

Abstract

The pharmacological inhibition of soluble epoxide hydrolase (sEH) is efficient for the treatment of inflammatory and pain-related diseases. Numerous potent sEH inhibitors (sEHIs) present adamantyl or phenyl moieties, such as the clinical candidates AR9281 or EC5026. Herein, in a new series of sEHIs, these hydrophobic moieties have been merged in a benzohomoadamantane scaffold. Most of the new sEHIs have excellent inhibitory activities against sEH. Molecular dynamics simulations suggested that the addition of an aromatic ring into the adamantane scaffold produced conformational rearrangements in the enzyme to stabilize the aromatic ring of the benzohomoadamantane core. A screening cascade permitted us to select a candidate for an in vivo efficacy study in a murine model of cerulein-induced acute pancreatitis. The administration of 22 improved the health status of the animals and reduced pancreatic damage, demonstrating that the benzohomoadamantane unit is a promising scaffold for the design of novel sEHIs.
1. Introduction
In mammals, arachidonic acid, a polyunsaturated fatty acid, is metabolized by cyclooxygenases (COXs), lipoxygenases (LOXs), and cytochrome P450s (CYPs). The COX and LOX pathways lead mainly to the production of pro-inflammatory lipid mediators, such as prostaglandins and leukotrienes, and are pharmaceutically targeted.1 In contrast, the CYP pathway produces both pro- and anti-inflammatory lipid mediators, such as the pro-inflammatory 20-hydroxyeicosatetraenoic acid or the potent anti-inflammatory epoxyeicosatrienoic acids (EETs).2 However, the EETs are rapidly metabolized by the soluble epoxide hydrolase (sEH, EPHX2, EC 3.3.2.3) into the corresponding dihydroxyeicosatrienoic acids, which are less biologically active.3,4 The pharmacological inhibition of sEH in vivo stabilizes the concentration of EETs, reducing inflammatory and pain states, suggesting sEH as a pharmacological target for the treatment of inflammatory diseases.5,6
X-ray crystallographic studies revealed that sEH has an L-shaped active pocket with the catalytic residues situated at the corner. Although each side of the pocket (10 and 15 Å long) accepts a variety of functional groups, the entire pocket is essentially hydrophobic.7 Indeed, a number of very potent sEH inhibitors (sEHIs) feature lipophilic moieties such as adamantyl or phenyl groups (Figure 1), limiting their usefulness.5,6
Figure 1.

Structure of the three sEHIs that have entered human clinical trials.
Nevertheless, several compounds have been tested in human. AR9281, which was developed by Arête Therapeutics for the treatment of hypertension in diabetic patients, failed in a phase II clinical trial largely due to its poor pharmacokinetic properties, likely related with its adamantyl unit.8 GSK2256294, developed for chronic obstructive pulmonary disease by GlaxoSmithKline, has entered clinical trials for obese smokers and other indications such as subarachnoidal hemorrhage or diabetic patients with insulin resistance.9 More recently, taking the AR9281 failure into account, EicOsis has recently replaced the adamantyl moiety of AR9281 by an aromatic ring for its drug candidate EC5026, which has recently completed human phase 1a clinical trials for the treatment of neuropathic pain.10
Using urea-based sEHIs with lipophilic units of very different sizes, we recently found that the pocket of the sEH can accommodate polycycles of a quite diverse volume and that the replacement of the adamantane moiety by larger polycyclic rings, such as the diamantane moiety, may be better than the replacement by smaller ones. Indeed, urea 6, a diamantane analogue of the well-known sEHI 4, t-AUCB, and 5, t-TUCB, showed to be a subnanomolar inhibitor of the human sEH (hsEH) (Figure 2).11 Therefore, herein we are testing new sEH that merge the adamantyl and phenyl groups in a unique polycyclic scaffold.
Figure 2.

Structure and IC50 in hsEH of compounds 4–6.
2. Results and Discussion
2.1. Design and Synthesis of New sEHIs
Taking into account that both adamantane and aromatic ring moieties fit very well in the hydrophobic pocket of the sEH and that the replacement of adamantane by larger polycyclic rings seems to be a promising strategy to obtain more potent sEHIs, a novel series of compounds bearing the very versatile benzohomoadamantane scaffold as the hydrophobic moiety were designed and synthesized. This polycyclic, readily accessible,12−15 system features a homoadamantane unit fused with an aromatic ring and permits several chemical derivatizations in its structure (Figure 3). Potent sEHIs and optimum drug-like properties could be achieved by modifying the substituents in the benzohomoadamantane unit and/or the right-hand side (RHS) of the molecule (Figure 3).
Figure 3.
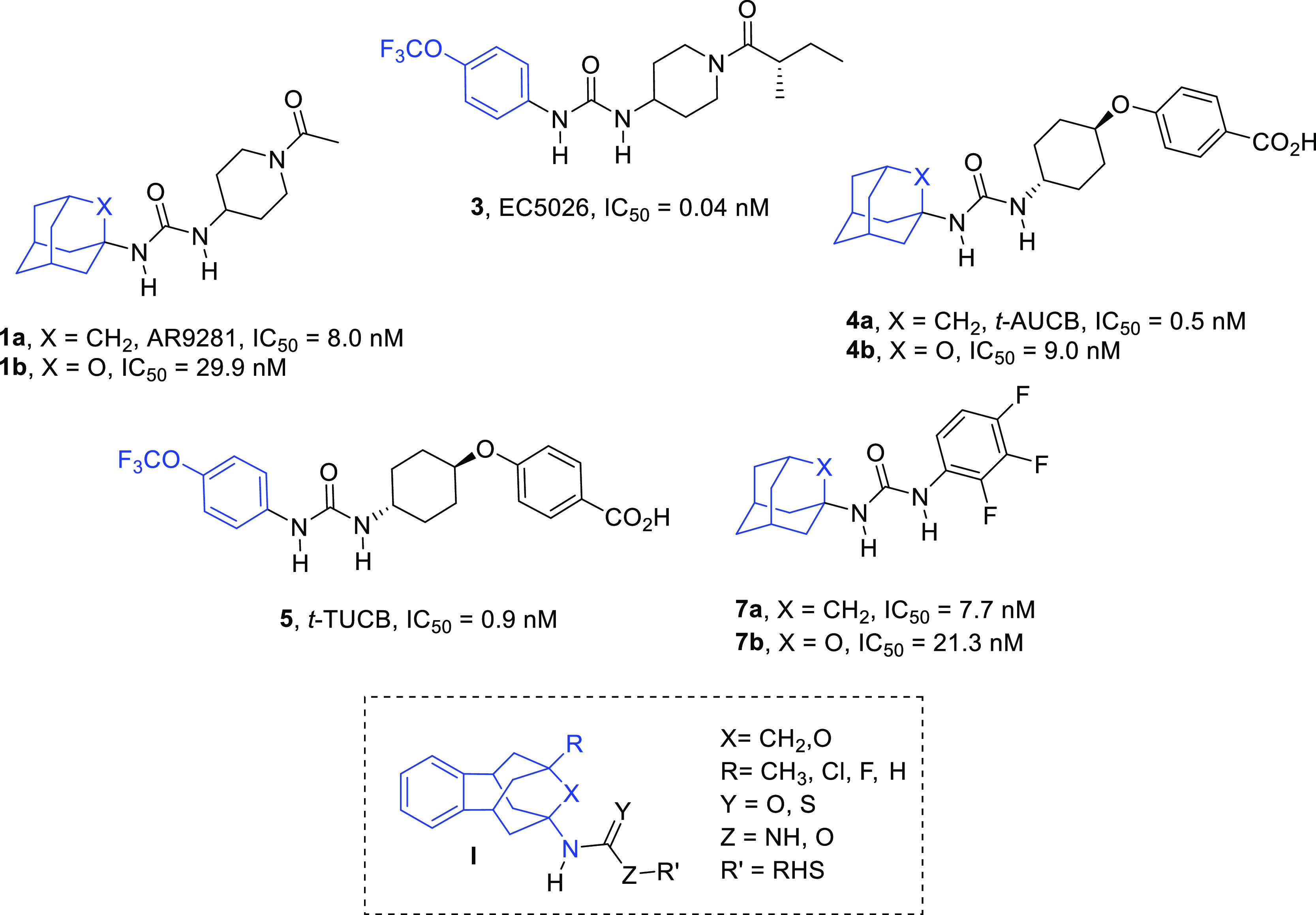
Known adamantyl- and phenyl-based sEHIs 1a, b, 3, 4a, b, 5, and 7a, b, and general structure, I, of the new sEHI reported in this work. (see below). For simplicity, only IC50 values obtained for the hsEH are reported.
Thioureas, carbamates and, particularly ureas are good pharmacophores for an sEHI.6 For this reason, thiourea 9, carbamate 11, and urea 13 were first synthesized in order to explore their relative potency as an sEHI and to select the more suitable pharmacophore for the novel polycyclic scaffold (Scheme 1). The three compounds were easily synthesized in low to moderate yields from known 9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine13 (II, R = CH3, X = CH2) and 4-(trifluoromethyl)phenyl isothiocyanate, 8, p-tolyl chloroformate, 10, and 2,3,4-trifluorophenyl isocyanate, 12, respectively (Scheme 1).
Scheme 1. Synthesis of New Compounds 9, 11, 13, 14, 16–18, and 20–23.

Reagents and conditions: (a) 4-(trifluoromethyl)phenyl isothiocyanate (8), Et3N, DCM, overnight (b) p-tolyl chloroformate (10), Et3N, DCM, overnight; (c) 2,3,4-trifluorophenyl isocyanate (12), DCM, overnight; (d) Triphosgene, sat. NaHCO3, DCM, 30 min; (e) 1-acetyl-4-aminopiperidine (15), DCM, overnight; (f) 4-((trans-4-aminocyclohexyl)oxy)benzoic acid hydrochloride20 (19), Et3N, DMF, overnight. See the Experimental Section and the Supporting Information for further details.
The inhibitory activities of the three compounds in hsEH were evaluated using a previously reported sensitive fluorescent-based assay (Table 1).16 While carbamate 11 was a very weak inhibitor (IC50 = 12.7 μM) and thiourea 9 displayed only a moderate inhibition (IC50 = 138 nM), urea 13 revealed as a very potent hsEHI (IC50 = 1 nM) (Table 1). The superior potency of the urea is in agreement with previous results in other series of sEHIs.17,18 For this reason, no further carbamates and thioureas derivatives were envisaged and the urea group was chosen as the main pharmacophore for the synthesis of further inhibitors.
Table 1. Inhibition of hsEH and Microsomal Stability Values of the New Benzohomoadamantane-Based sEHI.
| Cpd | hsEHaIC50 (nM) | microsomal stabilityb (h/m/r) |
|---|---|---|
| 9 | 138 | NDc |
| 11 | >10,000 | ND |
| 7 | 7.7 | 79/77/81 |
| 13 | 1.0 | 7/0.2/ND |
| 14 | 20 | 77/23/33 |
| 1, AR9281 | 8.0 | 72/100/87 |
| 16 | 3.1 | 1/0.5/ND |
| 17 | 941 | ND |
| 18 | >10,000 | ND |
| 4, t-AUCB | 0.5 | 94/92/46 |
| 20 | 0.9 | 70/10/2 |
| 21 | 28 | 90/83/ND |
Reported IC50 values are the average of three replicates. The fluorescent assay as performed here has a standard error between 10 and 20% suggesting that differences of twofold or greater are significant. Because of the limitations of the assay, it is difficult to distinguish among potencies <0.5 nM.16
Percentage of the remaining compound after 60 min of incubation with human, mice, and rat microsomes obtained from Tebu–Xenotech in the presence of NADP at 37 °C.
ND: not determined.
Having found that this novel scaffold may successfully replace the adamantane and/or the phenyl group found in known sEHIs, a series of benzohomoadamantane derivatives related with the potent sEHI AR9281, t-AUCB, t-TUCB, EC5026, and 7 were synthesized in order to explore their potency and DMPK properties. Of note, a very recent work has described that the replacement of a methylene unit of the adamantane moiety by an oxygen atom led to more soluble compounds while only slightly reducing the inhibitory activity against the sEH (e.g., 1b, 4b, and 7b in Figure 3).19 In this sense, an oxygen atom was introduced in the benzohomoadamantane scaffold in order to explore whether a similar trend was also followed within this new family of sEHIs (Figure 3 and Scheme 1).
The synthesis of the new sEHI started from the suitably substituted benzohomoadamantane amines of general structure II.12−15 Thus, the synthesis of urea 14 involved the reaction of 5-methyl-1,5,6,7-tetrahydro-1,5:3,7-dimethanobenzo[e]oxonin-3(2H)-amine12 (II, R = CH3, X = O) with 2,3,4-trifluorophenylisocyanate, 12, in dichloromethane (Scheme 1). For the obtention of the piperidine derivatives, we first prepared the isocyanate of the corresponding polycyclic amine II (II, R = CH3, X = CH2;13II, R = CH3, X = O;12II, R = H, X = O12) by reaction with triphosgene and saturated the aqueous solution of NaHCO3. Once the desired isocyanate of general structure III (III, R = CH3, X = CH2; III, R = CH3, X = O; III, R = H, X = O) was obtained, it was reacted with 1-acetyl-4-aminopiperidine 15 in dichloromethane to obtain ureas 16–18 in moderate overall yields (Scheme 1). Finally, the t-AUCB analogues 20–23 were obtained in low to moderate yields by the reaction, in the presence of triethylamine, of the corresponding isocyanate, obtained from II (II, R = CH3, X = CH2;13II, R = CH3, X = O;12II, R = Cl, X = CH2;15II, R = F, X = CH215) and triphosgene, in DMF with 4-((trans-4-aminocyclohexyl)oxy)benzoic acid, 19, prepared as previously reported20 (Scheme 1).
2.2. sEH Inhibition and DMPK Assays
The potency of the new compounds as inhibitors of the hsEH was tested using a previously reported sensitive fluorescent-based assay.16 Gratifyingly, the potency of the new benzohomoadamantane ureas was in the same range as that of diphosgene their corresponding adamantane-based analogues (compare 13vs7, 16vs AR9281, and 20vs t-AUCB) (Table 1). The comparison of the compounds presenting a methylene unit in the polycyclic scaffold with their analogues featuring an oxygen atom (13vs14, 16vs17, and 20vs21, Scheme 1 and Table 1) showed that, in all cases, the compound bearing an oxygen atom was less potent; these results are in line with those previously found within the adamantane series of sEHIs.19 Interestingly, the AR9281 analogues 16 and 17 showed the largest difference (300-fold difference, Table 1). Also, the replacement of the methyl group at the R position of the polycyclic scaffold by a hydrogen atom in the AR9281 analogues produced a dramatic drop of the inhibitory activity (17vs18, >10-fold decrease in potency, Table 1).
