

Abstract
The cell nucleus is best known as the container of the genome. Its envelope provides a barrier for passive macromolecule diffusion, which enhances the control of gene expression. As its largest and stiffest organelle, the nucleus also defines the minimal space requirements of a cell. Internal or external pressures that deform a cell to its physical limits cause a corresponding nuclear deformation. Evidence is consolidating that the nucleus, in addition to its genetic functions, serves as a physical sensing device for critical cell body deformation. Nuclear mechanotransduction allows cells to adapt their acute behaviors, mechanical stability, paracrine signaling, and fate to their physical surroundings. This review summarizes the basic chemical and mechanical properties of nuclear components, and how these properties are thought to be utilized for mechanosensing.
Keywords: mechanotransduction, nucleus, lamina, chromatin, nuclear membrane, laminopathy, progeria
“It is only with the nucleus that a cell can see rightly…”
— after Antoine de Saint-Exupéry
1. INTRODUCTION
Cells must sense their physical microenvironment to properly adapt to it and to function within it, either through short-term changes in morphology and behavior or long-term cell fate decisions. To this end, mechanical cues from the cell exterior and interior are converted into chemical signals inside the cell in a process called mechanotransduction.
Intuitively, this conversion first occurs where the cell intersects with its physical environment, that is, at the plasma membrane (PM). The PM houses many mechanosensitive proteins including adhesion molecules (integrins, cadherins, etc.), and mechanosensitive ion channels (MSCs), such as Piezo1 (Ranade et al. 2015). MSCs at the PM convert membrane stretch into ion influx to regulate cell volume homeostasis, neuronal growth cone navigation, sensing of bone load, and many other mechanotransduction processes. Another classic example for cell surface mechanotransduction is the augmentation of substrate adhesion by mechanical force. For instance, hydrodynamic shear from the blood stream promotes the adhesion of leukocytes to vessel walls during inflammation. Tensile force stabilizes adhesions by promoting stretch-dependent molecular interactions, for example, between the cytoplasmic adhesion complex members talin and vinculin, or between an adhesion molecule and its extracellular ligand (Sun et al. 2016). This force can also come from contractile actomyosin fibers within the cell. Coupling those fibers to the nucleus enhances cell contractility and motility on soft 2D or 3D substrates (Graham et al. 2018).
As counterweight to the cell surface, the nucleus contributes to tensile force generation and is, at the same time, deformed by it. This deformation is measured by nuclear mechanotransduction pathways that inform the cell about the physical properties of its local microenvironment. A key concept of nuclear mechanotransduction is that the nucleus provides the cell with a second sensory surface that complements cell surface mechanosensing. Cell surface mechanotransduction can render the entire cell, including its most peripheral sites (e.g., neuronal growth cones) mechanosensitive, regardless of whether a cell or cell-fragment is nucleated or not (e.g., erythrocytes, platelets). Nuclear mechanotransduction is selectively triggered by strong cell body deformation. In principle, these two modes of mechanotransduction allow a nucleated cell to distinguish whether forces act on its periphery or center.
Besides perinuclear actin fiber contraction (Khatau et al. 2009), other possible physiological causes of nuclear deformation include muscle contraction, hydrodynamic force from the blood or interstitial fluid flow, or a decrease of cytoplasmic macromolecule concentration, for instance, after hypo-osmotic shock or PM-permeabilization during necrotic cell death. Histone modifications or cell death can also cause nuclear swelling through decreasing chromatin compaction and integrity. Whether and how these cues trigger nuclear mechanotransduction likely depends on their magnitude and timescale and is only beginning to be unraveled.
Nuclear mechanotransduction activates enzymatic or transcriptional circuits that mediate behavioral changes, mechanical adaptation, and cell fate decisions. For example, nuclear compression during confined cell migration leads to cortical contraction and PM-blebbing, which may help cells to traverse narrow tissue channels or pores (Venturini et al. 2020, Lomakin et al. 2020). Through nuclear swelling, zebrafish larvae detect epithelial wounds by a drop of interstitial osmotic pressure that alerts distant neutrophils to the injury site (Enyedi et al. 2013, Enyedi et al. 2016). Furthermore, cells can adapt their nuclear stiffness to substrate stiffness, likely in part by sensing nuclear deformation (Swift et al. 2013).
