

Abstract
Potassium channels facilitate and regulate physiological processes as diverse as electrical signaling, ion, solute and hormone secretion, fluid homeostasis, hearing, pain sensation, muscular contraction, and the heartbeat. Potassium channels are each formed by either a tetramer or dimer of pore-forming α subunits that co-assemble to create a multimer with a K+-selective pore that in most cases is capable of functioning as a discrete unit to pass K+ ions across the cell membrane. The reality in vivo, however, is that the potassium channel α subunit multimers co-assemble with ancillary subunits to serve specific physiological functions. The ancillary subunits impart specific physiological properties that are often required for a particular activity in vivo; in addition, ancillary subunit interaction often alters the pharmacology of the resultant complex. In this chapter the modes of action of ancillary subunits on K+ channel physiology and pharmacology are described and categorized into various mechanistic classes.
Keywords: K2P, KCNE, KCNQ, Kir, Kv7, Long QT syndrome, voltage-gated potassium channel
1. Introduction
The potassium channels are one of the most numerous and diverse groups of ion channels, reflecting their many roles within specialized cell types in a range of tissues. The diversity is generated by five main mechanisms. First there are a high number of K+ channel pore-forming α subunit isoforms – for example, the voltage-gated potassium (Kv) channel family numbers forty in the human genome. Second, K+ channels are composed of either tetrameric (the majority) or dimeric (the K2P channels) assemblies of α subunits. In turn, in many K+ channel subfamilies, different α subunit isoforms can heteromultimerize to generate channels with properties distinct from homomeric relatives. Third, most K+ channels are composed not just of α subunits, but also of ancillary subunits, that co-assemble with the α subunits to impart often unique physiological and pharmacological properties. Fourth, many α and ancillary subunits exhibit splice variation with consequent altered properties, and fifth, further added functional complexity arises from post-translational modifications that can dynamically alter activity, which can also include the modulatory effects of signaling cascades. This great diversity of molecular identity and composition of K+ channels gives rise as one might expect to a wide functional diversity when it comes to the currents generated.
The potassium ion-selective channels can be divided by sequence and structural delineations into three to five families: the voltage-gated potassium (Kv) channels, the inward rectifier (Kir) channels, the two-pore domain potassium (KCNK or K2P) channels, and two families of calcium-activated potassium (KCa) channels (SK/IK and BK) (Atkinson et al. 1991; Fakler et al. 1995; Goldstein et al. 1996; Hille et al. 1999; Ketchum et al. 1995; Kubo et al. 1993; MacKinnon 2003; Tempel et al. 1988; Tempel et al. 1987). Ca2+-activated K+ channels sometimes are combined with Kv channels into a larger family of 6-transmembrane domain K+ channels.
The Kir channels comprise tetramers of α subunits each containing two transmembrane (TM) domains and a single pore (P) loop; in contrast, mammalian K2P channels form as dimers of 4-TM subunits each with two P-loops. Kv channels form as tetramers of 6-TM α subunits each with a P-loop. SK/IK architecture resembles that of Kv channels, while BK channels contain an extra (7th) TM domain at the N-terminal end. Aside from the extra TM domain of BK channels, the N-terminal end of which is exposed to the extracellular side of the plasma membrane, all K+ channel α subunit N- and C-termini are instead exposed at the intracellular face of the cell membrane.
For the purposes of this chapter, ancillary subunits are defined as proteins for which the principal activity is to regulate specific ion channels. Therefore, other proteins involved in generalized protein trafficking or localization, or post-translational modifications such as kinases and phosphorylases, are generally not included (although specific examples are described where informative).
Rather than a comprehensive list based on the different channel or ancillary subunit classes (which has been described in, e.g., (Abbott 2014, 2015, 2016b, c; Pongs and Schwarz 2010) (Gonzalez-Perez and Lingle 2019)), here the focus is on the different modes of regulation of K+ channel function and pharmacology and, accordingly, this chapter is organized based on the modes of action of various ancillary subunits to give an overview, using examples, of how they regulate channel function and pharmacology.
Effects on channel function are separated into:
obligate, essential for channel activity
not required for channel function but essential for a known physiological role
inhibitory to channel activity.
Effects on channel pharmacology are separated into:
drug sensitization by providing a novel binding site absent on the channel α subunit
drug sensitization by increasing the affinity of a small molecule that also binds to and modulates α subunit-only channels
drug desensitization by decreasing the affinity of a small molecule that binds to and modulates α subunit-only channels.
2. Ancillary subunit modulation of K+ channel function
2.1. Obligate ancillary subunit interactions, essential for channel activity
Perhaps the best known of the obligate ancillary subunits among the K+ channel regulators are the sulfonylurea (SUR) receptors, essential components of a specific class of inward rectifier potassium (Kir) channels. Inwardly rectifying potassium (Kir) channel α subunits exhibit a relatively simple architecture of two TM domains and a P-loop, organized into tetramers to form functional channels (in some cases with the necessity of additional regulatory subunits – see below). Inward rectification of Kir channels, which tends to limit outward versus inward K+ current under experimental conditions across the entire measurable voltage range, is caused by blockade by specific intracellular molecules that limit outward current. The most important of these molecules are Mg2+ ions and polyamines (of which spermine is particularly potent) (Kubo et al. 1993; Nishida and MacKinnon 2002; Ruppersberg 2000).
SURs are TM proteins that co-assemble via their cytoplasmic domains in a 4:4 stoichiometry with Kir6.x α subunits to form KATP channels, which sense cellular metabolic state, coupling this to cellular excitability (Chan et al. 2003). KATP channels are essential for regulation of insulin secretion in pancreatic β-cells, and for modulating cardiac myocyte and neuronal excitability, and vascular smooth muscle tone, among other functions. SURs are a form of ATP binding cassette (ABC) transporter that no longer transports nutrients and other molecules into cells, and instead now serves as a regulatory subunit for KATP channels. While Kir6.x α subunits directly bind ATP (in the absence of Mg2+) to inhibit KATP channels, binding of Mg2+-adenosine nucleotides (ATP or ADP) to SURs activates KATP channels. Thus, KATP channels sense changes in the ATP:ADP ratio, with a decrease in this ratio activating the channel and thereby suppressing excitability (because positive to the K+ equilibrium potential, which is generally around −65 to −80 mV under physiological conditions, K+ diffuses out of the cell when K+ channel pores open – hyperpolarizing the cell). SURs are essential to Kir6 channel function because in their absence, Kir6.1 or 6.2 α subunits fail to reach the cell surface and are therefore nonfunctional. This is because of the RKR motif, an endoplasmic reticulum (ER) retention signal that lies within each of the Kir6 and SUR subunits (Zerangue et al. 1999), which ensures ER retention of the homomeric subunits. Co-expression of the two subunit types permits each to reciprocally mask the RKR motifs of the other, permitting maturation and surface trafficking of the entire KATP channel complex. The Kir6.2 ER retention signal lies within the last 26 residues of the distal C-terminus; accordingly, deletion of this portion of the α subunit permits surface expression and channel activity (Tucker et al. 1997; Zerangue et al. 1999). Thus, SURs are essential modulators in KATP channels by virtue of an ER retention motif that has evolved to prevent the α subunits from reaching the cell surface alone. For in-depth coverage of physiology and pharmacology of KATP channels please see Chapters 10 (Cui et all 2021) and 11 (Li et al. 2021).
2.2. Ancillary subunit interactions not necessary for channel function but essential for a known physiological role
2.2.1. KCNE1 effects on KCNQ1 function
Ancillary subunits bearing a single TM segment are a common theme among various K+ channel types and are typically not required for channel function per se but can be essential for a known physiological role. These often-diminutive subunits can exert profound effects on K+ channel function, from early maturation and trafficking all the way to function at the cell surface and endocytosis for recycling or degradation. The KCNE subunits are a very highly studied class and their roles as examples in the various categories of α subunit-ancillary subunit interactions are discussed in detail throughout this chapter.
The KCNE subunits are single-pass TM proteins best known for their regulation of Kv channel α subunits, but also able to regulate other ion channel classes (Abbott and Goldstein 1998; Abbott et al. 2001b; Abbott et al. 1999). The first member of this gene family, which has 5 members in the human genome, was cloned in 1988 by expression cloning of rat kidney RNA fractions in the Xenopus laevis oocyte expression system (Takumi et al. 1988). The founding member was termed IsK or MinK (minimal potassium channel), and is now more commonly referred to as KCNE1, similar to its gene name, KCNE1. KCNE1 was first thought to generate a slow-activating Kv current on its own but it was subsequently realized that KCNE1 co-assembles with endogenous Xenopus oocyte KCNQ1 to augment its current, also slowing KCNQ1 activation. Co-assembly of KCNE1 with human KCNQ1 recapitulates properties of human cardiac IKs (slow-activating K+ current), a Kv current that is important in human ventricular myocyte repolarization (Barhanin et al. 1996; Sanguinetti et al. 1996). Mutations in either KCNQ1 or KCNE1 cause inherited variants of long QT syndrome, a surface electrocardiogram abnormality that reflects a delay in ventricular cardiac myocyte repolarization and can lead to the potentially life-threatening torsades de pointe arrhythmia, which can in turn degenerate intro ventricular fibrillation and sudden cardiac death (Splawski et al. 1997a; Splawski et al. 1997b; Wang et al. 1996) (see also chapter 4; Seebohm 2021).
KCNQ1 is capable of forming functional Kv channels in the absence of KCNE subunits (Barhanin et al. 1996; Sanguinetti et al. 1996), although it requires calmodulin (CaM) for folding and assembly, so in this manner CaM could be considered as an essential regulatory subunit for KCNQ1 (and in fact all the KCNQ isoforms, 1–5) (Ghosh et al. 2006; Shamgar et al. 2006; Sun and MacKinnon 2017; Wiener et al. 2008). However, thus far no native currents have been discovered that are generated by KCNQ1 in the absence of KCNE subunits. Functional ventricular IKs channels contain four KCNQ1 α subunits and also require KCNE1 in the complex; thus, KCNE1 is not absolutely required for KCNQ1 channel function but is essential for a known physiological role of KCNQ1. The stoichiometry of KCNE1 in complexes with 4 KCNQ1 α subunits is still under discussion, with some contending variable stoichiometry (between 1 and 4 KCNE1 subunits) and others contending there is a fixed stoichiometry of 4:2 (KCNQ1:KCNE1) (Morin and Kobertz 2008; Nakajo et al. 2010; Plant et al. 2014; Wang and Kass 2012).
In addition to slowing KCNQ1 activation 5–10-fold, KCNE1 shifts the KCNQ1 voltage dependence of activation towards more positive voltages and increases its unitary conductance several fold (Barhanin et al. 1996; Sanguinetti et al. 1996; Sesti and Goldstein 1998) (Figure 1). KCNE1 also eliminates KCNQ1 inactivation - a process that occurs in most Kv channels (but not in other members of the KCNQ/Kv7 family), in which the channel remains “open”, but the inactivation gate is closed, so no current passes, despite the membrane being depolarized. Inactivation in homomeric KCNQ1 channels is typically difficult to observe during a depolarizing pulse because the activation process is slow and is followed in turn by moderate inactivation, causing an equilibrium to be reached at the macroscopic (whole-cell) scale usually with little noticeable current decay. The inactivation process becomes evident, however, during a subsequent repolarizing tail pulse, in which a current “hook” caused by recovery from inactivation is observed for KCNQ1 but not for KCNQ1-KCNE1 channels (Tristani-Firouzi and Sanguinetti 1998).
Figure 1. Regulation of KCNQ1 activity by KCNE1 and KCNE3.
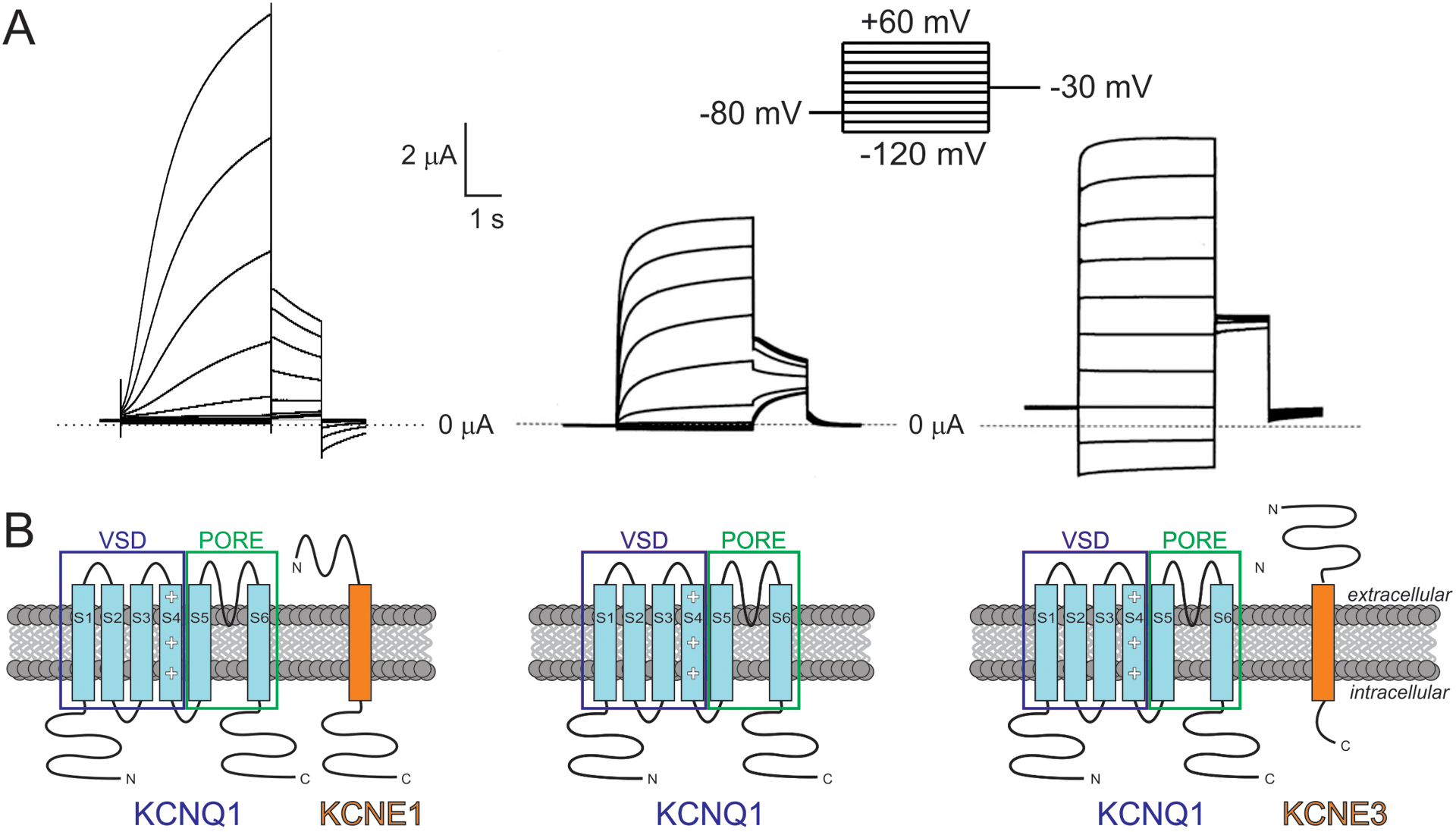
A. Representative electrophysiological recordings from Xenopus oocytes expressing KCNQ1 alone (center), or with KCNE1 (left) or KCNE3 (right). Voltage protocol inset.
B. Subunit topologies shown beneath corresponding traces from A. VSD, voltage-sensing domain. Adapted from (Abbott 2017b).
