

Keywords: chronic liver disease, exercise, muscle protein synthesis, nutrition, sarcopenia
Abstract
Sarcopenia, a condition of low muscle mass, quality, and strength, is commonly found in patients with cirrhosis and is associated with adverse clinical outcomes including reduction in quality of life, increased mortality, and posttransplant complications. In chronic liver disease (CLD), sarcopenia is most commonly defined through the measurement of the skeletal muscle index of the third lumbar spine. A major contributor to sarcopenia in CLD is the imbalance in muscle protein turnover, which likely occurs due to a decrease in muscle protein synthesis and an elevation in muscle protein breakdown. This imbalance is assumed to arise due to several factors including accelerated starvation, hyperammonemia, amino acid deprivation, chronic inflammation, excessive alcohol intake, and physical inactivity. In particular, hyperammonemia is a key mediator of the liver-gut axis and is known to contribute to mitochondrial dysfunction and an increase in myostatin expression. Currently, the use of nutritional interventions such as late-evening snacks, branched-chain amino acid supplementation, and physical activity have been proposed to help the management and treatment of sarcopenia. However, little evidence exists to comprehensively support their use in clinical settings. Several new pharmacological strategies, including myostatin inhibition and the nutraceutical Urolithin A, have recently been proposed to treat age-related sarcopenia and may also be of use in CLD. This review highlights the potential molecular mechanisms contributing to sarcopenia in CLD alongside a discussion of existing and potential new treatment strategies.
INTRODUCTION
Loss of skeletal muscle mass, quality, and strength, defined as sarcopenia, is a key component of malnutrition in cirrhosis (1, 2). The term “sarcopenia” was first proposed in 1989 to describe an age-related loss of muscle mass (3) but is now widely used to describe a loss of muscle mass and strength that occurs due to causes other than aging, including chronic inflammatory disease states (4). In contrast, the term cachexia is also used to describe a metabolic syndrome, which is associated with loss of muscle mass, with or without the presence of fat loss (5). Although the term cachexia has been linked with malnutrition within liver cirrhosis, the vast range of literature chooses to focus on the incorporation of the term sarcopenia. The prevalence of sarcopenia within patients with chronic liver disease (CLD) is estimated at ∼25%–70%, with higher rates identified among males (6). The presence of sarcopenia imposes significant clinical complications and has been associated with increased mortality (7), reduced quality of life (QoL) (8), increased length of hospital stays (9), and development of complications such as infections, both pre- and postliver transplant (LT) (10). Ultimately, sarcopenia inflicts a significant economic burden with increased health-care costs in patients with sarcopenic awaiting LT in comparison with patients with no sarcopenia (11).
Despite the known clinical problems associated with sarcopenia, the mechanisms that contribute to the development of this condition are still poorly understood. Current hypotheses suggest that a state of accelerated catabolism (12), hyperammonemia (13), and physical inactivity (14) may underpin impairments in muscle protein turnover. Specifically, these factors are thought to lead to a decrease in muscle protein synthesis (MPS) and an increase in muscle protein breakdown (MPB), although conclusive evidence is lacking (15). It is thus perhaps unsurprising that there are limited effective therapies to counteract sarcopenia in patients with CLD (15). To date, most treatment options have focused on supporting muscle anabolism through the use of physical activity (16) and nutritional interventions such as late-evening snacks (LES) and branched-chain amino acid (BCAA) supplementation (17). Therefore, this review provides an overview of the current understanding of sarcopenia in CLD, with a particular focus on liver cirrhosis and the molecular mechanisms potentially involved. We will also discuss the merits of current and emerging interventions to target sarcopenia.
DIAGNOSIS OF SARCOPENIA IN CHRONIC LIVER DISEASE
In age-related sarcopenia, the criteria for diagnosis have been agreed in Europe by the European Working Group on Sarcopenia in Older People (EWGSOP) (4). These criteria, updated in 2019, now include low muscle strength as the primary criterion, with low muscle quantity or quality and low physical performance as additional qualifying criteria (1). The presence of all three defines severe sarcopenia. Within CLD, there are currently no standardized definitions or cutoff values specific to liver-related sarcopenia (18). This is likely a consequence of the significant heterogeneity in the methods utilized to assess and define sarcopenia (Table 1) (18). This variability generates a major challenge and a degree of uncertainty that limits the assessment of sarcopenia in routine clinical care (19). Furthermore, many retrospective studies that have investigated the prevalence of sarcopenia only utilize one measure of sarcopenia, most commonly the measurement of muscle mass (10), potentially underestimating the prevalence and severity in this patient group.
Table 1.
Overview of modes used to assess sarcopenia in CLD
| Mode of Assessment | |
|---|---|
| Prescreen assessment | SARC-F Questionnaire |
| Muscle mass | MAMC, DEXA, BIA, Ultrasound, CT, MRI |
| Muscle strength | HGS, isokinetic dynamometry |
| Physical performance | SPPB, gait speed, Timed Up and Go, 6MWD, CPET, LFI |
BIA, bioelectrical impedance analysis; CLD, chronic liver disease; CPET, cardiopulmonary exercise test; CT, computed tomography; DEXA, dual-energy X-ray absorptiometry; HGS, handgrip strength; LFI, liver frailty index; MAMC, midarm muscle circumference; MRI, magnetic resonance imaging; 6MWD, 6-minute walk distance; SARC-F, simple five-item questionnaire; SPPB, short physical performance battery.
The most common measure of muscle mass in patients with CLD is the skeletal muscle mass index (SMI) of the third lumbar vertebrae (L3), imaged through computed tomography (CT) (15). Although SMI incorporates the measure of the psoas, paraspinal, and abdominal muscles at L3, the measurement of psoas muscle index (PMI) also obtained through CT imaging may be used (20, 21). Although CT scans are expensive and irradiating, abdominal imaging is often completed in the routine assessment of transplant candidates and cancer screening (7, 20), thereby allowing identification of sarcopenia without the need for additional imaging. However, the application of SMI in a clinical setting remains limited due to a lack of standardized definitions and sex-specific cutoff values. Nonetheless, recent work by Carey et al. (22) sought to generate specific SMI cutoff values for sarcopenia in patients with CLD. The authors reported these cutoffs as <50 cm2/m2 and <39 cm2/m2 in men and women with CLD, respectively. Importantly, these cutoff values were shown to correlate with LT waitlist mortality (22) and are considered to be the most robust definition of sarcopenia within patients with CLD to date (18). In contrast, when compared with the use of SMI, the use of PMI was unable to identify LT list patients with a higher mortality risk (20).
As mentioned earlier, the latest EWGSOP update shows appreciation of the importance of muscle strength and physical performance (1). As a result, the use of self-reported questionnaires that evaluate aspects of sarcopenia such as the simple five-item questionnaire (SARC-F) can be used to screen for the risk of sarcopenia development (23). The SARC-F incorporates questions that evaluate an individual’s muscle strength, ability to walk, rise from chairs, climb stairs, and the incidence of falls and has shown to be a valid assessment of sarcopenia risk (23). Additionally, high SARC-F scores have been more frequently identified with the progression of CLD (24). After the risk of sarcopenia has been identified, further assessment of muscle strength should be conducted. The use of handgrip strength (HGS) assessment has been identified as a key, reproducible measure of sarcopenia and is now typically incorporated in clinical assessments (25). HGS forms part of the recommended diagnostic criteria for patients with liver failure in both the European Association for the Study of Liver (EASL) and the European Society for Clinical Nutrition and Metabolism (ESPEN) guidelines (25, 26). HGS is seen as a valuable tool as it is a quick, validated, and cost-effective measure of muscle strength, which can be conducted at the bedside in patients with CLD, and correlates with mortality (27). In addition to muscle strength, measures of physical performance, and muscle function, including the 6-min walk distance (6MWD) (28), the short physical performance battery test (SPPB) (29), and gait speed (30) are all practical as a measure of sarcopenia in CLD. In patients with cirrhosis awaiting liver transplantation, a distance of <250 m achieved within the 6MWD (28) and each 1-unit decrease in SPPB score (29) are associated with an increase in mortality risk, while a reduced gait speed is associated with an increased risk of complications, such as hepatic encephalopathy (HE), gastrointestinal bleeding, and infections (30).
CLINICAL IMPACT OF SARCOPENIA WITHIN CHRONIC LIVER DISEASE
Sarcopenia has consistently been identified as a predictor of mortality risk, both pre-LT (7, 31) and post-LT (21). It has been shown that the addition of sarcopenia to the model for end-stage liver disease (MELD) scoring system improves the predictive accuracy of mortality, particularly in patients with MELD scores below 15 (31). Sarcopenia is also associated with several poor clinical outcomes including a risk of decompensation (32), HE (33), increased risk of infection (i.e., sepsis) (6, 7, 10), duration of mechanical ventilation, duration of hospital admission, and length of admission to intensive care post-LT (9, 34). It is therefore unsurprising that patients with sarcopenic have increased hospital costs in comparison to those associated with nonsarcopenic patients (11).
Although sarcopenia has clear implications for overall health outcomes, it is also associated with a reduction in QoL in patients with liver cirrhosis (8). Recent research found that patients with CLD with “presarcopenia,” defined as a loss of muscle mass without subsequent decreases in muscle strength and performance (4), experience greater reductions in QoL in comparison with patients with CLD without sarcopenia (8). One of the most frequently reported symptoms of CLD is sleep disturbance (35), which can have a large impact upon many aspects of QoL including fatigue (36). Sleep disturbances are often associated with the presence of HE and are believed to cause CLD owing to several mechanisms, which include alterations in the metabolism of melatonin and glucose (37) and disturbances to the intestinal microbiome (36). However, it is largely unclear whether the poor QoL identified within patients with CLD is because of muscle loss or functional disabilities (8). Furthermore, it is possible that factors such as clinical characteristics, disease severity, and cirrhosis-specific complications also contribute to reductions in QoL (38).
Although LTs are thought to reverse complications such as ascites and portal hypertension, sarcopenia may remain, or even worsen, post LT (39, 40). Indeed, previous research identified the presence of post-LT sarcopenia in both individuals with and without pre-LT sarcopenia, whereas only 6.1% of patients experienced a reversal of sarcopenia post LT (40). Alongside changes in muscle mass, patients often experience an increase in body weight post LT (39). This is largely attributed to an increase in fat mass (41), and a slower, incomplete restoration of muscle mass (39). These changes are thought to closely resemble the onset of sarcopenic-obesity, identified frequently in nonalcoholic fatty liver disease (NAFLD) (42). Collectively, current data highlight the need for better nutritional and functional assessment and the implementation of interventions before and after LT to reduce sarcopenia.
MECHANISMS OF MUSCLE LOSS IN CHRONIC LIVER DISEASE
Alterations in Muscle Protein Turnover and Anabolic Resistance
Skeletal muscle protein exists in a constant state of turnover, with simultaneous synthesis and breakdown (43). Alterations in muscle protein turnover occur daily owing to a number of environmental stimuli, such as the ingestion of dietary protein (44) and physical activity (45). These alterations in muscle protein turnover contribute to changes in net protein balance (NPB), whereby muscle loss may occur when MPB is greater than MPS, and muscle growth may occur when MPS is greater than MPB (43). Although protein ingestion alone can be sufficient to maintain muscle mass within young, healthy individuals (43), the combination of exercise and protein ingestion can synergistically augment MPS (46) (Fig. 1).
Figure 1.

Schematic representation of muscle protein turnover in response to anabolic stimuli (protein feeding with/without exercise) in healthy and chronic liver disease (CLD). A: we hypothesize that the primary reason for muscle loss in patients with CLD is blunted muscle protein synthesis (MPS) in response to protein ingestion that coincides with an increase in muscle protein breakdown (MPB). This likely equates to a reduction in net protein balance (NPB) within patients with CLD, in response to proposed alterations in muscle protein turnover. B: exercise in combination with protein ingestion may however partially restore the MPS response within patients with CLD patients.
Muscle protein turnover is regulated by several key molecular pathways, including the mechanistic target of rapamycin complex one (mTORC1) signaling cascade, satellite cell (SC) signaling, and the ubiquitin-proteasome pathway (UPP) (Fig. 2). mTORC1 is a critical serine/threonine protein kinase (47), which is activated by many factors including amino acids, growth factors, e.g., IGF-1, energy status, and mechanical stress (48). This subsequently leads to the phosphorylation of two key effectors eIF4E-binding protein 1 (4EBP1) and protein S6 kinase 1 (p70S6K1) (47). Additionally, SC plays a key role in the growth, repair, and regeneration of muscle fibers and is regulated by several growth factors such as IL-6, IGF-1, and myostatin (49–51). Several factors contribute to MPB including inflammation, inactivity, mitochondrial dysfunction, and myostatin. Myostatin is thought to inhibit MPS through the inhibition of the IGF/phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mTORC1 pathway and SCs (51–53). Additionally, myostatin has been suggested to activate the UPP, contributing to an increase in MPB, resulting in negative protein balance (52). However, the activation of Akt and inhibition of the transcription factor Forkhead box O (FOXO) can prevent the activation of the ubiquitin ligases, muscle RING finger-1 (MuRF-1), and muscle atrophy box (MAFbx) (54).
Figure 2.

Molecular regulation of muscle protein turnover. Muscle protein synthesis is regulated by several signals including energy status, amino acids, and growth factors, for example, insulin growth factor-1 (IGF-1). This leads to the activation of phosphoinositide 3-kinase (PI3K). Protein kinase B (Akt) regulates muscle protein turnover through the activation of mammalian target of rapamycin complex 1 (mTORC1) and inhibition of Forkhead box O (FOXO). In turn, the activation of mTORC1 leads to the phosphorylation of translation initiation factor 4E-binding protein 1 (4EBP1) and ribosomal protein S6 kinase (p70S6K) that contribute to muscle protein synthesis. Muscle protein breakdown can be activated by inflammatory cytokines, for example, tumor necrosis factor-α (TNF-α), which subsequently leads to the activation of nuclear factor-κB (NF-κB), myostatin, and the ubiquitin ligase Muscle RING finger-1 (MuRF-1). Furthermore, activation of the ubiquitin proteasome pathway through FOXO can increase MuRF-1 and muscle atrophy-box (MAFbx).