To understand how cells sense nuclear deformation and convert it into enzymatic or transcriptional activity, knowledge of the physical and chemical properties of the three main nuclear components (Section 2) —the nuclear membrane (NM), chromatin and the nuclear lamina—is crucial (Figure 1). Owing to their intricate mechanical connections (Section 3), perturbing one of these components affects all the others in complex ways, as evidenced by the respective disease phenotypes, such as Hutchinson-Gilford progeria and Emery-Dreifuss muscular dystrophy. These diseases preferentially affect mechanically strained tissues—often the bones, skeletal muscle, and the heart—and may reflect a structural weakening of the nucleus that promotes DNA damage, cell death, and senescence. Alternatively, the disruption of transcriptional regulation and nuclear mechanotransduction might be involved, or all of these factors.
Figure 1.

Components of nuclear mechanotransduction. Schematic overview (left panel) and zoomed-in section of the nuclear envelope (left panel, black box & right panel), with INM and ONM, chromatin (turquoise and gray), and the nuclear lamina (yellow and purple). LADs and the LINC are depicted in the zoomed-in view. Nuclear pore complexes (blue), the endoplasmic reticulum, and the invaginations of the nucleoplasmic reticulum are depicted in the overview panel. Abbreviations: BAF, barrier-to-autointegration factor; F-actin, fibrous actin; INM, inner nuclear membrane; LAD, lamina-associated domain; LINC, linker of nucleoskeleton and cytoskeleton complex; ONM, outer nuclear membrane; ER, endoplasmic reticulum; NR, nucleoplasmic reticulum; P, phosphorylated; SUN-1/2, Sad1p/UNC-84 1/2.
Like mechanotransduction at the cell surface, nuclear mechanotransduction involves the conversion of tensile force into altered molecular interactions (Figure 2A). To this end, cytoskeletal forces pull and squeeze on the nucleus to stretch and flatten it. Alternatively, chromatin-swelling or colloid osmotic pressure inflate the nucleus (Figure 2B). The resulting tension in the network of coupled nuclear components is partially relaxed by structural changes (e.g., unfolding, stretching, etc.) in the associated lipid bilayers, proteins, or DNA/chromatin. These events activate MSCs and modulate molecular interactions by masking/unmasking of binding epitopes (Figure 2A). The cell biological effects (Section 4) of these subnanometer, structural changes include enzyme binding to the NM, transcription factor redistribution between the cytoplasm and the nucleus, protein redistribution between the inner and outer NM, as well as transcriptional activation or repression (partially depicted in Figure 2B).
Figure 2.