Considering the metabolism liability of the adamantane and adamantane-related scaffolds,21,22 we evaluated the in vitro stability in human, mice, and rat microsomes of the new ureas bearing the benzohomoadamantane moiety (Table 1). Within the trifluorophenyl series, the substitution of the adamantane nucleus by the benzohomoadamantane scaffold showed an important decrease of the microsomal stability (7vs13, Table 1). By contrast, in the corresponding oxa-analogue, 14, the stability seemed to be restored in human, but marginally in mice and rat microsomes (13vs14, Table 1). Moreover, the analogue of AR9281, 16, presented very high metabolic liability in all three species, as less than 1% of the compound remains after being incubated with microsomes for 60 min (AR9281 vs16, Table 1). Finally, within the t-AUCB series, the replacement of the adamantane moiety by the benzohomoadamantane scaffold led to similar stability in human microsomes but to lower stability in mice and rat microsomes. Interestingly, in the oxa-analogue 21, the stability was also maintained in mice microsomes (20 and 21vs t-AUCB, Table 1). Although it seems that the ureas presenting the oxa-benzohomoadamantane moiety were more stable in microsomes, taking into account that all these derivatives (14, 17, 18, and 21) were considerably less potent, this oxa-polycyclic scaffold was abandoned and only the ureas featuring the benzohomoadamantane core were further evaluated.
Overall, the t-AUCB family of compounds presented the most favorable properties in terms of potency and microsomal stability, and this series was selected for further optimization. As the adamantane nucleus contributes to the high lipophilicity of the known sEHI that compromises the solubility of these compounds, we next measured the solubility of the selected t-AUCB series in a 1% DMSO: 99% PBS buffer solution. As expected, the solubility decreases from the adamantane-based t-AUCB to the benzohomoadamantane analogue 20 (Table 2) likely due to the increase of carbon atoms from the adamantane nucleus (10 atoms) to the new polycyclic scaffold (16 atoms). Taking this into account, novel substitutions in the R position of the benzohomoadamantane scaffold were explored toward improving solubility while maintaining or enhancing both potency and microsomal stability of 20. Thus, the methyl group of 20 was replaced by chlorine and fluorine atoms, leading to compounds 22 and 23, respectively. The potency, microsomal stability, solubility, and permeability of both compounds were assessed in order to explore their properties (Table 2).
Table 2. IC50 in Human, Murine, and Rat sEH, Microsomal Stability, Solubility, and Permeability Values of the t-AUCB Related Compounds.
| permeability (Caco-2) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| sEH IC50 (nM)a |
Papp (nm/s) |
||||||||||
| Cpd | human | murine | rat | microsomal stabilityb (h/m/r) | solubilityc (μM) | A→B | B→A | ERd | LD50e(μM) | IC50 hLOX-5f(μM) | IC50 hCOX-2g(μM) |
| 4, t-AUCB | 0.5 | 1.7 | 8.0h | 94/92/46 | 25 | 1.9 | 210.3 | 111 | NDi | ND | ND |
| 20 | 0.9 | 9.9 | 0.4 | 70/10/2 | 4 | 10 | 123.7 | 12.4 | >100 | >100 | >10 |
| 22 | 0.4 | 0.4 | 0.4 | 89/29/52 | 13 | 21.5 | 46.6 | 2.1 | >100 | >100 | >10 |
| 23 | 0.5 | 0.5 | 0.4 | 77/36/60 | 7 | 0.9 | 219.1 | 243.9 | >100 | >100 | >10 |
Reported IC50 values are the average of three replicates. The fluorescent assay as performed here has a standard error between 10 and 20%, suggesting that differences of twofold or greater are significant. Because of the limitations of the assay, it is difficult to distinguish among potencies <0.5 Nm.16
Percentage of remaining compound after 60 min of incubation with human, mice, and rat microsomes obtained from Tebu–Xenotech in the presence of NADP at 37 °C.
Solubility in a 1% DMSO: 99% PBS buffer solution, see the Experimental Section for details.
The efflux ratio was calculated as ER = (Papp B → A)/(Papp A → B). See the Experimental Section for further details.
sEHI cytotoxicity tested by propidium iodide staining after 24 h of incubation in SH-SY5Y cells. See the Experimental Section for further details.
IC50 in human LOX-5 (hLOX-5). See the Experimental Section for further details.
IC50 in human COX-2 (hCOX-2) performed by Eurofins (catalogue reference 4186).
Taken from ref (24).
ND: Not determined.
Satisfactorily, the evaluation of the inhibition activity against the hsEH showed that both compounds 22 and 23 were slightly more potent than 20, with IC50 values in the same range as t-AUCB. Of note, 22 and 23 presented the same potency inhibiting the human, murine, and rat enzymes, while t-AUCB was three- and twenty-fold less potent with the murine and the rat enzymes, respectively (Table 2).
Regarding metabolic stability, compounds 22 and 23 presented better stabilities than 20 (Table 2). Furthermore, the experimental solubility values of these new halogenated compounds were determined. In line with a previous work with adamantane derivatives,23 the solubility increases when the methyl group is replaced by a halogen atom (compare 20vs22 and 23, Table 2), particularly for the chlorinated compound 22.
The Caco-2 cell permeability model was used in order to evaluate the permeability of the compounds. Apparent permeability values (Papp) were determined from the amount permeated through the Caco-2 cell membranes at both apical-basolateral (A-B) and basolateral-apical (B-A) direction. Gratifyingly, compounds 20 and 22 presented higher permeability values than t-AUCB, 22 being the one that presented the best profile (Table 2). Moreover, the cytotoxicities of 20, 22, and 23 were evaluated in SH-SY5Y cells by propidium iodide staining after 24 h of incubation. None of the compounds showed to be cytotoxic at the highest concentration tested (100 μM).
Finally, inhibitors 20, 22, and 23 were tested for selectivity against hCOX-2 and hLOX-5, two enzymes involved in the AA cascade. Neither 20 nor the halogenated analogues 22 and 23 significantly inhibited these enzymes (see Table 2).
Next, considering the best permeability of 20 and 22, both compounds were selected for CYPs and hERG inhibition assays. Cytochromes P450 (CYP) inhibition was evaluated using human recombinant cytochrome P450 enzymes CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A, through a fluorescence-detection method. These assays were of great interest not only for the detection of possible drug–drug interactions but also in terms of selectivity as EETs are formed by several cytochrome P450 isoforms, particularly CYP2C19. Satisfactorily, the tested compounds did not significantly inhibit the evaluated cytochromes. We considered acceptable IC50 values around 2 μM in CYP2C19 taking into account that both compounds presented 2000-fold more potency inhibiting the sEH.
Regarding the hERG inhibition assay, both compounds inhibited the channel only 4 and 44% at 10 μM, respectively (Table 3). Overall, both compounds presented high potency inhibiting human, murine, and rat enzymes and did not significantly inhibit either cytochromes or hERG. Considering the biological profiling of the new sEH inhibitors, compounds 20 and 22 were selected as the candidates for the in vivo studies.
Table 3. Inhibition (Expressed as % of Inhibition at 10 μM or IC50) of Recombinant Human Cytochromes P450 Enzymes and Inhibition of the hERG Channel (Expressed as % of Inhibition at 10 μM).
| cytochrome
inhibitiona |
|||||||
|---|---|---|---|---|---|---|---|
| CYP 1A2 | CYP 2C9 | CYP 2C19 (μM) | CYP 2D6 | CYP
3A4b |
|||
| Cpd | (BFC) | (DBF) | hERG channel inhibition (% at 10 μM) | ||||
| 20 | 1 ± 2 | 17 ± 3 | 1.9 | 1 ± 1 | 2 ± 2 | 14 ± 1 | 4 |
| 22 | 14 ± 4 | 31 ± 3 | 2.2 | 11 ± 3 | 1 ± 2 | 52 ± 4 | 44 |
The cytochrome inhibition was tested at 10 μM. IC50 was calculated for those compounds that presented >50% of inhibition.
For the study of CYP3A4, two different substrates were used: benzyloxytrifluoromethylcoumarin (BFC) and dibenzylfluorescein (DBF).
2.3. In silico Study: Molecular Basis of Benzohomoadamantane-Derived Soluble Epoxide Hydrolase Inhibitors
The incorporation of an aromatic ring into the adamantane scaffold can potentially impact the orientation and molecular interactions of benzohomodamantane sEHIs compared to adamantane derivatives. To unravel how bulky benzohomodamantane ureas are accommodated in the active site of sEH and to understand the molecular basis of their inhibitory mechanism, molecular dynamics (MD) simulations were performed for compounds t-AUCB, 20, 22, and 23. The MD simulations revealed that the addition of an aromatic ring onto the adamantane scaffold of t-AUCB triggers conformational rearrangements in the active site and adjacent regions to stabilize the benzohomoadamantane scaffold. These interactions, together with a network of hydrogen bonds and hydrophobic interactions with the urea and benzoic acid moieties, are key for retaining the inhibitors in the active site.
First, we explored the preferred binding mode of the selected sEHI and the flexibility of the active site of the sEH–inhibitor complex. The L-shaped active site pocket of sEH consists of three regions: the left-hand side (LHS) and the RHS pockets and a central narrow channel defined by catalytic residues Asp335, Tyr383, and Tyr466 that connects the LHS and RHS hydrophobic cavities (see Figure 4).25 Previously, we showed that the active site of EHs present high plasticity.19,26 Available X-ray structures of sEH in complex with adamantyl ureas indicate that the adamantane scaffold can occupy both LHS and RHS pockets.27 In the case of t-AUCB (PDB: 5AM3), the inhibitor is orientated with the benzoic acid group occupying the RHS, while adamantane sits in the LHS (see Figure 4). To corroborate that this is also the preferred orientation for benzohomoadamantane derivatives, we carried out molecular docking calculations for compounds 20, 22, and 23 (see the Experimental Section for details). All the binding poses featuring the urea moiety interacting with Asp335 oriented the benzohomoadamantane scaffold in the LHS and the benzoic acid in the RHS, as observed for t-AUCB. The LHS pocket presents enough space to accommodate the bulky benzohomoadamantane scaffold (see Figure 4). To evaluate the stability and molecular interactions of the inhibitor in the active site of sEH, we carried out three replicas of 250 ns MD simulations for t-AUCB, 20, 22, and 23, starting from this orientation, that is the benzohomoadamantane occupying the LHS pocket. All inhibitors show considerable stability and no sign of unbinding or significant reorientations are observed along the MD simulation time. To evaluate the impact of the inhibitors on the active site conformational plasticity, we monitored the changes on the active site volume along the MD simulations (see Figures 4b and S1) using the POcket Volume MEasurer (POVME, see the Supporting Information for details).28 As observed previously, in the apo state, the total volume encompassing LHS, RHS, and the central channel displays wide fluctuations from 70 to 700 Å3 (average volume 290 ± 133 Å3).19 The size of the LHS pocket is mainly determined by flexible Ile363, Ile375, Met469, and Asn472 side chains, while the shape of the RHS pocket fluctuates with frequent side chain conformational changes of Leu408, Met419, and Phe497 residues (see Figure S1). When t-AUCB, 20, 22, and 23 compounds are bound in the active site, an expansion of the active site volume with respect to the average apo value is observed, which becomes stable at around 330–400 Å3 (see Figures 4b,c and S1). The average volumes determined for the last 150 ns of each MD simulation are 335 ± 33, 396 ± 37, 351 ± 25, and 356 ± 32 Å3 for t-AUCB, 20, 22, and 23, respectively. As expected, benzohomadamantane inhibitors show wider active sites than t-AUCB,compound 20, with a bulkier methyl group, being the one with the larger volume. The analysis of the root-mean-square fluctuation (RMSF) from the MD simulations shows that some active site residues become more rigid in the presence of inhibitors (see Figure S2). All inhibitors are able to restrict the conformational plasticity of the active site, indicating that they are tightly bound (see the population shift toward narrow distributions of active site volumes in Figure S1).
Figure 4.
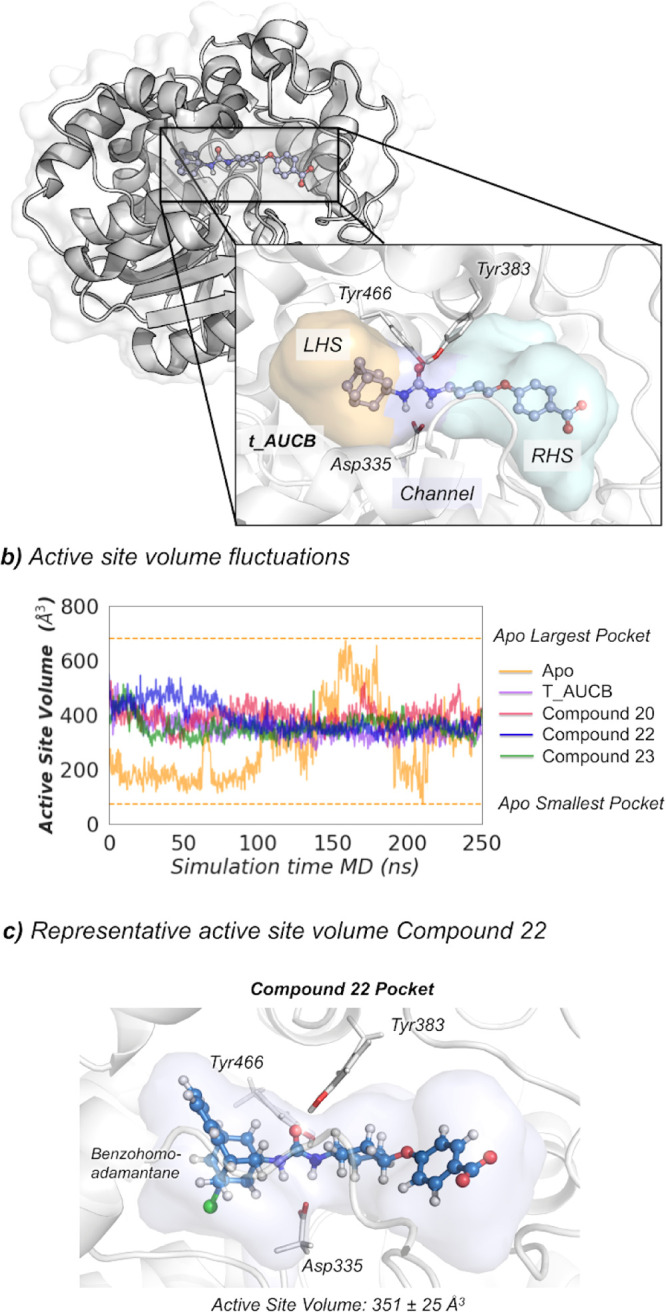
(a) Representation of the sEH structure (PDB: 5AM3), active site catalytic residues (nucleophilic Asp335, Tyr383, Tyr466), and t-AUCB inhibitor. The LHS pocket is colored in orange, the RHS pocket is colored in cyan, and the central channel in purple. (b) Plot of the fluctuations of the active site volume for the apo state (orange line, 290 ± 133 Å3), t-AUCB (purple line, 335 ± 33 Å3), compound 20 (red line, 396 ± 37 Å3), compound 22 (blue line, 351 ± 25 Å3), and compound 23 bound (green line, 356 ± 32 Å3) along a representative 250 ns MD simulation trajectory. The average volumes are calculated for the last 150 ns of the MD simulation. (c) Representative sEH structure with the active site volume obtained from MD simulations of compound 22.
