a), consistent with the idea that a discrete GPCR–GRK complex forms in solution before addition of crosslinker. Two major crosslinked regions between Rho* and GRK1 were identified (Fig. 4b–d, Supplementary Table 2). The first was between Rho*-Lys3118.48 and GRK1-Lys479 in the AST (Fig. 4b). The second was between two lysines in the Rho* C-terminus with the phosphoacceptor binding cleft of the GRK1 kinase domain. Rho*-Lys325 crosslinks with GRK1-Lys395 in the large lobe (Fig. 4b), whereas Rho*-Lys339 also crosslinks with Lys325 and eight additional sites (Fig. 4c, d, Supplementary Table 2). Rho*-Lys339 is positioned between Ser338 and Ser343 (Fig. 4c, d), which are the primary phosphorylation sites in the C-terminus of Rho*41. When Ser338 is modeled at the phosphoacceptor site of the kinase domain42, Lys339 can reach six out of the nine lysines in GRK1 identified by CLMS (Fig. 4c). Modeling instead Rho*-Ser343 at the active site places Rho*-Lys339 within range of the remainder of the GRK1 lysines identified (Fig. 4d). Thus, there is a high degree of dynamics in the receptor C terminus, which is consistent with its absence in our reconstructions and with the ability of Rho*-bound GRK1 to phosphorylate multiple sites in the receptor C-terminus.
Figure 4. CLMS confirms the cryo-EM structure and reveals dynamics in the receptor C-terminal tail.
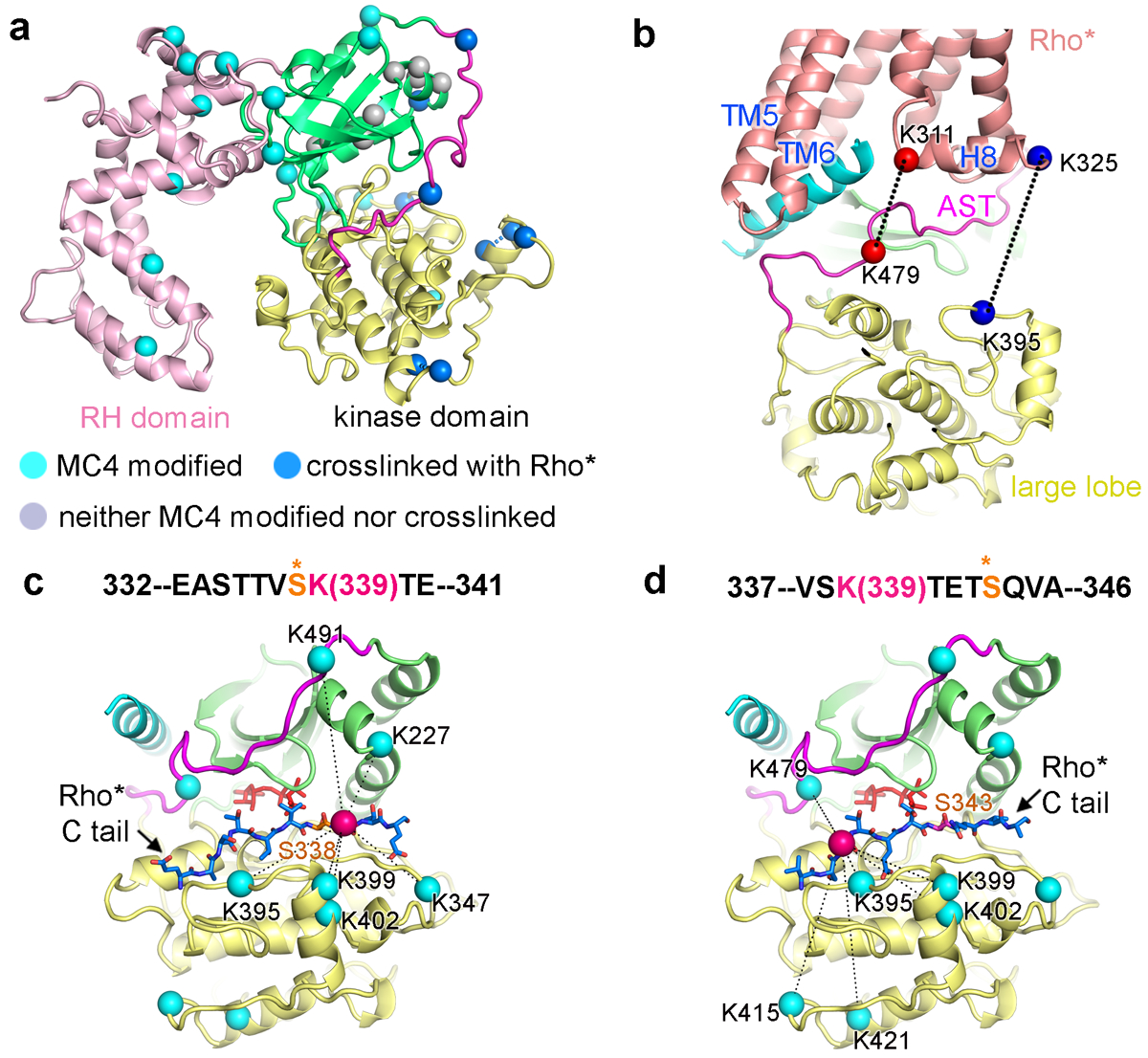
a) Lysine residues of GRK1 that crosslink with Rho* are indicated by blue Cα atoms, those that are dead-end modified are colored cyan (suggesting solvent accessibility), and those that are unreactive are colored grey (suggesting protection). b) Two prominent pairs of crosslinked lysines between Rho* and the kinase domain of GRK1 are highlighted with spheres connected by dashed lines. c, d) The C terminus of Rho* was modeled bounded to the peptide binding channel of GRK1 based on PDB entry 1O6L42. Rho*-Lys339 Cα is drawn as a red sphere, whereas the Cα atoms of lysines on GRK1 that crosslink with it are cyan. ATP is colored orange. c) Rho*-Ser338 in the phosphoacceptor site. d) Rho*-Ser343 in the phosphoacceptor site.
Discussion
Our Rho*–GRK1 reconstructions demonstrate that the few specific interactions formed in the Rho*–GRK1 interface involve sites highly conserved in both GRKs and most Class A and B1 GPCRs. Our data also indicate that GPCRs activate GRKs by coalescing the αN and AST regions into a bridge that spans the small and large lobes of the kinase domain and stabilizes it in a more closed, catalytically competent state. The unique conformation adopted by Rho* when it binds GRK1 relative to Gα or arrestin has possible implications for functional selectivity that would be imposed by ligands in other GPCRs. The fact that the GRK1 residues we observe to interact with Rho* are conserved, but not invariant, affords an opportunity for receptors to select for the GRKs that phosphorylate them, and hence for the installation of unique barcodes43 in their cytoplasmic domains that could lead to different downstream effects.
Finally, our work provides new insights into the overall process by which GRK phosphorylation of activated GPCRs occurs. In our current model, the αN helix of GRK1 is intrinsically disordered until it binds to activated GPCRs in a form of “molecular fly-casting”44 (Fig. 5). The fact that robust crosslinking to Rho* was not observed in the presence of either ATP or ADP suggests that GPCRs are optimized to bind GRKs in a conformation optimized for phosphotransfer, but not for ATP or ADP binding. A fly-casting mechanism however would allow for a partially dissociated state of the GRK wherein exchange of ADP for ATP could readily occur and enable further rounds of phosphorylation. Future studies are needed to validate this model and to examine how GRK2 and GRK4 subfamily members interact with receptors from the various GPCR classes, especially those lacking the key interaction signatures defined by the Rho*–GRK1 complex.
Figure 5. A generalized model for GRK activation by anionic lipids and activated GPCRs.
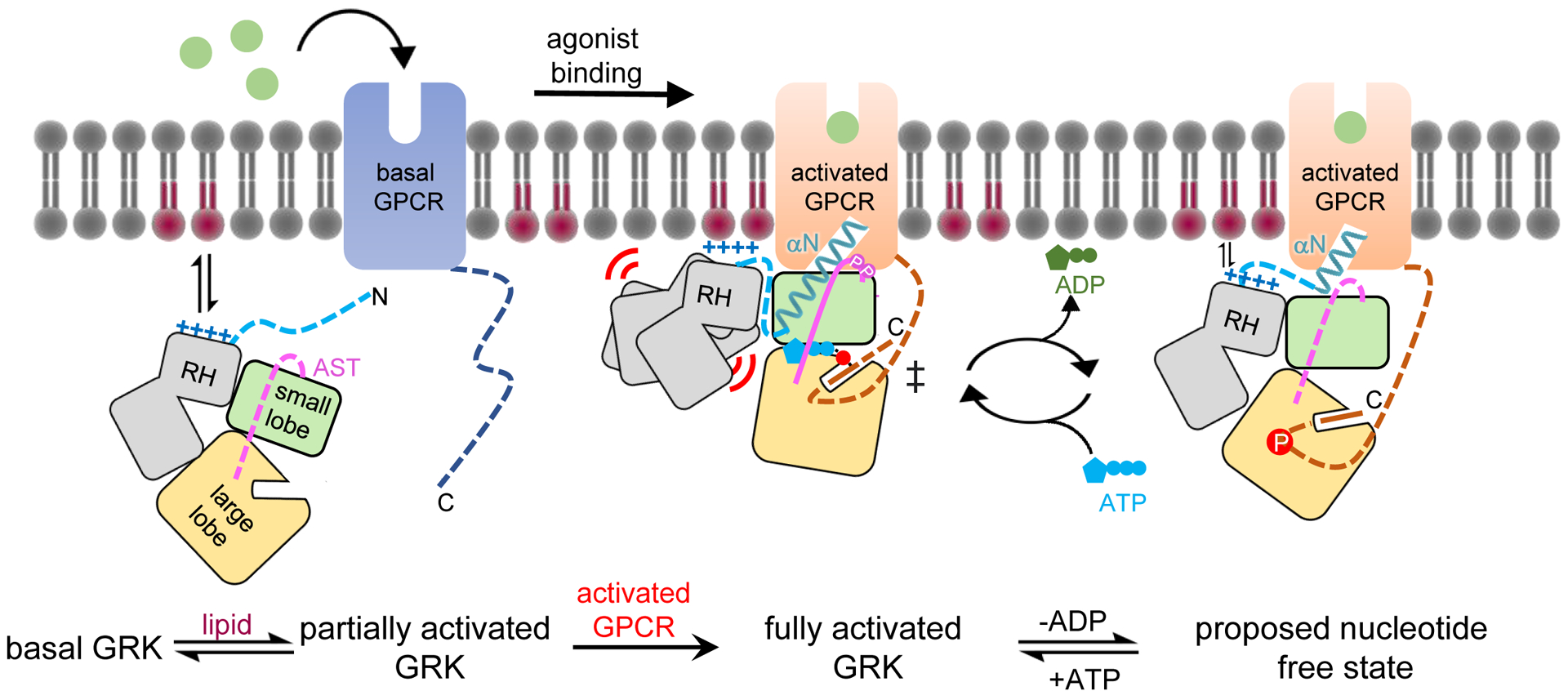
In its basal state, the GRK kinase domain is in an open conformation with its small and large lobes engaged with both lobes of the RH domain. The N terminus and portions of the AST in the GRK are disordered. A basic region adjacent to the receptor interface (α0 helix region in GRK1) promotes membrane association. Upon receptor activation, the GRK N terminus forms a helix (αN) that docks into the cytoplasmic cleft of the activated GPCR. αN also packs against the small lobe and AST to allosterically trigger kinase domain closure, aligning ATP with the phosphoacceptor in the C tail of GPCR in a transition state-like complex. GRKs may need to at least partially dissociate from the receptor to efficiently exchange adenine nucleotides, and in this state αN and the large lobe could remain bound to the cytoplasmic cleft and the phosphorylated tail of the receptor, respectively, to facilitate additional rounds of phosphorylation. In GRK1, and perhaps in the closely related GRK4 subfamily of GRKs, receptor binding also induces conformation changes that displace the RH domain. This could serve to strengthen membrane interactions mediated by the RH domain and its associated loops, including the α0 helix. GRK2 and GRK3 lack an analogous α0 helix and there is evidence that their RH domains may not be as dynamic in receptor complexes. Pink and red circles indicate GRK autophosphorylation sites and Rho* phosphosites, respectively.
Methods:
Expression and purification of GRK1.