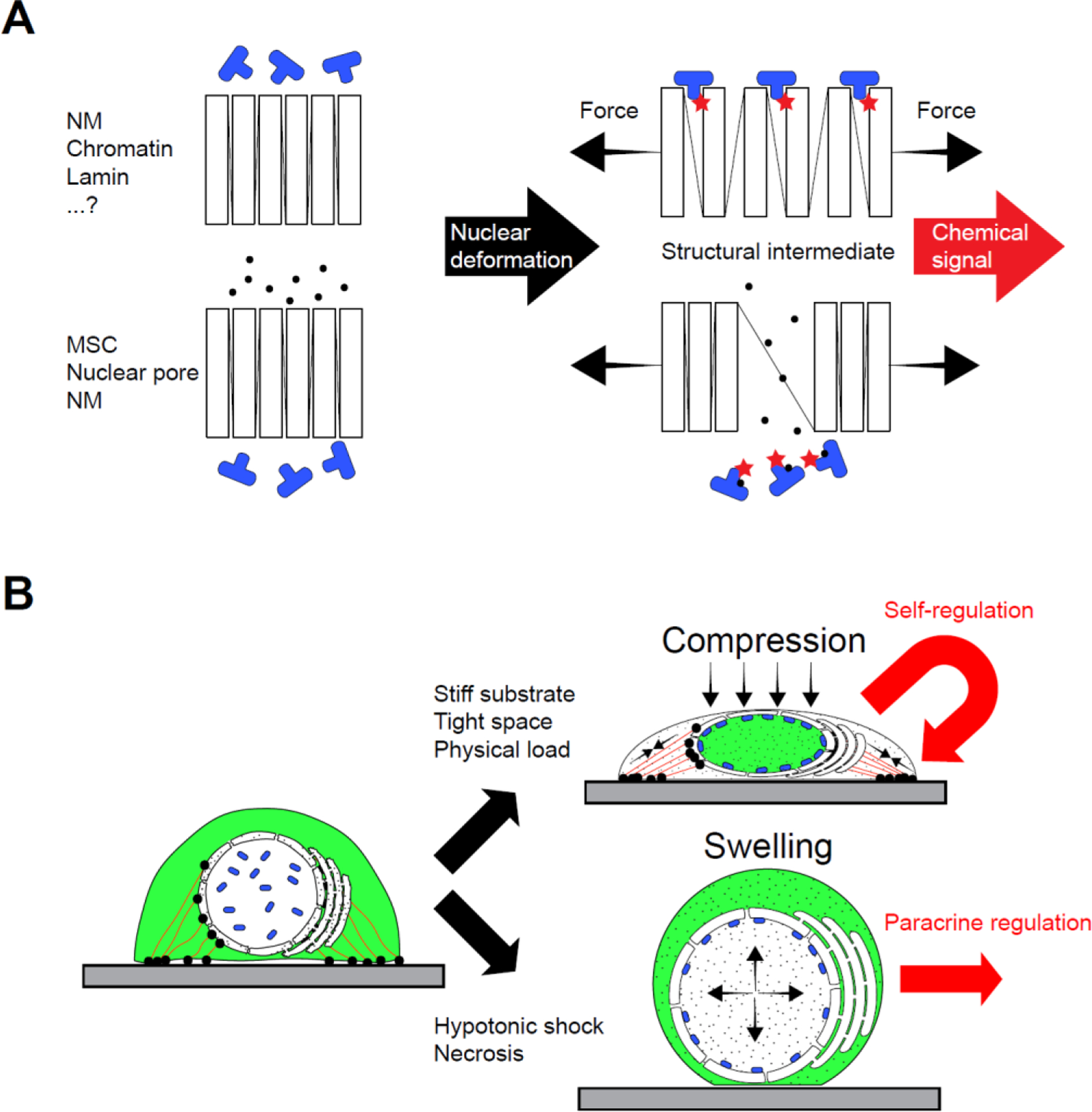
Mechanisms of nuclear mechanotransduction. (A) Molecular level: Deformation of the nucleus causes subnanometer structural changes in the NM lipid bilayers, integral NM proteins (channel- & pore-proteins), chromatin, lamins, and other deformable molecular complexes (conceptualized by boxes connected with lines) that unmask new interaction sites (void between boxes) and change NM permeability to ions and proteins (black dots). Binding/activation of enzymes and transcription factors (blue shapes) convert these structural changes into chemical signals (red stars). (B) Cellular level: the structural changes in nuclear components promote the redistribution of transcriptional regulators (green) between the cyto- and nucleoplasm, and lipid enzymes (blue shapes) between the nucleoplasm and the NM, as well as the efflux of Ca2+ (black sprinkles) from the endoplasmic reticulum through MSCs. For simplicity, protein redistribution within the NM and to chromatin is not depicted in this scheme. Red lines, contractile actomyosin fibers. Black circles, focal adhesions and LINC complexes. Abbreviations: NM, nuclear membrane; MSCs, mechanosensitive ion channels.