It is hypothesized that those suffering from CLD have a dysregulated muscle protein turnover that underpins the progression of sarcopenia (Fig. 1). Indeed, initial studies investigating muscle protein turnover in patients with cirrhosis suggested that protein synthesis may be lower in comparison with healthy controls when using arteriovenous (AV) balance and whole body tracer methodologies (55–57). However, for whole body protein breakdown (WbPB), previous work has yielded conflicting results, with different studies reporting that MPB rates in patients with cirrhosis increased, decreased, or even remained unaltered (57–60). These inconsistent findings are likely due to a range of factors including differences in methodology and patient characteristics such as age, severity of CLD, and disease etiology (15).
However, it is known that in age-related sarcopenia, a phenomenon known as muscle “anabolic resistance” is present, referring to a blunted MPS response to the ingestion of amino acids (61), or exercise (62), in comparison with young individuals. It has been proposed that patients with cirrhosis experience a similar state of muscle anabolic resistance (17), but to the best of our knowledge, no study has investigated the MPS response to the ingestion of protein or exercise stimuli in patients with CLD. The absence of such studies may be due to concerns relating to obtaining muscle biopsy samples from patients with CLD, owing to the perceived elevated risks related to platelet dysfunction, coagulopathy, and thrombocytopenia (63). However, more recently, muscle biopsies have been shown to be safe in patients with Child-Pugh class A cirrhosis (63), which will therefore enable a greater understanding of the dysregulation in muscle protein turnover that underpins muscle wasting in patients with CLD.
Hyperammonemia
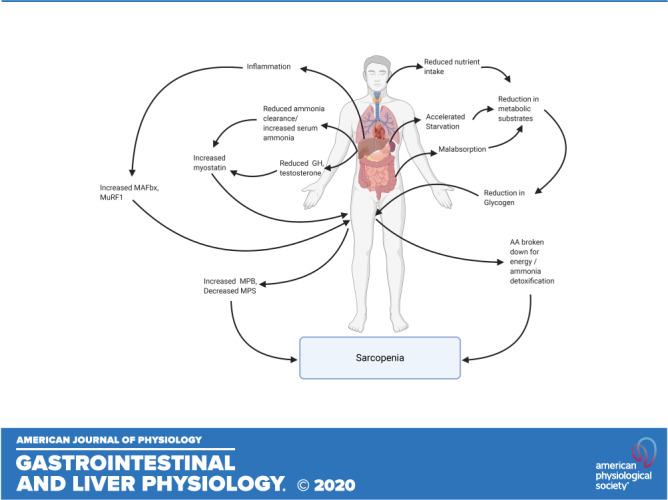
Hyperammonemia is a consistent abnormality in patients with cirrhosis caused by hepatocellular dysfunction, portosystemic shunting, and impaired ureagenesis, which in turn results in the increased concentration of ammonia within skeletal muscle (64, 65). The increased uptake of ammonia by skeletal muscle has been proposed as a protective mechanism in CLD, with the aim of preventing ammonia neurotoxicity (13, 64). The specific mechanism driving the increased uptake of ammonia is currently unknown, although it has been suggested that the expression of the ammonia transporters, Rh B glycoprotein (Rhbg) and Rh C glycoprotein (Rhcg) may both play a role (66). However, the resultant accumulation of ammonia is not without consequence and may contribute to the development of sarcopenia (13, 67, 68) (Fig. 2) thus reducing the muscle mass available to remove excess ammonia.
Mechanistically, in vitro experiments have shown that hyperammonemia-induced activation of NF-κB is associated with an increase in myostatin expression, an inhibitor of myogenesis, and a reduction in myotube diameter (13). In patients with cirrhosis, skeletal muscle hyperammonemia was also associated with an increase in myostatin expression and activation of NF-κB, resulting in impaired MPS, increased autophagy, and a reduction in skeletal muscle mass (13, 68–70). Therefore, hyperammonemia is considered a key regulator in the liver-muscle axis (13).
The elevated myostatin expression in patients with cirrhosis is thought to be one of the key drivers of muscle anabolic resistance (70), as it is associated with a reduction in p70S6K, s6 protein, and 4EBP1 protein content, indicating an impaired activation of the mTORC1 pathway that is critical in the regulation of MPS (63). This result is consistent with in vivo data, suggesting that myostatin inhibits MPS via impaired Akt signaling and subsequently reduces MPS via the Akt/mTOR/p70S6K pathway (53). Myostatin may also influence MPS through the impairment of SC function. In a portacaval anastomosis (PCA) rat model, myostatin expression was threefold higher than in control rats and was associated with a decline in SC function mediated via a reduction in myogenic transcription factors myoD, myf5, and myogenin (67, 71). In addition, hyperammonemia-induced upregulation of myostatin may contribute to a reduction in skeletal muscle mass through an increase in MPB. Patients with cirrhosis have been shown to exhibit increased WbPB, alongside an increase in the expression of myostatin, Beclin1, P62 degradation, and LC3 lipidation, representing an increase in autophagy compared with healthy adults (63). However, this increase in WbPB and autophagy occurred with no change in ubiquitinated proteins or MuRF1 mRNA at baseline, or after BCAA/leucine ingestion in healthy control and patients with cirrhosis (63, 68, 71). Collectively, this suggests that hyperammonemia-induced increases in MPB may largely occur through changes in autophagy rather than the UPP.
Hyperammonemia may also contribute to a decrease in MPS through mitochondrial dysfunction. Anaplerosis is the first reaction in the tricarboxylic acid (TCA) cycle, whereby glutamine and glutamate are converted to α-ketoglutarate (α-KG) and ammonia (72). This reaction is catalyzed by the enzyme glutamate dehydrogenase (GDH) (72), and under physiological conditions, this reaction is favored as GDH has a low affinity for ammonia, resulting in the conversion of glutamate to α-KG (73). However, owing to the high concentrations of ammonia present in the skeletal muscle of patients with cirrhosis; it is likely that cataplerosis, that is, the removal of TCA intermediates (72), may be favored and in turn cause a reduction in α-KG availability and impaired mitochondrial function (13, 15). The consequence of reduced TCA cycle intermediates is a reduction in ATP synthesis (74), which may contribute to a reduction in MPS, as translation initiation is an energy-intensive process (15). However, hyperammonemia has also been found to increase reactive oxygen species (ROS) and oxidative damage in both rats and humans (74). These data suggest that ROS may overwhelm the adaptive antioxidant defense systems and in turn lead to muscle loss and tissue injury (74). Thus, hyperammonemia may influence mitochondrial functioning through impairments in TCA cycle metabolism, oxidative damage, and, consequently, a decrease in cellular energy status and subsequently MPS. However, this remains to be clearly demonstrated.
Energy Expenditure and Amino Acid Availability in Chronic Liver Disease
Cirrhosis is associated with an accelerated state of starvation (12). An overnight fast has been shown to accelerate fat oxidation, gluconeogenesis, ketogenesis, and a catabolic state in patients with cirrhosis compared with healthy individuals (12, 75). Because of the increased gluconeogenesis, amino acids are often utilized as an energy source (12, 76), resulting in a low concentration of skeletal muscle BCAA in patients with cirrhosis (77). In response to a state of cellular stress, such as amino acid deprivation, an integrated stress response (ISR) is activated to promote the restoration of cellular homeostasis (78) (Fig. 3). In a state of amino acid deficiency, the ISR is mediated through the activation of general control nondepressed 2 (GCN2), an amino acid deficiency sensor (79). This leads to the phosphorylation of eukaryotic initiation factor 2 (eIF2α), which subsequently, causes a reduction in global protein synthesis and an increase in activating transcription factor 4 (ATF4) mRNA, thus reducing the requirement for amino acids (79, 80). The activation of ATF4 leads to the inhibition of mTORC1 signaling and promotes autophagy in an attempt to restore and preserve levels of amino acids (78). Once this balance has been restored, ATF4 signaling contributes to the dephosphorylation of eIF2α, restoring normal levels of protein synthesis (81).
Figure 3.

The proposed molecular alterations that contribute to ammonia-induced changes in muscle protein turnover. Ammonia enters the skeletal muscle through the Rhcg/Rhbg receptors. Subsequently, ammonia contributes to mitochondrial dysfunction, an impaired integrated stress response, and activation of myostatin. These contribute to an impairment in protein turnover and sarcopenia. 4EBP1, translation initiation factor 4E-binding protein 1; eIF2α, eukaryotic initiation factor 2α; IGF-1, insulin growth factor 1; MAFbx, muscle atrophy-box; mTORC1, mammalian target of rapamycin complex 1; MuRF1, muscle RING finger-1; NF-κB, nuclear factor-κB; PI3K, phosphoinositide 3-kinase; p70S6K, ribosomal protein S6 kinase; Rhbg, Rh B glycoprotein; Rhcg, Rh C glycoprotein; ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α; TCA cycle, tricarboxylic acid cycle; α-KG, α ketoglutarate.
Patients with cirrhosis display increased GCN2 activation and eIF2α phosphorylation alongside a reduction in mTORC1 signaling, similar to the ISR seen in response to intracellular amino acid deficiency (63, 69). However, hyperammonemia induces a state of cellular stress, which impairs the ISR by preventing an increase in ATF4 mRNA (69). This failure to induce an increase in ATF4 mRNA may contribute to a further decline in MPS and an increase in autophagy, owing to the inability to terminate the ISR and return to normal levels of protein synthesis (69, 78). Therefore, patients with cirrhosis have been suggested to experience an adaptive ISR, in which a second pathway is activated in response to increased ammonia concentrations (69) (Fig. 3). This second signaling pathway appears to be mediated by SLC7A5/LAT1, an amino acid transporter, which is increased in patients with cirrhosis (63, 69). SLC7A5/LAT1 appears to act as an amino acid exchanger, resulting in an increase in l-leucine uptake (63, 69). This increase in l-leucine concentration is thought to occur to utilize this amino acid to generate acetyl-CoA within the mitochondria, to generate an increase in energy output (82). Under normal conditions, the transport of leucine relies on glutamine transport via the glutamine exchanger SLC38A2 (83), but under a state of hyperammonemia, glutamine is primarily utilized for the detoxification of ammonia (69, 82). Taken collectively, the activation of the ISR in patients with cirrhosis results in an impairment in mTORC1 signaling and an increase in autophagy (63, 69), which may contribute to the development of sarcopenia.
Alcohol and Chronic Liver Disease
Both alcohol intake and CLD are believed to contribute to sarcopenia in alcohol-related liver disease (ArLD) (84). Therefore, it is often challenging to distinguish between the specific effects of CLD and that of alcohol. Although ethanol is primarily metabolized within the liver and the brain, it can also be metabolized within skeletal muscle (85, 86). ArLD is frequently associated with a reduction in skeletal muscle mass, alongside alterations in muscle protein turnover (86) and ethanol can inhibit mTORC1 stimulation (87). Furthermore, excessive alcohol intake has been linked to increased myostatin, which is assumed to mediate impairments in MPS (86, 88). Nonetheless, markers of the UPP remain unaltered in animal models of ArLD and are decreased in human ArLD patients, suggesting that autophagy is likely responsible for an increase in MPB (86). Similar to in hyperammonemia, ethanol may also contribute to reductions in MPS through impairment of mitochondrial function, resulting in the generation of ROS and activation of autophagy (89). In turn, the above may impair MPS through a reduction in ATP generation and mRNA translation (84).
Abnormalities in Endocrine Function
CLD is associated with several endocrine abnormalities that could contribute to sarcopenia, including low serum testosterone (90). In male patients with cirrhosis, low levels of testosterone may arise because of changes in the hypothalamic-pituitary-gonadal axis (91). This can lead to a decrease in testosterone production and an increase in aromatase activity, an enzyme responsible for the conversion of testosterone to estradiol, similar to that seen in aging males (92) and PCA rats (93). PCA rats exhibit lower levels of testosterone alongside a reduced growth rate, attributed to a reduction in food intake and efficiency, calculated as the increase in body weight per gram of food eaten (93). Inhibition of aromatase in this model led to an increase in testosterone and improved body weight, alongside improvements in food intake and efficiency (93). However, further research is required in patients with cirrhosis to confirm this relationship.
CLD is also associated with an increase in the secretion of growth hormone (GH) and a reduction in serum IGF-1 concentration (94), a change also seen in human aging. IGF-1 can promote muscle growth through the activation of the Akt/PI3K/mTORC1 signaling cascade, which leads to an increase in MPS (95, 96). Furthermore, IGF-1 also contributes to a reduction in muscle atrophy through the activation of Akt and inhibition of the transcription factor FOXO, in turn preventing the activation of the ubiquitin ligases MuRF-1 and MAFbx (54). GH has been associated with an increase in myostatin expression and impairments in IGF-1 signaling and therefore may further contribute to impairments in MPS (94, 97). Again, the relative contribution of changes in the GH/IGF-1 axis to sarcopenia in CLD remains poorly understood (15).
Nonalcoholic Fatty Liver Disease and Obesity
NAFLD is currently predicted to be the most common cause of CLD, with an estimated global prevalence of ∼25% (98). NAFLD is an umbrella term used to describe the spectrum of disease from nonalcoholic fatty liver (NAFL), identified through the presence of ≥5% steatosis, to nonalcoholic steatohepatitis (NASH) and cirrhosis (99, 100). In contrast, progression to NASH is characterized by the presence of hepatic steatosis alongside inflammation and hepatocellular injury with or without the presence of fibrosis, this, in turn, may lead to the development of cirrhosis and hepatocellular carcinoma (HCC) (99, 100). NAFLD is a growing cause of CLD, largely attributed to the increase in the incidence of obesity (101). However, obesity within this cohort may occur alongside the development of sarcopenia (sarcopenic obesity) (102). There are several risk factors associated with the development of both NAFLD and sarcopenia, with physical inactivity, insulin resistance, and chronic inflammation believed to play key roles (103). It is therefore unsurprising that NAFLD and sarcopenia are typically associated with increased sedentary behavior, alongside a reduction in physical activity (104, 105). Unfortunately, this physical inactivity likely contributes to an increased risk of metabolic syndrome due to an increase in visceral fat accumulation and insulin resistance (106).