The mechanism for the dramatic slowing of KCNQ1 activation imposed by KCNE1 is debated. Some studies suggest that KCNE1 directly slows KCNQ1 pore opening, whereas others favor KCNE1 slowing activation of the voltage-sensing domain (VSD) and thus indirectly slowing pore opening. Various lines of evidence suggest that KCNQ1 channels can, unusually for Kv channels, open when only one of the four VSDs is activated; in contrast KCNQ1-KCNE1 pore opening may require multiple, and even all four VSDS to activate – similar to the conventional model for Kv channels outside the KCNQ family (Gofman et al. 2012; Nakajo and Kubo 2007; Osteen et al. 2012; Osteen et al. 2010a; Osteen et al. 2010b; Ruscic et al. 2013). Interaction of KCNE1 with the VSD of KCNQ1 could also impose the positive shift in voltage dependence of activation. This might occur by modifying the manner in which S4 (the positively charged segment within the VSD that is primarily responsible for responding to changes in membrane potential) “senses” stabilizing acidic residues in other, nearby KCNQ1 TM domains (Strutz-Seebohm et al. 2011; Wu et al. 2010a; Wu et al. 2010b).
The multiple gating effects of KCNE1 on KCNQ1 suggest a tight physical coupling to multiple KCNQ1 domains and this is borne out by structural models (built from NMR structural analysis of KCNE1, high-resolution structures of other channels and incorporating mutagenesis results from functional studies of KCNQ1-KCNE1 complexes) showing KCNE1 lying in a groove between the pore module and VSD, where it can impact multiple functionally important gating processes (Figure 2A) (Sahu et al. 2014; Smith et al. 2007; Vanoye et al. 2009). Although no direct structure determination exists for KCNQ1-KCNE1 complexes, the structures of KCNQ1-CaM and KCNQ1-KCNE3-CaM have been solved and they also support the positioning of KCNEs in the aforementioned groove (Figure 2B) (Sun and MacKinnon 2017, 2020).
Figure 2. KCNQ1-KCNE1 model and KCNQ1-KCNE3-CaM structure.
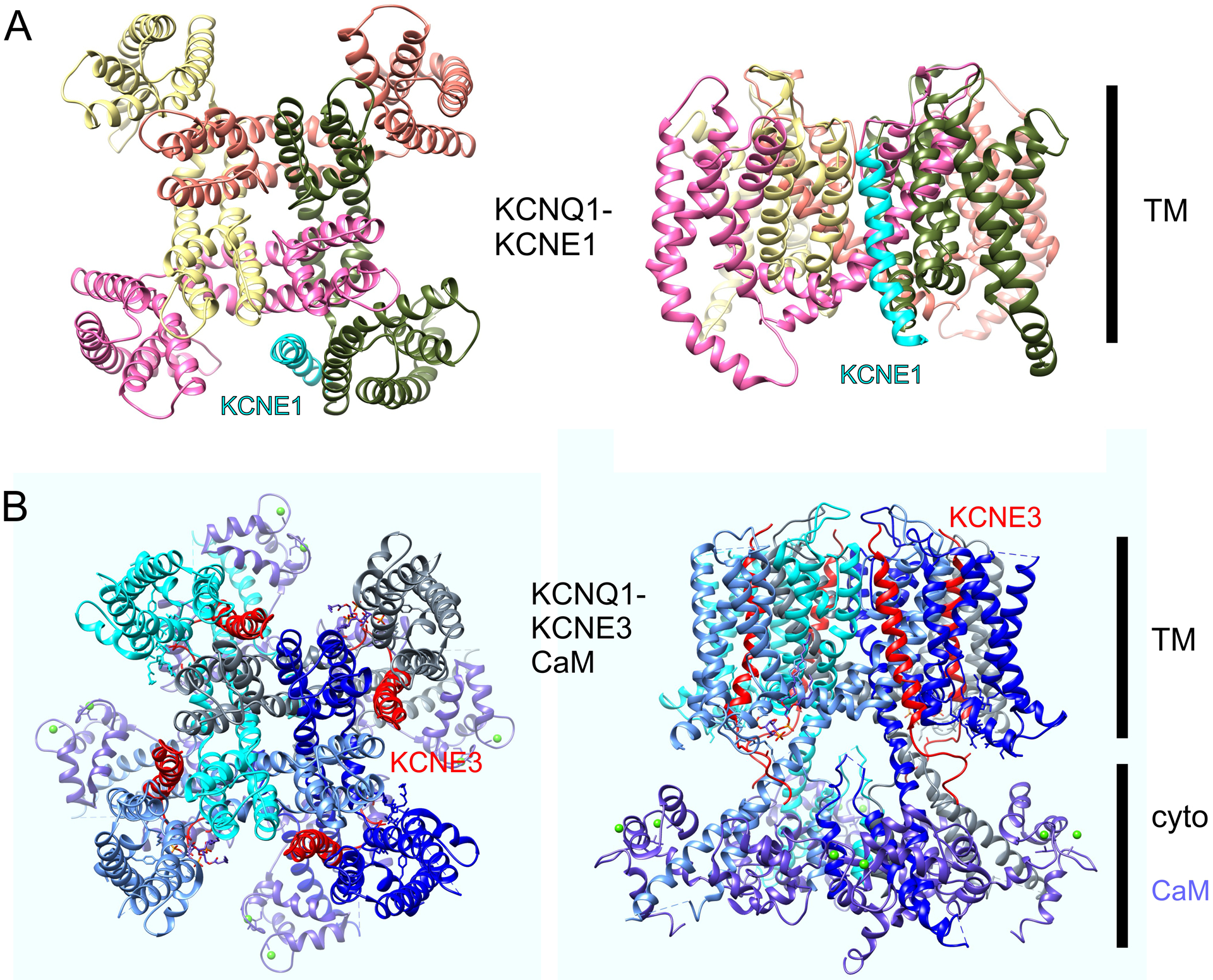
A. Structural model of KCNQ1-KCNE1 - image plotted from coordinates from (Kang et al. 2008).
B. High-resolution cryo-EM structure of KCNQ1-KCNE3-CaM – image plotted from coordinates from (Sun and MacKinnon 2020).
2.2.2. KCNE1 effects on KCNQ1 trafficking
KCNQ1 α subunits require CaM for folding and assembly, prerequisites for trafficking to the cell surface to become functional channels (Ghosh et al. 2006; Shamgar et al. 2006; Sun and MacKinnon 2017; Wiener et al. 2008). Once at the plasma membrane, as for all membrane proteins, KCNQ1 channels have a finite lifetime at the cell surface and must be recycled at some point either to be returned to the cell surface when needed again, or degraded. Both KCNQ1 and KCNQ1-KCNE1 channels are internalized by Nedd4/Nedd4-like and Rab-5 processes (Jespersen et al. 2007; Krzystanek et al. 2012). There is also an alternative, KCNE1-dependent internalization process, in which KCNE1 orchestrates clathrin-mediated endocytosis of KCNQ1-KCNE1 complexes, either when heterologously expressed in immortal cell lines, or when studying native IKs channels in guinea-pig ventricular myocytes. This internalization requires the dynamin GTPase, which pinches off the clathrin-lined vesicle from the plasma membrane so that it can be internalized along with membrane-associated proteins in the vesicle. Clathrin-mediated endocytosis of KCNQ1-KCNE1 can be disrupted by mutagenesis of either dynamin (the K44A dominant-negative dynamin mutation increases IKs current density by blocking KCNQ1-KCNE1 internalization) or KCNE1. Three motifs are necessary and sufficient for KCNE1-facilitated KCNQ1-KCNE1 internalization. First KCNE1 contains a DPFNVY motif in the C-terminal domain (residues 76–81) similar to a motif that is highly conserved in G-protein coupled receptors (GPCRs), (D/N)PX2–3Y, which is implicated in GPCR internalization; the FNVY motif within this stretch in KCNE1 is also similar to the YXXϕ motif required in other clathrin-internalized proteins for binding of the AP-2 adaptor complex, which helps to internalize cargo for endocytosis (and also present in KCNE2 and KCNE3). Accordingly, mutagenic disruption of this motif in KCNE1 reduces KCNQ1-KCNE1 internalization by about half. KCNE1 also contains a consensus SH3-binding domain (PSP) in the distal C-terminus, mutation of which blocks over half the KCNQ1-KCNE1 internalization (Xu et al. 2009). Finally, S102 in KCNE1 is a consensus PKC phosphorylation site, and had been shown in a prior study to mediate PKC regulation of IKs current magnitude by an unknown mechanism (Varnum et al. 1993; Zhang et al. 1994). S102 is conserved in all 5 KCNE proteins; mutation of S102 to alanine also blocked around half of KCNQ1-KCNE1 channel internalization. Mutation of all three sites simultaneously eliminated clathrin-mediated endocytosis of the channel. Further studies showed that PKC dynamically regulated IKs by inducing KCNQ1-KCNE1 endocytosis via S102 phosphorylation (Kanda et al. 2011c). Because PKC phosphorylation is a dynamic post-translational modification, this process could potentially act to dynamically alter IKs current properties in vivo as needed; in addition to decreasing current magnitude, selective removal by this process of solely KCNQ1-KCNE1 complexes could alter IKs gating kinetics by leaving at the membrane homomeric KCNQ1 channels lacking KCNE1 (Kanda et al. 2011c).
2.2.3. Effects of KCNE2 on KCNQ1 function and trafficking
Over a decade after the discovery of KCNE1, we and others discovered the remaining four members of the family, KCNE2–5, also named MinK-related peptides (MiRPs) 1–4; KCNE5 was originally named KCNE1L (KCNE1-like) (Abbott et al. 1999; Piccini et al. 1999). KCNE2–5 also all regulate KCNQ1, but with widely different outcomes compared to KCNE1, and for different physiological purposes (Abbott 2014).
KCNE2 confers upon KCNQ1 constitutive activation in the physiological voltage range, together with much-reduced current density compared to homomeric KCNQ1 – giving a shallow but linear IV curve, in contrast to the outward rectifying properties displayed by KCNQ1 or KCNQ1-KCNE1 (Tinel et al. 2000). The mechanism for reduced current has not been investigated at the single-channel level but there may be a contribution from altered trafficking due to KCNE2 interaction (Hu et al. 2019). KCNQ1-KCNE2 complexes serve essential functions in a range of non-excitable epithelial cell types, including in gastric parietal cells, thyroid epithelial and choroid plexus epithelial cells (Roepke et al. 2006; Roepke et al. 2011a; Roepke et al. 2009; Roepke et al. 2010). In parietal cells, KCNQ1-KCNE2 channels facilitate gastric acid secretion by the gastric H+/K+-ATPase by providing a conduit through which K+ ions can return to the stomach lumen to feed the ATPase to ensure K+/proton exchange through it to acidify the oxyntic pits. Highlighting the importance of KCNE2 in gastric complexes with KCNQ1, germline deletion of Kcne2 in mice results in complete abolition of gastric acid secretion and ultimately development of gastritis cystica profunda and gastric neoplasia, likely due to both bacterial overgrowth and inflammation because of the lack of gastric acid secretion. There are also separate effects of Kcne2 depletion on gastric cellular proliferation. Reduced gastric KCNE2 expression is similarly associated with human gastritis cystica profunda, gastric neoplasia and resistance to 5-fluorouracil-based chemotherapy or with gastric tumor proliferation outpacing this therapy (Abbott and Roepke 2016; Li et al. 2016; Roepke et al. 2006; Roepke et al. 2011a; Roepke et al. 2009; Roepke et al. 2010).
In addition to permitting KCNQ1 to remain open (and non-inactivated) at the moderately polarized membrane potentials in the parietal cells (−20 to −40 mV), KCNE2 bestows at least two other crucial properties on KCNQ1 for its gastric role. First, homomeric KCNQ1 (and KCNQ1-KCNE1) channels are inhibited by extracellular low pH, and therefore would be nonfunctional when at the apical side of the parietal cell and facing the stomach lumen, where they are required to ensure K+ return to the lumen. KCNE2 reverses this property such that KCNQ1-KCNE2 activity is augmented by extracellular low pH. This switch in pH sensitivity requires KCNE2 residues in its extracellular N-terminus and N-terminal portion of the TM segment, and is not observed in KCNQ1-KCNE3 channels, which are constitutively active (see below) yet pH-insensitive (Heitzmann et al. 2007).
Second, in the absence of KCNE2, KCNQ1 traffics to the basolateral membrane instead of the apical membrane of parietal cells, where it cannot serve its function of returning K+ to the stomach lumen. Using single- and double knockouts of the Kcne2 and Kcne3 genes in mice, we showed that in the absence of Kcne2, Kcne3 – normally more highly expressed in the colon and intestine than in the stomach – is upregulated in the stomach and hijacks Kcnq1, taking it to the basolateral side of parietal cells. Knockout of both Kcne2 and Kcne3 restored Kcnq1 to the apical side, but the mice still lacked gastric acid secretion – likely because lacking in Kcne2 their gastric Kcnq1 channels were inhibited by low pH (Roepke et al. 2011b).
KCNE2 is not required for KCNQ1 channel function per se but is essential for several of its known physiological roles in the stomach, thyroid and choroid plexus. Germline deletion of Kcne2 in mice also causes hypothyroidism, because Kcnq1-Kcne2 complexes are required at the basolateral side of thyroid epithelial cells for efficient function of the sodium-iodide symporter (NIS). Without Kcne2, NIS is unable to sequester sufficient iodide into thyroid cells, an essential ion for production of thyroid hormone, at key times in the mouse life cycle, including in early development, during gestation and lactation, and in old age (Roepke et al. 2009). We found that this was an uptake defect and not a storage defect, and that the effects of Kcne2 deletion on thyroid iodide uptake deficiency could be recapitulated using chromanol 293B, a blocker of KCNQ1-containing channels, in vitro and in vivo (Purtell et al. 2012). Studies using positron emission tomography to track I124 showed that lactating dams could not produce sufficient milk to feed their pups, and that the iodide that did reach their pups via milk was inefficiently sequestered by their thyroid glands, leading to developmental delay, cardiac hypertrophy and alopecia. These defects could be prevented by surrogacy of Kcne2 knockout pups with wild-type dams (and vice-versa), indicating a prominent role for maternal Kcne2 status in the pups’ phenotype (Roepke et al. 2009). While the molecular mechanism underlying the need for Kcnq1-Kcne2 channels to facilitate efficient iodide uptake by NIS is not fully understood, it is not simply a requirement for maintenance of a whole-cell transmembrane electrochemical gradient to permit sodium-dependent solute transporter function, as the sodium-coupled monocarboxylate transporter (SMCT) was able to function normally in the absence of Kcne2 (Purtell et al. 2012).
Kcne2 deletion also causes increased handling-related seizures in mice, increased susceptibility to seizures initiated by the chemoconvulsant pentylene tetrazole (PTZ), and reduced depressive/despair-type behavior – each suggesting increased excitability in the nervous system. These changes can be traced to another role of Kcne2 complexes, in the apical membrane of the choroid plexus epithelium. As the primary site of cerebrospinal fluid (CSF) production and secretion, the choroid plexus plays an important role in regulating the environment of the nervous system. Kcne2 deletion in mice uncovered its regulation of two different Kv channel isoforms in the choroid plexus, Kcnq1 and Kcna3 (Kv1.3). Compared to wild-type choroid plexus epithelia cells, Kcne2 knockout cells exhibited increased XE991-sensitive outward current but reduced XE991-sensitive inward current at hyperpolarized membrane potentials. This profile matches what one would expect for loss of Kcne2 from Kcnq1 complexes, XE991 being a relatively KCNQ-specific inhibitor; KCNE2 being known to decrease KCNQ1 outward current but permit its constitutive activation at hyperpolarized voltages. In addition, Kcne2 deletion increased Margatoxin (MgTx)-sensitive currents, while leaving Dendrotoxin-sensitive currents unchanged, indicative of altered Kcna3 (Kv1.3) but not Kcnq1 currents, respectively. Accordingly, in heterologous co-expression experiments, human KCNE2 was found to increase KCNA3 current but not alter KCNA1 current. Co-assembly of mouse Kcne2 with Kcnq1 and Kcne3 in choroid plexus epithelium was also supported by co-localization and co-immunoprecipitation experiments (Abbott et al. 2014; Roepke et al. 2011a).