Owing to their direct genetic connection to laminopathic and envelopathic disease, lamina-related mechanisms currently constitute the best-characterized branch of nuclear mechanotransduction, as documented by many excellent reviews (Cho et al. 2017, Janin et al. 2017, Maurer & Lammerding 2019, Méjat & Misteli 2010). The epigenetic regulation of chromatin has always attracted a lot of interest, and now a number of studies (Miroshnikova et al. 2019, Stephens et al. 2019) outline its intriguing links to nuclear mechanics and mechanotransduction. Most recently, nuclear membrane mechanotransduction (Enyedi & Niethammer 2017) has entered the field and is yet scarcely covered by scholarly reviews. Hence, I will give here some additional background on this latest branch besides providing a cross section through the field.
2. COMPONENTS OF NUCLEAR MECHANOTRANSDUCTION
2.1. The Nuclear Membrane
Just as the PM did earlier (Diz-Muñoz et al. 2013, Gauthier et al. 2012, Le Roux et al. 2019), the NM with its attached ER is emerging as an important sensor and transducer of mechanical signals. Yet the properties that allow for this function are still little understood and only covered by a few reviews (Agrawal & Lele 2019, Enyedi & Niethammer 2017).
2.1.1. Structural features.
The NM encloses the chromosomes and hinders the free diffusion of macromolecules between the nucleoplasm and cytoplasm. It allows the control of gene expression by regulating the access of transcription factors to and the export of messenger RNAs from the nucleus (Carmo-Fonseca 2002). In addition, an intact NM protects the genome from cytoplasmic factors that damage DNA (Maciejowski et al. 2015) and serves as a site for lipid synthesis and metabolism (Haider et al. 2018, Romanauska & Köhler 2018).
The NM is formed by two lipid bilayers, the inner nuclear membrane (INM) and outer nuclear membrane (ONM), that are separated by a ~30–50-nm-wide intermembrane space and are fused at hundreds of sites to form ~9-nm-wide pores. The complex, perforated topology of the NM has been likened to a donut with many holes, called an ultradonut (Torbati et al. 2016). The INM and ONM can form deep nucleoplasmic invaginations, the so-called nucleoplasmic reticulum (Drozdz & Vaux 2017, Fricker et al. 1997), and the ONM is contiguous with the rough ER (Figure 1, left panel). The nucleoplasmic reticulum and ER form a large surface reservoir for accommodating nuclear shape changes during swelling or compression. The INM-ONM fusion sites are lined by multiprotein nuclear pore complexes (NPCs) that control the nuclear transport of >50 kD macromolecules but not of smaller solutes such as ions, peptides, and amino acids. Through its embedded proteins, the NM connects to the nuclear lamina and the chromosomes underneath and to the extranuclear cytoskeleton, including the perinuclear actin cap (Khatau et al. 2009). These connections are pivotal for nuclear envelope (NE) integrity and shape regulation as well as for genome organization and force transmission from the ECM into the nucleus.
2.1.2. Lipid composition.
After biochemical organelle purification, the lipid composition of the NM largely resembles that of the ER. The NM contains ~1.5 x more zwitterionic phosphatidylcholines (PCs) than does the PM (Khandwala & Kasper 1971, van Meer et al. 2008), which is rich in lysophosphatidylcholine (LPC), sphingomyelin (SM), and cholesterol. PCs from animals and yeast often have unsaturated acyl side chains (Khandwala & Kasper 1971) including esterified arachidonic acid (AA). The acyl chains of SMs and LPCs are frequently saturated. With cholesterol acting as molecular glue between the phospholipid headgroups, this allows for the dense lipid packing of the PM, which enhances its barrier capabilities. By contrast, the NM is softer and more fluid than the PM. Whereas peripheral protein adsorption to the PM largely relies on electrostatic interactions, the looser lipid packing of the NM favors hydrophobic insertion into lipid-packing defects (LPDs) (Bigay & Antonny 2012).