A state of insulin resistance may directly lead to the development of sarcopenia through an increase in MPB and a decrease in MPS. Insulin resistance in adipose tissue stimulates lipolysis and the release of free fatty acids (FFAs) to the liver (107, 108). In turn, FFA inhibits IGF-1 signaling, which subsequently impairs PI3K/Akt signaling (109) and reduces MPS (110) (Fig. 4). Impairments in Akt signaling increase FOXO1 phosphorylation, which activates the UPP, leading to an increase in MAFbx and MuRF1 expression (109, 110). Combined, these alterations in protein turnover are thought to contribute to the muscle loss identified in type 2 diabetes (110). Therefore, increased insulin resistance may cause similar effects in patients with CLD with a NAFLD etiology. Insulin resistance also leads to the development of hyperinsulinemia within hepatocytes, which can contribute to a decrease in gluconeogenesis, an increase in lipogenesis, and inhibition of β-oxidation (107, 108). This leads to the accumulation of lipids within liver and muscle, termed myosteatosis (108, 110). Obesity and a reduction in muscle quality, calculated as the ratio of muscle strength to muscle mass, are often associated with the development of insulin resistance and a reduction in muscle mass and strength (111, 112). Furthermore, sarcopenia likely contributes to the development of insulin resistance, independent of obesity, as skeletal muscle is the primary tissue facilitating the uptake of glucose mediated by insulin (113). This suggests that the pathology of NAFLD and that of sarcopenia are closely intertwined (107).
Figure 4.

The molecular regulation of muscle protein synthesis and muscle protein breakdown in chronic liver disease (CLD). The mammalian target of rapamycin complex 1 (mTORC1) can be activated in response to the activation of insulin growth factor-1 (IGF-1), which leads to the activation of protein kinase B (Akt), and inhibited through general control nondepressed 2 (GCN2), activated by a reduction in amino acids (AAs) and ammonia. mTORC1 activation leads to the activation of translation initiation factor 4E-binding protein 1 (4EBP1) and ribosomal protein S6 kinase (p70S6K). Akt regulates FOXO, and the subsequent activation of muscle atrophy-box (MAFbx) and Muscle Ring Finger-1 (MuRF-1). Inflammatory cytokines, for example, tumor necrosis factor-α (TNF-α) and nuclear factor-κB (NF-κB) activate, leading to the activation of myostatin. Myostatin results in an increase in autophagy and inhibition of satellite cells. FOXO, Forkhead box O.
Chronic inflammation and oxidative damage are also important factors associated with the development of CLD (114) and sarcopenia (115). NAFLD is associated with an accumulation of FFAs and increased adiposity, which contribute to an increase in proinflammatory cytokines (116–118). Adipose tissue contains immune cells such as macrophages and also senescent cells, which are highly proinflammatory (119). In addition, adipose tissue produces its own cytokines, termed adipokines, which can also contribute to an increased inflammatory status (120). One key cytokine, TNF-α, appears to have a significant role in the impairment of muscle protein turnover within CLD (121, 122). In rats, TNF-α impairs MPS through a reduction in the phosphorylation of key signaling proteins involved in the mTORC1 pathway, such as eIF-4E (121). In addition, in vitro work has shown that TNF-α may increase MPB through an increase in UPP activity, as shown by an increase in atrogin-1 expression (122, 123). Further in vitro work has shown that TNF-α-induced MPB is thought to arise through an increase in type 1 TNF- receptor (TNFR-1) binding and consequently an increase in ROS, which leads to the subsequent activation of NF-κB (122, 124). An increase in NF-κB signaling enhances protein degradation through the activation of the UPP, reduction of myogenesis, and a further increase in inflammation (122, 125). This increase in proinflammatory cytokines may further contribute to the development of NAFLD and muscle loss (115, 126). Together, this suggests that an increase in circulating proinflammatory cytokines, in particular TNF-α, may contribute to a reduction in MPS and an increase in MPB.
Adiponectin
Adiponectin is an adipokine that plays an integral role in the regulation of glucose metabolism and oxidation of fatty acids in an insulin-dependent manner (127). In skeletal muscle, adiponectin binds to adiponectin receptor I (AdipoR1), which is responsible for the regulation of insulin sensitivity and fatty acid oxidation (128, 129). Therefore, a reduction in circulating levels of adiponectin as seen in obesity (130) may contribute to impairments in muscle protein turnover because of insulin resistance (131). In obese individuals, myostatin expression is positively associated with a state of insulin resistance (132). Therefore, this increase in myostatin is believed to contribute to muscle wasting through impairments in mTORC1 signaling and increased autophagy, as discussed previously (13, 53, 68). Therefore, it is plausible to suggest that cross talk between myostatin and adiponectin exists and warrants further investigation in CLD and NAFLD (107, 131).
Vitamin D
Vitamin D deficiency has been shown to contribute to insulin resistance, metabolic syndrome, and NAFLD (133, 134). Evidence from animal models of NAFLD suggests that rats fed a vitamin D-deficient Western diet experienced an increase in NAFLD activity, inflammation, and oxidative stress in comparison with those fed a vitamin D-replete Western diet (135). Thus, vitamin D deficiency may exacerbate NAFLD severity partly through an inflammatory-mediated mechanism (134). Vitamin D has also been suggested to contribute to an increased risk of age-related sarcopenia (136). In older individuals, low levels of vitamin D are independently associated with a greater risk of sarcopenia (136) and is thought to contribute to muscle loss through several mechanisms including a reduction in MPS, an increase in muscle atrophy, and mitochondrial dysfunction (137). However, it is currently unclear whether low 25-hydroxyvitamin D (25(OH)D) levels are a cause or a consequence of age-related sarcopenia (138). As patients with CLD experience vitamin D deficiency, similar to that identified within older sarcopenic individuals, it is possible that low levels of vitamin D contribute to the pathology of sarcopenia within these patients. However, patients with CLD often experience several fat-soluble vitamin deficiencies, including vitamin A, D, E, and K deficiencies, which are affected by malabsorption, impaired synthesis, and storage (139).
MANAGEMENT OF SARCOPENIA IN CLD: CURRENT AND EMERGING INTERVENTIONS
Although many complications associated with CLD such as ascites and encephalopathy can be treated by undergoing an LT, sarcopenia may develop, or be sustained, in patients post-LT (39, 40). Sarcopenia is commonly associated with adverse complications, but there are currently limited data, showing that improvements in muscle mass and strength improve survival both, before and after LT (15). Therefore, the management of sarcopenia in CLD and the treatment thereof is an urgent clinical priority. Inevitably, sarcopenia management in CLD requires a multifaceted approach including nutrition, exercise, and psychological and pharmacological interventions (25).
Nutrition
Patients with cirrhosis experience a persistent state of accelerated starvation, enhancing catabolism, in addition to a state of muscle anabolic resistance, resulting in blunted metabolic responses to anabolic stimuli such as nutritional intake (17). Furthermore, the daily intake of total calories, protein, and carbohydrates is consistently reported to be inadequate in ∼85%–95% of those undergoing assessment for LT, exacerbating malnutrition and more specifically, sarcopenia (140). International guidelines from the EASL and ESPEN suggest that nutritional interventions should include the optimal energy intake of 25–35 kcal/kg of dry body weight per day in well-compensated patients with cirrhosis, with a target protein intake of 1.2–1.5 g/kg of body weight per day (25, 26). However, stable patients with obesity should consider adopting a hypocaloric diet consisting of a 500–800 kcal/day caloric deficit while maintaining an optimal consumption of protein to promote the simultaneous reduction in adipose and maintenance of muscle mass, in conjunction with an exercise regime (25). In contrast, patients with decompensated cirrhosis require both an increased energy and protein intake, with an energy target of 35–40 kcal/kg and a protein target of 1.5–2 g/kg, respectively (25, 26).
To achieve the recommended energy intake, the consumption of more frequent meals and a late-evening snack (LES), consisting of 50 g of carbohydrate, are often recommended in patients with CLD owing to their accelerated state of starvation (25, 75). This accelerated catabolism may in turn contribute to the development of sarcopenia (17). Therefore, patients are advised to avoid prolonged periods of fasting through the ingestion of three main meals and three snacks (25). Because the most prolonged period of fasting occurs at night, an increased importance has been placed on the consumption of an LES and breakfast. The ingestion of a high-protein (21 g), high-calorie (500 kcal, 30% of daily calorie intake) breakfast has been shown to have beneficial effects on cognitive function (141).
Additionally, the consumption of BCAA as an LES has been shown to have several benefits, including improvements in QoL, nitrogen balance, respiratory quotient, carbohydrate utilization, and lean body mass (17, 142, 143). Recently, the use of BCAA supplementation as an LES in patients with cirrhosis has also been shown to improve HGS over a 24-wk intervention period (144). However, no change in body mass index (BMI) or lean body mass was identified, contradicting previous findings. The positive outcomes identified in response to short-term BCAA LES ingestion may be due to an increase in lipid utilization, amino acid availability, and reduction in gluconeogenesis, derived from the breakdown of protein in skeletal muscle (2, 17). This is consistent with indirect measures of MPS and MPB, which have shown an increase in nitrogen balance, alongside a reduction in urinary 3-methylhistidine (3-MH), a marker of MPB (17). This suggests that the ingestion of an LES and breakfast should be utilized to extend the time patients spend in a postprandial fed state, with implications for muscle mass maintenance. Although the consumption of BCAA as an LES is considered to be a safe and cost-effective intervention, potential adverse effects include esophageal reflux, increased insulin resistance, and disturbances in sleep cycle (17); therefore, the use of BCAA must be conducted with caution. As a result, future research should aim to investigate the ideal composition of LES, as there is little research comparing high carbohydrate foods against BCAA, alongside the effectiveness of LES and breakfast consumption at a molecular level.
It is important to note that malnourished patients with cirrhosis may be unable to achieve an adequate oral dietary intake of protein-rich whole foods often due to sickness and/or nausea. Therefore, to conserve lean body mass, the use of additional protein supplementation offers a practical method to allow individuals to reach the enhanced protein requirements in CLD (1.2 g to 1.5 g/kg of body weight per day) (25, 26). Protein intake can be achieved through the ingestion of whole foods, including both animal- and plant-based sources of protein (26). However, animal proteins are not recommended as a suitable protein source for patients with CLD, as they are rich in aromatic amino acids (AAAs). AAAs are not metabolized by the skeletal muscle and therefore may contribute to an increase in HE severity, due to the increase in associated ammonia (15, 145, 146). In comparison, vegetable proteins are rich in BCAA and may have a beneficial effect by removing one mole of ammonia per mole of BCAA (15). Nonetheless, adequate protein intake can still be difficult to achieve in patients with CLD and thus BCAA supplements can be utilized (63, 147). BCAA, in particular the essential amino acid leucine, is a potent stimulus for MPS, identified within both young (148) and older individuals (149). BCAA contributes to an increase in anabolic signaling through mTORC1 and its downstream targets 4EBP1 and p70S6K (150). Similarly, in patients with cirrhosis, supplementation of a BCAA solution rich in leucine (7.5 g leucine, 3.75 g l-isoleucine, and 3.75 g l-valine) has been shown to inhibit GCN2 activity, improve mTORC1 signaling, and decrease markers of autophagy, in comparison to presupplementation values (63). However, no change in myostatin expression or MPS was identified in response to the ingestion of BCAA (63). The authors later expanded upon these findings and found that the in vitro treatment of l-leucine can reverse the hyperammonemia-induced suppression of mTOR signaling and MPS, through the inhibition of GCN2 activity (69). However, further research investigating the effect of BCAA, in particular leucine, in this cohort is required due to the small sample size utilized in a previous mechanistic work (63).
Although BCAA supplementation appears to have beneficial molecular effects, previous research has shown that this approach did not improve intramuscular adipose tissue, or skeletal muscle mass, in patients with cirrhosis when given in the absence of exercise in patients who exhibited no changes in metabolic parameters (147). However, a decrease in adipose accumulation and maintenance of skeletal muscle mass was identified in patients who experienced improved glucose sensitivity (147). Work by Román et al. (151) demonstrated an increase in lower thigh circumference when 10 g/day of leucine was combined with 12 wk of moderate-intensity aerobic exercise training (AET), whereas no change was identified with the ingestion of leucine alone. This suggests that patients with CLD may experience a blunted MPS response to protein intake. Furthermore, this highlights the potential benefits of a combined nutritional and exercise intervention within patients with CLD. It is possible that resistance exercise training (RET), in combination with protein ingestion, may improve MPS within patients with CLD, as described in Fig. 1B. However, further research, which investigates the molecular adaptation in response to protein ingestion and exercise, is required within this population.
Following the optimization of oral intake and oral nutritional supplementation, the use of enteral or parenteral nutrition should be considered to counteract malnutrition and sarcopenia (25). Enteral feeding has been utilized in malnourished patients with cirrhosis admitted to hospital; however, studies have reported mixed results (152). ESPEN guidance strongly recommends the use of enteral feeding in those unable to meet nutritional requirements, with no increased adverse events identified (26). Previous work has shown improved clinical outcomes and a reduction in mortality, supporting the use of enteral feeding within patients with liver cirrhosis (153). Additionally, enteral feeding has been shown to improve nitrogen balance (154). Data on parental nutrition are less established and are only recommended as a method of nutrition when oral and enteral routes have been exhausted. The data regarding the composition of parental nutrition present conflicting results in patients with cirrhosis; however, it is likely to have a beneficial impact during prolonged periods of poor oral intake and improve the mental state in those with severe HE (155). However, in reality, added complications of total parental nutrition-induced liver damage and line-related complications, such as sepsis, make many clinicians hesitant to advocate parenteral nutrition in patients with CLD (156).
Exercise
Exercise is known to play an integral part in improving muscle mass and function in older patients with sarcopenia (157) and many other chronic diseases [e.g., cardiovascular, metabolic, pulmonary, and cancer (158)]. However, the safety of exercise interventions in treating frailty and sarcopenia in patients with CLD is unclear (16). Specifically, there are many considerations to be made including the potential for acute increases in ammonia, elevation of portal pressure, and potential risk of variceal bleeding during exercise (159, 160).