Full length bovine GRK1 (residues 1–561) has a farnesylation site at its C-terminus which limits its expression in insect cells. We therefore used truncated GRK1 (residues 1–535, hereafter referred to simply as GRK1). This variant exhibits slightly decreased kinase activity toward receptor relative to wild-type36. A His6-tag was added to the C terminus of GRK1 for metal affinity purification45. The mutagenesis and cloning of GRK1 was described previously35. We used the Bac-to-Bac baculovirus expression system (Invitrogen) to express GRK1 in insect cells. For purification, freshly prepared or thawed cell pellets were resuspended in cold lysis buffer (Buffer A) containing 20 mM HEPES (pH 8.0), 400 mM NaCl, 2 mM DTT, 0.1 mM PMSF and leupeptin, lima bean trypsin protease inhibitor. The cells were resuspended using a dounce homogenizer and then lysed using an Avestin C3 emulsifier. The lysed cells were centrifuged in a Beckman Ti-45 rotor at 45,000 rpm for 45 minutes. The supernatant was combined and filtered through a glass filter and loaded onto a 2 ml home-packed Ni2+-NTA column pre-equilibrated with Buffer A. The column was then washed with 50 ml Buffer A, followed by 50 ml Buffer A plus 20 mM imidazole. The bound protein was eluted in ~2 ml fractions with Buffer A plus 200 mM imidazole. The purity of GRK1535-His6 after this step was ~95% as revealed by Coomassie blue staining of samples assessed via SDS-PAGE. The fractions were pooled and concentrated with a 50 kDa cutoff 50 ml centrifugal concentrator (Amicon® Ultra) to ~1–2 ml. Then, 0.5 ml of the concentrated sample was injected onto a Superdex 200 Increase column (GE healthcare) equilibrated with buffer containing 20 mM HEPES (pH 8.0), 150 mM NaCl, and 0.5 mM TCEP. The peak fractions were pooled and concentrated to ~10 mg/ml, flash frozen in liquid nitrogen and stored at -80 °C for future use. We produced GRK1- V9A, V10A, N12A, S5E, EE(S488E/T489E), DD(S488D/T489D) and S5E/EE using the same procedure. For GRK1-ΔN26, ΔN23 and 5A (R31A/R33A/K34A/R38A/K40A) that exhibited decreased expression level, an extra ion exchange step using a 1 ml HiTrap Q column (GE Healthcare) was performed after meta affinity purification. For this, protein eluted from Ni2+-NTA column was diluted 10-fold with 20 mM HEPES, 2 mM DTT before loading to Q column. GRK1 variants were eluted at ~100 mM NaCl.
Preparation of ROS from bovine retinas.
All procedures were conducted in a dark room under red light as described previously48. Briefly, ~100 retinas were resuspended with ~100 ml of 45% (w/v) sucrose in Buffer B containing 70 mM phosphate buffer (pH 7.0), 1 mM MgCl2, 0.1 mM EDTA and were shaken vigorously by hand for ~3 minutes, and then the sample was centrifuged at 5000 rpm for 5 minutes. The supernatant was filtered through four layers of cheesecloth, diluted 1:1 with Buffer B and centrifuged at 13,000 rpm for 10 minutes. The pellet was then resuspended with 25.5% sucrose and overlaid on top of two sucrose layers containing 14 ml of 32.25% sucrose with 13 ml of 27.125% sucrose. The sucrose gradient was centrifuged in a swinging bucket rotor (SW-27) at 18,000 rpm for 90 minutes. ROS settled between the two layers and were collected using a syringe. The collected ROS were washed twice with Buffer B to remove sucrose, incubated with 5 M urea for 10 minutes, and then washed three times with 20 mM HEPES (pH 8.0), 2 mM MgCl2. The ROS were aliquoted and stored at −80 °C for future use.
Preparation of hyper-phosphorylated rhodopsin.
2 μM of ROS was incubated with 100 nM GRK1 under ambient light at room temperature for one hour in a buffer containing 50 mM HEPES (pH 8.0), 500 μM ATP, and 10 mM MgCl2. An γ-P32-ATP standard curve estimated 6–8 phosphates incorporated per rhodopsin, indicating full saturation given that there are only 7 possible sites in the cytoplasmic tail of the receptor. After phosphorylation, the membrane was washed twice with 20 mM HEPES (pH 8.0) and 100 mM NaCl and then incubated with ten-fold molar excess of 11-cis retinal in the dark at room temperature for 1 hour for the purpose of regeneration.
Preparation of Asp-N treated rhodopsin.
0.5 mg of ROS was incubated with 0.4 μg of Asp-N protease (Roche) overnight at room temperature in the dark. Asp-N treated ROS was washed twice with buffer containing 20 mM HEPES (pH 8.0) and 100 mM NaCl before detergent reconstitution.
Rhodopsin reconstitution in detergent micelles.
500 μl of 5 mM LMNG was added to 100 μg of c8-PI(4,5)P2 powder to make 5% (molar ratio) c8-PI(4,5)P2:LMNG micelles. Rhodopsin reconstitution was performed under dim light in a dark room. 400 μl of the c8-PI(4,5)P2/LMNG solution was used to dissolve 0.5 mg of rhodopsin in ROS membrane. After incubation on ice for 30 minutes, the solution was centrifuged at 13,000 rpm for 10 minutes to pellet insoluble material. The supernatant containing rhodopsin solubilized with c8-PI(4,5)P2/LMNG was isolated and its concentration assessed using a Bradford assay. The same procedure was followed to reconstitute rhodopsin with LMNG alone.
Rhodopsin reconstitution in nanodiscs.
All lipids used for nanodisc were purchased from Avanti and dissolved in the form of chloroform. 40% POPS or POPG were mixed with 60% POPC and the chloroform was dried under a gentle stream of N2. The glass vials containing the mixed lipids were kept in a desiccator overnight to get rid of any residual chloroform. CHAPS was added to dissolve the lipids till it became a clear solution. Lipids, MSP1D1 and Rho were mixed at the ratio of 360: 6: 1 in the dark for 30 minutes. Biobeads were added to the mix and incubate overnight. The reconstituted Rho was loaded onto a Superdex 200 increase column in buffer containing 20 mM HEPES (pH 8.0) and 100 mM NaCl under the light. The peak fractions containing both empty nanodiscs or nanodiscs with rhodopsin were pooled and concentrated using a 100 kDa cutoff 50 ml centrifugal concentrator (Amicon® Ultra). The amount of rhodopsin reconstituted in nanodiscs were determined using Coomassie staining with known amount of rhodopsin as standard.
Crosslinker screening.
To screen for conditions that covalently capture a Rho*–GRK1 complex, 10 μM GRK1 and 10 μM rhodopsin reconstituted with c8-PI(4,5)P2/LMNG was mixed in the absence or presence of various adenosine analogs (1 mM AMP, 1 mM ADP, 1 mM AMP-PNP, 1 mM ATP, or 0.8 mM Sgv) and incubated in dark for 20 minutes on ice. Crosslinkers were dissolved with buffer containing 50 mM HEPES (pH 8.0) and 10 mM MgCl2 immediately before use and added to the reaction. The samples were exposed to ambient light on ice for 30 minutes to activate rhodopsin and SDS sample buffer was used to quench the reaction. The crosslinking result was assessed with Coomassie blue staining of proteins separated by 10% SDS-PAGE. We screened seven crosslinkers: BS3, BS(PEG)5, BM(PEG)2, BM(PEG)3 (ThermoFisher) and PC4, DC423 and MC424 (provided by the Andrews lab, U Michigan). Both DC4 and MC4 successfully crosslinked rhodopsin and GRK1 in the presence of Sgv.
Testing of conditions necessary for crosslinking.
The same experiment used in the crosslinker screening was used to assess the effect of receptor activation and GRK ligands on crosslinking in the presence of 2 mg/ml MC4. To assess the light dependence of crosslinking, one reaction was kept in the dark throughout and was compared with a reaction pre-exposed to light for 30 minutes. To test the effect of an inhibitor that traps GRK1 in an inactive state, 10 μM GRK1, 10 μM rhodopsin and 0.4 mM Sgv were incubated along with increasing concentrations of CCG224062 (from 0.01 mM to 0.63 mM) for 20 minutes in the dark before light activation. All crosslinking experiments were repeated at least three times. For quantification, the density of crosslinked Rho*–GRK1 complex was normalized to the combined density of Rho* and GRK1 without crosslinker as an approximation of crosslinking efficiency and was used to compare between groups.
The purification of the crosslinked Rho*–GRK1 complex.
Although c8-PI(4,5)P2 did not significantly enhance the crosslinking between Rho* and GRK1 (Supplementary Fig. 10e), it was kept in the reaction for complex purification. A crosslinking reaction with total volume 5.0 ml was set up as described above. 50 mM Tris (pH 8.0) was added to quench the reaction. The reaction mix was loaded to a 1 ml HiTrap Q column (GE Healthcare) equilibrated with 20 mM HEPES (pH 8.0), 100 mM NaCl, 0.2 mM MgCl2, 0.2 mM LMNG and 20 μM Sgv. GRK1 and crosslinked Rho*–GRK1 complex were eluted over a gradient of 100 to 200 mM NaCl. Rhodopsin and self-crosslinked rhodopsin eluted at 500 mM NaCl. The fractions containing mainly crosslinked Rho*–GRK1 complex and some GRK1 were pooled together and concentrated with a 100 kDa cutoff 50 ml concentrator (Amicon® Ultra) to ~400 μl. The concentrated sample was loaded onto a Superdex 200 Increase column equilibrated with 20 mM HEPES (pH 8.0), 100 mM NaCl, 0.2 mM MgCl2, 0.2 mM LMNG and 20 μM Sgv. The crosslinked Rho*–GRK1 complex eluted at a volume consistent with a 150 kDa globular protein and was pooled for EM and CLMS analysis. The yellow color of the final sample was consistent with the retention of all-trans retinal in the sample, and also showed an absorption peak at 380nM, indicating Rho* is in the Meta II state (Fig. 1f).
Michaelis-Menten kinetic analysis.
In Fig. 1b and Extended Data Fig. 1a, Km and Vmax for Rho* were determined by varying Rho* concentrations from 0.05 μM to 6 μM for ROS and from 0.02 μM to 2.5 μM for rhodopsin reconstituted in LMNG or c8-PI(4,5)P2/LMNG. The lower concentration range used for rhodopsin reconstituted with detergent was necessary because GRK1 kinase activity is inhibited by detergent at high concentrations. The reactions contain 50 nM GRK1, 20 μM ATP (500 Ci/mol) and 50 mM HEPES (pH 8.0), 10 mM MgCl2. Km and Vmax for ATP were determined by varying ATP concentrations from ~0.16 μM to 20 μM (500 Ci/mol) in reactions containing 50 nM GRK1, 1 μM rhodopsin and 50 mM HEPES (pH 8.0) and 10 mM MgCl2. Rhodopsin was activated under an ambient light for 1 minute prior to the reaction. The reaction proceeded for 2 minutes at room temperature and was quenched with SDS sample buffer. Rho* and GRK1 were separated by SDS-PAGE and the extent of phosphorylation was assessed using image analysis with the Quantity One program (BioRad) on phosphor screens. Plots of initial velocity versus concentration of the varied substrates were fit to the Michaelis-Menten equation using GraphPad prism. For Extended Data Fig. 1b, the same procedure was performed as described above except that the range of ROS was varied from 0.15 μM to 18 μM when determining the Km and Vmax for Rho*. When comparing how nanodiscs affect GRK1 kinetics, lower amount of Rho* was used. For Extended Data Fig. 8c–e, the same procedure was performed as described above except that the reaction contains 0.1 μM rhodopsin instead of 1 μM when determining the Km and Vmax for ATP.
Analysis of tubulin phosphorylation by GRK1 and mutants.
The phosphorylation reactions contained 5 μM of tubulin (PurSolutions), 50 nM GRK1, 5 μM ATP and 50 mM HEPES (pH 8.0) and 10 mM MgCl2. The reaction was quenched at 2, 5, 10, 20 and 30 minutes with SDS sample buffer. Tubulin and GRK1 were separated by SDS-PAGE and the extent of phosphorylation was assessed using image analysis with the Quantity One program (BioRad) on phosphor screens. This assay was repeated three times.
Preparation of biotinylated GRK1.
An Avi-tag was added to GRK1 C terminus after the His6-tag. The same procedures used to produce GRK1 as described above were used to generate Avi-tagged GRK1. BirA500 kit (Avidity) was utilized to biotinylate Avi-tagged GRK1. Briefly, 2mg of Avi-tagged GRK1 was incubated with 8 μl of biotin ligase, 80 μl of biomix A and 80 μl of biomix B supplied from the kit overnight on ice. The biotinylated GRK1 was loaded onto a Superdex 200 Increase column with buffer containing 20 mM HEPES (pH 8.0), 100 mM NaCl and 0.2 mM TCEP. The monomeric peak was collected and concentrated using a 50 kDa cutoff 50 ml concentrator (Amicon® Ultra) to ~10 mg/ml. The biotinylation efficiency was estimated to be close to 100% by pull-down experiments using Streptavidin magnetic beads.
Phage display selections.
Sgv-bound biotinylated GRK1 was used as target for phage display selection, performed at 4 °C according to published protocols49 in a selection buffer supplemented with 10 μM Sgv. In brief, in the first round, 200 nM of target was immobilized on 250 μl of magnetic beads. Then, 100 μl of a phage library E50 containing 1012–1013 virions were added to the Streptavidin beads and incubated for 30 minutes. The resuspended beads containing bound virions were washed extensively and then used to infect freshly grown log phase E. coli XL1-Blue cells. Phages were amplified overnight in 2xYT media with 50 μg/ml ampicillin and 109 p.f.u./ml of M13-KO7 helper phage. To increase the stringency of selection, three additional rounds of sorting were performed with decreasing the target concentration in each round (second round: 50 nM, third round: 10 nM and fourth round: 10 and 5 nM) using the amplified pool of virions of the preceding round as the input. Selection from second to fourth rounds was done on a Kingfisher instrument using a solution capture method where the target was premixed with the amplified phage pool and then Streptavidin beads were added to the mixture. From the second round onwards, the bound phages were eluted using 0.1 M glycine (pH 2.7). This elution technique often risks the elution of non-specific and Streptavidin binders, which tend to overpopulate the amplified phage pool thereby reducing the chance to obtain the desired specific clones. To eliminate them, the precipitated virions from the second round onwards were negatively selected against 100 μl of Streptavidin beads before adding to the target. The pre-cleared phage pool was then used as an input for the selection.