According to molecular dynamics simulations, LPDs are subnanometer voids (~20–100 Å2) (Vanni et al. 2014) that emerge between lipid headgroups and facilitate the insertion of protruding hydrophobic protein residues into the bilayer core (Vanni et al. 2013). Lipids with conical shape (e.g., due to unsaturated and kinked acyl chains or small headgroups), positive curvature as well as membrane stretch increase lipid headgroup spacing and give rise to LPDs (Pinot et al. 2018, Vanni et al. 2014). LPD sensing has been best studied for proteins that mediate vesicle trafficking and contain amphipathic lipid-packing sensor (ALPS) motifs (Bigay & Antonny 2012). At the NM, LPDs are thought to mediate the sensing of NM stretch or curvature and the surveillance of elastic curvature stresses caused by excess conical lipids (Enyedi et al. 2016, Haider et al. 2018, Hetzer 2010, Lomakin et al. 2020, Pinot et al. 2018, Venturini et al. 2020). LPD sensing may be more widespread among peripheral membrane proteins than currently known: A similar functionality was described for the conserved domain 2 (C2) and polycystin-1, lipoxygenase, alpha-toxin domains as well as for lipidated proteins (Enyedi et al. 2016, Hatzakis et al. 2009). Whether ALPS- and C2-like domains distinguish stretch- from curvature-induced LPDs is unknown. Owing to its high conical lipid content, the INM seems particularly suited for LPD-mediated membrane mechanotransduction. In yeast, fluorescent biosensor imaging indicated the specific enrichment of diacylglycerol (DAG) on the INM (Romanauska & Köhler 2018). How this fast-diffusing lipid is retained on the INM is unclear. Conical-shaped DAG loosens lipid packing and could prime the INM for LPD formation during stretch. Data suggest that LPD sensing on the INM by nuclear choline phosphate cytidylyltransferase regulates PC synthesis (Haider et al. 2018). In addition, LPDs may target NPC proteins with amphipathic helical domains (e.g., NUP133 and NUP153) to regions of high positive curvature at nuclear pores (Hetzer 2010) and inflammatory enzymes to the stretched INM upon cell swelling or compression (Enyedi et al. 2016, Lomakin et al. 2020, Venturini et al. 2020).
2.1.3. Protein composition.
Per the Human Protein Atlas, >250 proteins are constitutively associated with nuclear membranes (Uhlén et al. 2005). Those comprise membrane-embedded or peripherally attached NPC proteins. Then there are the INM proteins, including the Lamin B receptor (LBR); the LAP2, Emerin, and MAN1 (LEM) family proteins; and the Sad1p/UNC-84 (SUN) family proteins. LEM proteins connect the INM to the underlying lamina and chromosomes. The SUN proteins engage with ONM-embedded Klarsicht, ANC-1, Syne Homology (KASH)-domain proteins in the intermembrane space to link the nucleoskeleton and cytoskeleton (Tapley & Starr 2013) and to regulate INM-ONM spacing (Crisp et al. 2006). B-type lamins associate with the INM via farnesyl anchors in a strain- and curvature-dependent manner (Nmezi et al. 2019). Lack of Lamin B farnesylation disrupts the spatial organization and stability of the nuclear lamina. By contrast, pathological INM interactions of Lamin A (Cao et al. 2007, Dechat et al. 2007, Hennekes & Nigg 1994) caused by a failure to remove the farnesyl group from the Lamin A precursor peptide lead to lobulated nuclei, laminar thickening, and the loss of peripheral heterochromatin. These cellular phenotypes are associated with premature aging, restricted dermopathy, and mandibuloacral dysplasia in humans (Cenni et al. 2018, De Sandre-Giovannoli et al. 2003, Eriksson et al. 2003, Navarro et al. 2004) as well as bone fractures and muscle weakness in mice (Bergo et al. 2002). INM proteins are selectively retained on the INM by lamina and chromatin interactions (Boni et al. 2015, Guo et al. 2014, Ungricht et al. 2015, Vaughan et al. 2001). Lamin A deficiency and mechanical straining of cells were reported to cause the loss of Emerin from the INM (Ho et al. 2013, Le et al. 2016). Finally, there are the KASH-domain-containing Nesprins, which directly and indirectly connect to fibrous actin (F-actin) (Nesprin-1/2), microtubules (Nesprin-3 via BPAG1 or MACF and Nesprin-4 via kinesin-1), or intermediate filaments (Nesprin-3 via plectin) in the cytoplasm (Crisp et al. 2006, Wilhelmsen et al. 2005, Q. Zhang et al. 2002) and with SUN-domain proteins in the intermembrane space. SUN-KASH protein complexes are found in all eukaryotes, including plants (Goswami et al. 2020), and are commonly referred to as LINC (linker of nucleoskeleton and cytoskeleton) complexes.
