

Abstract
MYC is a prolific proto-oncogene driving the malignant behaviors of numerous common cancers, yet potent and selective cell-permeable inhibitors of MYC remain elusive. In order to ultimately realize the goal of therapeutic MYC inhibition in cancer, we have initiated discovery chemistry efforts aimed at inhibiting MYC translation. Here we describe a series of conformationally stabilized synthetic antisense oligonucleotides designed to target MYC mRNA (MYCASOs). To support bioactivity, we designed and synthesized this focused library of MYCASOs incorporating locked nucleic acid (LNA) bases at the 5’- and 3’-ends, a phosphorothioate backbone, and internal DNA bases. Treatment of MYC-expressing cancer cells with MYCASOs leads to a potent decrease in MYC mRNA and protein levels. Cleaved MYC mRNA in MYCASO-treated cells is detected with a sensitive 5’ Rapid Amplification of cDNA Ends (RACE) assay. MYCASO treatment of cancer cell lines leads to significant inhibition of cellular proliferation while specifically perturbing MYC-driven gene expression signatures. In a MYC-induced model of hepatocellular carcinoma, MYCASO treatment decreases MYC protein levels within tumors, decreases tumor burden, and improves overall survival. MYCASOs represent a new chemical tool for in vitro and in vivo modulation of MYC activity, and promising therapeutic agents for MYC-addicted tumors.
Introduction
The transcription factor MYC is among the most activated genes in cancer. MYC deregulation can occur genetically, epigenetically, and post-transcriptionally through a wide variety of mechanisms (1). MYC mRNA can be transcriptionally upregulated by genomic copy number amplification (2), by activation of trans factors such as mutant NOTCH1 in T-cell leukemia (3), and by chromosomal rearrangement of the MYC locus to enhancers within the IgH or TCR loci (4,5). Functionally MYC heterodimerizes with the transcription factor MAX and together they bind to the genome via E-box motifs, leading to the recruitment of histone acetyltransferases and other chromatin regulators (6,7). With greater than 10 000 binding sites throughout the human genome, MYC coordinates a vast transcriptional regulatory network that controls the hallmark gene expression programs responsible for cancer cell proliferation, growth, metabolism, and evasion from apoptosis (8).
While deregulated MYC can dramatically alter cellular transcriptional programs, the precise mechanism for its oncogenic activity remains an area of intense investigation. Evidence suggests a model of oncogenic MYC as a global transcriptional amplifier that when highly expressed engages all active promoters and enhancers in a cancer cell (9). Others have proposed a model of oncogenic MYC that activates and represses transcription of discrete gene sets (10,11). Nevertheless, MYC-driven pleiotropic transcriptional changes act to potently transform cells to an oncogenic phenotype. Indeed, tumor cells with high levels of MYC are ‘addicted’ to its expression (12). Experiments modelling systemic MYC inhibition with engineered dominant negative constructs in mice have revealed that directly blocking MYC binding to MAX leads to potent tumor regression with an evident therapeutic window, suggesting that pharmacologic MYC inhibition may be a viable cancer therapeutic strategy (13). Yet realizing MYC-directed therapeutics has been a challenge due in part to its intrinsically disordered structure that has historically been difficult to disrupt with small molecules (14). While some compounds have been identified that block MYC/MAX dimerization, to date these chemical tools exhibit low potency, poor selectivity, and a lack of antitumor efficacy in vivo (15–20). More recently the development of advanced small molecule and peptide MYC/MAX dimerization inhibitors with apparent in vivo antitumor activity, and small molecule MAX stabilizers, has expanded opportunities for anti-MYC therapeutics (21–23).
To further advance the toolbox of MYC-directed agents, we investigated an orthogonal strategy to target MYC by inhibiting MYC mRNA translation with antisense oligonucleotides (ASOs). ASOs targeting the MYC translational start site (the first 15 bases of the MYC mRNA coding sequence) have previously been investigated. In these pioneering studies, ASO treatment resulted in inhibition of MYC protein expression in human T lymphocytes and promyelocytic leukemia cells (24–26). Subsequently, phosphorothioate bonds were introduced between nucleosides to enhance stability (27). These improved compounds inhibited the growth of human melanoma cells in vitro through induction of apoptosis and with evident efficacy in a murine melanoma model (27). An investigational ASO was translated to human clinical investigation by Inex Pharmaceuticals for the treatment of solid tumors and lymphoma. However, it was subsequently discovered that the anti-tumor effects of this ASO was likely due to immune stimulatory activity of a CpG motif within the molecule (28).
Advances in the chemistry of ASOs create a new opportunity to revisit this therapeutic strategy. Second generation ASOs incorporate 2’-alkyl ribose modifications, including 2’-O-methyl or 2’-O-methoxyethyl moieties, to enhance nuclease resistance and increase binding affinity (29,30). Third generation ASOs incorporate chemical modifications elsewhere in the sugar ring to further enhance target affinity and stability. This includes the locked nucleic acid (LNA), a conformationally restricted nucleotide containing a 2’-O,4’-C-methylene bridge, which serves to lock the sugar into a C3’-endo, or northern, conformation. Incorporation of LNA bases into ASOs increases nuclease resistance and dramatically improves affinity for target mRNA (31,32). Many second and third generation ASOs also employ a ‘gapmer’ design with a central gap region of 2’-deoxynucleotides flanked on both sides by several modified nucleotides. This allows for the recruitment of RNase H, the endogenous cellular enzyme responsible for DNA/RNA duplex recognition and target mRNA cleavage (29,33,34).
Here we describe the synthesis and characterization of 23 distinct LNA/DNA gapmer ASO molecules that effectively ‘scan’ the entire length of the human MYC mRNA coding region. These MYC-targeting ASOs (MYCASOs) potently and selectively decrease MYC expression in a variety of MYC-expressing cancer cell lines from distinct lineages. This potent MYC knockdown is associated with decreased cancer cell viability and proliferation. We also establish an RNase H-mediated mechanism of MYC mRNA degradation, use comprehensive transcriptomic profiling to decipher global MYCASO effects on cellular gene expression signatures, and use intermittent dosing in a murine tumor model to establish proof of concept for MYCASOs as a potential strategy for MYC-targeted therapy.
Results
MYCASOs potently decrease MYC protein expression in cancer cell lines
Our MYCASO library includes 23 distinct antisense oligonucleotides that target selected sites along the coding region of the full-length mature human MYC mRNA transcript (Figure 1a and 1b). MYCASOs are single-stranded 16-mers, a size shown to be optimal both for sequence-specific target knockdown and for cellular penetration (35). Notably, MYCASOs do not contain CpG dinucleotides or polyguanosines shown to be problematic with previous anti-MYC ASO investigation (28,36,37). We incorporated specific chemical features shown to improve ASO activity. The first and last three nucleotides on each MYCASO are comprised of LNA bases, and all bases are linked with phosphorothioate bonds. We also synthesized a LNA-containing gapmer oligonucleotide of randomized bases previously described as an effective non-targeted control in ASO studies (herein referred to as NT-ASO) (38).
Figure 1: Design and characterization of a library of MYC-targeting antisense oligonucleotides.
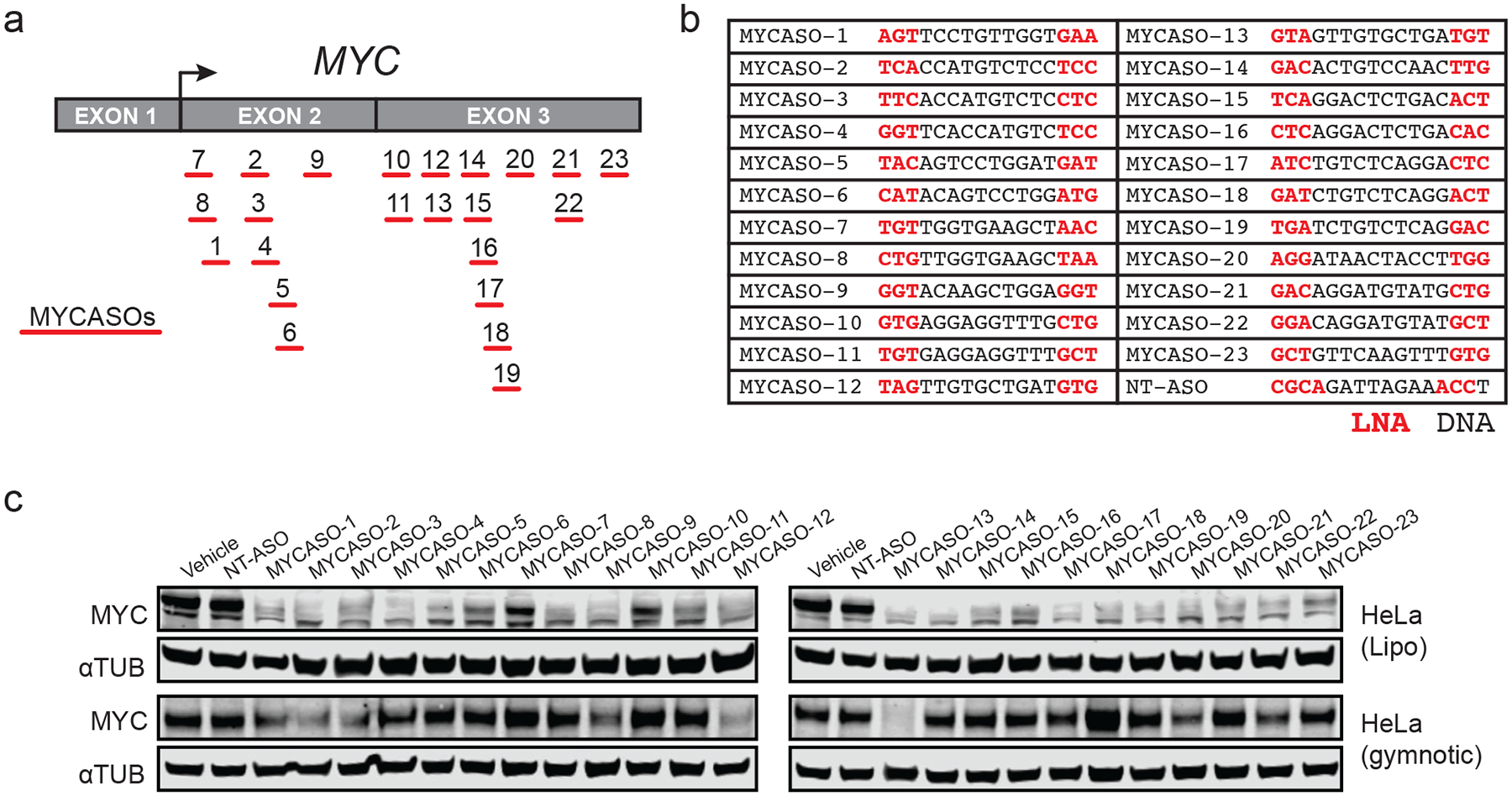
(a) Schematic of the MYCASO library with approximate seed site locations of each of 23 oligonucleotides along the MYC transcript. Arrow represents the major translational start site. (b) Sequences of MYCASOs and NT-ASO used as control. Locations of LNA bases are highlighted in red. (C) Immunoblotting for MYC and alpha-tubulin in HeLa cells. Top panel shows whole cell extracts from HeLa cells treated with 10 nM MYCASO formulated in Lipofectamine 2000 (Lipo) for 24 hours. Bottom panel shows HeLa cells with 10 μM gymnotic-treated MYCASO for 72 hours.