Single-point phage ELISA.
All ELISA experiments were performed at 4°C in 96-well plates coated with 50 μl of 2 μg/ml neutravidin in Na2CO3 buffer (pH 9.6) and subsequently blocked by 0.5% BSA in PBS. A single-point phage ELISA was used to rapidly screen the binding of the obtained Fab fragments in phage format. Colonies of E.coli XL1-Blue harboring phagemids from 4th round of selection were inoculated directly into 500 μl of 2xYT broth supplemented with 100 μg/ml ampicillin and M13-KO7 helper phage. The cultures were grown overnight at 37 °C in a 96-deep-well block plate. HBS with 0.05% (v/v) Tween-20 supplemented with 10 μM Sgv was used in ELISA. Culture supernatants containing Fab phage were diluted tenfold in HBST buffer in presence of 10 μM Sgv. After 15 minutes of incubation, the mixtures were transferred to ELISA plates that were incubated with 30 nM biotinylated GRK1 in experimental wells and with buffer in control wells for 15 minutes. The ELISA plates were incubated with the phage for another 15 min and then washed with ELISA buffer. The washed ELISA plates were incubated with a 1:1 mixture of mouse anti-M13 monoclonal antibody (cat: 27-9420-01, GE, 1:5,000 dilution in ELISA buffer) and peroxidase conjugated goat anti-mouse IgG (cat: 115-035-003, Jackson Immunoresearch, 1:5000 dilution in ELISA buffer) for 30 minutes. The plates were again washed, developed with TMB substrate and then quenched with 1.0 M HCl, and the absorbance at 450 nm was determined. The background binding of the phage was monitored by the absorbance from the control wells.
Sequencing, cloning, overexpression and purification of Fab fragments.
From phage ELISA, clones (selected based on a high ratio of ELISA signal of target binding to background) were sequenced at the DNA Sequencing Facility at the University of Chicago. Unique clones were sub-cloned in pRH2.2, an IPTG inducible vector for expression of Fabs in E. coli. E. coli C43 cells were transformed with sequence-verified clones of Fab fragments in pRH2.2. Fab fragments were grown in TB autoinduction media with 100 μg/ml ampicillin overnight at 30 °C. Harvested cells were kept frozen at −80 °C until use. Frozen pellets were re-suspended in PBS supplemented with 1 mM PMSF and 1 μg/ml DNaseI. The suspension was lysed by ultrasonication. The cell lysate was incubated at 65 °C for 30 minutes to eliminate of any undesired proteolyzed fragments of the Fab produced during overexpression. Heat-treated lysate was then cleared by centrifugation, filtered through 0.22 μm filter and loaded onto a HiTrap Protein L 5-mL column pre-equilibrated with lysis buffer (20 mM HEPES buffer, pH 7.5, 500 mM NaCl). The column was washed with 10 column volumes of lysis buffer followed by elution of Fab fragments with elution buffer (0.1 M acetic acid). Fractions containing protein were directly loaded onto a Resource S 1-mL column pre-equilibrated with buffer A (50 mM sodium acetate, pH 5.0) followed by washing with 10 column volumes wash with buffer A. Fab fragments were eluted with a linear gradient 0–50% of buffer B (50 mM sodium acetate, pH 5.0, 2.0 M NaCl). Affinity and ion-exchange chromatography were performed using an automated program on ÄKTA explorer system. Purified Fab fragments were dialyzed overnight against 20 mM HEPES (pH 7.4) 150 mM NaCl. The quality of purified Fab fragments was analyzed by SDS–PAGE.
Multipoint Protein ELISA for EC50 determination.
Multipoint ELISA was performed at 4°C to estimate the affinity of the Fabs to GRK1. HBS with 0.05% (v/v) Tween-20 supplemented with 10 μM Sgv was used as ELISA buffer. 30 nM of Sgv-bound biotinylated GRK1 immobilized on a neutravidin coated ELISA plate was incubated with 3-fold serial dilutions of the purified Fabs starting from 4 μM for 20 minutes. The plates were washed, and the bound GRK1-Fab complexes were incubated with a secondary HRP-conjugated Pierce recombinant protein L (cat: 32420, Thermofisher, 1:5000 dilution in ELISA buffer) for 30 minutes. The plates were again washed, developed with TMB substrate and quenched with 1.0 M HCl, and absorbance at 450 nm was measured. To determine the affinities, the data were fitted in a dose response sigmoidal function in GraphPad PRISM and EC50 values were calculated.
Cryo-EM sample preparation and image acquisition.
Purified Rho*–GRK1S5E/EE complex at ~0.3mg/ml was incubated with 1.2 molar excess of Fab1 or Fab6 for 20 minutes before freezing. Grids of Rho*–GRK1, Rho*–GRK1S5E/EE, Rho*–GRK1S5E/EE–Fab1 or Rho*–GRK1S5E/EE−Fab6 were prepared using the same procedure. Sample at ~0.3 mg/ml was applied onto glow-discharged 300 mesh grids (Quantifoil R1.2/1.3) and plunge-frozen into liquid ethane using a FEI Vitrobot Mark IV (Thermo Fisher Scientific). Data was collected on a Titan Krios electron microscope operated at 300kV using Leginon51 with a nominal magnification of 81,000x (resulting in a calibrated physical pixel size of 1.08 Å/pixel) and a defocus range of 1.2 – 2.5 μm. The images were recorded on a K3 summit electron direct detector in super-resolution mode at the end of a GIF-Quantum energy filter operated with a slit width of 20 eV. A dose rate of 20 e−/pix/s and an exposure time of 3.12 seconds were used, generating 40 movie frames with a total dose of ~54 electrons per Å2.
EM data processing.
For Rho*–GRK1 complex reconstruction, the movie frames were imported to RELION-352. Movie frames were aligned using MotionCor253 with binning factor of 2. Contrast transfer function (CTF) parameters were estimated using Gctf54. Laplacian-of-Gaussian picking was used to pick about 2.68 million particles, which were extracted from the dose weighted micrographs. The dataset was split into batches for multiple rounds of 2D classifications to exclude bad particles that fell into 2D averages with poor features. After this cleaning step, 705,966 particles remained. Particles from different views were used to generate an initial model in cryoSPARC55. Further 3D classification with three classes was performed to select the class with resolvable secondary structure features. The selected 183,717 particles were used for final 3D refinement in RELION-352, converging at 7 Å global resolution, as determined by the Fourier shell correlation (FSC) criterion in RELION-3 PostProcess52. The data processing workflow is shown in Extended Data Fig. 2. For Rho*–GRK1S5E/EE, Rho*–GRK1S5E/EE–Fab1 and Rho*–GRK1S5E/EE−Fab6 complex, the movie frames were first motion-corrected using MotionCor253 with binning factor of 2 and then imported to cryoSPARC55. CTF parameters were estimated using patch CTF. Blob picker was used to pick from 100 micrographs and the selected particles were processed using 2D classification. The good 2D classes were selected and used as templates for template picker to pick particles from all micrographs. The picked particles were extracted from the dose weighted micrographs. Multiple rounds of 2D classifications were performed to exclude bad particles that fell into 2D averages with poor features. The remaining particles from different views were used to generate an initial model in cryoSPARC55. For Rho*–GRK1S5E/EE, particles were imported into RELION-352 and further 3D classification with six classes was performed to select the class with resolvable secondary structure features. The selected 132,721 particles were used for final 3D refinement in RELION-352, converging at 5.8 Å global resolution, as determined by the FSC criterion in RELION-3 PostProcess52. The data processing workflow is shown in Extended Data Fig. 3. For Rho*–GRK1S5E/EE–Fab1 or Rho*–GRK1S5E/EE−Fab6 complex, heterogenous refinement with three classes was performed to select the class with resolvable secondary structure features in cryoSPARC55. The selected particles were used for homogenous refinement, followed by non-uniform refinement, converging at ~4 Å global resolution, as determined by the FSC criterion in cryoSPARC55. The data processing workflow is shown in Extended Data Fig. 4,5. Directional FSC was performed by online 3DFSC server to evaluate the resolution in different directions56. A number of factors prevented further improvement of the current resolution, including the small size and intrinsic flexibility of the complex, as well as the preferred orientations.
Principal component (PC) analysis.
PC analysis of GRK kinase domain conformations31,57 was recalculated based on an updated data set, as follows. Four GRK2 structures published since the last analysis (PDB entries 5UVC, 5UUU, 6C2Y, and 6U7C) were added to the previous PKA (201 structures) and GRK (59 structures) data set, and the kinase domains of the structures were aligned using the structurally invariant core residues identified previously31. PCs were calculated based on equivalent kinase domain Cα atoms of all structures excluding the new Rho*-bound GRK1 structures generated by MDFF and the recent GRK5 −Ca2+·CaM structure (PDB entry 6PJX). All structures (including the new models and 6PJX) were then projected into the subspace spanned by the two PCs that captured the largest structural variance (i.e., PC1 and PC2, which in total capture 77.2% of total variance) for inter-structure comparisons. Each PC axis represents a linear combination of Cartesian coordinates (in Å), where the origin represents the mean conformation of all structures used to compute PCs.
Negative stain EM.
To prepare the grids for negative stain EM, 4 μl of Rho*–GRK1 complex was added to a glow discharged 400-mesh copper grid covered with carbon-coated collodion film (EMS, Hatfield, PA). Grids were washed in one drop of water, stained in three drops of uranyl formate (0.75%) (EMS, Hatfield, PA) and air dried. Samples were visualized on a Tecnai T12 electron microscope (FEI, Hillsboro, OR) at an acceleration voltage of 120 kV under low-dose conditions. Images were taken at a magnification of 71,138× at a defocus value of −1.4 μm and recorded on a Gatan US4000 CCD camera (Gatan, Pleasanton, CA). Images were converted to mixed raster content format and binned (2×2 pixels), resulting in final images with a pixel size of 4.16 Å/pixel at the specimen level. A total of 18,227 particle projections were excised using Boxer (part of the EMAN 2.1 software suite)58 from 80 micrographs. Boxed out particle projections were subjected to two-dimensional reference-free alignment and classification using ISAC59. The particles from ISAC 2D classification were further cleaned using RELION52 and resulted in 29 good class averages with 15,056 particle projections.
Extended Data
Extended Data Figure 1. Activation of GRK1 by anionic lipids.
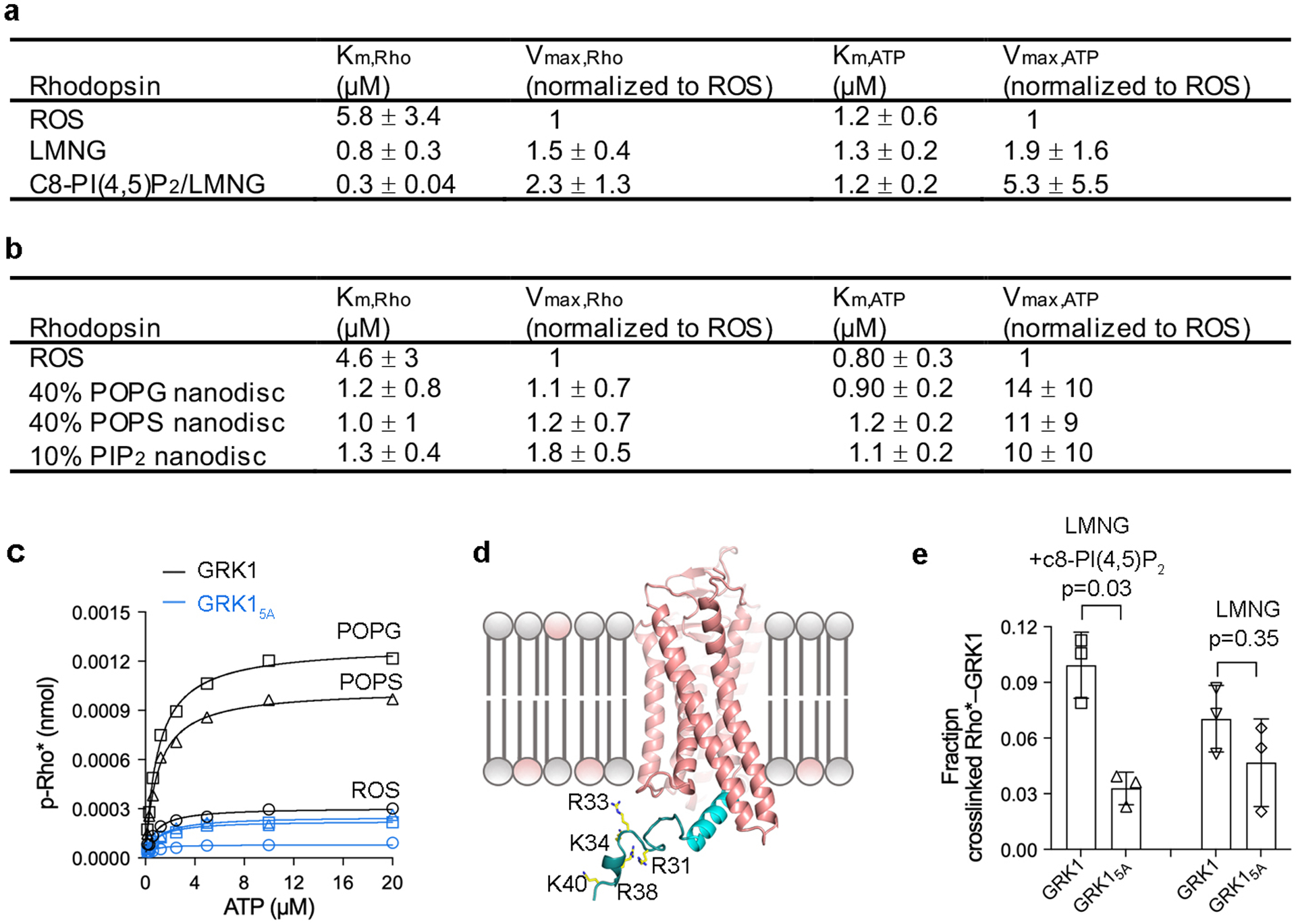
a) Kinetic analysis of GRK1 phosphorylation of Rho* in ROS or LMNG or LMNG/c8-P(4,5)IP2. Values shown represent mean ± standard deviation, n=4. Reactions were performed in 50 mM HEPES (pH 8.0), 10 mM MgCl2 for 2 minutes at room temperature. b) Kinetic analysis of GRK1 phosphorylation of Rho* in ROS or POPC nanodiscs containing 40% POPG or 40% POPS or 10% PIP2. Values represent mean ± standard deviation, n=3. Reactions were performed in 50 mM HEPES (pH 8.0), 10 mM MgCl2 for 2 minutes at room temperature. c) Representative Michaelis-Menten kinetics measurement varying ATP. Rho* was reconstituted with POPC nanodiscs containing 40% POPG (black squares) or 40% POPS (black triangles) and compared with Rho* in ROS (black circles). GRK15A activity was greatly diminished relative to GRK1 under these same conditions (blue symbols), but still somewhat responsive to anionic lipids. d) Approximate location of the five positively charged residues places them close to the lipid bilayer. e) Crosslinking yield, corresponding to the amount of complex formed divided by the sum of the input GRK1 and Rho*, of GRK15A relative to GRK1 is significantly reduced in the presence of c8-PI(4,5)P2 but not in its absence. The crosslinking level of GRK15A was compared with GRK1 using two-sided t-test (n=3) in the presence or absence of c8-PI(4,5)P2. For gel source data, see Supplementary Fig. 10f.