Akin to the PM, the NM scaffolds transmembrane proteins that mechanically couple cytoskeletal fibers on one side to a filament network on the other. Whereas very little is known about the physicochemical signaling properties of the NM, PM mechanotransduction has been studied for a while (Diz-Muñoz et al. 2013). Despite all of the differences between the PM and the NM (e.g., lipid packing, single bilayer versus double bilayer, and others), making analogies between the two structures can help us to think about the NM as a conduit for mechanical signals. Of central importance is the notion that integral membrane proteins immobilized by an underlying matrix act as obstacles to lipid flow or diffusion (Nakada et al. 2003, Niemelä et al. 2010). The PM, and perhaps regions of the NM, may form so-called picket fences (Kusumi et al. 2012) that limit flow between (relaxed) regions of high lateral lipid pressure to (tense) regions of low lateral lipid pressure (Shi et al. 2018). With a phospholipid-to-protein weight ratio of ~0.2–0.5, the bulk protein content of the NM is comparable to that of the PM (~0.3–0.8) (Brotherus & Renkonen 1977, Franke et al. 1970, Khandwala & Kasper 1971, Pfleger et al. 1968). Hence, NM picket fences may plausibly exist and may control INM tension through limiting lipid flow from the connected ER. In line with this idea, the INM protein Lem2 was shown to act as a valve for lipid flow between the ER and NM in yeast (Kume et al. 2019).
2.1.4. The nuclear membrane under load.
When the nucleus is osmotically swollen or mechanically squeezed, NE-area fluctuations and nuclear invaginations disappear, and its surface becomes smooth. These morphological changes are often interpreted as signs of NM tension (Dahl et al. 2004, Lomakin et al. 2020). Both cell swelling and compression cause NM tension and promote the adsorption of nucleoplasmic proteins to the INM (Figure 2B). So far, INM-stretch sensing has been demonstrated for two lipid enzymes, cytoplasmic phospholipase A2 (cPLA2) and 5-lipoxygenase, which may detect LPDs on the INM via their C2- and PLAT- domains (Enyedi et al. 2013, 2016; Lomakin et al. 2020; Riegman et al. 2020; Venturini et al. 2020). NM stretch downstream of cell swelling and compression can considerably differ in duration and amplitude. Unlike compression-induced NM stretch, osmotic NM stretch is intrinsically limited by regulatory volume decrease mechanisms (Hoffmann et al. 2009), which rapidly shrink intact cells and their nuclei back to their original size and tension, sometimes within a minute (Enyedi et al. 2016).
Membrane phospholipids stick tightly together to reduce the exposure of their hydrophobic acyl chains to water, which is energetically unfavorable. Through increasing the distance of lipid molecules, stretch can extend the area of a membrane only by ~5–10% without rupturing it (Needham & Nunn 1990, Hallett et al. 1993). Hence, this process alone cannot explain the often much larger nuclear surface increases observed during osmotic swelling or cell compression (Dahl et al. 2004, Enyedi et al. 2016, Lomakin et al. 2020). Where does this surface come from? Thermal undulations and the nucleoplasmic reticulum are likely sources. Once these reservoirs are depleted, the NM becomes tense. From work on the PM and artificial membranes, we know that membrane tension itself is a key regulator of membrane surface area (Morris & Homann 2001). Tension promotes vesicle fusion to the PM or giant unilamellar vesicles (Deshpande et al. 2019, Gauthier et al. 2012, Staykova et al. 2011). Within contiguous membrane systems, local tension can initiate lipid flow from areas of low to areas of high tension. Such flows have been observed between the growth cone and cell body of neurons (Dai & Sheetz 1995) and between Golgi membranes and the ER (Sciaky et al. 1997). According to current theory, lipid exchange between a tense INM and a relaxed ONM/ER should depend on the INM-ONM fusion pore radius and the lateral permeability of the membrane for lipids, which depend on the area fraction occupied by immobile membrane proteins and their radius (Chizmadzhev et al. 1999, Lamparter & Galic 2020). Hence, the bulk function of lamina- or chromatin-bound INM proteins as potential membrane flow valves deserve attention.