Nonetheless, to date, 11 studies have investigated the impact of exercise in patients with compensated and decompensated cirrhosis. These interventions reported improvements in QoL and key components of physical frailty demonstrated by improvements in muscle mass and function (161), V̇O2 peak (162), and 6MWD (163). Exercise is typically divided into two predominant categories: AET that induces improvements in cardiovascular function and RET that is associated with improvements in muscle mass and strength (164). In patients with cirrhosis, two studies aimed to investigate the impact of 8 wk of AET (162, 163). Zenith et al. (162) identified that 8 wk of supervised moderate-intensity AET induced significant improvements in quadriceps muscle thickness, muscle fatigue, and VO2 peak in patients with cirrhosis. Similarly, Kruger et al. (163) showed that a partially supervised 8-wk moderate-to-high intensity AET program also induced a significant increase in thigh muscle thickness and exercise capacity (identified as an increase in 6MWD) in patients with cirrhosis, in comparison with a control group receiving standard care. Importantly, no adverse events were identified throughout the duration of these interventions, suggesting that AET can be carried out safely in patients with cirrhosis (162, 163).
Despite the known benefits of RET on muscle mass and strength (157, 164), little research has been conducted in patients with CLD. However, Aamann et al. (161) recently conducted a 12-wk RET program consisting of seven whole body exercises that gradually progressed from 15 to 8 repetitions along with increasing load in patients with compensated cirrhosis. Improvements in whole body lean mass and strength of the knee extensors were observed in the RET group (161). However, no significant differences in QoL, assessed by short-form 36 (SF-36), were identified between trained and untrained individuals (161). This may be attributed to the absence of any significant effect of RET on physical function as measured by 6MWD. Additionally, it is currently unclear whether RET reduces the incidence of complications associated with cirrhosis, which could explain the failure to improve QoL (161).
Although only a limited number of studies have investigated the use of RET in patients with CLD, several have combined the use of AET and RET in patients with cirrhosis awaiting transplantation (165, 166). Debette-Gratien et al. (165) conducted a 12-wk adapted physical activity program which consisted of AET and RET twice weekly in 13 patients with cirrhosis awaiting LT. Following training, there was a significant increase in knee extensor strength, maximal power, and 6MWD. This result is in contrast to results of Aamann et al.’s study (161) who identified no such changes in 6MWD in response to the participation of RET alone. Therefore, the improvements in 6MWD identified within this study may be largely attributed to AET within a combined intervention. More recently, work by Williams et al. (166) investigated the effectiveness of a 12-wk home-based intervention consisting of daily step targets and functional body weight resistance exercises in 20 patients awaiting LT. The authors identified an improvement in SPPB scores, which suggests that patients experienced an improvement in overall function, which may be reflected through improvements in mobility. These findings suggest that RET improves skeletal muscle mass and strength. However, in patients with CLD, a concurrent training program, consisting of both AET and RET, should be recommended to improve cardiorespiratory fitness and delay sarcopenia progression, identified through improvements in muscle mass, strength, and function (16, 18).
Pharmaceutical Approaches
Hormone replacement therapy.
As previously discussed, low levels of serum testosterone have been identified in male patients with cirrhosis, which often decline with the progression of CLD (90). Previous research has shown that testosterone treatment in patients with cirrhosis can induce favorable effects on body composition via a reduction in fat mass (167) in addition to increasing HGS (168). It is believed that increasing levels of testosterone can increase androgen receptor expression and in turn may induce an increase in cell growth through the regulation of differentiation and MPS (169). Testosterone treatment also upregulates IGF-1 levels and Akt signaling, which could lead to improvements in muscle growth through SC activation and proliferation (170, 171). Finally, testosterone treatment is known to suppress myostatin levels in skeletal muscle, preventing the activation of JNK, and therefore apoptosis (170). However, the use of testosterone in patients with cirrhosis is not clear cut. Although there appears to be no significant increase in adverse events from testosterone therapy, the long-term outcomes remain largely unclear (90). Therefore, clear evidence to support the safe and efficacious use of adjunct testosterone therapy in patients with cirrhosis remains limited.
In addition, a state of hepatic GH resistance is present in CLD, which may lead to the development of malnutrition (172). A pilot study investigating the effectiveness of 7 days of GH therapy in patients with cirrhosis identified improvements in serum levels of IGF-1 and insulin-like growth factor binding protein 3 (IGFBP-3) in comparison with patients in a placebo group (173). This increase in IGF-1 may contribute to improvements in muscle mass, through an increase in MPS (174) via the activation of the PI3K-mTORC1 pathway (175). However, GH is also associated with several negative side effects, such as the worsening of edema and ascites (172). As a result, the use of GH supplementation should be conducted with caution, and further research into the safety and mechanisms by which GH therapy may contribute to improvements in sarcopenia are required.
Ammonia-lowering therapy.
As previously discussed, hyperammonemia is an abnormality consistently identified in cirrhosis that has been associated with impairments in MPS and increased proteolysis (13, 68, 69). Ammonia-lowering measures have previously been used in both preclinical and clinical settings in the treatment of hepatic endotoxemia, a known complication associated with hyperammonemia (176), but few studies have investigated the benefit of ammonia-lowering strategies in the treatment of sarcopenia in CLD. In vitro, work has shown that although a state of hyperammonemia induces myotube atrophy, ammonia withdrawal can significantly reverse these effects, increasing myotube diameter and anabolic signaling and decreasing catabolic signaling (177). Similarly, in vivo responses to ammonia-lowering therapy in hyperammonemia PCA rats resulted in a significant increase in lean body mass, grip strength, and anabolic signaling, alongside a reduction in markers of autophagy and myostatin expression (177). One of the mechanisms used in this study was administration of rifaxamin, a drug increasingly used in patients with decompensated CLD to manage HE (178). Whether these patients experience reductions in sarcopenia is not known but is an area of considerable interest that could be examined clinically. Although these animal and cellular models suggest that ammonia-lowering therapies may have the potential for clinical implications in the reversal of sarcopenia, human studies are required to confirm these findings (25).
Emerging therapies.
Over recent years, several new potential therapies have been suggested for the treatment of sarcopenia in patients with CLD, including the use of l-carnitine (179), vitamin D, and zinc supplementation (25). In addition, several innovative therapies have also been utilized for the treatment of age-related sarcopenia, and chronic inflammatory disease states such as chronic obstructive pulmonary disease (COPD) including the use of activin receptor II (ActRII) blockers (180, 181) and the nutraceutical Urolithin A (UA) (182). Although this research has not specifically been conducted with patients with CLD, some of the underlying molecular mechanisms that contribute to the development of sarcopenia in these cohorts are likely similar to CLD. As a result, these treatments could provide a novel insight into therapies for the treatment of sarcopenia within CLD.
Cirrhosis is often associated with the development of carnitine deficiency (183). l-Carnitine is a quaternary amine responsible for the transport of long-chain acetylated fatty acids into the mitochondria for fatty acid oxidation, through binding to acetyl groups (184). This leads to the generation of energy, through the production of ATP (184). As previously discussed, a state of hyperammonemia leads to a reduction in ATP synthesis due to a reduction in TCA cycle intermediates (74) and subsequently translation initiation (15). Previous reports suggest that l-carnitine supplementation in patients with cirrhosis results in reductions in ammonia level (185), alongside improvements in muscle cramps (186) and fatigue (187). More recent work has shown that l-carnitine supplementation is associated with a suppression of muscle loss in patients with cirrhosis, particularly when higher dosages were utilized (179).
In addition, vitamin and micronutrient deficiencies in CLD are common and related to hepatic dysfunction, malabsorption, and inadequate dietary intake (25, 26). In particular, CLD is associated with vitamin D (188) and zinc deficiencies (189). Current guidelines set by the EASL recommend that all patients with CLD undergo assessment of plasma vitamin D (25(OH)D) levels, and those who are vitamin D-deficient (below 20 ng/mL) should receive supplementation with oral vitamin D (25). However, vitamin D supplementation in older individuals has shown little benefit for muscle strength, whereas significant improvements in mobility have been identified (190). Zinc supplementation in patients with cirrhosis has been shown to have beneficial effects, improving clinical outcomes such as a decrease in liver enzyme levels and improvements in hyperammonemia and liver function (191). This study, however, did not specifically investigate sarcopenia in CLD. Currently, there is little available evidence on the use of vitamin D and zinc supplementation in patients with sarcopenic CLD and, hence, the effectiveness of these micronutrients is largely unknown.
Previously, this review has discussed the role of increased levels of myostatin in the development of muscle wasting within CLD, which arises because of hyperammonemia (13, 70). Myostatin regulates muscle growth/size by acting on the anti-ActRIIB receptor, consequently inhibiting myoblast differentiation and anabolic mTORC1 signaling (53). Therefore, the inhibition of myostatin has the potential to treat sarcopenia in patients with CLD. Bimagrumab, a human antagonistic anti-ActRIIB receptor antibody, prevents the binding of myostatin and has been shown to promote muscle hypertrophy (192). Indeed, when employed in community-dwelling older adults, 16 wk of bimagrumab treatment resulted in improvements in muscle mass, strength, and gait speed when compared with a placebo (181). Similarly, 24 wk of bimagrumab treatment in patients with COPD led to improvements in lean body mass, thigh muscle volume, and a reduction in intermuscular and subcutaneous fat in comparison to a placebo (180). Despite the improvements in muscle mass, no improvements in physical function measured by 6MWD, Timed Up and Go Test, and HGS were found in this cohort. However, the use of myostatin inhibitors is associated with a number of adverse side effects including muscle spasms, diarrhea, and acne (180, 181). Although this therapy has not yet been investigated in patients with CLD, the available findings imply that this is a promising therapeutic option for the treatment of muscle atrophy.
Mitochondrial dysfunction has been shown to arise because of hyperammonemia (74) and alcohol consumption (89) and is thought to contribute to muscle wasting identified within patients with CLD. Therefore, improvements in mitochondrial functioning may serve as a potential therapeutic target within this cohort. UA is a naturally occurring metabolite, which is formed by the gut microbiota in response to the consumption of ellagitannin-rich food, such as pomegranates, raspberries, strawberries, and walnuts (193). UA has been reported to have several health benefits including the maintenance of mitochondrial biogenesis and respiratory capacity, through the activation of mitophagy in C2C12 skeletal muscle cells and Caenorhabditis elegans (194). In rodent models of aging, induction of mitophagy was identified alongside improvements in muscle function, measured by grip strength and voluntary running activity (194). In addition, UA has been reported to have anti-inflammatory and antioxidant properties (195). In sedentary older individuals, UA induced improvements in mitochondrial and cellular health, with no adverse side effects identified (182). Although no research surrounding the use of UA has been conducted in CLD, the studies to date suggest that UA supplementation may have beneficial effects on mitochondrial health through the activation of mitophagy, which may, in turn, lead to improvements in skeletal muscle function.
CONCLUSIONS
Sarcopenia is common in patients with CLD and is associated with several adverse outcomes including reductions in QoL, increases in mortality, and complications such as infections. Sarcopenia is most commonly defined through the assessment of L3 SMI; however, a lack of sex-specific cutoff values limits the applicability of this modality for the diagnosis of sarcopenia. CLD is characterized by a state of hyperammonemia, an increased catabolism, systemic inflammation, physical inactivity, and poor nutritional status, all of which contribute to the development of sarcopenia through alterations in muscle protein turnover. The therapeutics developed to treat sarcopenia should focus on modifying these potential risk factors and targeting the downstream regulators of muscle mass that they affect. The current literature suggests that patients should ingest 1.6 g/kg/day of protein and reduce the amount of time spent in a period of fasting through the ingestion of more frequent meals, including breakfast and an LES. BCAA supplementation may provide an additional benefit, particularly to those who cannot reach the recommended protein target through the ingestion of whole foods alone. In patients unable to reach the required calorie intake, enteral feeding should be considered. Regular RET and AET are safe and should be encouraged. Emerging pharmaceutical strategies include ammonia-lowering therapy, myostatin inhibition, and anabolic hormone therapy. Future research should aim to identify the molecular and metabolic pathways that contribute to the development of sarcopenia in CLD, which will allow for the creation of targeted therapies to manage and reverse this and also demonstrate improvements in morbidity and mortality within these cohorts.
GRANTS
This work is supported by the National Institute for Health Research Birmingham Biomedical Research Center (BRC-1215-20009) (to S. L. Allen).
DISCLOSURES
The views expressed are those of the authors and not necessarily those of the National Institute for Health Research or the Department of Health and Social Care. No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.L.A., J.I.Q., and L.B. conceived and designed research; S.L.A. prepared figures; S.L.A., J.I.Q., and L.B. drafted manuscript; S.L.A., J.I.Q., A.D., M.J.A., A.M.E., C.A.G., J.M.L., G.G.L., and L.B. edited and revised manuscript; S.L.A., J.I.Q., A.D., M.J.A., A.M.E., C.A.G., J.M.L., G.G.L., and L.B. approved final version of manuscript.