Extended Data Figure 2. Workflow of cryo-EM data processing and resolution analysis of Rho*–GRK1.
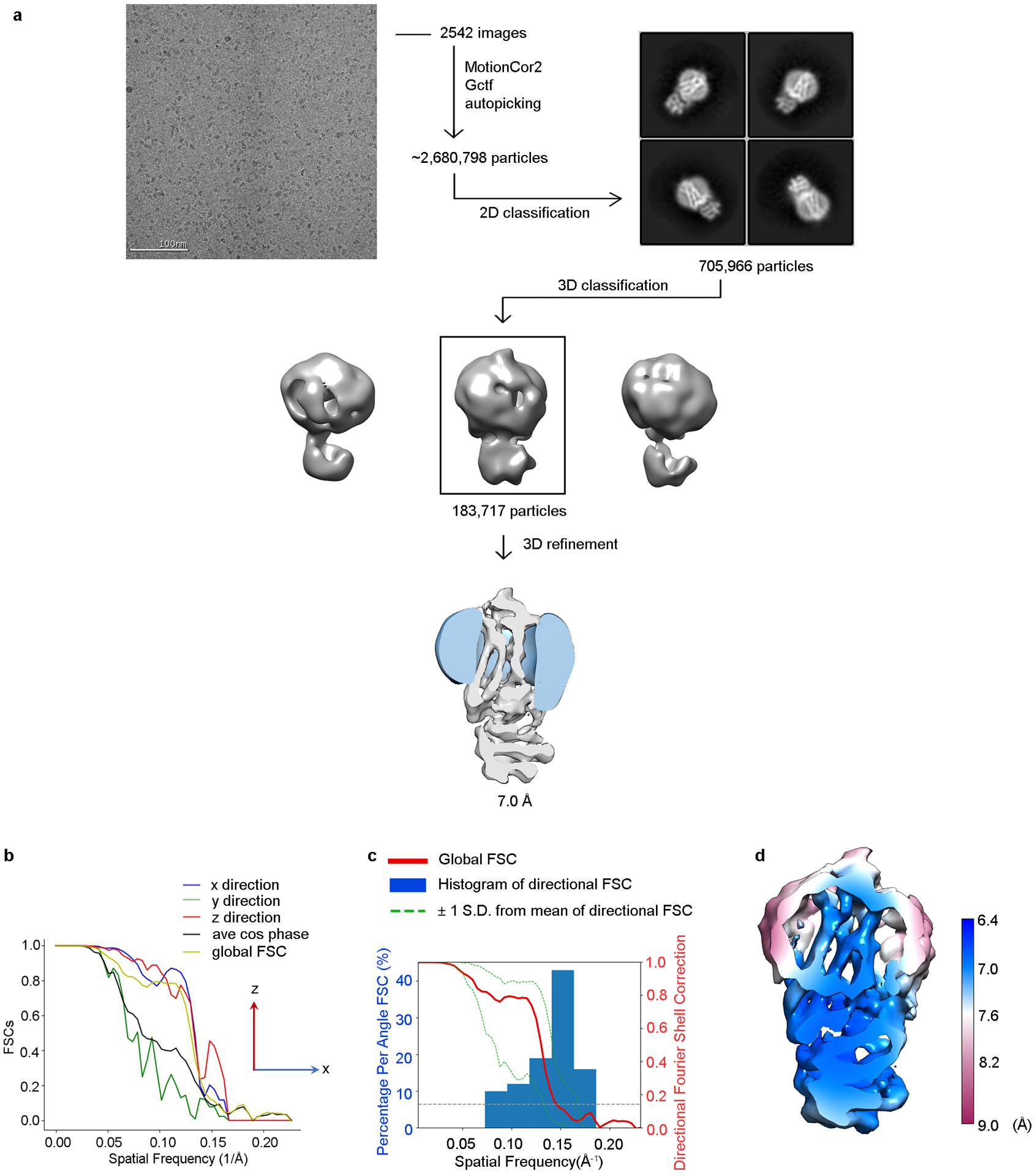
a) Representative raw cryo-EM micrograph from a total of 2,542. From these, 2.7 million particles were automatically picked in RELION-3 and were used to generate 2D class averages (shown are representative good classes). After screening out bad classes, 706 thousand particles remained. Three major classes from 3D classification were generated in RELION-3 using an initial model generated by cryoSPARC. Class 2 (183,717 particles) showed the best quality and was selected for 3D auto-refinement, resulting in a final map at a global resolution of 7.0 Å (FSC cut-off = 0.143). b) Directional FSC indicated that the resolutions in the x and z directions are similar to the global resolution, whereas the resolution in the y direction is lower due to less particles with this axis resolved. c) Plots of the global FSC together with the spread of directional resolution values defined by ±1 σ from the mean and a histogram of 100 such values evenly sampled over the 3D FSC. d) Local resolution map as estimated by RELION-3.
Extended Data Figure 3. Workflow of cryo-EM data processing and resolution analysis of Rho*–GRK1S5E/EE.
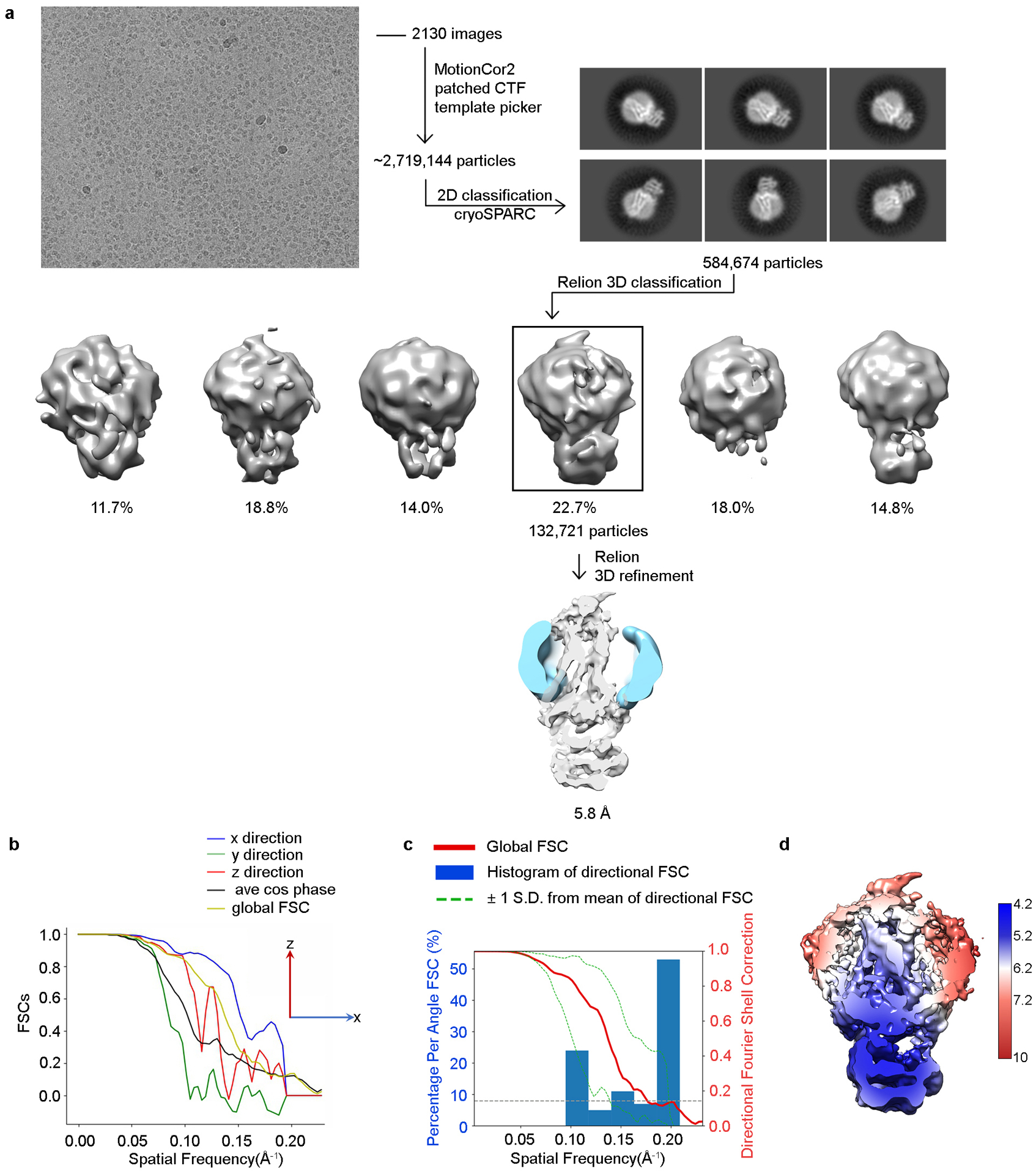
a) Representative raw cryo-EM micrograph from a total of 2,130. From these, 2.7 million particles were automatically picked in cryoSPARC and were used to generate 2D class averages (shown are representative good classes). After screening out bad classes, 585 thousand remaining particles were imported to RELION-3 and used to generate six major classes from 3D classification in RELION-3 using an initial model generated by cryoSPARC. Class 6 (132,721 particles) showed the best quality and was selected for 3D auto-refinement, resulting in a final map at a global resolution of 5.8 Å (FSC cut-off = 0.143). b) Directional FSC indicates that the resolutions in the x and z directions are similar to the global resolution, whereas the resolution in the y direction is lower. c) Plots of the global FSC together with the spread of directional resolution values defined by ±1 σ from the mean and a histogram of 100 such values evenly sampled over the 3D FSC. d) Local resolution map as estimated by RELION-3.
Extended Data Figure 4. Workflow of cryo-EM data processing and resolution analysis of Rho*–GRK1S5E/EE–Fab1.