To gain a deeper insight into the molecular basis of the inhibitory mechanism of benzohomoadamantane ureas, the non-covalent interactions between the selected inhibitors and the active site residues of sEH were analyzed with NCIplot on the most visited MD conformations (see Figures 5 and S3).29 First, we analyzed the interactions established in the RHS pocket and the central channel where all inhibitors share a common scaffold: the benzoic acid group. For t-AUCB, 20, 22, and 23 compounds, the carboxylate unit is stabilized by two hydrogen bonds with Ser412 and Ser415 that are located at the entrance of the RHS pocket, the interaction with Ser415 being more stable along the MD simulations (see Figure 5b and S3). The aromatic ring of the benzoic acid is further stabilized in the RHS pocket through CH···π interactions by the side chains of Trp525 and Phe497. The side chain of Phe497 transitions from the solvent to the active site to form a network of stable hydrophobic interactions that includes the benzoic acid group and the aromatic side chains of residues Trp525, Phe497, and catalytic Tyr383. Moreover, MD simulations show that four water molecules permanently occupy the RHS pocket in t-AUCB, 20, 22, and 23 (see Figures S4 and S5). These water molecules establish a network of interactions with the carboxylate group of the inhibitor and with residues Leu417, Ser407, Val416, Ser415, Ser412, and Lys495 that provides extra stabilization of the benzoic acid group in the RHS pocket. The urea moiety establishes hydrogen bonds with three catalytic residues: Asp335, Tyr383, and Tyr466 located in the central channel of the active site pocket. MD simulations show that the three hydrogen bonds remain significantly stable along the whole simulation time for all inhibitors with no significant differences (see Figure 5c). t-AUCB and 20 are able to retain a tighter hydrogen bond (below 3 Å) between the carbonyl of the urea and the OH of Tyr466 than halogenated compounds 22 and 23. These results indicate that all inhibitors remain stable in the active site pocket, forming similar interactions consistent with reported IC50 values. Previously, we have shown that less potent inhibitors displayed fluctuations in the interactions between the urea motif and the catalytic residues, shifting the ensemble toward longer distances.19 All inhibitors share a common scaffold on the RHS of the urea and MD simulations reported a similar behavior in terms of interactions and conformational dynamics in the RHS and central channel regions. The network of hydrogen bonds and π–π stacking interactions is key to retain the inhibitor in the active site.
Figure 5.
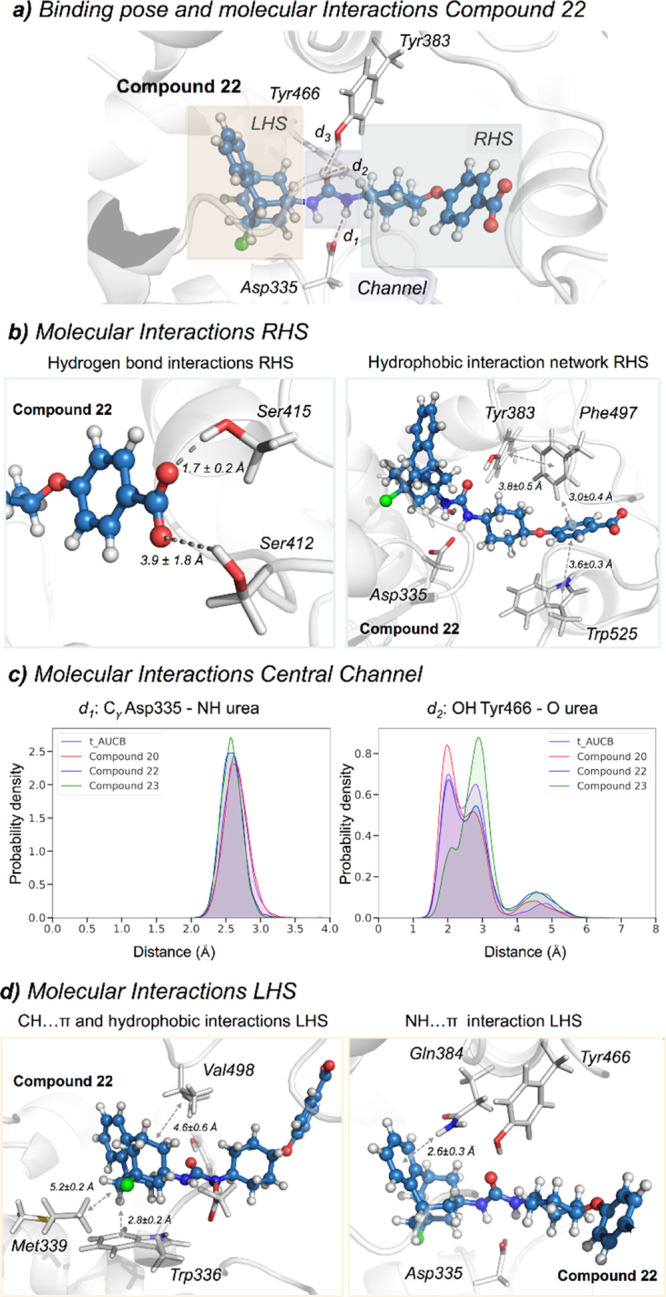
(a) Representative structure of 22 bound in the active site of sEH obtained from the most visited conformations along the MD simulations. The benzohomoadamantane moiety occupies the LHS pocket while the benzoic acid group lays in the RHS pocket. The central urea unit establishes hydrogen bonds with Asp335 (d1), Tyr466 (d2), and Tyr383 (d3). The PDB 5AM3 has been used as the starting point for all MD simulations. (b) Most relevant molecular interactions in the RHS. Average distances (in Å) obtained from the last 150 ns of MD simulations are represented. Hydrogen bonds between the oxygens of the carboxylate group of 22 and the hydrogen of the OH group of Ser412 and Ser415 are shown. The π–π stacking average distances are computed between the most proximal carbon atoms of each ring. The CH···π and hydrophobic interaction average distances are computed between the hydrogen atoms and the centroid of each aromatic ring (c) Histogram plots of the distance between the carboxylic group of the catalytic Asp335 and the amide groups of the inhibitor [d1(CγAsp335-NHINH)] and the distance between the carbonyl group of the urea inhibitor and the OH group of Tyr466 residue [d2(OHTyr466-OINH)] along the MD simulations of t-AUCB (purple), 20 (red), 22 (blue), and 23 (green). (d) Most relevant molecular interactions in the LHS. Average distances (in Å) obtained from the last 150 ns of MD simulations are represented. The CH···π interaction is calculated between the hydrogen of benzohomoadamantane unit and the centroid of the benzoid ring of Trp336. The NH···π interaction is monitored between the amide hydrogen of Gln384 and the center of the aromatic ring of the benzohomoadamantane scaffold.
Significant differences were observed in the LHS pocket, where the benzohomoadamantane scaffold is placed. For t-AUCB, 20, 22, and 23, the adamantane unit is mostly stabilized by the side chain of Trp336 through stable CH···π interactions (see Figure 5d and S3). In all cases, additional hydrophobic interactions with the side chains of Met339 and Val498 that wrap the adamantane in the LHS pocket are observed. The incorporation of an aromatic ring into the adamantane scaffold of t-AUCB induces a series of conformational rearrangements in the active site, which further stabilize both the adamantane and aromatic groups. In particular, the benzohomoadamantane scaffold is reoriented in the beginning of the MD simulations to position the aromatic ring toward the amide group of the side chain of Gln384 for establishing NH···π interactions that retain the benzohomoadamantane group fixed in the LHS pocket (see Figure 5d and S3). This interaction is observed in all MD simulations in the presence of 20, 22, and 23 inhibitors and, once formed, remains stable along the whole simulation time. In t-AUCB, the amide group of Gln384 forms a network of hydrogen bond interactions with the OH group of Tyr383 and the urea moiety that is partially disrupted in the presence of benzohomoadamantane. Additionally, Phe381 moves away from the LHS pocket to accommodate the aromatic rings of 20, 22, and 23, establishing frequent π-stacking interactions. Met468, that in the t-AUCB X-ray structure is pointing toward the solvent, moves toward the active site to establish hydrophobic interactions with the benzohomoadamantane moiety. The hydrophobic nature of the LHS pocket prevents the permanence of water molecules in this cavity (see Figure S5). However, transient entrance of one or two water molecules is frequently observed along the MD simulations of 20, 22, and 23 compounds. Finally, the symmetric adamantyl unit in t-AUCB freely rotates inside the LHS pocket, while the asymmetry introduced in the benzohomoadamantane scaffold limits its rotation inside the active site (see Figure S6). The strong and stable hydrophobic and NH···π interactions significantly reduce the rotation of the benzohomoadamantane moiety inside the LHS pocket (see Figure S6). This limited flexibility can pose some impediments in the binding pathway of benzohomoadamante derivatives.
A significant conformational rearrangement is observed for compounds 22 and 23 in the course of the 250 ns of the MD simulation. The active site loop (493–500 residues) containing Leu499 is significantly displaced from the reference X-ray structure (see Figures 6 and RMSF S2). To explore whether this conformational change can take place in longer time-scales also for t-AUCB and 20, we performed 500 ns of accelerated MD (aMD) simulations for all compounds.30 The analysis of aMD simulations confirms that this rearrangement occurs in the presence of all compounds (see Figure S7 and the Supporting Information for aMD details). This rearrangement includes the motion of the bulky Leu499 side chain that leaves the active site and the approximation of the carbonyl backbone of Val498 toward the benzohomoadamante moiety (see Figure 6b,c). Based on the analysis of NCIplot, the backbone of Val498 can establish dipole-induced dipole interactions generated by halogens F and Cl on the benzohomoadamantane scaffold. The computational mutagenesis of L499A and subsequent MD simulations on the L499A sEH variant showed that the loop is not perturbed along the simulation time, indicating that the displacement of the bulky side-chain of Leu499 can play a key role in accommodating the benzohomoadamantane scaffold. However, further investigations are required to explore the contribution of this conformational rearrangement in the thermodynamics and kinetics of inhibitor binding. Similar conformational changes in loops located at the vicinity of the active site have been described in other EH as key for substrate binding.26 The molecular insight gained from MD simulations paves the way toward the rational improvement of benzohomoadamantane scaffolds for enhanced inhibition.
Figure 6.

(a) Overlay of two representative structures of t-AUCB (purple, PDB 5AM3) and 22 (blue) bound in the active site of sEH. The image indicates the motion of the loop (493–500) containing Val498 and Leu499 with a black arrow. The loop is colored in purple for the X-ray t-AUCB conformation and in blue for the 22 conformation. The PDB 5AM3 have been used as the starting point for all MD simulations. (b) X-ray structure (PDB 5AM3), where Leu499 is pointing toward the adamantane moiety. (c) Most visited active site conformation with 22 bound, where Leu499 is displaced from the active site and Val498 carbonyl points toward the benzohomoadamantane moiety.
2.4. Pharmacokinetic Studies of Selected Compounds 20 and 22
The pharmacokinetic characterization of 20 and 22 was performed in male C57BL/6 mice by intraperitoneal administration of 3 mg/kg of each compound. As shown in Table 4, both compounds demonstrated good absorption and elimination characteristics. Notwithstanding, compound 22 presented a better profile than 20 considering its larger half-life (5.2 h), higher Cmax (3583 ng/mL) and AUC (23,328.12 h*ng/mL), and lowest clearance (0.13 L/h/kg). Considering its superior pharmacokinetic profile and its better potency and solubility (Tables 2 and 4), 22 was selected for conducting the in vivo efficacy study in the well-known murine model of cerulein-induced acute pancreatitis (AP).31−33
Table 4. Pharmacokinetic Parameters in the Male C57BL/6 Mouse for Compounds 20 and 22 after 3 mg/kg IP Administrationa.
| Cpd | dose (mg/Kg) | HL (h) | Tmax (h) | Cmax (ng/mL) | AUClast (h*ng/mL) | AUCINF (h*ng/mL) | Vd (L/Kg) | Cl (L/h/Kg) |
|---|---|---|---|---|---|---|---|---|
| 20 | 3 | 1.17 | 0.50 | 1610 | 2260 | 2323 | 2.18 | 1.29 |
| 22 | 3 | 5.2 | 2 | 3583 | 22543 | 23328 | 0.96 | 0.13 |
See the Experimental Section and Tables S2 and S3 and Figures S8 and S9 in the Supporting Information
2.5. In vivo Efficacy Study
AP is a potentially life-threatening gastrointestinal disease, and its incidence has been increasing over the last few decades. The onset of the disease is thought to be triggered by intra-acinar cell activation of digestive enzymes that results in interstitial edema, inflammation, and acinar cell death that often leads to a systemic inflammation response.31−34 The efficacy of the new sEHI 22 at 0.1 and 0.3 mg/kg was assessed in the cerulein-induced AP murine model. The experimental procedure for the in vivo efficacy study followed already published protocols.35
First, the health status of the animals was analyzed by monitoring their change in body weight along the experimental procedure. After food replacement (with the last cerulein injection), control animals gained some weight, and, as expected, it was not observed in animals receiving cerulein only. In contrast, animals treated with both doses (0.3 and 0.1 mg/kg) of 22 showed an increased body weight although only the group treated at 0.3 mg/kg reached statistical significance (p < 0.01 vs Cerulein group) (Figure 7).
Figure 7.

Percentage of body weight change at the end of the study vs t = 0 h. Effect of 12 consecutive administrations of cerulein (50 μg/kg, IP) and treatment with 22 (single dose, 0.3 or 0.1 mg/kg, IP) on C57BL/6 male mice body weight. Results are expressed as mean ± SEM (n = 3–9). *p < 0.05, **p < 0.01, ****p < 0.0001 vs Cerulein group (ANOVA-one way).
In addition, the concentrations of 22 in plasma and pancreatic tissue were measured 10 h post-administration. Taking into account that 22 is a subnanomolar inhibitor of the murine sEH, we confirmed that the administration of both doses produced enough plasma levels of compound 22 to inhibit the sEH (175 nM for the dosage of 0.3 mg/kg and 14 nM for the dosage of 0.1 mg/kg). Moreover, the compound concentration in pancreas was also analyzed showing 29.9 ng/g for the dosage of 0.3 mg/kg, confirming that 22 was able to reach the pancreatic tissue. However, no amount of 22 was detected in pancreatic tissues with the 0.1 mg/kg dose.
Finally, histologic analysis of pancreas was assessed in order to determine if treatment with 22 reduced the severity of the cerulein-induced pancreatitis. Pathologic changes were studied on H&E-stained pancreas sections (Figure 8A).
Figure 8.

Results of the histologic analysis of pancreas from mice treated with vehicle (control), cerulein, and cerulein plus either 0.1 or 0.3 mg/kg of compound 22. (A) Representative H&E-stained sections of the pancreas from the in vivo efficacy study. Arrows indicate inflammatory cells and edema. Bold arrows indicate intracellular vacuoles. (B–E) Histologic scoring of pancreatic tissues. (B), edema. (C), inflammatory cells (mononuclear and polymorphonuclear). (D), parenchymal atrophy. (E), total scoring (pancreatic parenchymal atrophy, vacuolar degeneration of cells, edema, hemorrhage, mononuclear inflammatory cells, mononuclear inflammatory cells, polimorfonuclear inflammatory cells and necrosis). *p < 0.05, **p < 0.01 and ***p < 0.001 vs control. #p < 0.05 vs cerulein.
As expected, the cerulein control group presents pancreatic damage representative of AP, including edema (Figure 8B), infiltration of inflammatory cells (Figure 8C), and parenchymal atrophy (Figure 8D). By contrast, treatment with both doses of 22 ameliorated cerulein-induced effects. The higher dose (0.3 mg/kg) more efficiently reversed the pancreatic damage, edema, and neutrophil infiltration (Figure 8 and Tables S4–S7 in the Supporting Information).