We first tested the capacity of each MYCASO to decrease MYC protein expression in MYC-overexpressing HeLa cells, derived from a cervical adenocarcinoma in which integration of the HPV-18 virus genome upstream of MYC amplifies its expression (39) (Figure 1c). Many cell types including cancer cell lines can take up ASOs in the absence of transfection reagents or other delivery formulations, and so we compared MYCASO effects using the transfection reagent Lipofectamine 2000 with that of gymnotic (unformulated, dissolved in saline) delivery (40,41). Most MYCASOs potently decrease MYC protein expression at 10 nM after 24 hours when delivered with lipid-based formulation (Figure 1c). With gymnotic-based delivery, only select MYCASOs decrease MYC protein expression when dosed at 10 μM for 72 hours revealing that structural or physicochemical properties of some ASOs preclude their uptake or target engagement in some cancer cell lines.
To investigate the activity of MYCASOs in other MYC-driven cell contexts, we treated with MYCASOs by gymnotic delivery three additional cancer cell lines representing distinct tumor types: MM1.S, a multiple myeloma cell line that harbors an IgH-MYC translocation that results in MYC overexpression driven by IgH enhancers (42); CUTLL1, a T-cell leukemia line that harbors a TCR-NOTCH1 rearrangement in which activated intracellular NOTCH1 transcriptionally upregulates MYC expression (43); and HLF cells, derived from a non-differentiated hepatoma, that have been shown to be highly dependent on MYC expression as part of the Broad Institute cancer cell line dependency mapping projects (44,45) (Figure 2a). Selected MYCASO treatment of CUTTL1, HLF, and MM1.S cells significantly decreased MYC protein expression when dosed at 10 μM for 72–120 hours. The dynamics of MYC protein knockdown varied between cell lines, likely because of different uptake dynamics (40). The molecules exhibiting the greatest average MYC knockdown activity across the four cell lines tested were MYCASO-3, MYCASO-9, MYCASO-12, and MYCASO-13 (Figure 2a, bottom). Because MYCASO-12 and MYCASO-13 target overlapping sites on the MYC mRNA, we chose to pursue further study of these compounds using three lead candidates: MYCASO-3, MYCASO-9, and MYCASO-13. Treatment with these three molecules led to decreased MYC protein and mRNA in both dose- and time-responsive manners in all four cancer cell lines (Figure 2b and 2c, Supplemental Figure 1a and 1b). Thus, our library of MYCASOs introduces novel and potent cell-active MYC-targeting agents.
Figure 2: MYCASOs decrease MYC protein expression in cancer cell lines.
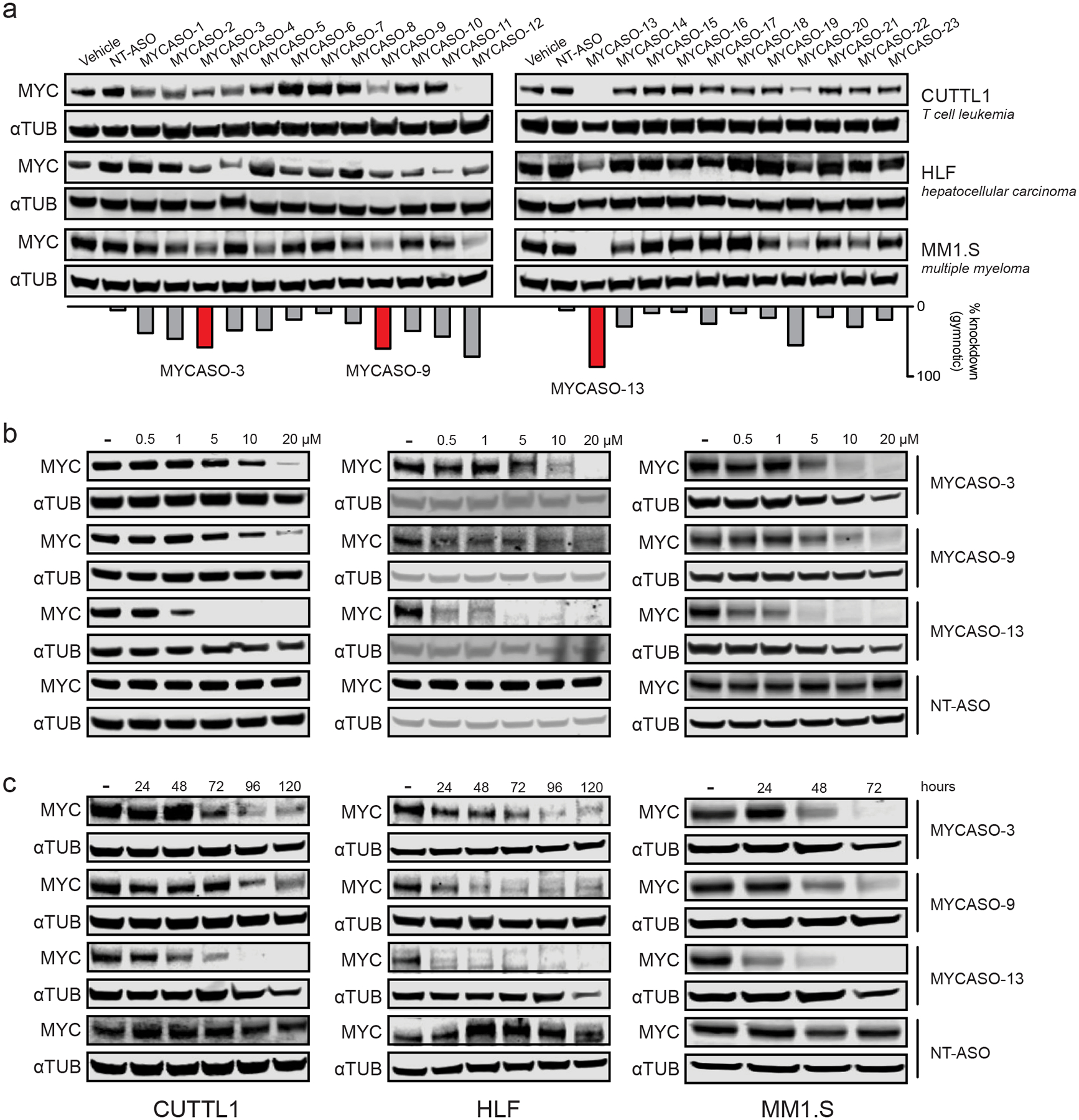
(a) Immunoblotting for MYC and alpha-tubulin in three MYC-expressing cancer cell lines after treatment with the full MYCASO library. Panels show CUTLL1 and MM1.S cells with 10 μM gymnotic-treated MYCASO for 72 hours. HLF cells were treated gymnotically at 10 μM for 120 hours. The bar graph shows average percent knockdown of MYC of gymnotic-treated cells across all four cell lines including HeLa. Highlighted MYCASOs (3, 9, and 13) were chosen for further study due to their superior knockdown activity and non-overlapping seed sites along the MYC mRNA. (b) Dose-proportional knockdown of MYC protein expression with MYCASO treatment. CUTLL1, MM1.S, and HLF cells were treated with 0, 0.5, 1, 5, 10, and 20 μM MYCASO (gymnotic) for 72 hours (CUTLL1 and MM1.S) or 120 hours (HLF). (c) Time-proportional knockdown of MYC protein expression with MYCASO treatment. CUTLL1 and HLF cells were treated with 10 μM MYCASO (gymnotic) for 24, 48, 72, 96, and 120 hours. MM1.S cells were treated with 10 μM MYCASO (gymnotic) for 24, 48, and 72 hours.
MYCASOs downregulate MYC expression through RNase H-mediated cleavage of MYC mRNA
We next investigated the mechanism by which MYCASOs mediate MYC knockdown. To determine MYCASO capability to induce MYC mRNA cleavage, we employed a 5’ Rapid Amplification of cDNA Ends (RACE) assay to capture MYC transcript fragments (Figure 3a). With this assay we observed MYC transcript fragments in RNA isolated from MYCASO-treated cells but not in vehicle-treated or NT-ASO-treated cells. Consistent with the mechanism of RNase H, the sizes of the fragments suggest cleavage of the MYC transcript at or near the sites of specific MYCASO binding with the dominant fragment generated by the more 3’-targeting MYCASO-13 smaller than those generated by MYCASO-3 and MYCASO-9. Interestingly, we observed multiple fragments following MYCASO-3 and MYCASO-9 treatment, which may reflect additional mechanisms by which ASOs mediate target mRNA cleavage outside their seed sites that complement the canonical RNase H1-mediated pathway (46).
Figure 3: MYCASOs downregulate MYC expression through RNase H1-mediated cleavage of MYC mRNA.
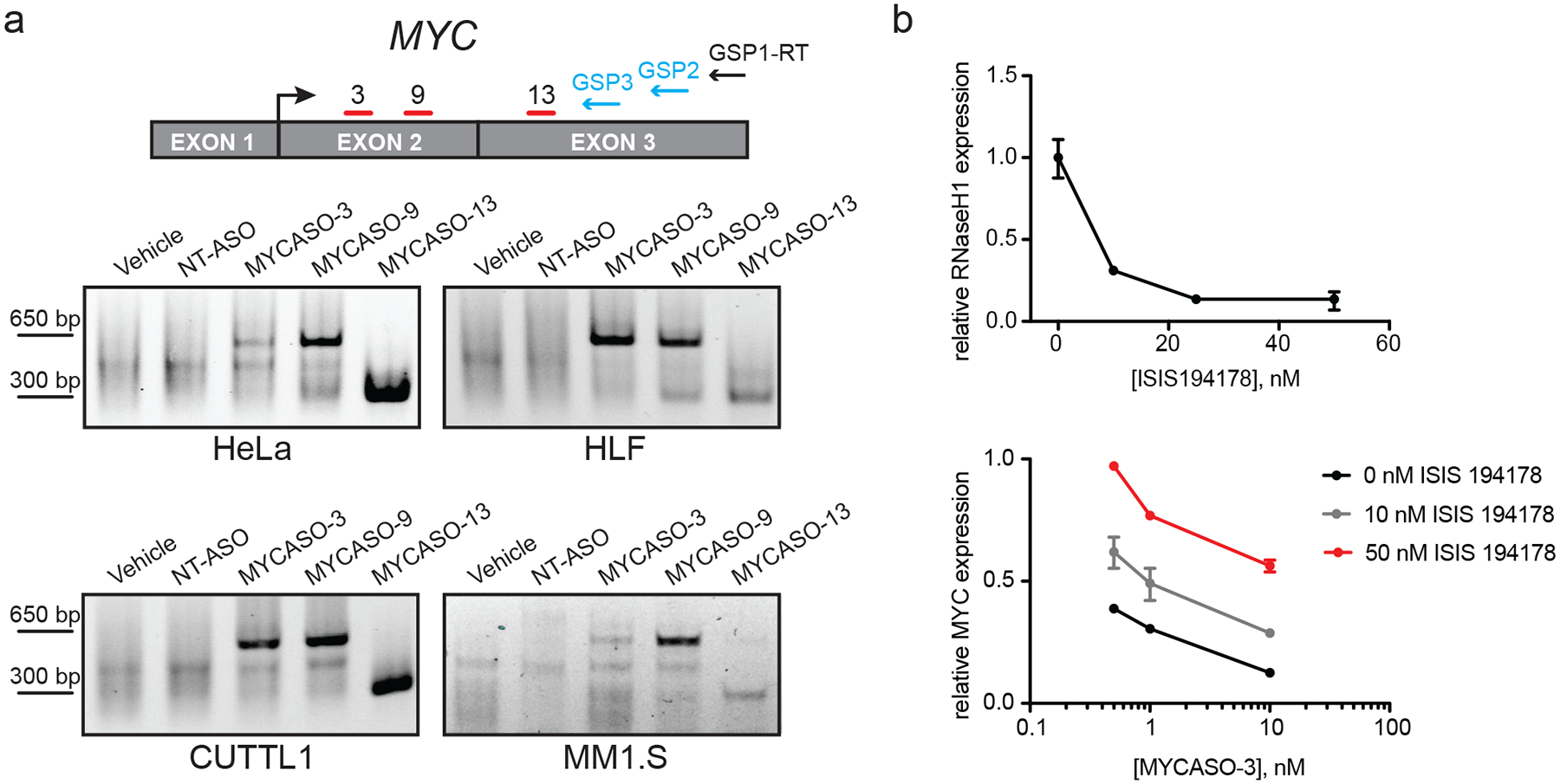
(a) MYCASOs induce cleavage of MYC mRNA. Top: schematic of 5’ Rapid Amplification of cDNA Ends (RACE) assay. Gene specific primers (GSPs) are used to capture and amplify MYC mRNA fragments from total cellular RNA for visualization on an agarose gel. Bottom: 5’ RACE PCR products following MYCASO treatment. MYCASO-treated samples contain fragments consistent with mRNA cleavage at MYCASO binding sites, whereas vehicle- and NT-ASO-treated cells produce no fragments. HeLa cells were treated with 10 nM MYCASO in Lipofectamine for 24 hours. CUTLL1, MM1.S, and HLF cells were treated with 10 μM MYCASO (gymnotic) for 72 hours (CUTLL1 and MM1.S) or 120 hours (HLF). (b) RNase H1 knockdown rescues MYCASO-mediated MYC mRNA knockdown. Top: RT-PCR for RNASEH1 in HeLa cells following treatment with an anti-RNASEH1 ASO (ISIS194178) in Lipofectamine for 24 hours normalized to vehicle-treated control. Bottom: RT-PCR for MYC in HeLa cells following co-transfection with the indicated doses of anti-RNASEH1 ASO and MYCASO-3 in Lipofectamine 2000 for 24 hours normalized to vehicle-treated control. Values represent quadruplicate means +/− SD.