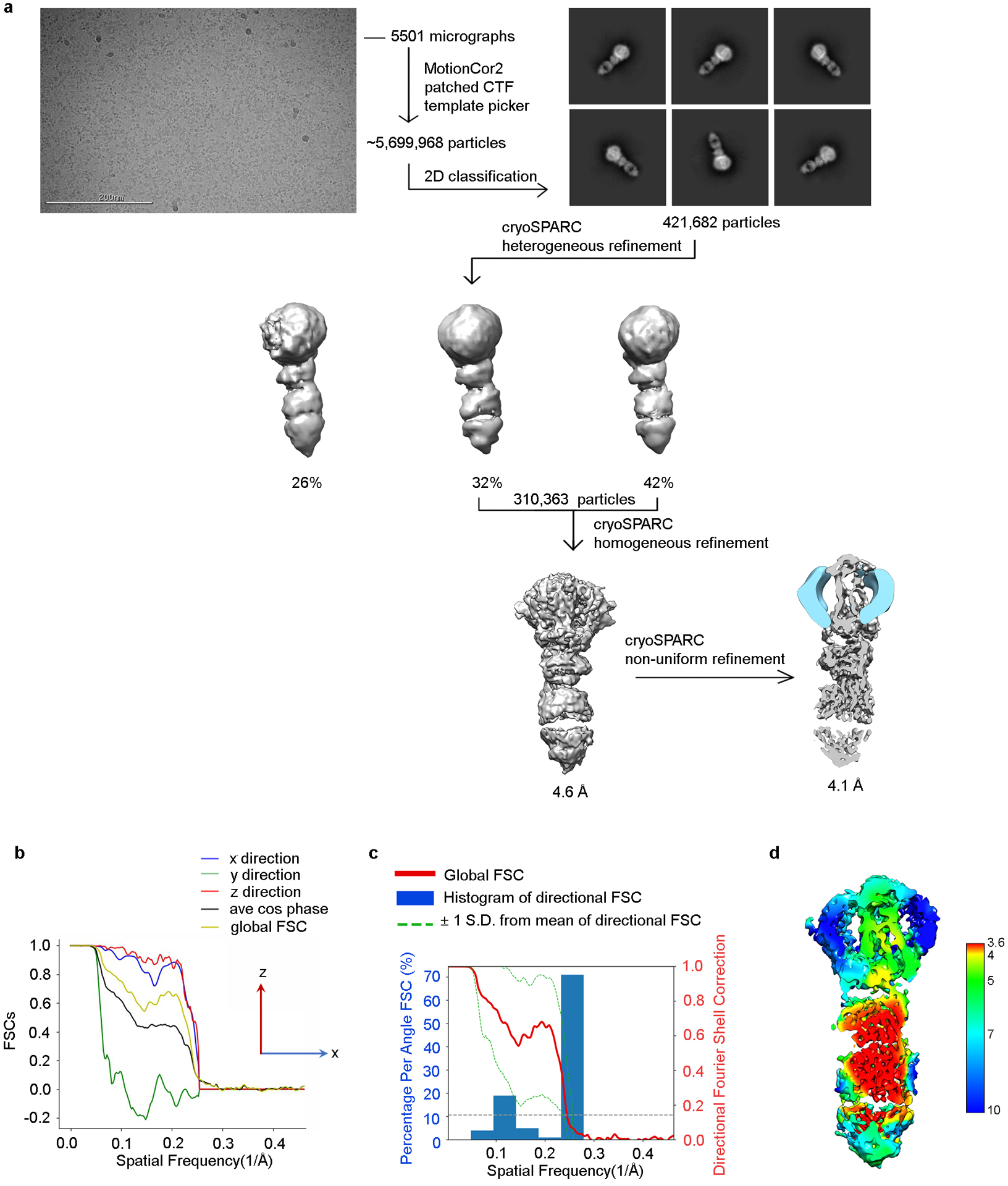
a) Representative raw cryo-EM micrograph from a total of 5,501. From these, 5.7 million particles were automatically picked in cryoSPARC and were used to generate 2D class averages (shown are representative good classes). After screening out bad classes, the remaining 422 thousand particles were further processed using heterogeneous refinement. Class 2 and Class 3 showed similar quality and were selected for homogeneous refinement and then non-uniform refinement in cryoSPARC, resulting in a final map at a global resolution of 4.1 Å (FSC cut-off = 0.143). b) Directional FSC indicates that the resolutions in the x and z directions are similar to the global resolution, whereas the resolution in the y direction was lower. c) Plots of the global FSC together with the spread of directional resolution values defined by ±1 σ from the mean and a histogram of 100 such values evenly sampled over the 3D FSC. d) Local resolution map as estimated by cryoSPARC.
Extended Data Figure 5. Flow chart of cryo-EM data processing and resolution analysis of Rho*–GRK1S5E/EE−Fab6.
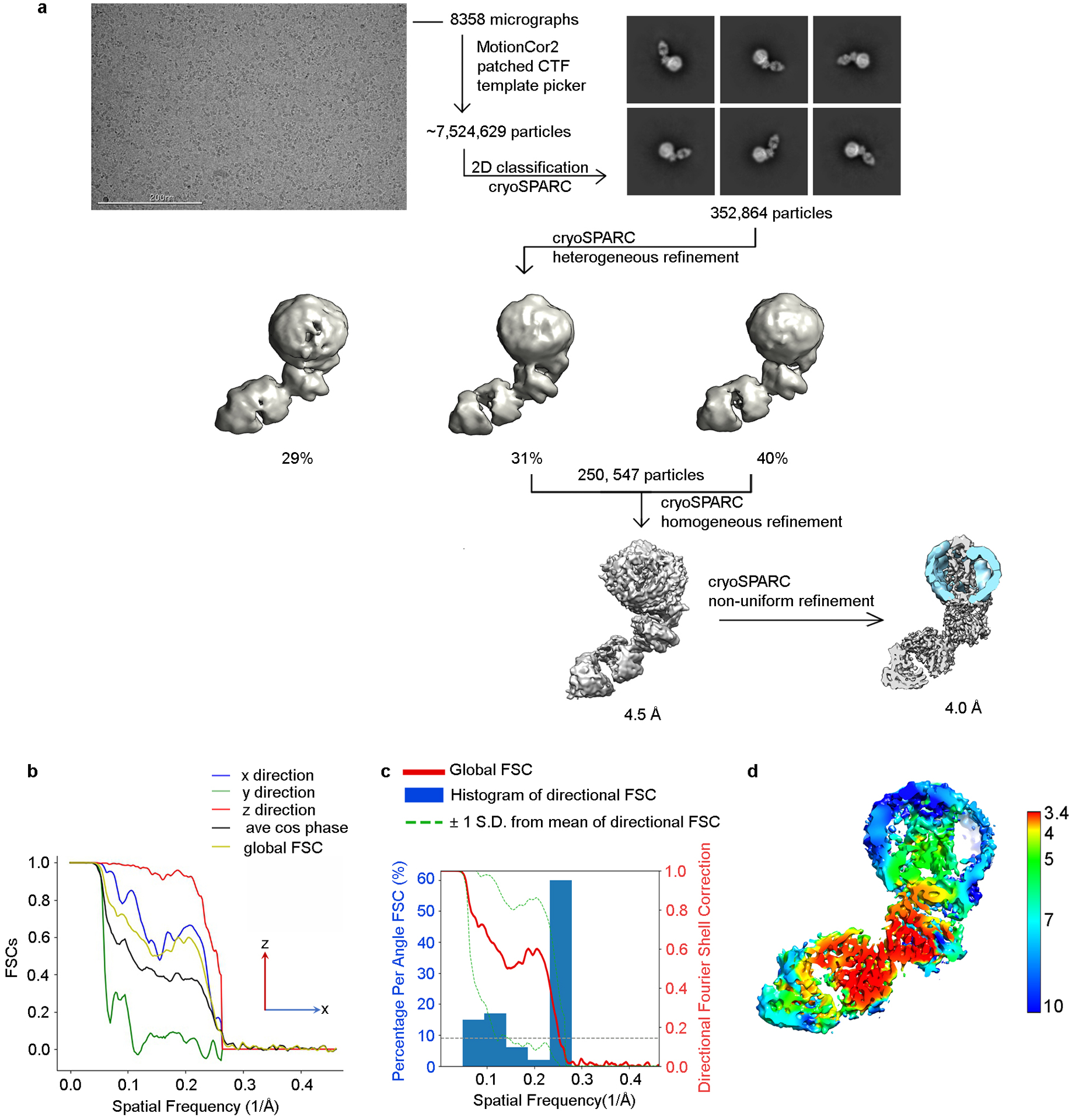
a) A representative raw cryo-EM micrograph from a total of 8,358. From these, 7.5 million particles were automatically picked in cryoSPARC and were used to generate 2D class averages (shown are representative good classes). After screening out bad classes, the remaining 353 thousand particles were further processed using heterogeneous refinement. Class 2 and Class 3 showed similar quality and were selected for homogeneous refinement and then non-uniform refinement in cryoSPARC. The resolution of the final map is estimated to be 4.0 Å. Global resolution was determined by FSC with a cut-off of 0.143. b) Directional FSC indicates that the resolutions in the x and z directions are similar to the globular resolution, whereas the resolution in the y direction was lower. c) Plots of the global FSC together with the spread of directional resolution values defined by ±1 σ from the mean and a histogram of 100 such values evenly sampled over the 3D FSC. d) Local resolution map as estimated by cryoSPARC.
Extended Data Figure 6. Assessment of ligand density, of the presence of all-trans retinal, and of the conformational heterogeneity of the GRK1 RH domain.
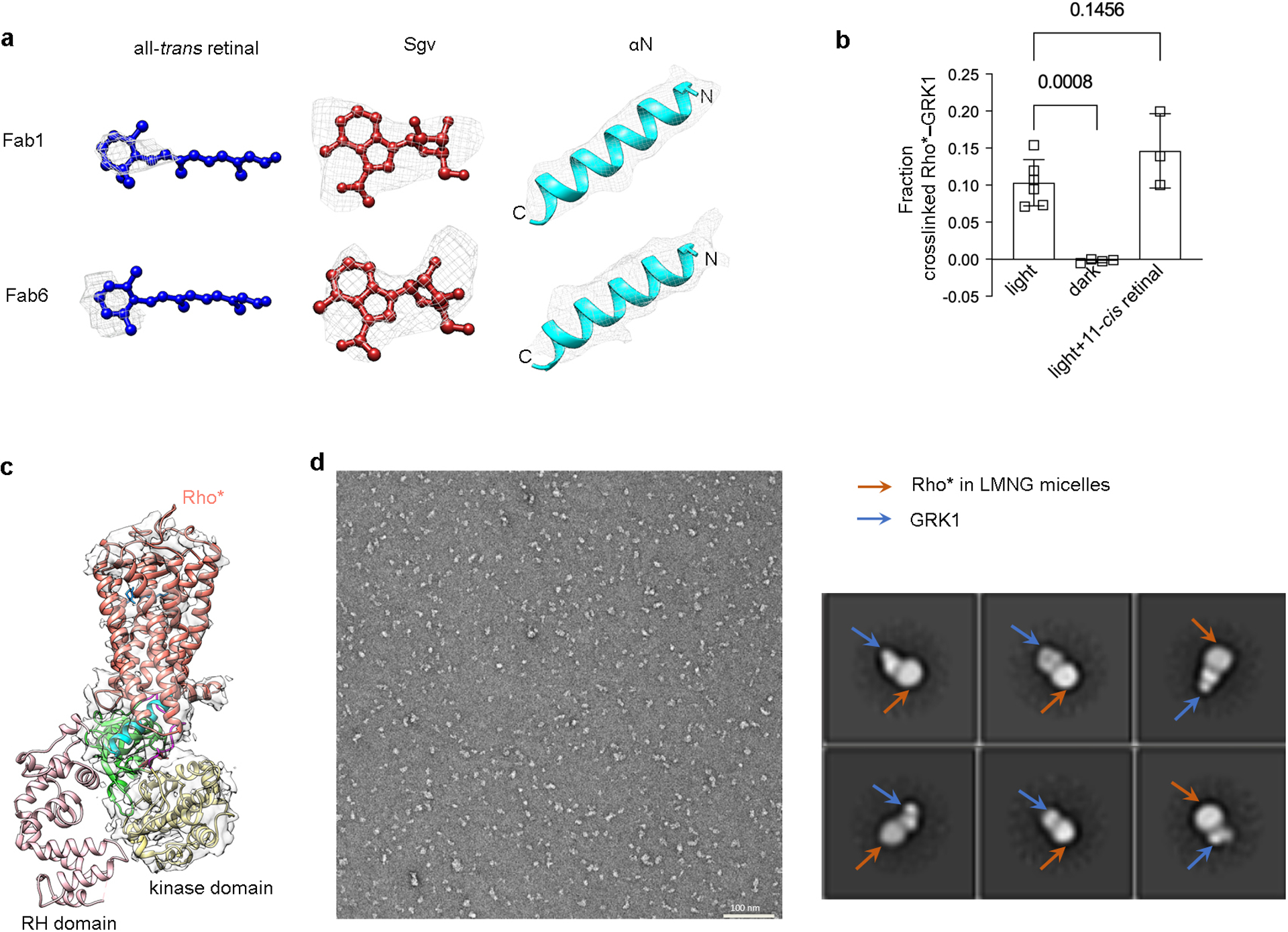
a) Electron density of all trans retinal, contoured at 17 σ (Fab1 map) and 13 σ (Fab6 map); Sgv, contoured at 24 σ (Fab1 map) and 26 σ (Fab6 map); αN, contoured at 21 σ (Fab1 map) and 25 σ (Fab6 map). c) Light and GRK ligand dependence of the crosslinking reaction between rhodopsin and GRK1. 11-cis retinal undergoes isomerization upon light exposure to all-trans retinal, which serves as a full agonist for rhodopsin. The crosslinking level of GRK1 with rhodopsin in the dark (n=4) and in the light with excess 11-cis retinal (n=3) were compared with that of GRK1 with rhodopsin in the light using one-way ANOVA followed by a Dunnett’s multiple comparison test. Symbols represent the mean value ± standard deviation. d) Density for the RH domain is not observed by cryo-EM in all our reconstructions. The map of Rho*–GRK1S5E/EE–Fab1 is shown here as an example. e) A representative negative stain EM micrograph of Rho*–GRK1 complex solubilized in LMNG (left) along with representative 2D averages (right), indicating heterogeneity in the bound GRK1. The smaller, variably positioned domain is interpreted as the RH domain.
Extended Data Figure 7. GRK αN interactions, kinase domain conformational changes, and comparison of the interactions between Rho* and its three principal downstream targets.