Kcne2 deletion hyperpolarized the choroid plexus epithelial cell membrane potential, as would be predicted from the above effects, and also increased CSF [Cl−], but a mechanism for increased seizure susceptibility was not immediately apparent (Roepke et al. 2011a). Metabolomics analysis subsequently revealed that Kcne2 deletion caused a reduction in CSF myo-inositol, and restoration of normal myo-inositol levels by mega-dosing in the water supply also restored normal seizure susceptibility and depressive behavior. The reason for reduced CSF myo-inositol was uncovered by further analysis – KCNQ1-KCNE2 channels negatively regulate activity of the SMIT1 sodium-dependent myo-inositol transporter, which is expressed at both the apical and basolateral membranes of the choroid plexus epithelium and which regulates movement of myo-inositol between the blood and CSF. Loss of Kcne2 in mice resulted in loss of the brake upon uptake of myo-inositol from the CSF back into the choroid plexus, suggesting a plausible mechanism for depleted CSF myo-inositol. This work represented the discovery of direct physical interaction of a K+ channel with a sodium-coupled solute transporter and has since led to many other examples of this type of interaction (Abbott 2016a; Abbott et al. 2014; Neverisky and Abbott 2015, 2017).
2.2.4. Effects of KCNE3 on KCNQ1 function
KCNE3, like KCNE2, induces constitutive activation in channels formed with KCNQ1, by greatly negative-shifting the voltage dependence of activation and thus dramatically reducing the time-and voltage dependence of the current across the physiological voltage range (Schroeder et al. 2000). This is achieved by KCNE3 locking the KCNQ1 voltage sensor in the activated state, thus indirectly locking open the pore (Nakajo and Kubo 2007; Panaghie and Abbott 2007). Unlike KCNE2, KCNE3 does not concurrently reduce overall current magnitude of KCNQ1, so the resultant channels are much easier to study and the majority of mechanistic analyses on loss of voltage dependence have been conducted in KCNQ1-KCNE3 complexes (reviewed in (Abbott 2016b)). The constitutive activation of KCNQ1-KCNE3 channels, which essentially provides a continuous K+ selective current that does not inactivate, permits the utility of KCNQ1-KCNE3 channels in non-excitable cells such as those in the colonic epithelium, where they regulate membrane potential and in turn, cAMP-stimulated chloride secretion (Schroeder et al. 2000), and in non-excitable cells of the airway (Grahammer et al. 2001) and mammary epithelia (vanTol et al. 2007).
KCNE3 also endows KCNQ1 with sensitivity to estrogen. Increased estrogen causes downregulation of KCNE3, which leaves homomeric KCNQ1 alone to function in the non-excitable colonic cell membrane without KCNE3 to lock open its voltage sensor and keep it activated. Consequently, during proestrus, when estrogen levels are high, female mammals exhibit water retention partly because KCNQ1 is less able than KCNQ1-KCNE3 to promote cAMP-stimulated chloride secretion. In addition, male rats have higher colonic crypt KCNE3 expression and enriched KCNQ1-KCNE3 channel versus homomeric KCNQ1, compared to female rats. Estrogen regulation of KCNE3 requires KCNE3-S82 because it requires a signaling cascade that includes phosphorylation of S82 by PKC (Alzamora et al. 2011; Kroncke et al. 2016; O’Mahony et al. 2009; Rapetti-Mauss et al. 2013). Phosphorylation of KCNE3-S82 is also required for KCNE3 to induce subthreshold activation in Kv3.4-KCNE3 complexes, which are important in skeletal muscle physiology (Abbott et al. 2001a; Abbott et al. 2006; King et al. 2017).
2.2.5. SK regulatory proteins and BK γ subunits
Separate from the classic Kv channels, the calcium-activated potassium channels have superficially similar architecture but different functional properties (Berkefeld and Fakler 2013; Berkefeld et al. 2010); see also Chapter 12 (Dudem et al 2021). The majority of calcium-activated potassium channels fall into one of two main groups. The BK (big conductance) calcium-activated potassium channels form one group, while the SK (small conductance) and IK (intermediate conductance) calcium-activated potassium channels fall into the other group. SK channels have a relatively small unitary conductance (~ 10 pS) and share somewhat similar tetrameric architecture and topology to that of Kv channels, with each α subunit containing 6 TM domains and a P-loop. The major difference to Kv channels is that the S4 segment of SK channels is only weakly charged (only two basic residues compared to seven for typical Kv channels) and SK channels are accordingly relatively insensitive to voltage, instead being activated by Ca2+. There are three SK channel α subunit isoforms (SK1–3), encoded by KCNN1–3, and the isoforms are expressed throughout the nervous system in higher animals, where they regulate neuronal excitability (especially the medium afterhyperpolarizing potential, mAHP) and participate in calcium signaling and synaptic plasticity (Bond et al. 1999).
SK channels are not known to possess any channel-specific ancillary subunits, but they participate in functionally essential interactions with generalized protein modulators calmodulin (CaM, which endows calcium sensitivity), casein kinase 2 (CK2, which phosphorylates T80 of channel-bound CaM) and protein phosphatase 2 (PP2A, which dephosphorylates CaM-T80). IK channels exhibit unitary conductances of ~35 pS; these are to date the least studied of the KCa channels. There is one IK isoform in the human genome, KCa3.1 (a.k.a. IKCa1, SK4), encoded by KCNN4. KCa3.1 exhibits similar TM topology and activation mechanism to that of the SK channels, including the voltage insensitivity and the dependence on CaM for Ca2+ binding and sensitivity. IK channels are expressed in epithelia, blood cells and in peripheral neurons (Lujan et al. 2009; Weatherall et al. 2010).
BK (also MaxiK or Slo) channels - encoded by KCNMA genes – belong in a different class to the SK and IK channels for several reasons. First, BK channels, as the name suggests, have a much larger unitary conductance, of 200–300 pS. Second, BK channels contain an additional TM helix, termed S0, at the N-terminal end, that distinguishes them from both SK/IK channels and from Kv channels and in fact all the classic voltage-dependent S4 family (repeating units of Nav and Cav channels each contain 6 TM segments). Third, BK channels possess intrinsic voltage sensitivity, via positive charges in their voltage sensing domain (VSD), primarily TM segments S2-S4. Fourth, BK channels also exhibit intrinsic Ca2+ sensitivity, with two types of Ca2+ binding site present on the channel α subunits themselves – high-affinity “regulator for conductance of potassium” (RCK) domains within the α subunit cytosolic C-terminal (RCK1 and RCK2) and a lower affinity binding site that binds Ca2+ (and other divalent metal ions such as Mg2+) located between the TM segments and the gating ring (Kshatri et al. 2018) (Vergara et al. 1998). Fifth, BK channels are regulated by channel-specific ancillary subunits. Specifically, BK channels are modulated by physical interaction with ancillary proteins termed β subunits, here referred to as BKβ subunits to distinguish them from Kvβ subunits and γ subunits (Li and Yan 2016).
The BK γ (gamma) subunits are represented as four isoforms (BKγ1–4, encoded by LRRC26, 52, 55 and 38, respectively) in the human genome. LRRC is an acronym for “leucine-rich repeat containing” and refers to the extracellular LRR domain. BKγ1 is widely expressed including in the brain, various glandular epithelia, aorta and mucosa; BKγ2 in skeletal muscle, testis placenta and sperm, BKγ3 also in sperm and in the brain, but also in the liver, spleen and olfactory bulb, and BKγ4 in muscle, testis and sperm, cerebellum, adrenal gland and thymus. They are each potent regulators of BK channel activation, shifting the voltage dependence of activation by −100 to −140 mV across the Ca2+ concentration range, such that the γ-containing BK channels are constitutively active at resting membrane potentials, even in the absence of elevated intracellular Ca2+, with approximately half activation at −80 mV in 100 μM intracellular free Ca2+ (Yan and Aldrich 2010). Each γ subunit also speeds activation and slows deactivation of BK channels, without inducing inactivation. It is thought that γ subunits exhibit variable stoichiometry, from 1–4 in a complex of 4 α subunits; increasing the number of γ subunits per complex increases the negative shift in voltage dependence of BK channel activation.
2.2.6. BK β subunits
Not as common as those possessing a single TM span, some ion channel regulatory subunits possess a 2-TM topology (Sun et al. 2012). In the K+ channels, the BKβ subunits are the exemplar, and they are another example of ancillary subunits that modify channel function to enable specific physiological roles but are not essential for channel activity per se (Castillo et al. 2015). The BKβ subunits each possess two TM domains, and there are four isoforms (BKβ1–4). BKβ1 and BKβ2 each shift the voltage dependence of human BK channel activation (with elevated intracellular Ca2+) by −70 and −50 mV, respectively; BKβ2 also induces inactivation that is absent from homomeric BK α subunit channels. BKβ3 hyperpolarizes murine BK channel activation across the Ca2+ concentration range (by −30 mV) but does not alter voltage dependence of human BK channel activation; however, BKβ3 induces inactivation in both human and murine BK. BKβ4 is able to differentially alter BK activation voltage dependence depending on Ca2+ levels: a 20-mV positive shift in low Ca2+, and a somewhat greater negative shift in high Ca2+; these effects are coupled with reduced activation and deactivation rate. BKβ1 and BKβ4 do not induce BK channel inactivation. As for γ subunits, β subunits are reported to exhibit variable stoichiometry, from 1–4 in a complex of 4 α subunits; increasing the number of BKβ in a channel increases their relative effect on the voltage dependence of BK activation (Castillo et al. 2015) (Figure 3).
Figure 3. Structure of human Slo1-β4 complex.
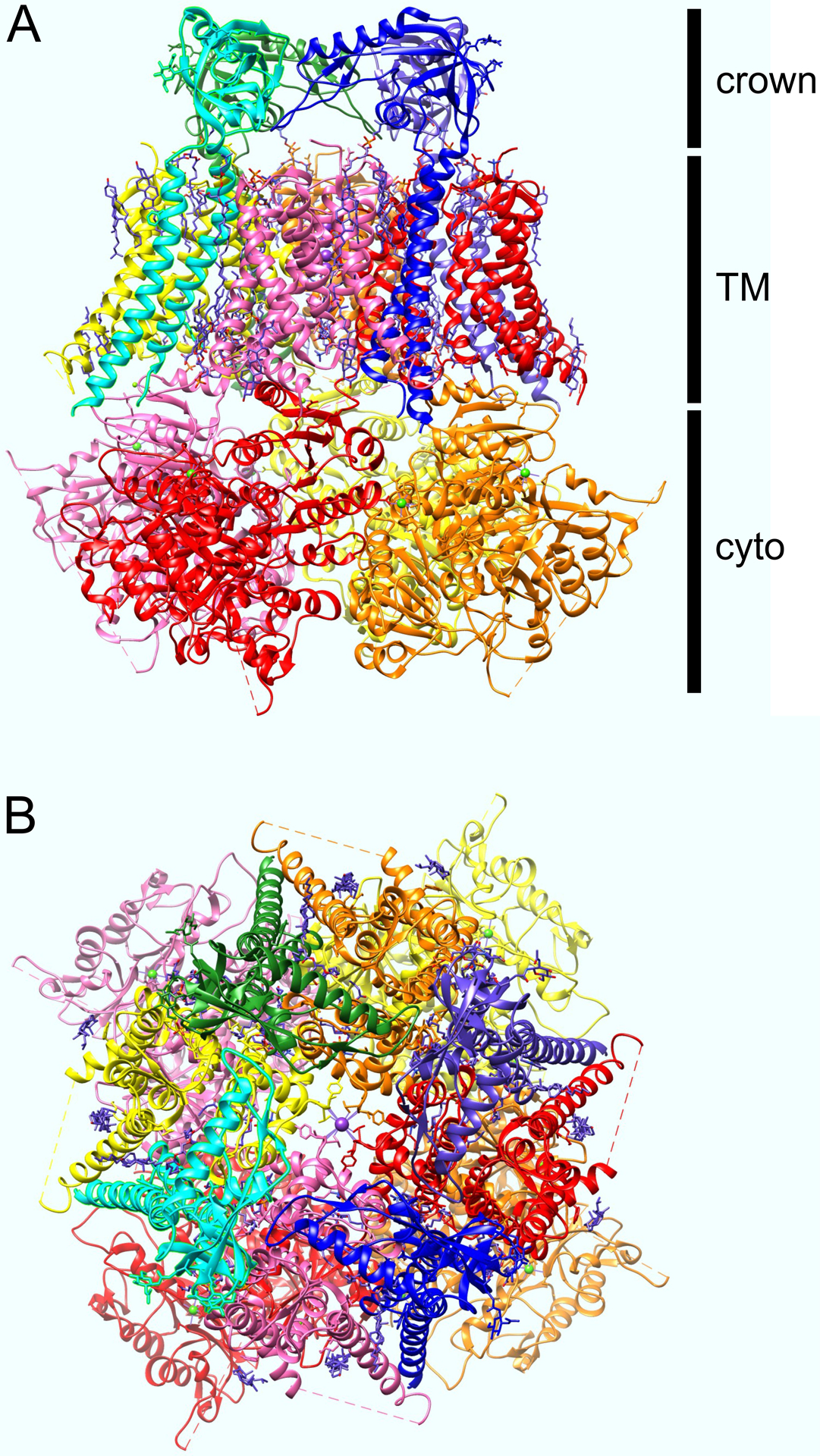
A. Side view of Ca2+-bound human Slo1-β4 complex, solved by cryo-EM, plotted from coordinates in (Tao and MacKinnon 2019). Slo1 (BK) α subunit colored pink, red, orange, yellow. B4 subunits colored purple, blue, cyan, green. Crown is an extracellular region formed from the β4 subunits.
B. View of Slo1-β4 complex as above but from extracellular face.
2.2.7. Cytosolic subunits that add novel functionality to Kv channels
An array of cytosolic ancillary subunits regulate K+ channels, typically by interaction with the cytosolic portions of the K+ channel and sometimes adding completely unique functionality not exhibited by the α subunits alone. One example is the Kvβ subunit family. Some but not all Kvβ subunits contain a fast inactivation domain that can plug the pore of slow-inactivating delayed rectifier α subunits to induce fast inactivation, that is, rapid current decay following voltage-dependent activation. The structural basis of Kvβ-mediated fast inactivation has been suggested by solving of the structure of the prokaryotic KcsA K+ channel containing tetrabutylantimony, an electron-dense quaternary ammonium ion that resembles the distal end of the inactivation peptide and adopts a deep pore binding site. In addition, solving of the octameric complex of Kvβ2 with the cytoplasmic tetramerization (T1) domain of KCNA1 suggested that lateral negatively charged fenestrations above this octameric structure could permit inactivation peptide access to the pore in the intact α subunit/β subunit complex (Gulbis et al. 2000; Zhou et al. 2001).
An earlier crystallographic analysis revealed that the Kvβ2 subunit displays structural homology to aldo-keto reductases, proteins that contain a bound NADP(+) co-factor (as does Kvβ2) and can sense the redox state of the cell (Gulbis et al. 1999). Further functional analyses have indeed shown that Kvβ2 can couple channel activity and other characteristics to the redox state of the cell, and that this occurs in vivo. For example, using Kcnab2 knockout mice for comparison, it was found that Kvβ2 is important for adequate expression of cardiac transient outward current (Ito, generated by Kv1.4, Kv4,2 and Kv4.3) and IK,slow (generated by Kv1.5) and that this ancillary subunit also coupled myocyte repolarization to metabolic state via its interaction with these Kv α subunits (Campomanes et al. 2002; Kilfoil et al. 2019).