For further evidence of RNase H-mediated transcript knockdown, we utilized a validated RNASEH1 ASO previously employed to establish the role of RNase H1 in targeted ASO activity (34). ASO-mediated decrease in RNASEH1 levels suppresses MYCASO-mediated knockdown of the MYC transcript suggesting that the decrease in MYC transcript and protein levels we observe with MYCASO treatment is achieved, at least in part, through RNase H-mediated cleavage of MYC mRNA (Figure 3b).
MYCASO treatment reduces proliferation of human cancer cells
Perturbation of MYC transcript levels in cancer cells with RNA interference and genetic manipulation leads to a loss of cell viability and survival both in vitro and in vivo (47). To assess the phenotypic effects of MYCASO treatment, we treated cell lines in both short-term and long-term cell proliferation and survival assays. MYCASO treatment is results in a decrease in cell viability when dosed gymnotically in CUTLL1, HLF, and MM1.S cancer cell lines. This decrease in viability is both dose-dependent as determined by cellular ATP content (Figure 4a), and time-dependent as determined via flow cytometry-enabled cell counting (Figure 4b). In MM1.S cells, this decrease in viability was coincident with an increase in apoptotic cells in MYCASO-treated cells as measured by Annexin V staining (Figure 4c). MYCASO treatment also leads to decreased cell viability when delivered to HeLa cells by lipid-based transfection (Figure 4d). To determine if decreased viability is a result of decreased MYC expression, we generated a HeLa cell line that stably, constitutively overexpresses a MYC construct containing silent mutations in the MYCASO-3, −9, and −13 seed sites, designed to abrogate MYCASO binding to the mutant MYC transcript while still producing wild-type MYC protein. Overexpression of this mutant MYC leads to a partial but significant rescue of MYCASO-induced cytotoxicity (Figure 4e and 4f). Lipid-based transfection of MYCASO was used for these experiments to efficiently deliver MYCASO to HeLa cells, for which gymnotic delivery was found to be inefficient (data not shown). Notably, a higher dose of MYCASO (100 nM) led to a more attenuated rescue effect of our MYC-expression system, suggesting that MYCASOs may lose some specificity at higher intracellular concentrations.
Figure 4: Phenotypic response to MYCASO treatment.
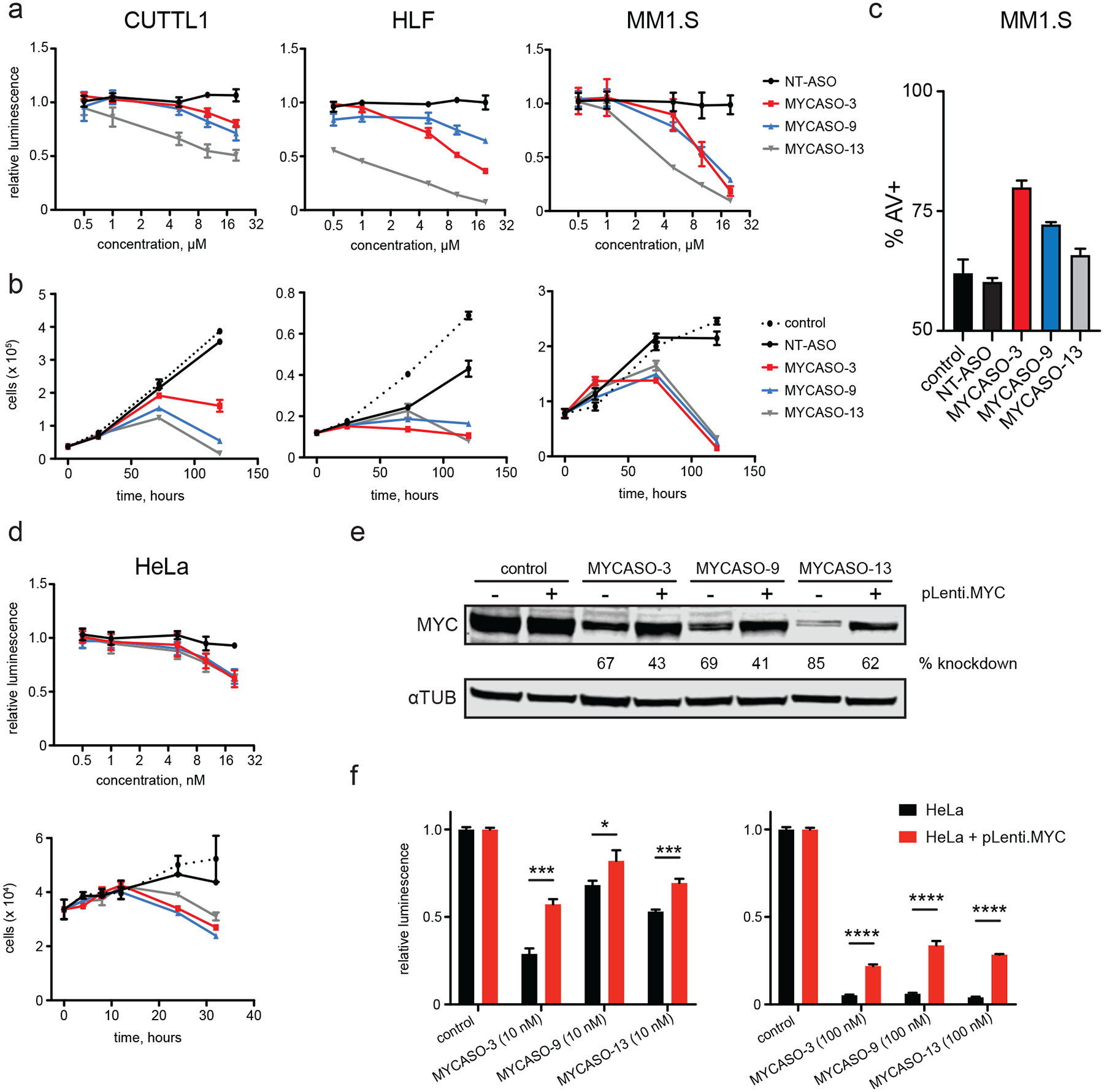
(a) Dose-proportional effect of MYCASO treatment on cellular viability as approximated by ATP-dependent luminescence normalized to vehicle-treated control. CUTLL1, MM1.S, and HLF cells were treated with MYCASO (gymnotic) for 72 hours (CUTLL1 and MM1.S) or 120 hours (HLF). Values represent quadruplicate means +/− SD. (b) Cell numbers over time with MYCASO treatment. Cells were stained using Guava ViaCount reagent and counted by a Guava flow cytometer. CUTLL1, MM1.S, and HLF cells were treated with 10 μM MYCASO (gymnotic). Values represent triplicate means +/− SD. (c) Annexin V (AV) positive MM1.S cells as measured by flow cytometry following 72 hours of treatment with 10 μM MYCASO or NT-ASO (gymnotic). Values represent triplicate means +/− SD. (d) Effects of MYCASO treatment on HeLa cells. Top panel shows dose-proportional effects on cellular viability as described above. HeLa cells were treated with MYCASO in Lipofectamine for 24 hours. Bottom panel shows cell numbers over time as described above. HeLa cells were treated with 10 nM MYCASO in Lipofectamine 2000. (e) Rescue of MYCASO-mediated MYC protein knockdown. Immunoblot for MYC and alpha-tubulin following MYCASO treatment (10 nM in Lipofectamine 2000 for 24 hours) in HeLa cells (pLenti.MYC –) and in a HeLa-derived cell line stably expressing a MYC construct containing silent mutations in MYCASO seed sites designed to abrogate MYCASO binding (pLenti.MYC +). (f) Rescue of MYCASO-mediated viability effects. Cellular viability as approximated by ATP-dependent luminescence following MYCASO treatment (10 nM (left) or 100 nM (right) in Lipofectamine 2000 for 24 hours) in HeLa and mutant MYC over-expressing cells normalized to vehicle-treated control. Values represent triplicate means +/− SD, asterisks represent p values based on t-test (* < 0.05, *** < 0.001, **** < 0.0001).