REFERENCES
- 1.Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyère O, Cederholm T, Cooper C, Landi F, Rolland Y, Sayer AA, Schneider SM, Sieber CC, Topinkova E, Vandewoude M, Visser M, Zamboni M; Writing Group for the European Working Group on Sarcopenia in Older People 2 (EWGSOP2), and the Extended Group for EWGSOP2. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing 48: 16–31, 2019. doi: 10.1093/ageing/afy169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Periyalwar P, Dasarathy S. Malnutrition in cirrhosis: contribution and consequences of sarcopenia on metabolic and clinical responses. Clin Liver Dis 16: 95–131, 2012. doi: 10.1016/j.cld.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg IH. Summary comments: epidemiological and methodological problems in determining nutritional status of older persons. Am J Clin Nutr 50: 1231–1233, 1989. doi: 10.1093/ajcn/50.5.1231. [DOI] [Google Scholar]
- 4.Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, Martin FC, Michel J-P, Rolland Y, Schneider SM, Topinkova E, Vandewoude M, Zamboni M. Sarcopenia: European consensus on definition and diagnosis: report of the European Working Group on Sarcopenia in Older People. Age Ageing 39: 412–423, 2010. doi: 10.1093/ageing/afq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, Jatoi A, Kalantar-Zadeh K, Lochs H, Mantovani G, Marks D, Mitch WE, Muscaritoli M, Najand A, Ponikowski P, Rossi Fanelli F, Schambelan M, Schols A, Schuster M, Thomas D, Wolfe R, Anker SD. Cachexia: a new definition. Clin Nutr 27: 793–799, 2008. doi: 10.1016/j.clnu.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Kim G, Kang SH, Kim MY, Baik SK. Prognostic value of sarcopenia in patients with liver cirrhosis: a systematic review and meta-analysis. PLoS One 12: e0186990, 2017. doi: 10.1371/journal.pone.0186990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montano-Loza AJ, Meza-Junco J, Prado CM, Lieffers JR, Baracos VE, Bain VG, Sawyer MB. Muscle wasting is associated with mortality in patients with cirrhosis. Clin Gastroenterol Hepatol 10: 166–173, 2012. doi: 10.1016/j.cgh.2011.08.028. [DOI] [PubMed] [Google Scholar]
- 8.Ohashi K, Ishikawa T, Imai M, Suzuki M, Hoshii A, Abe H, Koyama F, Nakano T, Ueki A, Noguchi H, Hasegawa E, Hirosawa S, Kobayashi M, Hirosawa H, Sato K, Munakata M, Yoshida T. Relationship between pre-sarcopenia and quality of life in patients with chronic liver disease: a cross-sectional study. Eur J Gastroenterol Hepatol 31: 1408–1413, 2019. [DOI] [PubMed] [Google Scholar]
- 9.Merli M, Giusto M, Gentili F, Novelli G, Ferretti G, Riggio O, Corradini SG, Siciliano M, Farcomeni A, Attili AF, Berloco P, Rossi M. Nutritional status: its influence on the outcome of patients undergoing liver transplantation. Liver Int 30: 208–214, 2010. doi: 10.1111/j.1478-3231.2009.02135.x. [DOI] [PubMed] [Google Scholar]
- 10.van Vugt JL, Levolger S, de Bruin RW, van Rosmalen J, Metselaar HJ, Ijzermans JN. Systematic review and meta-analysis of the impact of computed tomography-assessed skeletal muscle mass on outcome in patients awaiting or undergoing liver transplantation. Am J Transplant 16: 2277–2292, 2016. doi: 10.1111/ajt.13732. [DOI] [PubMed] [Google Scholar]
- 11.van Vugt JLA, Buettner S, Alferink LJM, Bossche N, de Bruin RWF, Darwish Murad S, Polak WG, Metselaar HJ, IJzermans JNM. Low skeletal muscle mass is associated with increased hospital costs in patients with cirrhosis listed for liver transplantation—a retrospective study. Transpl Int 31: 165–174, 2018. doi: 10.1111/tri.13048. [DOI] [PubMed] [Google Scholar]
- 12.Owen OE, Trapp VE, Reichard GAJ, Mozzoli MA, Moctezuma J, Paul P, Skutches CL, Boden G. Nature and quantity of fuels consumed in patients with alcoholic cirrhosis. J Clin Invest 72: 1821–1832, 1983. doi: 10.1172/JCI111142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu J, Thapaliya S, Runkana A, Yang Y, Tsien C, Mohan ML, Narayanan A, Eghtesad B, Mozdziak PE, McDonald C, Stark GR, Welle S, Naga Prasad SV, Dasarathy S. Hyperammonemia in cirrhosis induces transcriptional regulation of myostatin by an NF-kappaB-mediated mechanism. Proc Natl Acad Sci USA 110: 18162–18167, 2013. doi: 10.1073/pnas.1317049110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohashi K, Ishikawa T, Hoshi A, Suzuki M, Mitobe Y, Yamada E, Abeywickrama HM, Seki N, Koyama C, Aoki H, Koyama Y. Relationship between sarcopenia and both physical activity and lifestyle in patients with chronic liver disease. J Clin Med Res 10: 920–927, 2018. doi: 10.14740/jocmr3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dasarathy S, Merli M. Sarcopenia from mechanism to diagnosis and treatment in liver disease. J Hepatol 65: 1232–1244, 2016. doi: 10.1016/j.jhep.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams FR, Berzigotti A, Lord JM, Lai JC, Armstrong MJ. Review article: impact of exercise on physical frailty in patients with chronic liver disease. Aliment Pharmacol Ther 50: 988–1000, 2019. doi: 10.1111/apt.15491. [DOI] [PubMed] [Google Scholar]
- 17.Tsien C, McCullough AJ, Dasarathy S. Late evening snack: exploiting a period of anabolic opportunity in cirrhosis. J Gastroenterol Hepatol 27: 430–441, 2012. doi: 10.1111/j.1440-1746.2011.06951.x. [DOI] [PubMed] [Google Scholar]
- 18.Carey EJ, Lai JC, Sonnenday C, Tapper EB, Tandon P, Duarte-Rojo A, Dunn MA, Tsien C, Kallwitz ER, Ng V, Dasarathy S, Kappus M, Bashir MR, Montano-Loza AJ. A North American expert opinion statement on sarcopenia in liver transplantation. Hepatology 70: 1816–1829, 2019. doi: 10.1002/hep.30828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinclair M. Controversies in diagnosing sarcopenia in cirrhosis-moving from research into clinical practice. Nutrients 11: 2454, 2019. doi: 10.3390/nu11102454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebadi M, Wang CW, Lai JC, Dasarathy S, Kappus MR, Dunn MA, Carey EJ, Montano-Loza AJ, From the Fitness, Life Enhancement, and Exercise in Liver Transplantation (FLEXIT) Consortium. Poor performance of psoas muscle index for identification of patients with higher waitlist mortality risk in cirrhosis. J Cachexia Sarcopenia Muscle 9: 1053–1062, 2018. doi: 10.1002/jcsm.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Englesbe MJ, Patel SP, He K, Lynch RJ, Schaubel DE, Harbaugh C, Holcombe SA, Wang SC, Segev DL, Sonnenday CJ. Sarcopenia and mortality after liver transplantation. J Am Coll Surg 211: 271–278, 2010. doi: 10.1016/j.jamcollsurg.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carey EJ, Lai JC, Wang CW, Dasarathy S, Lobach I, Montano-Loza AJ, Dunn MA; for the Fitness, Life Enhancement, and Exercise in Liver Transplantation Consortium. A multicenter study to define sarcopenia in patients with end-stage liver disease. Liver Transpl 23: 625–633, 2017. doi: 10.1002/lt.24750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malmstrom TK, Miller DK, Simonsick EM, Ferrucci L, Morley JE. SARC-F: a symptom score to predict persons with sarcopenia at risk for poor functional outcomes. J Cachexia Sarcopenia Muscle 7: 28–36, 2016. doi: 10.1002/jcsm.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiraoka A, Nagamatsu K, Izumoto H, Yoshino T, Adachi T, Tsuruta M, Aibiki T, Okudaira T, Yamago H, Suga Y, Iwasaki R, Mori K, Miyata H, Tsubouchi E, Ninomiya T, Hirooka M, Abe M, Matsuura B, Hiasa Y, Michitaka K. SARC-F combined with a simple tool for assessment of muscle abnormalities in outpatients with chronic liver disease. Hepatol Res 50: 502–511, 2020. doi: 10.1111/hepr.13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.European Association for the Study of the Liver. European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu; European Association for the Study of the Liver. EASL Clinical Practice Guidelines on nutrition in chronic liver disease. J Hepatol 70: 172–193, 2019. doi: 10.1016/j.jhep.2018.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plauth M, Bernal W, Dasarathy S, Merli M, Plank LD, Schutz T, Bischoff SC. ESPEN guideline on clinical nutrition in liver disease. Clin Nutr 38: 485–521, 2019. doi: 10.1016/j.clnu.2018.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sinclair M, Chapman B, Hoermann R, Angus PW, Testro A, Scodellaro T, Gow PJ. Handgrip strength adds more prognostic value to the model for end-stage liver disease score than imaging-based measures of muscle mass in men with cirrhosis. Liver Transpl 25: 1480–1487, 2019. doi: 10.1002/lt.25598. [DOI] [PubMed] [Google Scholar]
- 28.Carey EJ, Steidley DE, Aqel BA, Byrne TJ, Mekeel KL, Rakela J, Vargas HE, Douglas DD. Six-minute walk distance predicts mortality in liver transplant candidates. Liver Transpl 16: 1373–1378, 2010. doi: 10.1002/lt.22167. [DOI] [PubMed] [Google Scholar]
- 29.Lai JC, Feng S, Terrault NA, Lizaola B, Hayssen H, Covinsky K. Frailty predicts waitlist mortality in liver transplant candidates. Am J Transplant 14: 1870–1879, 2014. doi: 10.1111/ajt.12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn MA, Josbeno DA, Tevar AD, Rachakonda V, Ganesh SR, Schmotzer AR, Kallenborn EA, Behari J, Landsittel DP, DiMartini AF, Delitto A. Frailty as tested by gait speed is an independent risk factor for cirrhosis complications that require hospitalization. Am J Gastroenterol 111: 1768–1775, 2016. doi: 10.1038/ajg.2016.336. [DOI] [PubMed] [Google Scholar]
- 31.Montano-Loza AJ, Duarte-Rojo A, Meza-Junco J, Baracos VE, Sawyer MB, Pang JX, Beaumont C, Esfandiari N, Myers RP. Inclusion of sarcopenia within MELD (MELD-Sarcopenia) and the prediction of mortality in patients with cirrhosis. Clin Transl Gastroenterol 6: e102, 2015. doi: 10.1038/ctg.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhanji RA, Carey EJ, Yang L, Watt KD. The long winding road to transplant: how sarcopenia and debility impact morbidity and mortality on the waitlist. Clin Gastroenterol Hepatol 15: 1492–1497, 2017. doi: 10.1016/j.cgh.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Bhanji RA, Moctezuma-Velazquez C, Duarte-Rojo A, Ebadi M, Ghosh S, Rose C, Montano-Loza AJ. Myosteatosis and sarcopenia are associated with hepatic encephalopathy in patients with cirrhosis. Hepatol Int 12: 377–386, 2018. doi: 10.1007/s12072-018-9875-9. [DOI] [PubMed] [Google Scholar]
- 34.DiMartini A, Cruz RJ, Jr, Dew MA, Myaskovsky L, Goodpaster B, Fox K, Kim KH, Fontes P. Muscle mass predicts outcomes following liver transplant. Liver Transpl 19: 1172–1180, 2013. doi: 10.1002/lt.23724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng JK, Hepgul N, Higginson IJ, Gao W. Symptom prevalence and quality of life patients with end-stage liver disease: a systematic review and meta-analysis. Palliat Med 33: 24–36, 2019. doi: 10.1177/0269216318807051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghabril M, Jackson M, Gotur R, Weber R, Orman E, Vuppalanchi R, Chalasani N. Most individuals with advanced cirrhosis have sleep disturbances, which are associated with poor quality of life. Clin Gastroenterol Hepatol 15: 1271–1278 e1276, 2017. doi: 10.1016/j.cgh.2017.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montagnese S, De Pitta C, De Rui M, Corrias M, Turco M, Merkel C, Amodio P, Costa R, Skene DJ, Gatta A. Sleep-wake abnormalities in patients with cirrhosis. Hepatology 59: 705–712, 2014. doi: 10.1002/hep.26555. [DOI] [PubMed] [Google Scholar]
- 38.Younossi Z, Henry L. Overall health-related quality of life in patients with end-stage liver disease. Clin Liver Dis (Hoboken) 6: 9–14, 2015. doi: 10.1002/cld.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dasarathy S. Posttransplant sarcopenia: an underrecognized early consequence of liver transplantation. Dig Dis Sci 58: 3103–3111, 2013. doi: 10.1007/s10620-013-2791-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsien C, Garber A, Narayanan A, Shah SN, Barnes D, Eghtesad B, Fung J, McCullough AJ, Dasarathy S. Post-liver transplantation sarcopenia in cirrhosis: a prospective evaluation. J Gastroenterol Hepatol 29: 1250–1257, 2014. doi: 10.1111/jgh.12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Everhart JE, Lombardero M, Lake JR, Wiesner RH, Zetterman RK, Hoofnagle JH. Weight change and obesity after liver transplantation: incidence and risk factors. Liver Transpl Surg 4: 285–296, 1998. doi: 10.1002/lt.500040402. [DOI] [PubMed] [Google Scholar]
- 42.El Sherif O, Dhaliwal A, Newsome PN, Armstrong MJ. Sarcopenia in nonalcoholic fatty liver disease: new challenges for clinical practice. Expert Rev Gastroeterol Hepatol 23: 1–9, 2020. [DOI] [PubMed] [Google Scholar]
- 43.Breen L, Phillips SM. Skeletal muscle protein metabolism in the elderly: interventions to counteract the ‘anabolic resistance’ of ageing. Nutr Metab 8, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bohe J, Low A, Wolfe RR, Rennie MJ. Human muscle protein synthesis is modulated by extracellular, not intramuscular amino acid availability: a dose-response study. J Physiol 552: 315–324, 2003. doi: 10.1113/jphysiol.2003.050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips SM, Tipton KD, Aarsland A, Wolf SE, Wolfe RR. Mixed muscle protein synthesis and breakdown after resistance exercise in humans. Am J Physiol Endocrinol Metab 273: E99–E107, 1997. doi: 10.1152/ajpendo.1997.273.1.E99. [DOI] [PubMed] [Google Scholar]