Another class of cytoplasmic subunits, the K+ channel interacting proteins (KChIPs) endows Kv channels with calcium sensitivity. KChIPs are best known for regulating Kv4 α subunits, inducing large increases in current magnitude upon co-expression; knockout of the cardiac KChIP isoform, KChIP2, in mice leads to loss of cardiac myocyte Ito because of impaired Kv4 channel activity. KChIPs increase the surface density of Kv4 α subunits, negative-shift the voltage dependence of activation, slow inactivation and speed recovery from inactivation. KChIPs are also important for regulating what are termed A-type current or ISA (fast-inactivating Kv4-generated currents) in the brain. KChIPs contain four EF-hand-like domains and are calcium-binding proteins related to neurocalcins and neuronal calcium sensor-1 (NCS-1). While there have been conflicting observations on the effects Ca2+ in Kv4-KChIP complex formation, stoichiometry, and gating, it does appear that changes in intracellular [Ca2+] can modulate Kv4-KChIP gating properties. Increasing intracellular Ca2+, but not Co2+, via patch pipette was found to increase ISA current magnitude, thought to be expressed by Kv4-KChIP complexes, in cultured rat cerebellar granule cells. In another study conducted using heterologous expression in HEK-293 cells concluded that direct binding of Ca2+ to KChIP2 EF hand motifs EF3 and EF4 likely accelerates inactivation recovery of Kv4.3/KChIP2 channels (Abbott 2017a; Abbott et al. 2007; Amadi et al. 2007; An et al. 2000; Bahring 2018; Bahring et al. 2001; Foeger et al. 2013; Gong et al. 2006; Takimoto and Ren 2002).
2.2.8. SUMO and 14-3-3 interactions with K2P channels
K2P channels are the molecular correlates of many potassium-selective leak currents in the cells of mammals and other organisms. K2P channels tend to exhibit linear current-voltage (IV) relationships with symmetrical [K+] on either side of the plasma membrane, while behaving as outward (open) rectifiers under physiological conditions (low K+ outside, high K+ inside the cell). K2P channels form as dimers each with 4 P-loops (two contributed from each subunit) and can form as heterodimers with unique properties (Figure 4). There are at least 18 K2P isoforms in the human genome, and most of them are categorized into four subfamilies: TALK, TASK, TREK and TWIK (Duprat et al. 2007; Goldstein et al. 2001; Goldstein et al. 1996; Ketchum et al. 1995; Kollewe et al. 2009; Mathie et al. 2010); see also Chapter 13 (Kamuene et al 2021).
Figure 4. Structure of human K2P1 channel.
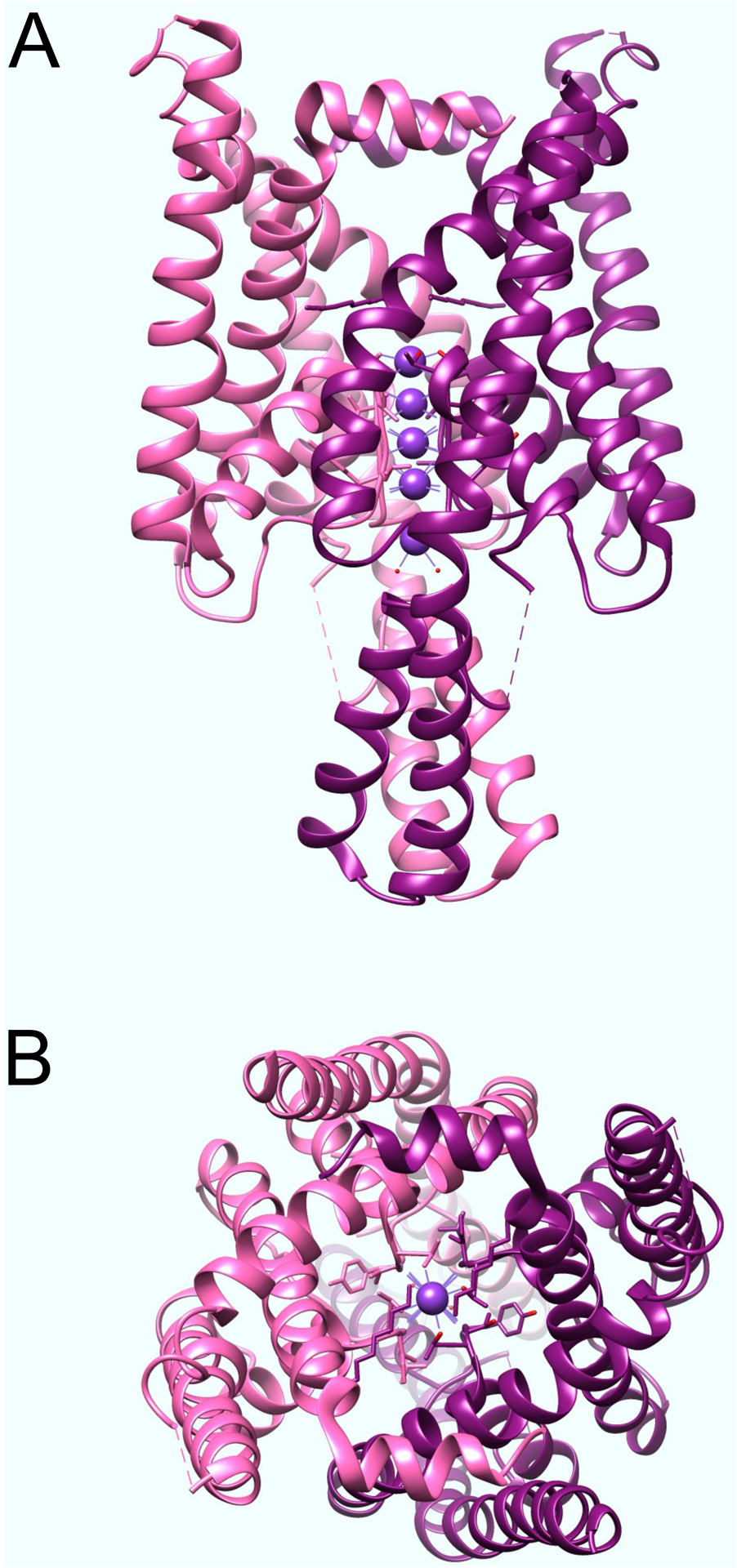
A. Side view of human K2P1 channel, solved by X-ray crystallography, plotted from coordinates in (Miller and Long 2012). The two α subunits are colored pink and purple.
B. View of human K2P1 channels as above but from extracellular face.
At the time of writing, K2P-specific ancillary subunits have yet to be uncovered; given that the first K2P α subunits were cloned a quarter century ago (in 1995), this suggests that K2P-specific regulatory subunits may be uncommon or absent. However, there are some notable examples of K2P channels being regulated by more general modulatory proteins. First, 14-3-3β (a cytoplasmic protein that forms as dimers and is composed of 9 α-helices per monomer in an anti-parallel organization) is required for maturation and expression of K2P3 and K2P9 at the cell surface. This is because β-COP, which belongs to the coatamer class of proteins, binds to K2P3 and K2P9 to prevent them from exiting the endoplasmic reticulum after translation. 14-3-3β binding to the K2Ps requires phosphorylation at a single residue on the channel C-terminus; once this occurs, β-COP binding is inhibited, permitting channel maturation and surface expression. 14-3-3 also serves to facilitate folding and maturation of other, non-channel proteins, using a similar mechanism (O’Kelly et al. 2002; O’Kelly and Goldstein 2008).
While 14-3-3 rescues K2P channel surface expression and activity, another protein – the small ubiquitin-related modifier protein (SUMO) – modifies K2P channels by silencing them when it is covalently attached to, e.g., K2P1 via interaction of the channel with the SUMO conjugating enzyme, Ubc-9 ligase. Removal of SUMO by the SUMO-specific protease, SENP1, restores K2P1 activity. Covalent modification by addition of SUMO occurs via a single residue, K2P1-K274 (Plant et al. 2010; Rajan et al. 2005). Before this discovery, SUMO was previously thought to not regulate proteins at the plasma membrane and instead perform solely nuclear functions; it is now known to regulate other plasma membrane proteins in addition to K2P1, including the cardiac KCNQ1-KCNE1 potassium channel and the neuronal and cardiac Kv2.1 potassium channel (Plant et al. 2011; Plant et al. 2014).
2.3. Ancillary subunit interactions inhibitory to channel activity
While it is not always completely understood why this occurs, some ancillary subunits strongly inhibit channel activity. KCNQ1 activity is strongly inhibited by the KCNE4 and KCNE5 subunits. KCNE4 is the largest of the KCNE subunits, with a recent N-terminal extension not previously detected bringing the total length of the long version of human KCNE4 (hKCNE4L) to 221 residues, still with a single TM segment. The shorter (170 amino acid) hKCNE4S isoform inhibits KCNQ1 by as much as 90% in heterologous co-expression studies in Xenopus laevis oocytes (and hKCNE4S also strongly inhibits KCNQ1 in COS cells), while hKCNE4L inhibits by only 40% in Xenopus oocytes (Abbott 2016d). The mechanistic studies of KCNE4 inhibition were all performed using KCNE4S. KCNE4S does not alter the surface expression of KCNQ1 protein, and the mechanism by which it inhibits KCNQ1 involves binding to both KCNQ1 and also directly to CaM, which is essential for KCNQ1 activity. A tetraleucine motif on the membrane-proximal region of the KCNE4 intracellular C-terminus is required for interaction with CaM. Further, KCNQ1 inhibition by KCNE4S is calcium-sensitive, with lower intracellular calcium levels decreasing inhibition. Thus, KCNE4, in concert with CaM, provides a mechanism to tune KCNQ1 current to intracellular calcium, and this may provide a reason for the existence of KCNE4 as a KCNQ1 inhibitory subunit (Abbott et al. 1999; Bendahhou et al. 2005; Ciampa et al. 2011; Grunnet et al. 2002; Grunnet et al. 2005; Lundquist et al. 2005; Manderfield et al. 2009; Manderfield and George 2008). KCNE4 also inhibits Kv1.1 and Kv1.3 channel activity (Grunnet et al. 2003).
KCNE5 also inhibits KCNQ1, shifting its voltage dependence by +140 mV such that it does not activate at physiologically relevant membrane potentials. The mechanism does not appear to be CaM-dependent and KCNE5 lacks the tetraleucine motif important for KCNE4 interaction with CaM. The physiological role of this inhibition is not known, although it is of note that both KCNE4 and KCNE5 can inhibit IKs (KCNQ1-KCNE1) complexes and all are expressed in human heart. KCNE4 is reportedly the highest expressed of all the KCNEs in human heart, which raises the question, still unanswered, of its role there – could it be to negatively regulate IKs, or render it calcium-sensitive? Perhaps KCNQ1 channels can gain or lose KCNE subunits while at the cell surface, or in endocytic pits, providing a dynamic mechanism for regulation of IKs kinetics, voltage dependence, and other properties (Angelo et al. 2002; Bendahhou et al. 2005; Piccini et al. 1999).
While KCNE1 and KCNE2 have opposite effects to one another on KCNQ1 (one slowing activation and increasing outward current, the other inducing constitutive activation and reducing outward current), they each exert a similar inhibitory effect on N-type inactivating (hereafter referred to as N-type) α subunits of the Kv1 and Kv3 (KCNA and KCNC) subfamilies. N-type subunits are fast-inactivating by virtue of a cytoplasmic “ball” domain at the end of the α subunit N-terminal domain, which plugs the pore from the intracellular side after channel activation, to induce inactivation, or block of the activated pore. This produces currents with rapid decay after rapid activation. In the Kv1 subfamily, Kv1.4 is the N-type subunit, while Kv1.1 and Kv1.2, for example, are delayed rectifier subunits (fast activating, slow-inactivating by a C-type mechanism involving pore collapse). In the Kv3 subfamily, Kv3.3 and Kv3.4 are the N-type subunits, while Kv3.1 and Kv3.2 are delayed rectifiers.
Within the same subfamily, co-expression of fast-inactivating N-type subunits with slow-inactivating delayed rectifier subunits produces heteromeric channels (if co-assembly is permissible) with intermediate inactivation kinetics (Coleman et al. 1999; Isacoff et al. 1990; Ruppersberg et al. 1990). This is because the rate of N-type inactivation is proportional to the number of N-type inactivation domains in the tetrameric complex, and any one of the up to 4 inactivation domains is necessary and sufficient to induce fast inactivation; a higher number increases the chance of this occurring in a given time period (Malysiak and Grzywna 2008). The rate of N-type inactivation is an important factor in determining the excitability and action potential frequency of cells such as neurons in which Kv1 and Kv3 α subunits are expressed. Therefore, it follows that co-assembly of N-type with delayed rectifier α subunits should be tightly regulated, to control inactivation rate. KCNE1 and KCNE2 were unexpectedly discovered to provide such a mechanism. When either of these subunits is co-expressed with any of the N-type subunits, Kv1.4, Kv3.3, or Kv3.4, it results in strong inhibition of channel activity. This is due to the KCNE subunits retaining the N-type subunits early in the secretory pathway, probably in the endoplasmic reticulum. Co-expression of KCNE1 or KCNE2 with the delayed rectifier subunits Kv1.1 or Kv3.1 did not result in inhibition. Crucially, triple expression of KCNE1 with both Kv1.1 and Kv1.4 prevented inhibition by KCNE1, permitting surface expression and channel activity of heteromeric Kv1.1-Kv1.4 complexes. A similar effect was observed with Kv3,1, which prevented intracellular retention of Kv3.4 by KCNE1 (and indeed prevented interaction of KCNE1 with Kv3.4). Furthermore, Kv3.2 was also able to rescue Kv3.4 from intracellular retention by KCNE1, while Kv2.1 – which can interact with KCNE1 but not Kv3.4 – was unable to rescue Kv3.4 from retention by KCNE1 (Kanda et al. 2011a, b). Thus, KCNE1 and KCNE2 can act as checkpoints that prevent surface expression of homomeric N-type channels and instead permit surface expression only of same-subfamily heteromeric N-type/delayed rectifier channels that have intermediate inactivation rates.
3. Ancillary subunit modulation of K+ channel pharmacology
3.1. Sensitization by providing a novel binding site absent from the channel α subunit, to directly impact channel function
Perhaps the most profound mode of pharmacomodulation by an ancillary subunit is when it introduces a completely novel site to endow sensitivity to a drug or other small molecule to which the α subunit-alone channel is insensitive. Here we divide these into two classes depending on whether the novel small molecule effect is on function or protein folding/assembly/trafficking. BKβ subunits, for example act in the first category, by providing a novel binding site and thereby sensitizing BK channels to activation by a range of steroids and steroid-like compounds. BKβ1 sensitizes BK channels to dehydrosoyasaponin-I (DHS-I), lithocholate, tamoxifen, 17β-estradiol and xenoestrogen; likewise, BKβ2 and BKβ4 sensitize BK channels to dehydroepiandrosterone and corticosterone, respectively. Several residues (T169, L172 and L173) in the BKβ1 TM2 segment form a steroid-sensing site essential for binding of, e.g., lithocholate. Neither homomeric BK α subunit channels, nor BK channels formed with any of the other β subunits, are activated by this steroid (Bukiya et al. 2007; Bukiya et al. 2009).
Sulfonylurea receptor (SUR) subunits participate in both forms of modulation. SURs are so-called because they bind sulphonylureas, which are used to treat type 2 diabetes mellitus and also some types of neonatal diabetes. Sulfonylurea binding to the SUR subunits inhibits KATP channels, thereby depolarizing pancreatic β-cells, which leads to voltage-gated calcium channel activation, and insulin secretion. Another important pharmacological role for SURs is in mediating the action of pharmacochaperones. This term describes drugs that can rescue KATP channel folding or assembly mutants, i.e., channels carrying mutations that prevent KATP channel surface expression, causing β-cell hyperexcitability and resultant congenital hyperinsulinism. Recent structural analyses utilizing cryo-electron microscopy (cryo-EM) showed that glibenclamide, repaglinide and carbamazepine bind within a common binding pocket located in the SUR1 subunit of Kir6.2-SUR1 channels, which are the pancreatic β-cell KATP channel isoform. Because this site is located close to the Kir6.2 N-terminus, binding of the drugs to the SUR receptor stabilizes the Kir6.2 N-terminus, providing a solid base for channel folding and assembly, rescuing the mutant KATP channel activity by facilitating their surface expression, even though the pharmacochaperones are also channel inhibitors (Martin et al. 2017; Martin et al. 2020; Martin et al. 2019) (Figure 5). Thus, SURs provide a novel binding site absent on the channel α subunit, to directly impact channel folding/assembly.