Selective perturbation of the MYC gene expression program by MYCASOs
To better understand the specificity and global effects of MYCASO treatment, we performed quantitative assessments of the global transcriptome using total mRNA sequencing. For these studies we focused on MYCASO-3, which showed both in vitro efficacy and a high degree of in vivo tolerability (discussed below). HeLa cells were treated with MYCASO-3 or NT-ASO by transfection with lipid-based reagent formulation, and samples were harvested at both 4 hours and 18 hours post-transfection. Lipid-based delivery was again used for these experiments to reliably assess the acute intracellular effects of MYCASO treatment. Vehicle treated cells were included as controls at both time points and we observed high concordance of mRNA expression between replicate samples (Supplemental Figure 2). In both NT-ASO and MYCASO-3 treated cells, significant effects on the transcriptome were observed (Figure 5a). Four hours of NT-ASO treatment induced downregulation of 771 genes and upregulation of 308 genes, while after 18 hours 4319 genes were downregulated and 4047 genes were upregulated (log2 fold change < −0.2, > 0.2, p-value < 0.01). With 4 hours of MYCASO-3 treatment we observed downregulation of MYC (p = 8.09e−16) in addition to 354 other genes; 166 genes were upregulated. With 18 hours of MYCASO-3 treatment, 3975 genes were downregulated; MYC ranked 23 in significance among downregulated genes (p = 5.18e−261); 4075 genes were had increased expression with MYCASO-3 treatment including CDKN1A, a gene negatively regulated by MYC (p = 6.32e−240) (48). While many genes are affected by NT-ASO treatment, MYC mRNA levels were not affected. The observed broad transcriptional effects of MYCASO-3 are consistent with the biology of MYC as prolific transcriptional control factor; moreover, the broad effects of NT-ASO treatment on the transcriptome emphasizes that ASO treatment in general can have consequences on mRNA expression irrespective of specific gene targeting.
Figure 5: Transcriptomic analysis of MYCASO-3 treated cells.
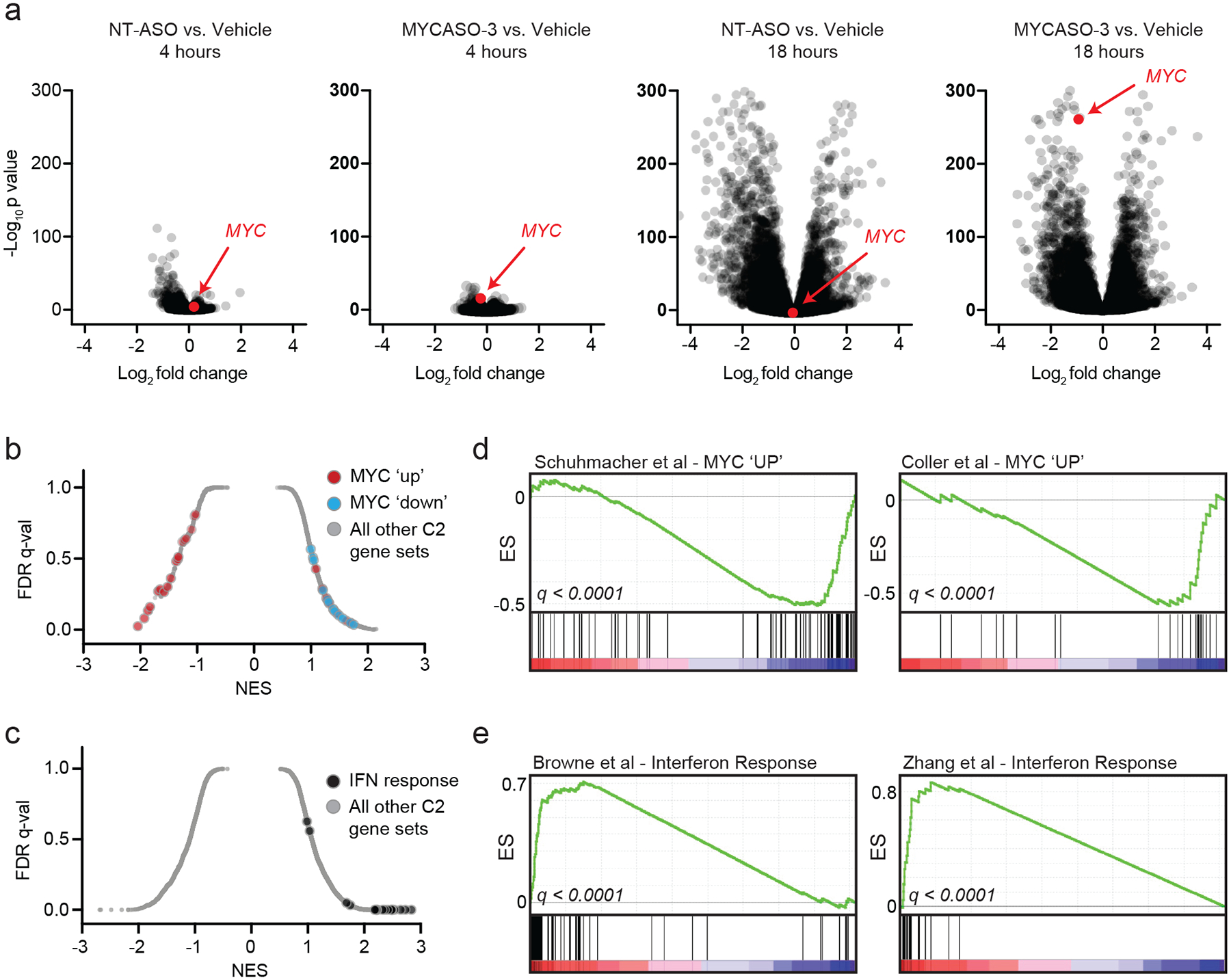
(a) RNA-sequencing was performed on HeLa cells transfected with NT-ASO or MYCASO-3 (10 nM) using Lipofectamine 2000 as delivery vehicle. Volcano plots of gene expression differences comparing NT-ASO and MYCASO-3 treated cells with vehicle after 4 and 18 hours. (b) Gene set enrichment analysis (GSEA) normalized enrichment scores for MYC-associated gene sets (red and blue) and the total C2 gene sets as part of MSigDB in MYCASO-3 treated cells. Differential expression analysis was performed with DESeq2 as described in Methods. (c) GSEA normalized enrichment scores for interferon (IFN)-associated gene sets and the total C2 gene sets as part of MSigDB in ASO-treated cells (NT-ASO and MYCASO-3 samples). Differential expression analysis was performed with DESeq2 as described in Methods. (d) GSEA signatures for two example gene sets of genes positively regulated by MYC (MYC ‘UP’). (e) GSEA signatures for two example gene sets of the interferon response.
To define transcriptional programs most affected by MYCASO-3 treatment specifically and ASO treatment generally we used gene set enrichment analysis (GSEA) to query for effects on all curated gene sets available in the Molecular Signatures Database (MSigDB ‘C2’, n = 3678 gene sets) (49). Global distributions of enrichment scores for MYCASO-3 treated samples are shown in Figure 5b, with 38 experimentally defined MYC-specific gene sets highlighted. Among the gene sets that are most negatively enriched with MYCASO-3 treatment are genes validated to be positively regulated by MYC (MYC ‘up’); 18 of 25 MYC ‘up’ gene sets had a negative enrichment score. Conversely, all gene sets associated with negative regulation by MYC (MYC ‘down’) were positively enriched with MYCASO-3 treatment. Next we assessed gene set enrichment scores across MSigDB for all ASO treated samples (MYCASO-3 and NT-ASO) compared to vehicle-treated control samples (Figure 5c). Nine of the top 12 enriched gene sets in ASO-treated samples were annotated as activated interferon signatures. Included in the MYC ‘up’ signatures found in MYCASO-3 treated samples are from studies from Coller et al. and Schuhmacher et al. (50,51) (Figure 5d); exemplary interferon signatures were described by Browne et al. and Zhang et al. (52,53) (Figure 5e). These studies reveal that while some unique gene expression programs may be activated non-specifically with ASO treatment, MYCASO-3 treatment has selective effects on both MYC and MYC-driven gene expression programs.
MYCASO treatment decreases tumor burden and prolongs survival in vivo
Motivated by selective anti-MYC effects of MYCASO treatment in vitro, we assessed the effects of MYCASO treatment in mice. To determine the tolerability of MYCASO treatments, C57BL/6J mice were dosed with vehicle (sterile saline), NT-ASO, MYCASO-3, MYCASO-9, or MYCASO-13. Two dosing regimens were tested with one cohort receiving 25 mg per kilogram by tail vein injection every three days. A lower dose cohort was also assessed, with mice receiving a single starting dose of 25 mg per kilogram, followed by maintenance dosing of 5 mg per kilogram every three days. To assess tolerability, mice were weighed at the time of each injection (Supplemental Figure 3). Notably, all doses were well tolerated over the course of 7 injections except for a high dose of MYCASO-13 which led to rapid weight loss in treated mice.
To determine if MYCASOs may have anti-tumor effects in vivo, we utilized an established model of MYC-induced hepatocellular carcinoma (54). In this model, FVB/N mice are hydrodynamically perfused via tail vein with both MYC-expressing and Sleeping Beauty transposase-expressing plasmids. Following perfusion hepatocytes are rapidly transformed and tumors form within 3 weeks; euthanasia is typically required around 6–10 weeks because of significant tumor burden. Tumors were established in a cohort of 25 mice, and five weeks following perfusion 10 mice were treated with MYCASO-3 or NT-ASO at a dose of 25 mg per kilogram every three days. Mice were treated for two weeks (a total of 5 injections). MYCASO-3 was well-tolerated with minimal effects on mouse weight (Supplemental Figure 4a). Five weeks post-perfustion we randomly selected and euthanized 6 mice and livers were harvested to assess pre-treatment tumor burden. Following treatment, 3 mice per treatment group were euthanized and livers were harvested to assess post-treatment tumor burden (Figure 6a). Notably, gross observation of MYCASO-3 treated tumors revealed decreased tumor volume compared to NT-ASO treated tumors (Figure 6b). Mice treated with NT-ASO had tumor burdens similar to untreated mice 7 weeks following perfusion, suggesting that in this model general ASO treatment does not have a significant effect on tumor growth (Supplemental Figure 4b,c). The remaining mice (7 per treatment group) were euthanized when moribund or until the end of observation, and livers were harvested to assess pharmacodynamic effects of MYCASO treatment. The liver weight to body weight ratios of mice treated with MYCASO-3 were similar to those of pretreatment mice, whereas NT-ASO treated mice had significantly increased liver weight to body weight ratios, suggesting that MYCASO-3 treatment reduces tumor burden (Figure 6c). MYCASO-3 treatment also led to a decrease in MYC protein expression compared to NT-ASO treatment (Figure 6d). Quantified assessments of MYC protein levels by immunohistochemistry revealed dark, MYC-positive staining in NT-ASO treated mice. MYCASO-3 treated mice showed smaller patches of MYC-positive tumors, with relatively less intense MYC expression when assessed visually and digitally (Figure 6e,f,g). MYCASO-3 treated tumors had fewer Ki67-positive cells, suggesting MYCASO-3 treatment slows tumor cell proliferation (Figure 6e). Notably, MYCASO-3 treatment significantly increased overall survival compared to NT-ASO treatment (Figure 6h). Taken together, our results suggest that MYCASO-3 has direct anti-MYC and anti-tumor effects in vivo, warranting future studies to assess biodynamic distribution and other pharmacokinetic parameters.
Figure 6: In vivo effects of MYASO-3 in a MYC-induced model of hepatocellular carcinoma.