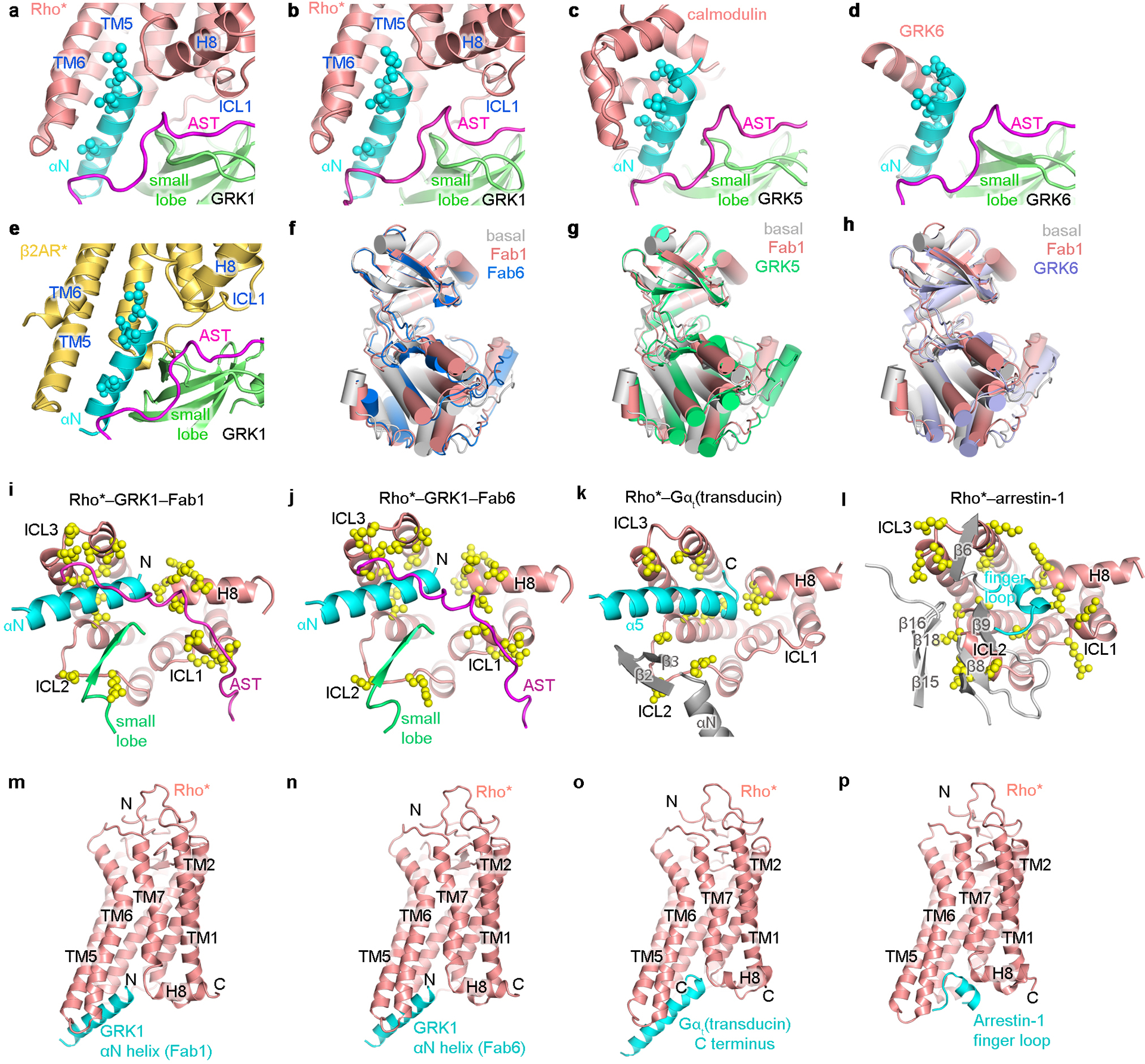
a-d) The GRK N terminus folds into a helix (αN) that packs against the small lobe and AST of the kinase domain, forming a docking side for either a-b) Rho* (GRK1 with Fab1 and Fab6 complexes, respectively), c) Ca2+·CaM (GRK5 in PDB entry 6PJX28), or d) a two-fold related crystal lattice contact (GRK6 in PDB entry 3NYN6). The interfaces shown are mediated by the same highly conserved hydrophobic residues (side chains shown with spheres). e) The cytoplasmic cleft and ICL1 of activated β2AR in its G protein bound conformation (PDB entry 3SN661) readily accommodates αN and AST of GRK1. f-h) The kinase domain small lobe from different “active” GRK structures were aligned to highlight differences in kinase domain closure in each. i-l) Comparison of downstream proteins bound to the cytoplasmic surface of Rho*. The side chains of residues interacting with i) GRK1 in the Fab1 complex (PDB entry 7MTA), j) GRK1 in the Fab6 complex (PDB entry 7MTB), k) Gαt (transducin) (PDB entry 6OYA14) and l) arrestin-1 (PDB entry 5W0P32) are shown as yellow spheres. Note that αN of GRK1 and α5 of Gα bind with opposite polarity. m, n) Cartoon representations of GRK1 αN helix docked to Rho* in the Fab1 and Fab6 models, respectively. o) Gαt C terminus bound to Rho* (PDB entry 6OYA14). p) Arrestin-1 finger loop bound to Rho* (PDB entry 5W0P32).
Extended Data Figure 8. Development of GRK1 autophosphorylation mimetic variants to aid in the structure determination of the Rho*–GRK1 complex.
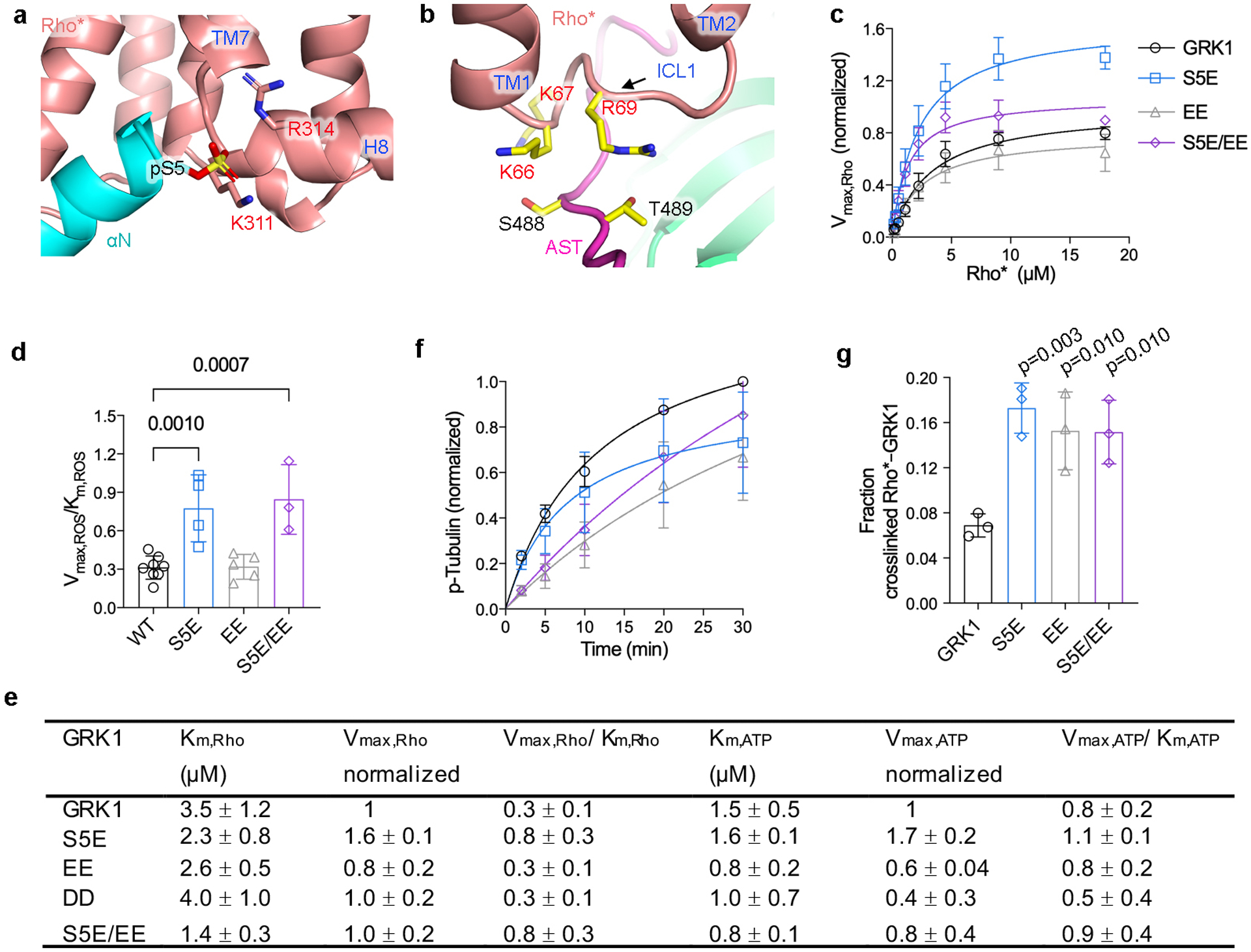
a) Interactions of GRK1 αN with the cytoplasmic cleft and H8 of Rho*. GRK1-Ser5 was modeled in a phosphorylated state to demonstrate proximity with Rho*-Lys311 and Arg314. b) GRK1 AST interaction with ICL1. some of the key participating residues are shown with stick side chains. Ser488 and Thr489 are autophosphorylation sites in GRK1. c-e) Kinetic analysis of GRK1 and phosphomimetic mutant of Rho* phosphorylation: S5E; S488E and T489E (EE); S488D and T489D (DD); S5E, S488E, and T489E (S5E/EE). One-way ANOVA followed by Dunnett’s multiple comparison test was carried out to compare each mutant with GRK1. Reactions were performed in 50 mM HEPES (pH 8.0), 10 mM MgCl2 for 2 minutes at room temperature. Data was normalized to the Vmax,Rho or Vmax,ATP of GRK1. For Rho kinetics, S5E, n=4; EE, n=5; DD, n=3; S5E/EE, n=3. For ATP kinetics, S5E, n=3; EE, n=3; DD, n=4; S5E/EE, n=4. f) Time courses of tubulin phosphorylation by GRK1 and variants are similar. Data was normalized to the phosphorylation level of tubulin by GRK1 at 30 minutes (n=3). g) Crosslinking yield of GRK1 variants relative to GRK1 (n=3). For gel source data, see Supplementary Fig. 10g. Error bars in panels above represent standard deviation from three or more experiments.
Extended Data Table 1.
GRK mutations and their impact on kinase activity towards Rho* or a soluble substrate.