Figure 5. Structure of Syrian hamster SUR1-rat Kir6.2 with glibenclamide and ATP bound.
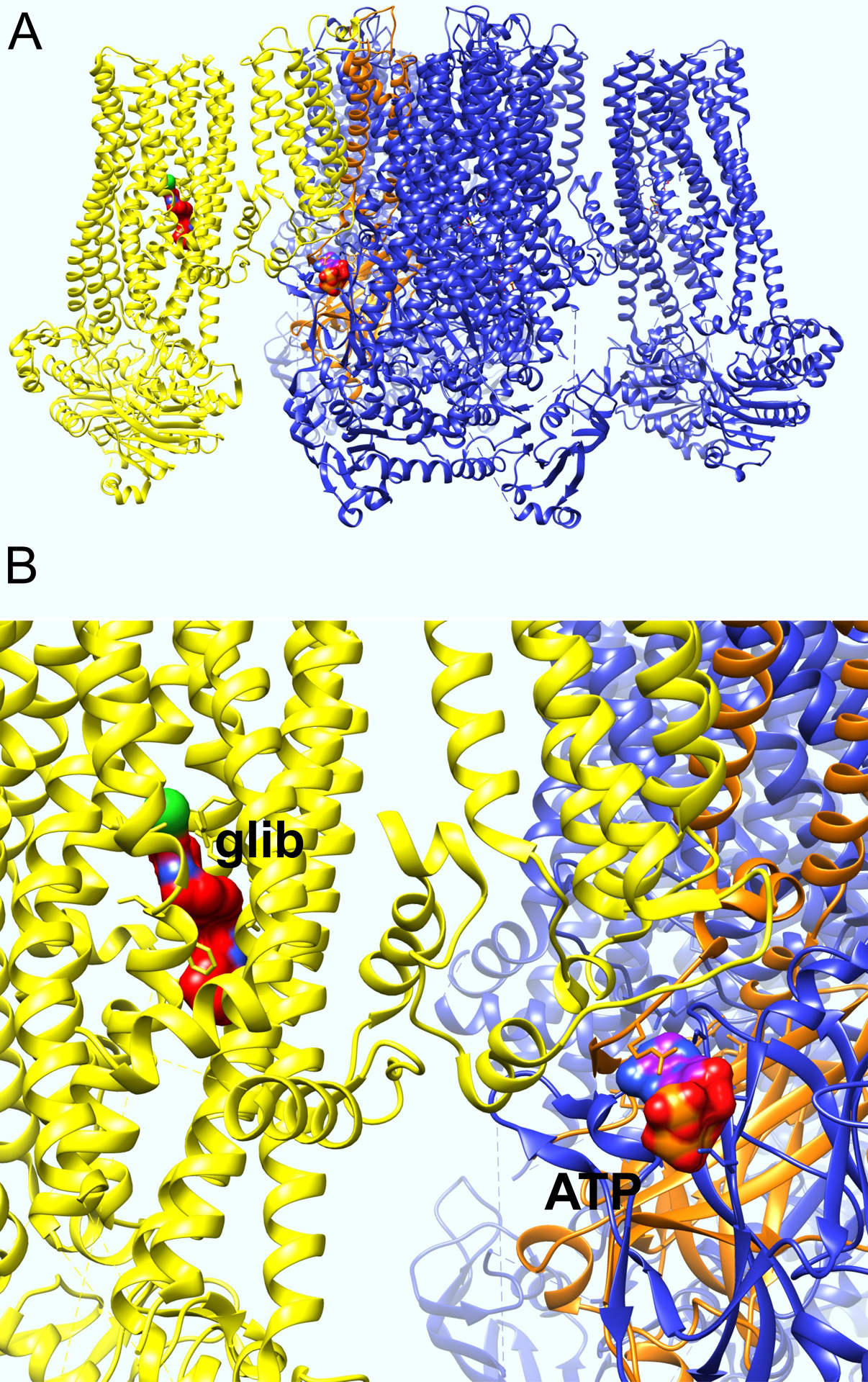
A. Side view of Syrian hamster SUR1-rat Kir6.2 with glibenclamide and ATP bound solver by cryo-EM, plotted from coordinates in (Martin et al. 2017). One SUR1 subunit is colored yellow, one Kir6.2 subunit is colored orange.
B. Close-up of structure in panel A, with glibenclamide (glib) and ATP labeled.
The Seebohm laboratory discovered a structural basis for drug sensitization of KCNQ1 by KCNE1, involving introduction of windows, or fenestrations, by KCNE1 that endow novel sensitivity of KCNQ1 to adamantane compounds. While KCNQ1-KCNE1 channels display an IC50 of 78.4 nM for inhibition by 2-(4-chlorophenoxy)-2-methyl-N-[5-[(methylsulfonyl)amino]tricyclo[3.3.1.13,7]dec-2-yl]-propanamide (also named JNJ303), homomeric KCNQ1 channels, or those formed by KCNQ1 with isoforms KCNE2–5, are completely insensitive to 1 μM JNJ303, which inhibits KCNQ1-KCNE1 by 80%. It is thought that rather than blocking the KCNQ1-KCNE1 pore, adamantane compounds instead stabilize a non-conducting closed state. By introducing a fenestration in the KCNQ1 pore, KCNE1 reportedly introduces a novel binding site that permits JNJ303 and other adamantane compounds to stabilize the closed state; hence, homomeric KCNQ1 is unaffected by these compounds (Wrobel et al. 2016). Pharmacological exploitation of this phenomenon has the potential to produce highly subtype-specific IKs modulators.
KCNQ channels can also form reciprocally regulating complexes with sodium-coupled solute transporters. In these cases, each protein can be considered as the others’ ancillary subunit (Abbott 2016a; Abbott et al. 2014; Neverisky and Abbott 2015, 2017). We found that KCNQ channels function as chemosensors to permit sodium-coupled myo-inositol transporters to respond to stimuli to which they are typically insensitive, including the neurotransmitter GABA, related metabolites β-hydroxybutyrate (an important ketone body) and the KCNQ2/3-targeting anticonvulsant, retigabine – all of which activate KCNQ2/3 channels, do not act on homomeric SMITs, but inhibit myo-inositol transport activity of SMITs in physical complexes with KCNQ2/3 channels (Manville and Abbott 2020). Reciprocally, SMITs alter the response of KCNQs to some of these molecules. KCNQ2/3 channels are the primary molecular correlate of the neuronal M-current, which regulates neuronal excitability. They are sensitive to GABA and β-hydroxybutyrate by virtue of a binding pocket on their KCNQ3 subunits formed by residues in the S4–5 linker region and S5 (Manville et al. 2018, 2020). Homomeric KCNQ channels are activated by both GABA and β-hydroxybutyrate, while homomeric KCNQ2 is insensitive to both. In contrast, when in complexes with SMIT1, KCNQ2 channels are still insensitive to GABA, but can sense and are able to facilitate inhibition of SMIT1 by β-hydroxybutyrate. Therefore, SMIT1 modulates KCNQ2 pharmacology and introduces a novel sensitivity of the channel α subunit to β-hydroxybutyrate (Manville and Abbott 2020). A related example is that for Kv4.3 (or Kv4.2) channel complexes to become sensitive to the drug NS5806, the KChiP3 ancillary subunit is required, because it provides the NS5806 binding site. In addition, the mode of action of NS5806 (potentiation or inhibition) depends on the presence of yet another ancillary subunit - DPP6 (Zhang et al. 2020).
3.2. Sensitization by increasing the affinity of a small molecule that also binds to and modulates α subunit-only channels
3.2.1. Modulation of KCNQ1 pharmacology by KCNEs
As described above, KCNQ1 is differentially regulated by a molecular toolkit of KCNE subunits to permit it to function in a wide range of cellular environments and overcome challenges such as extracellular acidification (in gastric parietal cells), lack of excitability (various epithelia cells) and diverse localization requirements (e.g., apical for KCNQ1-KCNE2 in parietal cells, basolateral for KCNQ1-KCNE3 in the lower digestive tract). KCNE subunits have long been known to alter the pharmacology of small molecules that modulate KCNQ1 (Panaghie and Abbott 2006). Therefore, there is the possibility that the differences in pharmacology of the different KCNQ1-KCNE complexes could one day be exploited to increase selectivity and target the desired tissue type while sparing others.
Unlike JNJ303, described in the previous section, Chromanol 293B is a classic inhibitor of both KCNQ1 and IKs, but it blocks with considerable differential sensitivity dependent on the presence of KCNE subunits. In particular, KCNE3 highly sensitizes KCNQ1 to inhibition by chromanol 293B, decreasing the IC50 100-fold from 65 to 0.5 μM; KCNQ1-KCNE1 complexes lie in the middle, with an IC50 of 15 μM. One might suspect that this effect could arise from the widely different gating attributes of the three channel types, in particular that KCNQ1-KCNE3 has a much higher open probability at more negative voltages than the others. However, it was found that KCNQ1-KCNE3-V72T channels, which exhibit time- and voltage-dependent activation more akin to KCNQ1-KCNE1 channels, showed similar chromanol 293B sensitivity to that of wild-type KCNQ1-KCNE3, thus suggesting a change in the drug binding site itself rather than solely an indirect effect linked to altered gating (Bett et al. 2006).
3.2.2. Wild-type and inherited mutant KCNE2 effects on hERG pharmacology
The hERG (human ether-à-go-go related gene product) potassium channel (a.k.a. Kv11.1; KCNH2), is essential for human ventricular myocyte repolarization. It generates IKr (the rapidly activating potassium current) in ventricular myocardium of humans, guineapigs, rabbits, rats, but is absent in mice. Inherited loss-of-function mutations in the KCNH2 gene that encodes hERG account for as many as 40–45% of sequenced inherited Long QT syndrome cases (termed LQT2 if they involve hERG), roughly equal to the proportion for KCNQ1 (LQT1), with sodium channel Nav1.5 gene SCN5A gain-of-function mutations (LQT3) reaching around 10%. The remaining small fraction of sequenced LQTS cases mostly involve ancillary subunits or other channels (Abbott 2013) (see also Chapter 4; Seebohm 2021).
hERG is regulated by both KCNE1 (originally termed MinK, IsK) and KCNE2 (MiRP1), although the relative importance of homomeric hERG, versus hERG-KCNE1 or hERG-KCNE2, channels in the myocardia of humans and the various other species is not fully understood (Abbott 2015; Abbott et al. 1999; McDonald et al. 1997). There is biochemical evidence for each type of heteromeric complex in cardiac tissue and also evidence from human genetics studies. While KCNE1 doubles hERG current by an unclear mechanism, KCNE2 reduces hERG current by 40%, partly by reducing unitary conductance. Other effects include speeding of deactivation, right-shifting of the voltage dependence of activation, reduction of sensitivity to external K+, and endowing hERG with a biphasic response to the inhibitor E-4031, more closely matching that of native IKr (Abbott 2015; Abbott et al. 1999; McDonald et al. 1997).
Inherited KCNE1 mutations are known to cause LQTS (the LQT5 form) but this is primarily considered to occur because of dysfunction of KCNQ1-KCNE1 (IKs) complexes in cardiac myocytes. A recessive inherited disorder, Jervell Lange-Nielsen syndrome (JLNS), is well known to cause both LQTS and sensorineural deafness. This reflects the importance of KCNQ1-KCNE1 channels in the inner ear, where they regulate K+ secretion into the endolymph. Without this channel functioning correctly, the inner ear develops abnormally resulting in deafness. Mutations in either KCNE1 or KCNQ1 cause JLNS; typically, both alleles carry a loss-of-function mutation to cause JLNS although this is not always the case (Schulze-Bahr et al. 1997; Tranebjaerg et al. 1993; Tyson et al. 1997). KCNQ1 has been found to form complexes with hERG in vitro and in vivo, a surprising result given that it is generally considered that Kv channels can only heteromultimerize with α subunits from their own subfamily (e.g., Kv1.1 with Kv1.4; Kv3.1 with Kv3.4). It is therefore possible that KCNE1 (or KCNE2) mutations could reduce myocyte repolarization reserve by disrupting IKs, IKr, or coupled IKs-IKr complexes (Organ-Darling et al. 2013; Ren et al. 2010).
For inherited KCNE2 gene variants, several associations between specific mutations and polymorphisms have been found to associate with LQTS, a caveat being that in most cases, a second insult is likely required to cause arrhythmia. hERG is known to be highly susceptible to inhibition by a wide range of small molecules, many of which are or were FDA-approved and in clinical use for disorders unrelated to cardiac function, e.g., antihistamines, antibiotics. Paradoxically, some Class I antiarrhythmics (main anti-arrhythmic action is sodium channel block) are also proarrhythmic in some settings, because they also block hERG channels. This has resulted in restricted use of some antiarrhythmics based on the diagnosis and susceptibility to acquired (drug-induced) arrhythmia, and also drugs being completely withdrawn from clinical use. Thus, avoiding hERG block is seen as an important step during drug development, to help avoid cardiotoxicity (Guo et al. 2009; Recanatini et al. 2005; Roy et al. 1996; Testai et al. 2004); see also Chapter 5; Su et al. 2021).
The relevance with respect to KCNE2 is that some mutations or even common polymorphisms in the KCNE2 gene can increase drug susceptibility of hERG-KCNE2 channels and therefore predispose to acquired arrhythmia (a combined inherited/acquired form of LQTS). There are several different categories of KCNE2 gene variant in this regard, three of which are found in gene variants that occur in a cluster on the extracellular N-terminal of KCNE2. The first is represented by the T8A polymorphism, which is present in around 1.6% of Caucasians studied in the United States and was not found in African-Americans. T8A does not alter channel function at baseline. However, it increases susceptibility of hERG-KCNE2 channels to block by the antibiotic sulfamethoxazole, shifting the Ki almost twofold, from 380 to 210 μg/ml. In addition, sulfamethoxazole only speeded deactivation of T8A, and not wild-type hERG-KCNE2 channels. T8A was discovered in a patient with sulfamethoxazole-induced arrhythmia and appears to predispose to this condition based on the clinical and cellular electrophysiology findings. Thus, T8A is a silent polymorphism at baseline, its effects on function only uncovered in the context of drug block (Abbott et al. 1999; Sesti et al. 2000). Interestingly, the mechanism for enhanced drug block was found to be loss of a consensus glycosylation site, preventing addition of a carbohydrate moiety that normally helps to shield hERG-KCNE2 channels from sulfamethoxazole block (Park et al. 2003).
These effects contrast with that of KCNE2-Q9E, which causes both altered hERG-KCNE2 channel function at baseline (+9 mV shift in the voltage dependence of activation and an 80% increase in the fast time constant of deactivation, with no change in the slow component) and threefold-increased sensitivity to the macrolide antibiotic, clarithromycin. The Q9E variant was originally discovered in an African-American woman who presented with clarithromycin-induced LQTS after receiving intravenous clarithromycin in hospital; Q9E was subsequently discovered to be present in 3% of African-Americans and absent from Caucasians in the US (Abbott et al. 1999; Ackerman et al. 2003).
A third gene variant in this N-terminal cluster, KCNE2-T10M, was found in a woman who had a history of episodes of auditory-induced syncope (a hallmark of KCNH2-linked LQTS) and who presented with prolonged QT interval and ventricular fibrillation with hypomagnesemia and hypocalcemia after running the New York Marathon. She also exhibited later ventricular fibrillation concurrent with hypokalemia. Other family members carried the variant and did not exhibit LQTS; the subject did not harbor mutations in KCNH2 or other known LQTS genes. In cellular electrophysiology experiments, we found that T10M reduced hERG-KCNE2 currents by up to 80%, left-shifts the voltage dependence of inactivation, slows inactivation and recovery from inactivation, but did not alter sensitivity to extracellular cations. It was concluded that the T10M variant is a loss-of-function mutation that is tolerable until overlapping with another insult, in this case perturbation of extracellular cations such as K+. Increased extracellular [K+] paradoxically augments hERG and hERG-KCNE2 outward currents, against driving force (up to 10 mM extracellular K+), because of beneficial interactions with the outer pore. Thus, the hypokalemia experienced by the patient, exacerbated by lower repolarization reserve due to the T10M variant, likely contributed to their arrhythmia (Gordon et al. 2008).