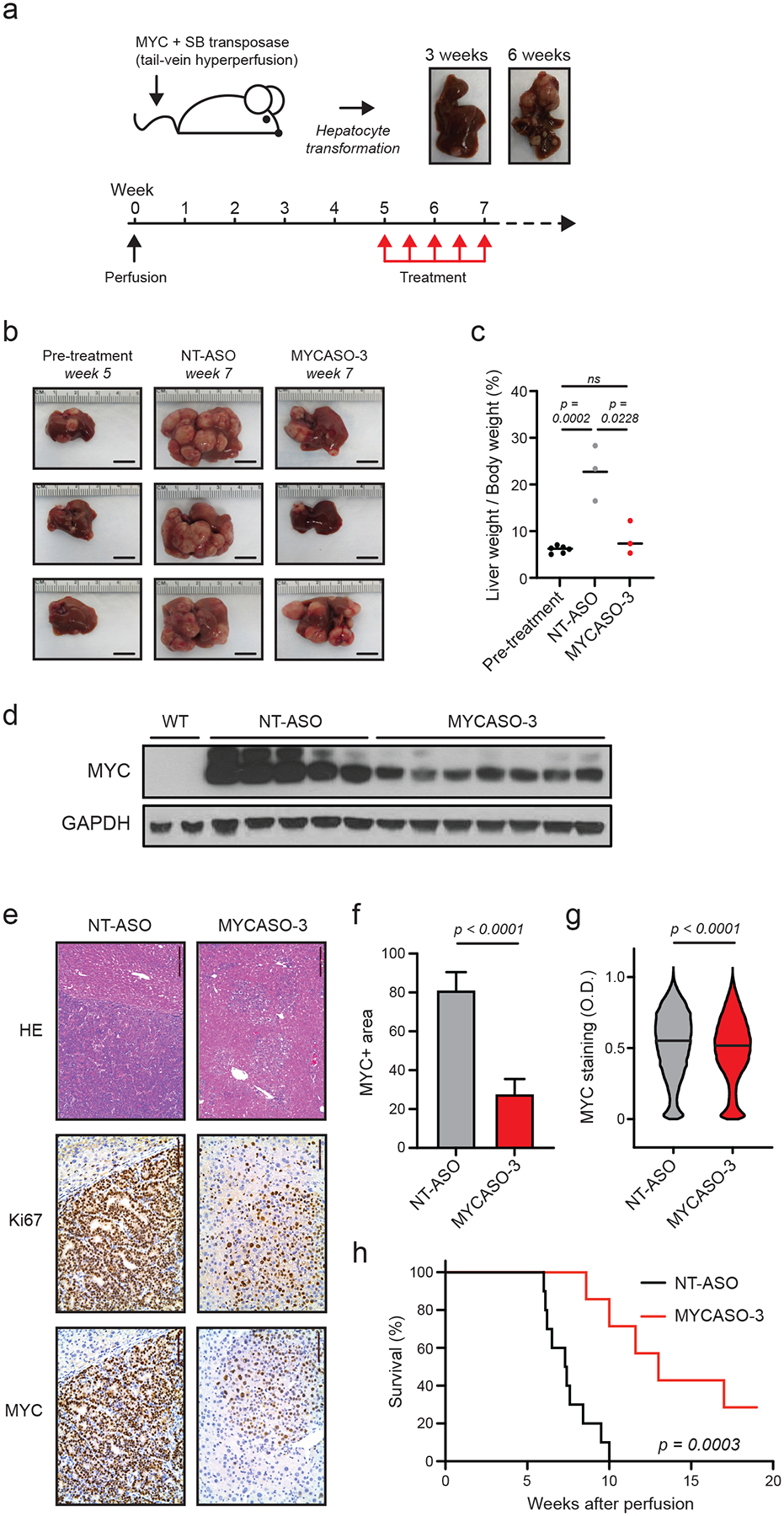
(a) Schematic of HCC tumor model used to assess MYCASO-3 effects. A plasmid expressing human MYC is co-injected with a plasmid expressing the Sleeping Beauty transposase by hydrodynamic injection. Liver tumor formation is observed by three weeks following perfusion, with large tumor masses observable by 6 weeks. MYCASO-3 was dosed at 25 mg/kg in mice beginning 5 weeks after perfusion, and dosing was performed every 3 days for a period of 2 weeks (a total of 5 doses). (b) Example harvested livers from pre-treatment (5 weeks after perfusion; n=6), NT-ASO treated (7 weeks after perfusion; n=3), and MYCASO-3 treated mice (7 weeks after perfusion; n=3). (c) Ratios of liver weight to body weight in pre-treatment (n=5), NT-ASO treated (n=3), and MYCASO-3 treated mice (n = 3). Bars represent mean values. Statistical analysis performed by t test. (d) MYC immunoblotting on extracts from wild-type (WT) liver tissue, tumor tissue from NT-ASO treated mice, and tumor tissue from MYCASO-3 treated mice. (e) HE and immunohistochemistry showing Ki67+ and MYC+ cells in livers of NT-ASO and MYCASO-3 treated mice. (f) Quantification of MYC+ area in (e). Error bars represent +/− SD of the mean of 8 separate tumor sections. Statistical analysis performed by t test. (g) Quantification of MYC+ cell staining in (e). MYC positivity grades on a per cell basis was assigned using automated Aperio Digital Pathology software. Bars represent median values. Statistical analysis performed by t test. (h) Survival of NT-ASO treated and MYCASO-3 treated mice over time (n=10). P value generated by log-rank Mantel-Cox test.
Discussion
As a historically ‘undruggable’ oncogene, MYC has proven exceptionally difficult to selectively inhibit with traditional pharmacology, making ASO-mediated targeting of MYC mRNA a promising strategy for development of MYC-directed therapeutics. The MYCASOs we describe here are LNA/DNA gapmer molecules that employ third-generation chemistry utilized for its ability to impart enhanced stability and target affinity (55–59). MYCASOs induce decreased MYC protein levels and MYC mRNA degradation, demonstrating the potential for MYC-directed ASOs to function as tools to assess anti-MYC effects in vitro and in vivo, and as potential therapeutics for MYC-addicted cancers.
We designed MYCASOs with target seed sites distributed along the mature MYC mRNA coding region, effectively ‘scanning’ the target mRNA. Delivery of each MYCASO into HeLa cells by transfection resulted in almost uniform knockdown of MYC. However, unformulated gymnotic delivery of MYCASOs in vitro resulted in variable MYC protein knockdown in all tested cell lines suggesting that cellular uptake is a significant factor in MYCASO activity. ASO uptake can be highly variable across cell types and has been shown to be sequence-dependent (40). Our observations with MYCASOs highlight the need to further understand mechanisms of ASO uptake, including identification of cell surface receptors that mediate internalization of ASO molecules, and both productive and non-productive mechanisms of ASO trafficking.
We also report here comprehensive transcriptomics by mRNA sequencing to decipher global MYCASO-specific and non-specific effects. Inclusion of a non-targeting control ASO allowed us to separate MYCASO-specific activity from a generalized ASO response, revealing that MYCASO treatment decreases MYC levels and MYC-associated gene expression signatures specifically, along with leading to generalized ASO responses such as activation of interferon pathways. Notably, MYC serves as a regulator of immune-related genes, promoting an immunosuppressive state in general in cells and tumors in which it is overexpressed, and suppressing interferon response in particular (60–62). Specific inhibition of MYC is expected to abrogate these immunosuppressive effects and lead to activation of immunostimulatory pathways, including interferon pathways. Thus, a generalized ASO effect may enhance the targeted anti-MYC effects of MYCASO treatment, although further studies will be required to fully understand the consquences of ASO-mediated inteferon responses as it relates to anti-tumor effects.
Despite demonstrating an anti-MYC effect in an established model of hepatocellular carcinoma, we recognize that efficacy in a murine model of this indication is likely bolstered by the known hepatotropism of this agent class. Yet the unique pharmacologic characteristics of MYCASOs may allow for use in cancers that arise in tissues that may be amenable to therapeutic oligonucleotides, including central nervous system tumors where intrathecal delivery is possible, or renal tumors. Further studies will be required to assess MYCASO profiles on hepatotoxicity and more general aspects of biodistribution to potentially nominate other oncologic indications for which they may prove useful agents. Further therapeutic development will also require consideration of immune-stimulatory effects of MYCASO treatment, and how this activity may cooperate with anti-MYC effects in tumors (possibly even favorably).
Recent studies have highlighted the capabilities of ASOs manufactured with next generation stabilizing chemistries of inducing anti-tumor effects in preclinical in vivo experiments and in patients (59). Moreover, while our studies here highlight the capabilities of MYCASOs without formulation agents, we predict that efficient MYCASO absorption, distribution, and metabolism kinetics could be greatly enhanced with either specialized nanoparticle or other optimized delivery modality. Notably, MYC deregulation is often a characteristic of hematologic malignancies, often with disease localized to the bone marrow. Recent evidence in mouse models of multiple myeloma suggest that bone marrow-targeting of ASOs may be feasible with possible therapeutic benefit, which may be enhanced by additional tissue-targeting methods (66). Enhanced anti-MYC efficacy may also be achieved by controlling the stereochemical conformation of phosphorothioate linkers in MYCASOs (67).
Oligonucleotide therapeutics including ASOs have been studied for several decades. Recent regulatory approvals of ASO molecules for use by patients suffering from rare diseases such as homozygous familial hypercholesterolemia, spinal muscular atrophy, and Duchenne muscular dystrophy provide invigoration for continued dedicated efforts to synthesize and study novel compounds of this class (68,69,70). MYCASOs represent an important addition to the anti-cancer toolbox of ASOs, with demonstrated anti-MYC effects in a variety of cell types. Further study and optimization of MYCASOs in cancer models will reveal their potential as therapeutics, and the clinical context for which they may provide the most benefit.
Materials and Methods
MYCASO synthesis
Synthesis of MYCASO oligonucleotides was performed as previously described (61). Briefly, the phosphoramidite approach was carried out on an OligoPilot automated synthesizer (GE Healthcare). Oligos were then cleaved from the solid support, purified by filtration followed by ion-exchange high-performance liquid chromatography. Material was then desalted by ultrafiltration, lyophilized and powder was stored at −20°C. Sequences and characteristics of all MYCASOs are listed in Supplemental Table 1.
Cell Culture
HeLa and HEK293T cells (ATCC) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM, + 4.5 g/L D-glucose, + L-Glutamine, Sigma) supplemented with 10% FBS. HLF cells (Broad Institute CCLE) were grown in DMEM with 5% FBS. CUTLL1 (generously provided by J. Aster, Brigham and Women’s Hospital) and MM1.S (ATCC) cells were grown in RPMI 1640 (+ L-Glutamine, Sigma) supplemented with 10% FBS. Cells were maintained at 37°C and 5% CO2. HeLa, HLF, and HEK293T cells were harvested by discarding used media, washing with PBS, adding 0.25% trypsin (Sigma) to just cover the vessel surface, placing cells at 37°C to detach, quenching trypsin with fresh media, and collecting the cell suspension. Semi-adherent MM1.S cells were harvested by both collecting media containing cells in suspension and detaching adherent cells using trypsin. Suspension CUTLL1 cells were harvested by collecting media. For passaging, cells were spun at 200×g for 3 minutes, resuspended in fresh medium, and seeded in new flasks. Cells were split 1:4 or 1:5 every 2–3 days.
MYCASO Treatments
Lyophilized MYCASO powders were suspended in 0.5% saline to a concentration of 5 mM as confirmed spectroscopically. The day before treatment, HeLa and HLF cells were seeded at a density of 100,000 cells/mL and CUTLL1 and MM1.S cells at 250,000 cells/mL; cells were then allowed to adhere and/or recover overnight. For gymnotic treatments, the saline solution was added directly to cell culture media. For transfection, MYCASOs were mixed with Lipofectamine 2000 in serum-free Opti-MEM (Sigma) and added to cell culture medium at a final concentration of 2.5 μL Lipofectamine 2000 / 1 mL medium.