- 46.Biolo G, Tipton KD, Klein S, Wolfe RR. An abundant supply of amino acids enhances the metabolic effect of exercise on muscle protein. Am J Physiol Endocrinol Metab 273: E122–E129, 1997. doi: 10.1152/ajpendo.1997.273.1.E122. [DOI] [PubMed] [Google Scholar]
- 47.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110: 163–175, 2002. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 48.Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 14: 133–139, 2013. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKay BR, De Lisio M, Johnston AP, O'Reilly CE, Phillips SM, Tarnopolsky MA, Parise G. Association of interleukin-6 signalling with the muscle stem cell response following muscle-lengthening contractions in humans. PLoS One 4: e6027, 2009. doi: 10.1371/journal.pone.0006027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKay BR, O'Reilly CE, Phillips SM, Tarnopolsky MA, Parise G. Co-expression of IGF-1 family members with myogenic regulatory factors following acute damaging muscle-lengthening contractions in humans. J Physiol 586: 5549–5560, 2008. doi: 10.1113/jphysiol.2008.160176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McKay BR, Ogborn DI, Bellamy LM, Tarnopolsky MA, Parise G. Myostatin is associated with age-related human muscle stem cell dysfunction. FASEB J 26: 2509–2521, 2012. doi: 10.1096/fj.11-198663. [DOI] [PubMed] [Google Scholar]
- 52.Lokireddy S, Mouly V, Butler-Browne G, Gluckman PD, Sharma M, Kambadur R, McFarlane C. Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin-proteasome pathway-mediated loss of sarcomeric proteins. Am J Physiol Cell Physiol 301: C1316–C1324, 2011. [Erratum in Am J Physiol Cell Physiol 307: C1152, 2014]. doi: 10.1152/ajpcell.00114.2011. [DOI] [PubMed] [Google Scholar]
- 53.Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol 296: C1258–C1270, 2009. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- 54.Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induces ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403, 2004. doi: 10.1016/S1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 55.Morrison WL, Bouchier IA, Gibson JN, Rennie MJ. Skeletal muscle and whole-body protein turnover in cirrhosis. Clin Sci (Lond) 78: 613–619, 1990. doi: 10.1042/cs0780613. [DOI] [PubMed] [Google Scholar]
- 56.Tessari P, Barazzoni R, Kiwanuka E, Davanzo GD, Pergola G, Orlando R, Vettore M,, Zanetti M,. M. Impairment of albumin and whole body postprandial protein synthesis in compensated liver cirrhosis. Am J Physiol Endocrinol Metab 282: E394–E311, 2002. [DOI] [PubMed] [Google Scholar]
- 57.Tessari P, Kiwanuka E, Vettore M, Barazzoni R, Zanetti M, Cecchet D, Orlando R. Phenylalanine and tyrosine kinetics in compensated liver cirrhosis: effects of meal ingestion. Am J Physiol Gastrointest Liver Physiol 295: G598–G604, 2008. doi: 10.1152/ajpgi.00355.2007. [DOI] [PubMed] [Google Scholar]
- 58.McCullough AJ, Mullen KD, Tavill AS, Kalhan SC. In vivo differences between the turnover rates of leucine and leucine’s ketoacid in stable cirrhosis. Gastroenterology 103: 571–578, 1992. doi: 10.1016/0016-5085(92)90849-T. [DOI] [PubMed] [Google Scholar]
- 59.Tessari P, Inchiostro S, Barazzoni R, Zanetti M, Orlando R, Biolo G, Sergi G, Pino A, Tiengo A. Fasting and postprandial phenylalanine and leucine kinetics in liver cirrhosis. Am J Physiol Endocrinol Metab 267: E140–E149, 1994. doi: 10.1152/ajpendo.1994.267.1.E140. [DOI] [PubMed] [Google Scholar]
- 60.Tessari P, Vettore M, Millioni R, Puricelli L, Orlando R. Effect of liver cirrhosis on phenylalanine and tyrosine metabolism. Curr Opin Clin Nutr Metab Care 13: 81–86, 2010. doi: 10.1097/MCO.0b013e32833383af. [DOI] [PubMed] [Google Scholar]
- 61.Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, Wackerhage H, Taylor PM, Rennie MJ. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J 19: 1–424, 2005. doi: 10.1096/fj.04-2640fje. [DOI] [PubMed] [Google Scholar]
- 62.Brook MS, Wilkinson DJ, Mitchell WK, Lund JN, Phillips BE, Szewczyk NJ, Greenhaff PL, Smith K, Atherton PJ. Synchronous deficits in cumulative muscle protein synthesis and ribosomal biogenesis underlie age-related anabolic resistance to exercise in humans. J Physiol 594: 7399–7417, 2016. doi: 10.1113/JP272857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsien C, Davuluri G, Singh D, Allawy A, Ten Have GA, Thapaliya S, Schulze JM, Barnes D, McCullough AJ, Engelen MP, Deutz NE, Dasarathy S. Metabolic and molecular responses to leucine-enriched branched chain amino acid supplementation in the skeletal muscle of alcoholic cirrhosis. Hepatology 61: 2018–2029, 2015. doi: 10.1002/hep.27717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olde Damink SW, Jalan R, Dejong CH. Interorgan ammonia trafficking in liver disease. Metab Brain Dis 24: 169–181, 2009. doi: 10.1007/s11011-008-9122-5. [DOI] [PubMed] [Google Scholar]
- 65.Shangraw RE, Jahoor F. Effect of liver disease and transplantation on urea synthesis in humans: relationship to acid-base status. Am J Physiol Gastrointest Liver Physiol 276: G1145–G1152, 1999. [DOI] [PubMed] [Google Scholar]
- 66.Takeda K, Takemasa T. Expression of ammonia transporters Rhbg and Rhcg in mouse skeletal muscle and the effect of 6-week training on these proteins. Physiol Rep 3: e12596, 2015. doi: 10.14814/phy2.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dasarathy S, Muc S, Hisamuddin K, Edmison JM, Dodig M, McCullough AJ, Kalhan SC. Altered expression of genes regulating skeletal muscle mass in the portacaval anastomosis rat. Am J Physiol Gastrointest Liver Physiol 292: G1105–G1113, 2007. doi: 10.1152/ajpgi.00529.2006. [DOI] [PubMed] [Google Scholar]
- 68.Qiu J, Tsien C, Thapalaya S, Narayanan A, Weihl CC, Ching JK, Eghtesad B, Singh K, Fu X, Dubyak G, McDonald C, Almasan A, Hazen SL, Naga Prasad SV, Dasarathy S. Hyperammonemia-mediated autophagy in skeletal muscle contributes to sarcopenia of cirrhosis. Am J Physiol Endocrinol Metab 303: E983–E993, 2012. doi: 10.1152/ajpendo.00183.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davuluri G, Krokowski D, Guan BJ, Kumar A, Thapaliya S, Singh D, Hatzoglou M, Dasarathy S. Metabolic adaptation of skeletal muscle to hyperammonemia drives the beneficial effects of l-leucine in cirrhosis. J Hepatol 65: 929–937, 2016. doi: 10.1016/j.jhep.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nishikawa H, Enomoto H, Ishii A, Iwata Y, Miyamoto Y, Ishii N, Yuri Y, Hasegawa K, Nakano C, Nishimura T, Yoh K, Aizawa N, Sakai Y, Ikeda N, Takashima T, Takata R, Iijima H, Nishiguchi S. Elevated serum myostatin level is associated with worse survival in patients with liver cirrhosis. J Cachexia Sarcopenia Muscle 8: 915–925, 2017. doi: 10.1002/jcsm.12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dasarathy S, Dodig M, Muc SM, Kalhan SC, McCullough AJ. Skeletal muscle atrophy is associated with an increased expression of myostatin and impaired satellite cell function in the portacaval anastamosis rat. Am J Physiol Gastrointest Liver Physiol 287: G1124–1130, 2004. doi: 10.1152/ajpgi.00202.2004. [DOI] [PubMed] [Google Scholar]
- 72.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277: 30409–30412, 2002. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- 73.Zaganas I, Pajęcka K, Wendel Nielsen C, Schousboe A, Waagepetersen HS, Plaitakis A. The effect of pH and ADP on ammonia affinity for human glutamate dehydrogenases. Metab Brain Dis 28: 127–131, 2013. doi: 10.1007/s11011-013-9382-6. [DOI] [PubMed] [Google Scholar]
- 74.Davuluri G, Allawy A, Thapaliya S, Rennison JH, Singh D, Kumar A, Sandlers Y, Van Wagoner DR, Flask CA, Hoppel C, Kasumov T, Dasarathy S. Hyperammonaemia-induced skeletal muscle mitochondrial dysfunction results in cataplerosis and oxidative stress. J Physiol 594: 7341–7360, 2016. doi: 10.1113/JP272796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakaya Y, Harada N, Kakui S, Okada K, Takahashi A, Inoi J, Ito S. Severe catabolic state after prolonged fasting in cirrhotic patients: effect of oral branched-chain amino-acid-enriched nutrient mixture. J Gastroenterol 37: 531–536, 2002. doi: 10.1007/s005350200082. [DOI] [PubMed] [Google Scholar]
- 76.Brosnan JT. Comments on metabolic needs for glucose and the role of gluconeogenesis. Eur J Clin Nutr 53: S107–S111, 1999. doi: 10.1038/sj.ejcn.1600748. [DOI] [PubMed] [Google Scholar]
- 77.Tajiri K, Shimizu Y. Branched-chain amino acids in liver diseases. WJG World J Gastroenterol 19: 7620–7629, 2013. doi: 10.3748/wjg.v19.i43.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep 17: 1374–1395, 2016. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108, 2000. doi: 10.1016/S1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 80.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167: 27–33, 2004. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2a. J Cell Biol 153: 1011–1022, 2001. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Holecek M, Kandar R, Sispera L, Kovarik M. Acute hyperammonemia activates branched-chain amino acid catabolism and decreases their extracellular concentrations: different sensitivity of red and white muscle. Amino Acids 40: 575–584, 2011. doi: 10.1007/s00726-010-0679-z. [DOI] [PubMed] [Google Scholar]
- 83.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136: 521–534, 2009. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dasarathy J, McCullough AJ, Dasarathy S. Sarcopenia in alcoholic liver disease: clinical and molecular advances. Alcohol Clin Exp Res 41: 1419–1431, 2017. doi: 10.1111/acer.13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Estonius M, Svensson S, Höög JO. Alcohol dehydrogenase in human tissues: localisation of transcripts coding for five classes of the enzyme. FEBS Lett 397: 338–342, 1996. doi: 10.1016/S0014-5793(96)01204-5. [DOI] [PubMed] [Google Scholar]
- 86.Thapaliya S, Runkana A, McMullen MR, Nagy LE, McDonald C, Naga Prasad SV, Dasarathy S. Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy 10: 677–690, 2014. doi: 10.4161/auto.27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steiner JL, Lang CH. Alcohol impairs skeletal muscle protein synthesis and mTOR signalling in a time-dependent manner following electrically stimulate muscle contraction. J Appl Physiol (1985 ) 117: 1170–1179, 2014. doi: 10.1152/japplphysiol.00180.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lang CH, Frost RA, Svanberg E, Vary TC. IGF-I/IGFBP-3 ameliorates alterations in protein synthesis, eIF4E availability, and myostatin in alcohol-fed rats. Am J Physiol Endocrinol Metab 286: E916–E926, 2004. doi: 10.1152/ajpendo.00554.2003. [DOI] [PubMed] [Google Scholar]
- 89.Bonet-Ponce L, Saez-Atienzar S, da Casa C, Flores-Bellver M, Barcia JM, Sancho-Pelluz J, Romero FJ, Jordan J, Galindo MF. On the mechanism underlying ethanol-induced mitochondrial dynamic disruption and autophagy response. Biochim Biophys Acta 1852: 1400–1409, 2015. doi: 10.1016/j.bbadis.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 90.Sinclair M, Grossmann M, Gow PJ, Angus PW. Testosterone in men with advanced liver disease: abnormalities and implications. J Gastroenterol Hepatol 30: 244–251, 2015. doi: 10.1111/jgh.12695. [DOI] [PubMed] [Google Scholar]
- 91.Thiel DH, Kumar S, Gavaler JS, Tarter RE. Effect of liver transplantation on the hypothalamic-pituitary-gonadal axis of chronic alcoholic men with advanced liver disease. Alcoholism Clin Exp Res 14: 478–481, 1990. doi: 10.1111/j.1530-0277.1990.tb00507.x. [DOI] [PubMed] [Google Scholar]
- 92.Kaufman JM, Vermeulen A. The decline of androgen levels in elderly men and its clinical and therapeutic implications. Endocr Rev 26: 833–876, 2005. doi: 10.1210/er.2004-0013. [DOI] [PubMed] [Google Scholar]
- 93.Dasarathy S, Mullen KD, Dodig M, Donofrio B, McCullough AJ. Inhibition of aromatase improves nutritional status following portacaval anastomosis in male rats. J Hepatol 45: 214–220, 2006. doi: 10.1016/j.jhep.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 94.Cuneo RC, Hickman PE, Wallace JD, Teh BT, Ward G, Veldhuis JD, Waters MJ. Altered endogenous growth hormone secretory kinetic and diurnal GH-binding protein profiles in adults with chronic liver disease. Clin Endocrinol 43: 265–275, 1995. doi: 10.1111/j.1365-2265.1995.tb02031.x. [DOI] [PubMed] [Google Scholar]
- 95.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3: 1014–1019, 2001. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 96.Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 3: 1009–1013, 2001. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 97.Liu W, Thomas SG, Asa SL, Gonzalez-Cadavid N, Bhasin S, Ezzat S. Myostatin is a skeletal muscle target of growth hormone anabolic action. J Clin Endo Metab 88: 5490–5496, 2003. doi: 10.1210/jc.2003-030497. [DOI] [PubMed] [Google Scholar]