3. Conclusions
The sEH has been identified as a suitable target for several inflammatory diseases. For this reason, many adamantane-based and aryl-based sEHIs have been designed and three selected compounds, AR9281, GSK2256294, and EC5026, have reached human clinical trials. In this work, we found that the aromatic and adamantane fragment can be merged, leading to the very versatile benzohomoadamantane scaffold. Therefore, three series of compounds replacing the adamantane moiety of AR9281, 7, and t-AUCB by this new polycycle have been synthesized and biologically evaluated. The in vitro profiling of these sEHIs (solubility, cytotoxicity, metabolic stability, CYP450s, hLOX-5, hCOX-2 and hERG inhibition) allowed to select a suitable candidate for an in vivo efficacy study in a murine model of AP, for which sEHIs showed effectiveness at ameliorating this condition.19,35,36 The administration of 22 improved the general health status of cerulein-induced AP mice and significantly reduced pancreatic damage. Hence, the benzohomoadamantane moiety emerges as a suitable hydrophobic scaffold for the design of novel sEHIs. The molecular insight provided by MD simulations indicated that sEH reshapes the active site pocket to stabilize the aromatic ring of the benzohomoadamantane scaffold. Due to the promising results obtained with compound 22, more research around benzohomoadamantane-based sEHIs for the treatment of inflammatory and pain-related diseases is currently ongoing.
4. Experimental Section
4.1. Chemical Synthesis
4.1.1. General Methods
Commercially available reagents and solvents were used without further purification unless stated otherwise. Preparative normal phase chromatography was performed on a CombiFlash Rf 150 (Teledyne Isco) with pre-packed RediSep Rf silica gel cartridges. Thin-layer chromatography was performed with aluminum-backed sheets with silica gel 60 F254 (Merck, ref 1.05554), and spots were visualized with UV light and 1% aqueous solution of KMnO4. Melting points were determined in open capillary tubes with a MFB 595010M Gallenkamp. 400 MHz 1H and 100.6 MHz 13C NMR spectra were recorded on a Varian Mercury 400 or on a Bruker 400 AVANCE III spectrometers. 500 MHz 1H and 125.7 13C NMR spectra were recorded on a Varian Inova 500 spectrometer. The chemical shifts are reported in ppm (δ scale) relative to internal tetramethylsilane, and coupling constants are reported in Hertz (Hz). Assignments given for the NMR spectra of selected new compounds have been carried out on the basis of DEPT, COSY 1H/1H (standard procedures), and COSY 1H/13C (gHSQC and gHMBC sequences) experiments. IR spectra were run on the PerkinElmer Spectrum RX I, PerkinElmer Spectrum TWO, or Nicolet Avatar 320 FT-IR spectrophotometers. Absorption values are expressed as wave-numbers (cm–1); only significant absorption bands are given. High-resolution mass spectrometry (HRMS) analyses were performed with an LC/MSD TOF Agilent Technologies spectrometer. The elemental analyses were carried out in a Flash 1112 series Thermofinnigan elemental microanalyzator (A5) to determine C, H, N, and S. The structure of all new compounds was confirmed by elemental analysis and/or accurate mass measurement, IR, 1H NMR and 13C NMR. The analytical samples of all the new compounds, which were subjected to pharmacological evaluation, possessed purity ≥95% as evidenced by their elemental analyses.
4.1.1.1. 1-(9-Methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)-3-(4-(trifluoromethyl)phenyl)thiourea (9)
To a solution of 9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (250 mg, 0.95 mmol) in DCM (2 mL), 1-isothiocyanato-4-(trifluoromethyl)benzene (193 mg, 0.95 mmol) and Et3N (287 mg, 2.84 mmol) were added. The reaction mixture was stirred at room temperature overnight and then the solvent was evaporated under vacuum. The residue was dissolved in EtOAc (30 mL) and water (20 mL) and the phases were separated. The aqueous phase was extracted with further EtOAc (2 × 30 mL). The combined organic phases were dried over anh. Na2SO4, filtered and concentrated under vacuum to obtain 369 mg of a yellow solid. The product was washed with Et2O to obtain thiourea 9 (188 mg, 46% yield) as a white solid, mp 158–159 °C. IR (NaCl disk): 3283, 2911, 2834, 1615, 1532, 1493, 1454, 1422, 1324, 120, 1166, 1124, 1067, 1015, 948, 909, 837, 759, 732, 697, 665 cm–1. 1H NMR (400 MHz, CDCl3): δ 0.95 (s, 3 H, C9–CH3), 1.57 [d, J = 13.6 Hz, 2 H, 10(13)-Hax], 1.67 (dd, J = 13.2 Hz, J′ = 6.0 Hz, 2 H, 10(13)-Heq], 2.00 (s, 2 H, 8-H), 2.31–2.40 [c.s., 4 H, 6(12)-H2], 3.12 [broad s, 2 H, 5(11)-H], 6.11 (s, 1 H, C7–NH), 7.05 [m, 2 H, 1(4)-H], 7.09 [m, 2 H, 2(3)-H], 7.28 [d, J = 8.2 Hz, 2 H, 2′(6′)-H], 7.64 [d, J = 8.2 Hz, 2 H, 3′(5′)-H], 7.81 (s, 1 H, C1′-NH). 13C NMR (100.5 MHz, CDCl3): δ 32.2 (CH3, C9–CH3), 33.9 (C, C9), 38.9 [CH2, C6(12)], 40.9 [CH, C5(11)], 41.1 [CH2, C10(13)], 47.5 (CH2, C8), 57.7 (C, C7), 123.7 (q, 1JC-F = 272 Hz, C, CF3), 123.8 [CH, C2′(6′)], 126.5 [CH, C2(3)], 127.2 [q, 3JC-F = 3.7 Hz, CH, C3′(5′)], 128.0 [CH, C1(4)], 128.1 (C, 2JC-F = 33.1 Hz, CH, C4′) 140.1 (C, C1′), 145.8 [C, C4a(11a)], 178.2 (C, CS).
4.1.1.2. p-Tolyl (9-Methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)carbamate (11)
To a solution of 9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (250 mg, 0.95 mmol) in DCM (2 mL), p-tolyl chloroformate (194 mg, 1.14 mmol) and Et3N (287 mg, 2.84 mmol) were added. The reaction mixture was stirred at room temperature overnight and then the solvent was evaporated under vacuum. The residue was dissolved in EtOAc (30 mL) and water (20 mL) and the phases were separated. The aqueous phase was extracted with further EtOAc (2 × 30 mL). The combined organic phases were dried over anh. Na2SO4, filtered, and concentrated under vacuum to obtain 300 mg of a yellow gum. Column chromatography (SiO2, hexane/ethyl acetate mixtures) gave carbamate 11 (46 mg, 14% yield) as a white solid, mp 114–115 °C. IR (NaCl disk): 3330, 3018, 2944, 2919, 2854, 1744, 1591, 1531, 1502, 1452, 1379, 1362, 1345, 1255, 1214, 1198, 1167, 1137, 1069, 1042, 1014, 987, 948, 900, 825, 757 cm–1. 1H NMR (400 MHz, CDCl3): δ 0.94 (s, 3 H, C9–CH3), 1.56 [d, J = 13.6 Hz, 2 H, 10(13)-Hax], 1.66 [dd, J = 13.6 Hz, J′ = 6.0 Hz, 2 H, 10(13)-Heq], 1.85 (s, 2 H, 8-H), 2.01 [d, J = 13.2 Hz, 2 H, 6(12)-Hax], 2.18 [dd, J = 13.2 Hz, J′ = 6.8 Hz, 2 H, 6(12)-Heq], 2.32 (s, 3 H, C4′-CH3), 3.10 [t, J = 5.6 Hz, 2 H, 5(11)-H], 4.92 (s, 1 H, NH), 6.97–7.00 [dm, J = 8.2 Hz, 2 H, 2′(6′)-H], 7.06 [cs, 2 H, 1(4)-H], 7.09 [c.s., 2 H, 2(3)-H], 7.12–7.14 [broad d, J = 8.2 Hz, 2 H, 3′(5′)-H]. 13C NMR (100.5 MHz, CDCl3): δ 20.8 (CH3, Ar-CH3), 32.2 (CH3, C9–CH3), 33.7 (C, C9), 39.3 [CH2, C6(12)], 40.9 [CH, C5(11)], 41.1 [CH2, C10(13)], 47.1 (CH2, C8), 53.8 (C, C7), 121.4 [CH, C2′(6′)], 126.3 [CH, C2(3)], 128.0 [CH, C1(4)], 129.6 [CH, C3′(5′)], 134.6 (C, C4′), 146.1 [C, C4a(11a)], 148.6 (C, C1′), 152.4 (C, CO).
4.1.1.3. 1-(9-Methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)-3-(2,3,4-trifluorophenyl)urea (13)
To a solution of 9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (193 mg, 0.73 mmol) in anh. DCM (6.5 mL) were added 2,3,4-trifluorophenyl isocyanate (105 mg, 0.61 mmol) and triethylamine (246 mg, 2.43 mmol) under a nitrogen atmosphere. The reaction mixture was stirred at room temperature overnight and the solvent was evaporated under vacuum. Column chromatography (SiO2, hexane/EtOAc mixture) of the crude and concentration under vacuum of the appropriate fractions gave urea 13 (38 mg, 13% yield) as a white solid, mp 206–207 °C. IR (ATR) ν: 3331, 2903, 2839, 1654, 1556, 1510, 1473, 1361, 1344, 1290, 1237, 1174, 1101, 1038, 1019, 1004, 800, 756, 690, 669, 625 cm–1. 1H NMR (500 MHz, CD3OD): δ 0.94 (s, 3 H, C9–CH3), 1.50 [d, J = 13.5 Hz, 2 H, 10(13)-Hax], 1.69 [m, 2 H, 10(13)-Heq], 1.77 (s, 2 H, 8-H), 2.10 [m, 2 H, 6(12)-Heq], 2.15 [d, J = 13 Hz, 2 H, 6(12)-Hax], 3.08 [tt, J = 6 Hz, J’ = 1.5 Hz, 2 H, 5(11)-H], 6.98 (m, 1 H, 5′-H), 7.04 [broad s, 4 H, 1(4)-H and 2(3)-H], 7.66 (m, 1 H, 6′-H). 13C NMR (125.7 MHz, CD3OD): δ 32.9 (CH3, C9–CH3), 34.6 (C, C9), 40.6 [CH2, C6(12)], 42.5 [CH, C5(11)], 42.5 [CH2, C10(13)], 49.0 (CH2, C8), 54.5 (C, C7), 112.2 (CH, dd, 2JC-F = 17.8 Hz, 3JC-F = 3.9 Hz, C5′), 116.6 (CH, C6′), 127.0 (C, dd, 2JC-F = 8.0 Hz, 3JC-F = 2.4 Hz Ar-C1′), 127.4 [CH, C2(3)], 129.0 [CH, C1(4)], 141.0 (C, dt, 1JC-F = 247.8 Hz, 2JC-F = 14.9 Hz, Ar-C3′), 143.6 (C, dd, 1JC-F = 245.7 Hz, 2JC-F = 12.8 Hz, Ar-C4′), 147.3 (C, dd, 1JC-F = 242.6 Hz, 2JC-F = 10.3 Hz, Ar-C2′), 147.6 [C, C4a(C11a)], 156.1(C, CO). MS (DIP), m/z (%); significant ions: 400 (M+, <1), 253 (19), 228 (14), 211 [(C16H19)+, 16], 172 (23), 155 (54), 149 (56), 148 (100), 147 (52), 143 (22), 141 (20), 129 (21), 128 (18), 115 (16).
4.1.1.4. 1-(5-Methyl-1,5,6,7-hexahydro-1,5:3,7-dimethanobenzo[e]oxonin-3(2H)-yl)-3-(2,3,4-trifluorophenyl)urea (14)
To a solution of 5-methyl-1,5,6,7-tetrahydro-1,5:3,7-dimethanobenzo[e]oxonin-3(2H)-amine hydrochloride (250 mg, 0.94 mmol) in anh. DCM (8.5 mL) were added 2,3,4-trifluorophenyl isocyanate (135 mg, 0.78 mmol) and triethylamine (316 mg, 3.13 mmol) under a nitrogen atmosphere. The reaction mixture was stirred at room temperature overnight and the solvent was evaporated under vacuo to furnish pure urea 14 as a white solid (205 mg, 54% yield), mp 257–259 °C. IR (ATR) ν: 3295, 3241, 3118, 2916, 2173, 1693, 1620, 1564, 1510, 1493, 1468, 1462, 1356, 1345, 1320, 1302, 1286, 1273, 1254, 1229, 1210, 1181, 1167, 1111, 1091, 1074, 1049, 1035, 1008, 999, 958, 906, 820, 812, 763, 646 cm–1. 1H NMR (400 MHz, DMSO-d6): δ 1.18 (s, 3 H, C5–CH3), 1.56 [d, J = 13.6 Hz, 2 H, 6(13)-Hb], 1.84 [m, 2 H, 6(13)-Ha], 1.97 [d, J = 13.2 Hz, 2 H, 2(12)-Hb], 2.20 [m, 2 H, 2(12)-Ha], 3.16 [t, J = 5.5 Hz, 2 H, 1(7)-H], 4.06 (s, 1 H, C3–NH), 7.14 (complex signal, 5 H, 8(11)-H, 9(10)-H, 5′-H), 7.84 (m, 1 H, 6′-H), 8.52 (broad s, 1 H, C1′-NH). 13C NMR (100.6 MHz, DMSO-d6): δ 31.1 (CH3, C5–CH3), 37.4 [CH2, C2(12)], 38.1 [CH2, C6(13)], 38.2 [CH, C1(7)], 73.4 (C, C5), 82.7 (C, C3), 111.6 (CH, dd, 2JC-F = 17.2 Hz, 3JC-F = 3.5 Hz, C5′), 114.3 (CH, broad s, C6′), 126.0 (C, dd, 2JC-F = 7.8 Hz, 3JC-F = 3.0 Hz, C1′), 126.5 [CH, C9(10)], 128.2 [CH, C8(11)], 139.0 (C, dd, 1JC-F = 246 Hz, 2JC-F = 15 Hz, C3′), 141.0 (C, dd, 1JC-F = 248 Hz, 2JC-F = 12 Hz, C4′), 144.7 (C, dd, 1JC-F = 241 Hz, 2JC-F = 11 Hz, C2′),145.5 [C, C7a(C11a)], 152.3 (C, CO). MS (DIP), m/z (%); significant ions: 402 (M+, 48), 171 (13), 170 (34), 169 (21), 157 (20), 156 (18), 155 (53), 154 (14), 153 (11), 148 (18), 147 [(C6H4F3N)+, 100], 146 (53), 145 (15), 143 (25), 142 (21), 141 (23), 131 (12), 130 (15), 129 (65), 128 (46), 127 (22), 116 (12), 115 (55), 91 (17), 84 (19), 83 (28), 71 (15), 70 (16), 69 (21). HRMS-ESI+m/z: [M + H]+ calcd for [C22H21F3N2O2+H]+, 403.1633; found, 403.1631.