| αN Helix | Rho* | soluble substrate | Ref | |
|---|---|---|---|---|
| ΔN19 | GRK1 | ---- † | --- | 29 |
| D2A | GRK1 | NC | NC | 62 |
| D3A | GRK2 | - | ND§ | 5,34 |
| D3K | GRK2 | ---- | NC | 34 |
| S5A | GRK1 | -- | ND | 45 |
| L6A | GRK1 | --- | + | 35 |
| L4A | GRK2 | -- | + | 5 |
| L3Q | GRK5 | -- | ND | 7 |
| L3A | GRK6 | - | NC | 6 |
| E7A | GRK1 | --- | + | 35 |
| E4A | GRK6 | - | - | 6 |
| T8A | GRK1 | - | ND | 45 |
| V9A | GRK1 | ---- | + | 35 |
| I6A | GRK6 | --- | NC | 6 |
| V10A | GRK1 | ---- | + | 35 |
| L7A/8A | GRK2 | ---- | ND | 34 |
| I6E/V7E | GRK6 | ---- | NC | 6 |
| A11E | GRK1 | - | - | 35 |
| N12A | GRK1 | ---- | --- | 35 |
| D10A | GRK2 | - | - | 5 |
| N9A | GRK6 | ---- | -- | 6 |
| T10P | GRK5 | -- | NC | 7 |
| F15A | GRK1 | ---- | ---- | 35 |
| L12A | GRK6 | ---- | - | 6 |
| I16A | GRK1 | -- | -- | 35 |
| M15A | GRK2 | NC | + | 5 |
| A18E | GRK1 | -- | -- | 35 |
| M17A | GRK2 | - | - | 5 |
| E18A | GRK2 | + | ND | 5 |
| AST region | Rho* | soluble substrate | Ref | |
| P473A | GRK2 | NC | ND | 63 |
| R474A | GRK2 | NC | ND | 5 |
| G475I | GRK2 | - | - | 5 |
| E476K | GRK2 | NC | ND | 63 |
| V476A | GRK1 | ---- | --- | 29 |
| N478A | GRK2 | NC | ND | 5 |
| A479S | GRK2 | NC | ND | 5 |
| A480S | GRK2 | NC | ND | 5 |
| N480A | GRK1 | + | ND | 29 |
| A482I | GRK2 | - | ND | 5 |
| Q482A | GRK1 | + | ND | 29 |
| D483A | GRK1 | - | ND | 29 |
| D484A | GRK2 | NC | ND | 63 |
| V484A | GRK1 | ---- | ---- | 29 |
| F487A | GRK1 | - | ND | 29 |
| S488E/T489E | GRK1 | NC | - | ¶ |
| S5E/S488E/T489E | GRK1 | + | NC | ¶ |
| E490K | GRK2 | NC | ND | 63 |
| G495A | GRK2 | NC | ND | 63 |
| L499D | GRK2 | NC | ND | 63 |
| Small lobe | Rho* | soluble substrate | Ref | |
| F190A | GRK1 | --- | ---- | 29 |
| F190H | GRK1 | + | ND | 29 |
| R191A | GRK1 | ---- | ---- | 29 |
| R190A | GRK6 | ---- | ND | 29 |
| V192A | GRK1 | NC | ND | 29 |
| R195A | GRK1 | - | ND | 29 |
| Q205A | GRK1 | + | ND | 29 |
| G210E | GRK1 | -- | ND | 29 |
| L212A | GRK1 | --- | --- | 29 |
| N268A | GRK1 | NC | ND | 29 |
| Large lobe | Rho* | soluble substrate | Ref | |
| H280A | GRK2 | NC | ND | 63 |
| Y274A | GRK1 | ---- | ---- | 29 |
| Y274F | GRK1 | + | ND | 29 |
| Y281A | GRK2 | - | - | 63 |
| H275A | GRK1 | + | ND | 29 |
| S284A | GRK2 | NC | ND | 63 |
| Q285A | GRK2 | NC | ND | 63 |
| Q285K | GRK2 | NC | ND | 63 |
| K395A | GRK2 | NC | ND | 63 |
| K397A | GRK2 | NC | ND | 63 |
| RH domain | Rho* | soluble substrate | Ref | |
| 5A | GRK1 | ---- | ND | ¶ |
| D164A | GRK1 | - | ND | 45 |
| L166K | GRK1 | - | ND | 45 |
| D164A/L166K | GRK1 | - | ND | 45 |
| L33N | GRK2 | NC | NC | 2 |
| E36A | GRK2 | NC | NC | 65,2 |
| V42E | GRK2 | -- | ND | 65 |
| Y46A | GRK2 | - | ND | 65 |
| R68A | GRK5 | + | ND | 66 |
| P61A/Q69A | GRK5 | = | ND | 66 |
| L66A/H38A | GRK5 | --- | ND | 66 |
| L66A/P73A | GRK5 | --- | ND | 66 |
| L66A/E514A | GRK5 | --- | ND | 66 |
| L66A/P37A | GRK5 | --- | ND | 66 |
| L66A/R69A | GRK6 | -- | ND | 66 |
| L66A/Q172A | GRK6 | ---- | ND | 66 |
| F166A/H38A | GRK5 | --- | ND | 66 |
| F166A/E514A | GRK5 | --- | ND | 66 |
| F166A/P61A | GRK5 | --- | - | 66 |
| F166A/L66A | GRK5 | --- | - | 66 |
| F166A/Q172A | GRK5 | --- | ND | 66 |
| Y166A/L176A | GRK6 | --- | ND | 66 |
| F166A/W173A | GRK5 | --- | - | 66 |
| Q172A/I176A | GRK5 | --- | - | 66 |
| Q172A/W173A | GRK6 | ---- | ND | 66 |
| E96A | GRK2 | NC | ND | 65 |
| R516A | GRK2 | NC | ND | 65 |
| E520A | GRK2 | -- | ND | 65 |
| E532A | GRK2 | NC | NC | 2 |
| L536N | GRK2 | NC | NC | 2 |
| L547N | GRK2 | NC | NC | 2 |
| P638D | GRK2 | -- | ND | 65 |
| V92A | GRK5 | ND | NC | 31 |
| V92L | GRK5 | ND | NC | 31 |
| V92M | GRK5 | ND | NC | 31 |
| K454A | GRK5 | ND | NC | 31 |
| R455A | GRK5 | ND | NC | 31 |
| K454A/R455A | GRK5 | ND | NC | 31 |
| I39E/I165E | GRK6 | NC | ND | 67 |
| I165E/F527D | GRK6 | NC | ND | 67 |
----, >100-fold reduction; ----, 20–100 fold reduction; ---, 10–20 fold reduction; --, 5–10 fold reduction; -, 3–5 fold reduction.
NC, no change.
ND, not determined.
+, 1–3 fold increase.
Mutation is characterized in the current paper.
Extended Data Table 2.
Cryo-EM data collection, refinement, and validation statistics.
| Rho*–GRK1 (ensemble1†) |
Rho*–GRK1S5E/EE (ensemble1) |
Fab1 (ensemble1) |
Fab6 (ensemble1) |
|
|---|---|---|---|---|
| Data collection and processing | ||||
| Magnification | 81,000 x | 81,000 x | 81,000 x | 81,000 x |
| Voltage (kV) | 300 | 300 | 300 | 300 |
| Electron exposure(e-/Å2) | ~54 | ~54 | ~54 | ~54 |
| Defocus range (μm) | 1.2 – 2.5 | 1.2 – 2.5 | 1.2 – 2.5 | 1.2 – 2.5 |
| Pixel size (Å) | 1.08 | 1.08 | 1.08 | 1.08 |
| Symmetry imposed | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 2,680,798 | 2,719,144 | 5,699,968 | 7,524,629 |
| Final particle images (no.) | 183,717 | 132,721 | 310,363 | 250,547 |
| Map resolution (Å) | 7.0 | 5.8 | 4.1 | 4.0 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 6 – 11 | 4.2–11 | 3.6–10 | 3.4–10 |
| Refinement with MDFF | ||||
| Initial model used(PDB entry) | Rho*: 3PQR; GRK1: a homology model of 3NYN, Fab models: SWISSMODEL68 | |||
| Model composition | ||||
| Non-hydrogen atoms | 5358 | 5363 | 8699 | 8707 |
| Residues | Protein: 670 | Protein: 670 | Protein:1108 | Protein: 1109 |
| Ligands | Sgv: 1 Ret: 1 | Sgv: 1 Ret: 1 | Sgv: 1 Ret: 1 | Sgv: 1 Ret: 1 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.019 | 0.019 | 0.143 | 0.019 |
| Bond angles (°) | 2.01 | 1.95 | 2.88 | 2.02 |
| Validation | ||||
| MolProbity score | 1.31 | 1.26 | 1.22 | 1.17 |
| Clash score | 0.48 | 0.48 | 0.24 | 0.18 |
| Poor rotamers (%) | 1.52 | 1.52 | 1.46 | 1.46 |
| Ramachandran Plot | ||||
| Favored (%) | 91.6 | 92.8 | 91.4 | 92.3 |
| Allowed (%) | 6.3 | 5.9 | 6.2 | 6.1 |
| Disallowed (%) | 2.1 | 1.4 | 2.4 | 1.6 |
The ensemble most similar to all other ensembles (lowest backbone RMSD over the Rho*–GRK1 complex residues) was used to generate statistics.
Supplementary Material
Acknowledgement
This work was supported by National Institutes of Health grants HL071818, HL122416 and CA221289 (to J.J.G.T.), GM117372 (to A.A.K.), GM095832, GM105942, GM109896, and GM105920 (to P.C.A.), by an American Heart Association Post-Doctoral Fellowship 19POST34450193 (to Q.C.), and National Science Foundation grant MCB-1517617 to X.Q. J.J.G.T. was also supported by the Walther Cancer Foundation. We thank the Purdue Cryo-EM Facility for equipment access and support.
Footnotes
Competing interests
All authors declare no conflict of interest.
Data availability.
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The structures of the four Rho*–GRK1 complexes: Rho*–GRK1, Rho*–GRK1S5E/EE, Rho*–GRK1S5E/EE–Fab1, and Rho*–GRK1S5E/EE–Fab6, and their associated data have been deposited into the Protein Data Bank under accession codes 7MT9, 7MT8, 7MTA, and 7MTB, and the Electron Microscopy Data Bank under accession codes EMD-23978, 23977, 23979, and 23980, respectively. CLMS data have been deposited to the ProteomeXchange Consortium via the PRIDE60 partner repository with dataset identifier PXD019215.
References:
- 1.Gurevich EV, Tesmer JJ, Mushegian A & Gurevich VV G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther 133, 40–69, doi: 10.1016/j.pharmthera.2011.08.001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cato MC et al. The open question of How GPCRs interact with GPCR kinase (GRKs). Biomolecules 11, 447 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komolov KE et al. Structural and Functional Analysis of a beta2-Adrenergic Receptor Complex with GRK5. Cell 169, 407–421 e416, doi: 10.1016/j.cell.2017.03.047 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He Y et al. Molecular assembly of rhodopsin with G protein-coupled receptor kinases. Cell Res 27, 728–747, doi: 10.1038/cr.2017.72 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beautrait A et al. Mapping the putative G protein-coupled receptor (GPCR) docking site on GPCR kinase 2: insights from intact cell phosphorylation and recruitment assays. J Biol Chem 289, 25262–25275, doi: 10.1074/jbc.M114.593178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boguth CA, Singh P, Huang CC & Tesmer JJ Molecular basis for activation of G protein-coupled receptor kinases. EMBO J 29, 3249–3259, doi: 10.1038/emboj.2010.206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noble B, Kallal LA, Pausch MH & Benovic JL Development of a yeast bioassay to characterize G protein-coupled receptor kinases. Identification of an NH2-terminal region essential for receptor phosphorylation. J Biol Chem 278, 47466–47476, doi: 10.1074/jbc.M308257200 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Palczewski K, Buczylko J, Lebioda L, Crabb JW & Polans AS Identification of the N-terminal region in rhodopsin kinase involved in its interaction with rhodopsin. J Biol Chem 268, 6004–6013 (1993). [PubMed] [Google Scholar]
- 9.Lefkowitz RJ Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) 190, 9–19, doi: 10.1111/j.1365-201X.2007.01693.x (2007). [DOI] [PubMed] [Google Scholar]
- 10.Brinks H & Koch WJ Targeting G protein-coupled receptor kinases (GRKs) in Heart Failure. Drug Discov Today Dis Mech 7, e129–e134, doi: 10.1016/j.ddmec.2010.07.007 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nogues L et al. G protein-coupled receptor kinases (GRKs) in tumorigenesis and cancer progression: GPCR regulators and signaling hubs. Semin Cancer Biol 48, 78–90, doi: 10.1016/j.semcancer.2017.04.013 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Kannan N, Haste N, Taylor SS & Neuwald AF The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc Natl Acad Sci U S A 104, 1272–1277, doi: 10.1073/pnas.0610251104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lodowski DT, Pitcher JA, Capel WD, Lefkowitz RJ & Tesmer JJ Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gbetagamma. Science 300, 1256–1262, doi: 10.1126/science.1082348 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Gao Y et al. Structures of the Rhodopsin-Transducin Complex: Insights into G-Protein Activation. Mol Cell 75, 781–790 e783, doi: 10.1016/j.molcel.2019.06.007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maeda S, Qu Q, Robertson MJ, Skiniotis G & Kobilka BK Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 364, 552–557, doi: 10.1126/science.aaw5188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang W et al. Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature 579, 303–308, doi: 10.1038/s41586-020-1953-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staus DP et al. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 579, 297–302, doi: 10.1038/s41586-020-1954-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato HE et al. Conformational transitions of a neurotensin receptor 1-Gi1 complex. Nature 572, 80–85, doi: 10.1038/s41586-019-1337-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pulvermuller A, Palczewski K & Hofmann KP Interaction between photoactivated rhodopsin and its kinase: stability and kinetics of complex formation. Biochemistry 32, 14082–14088, doi: 10.1021/bi00214a002 (1993). [DOI] [PubMed] [Google Scholar]
- 20.Pitcher JA, Freedman NJ & Lefkowitz RJ G protein-coupled receptor kinases. Annu Rev Biochem 67, 653–692, doi: 10.1146/annurev.biochem.67.1.653 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Kuhn H & Dreyer WJ Light dependent phosphorylation of rhodopsin by ATP. FEBS Lett 20, 1–6, doi: 10.1016/0014-5793(72)80002-4 (1972). [DOI] [PubMed] [Google Scholar]
- 22.Kuhn H, Cook JH & Dreyer WJ Phosphorylation of rhodopsin in bovine photoreceptor membranes. A dark reaction after illumination. Biochemistry 12, 2495–2502, doi: 10.1021/bi00737a020 (1973). [DOI] [PubMed] [Google Scholar]
- 23.Clifford-Nunn B, Showalter HD & Andrews PC Quaternary diamines as mass spectrometry cleavable crosslinkers for protein interactions. J Am Soc Mass Spectrom 23, 201–212, doi: 10.1007/s13361-011-0288-4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagen SE et al. Synthesis of CID-cleavable protein crosslinking agents containing quaternary amines for structural mass spectrometry. Org Biomol Chem 16, 8245–8248, doi: 10.1039/c8ob00329g (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayburt TH et al. Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J Biol Chem 286, 1420–1428, doi: 10.1074/jbc.M110.151043 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palczewski K, Kahn N & Hargrave PA Nucleoside inhibitors of rhodopsin kinase. Biochemistry 29, 6276–6282, doi: 10.1021/bi00478a024 (1990). [DOI] [PubMed] [Google Scholar]