Immunoblotting
Cells were plated in 6-well dishes, allowed to recover overnight, treated as described above, and harvested after a treatment time of 1 hour to 5 days. Harvested cells were placed on ice, centrifuged at 500×g at 4°C for 5 minutes, washed with cold PBS, centrifuged again, resuspended in cold RIPA buffer (Sigma) containing Halt Protease Cocktail Inhibitor (Sigma), and placed on ice. After 45 minutes, cells were centrifuged at 13,000×g at 4°C for 15 min. Protein concentration in the supernatant was determined via BCA assay. Western blotting was performed using the ThermoFisher Bolt system. Forty μg protein in Bolt Sample Buffer with 5% beta-mercaptoethanol was loaded per lane into Bolt 4–12% Bis-Tris gels. Gels were submerged in Bolt MES running buffer and run at 165 V for 35 min. Gels were transferred onto nitrocellulose membranes at 10 V for 60 minutes in Bolt Transfer Buffer. Membranes were blocked in Odyssey Blocking Buffer for 30 minutes at room temperature. Primary antibody incubation was done overnight in Odyssey Blocking Buffer at 4°C. Secondary antibody incubation was done for 1 hour at room temperature. Membranes were washed 3 times in TBS-T between antibodies. Primary antibodies: c-MYC (Santa Cruz N-262, 1:500); α-tubulin (Sigma T5168, 1:4,000). Secondary antibodies: LI-COR IRDye 800CW goat anti-rabbit (1:7,000); LI-COR IRDye 680RD goat anti-mouse (1:7,000). Membranes were imaged on a LI-COR Odyssey CLx using Image Studio software.
RT-PCR
Cells were seeded in 6-well dishes, allowed to recover overnight, treated as described above, and harvested after a treatment time of 1 hour to 5 days. Harvested cells were placed on ice, centrifuged at 500×g at 4°C for 5 minutes, washed with cold PBS, and centrifuged again. RNA was isolated using a QIAGEN RNeasy Mini Kit and eluted in RNAse-free water. cDNA synthesis was carried out using the SuperScript VILO cDNA synthesis kit (ThermoFisher): 1 μg RNA, 4 μL of 5X VILO™ Reaction Mix, and 2 μL of 10X SuperScript Enzyme Mix were diluted in DEPC-treated water to a final volume of 20 uL in 8-strip PCR tubes. Samples were placed in a thermal cycler that ran the following program: 25°C for 10 min., 42°C for 60 min., 85°C for 5 min. Each RT-PCR reaction consisted of 0.1 μL cDNA, 10 μL SYBR Select Master Mix, 1 μL of each primer at 10 μM, and nuclease-free water to a total volume of 20 μL in a MicroAmp Optical 384-well plate (Applied Biosystems). The following PCR protocol was run on an Applied Biosystems ViiA 7 RT-PCR instrument: 50°C for 2 minutes, 95°C for 2 minutes, 40 cycles of 95°C for 15 seconds followed by 60°C for 1 minute with a SYBR Green fluorescence measurement made after every cycle, followed by a melting curve measurement. Ct thresholds were determined automatically by the instrument software. Ct values were converted to transcript abundance relative to vehicle-treated controls.
5’ Rapid Amplification of cDNA Ends (RACE)
Cells were seeded in 6-well dishes, allowed to recover overnight, treated as described above, and harvested after a treatment time of 1 hour to 5 days. Harvested cells were placed on ice, centrifuged at 500×g at 4°C for 5 min., washed with cold PBS, and centrifuged again. RNA was isolated using a QIAGEN RNeasy Mini Kit and eluted in RNAse-free water. The 5’ RACE assay was carried out as described by Invitrogen using their 5’ RACE kit. All primers used are listed in Supplemental Table 2. A thermal cycler was used for all incubations.
First Strand cDNA Synthesis:
In 8-strip PCR tubes, 5 μg total RNA and 2.5 pmol gene specific primer 1 (GSP1) were diluted to 15.5 μL in DEPC-treated water, incubated for 10 minutes at 70°C, then chilled for 1 minutes on ice. Next, 2.5 μL 10X PCR buffer, 2.5 μL 25 mM MgCl2, 1 uL 10 nM dNTP mix, and 2.5 μL 0.1 M DTT were added to the sample, followed by a 1 minute incubation at 42°C. 1 μL SuperScript II RT was added to the reaction, followed by incubation at 42°C for 50 minutes, then 70°C for 15 min. 1 μL RNase mix was then added, followed by incubation at 37°C for 30 minutes. The cDNA reaction was purified using a Qiagen PCR purification kit and eluted in nuclease-free water.
TdT Tailing of cDNA:
The following components were added to an 8-strip PCR tube: 6.5 μL DEPC-treated water, 5 μL 5X tailing buffer, 2.5 μL 2 mM dCTP, and 10 μL cDNA sample. Reactions were incubated for 2.5 minutes at 94°C, then chilled for 1 minute on ice. 1 μL TdT was added, followed by incubation for 10 minutes at 37°C, then 10 minutes at 65°C.
PCR of dC-tailed cDNA:
The following components were added to an 8-strip PCR tube: 32 μL nuclease-free water, 5 μL 10X PCR buffer, 3 μL 25 mM MgCl2, 1 μL 10 mM dNTP mix, 2 μL nested gene specific primer 2 (GSP2, 10 μM), 2 μL Abridged Anchor Primer (AAP, 10 μM), 5 μL dC-tailed cDNA. 0.25 μL Taq DNA polymerase (5 units/μL) was added immediately before mixing the reaction and placing in a thermal cycler pre-equilibrated to 95°C to run the following PCR protocol: 95°C for 2 minutes; 30 cycles of 95°C for 15 seconds, 58°C for 15 seconds, and 68°C for 1 minute; and final extension at 68°C for 5 minutes.
Nested Amplification:
For improved visualization of PCR products, a second PCR reaction was carried out using another nested gene specific primer (GSP3) and the 5’ RACE abridged universal anchor primer (AUAP). The PCR products from the previous step were diluted 1:100 in TE buffer. The following components were added to an 8-strip PCR tube: 34 μL nuclease-free water, 5 μL 10X PCR buffer, 3 μL 25 mM MgCl2, 1 μL 10 mM dNTP mix, 1 uL GSP3 (10 mM), 1 μL AUAP, 5 μL diluted primary PCR product. 0.25 μL Taq DNA polymerase (5 units/μL) was added immediately before mixing the reaction and placing in a thermal cycler pre-equilibrated to 95°C to run the following PCR protocol: 95°C for 2 minutes; 30 cycles of 95°C for 15 seconds, 57°C for 15 seconds, and 68°C for 1 minutes; and final extension at 68°C for 5 minutes.
Gel Visualization of PCR Products:
10 μL PCR product was loaded onto an E-Gel 1.2% with Ethidium Bromide Agarose Gel (Invitrogen). Gels were run using an E-Gel iBase Power System (Invitrogen) set to program 1 for 20 min. 5 μL of E-Gel 1 kb Plus DNA Ladder (Invitrogen) was loaded into one lane per gel.
RNASEH1 Knockdown
HeLa cells were seeded in 6-well dishes at 250,000 cells/mL and allowed to recover overnight. The following day, cells were co-transfected with an anti-RNASEH1 ASO (36) (Supplemental Table 2) (TriLink) and MYCASO using Lipofectamine 2000 as described above. Cells were harvested after 24 hours for RT-PCR.
Viability and Apoptosis Assays
For viability assays, ATPlite substrate was reconstituted in buffer as described by PerkinElmer and diluted 1:1 in PBS. Cells were plated in white-bottom 96-well plates (PerkinElmer), allowed to recover overnight, and treated as described above for 1 hour to 5 days. Following treatment, 100 μL diluted ATPlite solution was added to each well, and luminescence was read using a PerkinElmer EnVision microplate reader. Apoptosis analysis was performed by staining with a Cy5-conjugated anti-Annexin V antibody (BioVision) and read out by flow cytometry on a Beckman CytoFLEX per the manufacturers instructions.
Growth-over-time Assays:
Growth-over-time assays were carried out using Guava ViaCount Reagent and a Guava easyCyte HT flow cytometer (Millipore). Briefly, cells were plated in 48-well plates, allowed to recover overnight, and treated as described above. At each time point following treatment, cells were harvested and mixed 1:1 with Guava ViaCount Reagent. 100 μL sample was plated in a clear 96-well plate, and the plate was loaded onto the flow cytometer. Each time point was performed in triplicate. The ViaCount program in the Guava software was used to count cells. Gating for live and dead cells was performed manually for each cell line.
Generation of mutant MYC-overexpressing cell line
Plasmid Design:
A mutant MYC cDNA construct was designed containing 17 silent mutations in the MYCASO-3, MYCASO-9, and MYCASO-13 seed sites and a T58A mutation, shown to impair MYC degradation by the proteasome. The construct was synthesized by GENEWIZ, LLC (South Plainfield, NJ, USA). The cDNA was cloned into the pLenti6.3/V5-DEST plasmid using Gateway Cloning. First, mutant MYC cDNA was amplified using custom primers containing Gateway attB sequences on either end to generate an attB-PCR product. The reverse primer includes a stop codon to prevent translation of C-terminal tags encoded in destination vectors. Next, a Gateway BP recombination reaction was carried out to generate an entry clone: 100 fmol mutant MYC attB-PCR product, 300 ng pDONR221 plasmid, and 4 μL 5X BP Clonase Reaction Buffer were diluted to 16 μL in TE buffer. 4 μL BP Clonase enzyme mix were added, and the reaction was incubated at room temperature for 1 hr. After completion of the reaction, 1 μL reaction mix was mixed into 50 μL DH5α competent cells (Invitrogen) on ice. The cell mixture was placed on ice for 30 minutes, heat-shocked at 42°C for 45 sec., and placed back on ice for 2 minutes. 500 μL of SOC media was added to the cells and they were placed in a shaker at 37°C for 45 minutes. The cell mixture was then spread on an agar plate containing 50 μg/mL Kanamycin and placed at 37°C overnight. The next day, individual bacterial colonies were picked from the agar plate and sequenced for inclusion of the mutant MYC sequence in the pDONR221 plasmid. A positive clone was grown up in 2 mL LB media containing 50 μg/mL Kanamycin shaking at 37°C overnight, then scaled up to 50 mL the next day and placed at 37°C shaking overnight again. The next day, the plasmid was isolated from bacterial cells using a Qiagen Plasmid Midi Kit. Next, a Gateway LR recombination reaction was carried out to generate the final expression clone: 300 ng mutant MYC entry clone, 300 ng pLenti6.3/V5-DEST plasmid, and 4 μL 5X LR Clonase Reaction Buffer were diluted to 16 μL in TE buffer. 4 μL LR Clonase enzyme mix were added, and the reaction was incubated at room temperature overnight. The next day, DH5α competent cells were transformed with the reaction mixture as described above and the bacterial cell mixture was spread on an agar plate containing 100 μg/mL Ampicillin and placed at 37°C overnight. Individual colonies were picked and sequenced for inclusion of the mutant MYC construct in the pLenti6.3/V5-DEST plasmid. A positive clone was grown up in LB media containing 100 μg/mL Ampicillin as described above, and the plasmid was isolated from bacterial cells using a Qiagen Plasmid Midi Kit. This plasmid will be referred to as pLenti6.3.mycASOmut.