4.1.1.5. 1-(1-Acetylpiperidin-4-yl)-3-(9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)urea (16)
To a solution of 9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (180 mg, 0.68 mmol) in DCM (3 mL) and saturated aqueous NaHCO3 solution (2 mL), triphosgene (102 mg, 0.34 mmol) was added. The biphasic mixture was stirred at room temperature for 30 min and then the two phases were separated and the organic layer was washed with brine (5 mL), dried over anh. Na2SO4, filtered, and evaporated under vacuum to obtain 1–2 mL of a solution of isocyanate in DCM. To this solution were added 1-(4-aminopiperidin-1-yl)ethan-1-one hydrochloride (122 mg, 0.68 mmol) and Et3N (138 mg, 1.36 mmol). The mixture was stirred overnight at room temperature, diluted with further DCM (10 mL), and washed with 2N NaOH solution (2 × 10 mL). Organics were dried over anh. Na2SO4, filtered, and concentrated under vacuum to obtain a yellow oil (232 mg). Column chromatography (SiO2, DCM/methanol mixtures) gave urea 16 as a white solid (143 mg, 53% yield). The analytical sample was obtained by crystallization from hot EtOAc (113 mg), mp 206–207 °C. IR (NaCl disk): 3359, 3065, 3016, 2938, 2906, 2860, 1644, 1620, 1555, 1493, 1452, 1360, 1344, 1319, 1267, 1228, 1212, 1136, 1090, 1049 cm–1. 1H NMR (400 MHz, CDCl3): δ 0.90 (s, 3 H, C9–CH3), 1.13 (dq, J = 12.0 Hz, J′ = 4.0 Hz, 1 H, 3′-Hax or 5′-Hax), 1.20 (dq, J = 12.0 Hz, J′ = 4.0 Hz, 1 H, 5′-Hax or 3′-Hax), 1.52 [d, J = 13.2 Hz, 2 H, 10(13)-Hax], 1.62 [dd, J = 6 Hz, J′ = 12.8 Hz, 2 H, 10(13)-Heq], 1.80 (s, 2 H, 8-H), 1.85 (m, 1 H, 3′-Heq or 5′-Heq), 1.93 [d, J = 12.8 Hz, 2 H, 6(12)-Hax], 2.01 (m, 1 H, 5′-Heq or 3′-Heq), 2.06 (s, 3 H, COCH3), 2.12 [dd, J = 12.8 Hz, J′ = 6.0 Hz, 2 H, 6(12)-Heq], 2.70 (m, 1 H, 6′-Hax or 2′-Hax), 3.02–3.14 [complex signal, 3 H, 5(11)-H, 2′-Hax or 6′-Hax], 3.68–3.78 (complex signal, 2 H, 4′-H, 2′-Heq or 6′-Heq), 4.41 (dm, J = 13.6 Hz, 1 H, 6′-Heq or 2′-Heq), 4.62–4.68 (complex signal, 2 H, C7–NH and C4′-NH), 7.02 [m, 2 H, 1(4)-H], 7.06 [m, 2 H, 2(3)-H]. 13C NMR (100.5 MHz, CDCl3): δ 21.4 (CH3, COCH3), 32.3 (CH3, C9–CH3), 32.4 (CH2, C3′ or C5′), 33.6 (CH2, C5′ or C3′), 33.7 (C, C9), 39.9 [CH2, C6(12)], 40.7 (CH2, C6′ or C2′), 41.1 [CH, C5(11)], 41.2 [CH2, C10(13)], 45.4 (CH2, C2′ or C6′), 46.7 (CH, C4′), 48.0 (CH2, C8), 53.4 (C, C7), 126.2 [CH, C2(3)], 128.0 [CH, C1(4)], 146.3 [C, C4a(11a)], 156.4 (C, NHCONH), 169.0 (C, COCH3).
4.1.1.6. 1-(1-Acetylpiperidin-4-yl)-3-(5-methyl-1,5,6,7-tetrahydro-1,5:3,7-dimethanobenzo[e]oxonin-3(2H)-yl)urea (17)
To a solution of 5-methyl-1,5,6,7-tetrahydro-1,5:3,7-dimethanobenzo[e]oxonin-3(2H)-amine hydrochloride (180 mg, 0.68 mmol) in DCM (3 mL) and saturated aqueous NaHCO3 solution (2 mL), triphosgene (102 mg, 0.34 mmol) was added. The biphasic mixture was stirred at room temperature for 30 min and then the two phases were separated and the organic one was washed with brine (5 mL), dried over anh. Na2SO4, filtered, and evaporated under vacuum to obtain 1–2 mL of a solution of isocyanate in DCM. To this solution were added 1-(4-aminopiperidin-1-yl)ethan-1-one hydrochloride (122 mg, 0.68 mmol) and Et3N (139 mg, 1.37 mmol). The mixture was stirred overnight at room temperature, diluted with further DCM (10 mL), and washed with 2N NaOH solution (2 × 10 mL). The organic layer was dried over anh. Na2SO4, filtered, and concentrated under vacuum to obtain a yellow residue (206 mg). Column chromatography (SiO2, DCM/methanol mixtures) furnished urea 17 as a white solid (135 mg, 49% yield). The analytical sample was obtained by crystallization from hot EtOAc (112 mg), mp 208–209 °C. IR (NaCl disk): 3357, 3054, 3012, 2969, 2926, 2853, 1646, 1611, 1546, 1492, 1450, 1358, 1324, 1268, 1222, 1156, 1101, 1088, 1035, 1212, 991, 947, 918, 900, 866, 829, 760, 733, 699 cm–1. 1H NMR (400 MHz, CDCl3): δ 1.26 (s, 3H, C9–CH3), 1.32–1.42 (complex signal, 2 H, 3′-Hax, 5′-Hax), 1.66–1.67 [complex signal, 4 H, 2-Hax, 12-Hax, 6(13)-Hax], 1.85–1.91 [m, 2 H, 6(13)-Heq], 1.97 (m, 1 H, 3′-Heq or 5′-Heq), 2.08 (m, 1 H, 5′-Heq or 3′-Heq), 2.10 (s, 3 H, COCH3), 2.24–2.34 (complex signal, 2 H, 2-Heq, 12-Heq), 2.95 (ddd, J = 3.2 Hz, J′ = 10.8 Hz, 1 H, 2′-Hax or 6′-Hax), 3.16–3.26 [complex signal, 3 H, 6′-Hax or 2′-Hax, 1(7)-H], 3.72 (m, 1 H, 2′-Heq or 6′-Heq), 3.89 (m, 1 H, 4′-H), 4.34 (m, 1 H, 2′-Heq or 6′-Heq), 4.78 (s, 1 H, C3–NH), 6.34 (d, J = 7.5 Hz, 1 H, 4′-NH), 7.09–7.15 (complex signal, 4 H, 8-H, 9-H, 10-H, 11-H). 13C NMR (100.5 MHz, CDCl3): δ 21.4 (CH3, COCH3), 31.5 (CH3, C5–CH3), 32.1 (CH2, C3′or 5′), 33.2 (CH2, C5′ or C3′), 37.4 (CH2, C2 or C12), 37.6 (CH2, C12 o C2), 38.4 [CH2, C6 (13)], 38.8 [CH, C1(7)], 40.2 (CH2, C6′ or C2′), 45.0 (CH2, C2′ or C6′), 46.5 (CH, C4′), 74.6 (C, C5), 82.6 (C, C3), 126.94 (CH, C9 or C10), 126.96 (CH, C10 or C9), 128.38 (CH, C8 or C11), 128.43 (CH, C11 or C8), 144.70 (C, C7a or C11a), 144.75 (C, C11a or C7a), 156.6 (CO, NHCONH), 168.9 (C, COCH3).
4.1.1.7. 1-(1-Acetylpiperidin-4-yl)-3-(1,5,6,7-tetrahydro-1,5:3,7-dimethanobenzo[e]oxonin-3(2H)-yl)urea (18)
To a solution of 1,5,6,7-tetrahydro-1,5:3,7-dimethanobenzo[e]oxonin-3(2H)-amine hydrochloride (300 mg, 1.19 mmol) in DCM (6.5 mL) and saturated aqueous NaHCO3 solution (6.3 mL), triphosgene (131 mg, 0.44 mmol) was added. The biphasic mixture was stirred at room temperature for 30 min and then the two phases were separated and the organic one was washed with brine (5 mL), dried over anh. Na2SO4, filtered, and evaporated under vacuum to obtain 1–2 mL of a solution of isocyanate in DCM. To this solution was added 1-(4-aminopiperidin-1-yl)ethan-1-one (203 mg, 1.43 mmol). The mixture was stirred overnight at room temperature and the solvent was evaporated under vacuum. Column chromatography (SiO2, DCM/methanol mixtures) gave urea 18 as a white solid (90 mg, 20% yield). The analytical sample was obtained by a crystallization from a hot ethyl acetate/mixture, mp 120–121 °C. IR (ATR): 3340, 2921, 1856, 1730, 1632, 1552, 1493, 1453, 1356, 1327, 1299, 1274, 1244, 1204, 1122, 1088, 1047, 1025, 993, 947, 970, 907, 801, 760, 729, 643 cm–1. 1H NMR (400 MHz, CDCl3): δ 1.33–1.45 (complex signal, 2 H, 3′-Hax, 5′-Hax), 1.68–1.84 [complex signal, 4 H, 2(12)-Hax, 6(13)-Hax], 1.97 (m, 1 H, 3′-Heq or 5′-Heq), 2.05–2.13 (complex signal, 4 H, 5′-Heq or 3′-Heq, COCH3), 2.21 [m, 2 H, 6(13)-Heq], 2.40 [m, 2 H, 2(12)-Heq], 2.87 (ddd, J = 11.2 Hz, J′ = 3.2 Hz, 1 H, 2′-Hax or 6′ H-ax), 3.13–3.25 [complex signal, 3 H, 6′-Hax or 2′-Hax, 1(7)-H], 3.74 (dm, J = 13.6 Hz, 1 H, 6′-Heq or 2′-Heq), 3.90 (m, 1 H, 4′-H), 4.40 (dm, J = 13.2 Hz, 2′-Heq or 6′-Heq), 4.52 (t, J = 5.6 Hz, 1 H, 5-H), 4.78 (s, 1 H, 3-NH), 6.14 (d, J = 7.6 Hz, 1 H, 4′-NH), 7.08–7.16 [complex signal, 4 H, 8-H, 9-H, 10-H, 11-H]. 13C NMR (100.5 MHz, CDCl3): δ 21.4 (CH3, COCH3), 32.2 (CH2, C3′ or C5′), 32.5 [CH2, C6(13)], 33.2 (CH2, C5′ or C3′), 38.0 (CH2, C2 or C12), 38.3 (CH2, C12 or C2), 38.65 (CH, C7 or C1), 38.69 (CH, C1 or C7), 40.4 (CH2, C2′ or C6′), 45.2 (CH2, C6′ or C2′), 46.7 (CH, C4′), 71.7 (CH, C5), 80.9 (C, C3), 126.91 (CH, C9 or C10), 126.93 (CH, C10 or C9), 128.47 (CH, C8 or C11), 128.52 (CH, C11 or C8), 145.0 (C, C7a or C11a), 145.1 (C11a or C7a), 156.5 (CO, NHCONH), 168.9 (CO, COCH3). HRMS: Calcd for [C25H31ClFN3O2+H]+; 460.2162; found, 460.2165.
4.1.1.8. 4-[((1r,4r)-4-(3-(9-Methyl-5,6,8,9,10,11-Hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)ureido)cyclohexyl)oxy]benzoic Acid (20)
To a solution of 9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (200 mg, 0.76 mmol) in DCM (3.5 mL) and saturated aqueous NaHCO3 solution (2.2 mL) was added triphosgene (113 mg, 0.38 mmol). The biphasic mixture was stirred at room temperature for 30 min and then the two phases were separated and the organic layer was washed with brine (5 mL), dried over anh. Na2SO4, filtered, and evaporated under vacuum to obtain 1–2 mL of a solution of isocyanate in DCM. To this solution were added 4-[((1r,4r)-4-aminocyclohexyl)oxy]benzoic acid hydrochloride (206 mg, 0.76 mg), Et3N (153 mg, 1.51 mmol), and DMF (5 mL). The mixture was stirred overnight at room temperature. The resulting suspension was evaporated, and the residue was suspended in DCM (20 mL) and washed with 2N HCl solution (2 × 10 mL). The resulting organic suspension was filtered, and the filtrate was dried over anh. Na2SO4, filtered, and concentrated under vacuum to give a white gum. Crystallization from hot EtOAc provided benzoic acid 20 as a white solid (55 mg, 16% yield), mp 182–183 °C. IR (NaCl disk): 3335, 2921, 2855, 1692, 1681, 1642, 1632, 1602, 1564, 1537, 1504, 1494, 1469, 1453, 1419, 1360, 1307, 1248, 1163, 1122, 1096, 1969 cm–1. 1H NMR (400 MHz, MeOD): δ 0.91 (s, 3 H, C9–CH3), 1.31 [m, 2 H, 3′(5′)-Hax], 1.47 [broad d, J = 13.2 Hz, 2 H, 10″(13″)-Hax], 1.53 [m, 2 H, 2′(6′)-Hax], 1.66 [dd, J = 12.8 Hz, J′ = 6.4 Hz, 2 H, 10’’(13″)-Heq], 1.70 (s, 2 H, 8″-H), 1.96 [m, 2 H, 3′(5′)-Heq], 2.00–2.11 [complex signal, 6 H, 2′(6′)-Heq, 6″(12″)-H2], 3.05 [t, J = 5.6 Hz, 2 H, 5″(11″)-H], 3.48 (m, 1 H, 4′-H), 4.38 (m, 1 H, 1′-H), 6.95 [m, 2 H, 3(5)-H], 7.02–7.03 [complex signal, 4 H, 1’’(2′)-H, 3″(4″)-H], 7.93 [m, 2 H, 2(6)-H]. 13C NMR (100.5 MHz, MeOD): δ 31.2 [CH2, C2′(6′)], 31.7 [CH2, C3′(5′)], 32.9 (CH3, C9″-CH3), 34.5 (C, C9″), 41.0 [CH2, C6″(12″)], 42.6 [CH, C5″(11″)], 42.6 [CH2, C10″(13″) or CH, C5″(11″)], 48.7 (CH, C4′), 49.3 (CH2, C8″), 54.1 (C, C7″), 76.0 (CH, C1′), 116.1 [CH, C3(5)], 123.8 (C, C1), 127.3 [CH, C2″(3″)], 128.9 [CH, C1″(4″)], 132.9 [CH, C2(6)], 147.7 [C, C4a″(11a″)], 159.5 (C, NHCONH), 163.3 (C, C4), 169.8 (C, CO2H).