2.1.5. Nuclear membrane damage.
Load on the NM and underlying structures during, for example, confined cell migration (Denais et al. 2016, Raab et al. 2016) can cause NM rupture, which is often but not always preceded by bleblike protrusions. Nuclear blebs are also observed in stationary cells with NE defects such as cancer cells or cells deficient in lamins or Emerin (Lammerding et al. 2005, 2006; Vargas et al. 2012). Interestingly, complete lamin deficiency suppresses nuclear blebs but not NM damage in mouse embryonic fibroblasts (Chen et al. 2018), supporting the idea that structural changes in the lamin network (Funkhouser et al. 2013) rather than lack of membrane support by lamins cause nuclear bleb formation. Nuclear blebs are enriched in Lamin A/C and decondensed chromatin (Shimi et al. 2011, Stephens et al. 2018). Drugs that promote chromatin decondensation enhance nuclear blebbing, whereas drugs that promote chromatin condensation decrease it (Stephens et al. 2018). It will be interesting to further test whether the lamin-dependent induction of protrusive euchromatin drives nuclear bleb formation. Confinement-induced NM rupture amplifies DNA damage. Such rupture is augmented by actomyosin contractility and rapidly repaired by components of the endosomal sorting complexes required for transport-III (ESCRT-III) machinery (Denais et al. 2016, Raab et al. 2016).
2.2. Chromatin
As molecular storage device for genetic information, chromatin is widely known and investigated. Less studied are its adaptive material properties that depend on epigenetic modifications and transcriptional activity, respectively. Its ability to either stabilize or to push on the nuclear envelop makes chromatin the perhaps most versatile component of nuclear mechanotransduction.
2.2.1. Structural features.
In human cells, the ~2-m-long genome (Piovesan et al. 2019) is packed into a nucleus of ~10–20-μm diameter. To achieve such a high degree of compaction, 146 bp of DNA wrap around an octamer of the core histones H2A, H2B, H3, and H4 to form the nucleosome. Nucleosomes are connected like beads on a string by linker DNA. This assembly further condenses into a 30-nm chromatin fiber that folds itself into higher-order chromosomal structures. In heterochromatin, DNA packing into nucleosomes is tight, which correlates with low levels of gene transcription. By contrast, euchromatin features loose nucleosomal packing and high transcriptional activity. Posttranslational histone and DNA modifications, such as acetylation, citrullination, and methylation, alter chromatin packing. How these epigenetic modifications regulate nuclear mechanics and vice versa is starting to be explored (Stephens et al. 2019).
Chromatin compaction is enhanced by histone-tail, polyamine, and cation interactions that counterbalance the negative charges of DNA. One Ca2+ binds to every ~12.5–20 nucleotides and one Mg2+ to every 20–30 nucleotides (Strick et al. 2001). Cation depletion, proteolytic digestion of histones, and limited nuclease treatment lead to the loss of rigidity and the swelling of isolated nuclei driven by the outward entropic forces of decondensed chromatin (Dahl et al. 2005, Mazumder et al. 2008, Shimamoto et al. 2017).