Three other variants, M54T, I57T and A116V, were found in individuals with procainamide-, oxatomide- and quinidine- induced LQTS, respectively (Abbott et al. 1999; Sesti et al. 2000). None of these mutations altered the affinity of hERG-KCNE2 channels to block by the respective drugs, but all impaired channel function at baseline (by ~30% in terms of peak tail current inhibition). These then would fall into the category, like T10M of KCNE2 variants that predispose to acquired arrhythmia by reducing repolarization reserve enough that superimposition of another IKr-impairing environmental agent (e.g., drug, electrolyte imbalance) causes pathogenic IKr reduction. We also identified M54T and I57T in subjects with LQTS with no known environmental IKr reducing factors, so it is possible that in rare cases a drug or other agent is not needed for KCNE2-associated LQTS. Overall, given its effects on hERG sensitivity to electrolytes, E-4031, sulfamethoxazole and clarithromycin, it is clear that KCNE2 (either in wild-type or mutant form) falls into the category of an ancillary subunit that modulates hERG sensitivity to drugs that also act on the homomeric channel (Abbott et al. 1999; Sesti et al. 2000).
3.2.3. G protein βγ (Gβγ) subunits
Aside from the SURs (described in section 2.1), the other most important Kir channel ancillary subunits are the membrane-associated G protein βγ (Gβγ) subunits. G protein-coupled inwardly rectifying potassium channels (GIRKs) are activated by a combination of phosphatidylinositol 4,5-bisphosphate (PIP2) and by GPCR stimulation, e.g., by neurotransmitters. GPCR stimulation causes Gβγ subunits to be released, dissociating from the Gα subunit. Gβγ subunits are then free to directly bind to and activate Kir3.1–3.4 (GIRK1–4) α subunits; the resultant heteromeric channel complexes are referred to as Kir3.x or GIRK channels. Kir3.1–3.3 are expressed predominantly in the nervous system, while Kir3.4 is primarily a cardiac subunit. The minor membrane phospholipid component but prominent signaling molecule, PIP2, is able to directly bind to and activate homomeric Kir3 channels in the absence of Gβγ subunits, but PIP2 is a much more potent activator of heteromeric Kir3-Gβγ channels. In contrast, Gβγ subunits cannot activate Kir3 channels in the absence of PIP2. Thus, Gβγ subunits regulate Kir3 channels by sensitizing them to PIP2, probably by increasing the affinity of Kir3 channels for PIP2 (Glaaser and Slesinger 2015; Hibino et al. 2010; Sadja et al. 2003; Yamada et al. 1998).
3.3. Desensitization by decreasing the affinity of a small molecule that binds to and modulates α subunit-only K+ channels
Receptor desensitization by ancillary subunits is an important consideration in channel pharmacology because it can potentially be leveraged to provide increased specificity if the α subunit-ancillary subunit expression and interaction profiles are well understood across different cell and tissue types. Several of the TM ancillary subunits desensitize their α subunit partners to pharmacological agents and other types of channel regulators, and we consider various key examples below.
The BKβ subunits can act to decrease the sensitivity of BK channels to some molecules that regulate homomeric BK α subunit channels. Thus, BKβ1, and more so BKβ2- β4, reduce BK channel sensitivity to block by charybodotoxin (ChTx) and/or Iberiotoxin (IbTx), with β4 inducing almost complete resistance. The extracellular loop of the BKβ subunits mediates the altered sensitivity to ChTx, specifically L90, Y91, T93 and E94 in β1, and a lysine-rich ring comprising K137, 141, 147 and 150 in β2 (Li and Yan 2016; Torres et al. 2014). In BKβ4, which is a more powerful protector against of toxin inhibition, K120, R121 and K125 together with some BK α subunit residues, form a shield against toxin blockade (Gan et al. 2008) (Figure 3).
The KCNE3 regulatory subunit, which regulates KCNQ1 in colonic epithelium, also regulates the Kv3.4 potassium channel α subunit, in skeletal muscle. An R83H gene variant in KCNE3 predisposes to human periodic paralysis and reduces the ability of Kv3.4-KCNE3 channels to activate at subthreshold membrane potentials, thereby shifting the membrane potential of C2C12 skeletal muscle cells more positive (Abbott et al. 2001a; Abbott et al. 2006). Mice with germline deletion of Kcne3 exhibit skeletal muscle downregulation of hERG (also a partner of KCNE3), Kv3.4 and the K2P channel, KCNK4 (TRAAK). Kcne3 knockout gastrocnemius muscle exhibits transcript remodeling and - consistent with increased oxidative metabolic activity - increased type IIa fast twitch oxidative muscle tissue. Kcne3 knockout also causes abnormal hind limb clasping upon tail suspension, loss of the normal biphasic decline in contractile force upon repetitive hind limb muscle stimulation, and impaired skeletal myoblast K+ currents, indicating a primary role for Kv3.4-KCNE3 channels in skeletal muscle (King et al. 2017). Strikingly, and as observed for the toxin response in BK channels containing BKβ subunits (see above), KCNE3 greatly desensitizes Kv3.4 channels to block by the sea anemone toxin, blood-depressing substance II (BDS-II), assessed by quantifying open probability at the single-channel level. KCNE3 reduced the BDS-II Ki of Kv3.4 30-fold, from 260 nM to 7.2 μM, recapitulating the BDS-II sensitivity of native C2C12 muscle cell currents (6.9 μM). It is likely that KCNE3 forms a protective shield that disrupts toxin binding to a site on the α subunit, as observed for BK channels (Abbott et al. 2001a).
Finally, as mentioned above, homomeric KCNQ3 channels are sensitive to and are activated by GABA and β-hydroxybutyrate (Manville et al. 2018, 2020). Heteromeric KCNQ2/3 channels are also sensitive and can transmit this sensitivity to physically coupled SMIT1 myo-inositol transporter, resulting in inhibition of transport activity. However, when KCNQ3 (in the absence of KCNQ2) forms physical complexes with SMIT1, the complex loses sensitivity to β-hydroxybutyrate, while retaining GABA sensitivity. An arginine in the S4–5 linker (R242 in KCNQ3) is required for communicating GABA binding to SMIT1 and is also known to be important for GABA binding; mutation of this residue also results in dramatic current inhibition upon SMIT1 binding to the channel. GABA induces a novel pore conformation in KCNQ3 and KCNQ5 that represents an activated state (GABA also binds to KCNQ2 and KCNQ4 but neither alters the pore conformation nor activates these two isoforms) and SMIT1 itself binds to the KCNQ pore module and induces a similar conformational shift (Manville and Abbott 2020). In sum, it appears that SMIT1 co-assembly alters the conformation of the S5/S4–5 binding site that coordinates binding of GABA, β-hydroxybutyrate and other small molecules such as retigabine.
SMIT1 also reduces sensitivity of KCNQ2/3 to the classic K+ channel pore-blocking quaternary ammonium ion, tetraethylammonium (TEA) (by fourfold, increasing the EC50 from 7 to 29 mM at 0 mV). SMIT1 also decreased KCNQ1 TEA sensitivity by fourfold at 0 mV (EC50 shifted from 29 to 121 mV) and reversed the voltage dependence of KCNQ1 inhibition (Manville et al. 2017).
Conclusions
The ion channel structural biology revolution that started with X-ray crystallography and that has now benefitted from rapid advances in cryo-EM is enabling structural biologists to solve high-resolution structures of ion channel complexes with both their α and ancillary subunits, and in some cases drugs and other regulatory small molecules bound. These data can be understood in the context of advances in our knowledge of the essential roles of ancillary subunits in specific tissues and physiological roles, and with understanding of the effects of ancillary subunits on native ion channel function, derived from cellular electrophysiology, transgenic mouse and human genetics studies. Emerging from this are realistic prospects of future drugs that achieve specificity by targeting specific α-ancillary subunit complexes while leaving others untouched.
Acknowledgements
GWA is grateful for financial support from the National Institutes of Health, National Institute of General Medical Sciences and National Institute of Neurological Disorders and Stroke (GM130377 to GWA).
References
- Abbott GW (2013) KCNE genetics and pharmacogenomics in cardiac arrhythmias: much ado about nothing? Expert Rev Clin Pharmacol 6: 49–60. doi: 10.1586/ecp.12.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW (2014) Biology of the KCNQ1 potassium channel. New Journal of Science 2014: 26. doi: 10.1155/2014/237431 [DOI] [Google Scholar]
- Abbott GW (2015) The KCNE2 K(+) channel regulatory subunit: Ubiquitous influence, complex pathobiology. Gene 569: 162–72. doi: 10.1016/j.gene.2015.06.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW (2016a) Channel-transporter complexes: an emerging theme in cell signaling. Biochem J 473: 3759–3763. doi: 10.1042/BCJ20160685C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW (2016b) KCNE1 and KCNE3: The yin and yang of voltage-gated K(+) channel regulation. Gene 576: 1–13. doi: 10.1016/j.gene.2015.09.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW (2016c) KCNE4 and KCNE5: K(+) channel regulation and cardiac arrhythmogenesis. Gene 593: 249–60. doi: 10.1016/j.gene.2016.07.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW (2016d) Novel exon 1 protein-coding regions N-terminally extend human KCNE3 and KCNE4. FASEB J 30: 2959–69. doi: 10.1096/fj.201600467R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW (2017a) beta Subunits Functionally Differentiate Human Kv4.3 Potassium Channel Splice Variants. Front Physiol 8: 66. doi: 10.3389/fphys.2017.00066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW (2017b) Chansporter complexes in cell signaling. FEBS Lett 591: 2556–2576. doi: 10.1002/1873-3468.12755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW, Butler MH, Bendahhou S, Dalakas MC, Ptacek LJ, Goldstein SA (2001a) MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 104: 217–31. [DOI] [PubMed] [Google Scholar]
- Abbott GW, Butler MH, Goldstein SA (2006) Phosphorylation and protonation of neighboring MiRP2 sites: function and pathophysiology of MiRP2-Kv3.4 potassium channels in periodic paralysis. FASEB J 20: 293–301. doi: 10.1096/fj.05-5070com [DOI] [PubMed] [Google Scholar]
- Abbott GW, Goldstein SA (1998) A superfamily of small potassium channel subunits: form and function of the MinK-related peptides (MiRPs). Q Rev Biophys 31: 357–98. [DOI] [PubMed] [Google Scholar]
- Abbott GW, Goldstein SA, Sesti F (2001b) Do all voltage-gated potassium channels use MiRPs? Circ Res 88: 981–3. [DOI] [PubMed] [Google Scholar]
- Abbott GW, Roepke TK (2016) KCNE2 and gastric cancer: bench to bedside. Oncotarget 7: 17286–7. doi: 10.18632/oncotarget.7921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA (1999) MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97: 175–87. [DOI] [PubMed] [Google Scholar]
- Abbott GW, Tai KK, Neverisky DL, Hansler A, Hu Z, Roepke TK, Lerner DJ, Chen Q, Liu L, Zupan B, Toth M, Haynes R, Huang X, Demirbas D, Buccafusca R, Gross SS, Kanda VA, Berry GT (2014) KCNQ1, KCNE2, and Na+-Coupled Solute Transporters Form Reciprocally Regulating Complexes That Affect Neuronal Excitability. Sci Signal 7: ra22. doi: 10.1126/scisignal.2005025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott GW, Xu X, Roepke TK (2007) Impact of ancillary subunits on ventricular repolarization. J Electrocardiol 40: S42–6. doi: 10.1016/j.jelectrocard.2007.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME (2003) Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc 78: 1479–87. doi: 10.4065/78.12.1479 [DOI] [PubMed] [Google Scholar]
- Alzamora R, O’Mahony F, Bustos V, Rapetti-Mauss R, Urbach V, Cid LP, Sepulveda FV, Harvey BJ (2011) Sexual dimorphism and oestrogen regulation of KCNE3 expression modulates the functional properties of KCNQ1 K(+) channels. J Physiol 589: 5091–107. doi: 10.1113/jphysiol.2011.215772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadi CC, Brust RD, Skerritt MR, Campbell DL (2007) Regulation of Kv4.3 closed state inactivation and recovery by extracellular potassium and intracellular KChIP2b. Channels (Austin) 1: 305–14. [DOI] [PubMed] [Google Scholar]
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ (2000) Modulation of A-type potassium channels by a family of calcium sensors. Nature 403: 553–6. doi: 10.1038/35000592 [DOI] [PubMed] [Google Scholar]
- Angelo K, Jespersen T, Grunnet M, Nielsen MS, Klaerke DA, Olesen SP (2002) KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys J 83: 1997–2006. doi: 10.1016/S0006-3495(02)73961-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson NS, Robertson GA, Ganetzky B (1991) A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science 253: 551–5. doi: 10.1126/science.1857984 [DOI] [PubMed] [Google Scholar]
- Bahring R (2018) Kv channel-interacting proteins as neuronal and non-neuronal calcium sensors. Channels (Austin) 12: 187–200. doi: 10.1080/19336950.2018.1491243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahring R, Dannenberg J, Peters HC, Leicher T, Pongs O, Isbrandt D (2001) Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. J Biol Chem 276: 23888–94. doi: 10.1074/jbc.M101320200 [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G (1996) K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384: 78–80. doi: 10.1038/384078a0 [DOI] [PubMed] [Google Scholar]
- Bendahhou S, Marionneau C, Haurogne K, Larroque MM, Derand R, Szuts V, Escande D, Demolombe S, Barhanin J (2005) In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc Res 67: 529–38. doi: 10.1016/j.cardiores.2005.02.014 [DOI] [PubMed] [Google Scholar]
- Berkefeld H, Fakler B (2013) Ligand-gating by Ca2+ is rate limiting for physiological operation of BK(Ca) channels. J Neurosci 33: 7358–67. doi: 10.1523/JNEUROSCI.5443-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Fakler B, Schulte U (2010) Ca2+-activated K+ channels: from protein complexes to function. Physiol Rev 90: 1437–59. doi: 10.1152/physrev.00049.2009 [DOI] [PubMed] [Google Scholar]
- Bett GC, Morales MJ, Beahm DL, Duffey ME, Rasmusson RL (2006) Ancillary subunits and stimulation frequency determine the potency of chromanol 293B block of the KCNQ1 potassium channel. J Physiol 576: 755–67. doi: 10.1113/jphysiol.2006.116012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond CT, Maylie J, Adelman JP (1999) Small-conductance calcium-activated potassium channels. Ann N Y Acad Sci 868: 370–8. doi: 10.1111/j.1749-6632.1999.tb11298.x [DOI] [PubMed] [Google Scholar]
- Bukiya AN, Liu J, Toro L, Dopico AM (2007) Beta1 (KCNMB1) subunits mediate lithocholate activation of large-conductance Ca2+-activated K+ channels and dilation in small, resistance-size arteries. Mol Pharmacol 72: 359–69. doi: 10.1124/mol.107.034330 [DOI] [PubMed] [Google Scholar]
- Bukiya AN, Vaithianathan T, Toro L, Dopico AM (2009) Channel beta2-4 subunits fail to substitute for beta1 in sensitizing BK channels to lithocholate. Biochem Biophys Res Commun 390: 995–1000. doi: 10.1016/j.bbrc.2009.10.091 [DOI] [PubMed] [Google Scholar]
- Campomanes CR, Carroll KI, Manganas LN, Hershberger ME, Gong B, Antonucci DE, Rhodes KJ, Trimmer JS (2002) Kv beta subunit oxidoreductase activity and Kv1 potassium channel trafficking. J Biol Chem 277: 8298–305. doi: 10.1074/jbc.M110276200 [DOI] [PubMed] [Google Scholar]