4.1.1.9. 4-[((1r,4r)-4-(3-(5-Methyl-1,5,6,7-tetrahydro-1,5:3,7-Dimethanobenzo[e]oxonin-3(2H)-yl)ureido)cyclohexyl)oxy]benzoic Acid (21)
To a solution of 5-methyl-1,5,6,7-tetrahydro-1,5:3,7-dimethanobenzo[e]oxonin-3(2H)-amine hydrochloride (200 mg, 0.75 mmol) in DCM (3.5 mL) and saturated aqueous NaHCO3 solution (2.2 mL) was added triphosgene (113 mg, 0.38 mmol). The biphasic mixture was stirred at room temperature for 30 min and then the two phases were separated and the organic layer was washed with brine (5 mL), dried over anh. Na2SO4, filtered, and evaporated under vacuum to obtain 1–2 mL of a solution of isocyanate in DCM. To this solution were added 4-[((1r,4r)-4-aminocyclohexyl)oxy]benzoic acid hydrochloride (206 mg, 0.76 mg), Et3N (153 mg, 1.52 mmol), and DMF (5 mL). The mixture was stirred overnight at room temperature. The resulting suspension was evaporated to obtain a white solid, which was suspended in DCM (20 mL) and washed with 2N HCl solution (2 × 10 mL). The resulting organic suspension was filtered to afford benzoic acid 21 as a white solid (200 mg, 54% yield), mp 220–222 °C. IR (NaCl disk): 3352, 2626, 1678, 1601, 1558, 1506, 1454, 1373, 1343, 1312, 1288, 1247, 1221, 1161, 1104, 1029, 997, 953, 776 cm–1. 1H NMR (400 MHz, DMSO): δ 1.14 (s, 3 H, C5–CH3), 1.30 [q, J = 11.6 Hz, 2 H, 3′(5′)-Hax], 1.41–1.54 (complex signal, 4 H, 2′(6′)-Hax, 6″(13″)-Hax], 1.76–1.87 [complex signal, 6 H, 3′(5′)-Heq, 2″(12″)-Hax, 6″(13″)-Heq], 2.01 [d, J = 9.2 Hz, 2 H, 2′(6′)-Heq], 2.19 [dd, J = 6 Hz, J′ = 12.8 Hz, 2 H, 2″(12″)-Heq], 3.12 [t, J = 6.0 Hz, 1″(7″)-H], 3.41 (m, 1 H, 4′-H), 4.45 (m, 1 H, 1′-H), 5.97 (d, J = 7.6 Hz, 1 H, 4′-NH), 6.11 (s, 1 H, 3″-NH), 7.01 [d, J = 8.4 Hz, 2 H, 3(5)-H], 7.08–7.14 [complex signal, 4 H, 8″(11″)-H, 9″(10″)-H], 7.85 [d, J = 8.4 Hz, 2 H, 2(6)-H], 12.55 (broad s, 1 H, COOH). 13C NMR (100.5 MHz, DMSO): δ 29.5 [CH2, C2′(6′)], 30.1 [CH2, C3′(5′)], 31.2 (CH3, C5–CH3), 37.5 [CH2, C2″(12″)], 38.3 [2 signals, CH2, C6″(13″), and CH, C1″(7″)], 46.6 (CH, C4′), 73.0 (C, C5″), 74.2 (CH, C1′), 82.2 (C, C3″), 115.1 [CH, C3(5)], 122.6 (C, C1), 126.4 [CH, C9″(10″)], 128.1 [CH, C8″(11″)], 131.4 [CH, C2(6)], 145.6 (C, C7a″(11a″)], 155.8 (C, NHCONH), 161.1 (C, C4), 167.0 (C, CO2H). HRMS calcd for [C29H35N2O5+H]+; 491.254, found; 491.254.
4.1.1.10. 4-[((1r,4r)-4-(3-(9-Chloro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)ureido)cyclohexyl)oxy]benzoic Acid (22)
To a solution of 9-chloro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (180 mg, 0.63 mmol) in DCM (3 mL) and saturated aqueous NaHCO3 solution (2 mL), triphosgene (69 mg, 0.23 mmol) was added. The biphasic mixture was stirred at room temperature for 30 min and then the two phases were separated and the organic one was washed with brine (3 mL), dried over anh. Na2SO4, filtered, and evaporated under vacuum to obtain 1–2 mL of a solution of isocyanate in DCM. To this solution were added DMF (4 mL), 4-(((1r,4r)-4-aminocyclohexyl)oxy)benzoic acid hydrochloride (171 mg, 0.63 mmol), and Et3N (127 mg, 1.26 mmol). The mixture was stirred overnight at room temperature and the solvent was then evaporated. The residue was dissolved in DCM (5 mL) and washed with 2N HCl (3 mL). The organic phase was dried over anh. Na2SO4, filtered, and evaporated under vacuum to obtain benzoic acid 22 (217 mg, 67% yield) as a yellow residue. The analytical sample was obtained by crystallization from hot ethyl acetate/pentane mixtures, mp 201–202 °C. IR (ATR): 3355, 3299, 2932, 2856, 1697, 1682, 1631, 1605, 1555, 1498, 1469, 1452, 1428, 1406, 1373, 1357, 1322, 1301, 1253, 1163, 1100, 1077, 1041, 1027, 1013, 977, 946, 905, 844, 804, 772, 753, 695, 643, 634, 608 cm–1. 1H NMR (400 MHz, MeOD): δ 1.32 [m, 2 H, 3′(5′)-Hax], 1.54 [m, 2 H, 2′(6′)-Hax], 1.93–2.02 [complex signal, 4 H, 3′(5′)-Heq, 6″(12″)-Hax], 2.03–2.15 [complex signal, 4 H, 2′(6′)-Heq, 10″(13″)-Hax], 2.15–2.24 [m, 2 H, 6″(12″)-Heq], 2.35–2.41 [m, 2 H, 10″(13″)-Heq], 2.43 (s, 2 H, 8″-H), 3.17 [t, J = 6.0 Hz, 2 H, 5″(11″)-H], 3.49 (m, 1 H, 4′-H), 4.37 (m, 1 H, 1′-H), 6.95 [d, J = 8.6 Hz, 2 H, 3(5)-H], 7.04–7.12 [complex signal, 4 H, 1″(4″)-H, 2″(3″)-H], 7.94 [d, J = 8.6 Hz, 2 H, 2(6)-H]. 13C NMR (100.5 MHz, MeOD): δ 31.1 [CH2, C2′(6′)], 31.6 [CH2, C3′(5′)], 40.0 [CH2, C6″(12″)], 42.7 [CH, C5″(11″)], 46.0 [CH2, C10″(13″)], 48.7 (CH, C4″), 52.1 (CH, C8″), 56.3 (C, C7″), 70.5 (C, C9″), 76.0 (CH, C1′), 116.1 [CH, C5(3)], 123.9 (C, C1), 127.9 [CH, C2″(3″)], 129.1 [CH, C1″(4″)], 132.9 [CH, C2(6)], 146.3 [C, C4a″(11a″)], 159.2 (C, NHCONH), 163.2 (C, C4), the signal from CO2H was not observed. HRMS: Calcd for [C29H33ClN2O4–H]−; 507.2056, found; 507.2057.
4.1.1.11. 4-[((1r,4r)-4-(3-(9-Fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)ureido)cyclohexyl)oxy]benzoic Acid (23)
To a solution of 9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (180 mg, 0.67 mmol) in DCM (3 mL) and saturated aqueous NaHCO3 solution (2 mL), triphosgene (74 mg, 0.25 mmol) was added. The biphasic mixture was stirred at room temperature for 30 min and then the two phases were separated and the organic one was washed with brine (3 mL), dried over anh. Na2SO4, filtered, and evaporated under vacuum to obtain 1–2 mL of a solution of isocyanate in DCM. To this solution were added DMF (4 mL), 4-[((1r,4r)-4-aminocyclohexyl)oxy]benzoic acid hydrochloride (182 mg, 0.67 mmol), and Et3N (136 mg, 1.34 mmol). The mixture was stirred overnight at room temperature and the solvent was then evaporated. The residue was dissolved in DCM (5 mL) and washed with 2N HCl (3 mL). The organic phase was dried over anh. Na2SO4, filtered, and evaporated under vacuum to obtain benzoic acid 23 (240 mg, 72% yield) as a yellow residue. The analytical sample was obtained by crystallization from hot ethyl acetate/pentane mixtures, mp 253–254 °C. IR (ATR): 3325, 2929, 2859, 1682, 1629, 1606, 1558, 1511, 1424, 1359, 1317, 1282, 1251, 1221, 1165, 1104, 1090, 1003, 938, 851, 772, 697, 642 cm–1. 1H NMR (400 MHz, MeOD): δ 1.31 [dq, J = 3.2 Hz, J′ = 13.2 Hz, 2 H, 3′(5′)-Hax], 1.54 [dq, J = 3.2 Hz, J′ = 12.8 Hz, 2 H, 2′(6′)-Hax], 1.81 [d, J = 12 Hz, 2 H, 10″(13″)-Hax)], 1.93–2.02 [complex signal, 4 H, 3′(5′)-Heq, 6″(12″)-Hax], 2.07–2.19 [complex signal, 8 H, 2′(6′)-Heq, 4-H, 6″(12″)-Heq, 10″(13″)-Heq], 3.23 [t, 2 H, J = 7.2 Hz, 5″(11″)-H], 3.49 (m, 1 H, 4′-H), 4.37 (m, 1 H, 1′-H), 6.95 [d, J = 8.8 Hz, 2 H, 3(5)-H], 7.09 [broad s, 4 H, 1″(4″)-H, 2″(3″)-H], 7.94 [d, J = 8.8 Hz, 2 H, 2(6)-H]. 13C NMR (100.5 MHz, MeOD): δ 31.1 [CH2, C2′(6′)], 31.6 [CH2, C3′(5′)], 40.4 [CH2, d, 4JC-F = 1.9 Hz, C6″(12″)], 41.1 [CH, d, 3JC-F = 13.3 Hz, C5″(11″)], 41.4 [CH2, d, 2JC-F = 20 Hz, C10″(13″)], 47.9 (CH2, d, 2JC-F = 17.9, C8″), 48.7 (CH, C4′), 57.6 (C, d, 3JC-F = 11.2 Hz, C7″), 76.0 (CH, C1′), 95.4 (C, d, 1JC-F = 177 Hz, C9″), 116.1 [CH, C3(5)], 123.9 (C, C1), 127.9 [CH, C2″(3″)], 129.1 [CH, C1″(C4″)], 132.8 [CH, C2(6)], 146.4 [C, C4a″(11a″)], 159.2 (C, NHCONH), 163.2 (C, C4), 169.8 (C, CO2H). HRMS: calcd for [C29H33FN2O4–H]−; 491.2352; found, 491.2334.
4.1.2. Determination of IC50 sEHIs in Human, Murine, and Rat Purified sEH
IC50 is the concentration of a compound that reduces the sEH activity by 50%. The IC50 values reported herein were determined using a fluorescent-based assay (CMNPC as substrate).16 The fluorescent assay was used with purified recombinant human, mouse, or rat sEH proteins. The enzymes were incubated at 30 °C with the inhibitors ([I]final = 0.4–100,000 nM) for 5 min in 100 mM sodium phosphate buffer (200 μL, pH 7.4) containing 0.1 mg/mL of BSA and 1% of DMSO. The substrate (CMNPC) was then added ([S]final = 5 μM). Activity was assessed by measuring the appearance of the fluorescent 6-methoxynaphthaldehyde product (λex = 330 nm, λem = 465 nm) every 30 s for 10 min at 30 °C on a SpectraMax M2 (Molecular Devices). Results were obtained by regression analysis from a linear region of the curve. All measurements were performed in triplicate and the mean is reported. t-TUCB, a classic sEHI, was run in parallel and the obtained IC50s were corroborated with reported literature values37 to validate the experimental results.
4.2. In silico Study
4.2.1. MD Simulation Details
The parameters for t-AUCB, 20, 22, and 23 for the MD simulations were generated within the ANTECHAMBER module of AMBER 1838 using the general AMBER force field (GAFF),39 with partial charges set to fit the electrostatic potential generated at the HF/6-31G(d) level by the RESP model.40 The charges were calculated according to the Merz–Singh–Kollman scheme41,42 using Gaussian 09.43
MD simulations of sEH were carried out using PDB 5AM3 (crystallized with t-AUCB)27 as a starting point. For the MD simulations in the apo state, the t-AUCB was removed from the active site. The benzohomoadamanatane derivatives corresponding to 20, 22, and 23 were manually prepared using the t-AUCB structure as a starting point. Molecular docking calculations using the standard parameters of the SwissDock web server were carried out to assess the preferred orientation of 20, 22, and 23.44,45 The coordinates of t-AUCB in PDB 5AM3 were a reference for placing compounds 20, 22, and 23 in molecular docking calculations. From these coordinates, conventional MD simulations were used to explore the conformational plasticity of sEH in the apo state and in the presence of t-AUCB, 20, 22, and 23 bound in the active site. All simulations were performed using the AMBER ff14SB force field.46 Amino acid protonation states were predicted using the H++ server (http://biophysics.cs.vt.edu/H++). The MD simulations have been carried with the following protonation of histidine residues: HIE146, HIE239, HIP251, HID265, HIP334, HIE420, HIE506, HIE513, HIE518, and HIP524.
Each system was immersed in a pre-equilibrated truncated octahedral box of water molecules with an internal offset distance of 10 Å. All systems were neutralized with explicit counterions (Na+ or Cl–). A two-stage geometry optimization approach was performed. First, a short minimization of the positions of water molecules with positional restraints on the solute by a harmonic potential with a force constant of 500 kcal mol–1 Å–2 was done. The second stage was an unrestrained minimization of all the atoms in the simulation cell. Then, the systems were gently heated in six 50 ps steps, increasing the temperature by 50 K each step (0–300 K) under constant-volume, periodic-boundary conditions, and the particle-mesh Ewald approach47 to introduce long-range electrostatic effects. For these steps, a 10 Å cutoff was applied to Lennard-Jones and electrostatic interactions. Bonds involving hydrogen were constrained with the SHAKE algorithm.48 Harmonic restraints of 10 kcal mol–1 were applied to the solute, and the Langevin equilibration scheme was used to control and equalize the temperature.49 The time step was kept at 2 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Each system was then equilibrated for 2 ns with a 2 fs timestep at a constant pressure of 1 atm (NPT ensemble). Finally, conventional MD trajectories at a constant volume and temperature (300 K) were collected. In total, there were three replicas of 250 ns MD simulations for sEH in the apo state and in the presence of t-AUCB, 20, 22, and 23, gathering a total of 3.75 μs of MD simulation time. Each MD simulation was clusterized based on active site residues, and the structures corresponding to the most populated clusters were used in the non-covalent interactions analysis. We monitored the presence of water molecules using the watershell function of the cpptraj MD analysis program.50
4.2.2. Microsomal Stability
The human, rat, and mice recombinant microsomes employed were purchased from Tebu–Xenotech. The compound was incubated at 37 °C with the microsomes in a 50 mM phosphate buffer (pH = 7.4) containing 3 mM MgCl2, 1 mM NADP, 10 mM glucose-6-phosphate, and 1 U/mL glucose-6-phosphate-dehydrogenase. Samples (75 μL) were taken from each well at 0, 10, 20, 40, and 60 min and transferred to a plate containing 4 °C 75 μL of acetonitrile and 30 μL of 0.5% formic acid in water were added for improving the chromatographic conditions. The plate was centrifuged (46,000g, 30 min) and supernatants were taken and analyzed by an ultraperformance liquid chromatograph–tandem mass spectrometer (Xevo-TQD, Waters) by employing a BEH C18 column and an isocratic gradient of 0.1% formic acid in water: 0.1% formic acid acetonitrile (60:40). The metabolic stability of the compounds was calculated from the logarithm of the remaining compounds at each of the time points studied.
4.2.3. Solubility
A 10 mM stock solution of the compound was serially diluted in 100% DMSO and 1 μL of this solution was added to a 384-well UV-transparent plate (Greiner) containing 99 μL of PBS. The plate was incubated at 37 °C for 2 h and the light scattering was measured in a Nephelostar Plus reader (BMG LABTECH). The data were fitted to a segmented linear regression for measuring the compound solubility.