- 27.Waldschmidt HV et al. Structure-Based Design, Synthesis, and Biological Evaluation of Highly Selective and Potent G Protein-Coupled Receptor Kinase 2 Inhibitors. J Med Chem 59, 3793–3807, doi: 10.1021/acs.jmedchem.5b02000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Komolov KE et al. Structure of a GRK5-Calmodulin Complex Reveals Molecular Mechanism of GRK Activation and Substrate Targeting. Mol Cell 81, 323–339 e311, doi: 10.1016/j.molcel.2020.11.026 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang CC, Yoshino-Koh K & Tesmer JJ A surface of the kinase domain critical for the allosteric activation of G protein-coupled receptor kinases. J Biol Chem 284, 17206–17215, doi: 10.1074/jbc.M809544200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madhusudan, Akamine P, Xuong NH & Taylor SS Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat Struct Biol 9, 273–277, doi: 10.1038/nsb780 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Yao XQ et al. Navigating the conformational landscape of G protein-coupled receptor kinases during allosteric activation. J Biol Chem 292, 16032–16043, doi: 10.1074/jbc.M117.807461 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou XE et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 170, 457–469 e413, doi: 10.1016/j.cell.2017.07.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farrens DL, Altenbach C, Yang K, Hubbell WL & Khorana HG Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274, 768–770, doi: 10.1126/science.274.5288.768 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Pao CS, Barker BL & Benovic JL Role of the amino terminus of G protein-coupled receptor kinase 2 in receptor phosphorylation. Biochemistry 48, 7325–7333, doi: 10.1021/bi900408g (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang CC, Orban T, Jastrzebska B, Palczewski K & Tesmer JJ Activation of G protein-coupled receptor kinase 1 involves interactions between its N-terminal region and its kinase domain. Biochemistry 50, 1940–1949, doi: 10.1021/bi101606e (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones Brunette AM, Sinha A, David L & Farrens DL Evidence that the Rhodopsin Kinase (GRK1) N-Terminus and the Transducin Galpha C-Terminus Interact with the Same “Hydrophobic Patch” on Rhodopsin TM5. Biochemistry 55, 3123–3135, doi: 10.1021/acs.biochem.6b00328 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Kelleher DJ & Johnson GL Characterization of rhodopsin kinase purified from bovine rod outer segments. J Biol Chem 265, 2632–2639 (1990). [PubMed] [Google Scholar]
- 38.Buczylko J, Gutmann C & Palczewski K Regulation of rhodopsin kinase by autophosphorylation. Proc Natl Acad Sci U S A 88, 2568–2572, doi: 10.1073/pnas.88.6.2568 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kunapuli P, Gurevich VV & Benovic JL Phospholipid-stimulated autophosphorylation activates the G protein-coupled receptor kinase GRK5. J Biol Chem 269, 10209–10212 (1994). [PubMed] [Google Scholar]
- 40.Premont RT, Koch WJ, Inglese J & Lefkowitz RJ Identification, purification, and characterization of GRK5, a member of the family of G protein-coupled receptor kinases. J Biol Chem 269, 6832–6841 (1994). [PubMed] [Google Scholar]
- 41.Ohguro H, Palczewski K, Ericsson LH, Walsh KA & Johnson RS Sequential phosphorylation of rhodopsin at multiple sites. Biochemistry 32, 5718–5724, doi: 10.1021/bi00072a030 (1993). [DOI] [PubMed] [Google Scholar]
- 42.Yang J et al. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol 9, 940–944, doi: 10.1038/nsb870 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Nobles KN et al. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal 4, ra51, doi: 10.1126/scisignal.2001707 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shoemaker BA, Portman JJ & Wolynes PG Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci U S A 97, 8868–8873, doi: 10.1073/pnas.160259697 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh P, Wang B, Maeda T, Palczewski K & Tesmer JJ Structures of rhodopsin kinase in different ligand states reveal key elements involved in G protein-coupled receptor kinase activation. J Biol Chem 283, 14053–14062, doi: 10.1074/jbc.M708974200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palczewski K et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745, doi: 10.1126/science.289.5480.739 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Pitcher JA et al. Feedback inhibition of G protein-coupled receptor kinase 2 (GRK2) activity by extracellular signal-regulated kinases. J Biol Chem 274, 34531–34534, doi: 10.1074/jbc.274.49.34531 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Papermaster DS Preparation of retinal rod outer segments. Methods Enzymol 81, 48–52, doi: 10.1016/s0076-6879(82)81010-0 (1982). [DOI] [PubMed] [Google Scholar]
- 49.Paduch M et al. Generating conformation-specific synthetic antibodies to trap proteins in selected functional states. Methods 60, 3–14, doi: 10.1016/j.ymeth.2012.12.010 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller KR et al. T cell receptor-like recognition of tumor in vivo by synthetic antibody fragment. PLoS One 7, e43746, doi: 10.1371/journal.pone.0043746 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suloway C et al. Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60, doi: 10.1016/j.jsb.2005.03.010 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, doi: 10.7554/eLife.42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332, doi: 10.1038/nmeth.4193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang K Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12, doi: 10.1016/j.jsb.2015.11.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296, doi: 10.1038/nmeth.4169 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Tan YZ et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat Methods 14, 793–796, doi: 10.1038/nmeth.4347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bouley R et al. Structural Determinants Influencing the Potency and Selectivity of Indazole-Paroxetine Hybrid G Protein-Coupled Receptor Kinase 2 Inhibitors. Mol Pharmacol 92, 707–717, doi: 10.1124/mol.117.110130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ludtke SJ, Baldwin PR & Chiu W EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128, 82–97, doi: 10.1006/jsbi.1999.4174 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Yang Z, Fang J, Chittuluru J, Asturias FJ & Penczek PA Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure 20, 237–247, doi: 10.1016/j.str.2011.12.007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perez-Riverol Y et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450, doi: 10.1093/nar/gky1106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rasmussen SG et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555, doi: 10.1038/nature10361 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu QM et al. The amino terminus with a conserved glutamic acid of G protein-coupled receptor kinases is indispensable for their ability to phosphorylate photoactivated rhodopsin. J Neurochem 73, 1222–1227, doi: 10.1046/j.1471-4159.1999.0731222.x (1999). [DOI] [PubMed] [Google Scholar]
- 63.Sterne-Marr R et al. GRK2 activation by receptors: role of the kinase large lobe and carboxyl-terminal tail. Biochemistry 48, 4285–4293, doi: 10.1021/bi900151g (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Komolov KE, Bhardwaj A & Benovic JL Atomic Structure of GRK5 Reveals Distinct Structural Features Novel for G Protein-coupled Receptor Kinases. J Biol Chem 290, 20629–20647, doi: 10.1074/jbc.M115.647297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lodowski DT et al. The role of G beta gamma and domain interfaces in the activation of G protein-coupled receptor kinase 2. Biochemistry 44, 6958–6970, doi: 10.1021/bi050119q (2005). [DOI] [PubMed] [Google Scholar]
- 66.Baameur F et al. Role for the regulator of G-protein signaling homology domain of G protein-coupled receptor kinases 5 and 6 in beta 2-adrenergic receptor and rhodopsin phosphorylation. Mol Pharmacol 77, 405–415, doi: 10.1124/mol.109.058115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lodowski DT, Tesmer VM, Benovic JL & Tesmer JJ The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J Biol Chem 281, 16785–16793, doi: 10.1074/jbc.M601327200 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Waterhouse A et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296–W303, doi: 10.1093/nar/gky427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Homan KT, Glukhova A & Tesmer JJ Regulation of G protein-coupled receptor kinases by phospholipids. Curr Med Chem 20, 39–46 (2013). [PubMed] [Google Scholar]
- 70.Song W, Yen HY, Robinson CV & Sansom MSP State-dependent Lipid Interactions with the A2a Receptor Revealed by MD Simulations Using In Vivo-Mimetic Membranes. Structure 27, 392–403 e393, doi: 10.1016/j.str.2018.10.024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palczewski K, Buczylko J, Kaplan MW, Polans AS & Crabb JW Mechanism of rhodopsin kinase activation. J Biol Chem 266, 12949–12955 (1991). [PubMed] [Google Scholar]
- 72.Benovic JL et al. Synthetic peptides of the hamster beta 2-adrenoceptor as substrates and inhibitors of the beta-adrenoceptor kinase. Br J Clin Pharmacol 30 Suppl 1, 3S–12S, doi: 10.1111/j.1365-2125.1990.tb05462.x (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Onorato JJ et al. Role of acidic amino acids in peptide substrates of the beta-adrenergic receptor kinase and rhodopsin kinase. Biochemistry 30, 5118–5125, doi: 10.1021/bi00235a002 (1991). [DOI] [PubMed] [Google Scholar]
- 74.Kim CM, Dion SB, Onorato JJ & Benovic JL Expression and characterization of two beta-adrenergic receptor kinase isoforms using the baculovirus expression system. Receptor 3, 39–55 (1993). [PubMed] [Google Scholar]
- 75.Choe HW et al. Crystal structure of metarhodopsin II. Nature 471, 651–655, doi: 10.1038/nature09789 (2011). [DOI] [PubMed] [Google Scholar]
- 76.Trabuco LG, Villa E, Mitra K, Frank J & Schulten K Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–683, doi: 10.1016/j.str.2008.03.005 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Benovic JL, Mayor F Jr., Somers RL, Caron MG & Lefkowitz RJ Light-dependent phosphorylation of rhodopsin by beta-adrenergic receptor kinase. Nature 321, 869–872, doi: 10.1038/321869a0 (1986). [DOI] [PubMed] [Google Scholar]
- 78.Ping YQ et al. Structures of the glucocorticoid-bound adhesion receptor GPR97-Go complex. Nature 589, 620–626, doi: 10.1038/s41586-020-03083-w (2021). [DOI] [PubMed] [Google Scholar]
- 79.Dhami GK & Ferguson SS Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacol Ther 111, 260–271, doi: 10.1016/j.pharmthera.2005.01.008 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Dhami GK et al. G Protein-coupled receptor kinase 2 regulator of G protein signaling homology domain binds to both metabotropic glutamate receptor 1a and Galphaq to attenuate signaling. J Biol Chem 279, 16614–16620, doi: 10.1074/jbc.M314090200 (2004). [DOI] [PubMed] [Google Scholar]
- 81.Iacovelli L et al. Regulation of group II metabotropic glutamate receptors by G protein-coupled receptor kinases: mGlu2 receptors are resistant to homologous desensitization. Mol Pharmacol 75, 991–1003, doi: 10.1124/mol.108.052316 (2009). [DOI] [PubMed] [Google Scholar]
- 82.Orban T et al. Substrate-induced changes in the dynamics of rhodopsin kinase (G protein-coupled receptor kinase 1). Biochemistry 51, 3404–3411, doi: 10.1021/bi300295y (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith JS, Lefkowitz RJ & Rajagopal S Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov 17, 243–260, doi: 10.1038/nrd.2017.229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weis WI & Kobilka BK The Molecular Basis of G Protein-Coupled Receptor Activation. Annu Rev Biochem 87, 897–919, doi: 10.1146/annurev-biochem-060614-033910 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reiter E, Ahn S, Shukla AK & Lefkowitz RJ Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol 52, 179–197, doi: 10.1146/annurev.pharmtox.010909.105800 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zoete V, Cuendet MA, Grosdidier A & Michielin O SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32, 2359–2368, doi: 10.1002/jcc.21816 (2011). [DOI] [PubMed] [Google Scholar]
- 88.Tajkhorshid E, Baudry J, Schulten K & Suhai S Molecular dynamics study of the nature and origin of retinal’s twisted structure in bacteriorhodopsin. Biophys J 78, 683–693, doi: 10.1016/S0006-3495(00)76626-4 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Phillips JC et al. Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802, doi: 10.1002/jcc.20289 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38, doi: 10.1016/0263-7855(96)00018-5 (1996). [DOI] [PubMed] [Google Scholar]
- 91.Gotze M et al. Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J Am Soc Mass Spectrom 26, 83–97, doi: 10.1007/s13361-014-1001-1 (2015). [DOI] [PubMed] [Google Scholar]
- 92.Kahraman A, Malmstrom L & Aebersold R Xwalk: computing and visualizing distances in cross-linking experiments. Bioinformatics 27, 2163–2164, doi: 10.1093/bioinformatics/btr348 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Iacobucci C et al. First Community-Wide, Comparative Cross-Linking Mass Spectrometry Study. Anal Chem 91, 6953–6961, doi: 10.1021/acs.analchem.9b00658 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D & Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods 14, 513–520, doi: 10.1038/nmeth.4256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kooistra AJ et al. GPCRdb in 2021: integrating GPCR sequence, structure and function. Nucleic Acids Res 49, D335–D343, doi: 10.1093/nar/gkaa1080 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bujacz A Structures of bovine, equine and leporine serum albumin. Acta Crystallogr D Biol Crystallogr 68, 1278–1289, doi: 10.1107/S0907444912027047 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Combe CW, Fischer L & Rappsilber J xiNET: cross-link network maps with residue resolution. Mol Cell Proteomics 14, 1137–1147, doi: 10.1074/mcp.O114.042259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The structures of the four Rho*–GRK1 complexes: Rho*–GRK1, Rho*–GRK1S5E/EE, Rho*–GRK1S5E/EE–Fab1, and Rho*–GRK1S5E/EE–Fab6, and their associated data have been deposited into the Protein Data Bank under accession codes 7MT9, 7MT8, 7MTA, and 7MTB, and the Electron Microscopy Data Bank under accession codes EMD-23978, 23977, 23979, and 23980, respectively. CLMS data have been deposited to the ProteomeXchange Consortium via the PRIDE60 partner repository with dataset identifier PXD019215.