- Castillo K, Contreras GF, Pupo A, Torres YP, Neely A, Gonzalez C, Latorre R (2015) Molecular mechanism underlying beta1 regulation in voltage- and calcium-activated potassium (BK) channels. Proc Natl Acad Sci U S A 112: 4809–14. doi: 10.1073/pnas.1504378112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KW, Zhang H, Logothetis DE (2003) N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J 22: 3833–43. doi: 10.1093/emboj/cdg376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampa EJ, Welch RC, Vanoye CG, George AL Jr. (2011) KCNE4 juxtamembrane region is required for interaction with calmodulin and for functional suppression of KCNQ1. J Biol Chem 286: 4141–9. doi: 10.1074/jbc.M110.158865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman SK, Newcombe J, Pryke J, Dolly JO (1999) Subunit composition of Kv1 channels in human CNS. J Neurochem 73: 849–58. doi: 10.1046/j.1471-4159.1999.0730849.x [DOI] [PubMed] [Google Scholar]
- Duprat F, Lauritzen I, Patel A, Honore E (2007) The TASK background K2P channels: chemo- and nutrient sensors. Trends Neurosci 30: 573–80. doi: 10.1016/j.tins.2007.08.003 [DOI] [PubMed] [Google Scholar]
- Fakler B, Brandle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP (1995) Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell 80: 149–54. [DOI] [PubMed] [Google Scholar]
- Foeger NC, Wang W, Mellor RL, Nerbonne JM (2013) Stabilization of Kv4 protein by the accessory K(+) channel interacting protein 2 (KChIP2) subunit is required for the generation of native myocardial fast transient outward K(+) currents. J Physiol 591: 4149–66. doi: 10.1113/jphysiol.2013.255836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan G, Yi H, Chen M, Sun L, Li W, Wu Y, Ding J (2008) Structural basis for toxin resistance of beta4-associated calcium-activated potassium (BK) channels. J Biol Chem 283: 24177–84. doi: 10.1074/jbc.M800179200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Nunziato DA, Pitt GS (2006) KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ Res 98: 1048–54. doi: 10.1161/01.RES.0000218863.44140.f2 [DOI] [PubMed] [Google Scholar]
- Glaaser IW, Slesinger PA (2015) Structural Insights into GIRK Channel Function. Int Rev Neurobiol 123: 117–60. doi: 10.1016/bs.irn.2015.05.014 [DOI] [PubMed] [Google Scholar]
- Gofman Y, Shats S, Attali B, Haliloglu T, Ben-Tal N (2012) How does KCNE1 regulate the Kv7.1 potassium channel? Model-structure, mutations, and dynamics of the Kv7.1-KCNE1 complex. Structure 20: 1343–52. doi: 10.1016/j.str.2012.05.016 [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Bockenhauer D, O’Kelly I, Zilberberg N (2001) Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci 2: 175–84. doi: 10.1038/35058574 [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Price LA, Rosenthal DN, Pausch MH (1996) ORK1, a potassium-selective leak channel with two pore domains cloned from Drosophila melanogaster by expression in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 93: 13256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong N, Bodi I, Zobel C, Schwartz A, Molkentin JD, Backx PH (2006) Calcineurin increases cardiac transient outward K+ currents via transcriptional up-regulation of Kv4.2 channel subunits. J Biol Chem 281: 38498–506. doi: 10.1074/jbc.M607774200 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez V, Lingle CJ (2019) Regulation of BK Channels by Beta and Gamma Subunits. Annu Rev Physiol 81: 113–137. doi: 10.1146/annurev-physiol-022516-034038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, Christini DJ, Ostrer H, Basson CT, Chung W, Abbott GW (2008) A KCNE2 mutation in a patient with cardiac arrhythmia induced by auditory stimuli and serum electrolyte imbalance. Cardiovasc Res 77: 98–106. doi: 10.1093/cvr/cvm030 [DOI] [PubMed] [Google Scholar]
- Grahammer F, Warth R, Barhanin J, Bleich M, Hug MJ (2001) The small conductance K+ channel, KCNQ1: expression, function, and subunit composition in murine trachea. J Biol Chem 276: 42268–75. doi: 10.1074/jbc.M105014200 [DOI] [PubMed] [Google Scholar]
- Grunnet M, Jespersen T, Rasmussen HB, Ljungstrom T, Jorgensen NK, Olesen SP, Klaerke DA (2002) KCNE4 is an inhibitory subunit to the KCNQ1 channel. J Physiol 542: 119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunnet M, Olesen SP, Klaerke DA, Jespersen T (2005) hKCNE4 inhibits the hKCNQ1 potassium current without affecting the activation kinetics. Biochem Biophys Res Commun 328: 1146–53. doi: 10.1016/j.bbrc.2005.01.071 [DOI] [PubMed] [Google Scholar]
- Grunnet M, Rasmussen HB, Hay-Schmidt A, Rosenstierne M, Klaerke DA, Olesen SP, Jespersen T (2003) KCNE4 is an inhibitory subunit to Kv1.1 and Kv1.3 potassium channels. Biophys J 85: 1525–37. doi: 10.1016/S0006-3495(03)74585-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbis JM, Mann S, MacKinnon R (1999) Structure of a voltage-dependent K+ channel beta subunit. Cell 97: 943–52. [DOI] [PubMed] [Google Scholar]
- Gulbis JM, Zhou M, Mann S, MacKinnon R (2000) Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science 289: 123–7. [DOI] [PubMed] [Google Scholar]
- Guo D, Klaasse E, de Vries H, Brussee J, Nalos L, Rook MB, Vos MA, van der Heyden MA, Ijzerman AP (2009) Exploring chemical substructures essential for HERG k(+) channel blockade by synthesis and biological evaluation of dofetilide analogues. ChemMedChem 4: 1722–32. doi: 10.1002/cmdc.200900203 [DOI] [PubMed] [Google Scholar]
- Heitzmann D, Koren V, Wagner M, Sterner C, Reichold M, Tegtmeier I, Volk T, Warth R (2007) KCNE beta subunits determine pH sensitivity of KCNQ1 potassium channels. Cell Physiol Biochem 19: 21–32. doi: 10.1159/000099189 [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366. doi: 10.1152/physrev.00021.2009 [DOI] [PubMed] [Google Scholar]
- Hille B, Armstrong CM, MacKinnon R (1999) Ion channels: from idea to reality. Nat Med 5: 1105–9. doi: 10.1038/13415 [DOI] [PubMed] [Google Scholar]
- Hu B, Zeng WP, Li X, Al-Sheikh U, Chen SY, Ding J (2019) A conserved arginine/lysine-based motif promotes ER export of KCNE1 and KCNE2 to regulate KCNQ1 channel activity. Channels (Austin) 13: 483–497. doi: 10.1080/19336950.2019.1685626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isacoff EY, Jan YN, Jan LY (1990) Evidence for the formation of heteromultimeric potassium channels in Xenopus oocytes. Nature 345: 530–4. doi: 10.1038/345530a0 [DOI] [PubMed] [Google Scholar]
- Jespersen T, Membrez M, Nicolas CS, Pitard B, Staub O, Olesen SP, Baro I, Abriel H (2007) The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc Res 74: 64–74. doi: 10.1016/j.cardiores.2007.01.008 [DOI] [PubMed] [Google Scholar]
- Kanda VA, Lewis A, Xu X, Abbott GW (2011a) KCNE1 and KCNE2 inhibit forward trafficking of homomeric N-type voltage-gated potassium channels. Biophys J 101: 1354–63. doi: 10.1016/j.bpj.2011.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda VA, Lewis A, Xu X, Abbott GW (2011b) KCNE1 and KCNE2 provide a checkpoint governing voltage-gated potassium channel alpha-subunit composition. Biophys J 101: 1364–75. doi: 10.1016/j.bpj.2011.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda VA, Purtell K, Abbott GW (2011c) Protein kinase C downregulates I(Ks) by stimulating KCNQ1-KCNE1 potassium channel endocytosis. Heart Rhythm 8: 1641–7. doi: 10.1016/j.hrthm.2011.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, Tian C, Sonnichsen FD, Smith JA, Meiler J, George AL Jr., Vanoye CG, Kim HJ, Sanders CR (2008) Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry 47: 7999–8006. doi: 10.1021/bi800875q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketchum KA, Joiner WJ, Sellers AJ, Kaczmarek LK, Goldstein SA (1995) A new family of outwardly rectifying potassium channel proteins with two pore domains in tandem. Nature 376: 690–5. doi: 10.1038/376690a0 [DOI] [PubMed] [Google Scholar]
- Kilfoil PJ, Chapalamadugu KC, Hu X, Zhang D, Raucci FJ Jr., Tur J, Brittian KR, Jones SP, Bhatnagar A, Tipparaju SM, Nystoriak MA (2019) Metabolic regulation of Kv channels and cardiac repolarization by Kvbeta2 subunits. J Mol Cell Cardiol 137: 93–106. doi: 10.1016/j.yjmcc.2019.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EC, Patel V, Anand M, Zhao X, Crump SM, Hu Z, Weisleder N, Abbott GW (2017) Targeted deletion of Kcne3 impairs skeletal muscle function in mice. FASEB J. doi: 10.1096/fj.201600965RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollewe A, Lau AY, Sullivan A, Roux B, Goldstein SA (2009) A structural model for K2P potassium channels based on 23 pairs of interacting sites and continuum electrostatics. J Gen Physiol 134: 53–68. doi: 10.1085/jgp.200910235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroncke BM, Van Horn WD, Smith J, Kang C, Welch RC, Song Y, Nannemann DP, Taylor KC, Sisco NJ, George AL Jr., Meiler J, Vanoye CG, Sanders CR (2016) Structural basis for KCNE3 modulation of potassium recycling in epithelia. Sci Adv 2: e1501228. doi: 10.1126/sciadv.1501228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzystanek K, Rasmussen HB, Grunnet M, Staub O, Olesen SP, Abriel H, Jespersen T (2012) Deubiquitylating enzyme USP2 counteracts Nedd4-2-mediated downregulation of KCNQ1 potassium channels. Heart Rhythm 9: 440–8. doi: 10.1016/j.hrthm.2011.10.026 [DOI] [PubMed] [Google Scholar]
- Kshatri AS, Gonzalez-Hernandez A, Giraldez T (2018) Physiological Roles and Therapeutic Potential of Ca(2+) Activated Potassium Channels in the Nervous System. Front Mol Neurosci 11: 258. doi: 10.3389/fnmol.2018.00258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Baldwin TJ, Jan YN, Jan LY (1993) Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature 362: 127–33. doi: 10.1038/362127a0 [DOI] [PubMed] [Google Scholar]
- Li N, Wu JX, Ding D, Cheng J, Gao N, Chen L (2017) Structure of a Pancreatic ATP-Sensitive Potassium Channel. Cell 168: 101–110 e10. doi: 10.1016/j.cell.2016.12.028 [DOI] [PubMed] [Google Scholar]
- Li Q, Yan J (2016) Modulation of BK Channel Function by Auxiliary Beta and Gamma Subunits. Int Rev Neurobiol 128: 51–90. doi: 10.1016/bs.irn.2016.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Cai H, Zheng W, Tong M, Li H, Ao L, Li J, Hong G, Li M, Guan Q, Yang S, Yang D, Lin X, Guo Z (2016) An individualized prognostic signature for gastric cancer patients treated with 5-Fluorouracil-based chemotherapy and distinct multi-omics characteristics of prognostic groups. Oncotarget. doi: 10.18632/oncotarget.7087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Maylie J, Adelman JP (2009) New sites of action for GIRK and SK channels. Nat Rev Neurosci 10: 475–80. doi: 10.1038/nrn2668 [DOI] [PubMed] [Google Scholar]
- Lundquist AL, Manderfield LJ, Vanoye CG, Rogers CS, Donahue BS, Chang PA, Drinkwater DC, Murray KT, George AL Jr. (2005) Expression of multiple KCNE genes in human heart may enable variable modulation of I(Ks). J Mol Cell Cardiol 38: 277–87. doi: 10.1016/j.yjmcc.2004.11.012 [DOI] [PubMed] [Google Scholar]
- MacKinnon R (2003) Potassium channels. FEBS Lett 555: 62–5. [DOI] [PubMed] [Google Scholar]
- Malysiak K, Grzywna ZJ (2008) On the possible methods for the mathematical description of the ball and chain model of ion channel inactivation. Cell Mol Biol Lett 13: 535–52. doi: 10.2478/s11658-008-0015-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderfield LJ, Daniels MA, Vanoye CG, George AL Jr. (2009) KCNE4 domains required for inhibition of KCNQ1. J Physiol 587: 303–14. doi: 10.1113/jphysiol.2008.161281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderfield LJ, George AL Jr. (2008) KCNE4 can co-associate with the I(Ks) (KCNQ1-KCNE1) channel complex. FEBS J 275: 1336–49. doi: 10.1111/j.1742-4658.2008.06294.x [DOI] [PubMed] [Google Scholar]
- Manville RW, Abbott GW (2020) Potassium channels act as chemosensors for solute transporters. Commun Biol 3: 90. doi: 10.1038/s42003-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville RW, Neverisky DL, Abbott GW (2017) SMIT1 Modifies KCNQ Channel Function and Pharmacology by Physical Interaction with the Pore. Biophys J 113: 613–626. doi: 10.1016/j.bpj.2017.06.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville RW, Papanikolaou M, Abbott GW (2018) Direct neurotransmitter activation of voltage-gated potassium channels. Nat Commun 9: 1847. doi: 10.1038/s41467-018-04266-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville RW, Papanikolaou M, Abbott GW (2020) M-Channel Activation Contributes to the Anticonvulsant Action of the Ketone Body beta-Hydroxybutyrate. J Pharmacol Exp Ther 372: 148–156. doi: 10.1124/jpet.119.263350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM, Kandasamy B, DiMaio F, Yoshioka C, Shyng SL (2017) Anti-diabetic drug binding site in a mammalian KATP channel revealed by Cryo-EM. Elife 6. doi: 10.7554/eLife.31054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM, Sung MW, Shyng SL (2020) Pharmacological chaperones of ATP-sensitive potassium channels: Mechanistic insight from cryoEM structures. Mol Cell Endocrinol 502: 110667. doi: 10.1016/j.mce.2019.110667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM, Sung MW, Yang Z, Innes LM, Kandasamy B, David LL, Yoshioka C, Shyng SL (2019) Mechanism of pharmacochaperoning in a mammalian KATP channel revealed by cryo-EM. Elife 8. doi: 10.7554/eLife.46417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A, Al-Moubarak E, Veale EL (2010) Gating of two pore domain potassium channels. J Physiol 588: 3149–56. doi: 10.1113/jphysiol.2010.192344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, Goldstein SA, Fishman GI (1997) A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature 388: 289–92. doi: 10.1038/40882 [DOI] [PubMed] [Google Scholar]
- Miller AN, Long SB (2012) Crystal structure of the human two-pore domain potassium channel K2P1. Science 335: 432–6. doi: 10.1126/science.1213274 [DOI] [PubMed] [Google Scholar]
- Morin TJ, Kobertz WR (2008) Counting membrane-embedded KCNE beta-subunits in functioning K+ channel complexes. Proc Natl Acad Sci U S A 105: 1478–82. doi: 10.1073/pnas.0710366105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, Kubo Y (2007) KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J Gen Physiol 130: 269–81. doi: 10.1085/jgp.200709805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, Ulbrich MH, Kubo Y, Isacoff EY (2010) Stoichiometry of the KCNQ1 - KCNE1 ion channel complex. Proc Natl Acad Sci U S A 107: 18862–7. doi: 10.1073/pnas.1010354107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neverisky DL, Abbott GW (2015) Ion channel-transporter interactions. Crit Rev Biochem Mol Biol 51: 257–67. doi: 10.3109/10409238.2016.1172553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neverisky DL, Abbott GW (2017) KCNQ-SMIT complex formation facilitates ion channel-solute transporter cross talk. FASEB J 31: 2828–2838. doi: 10.1096/fj.201601334R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, MacKinnon R (2002) Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell 111: 957–65. [DOI] [PubMed] [Google Scholar]
- O’Kelly I, Butler MH, Zilberberg N, Goldstein SA (2002) Forward transport. 14-3-3 binding overcomes retention in endoplasmic reticulum by dibasic signals. Cell 111: 577–88. [DOI] [PubMed] [Google Scholar]
- O’Kelly I, Goldstein SA (2008) Forward Transport of K2p3.1: mediation by 14-3-3 and COPI, modulation by p11. Traffic 9: 72–8. doi: 10.1111/j.1600-0854.2007.00663.x [DOI] [PubMed] [Google Scholar]