4.2.4. Permeability
The Caco-2 cells were cultured to confluency, trypsinized, and seeded onto a 96-filter transwell insert (Corning) at a density of ∼10,000 cells/well in DMEM cell culture medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and 1% penicillin/streptomycin. Confluent Caco-2 cells were subcultured at passages 58–62 and grown in a humidified atmosphere of 5% CO2 at 37 °C. Following an overnight attachment period (24 h after seeding), the cell medium was replaced with fresh medium in both the apical and basolateral compartments every other day. The cell monolayers were used for transport studies 21 days post seeding. The monolayer integrity was checked by measuring the transepithelial electrical resistance (TEER), obtaining values ≥500 Ω/cm.2 On the day of the study, after the TEER measurement, the medium was removed and the cells were washed twice with pre-warmed (37 °C) Hank’s balanced salt solution (HBSS) buffer to remove traces of medium. Stock solutions were made in dimethyl sulfoxide (DMSO) and further diluted in HBSS (final DMSO concentration 1%). Each compound and reference compounds (Colchicine, E3S) were all tested at a final concentration of 10 μM. For A → B directional transport, the donor working solution was added to the apical (A) compartment and the transport media as receiver working solution was added to the basolateral (B) compartment. For B → A directional transport, the donor working was added to the basolateral (B) compartment and transport media as receiver working solution was added to the apical (A) compartment. The cells were incubated at 37 °C for 2 h with gentle stirring.
At the end of the incubation, samples were taken from both donor and receiver compartments and transferred into 384-well plates and analyzed by ultraperformance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS). The detection was performed using an ACQUITY UPLC/Xevo TQD System. After the assay, Lucifer yellow was used to further validate the cell monolayer integrity, cells were incubated with LY 10 μM in HBSS for 1 h at 37 °C, obtaining permeability (Papp) values for LY of ≤10 nm/s, confirming the well-established Caco-2 monolayer.
4.2.5. Cytotoxicity in SH-SY5Y Cells
Cytotoxicity was evaluated in the human neuroblastoma SH-SY5Y cell line (ATCC number: CRL-2266). Cells were cultured in minimum essential medium/Ham’s-F12 (1:1, v/v) medium, supplemented with non-essential amino acids, 10% fetal bovine serum, 1 mM glutamine, and 50 μg/mL gentamycin (all reagents from Gibco, Invitrogen). For experiments, the cells were seeded at 3 × 105 cells/mL (100 μL/well) in 96-well plates (Nunc). After 24 h, the testing compounds were added concentrated to triplicate wells to obtain the final different concentrations up to 100 μM. Compounds were incubated for further 24 h. At termination, cytotoxicity was analyzed by the propidium iodide (PI) fluorescence stain assay. All compounds were tested in three independent experiments using different cell passages. The PI assay measures cell death. PI enters into the cells with damaged membranes and greatly increases the fluorescence by binding to DNA. PI reagent (Molecular Probes) at the final concentration of 7.5 μg/mL was added to the cells and incubated for 1 h. The resulting fluorescence was measured by a Gemini XPS Microplate reader (Millipore) at 530 nm excitation and 645 nm emission. The percentage of cell death induced by the treatments was calculated from the fluorescence of treated cells (Ft) relative to that of control cells (Fmin) and cells incubated with Triton X100 (Fmax) as the 0 and 100% cell death, respectively [% = ((Ft – Fmin)/(Fmax – Fmin)) × 100].
4.2.6. Inhibition of hLOX-5
AA and 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) were obtained from Sigma. Human recombinant LOX-5 was purchased from Cayman Chemical. For the determination of hLOX-5 activity, the method described by Pufahl et al. was followed.51 The assay solution consisted of 50 mM Tris (pH 7.5), 2 mM EDTA, 2 mM CaCl2, 3 μM AA, 10 μM ATP, 10 μM H2DCFDA, and 100 mU/well hLOX-5. For the enzyme inhibition studies, the compounds to be tested were added to the assay solution prior to AA and ATP and were preincubated for a period of 10 min at room temperature, after which AA and ATP were added. The enzymatic reaction was carried out for 20 min and terminated by the addition of 40 μL of acetonitrile. The fluorescence measurement, 485 nm excitation and 520 nm emission, was performed on a FLUOstar OPTIMA (BMG LABTECH, Offenburg, Germany.). The IC50 is defined as the concentration of compound that inhibits enzymatic activity by 50% over the untreated enzyme control.
4.2.7. Cytochrome P450 Inhibition Assay
The objective of this study was to screen the inhibition potential of the compounds using recombinant human cytochrome P450 enzymes CYP3A4 (BFC and DBF substrates) and probe substrates with fluorescent detection. Incubations were conducted in a 200 μL volume in 96-well microtiter plates (COSTAR 3915). The addition of the mixture buffer-cofactor (KH2PO4 buffer, 1.3 mM NADP, 3.3 mM MgCl2, 0.4 U/mL glucose-6-phosphate dehydrogenase), control supersomes, the Standard inhibitor Ketoconazole (Sigma K1003), and previously diluted compounds to plates was carried out by a liquid handling station (Zephyr Caliper). The plate was then preincubated at 37 °C for 5 min in a 100 μL volume, and reaction was initiated by the addition of a prewarmed enzyme/substrate (E/S) mix. The E/S mix contained buffer (KH2PO4), enzyme (CYP), substrate 7-benzyloxytrifluoromethyl coumarin (7-BFC), and dibenzylfluorescein (DBF) in a reaction volume of 200 μL. Reactions were terminated after various times depending on the substrate by addition of STOP solution (ACN/TrisHCl 0.5 M 80:20 (BFC) or 2 N NaOH for CYP3A4 (DBF). Fluorescence per well was measured using a fluorescence plate reader (Tecan M1000 pro) and percentage of inhibition was calculated.
4.2.8. hERG Inhibition Assay
The assay was carried out at a CHO cell line transfected with the hERG potassium channel. 72 h before the assay, 2500 cells were seeded on a 384-well black plate (Greiner 781091). The cell line was maintained at 37 °C in a 5% CO2 atmosphere for 24 h and at 30 °C in a 5% CO2 atmosphere for 48 h plus. hERG activity was measured by using the FluxorTM Potassium Ion Chanel Assay Kit (Thermo Fisher F10016). Medium was replaced for 20 μL of loading buffer and the cells were incubated for 60 min at RT, protected from direct light. After incubation, the loading buffer was replaced for the assay buffer and the compounds were incubated for 30 min at RT. 5 μL of a stimulus buffer was added to each well and the fluorescence was read (λex = 490 nm, λem = 525 nm) using an imaging plate reader system (FDSS7000EX, Hamamatsu) every second after the establishment of a baseline line.
4.3. Pharmacokinetic Study
24 male C57BL/6 mice (21 g approx.) were supplied by Envigo (Barcelona, Spain) (Ref 15131). During the experimental procedure, animals were identified with a permanent marker (tail code numbers). Plasma samples were obtained at 0, 0.25, 0.5, 1, 2, 4, 6, and 24 h post-dosing. Upon arrival, animals were housed in groups of 3 animals/cage in polycarbonate maintenance cages (type IIL; 365 × 207 × 140 mm, with a surface area of 530 cm2) with absorbent bedding (Lignocel, JRS). Animals were kept in an environmentally controlled room (ventilation of 10–15 air changes per hour, temperature of 22 ± 3 °C, and humidity of 35–70%) on a 12 h light/dark cycle. They underwent a period of at least 5 days of observation and acclimatization between the date of arrival and the start of the procedure. During this period, the animals were observed to check their general health state. A maintenance diet was supplied by Harlan Interfauna Ibérica S.L. (2014 Harlan Teklad Global Diets) and was provided to the animals ad libitum. Diet was analyzed by the manufacturer to detect possible contaminants. Tap water was supplied by CASSA (Servei d’Aigües de Sabadell) and was provided to the animals by bottles ad libitum. The animals were maintained in accordance with the European Directive for the Protection of Vertebrate Animals Used for Experimental and other Scientific Purposes (86/609/EU). Decree 214/1997 of 30th July. Ministry of Agriculture, Livestock, and Fishing of the autonomous government of Catalonia, Spain. Royal Decree 53/2013 of 1st February (Spain) Animal care including environmental and housing conditions conformed to the applicable standard operating procedures regarding laboratory animals of Draconis Pharma S.L. All the experimental procedures were approved by the Animal Experimentation Ethical Committee of Universitat Autònoma de Barcelona (procedure number: 3718) and by the Animal Experimentation Commission of the Generalitat de Catalunya (Catalan Government) (DAAM:9590). Formulations were prepared on the day of the study. Vehicle was 10% of 2-hydroxypropyl-β-cyclodextrin, (CAS 128446-35-5) Sigma-Aldrich (Ref.332607-25G). 21 mice were intraperitoneally administered with a single dose of 3 mg/kg of 22. The volume of administration was 10 mL/kg. Animals were weighed before each administration to adjust the required volume. Blood samples were collected at different times post administration: 0, 0.25, 0.5, 1, 2, 4, 6, and 24 h. Three mice were not administered and referred as t = 0. Blood samples (0.5–0.8 mL) were collected from anesthetized animals with isoflurane by cava vein puncture in tubes containing K2-EDTA 5%. Blood samples were centrifuged at 10,000 rpm for 5 min to obtain plasma that was stored at −20 °C until analysis. Analytical measurements were performed by LC–MS/MS. Pharmacokinetics parameters were calculated with Phoenix 64 (WinNonlin).
4.4. In vivo Efficacy Study
Forty-one male C57BL/6 mice (8-week-old; approximately 24 g) were supplied by Envigo (Barcelona, Spain) (Ref 16512). During the experimental procedure, the animals were identified with a permanent marker (tail code numbers). Upon arrival, the animals were housed in groups of 8–9 animals/cage in polysulfone maintenance cages (480 × 265 × 210 mm, with a surface area of 940 cm2) with wire tops and wood chip bedding. The animals were kept in an environmentally controlled room (ventilation, temperature 22 ± 2 °C, and humidity 35–65%) on a 12 h light/dark cycle. They underwent a period of 7 days of acclimatization between the date of arrival and the start of the procedure. During this period, the animals were observed to check their general health state. The maintenance diet was supplied by Harlan Interfauna Ibérica S.L. (2018 Harlan Teklad Global Diets). Diet was provided to the animals ad libitum, but they were fasted overnight before the first cerulein injection, and food was replaced after the last cerulein injection. Tap water was supplied by CASSA (Servei d’Aigües de Sabadell) ad libitum. The animals were maintained in accordance with the European Directive for the Protection of Vertebrate Animals Used for Experimental and other Scientific Purposes (86/609/EU). Decree 214/1997 of 30th July. Ministry of agriculture, livestock and fishing of the Autonomous Government of Catalonia, Spain. Royal Decree 53/2013 of 1st February (Spain). All the experimental procedures were approved by the Ethical Committee on human and animal experimentation (CEEAH) of Universitat Autònoma de Barcelona (UAB) (procedure number: 4107) and by the Animal Experimentation Commission of the Generalitat de Catalunya (Catalan Government) (DAAM: 10146). The test item was dissolved in vehicle 10% 2-hydroxypropyl-β-cyclodextrin (CAS 128446-35-5) Sigma-Aldrich (Ref 332607). The vehicle was prepared the day before and kept at 4 °C. Pancreatitis induction: Mice (n = 41) were weighed, identified by a distinct number at the base of the tail, and fasted overnight. Cerulein (cerulein and cerulein + 22 groups) (50 μg/kg, prepared in 0.9% NaCl) or vehicle (0.9% NaCl) (control group) were intraperitoneally injected (V = 5 mL/kg) 12 consecutive times, at 1 h intervals (h = 0–11). Food was replaced after the last injection. A satellite experiment was designed where animals (n = 3) were distributed in control, cerulein, and cerulein + 22-treated groups. Pancreatitis was induced by seven injections of cerulein (or vehicle in the control group) at 1 h intervals (h = 0–6). Treatments: the test item was administered intraperitoneally in one injection to 22_03 (0.3 mg/kg) and 22_01 (0.1 mg/kg) groups at 14 h after the first cerulein injection. Animals from the control and cerulein groups received vehicle administration (10% 2-hydroxypropyl-β-cyclodextrin) (V = 10 mL/kg). Extra groups were treated 2 h after the first cerulein injection: 22 (0.3 mg/kg), control, and cerulein groups (10% 2-hydroxypropyl-β-cyclodextrin). Study end: 24 h after the first cerulein injection, animals were weighed and anesthetized with isoflurane. Blood was collected from the vena cava in an Eppendorf containing K2-EDTA and centrifuged at 10,000 rpm for 5 min for plasma collection. Plasma was stored at −80 °C until analysis. The mice were sacrificed by cervical dislocation and the pancreases were dissected and weighed. Pancreases from three animals were frozen in liquid N2 and stored at −80 °C until analysis. Pancreases from five mice were sectioned and one part was placed in 10% formalin and sent to Anapath (Granada, Spain) for histology analysis and the other was immediately placed in RNAse-free eppendorfs, frozen in N2, and stored at −80 °C for gene expression assays.
4.4.1. Histologic Analysis
Pancreatic samples were treated with increasing grade alcohols and two xylol baths and embedded in paraffin. They were subsequently cut using a microtome and processed for staining. For the deparaffinization of the samples, two xylene baths (10 min) and three alcohols were used in decreasing solutions (100%, 90 and 70%) (5 min) and subsequently stained with hematoxylin (5 min) and eosin (5 min). During the dehydration process after staining with eosin, alcohols in increasing solution (70, 96, and 100%) and xylene were used again. Finally, the samples were mounted with DPX.
Histologic scoring of pancreatic sections was performed to grade the extent of pancreatic parenchymal atrophy, vacuolar degeneration of cells, edema, hemorrhage, mononuclear inflammatory cells, mononuclear inflammatory cells, polymorphonuclear inflammatory cells, and necrosis. The assigned scores were the following: 0 (no changes): when no lesions were observed or the observed changes were within normality; 1 (minimal): when changes were few but exceeded those considered normal; 2 (light): lesions were identifiable but with moderate severity; 3 (moderate): important injuries but they can still increase in severity; 4 (very serious): the lesions are very serious and occupy most of the analyzed tissue. The lesions were evaluated in the most affected lobes of all the pancreases. In the case of assessment of atrophy, it was determined based on the percentage of atrophied tissue as: 0 without atrophy; 1: 0–25% of atrophic parenchyma; 2: between 25 and 50%; 3: between 50 and 75% and 4: between 75 and 100%.