Lentiviral Production:
HEK293T cells were plated in a 10 cm dish and co-transfected with 600 ng pMD2.G (envelope plasmid), 1 μg psPAX2 (packaging plasmid), and 5 μg pLenti6.3.mycASOmut using Xfect Transfection Reagent (Clontech). After 48 hours, virus-containing media was collected, filtered through a 0.45-μM filter (Millipore), and concentrated using Lenti-X Concentrator (Clontech).
Lentiviral Transduction:
HeLa cells were plated in a 24-well dish at 60,000 cells/well. The next day, 10 μL of concentrated virus was added per well. Polybrene (Millipore) was added to half of the wells to a final concentration of 8 μg/mL. The plate was spun at 250×g for 1 hour at room temperature, then placed in an incubator at 37°C and 5% CO2. After 24 hours cells were harvested, pooled, and plated in fresh media. After 24 hours, Blasticidin was added to the media at a final concentration of 1 μg/mL. Cells were maintained in DMEM with 10% FBS + 1 μg/mL Blasticidin, refreshed every 2–3 days.
mRNA sequencing
HeLa cells were seeded in 6-well dishes, allowed to recover overnight, treated as described above, and harvested after treatment times of 0, 4, and 18 hours. Harvested cells were placed on ice, centrifuged at 500×g at 4°C for 5 min., washed with cold PBS, and centrifuged again. For each sample, 10 μL cells were mixed with 10 μL trypan blue dye, loaded onto a Countess cell counting chamber slide, and counted using a Countess II Automated Cell Counter. Each sample was adjusted to contain 400,000 cells. RNA was isolated using a QIAGEN RNeasy Mini Kit and eluted in RNAse-free water, and ERCC synthetic spike-ins were added per the manufacturer’s instructions (Ambion). RNA samples were quantified using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and RNA integrity was checked with Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA). RNA library preparations and sequencing reactions were conducted at GENEWIZ, LLC (South Plainfield, NJ, USA). RNA sequencing library preparation used the NEBNext Ultra RNA Library Prep Kit for Illumina by following manufacturer’s recommendations (NEB, Ipswich, MA, USA). Briefly, mRNA was first enriched with Oligod(T) beads. Enriched mRNAs were fragmented for 15 minutes at 94 °C. First strand and second strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3’ends, and universal adapter was ligated to cDNA fragments, followed by index addition and library enrichment with limited cycle PCR. Sequencing libraries were validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA), and quantified by using Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) as well as by quantitative PCR (Applied Biosystems, Carlsbad, CA, USA).
The sequencing libraries were multiplexed and clustered on two lanes of a flowcell. After clustering, the flowcell was loaded on the Illumina HiSeq instrument according to manufacturer’s instructions. The samples were sequenced using a 2×150 Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
For differential expression analysis, transcripts were quantified from raw paired-end FASTQs using Salmon 0.8.2 with automatic library type detection and default parameters. The index used was built from RefMrna sequences obtained from hg19 (Feb. 2009, GRCh37 (GCA_000001405.1)). Transcript-level counts were imported and merged to gene-level counts via the Tximport v1.4.0 Bioconductor package using a transcript-to-gene key generated from the UCSC RefGene database. DESeq2 v1.14 was used with single-factor design formula (“~ condition”) to find differentially expressed genes. Raw and processed data is available the in the Gene Expression Omnibus, GEO accession GSE183535.
To generate MYCASO-3 and ASO signatures, transcripts were quantified and merged to the gene-level as described above. Prior to analysis with DESeq2 v1.14, zero-hour samples were excluded and a three-factor design formula was used to test for expression changes attributable to the presence of a MYCASO-3 while controlling for treatment time as well as the presence of any ASO (MYCASO-3 or NT-ASO). We also used this approach to test for expression changes attributable to ASOs in general while controlling for time and the presence of MYC-targeted ASO. For each of these comparisons, all genes and associated Wald statistics were exported for use with the GSEA Preranked function.
In vivo tolerability study
Thirty-six 7-week old female C57BL/6 mice (Jackson Labs) were divided into nine treatment groups (n = 4 mice per group): Vehicle, NT-ASO (low), NT-ASO (high), MYCASO-3 (low), MYCASO-3 (high), MYCASO-9 (low), MYCASO-9 (high), MYCASO-13 (low), MYCASO-13 (high). Low dose treatment included an initial dose of 25 mg per kilogram, followed by maintenance dosing of 5 mg per kg every three days. High dose treatments were performed with 25 mg per kilogram doses every three days. ASOs were resuspended in 0.9% sterile saline solution prior to dosing. Body weight measurements were recorded for each mouse immediately prior to each dose. All treatments were administered via intravenous injection in the lateral tail vein over a period of 21 days (7 total injections per mouse). MYCASO tolerability study was performed at the Lurie Family Imaging Center, Dana-Farber Cancer Institute under an approved protocol by the Animal Care and Use Committee.
In vivo HCC study
Tumor formation, treatments:
MYC-induced HCC tumor models were performed as previously described (54,71). Briefly, 20 μg of plasmid encoding MYC and transposon were mixed with 0.8 μg plasmid encoding the Sleeping Beauty transposase in 2 mL PBS and injected into the lateral tail vein of 6- to 8- week old female FVB/N mice (Jackson Labs). Mice were randomly selected to receive five doses of MYCASO-3 (or NT-ASO control) five weeks following plasmid perfusion; no investigator blinding was performed. Mice were treated with a total of five doses, 25 mg/kg every three days. Following the final dose, three mice from each group were euthanized and liver samples were harvested for subsequent analysis. Remaining mice were observed and euthanized when predetermined survival endpoints were reached. This model and study was approved by the Committee for Animal Research at the University of California, San Francisco.
Histology, Immunohistochemistry, and Immunofluorescence:
Liver samples were fixed 24 hours with Zinc Formal-Fix (Thermo Shandon Ltd, Runcorn) at 4 °C before paraffin-embedding. Sections were done at 5 μm in thickness. For immunohistochemistry, sodium citrate buffer (10 mmol/L, pH 6.0) was utilized for antigen retrieving. Slides in sodium citrate buffer were heated in a microwave on high level for 10 minutes, followed by cooling down for 20 minutes. Subsequently, goat serum (5% diluted in PBS) and Avidin-Biotin blocking kit (Vector Laboratories, Burlingame, CA) were applied for blocking. Specimens were then incubated with designated primary antibody at 4 °C overnight. The primary antibodies against Ki-67 (Thermo Fisher Scientific; 1:150) and MYC (ab32072, 1:200) were used. The following day, 3% hydrogen peroxide was used to quench endogenous peroxidase activity for 10 minutes, followed by biotin-conjugated secondary antibody at a 1:500 dilution for 30 minutes at room temperature. The immunoreactivity was visualized with the Vectastain Elite ABC kit (Vector Laboratories) and 3,30-diaminobenzidine as the chromogen. Slides were counterstained with hematoxylin. Images were acquired using a Leica Aperio CS-O slide scanning microscope at 40x magnification. Image quantification was performed using the Multiplex IHC v2.0.1 module in Halo digital pathology software Indica Labs). Representative regions were chosen for analysis that avoided debris, folds and any other artifacts. Color deconvolution was performed to separate the color components of hematoxylin and DAB (MYC). The optical density (O.D.) of the DAB was determined for each cell object. The total number of cells in a region of interest ranged between 8,000–53,000 cells. Statistical comparison between the NT-ASO images and the MYCASO-3 images was performed with GraphPad Prism software. Quantification of MYC+ area was performed as described (71).
Protein extraction and Western blot analysis:
Protein extraction was conducted according to manufacturer’s protocol by using the M-PER mammalian protein extraction reagent (Cat. N. 78501; Thermo Fisher Scientific, Rockford, IL) and the Complete Protease Inhibitor Cocktail. Aliquots of 30 μg protein lysate were denatured by boiling in Tris-Glycine SDS Sample Buffer (Invitrogen, Grand Island, NY). Proteins were separated by SDS-PAGE gel and transfected onto nitrocellulose membranes by using RTA transfer kit (Cat. N. 1704271, Biorad, Hercules, CA). Membranes were blocked in 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h and incubated with specific primary antibodies. The primary antibodies against MYC (Abcam, ab32072, 1:5000) and Gapdh (Cell Signalling Technology, 2118, 1:5000) were used. Subsequently, a horseradish peroxidase-secondary antibody diluted 1:10,000 for 1 h was applied and was revealed with the Super Signal West Pico Chemiluminescent Substrate (Pierce Chemical Co., New York, NY). Image-J software (http://imagej.nih.gov/ij/) was used for densitometric analysis of the blots.
Statistical analysis:
GraphPad Prism version 6.0 (GraphPad Software Inc.) was used to evaluate statistical significance by the Tukey–Kramer test. Comparisons between two groups were performed with two-tailed unpaired t test. P values < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
We thank M. Gabay and D. Felsher (Stanford University), S. He, T. Look, E. Dhimolea, C. Mitsiades, and members of the Lurie Family Imaging Center (Dana-Farber Cancer Institute) for assistance with animal studies. This work was supported by a US National Cancer Institute (NCI) Pathway to Independence Award (K99/R00CA190861) (C.J.O), and institutional funding from DFCI, MGH, and the Broad Institute (J.E.B, C.J.O, T.G.), and the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute (D.G.A.).
Competing Interests
C.JO., R.B, Y.Z, and J.E.B. are listed as inventors in an issued patent related to the compounds described in this manuscript (WO2013123451A1). D.T.T. has received consulting fees from ROME Therapeutics, Foundation Medicine, Inc., NanoString Technologies, EMD Millipore Sigma, Pfizer, and Third Rock Ventures that are not related to this work. D.T.T. is a founder and has equity in ROME Therapeutics, PanTher Therapeutics and TellBio, Inc., which is not related to this work. D.T.T. receives research support from ACD-Biotechne, PureTech Health LLC, and Ribon Therapeutics, which was not used in this work. Dr. Ting’s interests were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict of interest policies. J.E.B. is now an executive and shareholder of Novartis and has been a founder and shareholder of SHAPE (acquired by Medivir), Acetylon (acquired by Celgene), Tensha (acquired by Roche), Syros, Regenacy and C4 Therapeutics.