- 98.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64: 73–84, 2016. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 99.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, Sanyal AJ. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67: 328–357, 2018. doi: 10.1002/hep.29367. [DOI] [PubMed] [Google Scholar]
- 100.European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), and European Association for the Study of Obesity EASO). EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. Obes Facts 9: 65–90, 2016. doi: 10.1159/000443344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pimpin L, Cortez-Pinto H, Negro F, Corbould E, Lazarus JV, Webber L, Sheron N. Burden of liver disease in Europe: epidemiology and analysis of risk factors to identify prevention policies. J Hepatol 69: 718–735, 2018. doi: 10.1016/j.jhep.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 102.Montano-Loza AJ, Angulo P, Meza-Junco J, Prado CM, Sawyer MB, Beaumont C, Esfandiari N, Ma M, Baracos VE. Sarcopenic obesity and myosteatosis are associated with higher mortality in patients with cirrhosis. J Cachexia Sarcopenia Muscle 7: 126–135, 2016. doi: 10.1002/jcsm.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jung KS, Kim SU, Yoon HJ, Yun YJ, Lee BW, Kang ES, Han KH, Lee HC, Bs C. Sarcopenia is associated with NAFLD independently of obesity and insulin resistance: nationwide surveys (KNHANES 2008-2011). J Hepatol 63: 486–493, 2015. doi: 10.1016/j.jhep.2015.02.051. [DOI] [PubMed] [Google Scholar]
- 104.Hallsworth K, Thoma C, Moore S, Ploetz T, Anstee QM, Taylor R, Day CP, Trenell MI. Non-alcoholic fatty liver disease is associated with higher levels of objectively measured sedentary behaviour and lower levels of physical activity than matched healthy controls. Frontline Gastroenterol 6: 44–51, 2015. doi: 10.1136/flgastro-2014-100432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sanchez-Sanchez JL, Manas A, Garcia-Garcia FAI, Carnicero JA, Walter S, Rodriguez-Manas L. Sedentary behaviour, physical activity, and sarcopenia among older adults in the TSHA: isotemporal substitution model. J Cachexia Sarcopenia Muscle 10: 188–198, 2019. doi: 10.1002/jcsm.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim TN, Yang SJ, Yoo HJ, Lim KI, Kang HJ, Song W, Seo JA, Kim SG, Kim NH, Baik SH, Choi DS, Choi KM. Prevalence of sarcopenia and sarcopenic obesity in Korean adults: the Korean sarcopenic obesity study. Int J Obes 33: 885–892, 2009. doi: 10.1038/ijo.2009.130. [DOI] [PubMed] [Google Scholar]
- 107.Bhanji RA, Narayanan P, Allen AM, Malhi H, Watt KD. Sarcopenia in hiding: the risk and consequence of underestimating muscle dysfunction in nonalcoholic steatohepatitis. Hepatology 66: 2055–2065, 2017. doi: 10.1002/hep.29420. [DOI] [PubMed] [Google Scholar]
- 108.Utzschneider KM, Kahn SE. Review: the role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab 91: 4753–4761, 2006. doi: 10.1210/jc.2006-0587. [DOI] [PubMed] [Google Scholar]
- 109.Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 147: 4160–4168, 2006. doi: 10.1210/en.2006-0251. [DOI] [PubMed] [Google Scholar]
- 110.Kalyani RR, Corriere M, Ferrucci L. Age-related and disease-related muscle loss: the effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol 2: 819–829, 2014. doi: 10.1016/S2213-8587(14)70034-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim TN, Park MS, Lim KI, Choi HY, Yang SJ, Yoo HJ, Kang HJ, Song W, Choi H, Baik SH, Choi DS, Choi KM. Relationships between sarcopenic obesity and insulin resistance, inflammation, and vitamin D status: the Korean Sarcopenic Obesity Study. Clin Endocrinol 78: 525–532, 2013. doi: 10.1111/j.1365-2265.2012.04433.x. [DOI] [PubMed] [Google Scholar]
- 112.Koster A, Ding J, Stenholm S, Caserotti P, Houston DK, Nicklas BJ, You T, Lee JS, Visser M, Newman AB, Schwartz AV, Cauley JA, Tylavsky FA, Goodpaster BH, Kritchevsky SB, Harris TB and Health ABCs. Does the amount of fat mass predict age-related loss of lean mass, muscle strength, and muscle quality in older adults? J Gerontol A Biol Sci Med Sci 66A: 888–895, 2011. doi: 10.1093/gerona/glr070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Srikanthan P, Hevener AL, Karlamangla AS. Sarcopenia exacerbates obesity-associated insulin resistance and dysglycemia: findings from the National Health and Nutrition Examination Survey III. PLoS One 5: e10805, 2010. doi: 10.1371/journal.pone.0010805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest 127: 55–64, 2017. doi: 10.1172/JCI88881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dalle S, Rossmeislova L, Koppo K. The role of inflammation in age-related sarcopenia. Front Physiol 8: 1045, 2017. doi: 10.3389/fpls.2017.01045, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic setatohepatitis: pathphysiology and clinical implications. Gastroenterology 142: 711–725, 2012. doi: 10.1053/j.gastro.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 117.Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 40: 185–194, 2004. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 118.Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, García-Ruiz C. Mitochondrial free cholesterol loading sensitises to TNF- and Fas-mediated steatohepatitis. Cell Metab 4: 185–198, 2006. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 119.Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, Kirkland JL. Fat tissue, aging, and cellular senescence. Aging Cell 9: 667–684, 2010. doi: 10.1111/j.1474-9726.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 92: 347–355, 2004. . doi: 10.1079/BJN20041213. [DOI] [PubMed] [Google Scholar]
- 121.Lang CH, Frost RA, Nairn AC, MacLean DA, Vary TC. TNF-alpha impairs heart and skeletal muscle protein synthesis by altering translation initiation. Am J Physiol Endocrinol Metab 282: E336–E247, 2002. doi: 10.1152/ajpendo.00366.2001. [DOI] [PubMed] [Google Scholar]
- 122.Reid MB, Li Y-P. Tumor necrosis factor-α and muscle wasting: a cellular perspective. Respir Res 2: 269–272, 2001. doi: 10.1186/rr67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 19: 362–370, 2005. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li YP, Schwartz RJ, Waddell ID, Holloway BR, Reid MB. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. FASEB J 12: 871–880, 1998. doi: 10.1096/fasebj.12.10.971, . doi:. [DOI] [PubMed] [Google Scholar]
- 125.Thoma A, Lightfoot AP. NF-kB and inflammatory cytokine signalling: role in skeletal muscle atrophy. Adv Exp Med Biol 1088: 267–279, 2018. doi: 10.1007/978-981-13-1435-3_12. [DOI] [PubMed] [Google Scholar]
- 126.Wree A, Kahraman A, Gerken G, Canbay A. Obesity affects the liver—the link between adipocytes and hepatocytes. Digestion 83: 124–133, 2011. doi: 10.1159/000318741. [DOI] [PubMed] [Google Scholar]
- 127.Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care 26: 2442–2450, 2003. doi: 10.2337/diacare.26.8.2442. [DOI] [PubMed] [Google Scholar]
- 128.Combs TP, Marliss EB. Adiponectin signaling in the liver. Rev Endocr Metab Disord 15: 137–147, 2014. doi: 10.1007/s11154-013-9280-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ruan H, Dong LQ. Adiponectin signaling and function in insulin target tissues. J Mol Cell Biol 8: 101–109, 2016. doi: 10.1093/jmcb/mjw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu T, Xu L, Chen FH, Zhou YB. Association of serum vitamin D level and nonalcoholic fatty liver disease: a meta-analysis. Eur J Gastronenterol Hepatol 32: 140–147, 2020. doi: 10.1097/MEG.0000000000001486. [DOI] [PubMed] [Google Scholar]
- 131.Dasarathy S. Is the adiponectin-AMPK-mitochondrial axis involves in progression of nonalcoholic fatty liver disease? Hepatology 60: 22–25, 2014. doi: 10.1002/hep.27134, . doi: 10.1002/hep.27134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Amor M, Itariu BK, Moreno-Viedma V, Keindl M, Jürets A, Prager G, Langer F, Grablowitz V, Zelda M, Stulnig TM. Serum myostatin is upregulated in obesity and correlates with insulin resistance in humans. Exp Clin Endocrinol Diabetes 127: 550–556, 2019. doi: 10.1055/a-0641-5546. [DOI] [PubMed] [Google Scholar]
- 133.Barchetta I, De Bernardinis M, Capoccia D, Baroni MG, Fontana M, Fraioli A, Morini S, Leonetti F, Cavallo MG. Hypovitaaminosis D is independently association with metabolic syndrome in obese patients. PLoS One 8: e68689, 2013. doi: 10.1371/journal.pone.0068689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Eliades M, Spyrou E, Agrawal N, Lazo M, Brancati FL, Potter JJ, Koteish AA, Clark JM, Guallar E, Hernaez R. Meta-analysis: vitamin D and non-alcoholic fatty liver disease. Aliment Pharmacol Ther 38: 246–254, 2013. doi: 10.1111/apt.12377. [DOI] [PubMed] [Google Scholar]
- 135.Roth CL, Elfers CT, Figlewicz DP, Melhorn SJ, Morton GJ, Hoofnagle A, Yeh MM, Nelson JE, Kowdley KV. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and Toll-like receptor activation. Hepatology 55: 1103–1111, 2012. doi: 10.1002/hep.24737. [DOI] [PubMed] [Google Scholar]
- 136.Visser M, Deeg DJ, Lips P. and Longitudinal Aging Study A. Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (sarcopenia): the Longitudinal Aging Study Amsterdam. J Clin Endocrinol Metab 88: 5766–5772, 2003. doi: 10.1210/jc.2003-030604. [DOI] [PubMed] [Google Scholar]
- 137.Dzik KP, Kaczor JJ. Mechanisms of vitamin D on skeletal muscle function: oxidative stress, energy metabolism and anabolic state. Eur J Appl Physiol 119: 825–839, 2019. doi: 10.1007/s00421-019-04104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kim MK, Baek KH, Song KH, Kang M, Park CY, Lee WY, Oh KW. Vitamin D deficiency is associated with sarcopenia in older Koreans, regardless of obesity: the Fourth Korea National Health and Nutrition Examination Surveys (KNHANES IV). J Clin Endocrinol Metab 96: 3250–3256, 2011. doi: 10.1210/jc.2011-1602. [DOI] [PubMed] [Google Scholar]
- 139.Kozeniecki M, Ludke R, Kerner J, Patterson B. Micronutrients in liver disease: roles, risk factors for deficiency, and recommendations for supplementation. Nutr Clin Pract 35: 50–62, 2020. doi: 10.1002/ncp.10451. [DOI] [PubMed] [Google Scholar]
- 140.Palmese F, Bolondi I, Giannone FA, Zaccherini G, Tufoni M, Baldassarre M, Caraceni P. The analysis of food intake in patients with cirrhosis waiting for liver transplantation: a neglected step in the nutritional assessment. Nutrients 11: 2462, 2019. doi: 10.3390/nu11102462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vaisman N, Katzman H, Carmiel-Haggai M, Lusthaus M, Niv E. Breakfast improves cognitive function in cirrhotic patients with cognitive impairment. Am J Clin Nutr 92: 137–140, 2010. doi: 10.3945/ajcn.2010.29211. [DOI] [PubMed] [Google Scholar]
- 142.Guo YJ, Tian ZB, Jiang N, Ding XL, Mao T, Jing X. Effects of late evening snack on cirrhotic patients: a systematic review and meta-analysis. Gastroenterol Res Pract 9189062, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Plank LD, Gane EJ, Peng S, Muthu C, Mathur S, Gillanders L, McIlroy K, Donaghy AJ, McCall JL. Nocturnal nutritional supplementation improves total body protein status of patients with liver cirrhosis: a randomized 12-month trial. Hepatology 48: 557–566, 2008. doi: 10.1002/hep.22367. [DOI] [PubMed] [Google Scholar]
- 144.Uojima H, Sakurai S, Hidaka H, Kinbara T, Sung JH, Ichita C, Tokoro S, Masuda S, Sasaki A, Koizumi K, Egashira H, Kako M, Kobayashi S. Effect of branched-chain amino acid supplements on muscle strength and muscle mass in patients with liver cirrhosis. Eur J Gastroenterol Hepatol 29: 1402–1407, 2017. doi: 10.1097/MEG.0000000000000968. [DOI] [PubMed] [Google Scholar]
- 145.Nguyen DL, Morgan T. Protein restriction in hepatic encephalopathy is appropriate for selected patients: a point of view. Hepatol Int 8: 447–451, 2014. doi: 10.1007/s12072-013-9497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Uribe M, Márquez MA, Ramos GG, Ramos-Uribe MH, Vargas F, Villalobos A, Ramos C. Treatment of chronic portal—systemic encephalopathy with vegetable and animal protein diets. A controlled crossover study. Digest Dis Sci 27: 1109–1116, 1982. doi: 10.1007/BF01391449. [DOI] [PubMed] [Google Scholar]
- 147.Kitajima Y, Takahashi H, Akiyama T, Murayama K, Iwane S, Kuwashiro T, Tanaka K, Kawazoe S, Ono N, Eguchi T, Anzai K, Eguchi Y. Supplementation with branched-chain amino acids ameliorates hypoalbuminemia, prevents sarcopenia, and reduces fat accumulation in the skeletal muscles of patients with liver cirrhosis. J Gastroenterol 53: 427–437, 2018. doi: 10.1007/s00535-017-1370-x. [DOI] [PubMed] [Google Scholar]
- 148.Churchward-Venne TA, Breen L, Di Donato DM, Hector AJ, Mitchell CJ, Moore DR, Stellingwerff T, Breuille D, Offord EA, Baker SK, Phillips SM. Leucine supplementation of a low-protein mixed macronutrient beverage enhances myofibrillar protein synthesis in young men: a double-blind, randomized trial. Am J Clin Nutr 99: 276–286, 2014. doi: 10.3945/ajcn.113.068775. [DOI] [PubMed] [Google Scholar]