- O’Mahony F, Thomas W, Harvey BJ (2009) Novel female sex-dependent actions of oestrogen in the intestine. J Physiol 587: 5039–44. doi: 10.1113/jphysiol.2009.177972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organ-Darling LE, Vernon AN, Giovanniello JR, Lu Y, Moshal K, Roder K, Li W, Koren G (2013) Interactions between hERG and KCNQ1 alpha-subunits are mediated by their COOH termini and modulated by cAMP. Am J Physiol Heart Circ Physiol 304: H589–99. doi: 10.1152/ajpheart.00385.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Barro-Soria R, Robey S, Sampson KJ, Kass RS, Larsson HP (2012) Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc Natl Acad Sci U S A 109: 7103–8. doi: 10.1073/pnas.1201582109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Gonzalez C, Sampson KJ, Iyer V, Rebolledo S, Larsson HP, Kass RS (2010a) KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc Natl Acad Sci U S A 107: 22710–5. doi: 10.1073/pnas.1016300108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Sampson KJ, Kass RS (2010b) The cardiac IKs channel, complex indeed. Proc Natl Acad Sci U S A 107: 18751–2. doi: 10.1073/pnas.1014150107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G, Abbott GW (2006) The impact of ancillary subunits on small-molecule interactions with voltage-gated potassium channels. Curr Pharm Des 12: 2285–302. [DOI] [PubMed] [Google Scholar]
- Panaghie G, Abbott GW (2007) The role of S4 charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J Gen Physiol 129: 121–33. doi: 10.1085/jgp.200609612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KH, Kwok SM, Sharon C, Baerga R, Sesti F (2003) N-Glycosylation-dependent block is a novel mechanism for drug-induced cardiac arrhythmia. FASEB J 17: 2308–9. doi: 10.1096/fj.03-0577fje [DOI] [PubMed] [Google Scholar]
- Piccini M, Vitelli F, Seri M, Galietta LJ, Moran O, Bulfone A, Banfi S, Pober B, Renieri A (1999) KCNE1-like gene is deleted in AMME contiguous gene syndrome: identification and characterization of the human and mouse homologs. Genomics 60: 251–7. doi: 10.1006/geno.1999.5904 [DOI] [PubMed] [Google Scholar]
- Plant LD, Dementieva IS, Kollewe A, Olikara S, Marks JD, Goldstein SA (2010) One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc Natl Acad Sci U S A 107: 10743–8. doi: 10.1073/pnas.1004712107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Dowdell EJ, Dementieva IS, Marks JD, Goldstein SA (2011) SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J Gen Physiol 137: 441–54. doi: 10.1085/jgp.201110604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Xiong D, Dai H, Goldstein SA (2014) Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proc Natl Acad Sci U S A 111: E1438–46. doi: 10.1073/pnas.1323548111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongs O, Schwarz JR (2010) Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev 90: 755–96. doi: 10.1152/physrev.00020.2009 [DOI] [PubMed] [Google Scholar]
- Purtell K, Paroder-Belenitsky M, Reyna-Neyra A, Nicola JP, Koba W, Fine E, Carrasco N, Abbott GW (2012) The KCNQ1-KCNE2 K(+) channel is required for adequate thyroid I(−) uptake. FASEB J 26: 3252–9. doi: 10.1096/fj.12-206110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA (2005) Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 121: 37–47. doi: 10.1016/j.cell.2005.01.019 [DOI] [PubMed] [Google Scholar]
- Rapetti-Mauss R, O’Mahony F, Sepulveda FV, Urbach V, Harvey BJ (2013) Oestrogen promotes KCNQ1 potassium channel endocytosis and postendocytic trafficking in colonic epithelium. J Physiol 591: 2813–31. doi: 10.1113/jphysiol.2013.251678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recanatini M, Poluzzi E, Masetti M, Cavalli A, De Ponti F (2005) QT prolongation through hERG K(+) channel blockade: current knowledge and strategies for the early prediction during drug development. Med Res Rev 25: 133–66. doi: 10.1002/med.20019 [DOI] [PubMed] [Google Scholar]
- Ren XQ, Liu GX, Organ-Darling LE, Zheng R, Roder K, Jindal HK, Centracchio J, McDonald TV, Koren G (2010) Pore mutants of HERG and KvLQT1 downregulate the reciprocal currents in stable cell lines. Am J Physiol Heart Circ Physiol 299: H1525–34. doi: 10.1152/ajpheart.00479.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TK, Anantharam A, Kirchhoff P, Busque SM, Young JB, Geibel JP, Lerner DJ, Abbott GW (2006) The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J Biol Chem 281: 23740–7. doi: 10.1074/jbc.M604155200 [DOI] [PubMed] [Google Scholar]
- Roepke TK, Kanda VA, Purtell K, King EC, Lerner DJ, Abbott GW (2011a) KCNE2 forms potassium channels with KCNA3 and KCNQ1 in the choroid plexus epithelium. FASEB J 25: 4264–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TK, King EC, Purtell K, Kanda VA, Lerner DJ, Abbott GW (2011b) Genetic dissection reveals unexpected influence of beta subunits on KCNQ1 K+ channel polarized trafficking in vivo. FASEB J 25: 727–36. doi: 10.1096/fj.10-173682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TK, King EC, Reyna-Neyra A, Paroder M, Purtell K, Koba W, Fine E, Lerner DJ, Carrasco N, Abbott GW (2009) Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat Med 15: 1186–94. doi: 10.1038/nm.2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TK, Purtell K, King EC, La Perle KM, Lerner DJ, Abbott GW (2010) Targeted deletion of Kcne2 causes gastritis cystica profunda and gastric neoplasia. PLoS One 5: e11451. doi: 10.1371/journal.pone.0011451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Dumaine R, Brown AM (1996) HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation 94: 817–23. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP (2000) Intracellular regulation of inward rectifier K+ channels. Pflugers Arch 441: 1–11. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Schroter KH, Sakmann B, Stocker M, Sewing S, Pongs O (1990) Heteromultimeric channels formed by rat brain potassium-channel proteins. Nature 345: 535–7. doi: 10.1038/345535a0 [DOI] [PubMed] [Google Scholar]
- Ruscic KJ, Miceli F, Villalba-Galea CA, Dai H, Mishina Y, Bezanilla F, Goldstein SA (2013) IKs channels open slowly because KCNE1 accessory subunits slow the movement of S4 voltage sensors in KCNQ1 pore-forming subunits. Proc Natl Acad Sci U S A 110: E559–66. doi: 10.1073/pnas.1222616110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadja R, Alagem N, Reuveny E (2003) Gating of GIRK channels: details of an intricate, membrane-delimited signaling complex. Neuron 39: 9–12. doi: 10.1016/s0896-6273(03)00402-1 [DOI] [PubMed] [Google Scholar]
- Sahu ID, Kroncke BM, Zhang R, Dunagan MM, Smith HJ, Craig A, McCarrick RM, Sanders CR, Lorigan GA (2014) Structural investigation of the transmembrane domain of KCNE1 in proteoliposomes. Biochemistry 53: 6392–401. doi: 10.1021/bi500943p [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT (1996) Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384: 80–3. doi: 10.1038/384080a0 [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R, Jentsch TJ (2000) A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403: 196–9. doi: 10.1038/35003200 [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Rubie C, Hordt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H (1997) KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat Genet 17: 267–8. doi: 10.1038/ng1197-267 [DOI] [PubMed] [Google Scholar]
- Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, Priori SG, Roden DM, George AL Jr., Goldstein SA (2000) A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci U S A 97: 10613–8. doi: 10.1073/pnas.180223197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti F, Goldstein SA (1998) Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J Gen Physiol 112: 651–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamgar L, Ma L, Schmitt N, Haitin Y, Peretz A, Wiener R, Hirsch J, Pongs O, Attali B (2006) Calmodulin is essential for cardiac IKS channel gating and assembly: impaired function in long-QT mutations. Circ Res 98: 1055–63. doi: 10.1161/01.RES.0000218979.40770.69 [DOI] [PubMed] [Google Scholar]
- Smith JA, Vanoye CG, George AL Jr., Meiler J, Sanders CR (2007) Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry 46: 14141–52. doi: 10.1021/bi701597s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT (1997a) Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med 336: 1562–7. doi: 10.1056/NEJM199705293362204 [DOI] [PubMed] [Google Scholar]
- Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT (1997b) Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet 17: 338–40. doi: 10.1038/ng1197-338 [DOI] [PubMed] [Google Scholar]
- Strutz-Seebohm N, Pusch M, Wolf S, Stoll R, Tapken D, Gerwert K, Attali B, Seebohm G (2011) Structural basis of slow activation gating in the cardiac I Ks channel complex. Cell Physiol Biochem 27: 443–52. doi: 10.1159/000329965 [DOI] [PubMed] [Google Scholar]
- Sun J, MacKinnon R (2017) Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 169: 1042–1050 e9. doi: 10.1016/j.cell.2017.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, MacKinnon R (2020) Structural Basis of Human KCNQ1 Modulation and Gating. Cell 180: 340–347 e9. doi: 10.1016/j.cell.2019.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zaydman MA, Cui J (2012) Regulation of Voltage-Activated K(+) Channel Gating by Transmembrane beta Subunits. Front Pharmacol 3: 63. doi: 10.3389/fphar.2012.00063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto K, Ren X (2002) KChIPs (Kv channel-interacting proteins)--a few surprises and another. J Physiol 545: 3. doi: 10.1113/jphysiol.2002.033993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi T, Ohkubo H, Nakanishi S (1988) Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science 242: 1042–5. [DOI] [PubMed] [Google Scholar]
- Tao X, MacKinnon R (2019) Molecular structures of the human Slo1 K(+) channel in complex with beta4. Elife 8. doi: 10.7554/eLife.51409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel BL, Jan YN, Jan LY (1988) Cloning of a probable potassium channel gene from mouse brain. Nature 332: 837–9. doi: 10.1038/332837a0 [DOI] [PubMed] [Google Scholar]
- Tempel BL, Papazian DM, Schwarz TL, Jan YN, Jan LY (1987) Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science 237: 770–5. [DOI] [PubMed] [Google Scholar]
- Testai L, Bianucci AM, Massarelli I, Breschi MC, Martinotti E, Calderone V (2004) Torsadogenic cardiotoxicity of antipsychotic drugs: a structural feature, potentially involved in the interaction with cardiac HERG potassium channels. Curr Med Chem 11: 2691–706. [DOI] [PubMed] [Google Scholar]
- Tinel N, Diochot S, Borsotto M, Lazdunski M, Barhanin J (2000) KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J 19: 6326–30. doi: 10.1093/emboj/19.23.6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres YP, Granados ST, Latorre R (2014) Pharmacological consequences of the coexpression of BK channel alpha and auxiliary beta subunits. Front Physiol 5: 383. doi: 10.3389/fphys.2014.00383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranebjaerg L, Samson RA, Green GE (1993) Jervell and Lange-Nielsen Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K (eds) GeneReviews(R), Seattle (WA) [Google Scholar]
- Tristani-Firouzi M, Sanguinetti MC (1998) Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J Physiol 510 (Pt 1): 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM (1997) Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 387: 179–83. doi: 10.1038/387179a0 [DOI] [PubMed] [Google Scholar]
- Tyson J, Tranebjaerg L, Bellman S, Wren C, Taylor JF, Bathen J, Aslaksen B, Sorland SJ, Lund O, Malcolm S, Pembrey M, Bhattacharya S, Bitner-Glindzicz M (1997) IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Mol Genet 6: 2179–85. [DOI] [PubMed] [Google Scholar]
- Vanoye CG, Welch RC, Daniels MA, Manderfield LJ, Tapper AR, Sanders CR, George AL Jr. (2009) Distinct subdomains of the KCNQ1 S6 segment determine channel modulation by different KCNE subunits. J Gen Physiol 134: 207–17. doi: 10.1085/jgp.200910234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- vanTol BL, Missan S, Crack J, Moser S, Baldridge WH, Linsdell P, Cowley EA (2007) Contribution of KCNQ1 to the regulatory volume decrease in the human mammary epithelial cell line MCF-7. Am J Physiol Cell Physiol 293: C1010–9. doi: 10.1152/ajpcell.00071.2007 [DOI] [PubMed] [Google Scholar]
- Varnum MD, Busch AE, Bond CT, Maylie J, Adelman JP (1993) The min K channel underlies the cardiac potassium current IKs and mediates species-specific responses to protein kinase C. Proc Natl Acad Sci U S A 90: 11528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP (1998) Calcium-activated potassium channels. Curr Opin Neurobiol 8: 321–9. [DOI] [PubMed] [Google Scholar]
- Wang M, Kass RS (2012) Stoichiometry of the slow I(ks) potassium channel in human embryonic stem cell-derived myocytes. Pediatr Cardiol 33: 938–42. doi: 10.1007/s00246-012-0255-2 [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT (1996) Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12: 17–23. doi: 10.1038/ng0196-17 [DOI] [PubMed] [Google Scholar]
- Weatherall KL, Goodchild SJ, Jane DE, Marrion NV (2010) Small conductance calcium-activated potassium channels: from structure to function. Prog Neurobiol 91: 242–55. doi: 10.1016/j.pneurobio.2010.03.002 [DOI] [PubMed] [Google Scholar]
- Wiener R, Haitin Y, Shamgar L, Fernandez-Alonso MC, Martos A, Chomsky-Hecht O, Rivas G, Attali B, Hirsch JA (2008) The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J Biol Chem 283: 5815–30. doi: 10.1074/jbc.M707541200 [DOI] [PubMed] [Google Scholar]
- Wrobel E, Rothenberg I, Krisp C, Hundt F, Fraenzel B, Eckey K, Linders JT, Gallacher DJ, Towart R, Pott L, Pusch M, Yang T, Roden DM, Kurata HT, Schulze-Bahr E, Strutz-Seebohm N, Wolters D, Seebohm G (2016) KCNE1 induces fenestration in the Kv7.1/KCNE1 channel complex that allows for highly specific pharmacological targeting. Nat Commun 7: 12795. doi: 10.1038/ncomms12795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Delaloye K, Zaydman MA, Nekouzadeh A, Rudy Y, Cui J (2010a) State-dependent electrostatic interactions of S4 arginines with E1 in S2 during Kv7.1 activation. J Gen Physiol 135: 595–606. doi: 10.1085/jgp.201010408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Pan H, Delaloye K, Cui J (2010b) KCNE1 remodels the voltage sensor of Kv7.1 to modulate channel function. Biophys J 99: 3599–608. doi: 10.1016/j.bpj.2010.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Kanda VA, Choi E, Panaghie G, Roepke TK, Gaeta SA, Christini DJ, Lerner DJ, Abbott GW (2009) MinK-dependent internalization of the IKs potassium channel. Cardiovasc Res 82: 430–8. doi: 10.1093/cvr/cvp047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Inanobe A, Kurachi Y (1998) G protein regulation of potassium ion channels. Pharmacol Rev 50: 723–60. [PubMed] [Google Scholar]
- Yan J, Aldrich RW (2010) LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature 466: 513–6. doi: 10.1038/nature09162 [DOI] [PubMed] [Google Scholar]
- Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22: 537–48. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhang H, Wang C, Wang Y, Zou R, Shi C, Guan B, Gamper N, Xu Y (2020) Auxiliary subunits control biophysical properties and response to compound NS5806 of the Kv4 potassium channel complex. FASEB J 34: 807–821. doi: 10.1096/fj.201902010RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, Jurkiewicz NK, Folander K, Lazarides E, Salata JJ, Swanson R (1994) K+ currents expressed from the guinea pig cardiac IsK protein are enhanced by activators of protein kinase C. Proc Natl Acad Sci U S A 91: 1766–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R (2001) Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 411: 657–61. doi: 10.1038/35079500 [DOI] [PubMed] [Google Scholar]