Acknowledgments
This work was funded by the Spanish Ministerio de Economía, Industria y Competitividad (grants SAF2017-82771-R to S.V., RTI2018-093999-B-100 to M. V.-C., RTI2018-093955-B-C21 to M.I.R.-F., SAF2016-77703-C2 to M.P. and C.S., PGC2018-102192-B-I00 to S.O. and RTI2018-101032-J-I00 to F.F.), the Spain EIT Health (Proof of concept 2016), the European Regional Development Fund (ERDF), the Xunta de Galicia (ED431G 2019/02 and ED431C 2018/21), the Fundació Bosch i Gimpera, Universitat de Barcelona (F2I grant), the Generalitat de Catalunya (2017 SGR 106, 2017 SGR 124 and 2017 SGR 1707), the European Research Council (ERC-2015-StG-679001-NetMoDEzyme to S.O.), and the European Community (MSCA-IF-2014-EF-661160-MetAccembly to F.F.). S.C. and E.V. acknowledge the PhD fellowships from the Universitat de Barcelona (APIF grant) and the Institute of Biomedicine of the University of Barcelona (IBUB), respectively. Partial support was provided by NIH-NIEHS River Award R35 ES03443, NIH-NIEHS Superfund Program P42 ES004699, NINDS R01 DK107767, and NIDDK R01 DK103616 to B.D.H. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
Abbreviations USED
- AP
acute pancreatitis
- ATR
attenuated total reflectance
- BEH
ethylene bridged hybrid
- BFC
benzyloxytrifluoromethylcoumarin
- CER
cerulein
- COXs
cyclooxygenases
- CYPs
cytochrome P450s
- DBF
dibenzylfluorescein
- DMPK
drug metabolism pharmacokinetics
- EETs
epoxyeicosatrienoic acids
- EtOAc
ethyl acetate
- H&E
hematoxylin and eosin
- hsEH
human soluble epoxide hydrolase
- LHS
left-hand side
- LOXs
lipoxygenases
- ND
not determined
- RHS
right-hand side
- sEH
soluble epoxide hydrolase
- sEHI
soluble epoxide hydrolase inhibitors.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01601.
1H and 13C NMR spectra and elemental analysis data of the new compounds, HPLC trace for compound 22, representation of the active site volume fluctuations, non-covalent interactions between the inhibitors and the active site residues of sEH, analysis of active site water molecules in MD simulations, plots of the dihedral angle that describes the rotation of the benzohomoadamantane and adamantane moieties in the left-hand-side pocket of the sEH active site along the MD simulations, overlay of the X-ray and representative snapshot from the apo state and inhibitor-bound aMD simulations, pharmacokinetic data of compounds 20 and 22, and histologic scoring of pancreatic tissues of mice treated with 22 (PDF).
Molecular formula strings and data (CSV)
Author Contributions
S.V. conceived the idea. S.C. and E.V. synthesized and chemically characterized the compounds. J.P.-D. and M.V-C. designed and carried out the in vivo experiments. C.M. and B.D.H. performed the determination of the IC50 in human, murine, and rat sEH. F.F., C.C.-T., and S.O. performed the MD calculations. M.I.L. and J.M.B. carried out DMPK studies. R.C., C.G.-F., M.P., and C.S. performed cytotoxicity studies. C.P. and M.I.R.-F. performed the hLOX-5 studies. S.C., S.O., C.M., B.D.H., F.F., and S.V. analyzed the data. S.C., C.C.-T., F.F., and S.V. wrote the manuscript with feedback from all the authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): S.C., E.V. and S.V. are inventors of the Universitat de Barcelona patent application on sEH inhibitors WO2019/243414.C.M. and B.D.H. are inventors of the University of California patents on sEH inhibitors licensed to EicOsis.
Notes
S.C., E.V., and S.V. are inventors of the Universitat de Barcelona patent application on sEH inhibitors WO2019/243414. C.M. and B.D.H. are inventors of the University of California patents on sEH inhibitors licensed to EicOsis. None of the other authors has any disclosures to declare.
Supplementary Material
References
- Merirer K.; Steinhilber D.; Proschak E. Inhibitors of the arachidonic acid cascade: interfering with multiple pathways. Basic Clin. Pharmacol. Toxicol. 2014, 114, 83–91. 10.1111/bcpt.12134. [DOI] [PubMed] [Google Scholar]
- Kaspera R.; Totah R. A. Epoxyeicosatrienoic acids: formation, metabolism and potential role in tissue physiology and pathology. Expert Opin. Drug Metab. Toxicol. 2009, 5, 757–771. 10.1517/17425250902932923. [DOI] [PubMed] [Google Scholar]
- Morisseau C.; Hammock B. D. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 37–58. 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris T. R.; Hammock B. D. Soluble epoxide hydrolase: gene structure, expression and deletion. Gene 2013, 526, 61–74. 10.1016/j.gene.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C.-P.; Zhang X.-Y.; Morisseau C.; Hwang S. H.; Zhang Z.-J.; Hammock B. D.; Ma X.-C. Discovery of soluble epoxide hydrolase inhibitors from chemical synthesis and natural products. J. Med. Chem. 2021, 64, 184–215. 10.1021/acs.jmedchem.0c01507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H. C.; Hammock B. D. Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J. Med. Chem. 2012, 55, 1789–1808. 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano Y.; Yamaguchi T.; Tanabe E. Structural insights into binding of inhibitors to soluble epoxide hydrolase gained by fragment screening and X-ray crystallography. Bioorg. Med. Chem. 2014, 22, 2427–2434. 10.1016/j.bmc.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Chen D.; Whitcomb R.; MacIntyre E.; Tran V.; Do Z. N.; Sabry J.; Patel D. V.; Anandan S. K.; Gless R.; Webb H. K. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol. 2012, 52, 319–328. 10.1177/0091270010397049. [DOI] [PubMed] [Google Scholar]
- Lazaar A. L.; Yang L.; Boardley R. L.; Goyal N. S.; Robertson J.; Baldwin S. J.; Newby D. E.; Wilkinson I. B.; Tal-Singer R.; Mayer R. J.; Cheriyan J. Pharmacokinetics, pharmacodynamics ans adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol. 2016, 81, 971–979. 10.1111/bcp.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammock B. D.; McReynolds C. B.; Wagner K.; Buckpitt A.; Cortes-Puch I.; Croston G.; Lee K. S. S.; Yang J.; Schmidt W. K.; Hwang S. H. Movement to the clinic of soluble epoxide hydrolase inhibitor EC5026 as an analgesic for neuropathic pain and for use as a nonaddictive opioid alternative. J. Med. Chem. 2021, 64, 1856. 10.1021/acs.jmedchem.0c01886. [DOI] [PMC free article] [PubMed] [Google Scholar]; in press
- Codony S.; Valverde E.; Leiva R.; Brea J.; Isabel Loza M.; Morisseau C.; Hammock B. D.; Vázquez S. Exploring the size of the lipophilic unit of the soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. 2019, 27, 115078. 10.1016/j.bmc.2019.115078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque M. D.; Camps P.; Torres E.; Valverde E.; Sureda F. X.; López-Querol M.; Camins A.; Prathalingam S. R.; Kelly J. M.; Vázquez S. New oxapolycyclic cage amines with NMDA receptor antagonist and trypanocidal activities. Bioorg. Med. Chem. 2010, 18, 46–57. 10.1016/j.bmc.2009.11.017. [DOI] [PubMed] [Google Scholar]
- Torres E.; Duque M. D.; López-Querol M.; Taylor M. C.; Naesens L.; Ma C.; Pinto L. H.; Sureda F. X.; Kelly J. M.; Vázquez S. Synthesis of benzopolycyclic cage amines: NMDA receptor antagonist, trypanocidal and antiviral activities. Bioorg. Med. Chem. 2012, 20, 942–948. 10.1016/j.bmc.2011.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde E.; Sureda F. X.; Vázquez S. Novel benzopolycliclic amines with NMDA receptor antagonist activity. Bioorg. Med. Chem. 2014, 22, 2678–2683. 10.1016/j.bmc.2014.03.025. [DOI] [PubMed] [Google Scholar]
- Barniol-Xicota M.; Escandell A.; Valverde E.; Julián E.; Torrents E.; Vázquez S. Antibacterial activity of novel benzopolycyclic amines. Bioorg. Med. Chem. 2015, 23, 290–296. 10.1016/j.bmc.2014.11.041. [DOI] [PubMed] [Google Scholar]
- Morisseau C.; Hammock B. D. Measurement of soluble epoxide hydrolase (sEH) activity. Curr. Protoc. Toxicol. 2007, 33, 4.23.1–4.23.18. 10.1002/0471140856.tx0423s33. [DOI] [PubMed] [Google Scholar]
- Burmistrov V.; Morisseau C.; Pitushkin D.; Karlov D.; Fayzullin R. R.; Butov G. M.; Hammock B. D. Adamantyl thioureas as soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 2302–2313. 10.1016/j.bmcl.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C.; Goodrow M. H.; Dowdy D.; Zheng J.; Greene J. F.; Sanborn J. R.; Hammock B. D. Potent urea and carbamate inhibitors of soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 8849–8854. 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codony S.; Pujol E.; Pizarro J.; Feixas F.; Valverde E.; Loza M. I.; Brea J. M.; Saez E.; Oyarzabal J.; Pineda-Lucena A.; Pérez B.; Pérez C.; Rodríguez-Franco M. I.; Leiva R.; Osuna S.; Morisseau C.; Hammock B. D.; Vázquez-Carrera M.; Vázquez S. 2-Oxaadamant-1-yl ureas as soluble epoxide hydrolase inhibitors: in vivo evaluation in a murine model of acute pancreatitis. J. Med. Chem. 2020, 63, 9237–9257. 10.1021/acs.jmedchem.0c00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammock B. D.; Hwang S. H.; Wecksler A. T.; Morisseau C.. Sorafenib derivatives as soluble epoxide hydrolase inhibitors. WO 2012112570 A1, Aug 23, 2012.
- Liu J.-Y.; Tsai H.-J.; Morisseau C.; Lango J.; Hwang S. H.; Watanabe T.; Kim I.-H.; Hammock B. D. In vitro and in vivo metabolism of N-adamantyl substituted urea-based soluble epoxide hydrolase inhibitors. Biochem. Pharmacol. 2015, 98, 718–731. 10.1016/j.bcp.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodek P.; Janscák P.; Anzenbacher P.; Burkhard J.; Janků J.; Vodicka L. Metabolism of diamantane by rat liver microsomal cytochromes P-450. Xenobiotica 1988, 18, 1109–1118. 10.3109/00498258809042233. [DOI] [PubMed] [Google Scholar]
- Burmistrov V.; Morisseau C.; Harris T. R.; Butov G.; Hammock B. D. Effects of adamantane alterations on soluble epoxide hydrolase inhibition potency, physical properties and metabolic stability. Bioorg. Chem. 2018, 76, 510–527. 10.1016/j.bioorg.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S. H.; Tsai H.-J.; Liu J.-Y.; Morisseau C.; Hammock B. D. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J. Med. Chem. 2007, 50, 3825–3840. 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R.; Hovan L.; Tribello G. A.; Cossins B. P.; Estarellas C.; Gervasio F. L. Combining machine learning and enhanced sampling techniques for efficient and accurate calculations of absolute binding free energies. J. Chem. Theory Comput. 2020, 16, 4641–4654. 10.1021/acs.jctc.0c00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Hervás E.; Casadevall G.; Garcia-Borràs M.; Feixas F.; Osuna S. Epoxide hydrolase conformational heterogeneity for the resolution of bulky pharmacologically relevant epoxide substrates. Chem.—Eur J. 2018, 24, 12254–12258. 10.1002/chem.201801068. [DOI] [PubMed] [Google Scholar]
- Öster L.; Tapani S.; Xue Y.; Käck H. Successful generation of structural information for fragment-based drug discovery. Drug Discovery Today 2015, 20, 1104–1111. 10.1016/j.drudis.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; Votapka L.; Sørensen J.; Amaro R. E. POVME 2.0: An enhanced tool for determining pocket shape and volume characteristics. J. Chem. Theory Comput. 2014, 10, 5047–5056. 10.1021/ct500381c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-García J.; Johnson E. R.; Keinan S.; Chaudret R.; Piquemal J.-P.; Beratan D. N.; Yang W. NCIPLOT: a program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. 10.1021/ct100641a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelberg D.; de Oliveira C. A. F.; McCammon J. A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007, 127, 155102. 10.1063/1.2789432. [DOI] [PubMed] [Google Scholar]
- Yadav D.; Lowenfels A. B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. 10.1053/j.gastro.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsmark C. E.; Swaroop Vege S.; Wilcox C. M. Acute pancreatitis. N. Engl. J. Med. 2016, 375, 1972–1981. 10.1056/nejmra1505202. [DOI] [PubMed] [Google Scholar]
- Krishna S. G.; Kamboj A. K.; Hart P. A.; Hinton A.; Conwell D. L. The changing epidemiology of acute pancreatitis hospitalizations: a decade of trends and the impact of chronic pancreatitis. Pancreas 2017, 46, 482–488. 10.1097/mpa.0000000000000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afghani E.; Pandol S. J.; Shimosegawa T.; Sutton R.; Wu B. U.; Vege S. S.; Gorelick F.; Hirota M.; Windsor J.; Lo S. K.; Freeman M. L.; Lerch M. M.; Tsuji Y.; Melmed G. Y.; Wassef W.; Mayerle J. Acute pancreatitis-progress and challenges: a report on an international symposium. Pancreas 2015, 44, 1195–1210. 10.1097/mpa.0000000000000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettaieb A.; Chahed S.; Bachaalany S.; Griffey S.; Hammock B. D.; Haj F. G. Soluble epoxide hydrolase pharmacological inhibition ameliorates experimental acute pancreatitis in mice. Mol. Pharmacol. 2015, 88, 281–290. 10.1124/mol.114.097501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettaieb A.; Chahed S.; Tabet G.; Yang J.; Morisseau C.; Griffey S.; Hammock B. D.; Haj F. G. Effects of soluble epoxide hydrolase deficiency on acute pancreatitis in mice. PLoS One 2014, 9, e113019 10.1371/journal.pone.0113019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K.; Inceoglu B.; Dong H.; Yang J.; Hwang S. H.; Jones P.; Morisseau C.; Hammock B. D. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. Eur. J. Pharmacol. 2013, 700, 93–101. 10.1016/j.ejphar.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. I.; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Gilson M. K.; Gohlke H.; Goetz A. W.; Greene D.; Harris R.; Homeyer N.; Izadi S.; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Mermelstein D. J.; Merz K. M.; Miao Y.; Monard G.; Nguyen C.; Nguyen H.; Omelyan I.; Onufriev A.; Pan F.; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Smith J.; Salomon-Ferrer R.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER 2018; University of California: San Francisco, 2018.
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W.; Kollman P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. 10.1021/j100142a004. [DOI] [Google Scholar]
- Besler B. H.; Merz K. M. Jr.; Kollman P. A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. 10.1002/jcc.540110404. [DOI] [Google Scholar]
- Singh U. C.; Kollman P. A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. 10.1002/jcc.540050204. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision A.02; Gaussian, Inc.: Pittsburgh, PA, 2009.
- Grosdidier A.; Zoete V.; Michielin O. SwissDock a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosdidier A.; Zoete V.; Michielin O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. 10.1002/jcc.21797. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagui C.; Darden T. A. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 155–179. 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Wu X.; Brooks B. R. Self-guided Langevin dynamics simulation method. Chem. Phys. Lett. 2003, 381, 512–518. 10.1016/j.cplett.2003.10.013. [DOI] [Google Scholar]
- Roe D. R.; Cheatham T. E. III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Pufahl R. A.; Kasten T. P.; Hills R.; Gierse J. K.; Reitz B. A.; Weinberg R. A.; Masferrer J. L. Development of a fluorescence-based enzyme assay of human 5-lipoxygenase. Anal. Biochem. 2007, 364, 204–212. 10.1016/j.ab.2007.02.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.