- 149.Wilkinson DJ, Bukhari SSI, Phillips BE, Limb MC, Cegielski J, Brook MS, Rankin D, Mitchell WK, Kobayashi H, Williams JP, Lund J, Greenhaff PL, Smith K, Pj A. Effects of leucine-enriched essential amino acid and whey protein bolus dosing upon skeletal muscle protein synthesis at rest and after exercise in older women. Clin Nutr, 2017. pii: S0261-5614: 31340–31347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Atherton PJ, Smith K, Etheridge T, Rankin D, Rennie MJ. Distinct anabolic signalling response to amino acids in C2C12 skeletal muscle cells. Amino Acids 38: 1533–1539, 2010. doi: 10.1007/s00726-009-0377-x. [DOI] [PubMed] [Google Scholar]
- 151.Romàn E, Torrades MT, Nadal MJ, Càdenas G, Nieto JC, Vidal S, Juàrez C, Guarner C, Córdoba J, Soriano G. Randomised pilot study: effects of an exercise programme and leucine supplementation in patients with cirrhosis. Dig Dis Sci 59: 1966–1975, 2014. doi: 10.1007/s10620-014-3086-6. [DOI] [PubMed] [Google Scholar]
- 152.Fialla AD, Israelsen M, Hamberg O, Krag A, Gluud LL. Nutritional therapy in cirrhosis or alcoholic hepatitis: a systematic review and meta-analysis. Liver Int 35: 2072–2078, 2015. doi: 10.1111/liv.12798. [DOI] [PubMed] [Google Scholar]
- 153.Shergill R, Syed W, Rizvi SA, Singh I. Nutritional support in chronic liver disease and cirrhotics. World J Hepatol 10: 685–694, 2018. doi: 10.4254/wjh.v10.i10.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Koretz RL, Avenell A, Lipman TO. Nutritional support for liver disease. Cochrane Database Syst Rev, 2012. CD008344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Plauth M, Cabre E, Campillo B, Kondrup J, Marchesini G, Schutz T, Shenkin A, Wendon J. and ESPEN Guidelines on Parenteral Nutrition: Hepatology. Clin Nutr 28: 436–444, 2009. doi: 10.1016/j.clnu.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 156.Buchman AL, Iyer K, Fryer J. Parenteral nutrition-associated liver disease and the role for isolated intestine and intestine/liver transplantation. Hepatology 43: 9–19, 2006. doi: 10.1002/hep.20997. [DOI] [PubMed] [Google Scholar]
- 157.Peterson MD, Rhea MR, Sen A, Gordon PM. Resistance exercise for muscular strength in older adults: a meta-analysis. Ageing Res Rev 9: 226–237, 2010. doi: 10.1016/j.arr.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pedersen BK, Saltin B. Exercise as medicine—evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand J Med Sci Sports 25, Suppl 3: 1–72, 2015. doi: 10.1111/sms.12581. [DOI] [PubMed] [Google Scholar]
- 159.Dietrich R, Bachmann C, Lauterburg BH. Exercise-induced hyperammonemia in patients with compensated chronic liver disease. Scand J Gastroenterol 25: 329–334, 1990. doi: 10.3109/00365529009095494. [DOI] [PubMed] [Google Scholar]
- 160.Garcia-Pagan JC, Santos C, Barbera JA, Luca A, Roca J, Rodriguez-Roisin R, Bosch J, Rodes J. Physical exercise increases portal pressure in patients with cirrhosis and portal hypertension. Gastroenterology 111: 1300–1306, 1996. doi: 10.1053/gast.1996.v111.pm8898644. [DOI] [PubMed] [Google Scholar]
- 161.Aamann L, Dam G, Borre M, Drljevic-Nielsen A, Overgaard K, Andersen H, Vilstrup H, Aagaard NK. Resistance training increases muscle strength and muscle size in patients with liver cirrhosis. Clin Gastroenterol Hepatol 18: 1179–1187.e1176, 2020. doi: 10.1016/j.cgh.2019.07.058. [DOI] [PubMed] [Google Scholar]
- 162.Zenith L, Meena N, Ramadi A, Yavari M, Harvey A, Carbonneau M, Ma M, Abraldes JG, Paterson I, Haykowsky MJ, Tandon P. Eight weeks of exercise training increases aerobic capacity and muscle mass and reduces fatigue in patients with cirrhosis. Clin Gastroenterol Hepatol 12: 1920–1926 e1922, 2014. doi: 10.1016/j.cgh.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 163.Kruger C, McNeely ML, Bailey RJ, Yavari M, Abraldes JG, Carbonneau M, Newnham K, DenHeyer V, Ma M, Thompson R, Paterson I, Haykowsky MJ, Tandon P. Home exercise training improves exercise capacity in cirrhosis patients: role of exercise adherence. Sci Rep 8: 99, 2018. doi: 10.1038/s41598-017-18320-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pollock ML, Franklin BA, Balady GJ, Chaitman BL, Fleg JL, Fletcher B, Limacher M, Pina IL, Stein RA, Williams M, Bazzarre T. AHA Science Advisory. Resistance exercise in individuals with and without cardiovascular disease: benefits, rationale, safety, and prescription: an advisory from the Committee on Exercise, Rehabilitation, and Prevention, Council on Clinical Cardiology, American Heart Association; Position paper endorsed by the. Am Coll Sports Med Circul 101: 828–833, 2000. [DOI] [PubMed] [Google Scholar]
- 165.Debette-Gratien M, Tabouret T, Antonini MT, Dalmay F, Carrier P, Legros R, Jacques J, Vincent F, Sautereau D, Samuel D, Loustaud-Ratti V. Personalized adapted physical activity before liver transplantation: acceptability and results. Transplantation 99: 145–150, 2015. doi: 10.1097/TP.0000000000000245. [DOI] [PubMed] [Google Scholar]
- 166.Williams FR, Vallance A, Faulkner T, Towey J, Kyte D, Durman S, Johnson J, Holt A, Perera MT, Ferguson J, Armstrong MJ. Home-based exercise therapy in patients awaiting liver transplantation: protocol for an observational feasibility trial. BMJ Open 8: e019298, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sinclair M, Grossmann M, Hoermann R, Angus PW, Gow PJ. Testosterone therapy increases muscle mass in men with cirrhosis and low testosterone: a randomised controlled trial. J Hepatol 65: 906–913, 2016. doi: 10.1016/j.jhep.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 168.Yurci A, Yucesoy M, Unluhizarci K, Torun E, Gursoy S, Baskol M, Guven K, Ozbakir O. Effects of testosterone gel treatment in hypogonadal men with liver cirrhosis. Clin Res Hepatol Gastroenterol 35: 845–854, 2011. doi: 10.1016/j.clinre.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 169.Sinha-Hikim I, Taylor WE, Gonzalez-Cadavid NF, Zheng W, Bhasin S. Androgen receptor in human skeletal muscle and cultured muscle satellite cells: up-regulation by androgen treatment. J Clin Endocrinol Metab 89: 5245–5255, 2004. doi: 10.1210/jc.2004-0084. [DOI] [PubMed] [Google Scholar]
- 170.Kovacheva EL, Hikim AP, Shen R, Sinha I, Sinha-Hikim I. Testosterone supplementation reverses sarcopenia in aging through regulation of myostatin, c-Jun NH2-terminal kinase, Notch, and Akt signaling pathways. Endocrinology 151: 628–638, 2010. doi: 10.1210/en.2009-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhao Y, Zhao W, Pan J, Wu Q, Zhang Y, Bauman WA, Cardozo CP. Identification of androgen response elements in the insulin-like growth factor I upstream promoter. Endocrinology 148: 2984–2993, 2007. doi: 10.1210/en.2006-1653. [DOI] [PubMed] [Google Scholar]
- 172.Wallace JD, Abbott-Johnson WJ, Crawford DH, Barnard R, Potter JM, Cuneo RC. GH treatment in adults with chronic liver disease: a randomized double-blind, placebo-controlled, cross-over study. J Clin Endocrinol Metab 87: 2751–2759, 2002. doi: 10.1210/jcem.87.6.8548. [DOI] [PubMed] [Google Scholar]
- 173.Donaghy A, Ross R, Wicks C, Hughes SC, Holly J, Gimson A, Williams R. Growth hormone therapy in patients with cirrhosis: a pilot study of efficacy and safety. Gastroenterology 113: 1617–1622, 1997. doi: 10.1053/gast.1997.v113.pm9352864. [DOI] [PubMed] [Google Scholar]
- 174.Velloso CP. Regulation of muscle mass by growth hormone and IGF-1. Br J Pharmacol 154: 557–568, 2008. doi: 10.1038/bjp.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bolster DR, Jefferson LS, Kimball SR. Regulation of protein synthesis associated with skeletal muscle hypertrophy by insulin-, amino acid- and exercise-induced signalling. Proc Nutr Soc 63: 351–356, 2004. doi: 10.1079/PNS2004355. [DOI] [PubMed] [Google Scholar]
- 176.Rose CF. Ammonia-lowering strategies for the treatment of hepatic encephalopathy. Clin Pharmacol Ther 92: 321–331, 2012. doi: 10.1038/clpt.2012.112. [DOI] [PubMed] [Google Scholar]
- 177.Kumar A, Davuluri G, Silva RNE, Engelen M, Ten Have GAM, Prayson R, Deutz NEP, Dasarathy S. Ammonia lowering reverses sarcopenia of cirrhosis by restoring skeletal muscle proteostasis. Hepatology 65: 2045–2058, 2017. doi: 10.1002/hep.29107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bass NM, Mullen KD, Sanyal AJ, Poordad F, Neff G, Leevy CB, Sigal S, Sheikh MY, Beavers K, Frederick T, Teperman L, Hillebrand D, Huang S, Merchant K, Shaw A, Bortey E, Forbes WP. Rifaximin treatment in hepatic encephalopathy. N Engl J Med 362: 1071–1081, 2010. doi: 10.1056/NEJMoa0907893. [DOI] [PubMed] [Google Scholar]
- 179.Hiramatsu A, Aikata H, Uchikawa S, Ohya K, Kodama K, Nishida Y, Daijo K, Osawa M, Teraoka Y, Honda F, Inagaki Y, Morio K, Morio R, Fujino H, Nakahara T, Murakami E, Yamauchi M, Kawaoka T, Miki D, Tsuge M, Imamura M, Tanaka J, Chayama K. Levocarnitine use is associated with improvement in sarcopenia in patients with liver cirrhosis. Hepatol Commun 3: 348–355, 2019. doi: 10.1002/hep4.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Polkey MI, Praestgaard J, Berwick A, Franssen FME, Singh D, Steiner MC, Casaburi R, Tillmann HC, Lach-Trifilieff E, Roubenoff R, Rooks DS. Activin type II receptor blockade for treatment of muscle depletion in chronic obstructive pulmonary disease. A randomized trial. Am J Respir Crit Care Med 199: 313–320, 2019. doi: 10.1164/rccm.201802-0286OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rooks D, Praestgaard J, Hariry S, Laurent D, Petricoul O, Perry RG, Lach-Trifilieff E, Roubenoff R. Treatment of sarcopenia with bimagrumab: results from a phase II, randomized, controlled, proof-of-concept study. J Am Geriatr Soc 65: 1995–2017, 1988. [DOI] [PubMed] [Google Scholar]
- 182.Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, Auwerx J, Singh A, Rinsch C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab 1: 595–603, 2019. doi: 10.1038/s42255-019-0073-4. [DOI] [PubMed] [Google Scholar]
- 183.Rudman D, Sewell CW, Ansley JD. Deficiency of carnitine in cachetic cirrhotic patients. J Clin Invest 60: 716–723, 1977. doi: 10.1172/JCI108824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation. Biochim Biophys Acta 1863: 2422–2435, 2016. doi: 10.1016/j.bbamcr.2016.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shiraki M, Shimizu M, Moriwaki H, Okita K, Koike K. Carnitine dynamics and their effects on hyperammonemia in cirrhotic Japanese patients. Hepatol Res 47: 321–327, 2017. doi: 10.1111/hepr.12750. [DOI] [PubMed] [Google Scholar]
- 186.Nakanishi H, Kurosaki M, Tsuchiya K, Nakakuki N, Takada H, Matsuda S, Gondo K, Asano Y, Hattori N, Tamaki N, Suzuki S, Yasui Y, Hosokawa T, Itakura J, Takahashi Y, Izumi N. L-carnitine reduces muscle cramps in patients with cirrhosis. Clin Gastroenterol Hepatol 13: 1540–1543, 2015. doi: 10.1016/j.cgh.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 187.Malaguarnera M, Vacante M, Giordano M, Pennisi G, Bella R, Rampello L, Malaguarnera M, Li Volti G, Galvano F. Oral acetyl-L-carnitine therapy reduces fatigue in overt hepatic encephalopathy: a randomized, double-blind, placebo-controlled study. Am J Clin Nutr 93: 799–808, 2011. doi: 10.3945/ajcn.110.007393. [DOI] [PubMed] [Google Scholar]
- 188.Venu M, Martin E, Saeian K, Gawrieh S. High prevalence of vitamin A deficiency and vitamin D deficiency in patients evaluated for liver transplantation. Liver Transpl 19: 627–633, 2013. doi: 10.1002/lt.23646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Stamoulis I, Kouraklis G, Theocharis S. Zinc and the liver: an active interaction. Dig Dis Sci 52: 1595–1612, 2007. doi: 10.1007/s10620-006-9462-0. [DOI] [PubMed] [Google Scholar]
- 190.Rosendahl-Riise H, Spielau U, Ranhoff AH, Gudbrandsen OA, Dierkes J. Vitamin D supplementation and its influence on muscle strength and mobility in community-dwelling older persons: a systematic review and meta-analysis. J Hum Nutr Diet 30: 3–15, 2017. [Erratum in J Hum Nutr Diet: 825–826]. doi: 10.1111/jhn.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Matsuoka S, Matsumura H, Nakamura H, Oshiro S, Arakawa Y, Hayashi J, Sekine N, Nirei K, Yamagami H, Ogawa M, Nakajima N, Amaki S, Tanaka N, Moriyama M. Zinc supplementation improves the outcome of chronic hepatitis C and liver cirrhosis. J Clin Biochem Nutr 45: 292–303, 2009. doi: 10.3164/jcbn.jcbn08-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, Brachat S, Rivet H, Koelbing C, Morvan F, Hatakeyama S, Glass DJ. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol 34: 606–618, 2014. doi: 10.1128/MCB.01307-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Garcia-Munoz C, Vaillant F. Metabolic fate of ellagitannins: implications for health, and research perspectives for innovative functional foods. Crit Rev Food Sci Nutr 54: 1584–1598, 2014. doi: 10.1080/10408398.2011.644643. [DOI] [PubMed] [Google Scholar]
- 194.Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Felix AA, Williams EG, Jha P, Lo Sasso G, Huzard D, Aebischer P, Sandi C, Rinsch C, Auwerx J. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med 22: 879–888, 2016. doi: 10.1038/nm.4132. [DOI] [PubMed] [Google Scholar]
- 195.Tomas-Barberan FA, Gonzalez-Sarrias A, Garcia-Villalba R, Nunez-Sanchez MA, Selma MV, Garcia-Conesa MT, Espin JC. Urolithins, the rescue of “old” metabolites to understand a “new” concept: metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status. Mol Nutr Food Res 61, 2017. [DOI] [PubMed] [Google Scholar]