References
- 1.Kalkat M, Melo JD, Hickman KA, Lourenco C, Redel C, Resetca D, et al. MYC Deregulation in Primary Human Cancers. Genes-basel. 2017;8(6):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yashiro-Ohtani Y, Wang H, Zang C, Arnett KL, Bailis W, Ho Y, et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc National Acad Sci. 2014;111(46):E4946–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cory S. Activation Of Cellular Oncogenes in Hemopoietic Cells by Chromosome Translocation. Adv Cancer Res. 1986;47:189–234. [DOI] [PubMed] [Google Scholar]
- 5.Croce CM, Nowell PC. Molecular Genetics of Human B Cell Neoplasia. Adv Immunol. 1986;38:245–74. [DOI] [PubMed] [Google Scholar]
- 6.Cheng S-WG, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet. 1999;22(1):102–5. [DOI] [PubMed] [Google Scholar]
- 7.McMahon SB, Wood MA, Cole MD. The Essential Cofactor TRRAP Recruits the Histone Acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20(2):556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang CV. MYC, Metabolism, Cell Growth, and Tumorigenesis. Csh Perspect Med. 2013;3(8):a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell. 2012;151(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sabò A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature. 2014;511(7510):488–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature. 2014;511(7510):483–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felsher DW, Bishop JM. Reversible Tumorigenesis by MYC in Hematopoietic Lineages. Mol Cell. 1999;4(2):199–207. [DOI] [PubMed] [Google Scholar]
- 13.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455(7213):679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koehler AN. A complex task? Direct modulation of transcription factors with small molecules. Curr Opin Chem Biol. 2010;14(3):331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc National Acad Sci. 2002;99(6):3830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, et al. In Vitro Cytotoxicity and In Vivo Efficacy, Pharmacokinetics, and Metabolism of 10074-G5, a Novel Small-Molecule Inhibitor of c-Myc/Max Dimerization. J Pharmacol Exp Ther. 2010;335(3):715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiessling A, Wiesinger R, Sperl B, Berg T. Selective Inhibition of c-Myc/Max Dimerization by a Pyrazolo[1,5-a]pyrimidine. Chemmedchem. 2007;2(5):627–30. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, et al. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther. 2007;6(9):2399–408. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Chauhan J, Hu A, Pendleton K, Yap JL, Sabato PE, et al. Disruption of Myc-Max Heterodimerization with Improved Cell-Penetrating Analogs of the Small Molecule 10074-G5. Oncotarget. 2013;4(6):936–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK, Janda KD. A credit-card library approach for disrupting protein–protein interactions. Bioorgan Med Chem. 2006;14(8):2660–73. [DOI] [PubMed] [Google Scholar]
- 21.Beaulieu M-E, Jauset T, Massó-Vallés D, Martínez-Martín S, Rahl P, Maltais L, et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci Transl Med. 2019. March 20;11(484):eaar5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Struntz NB, Chen A, Deutzmann A, Wilson RM, Stefan E, Evans HL, et al. Stabilization of the Max Homodimer with a Small Molecule Attenuates Myc-Driven Transcription. Cell Chem Biol. 2019. March 26;26(5):1 37. [DOI] [PubMed] [Google Scholar]
- 24.Heikkila R, Schwab G, Wickstrom E, Loke SL, Pluznik DH, Watt R, et al. A c-myc antisense oligodeoxynucleotide inhibits entry into S phase but not progress from G0 to G1. Nature. 1987;328(6129):445–9. [DOI] [PubMed] [Google Scholar]
- 25.Holt JT, Redner RL, Nienhuis AW. An oligomer complementary to c-myc mRNA inhibits proliferation of HL-60 promyelocytic cells and induces differentiation. Mol Cell Biol. 1988;8(2):963–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wickstrom EL, Bacon TA, Gonzalez A, Freeman DL, Lyman GH, Wickstrom E. Human promyelocytic leukemia HL-60 cell proliferation and c-myc protein expression are inhibited by an antisense pentadecadeoxynucleotide targeted against c-myc mRNA. Proc National Acad Sci. 1988;85(4):1028–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leonetti C, Agnano ID, Lozupone F, Valentini A, Geiser T, Zon G, et al. Antitumor Effect of c-myc Antisense Phosphorothioate Oligodeoxynucleotides on Human Melanoma Cells In Vitro and in Mice. Jnci J National Cancer Inst. 1996;88(7):419–29. [DOI] [PubMed] [Google Scholar]
- 28.Mui B, Raney SG, Semple SC, Hope MJ. Immune stimulation by a CpG-containing oligodeoxynucleotide is enhanced when encapsulated and delivered in lipid particles. Journal of Pharmacology and Experimental Therapeutics [Internet]. 2001;3(298):1185–92. Available from: https://jpet.aspetjournals.org/content/298/3/1185.short [PubMed] [Google Scholar]
- 29.Chan JH, Lim S, Wong WF. ANTISENSE OLIGONUCLEOTIDES: FROM DESIGN TO THERAPEUTIC APPLICATION. Clin Exp Pharmacol P. 2006;33(5–6):533–40. [DOI] [PubMed] [Google Scholar]
- 30.Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol. 2017;35(3):238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koshkin AA, Singh SK, Nielsen P, Rajwanshi VK, Kumar R, Meldgaard M, et al. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. 1998;54(14):3607–30. [Google Scholar]
- 32.Singh SK, Koshkin AA, Wengel J, Nielsen P. LNA (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chem Commun. 1998;0(4):455–6. [Google Scholar]
- 33.Bennett CF, Swayze EE. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu Rev Pharmacol. 2010;50(1):259–93. [DOI] [PubMed] [Google Scholar]
- 34.Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J Biol Chem. 2004;279(17):17181–9. [DOI] [PubMed] [Google Scholar]
- 35.Koch T, Orum H. Locked Nucleic Acid. In: Crooke ST, editor. Antisense Drug Technology [Internet]. Second. CRC Press; 2007. p. 519–564. Available from: https://www.routledge.com/Antisense-Drug-Technology-Principles-Strategies-and-Applications-Second/Crooke/p/book/9780849387968 [Google Scholar]
- 36.Burgess TL, Fisher EF, Ross SL, Bready JV, Qian YX, Bayewitch LA, et al. The antiproliferative activity of c-myb and c-myc antisense oligonucleotides in smooth muscle cells is caused by a nonantisense mechanism. Proc National Acad Sci. 1995;92(9):4051–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coulis CM, Lee C, Nardone V, Prokipcak RD. Inhibition of c-myc Expression in Cells by Targeting an RNA-Protein Interaction Using Antisense Oligonucleotides. Mol Pharmacol. 2000;57(3):485–94. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Castaneda S, Dumble M, Wang M, Mileski M, Qu Z, et al. Reduced Expression of the Androgen Receptor by Third Generation of Antisense Shows Antitumor Activity in Models of Prostate Cancer. Mol Cancer Ther. 2011;10(12):2309–19. [DOI] [PubMed] [Google Scholar]
- 39.Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500(7461):207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crooke ST, Wang S, Vickers TA, Shen W, Liang X. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. 2017;35(3):230–7. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Qu Z, Kim S, Shi V, Liao B, Kraft P, et al. Down-modulation of cancer targets using locked nucleic acid (LNA)-based antisense oligonucleotides without transfection. Gene Ther. 2011;18(4):326–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc National Acad Sci. 2000;97(1):228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palomero T, Barnes KC, Real PJ, Bender JLG, Sulis ML, Murty VV, et al. CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to γ-secretase inhibitors. Leukemia. 2006;20(7):1279–87. [DOI] [PubMed] [Google Scholar]
- 44.Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data. 2014;1(1):140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell. 2017;170(3):564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castanotto D, Lin M, Kowolik C, Wang L, Ren X-Q, Soifer HS, et al. A cytoplasmic pathway for gapmer antisense oligonucleotide-mediated gene silencing in mammalian cells. Nucleic Acids Res. 2015;43(19):9350–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell. 2011;146(6):904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Claassen GF, Hann SR. A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor β-induced cell-cycle arrest. Proc National Acad Sci. 2000;97(17):9498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. P Natl Acad Sci Usa. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc National Acad Sci. 2000;97(7):3260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schuhmacher M, Kohlhuber F, Hölzel M, Kaiser C, Burtscher H, Jarsch M, et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 2001;29(2):397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Browne EP, Wing B, Coleman D, Shenk T. Altered Cellular mRNA Levels in Human Cytomegalovirus-Infected Fibroblasts: Viral Block to the Accumulation of Antiviral mRNAs. J Virol. 2001;75(24):12319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang W, Yang H, Kong X, Mohapatra S, Juan-Vergara HS, Hellermann G, et al. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat Med. 2005;11(1):56–62. [DOI] [PubMed] [Google Scholar]
- 54.Chow EK, Fan L, Chen X, Bishop JM. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology. 2012;56(4):1331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chery J, Petri A, Wagschal A, Lim S-Y, Cunningham J, Vasudevan S, et al. Development of Locked Nucleic Acid Antisense Oligonucleotides Targeting Ebola Viral Proteins and Host Factor Niemann-Pick C1. Nucleic Acid Ther. 2018;28(5):273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klar R, Michel S, Schell M, Hinterwimmer L, Zippelius A, Jaschinski F. A highly efficient modality to block the degradation of tryptophan for cancer immunotherapy: locked nucleic acid-modified antisense oligonucleotides to inhibit human indoleamine 2,3-dioxygenase 1/tryptophan 2,3-dioxygenase expression. Cancer Immunol Immunother. 2020;69(1):57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le BT, Adams AM, Fletcher S, Wilton SD, Veedu RN. Rational Design of Short Locked Nucleic Acid-Modified 2′-O-Methyl Antisense Oligonucleotides for Efficient Exon-Skipping In Vitro. Mol Ther - Nucleic Acids. 2017;9:155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ross SJ, Revenko AS, Hanson LL, Ellston R, Staniszewska A, Whalley N, et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med. 2017;9(394):eaal5253. [DOI] [PubMed] [Google Scholar]
- 59.Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7(314):314ra185–314ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Casey SC, Baylot V, Felsher DW. MYC: Master Regulator of Immune Privilege. Trends Immunol. 2017;38(4):298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schlee M, Hölzel M, Bernard S, Mailhammer R, Schuhmacher M, Reschke J, et al. c-MYC activation impairs the NF-κB and the interferon response: Implications for the pathogenesis of Burkitt’s lymphoma. Int J Cancer. 2007;120(7):1387–95. [DOI] [PubMed] [Google Scholar]
- 62.Schlee M, Schuhmacher M, Hölzel M, Laux G, Bornkamm GW. c-MYC Impairs Immunogenicity of Human B Cells. Adv Cancer Res. 2007;97:167–88. [DOI] [PubMed] [Google Scholar]
- 63.Harrington EA, Bennett MR, Fanidi A, Evan GI. c-Myc-induced apoptosis in fibroblasts is inhibited by specific cytokines. Embo J. 1994;13(14):3286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene. 1991;6(10):1915–22. [PubMed] [Google Scholar]
- 65.Evan G, Harrington E, Fanidi A, Land H, Amati B, Bennett M. Integrated control of cell proliferation and cell death by the c-myc oncogene. Philosophical Transactions Royal Soc Lond Ser B Biological Sci. 1994;345(1313):269–75. [DOI] [PubMed] [Google Scholar]
- 66.Mondala PK, Vora AA, Zhou T, Lazzari E, Ladel L, Luo X, et al. Selective antisense oligonucleotide inhibition of human IRF4 prevents malignant myeloma regeneration via cell cycle disruption. Cell Stem Cell. 2021;28:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iwamoto N, Butler DCD, Svrzikapa N, Mohapatra S, Zlatev I, Sah DWY, et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat Biotechnol. 2017;35(9):845–51. [DOI] [PubMed] [Google Scholar]
- 68.Parham JS, Goldberg AC. Mipomersen and its use in familial hypercholesterolemia. Expert Opin. Pharmacother 2019;20(2):127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoy SM. Nusinersen: First global approval. Drugs 2017;77(4):473–79. [DOI] [PubMed] [Google Scholar]
- 70.Dowling JJ. Eteplirsen therapy for Duchenne muscular dystrophy: skipping to the front of the line. Nat. Rev. Neuro 2016;12(12):675–76. [DOI] [PubMed] [Google Scholar]
- 71.Shang R, Song X, Wang P, Zhou Y, Lu X, Wang J, et al. Cabozantinib-based combination therapy for the treatment of hepatocellular carcinoma. Gut 2020;0:1–12.doi.1136/gutjnl-2020–320716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.