Depending on its condensation state, chromatin can cause or protect against nuclear deformation. Chromatin decondensation drives nuclear blebbing, swelling, and rupture in NETosisneutrophil extracellular trap activation and release). During this type of cell death, white blood cells release their chromatin into the extracellular space to trap bacteria within it (Neubert et al. 2018, Papayannopoulos et al. 2010, Thiam et al. 2020, Wang et al. 2009). NETotic chromatin decompaction involves the digestion and hypercitrullination of histones by neutrophil elastase and peptidylarginine deiminase 4, respectively (Papayannopoulos et al. 2010, Wang et al. 2009).
2.2.2. Chromatin under load.
In addition to the nuclear lamina, condensed heterochromatin also enhances the structural stability of the NM. In Schizosaccharomyces pombe cells, which do not have lamins, the tethering of chromatin to integral INM proteins dampens microtubule-induced NM deformation (Schreiner et al. 2015). When HeLa cell nuclei were stretched between two micropipettes, condensed chromatin resisted small (<3 μm) and the lamina large (>3 μm) extensions (Stephens et al. 2017). The ability of chromatin to resist nuclear deformation was enhanced by histone methylation and decreased by histone acetylation (Schreiner et al. 2015, Stephens et al. 2018). Histone methylation reduced shape defects in lamin-mutant or -deficient cells, suggesting that those were due at least partly to peripheral chromatin decompaction (Stephens et al. 2018). Overexpression of the nucleosome-binding protein HGMN5 caused the loss of heterochromatin and nuclear blebbing. HGMN5-overexpressing mice developed heart defects similar to Lamin A/C–deficient animals (Furusawa et al. 2015). Although HGMN5-dependent chromatin decondensation was observable in newborn HGMN5 animals, the nuclear phenotypes only appeared during adulthood when heart activity is high, suggesting that they emerged under mechanical stress. Embryonic HGMN5 and wild-type mice showed similar gene expression profiles, arguing that the HGMN5 phenotype reflects the structural rather than the transcriptional consequences of heterochromatin loss (Furusawa et al. 2015). Altogether, these studies point to a primordial nucleoskeletal function of peripheral heterochromatin that is conserved from yeast to mammals.
Within cells, chromatin seems to adapt its transcriptional activity and rheological properties to mechanical stress. Namely, the cyclic straining of skin epidermis progenitor cells increased the histone H3K27me3 methylation of gene-rich chromatin regions, which is associated with transcriptional repression. Conversely, strain decreased the H3K9me3 methylation of gene-poor peripheral heterochromatin. The loss of H3K9me3 from peripheral heterochromatin was associated with higher heterochromatin mobility and nuclear softening (Le et al. 2016, Nava et al. 2020). Chromatin fluidization required intracellular Ca2+ release through stretch-sensitive Piezo1 channels downstream of nuclear deformation. Blocking chromatin-dependent nuclear softening increased strain-induced DNA damage (Nava et al. 2020). Evidence for chromatin remodeling was also observed in immune cells that migrate through narrow pores (Jacobson et al. 2018, Wang et al. 2018).
2.2.3. Chromatin damage.
Damage-inducing chromatin distortion may occur when cells migrate through narrow pores or tissue channels or are repeatedly stretched. Cells with a weak lamina (e.g., some cancer cells) or with noncompliant heterochromatin rheology are prone to mechanically induced DNA damage and cell cycle arrest (Chen et al. 2018; Denais et al. 2016; Irianto et al. 2016, 2017; Nava et al. 2020; Raab et al. 2016; Xia et al. 2018, 2019). Possible causes for confinement-induced DNA damage remain disputed. NM rupture likely plays an important role (Cho et al. 2019, Denais et al. 2016, Raab et al. 2016), perhaps through allowing the leakage of cytoplasmic nucleases into (Maciejowski et al. 2015) or DNA damage–repair factors out of (Xia et al. 2018) ruptured nuclei. But nuclear deformation can also cause DNA damage in the absence of obvious NE rupture (Denais et al. 2016, Nava et al. 2020, Shah et al. 2020). Spatial exclusion (squeezing out) of DNA repair factors from compressed chromatin may facilitate damage in such cases (Irianto et al. 2016, 2017). The contributions of replication-dependent damage have been also considered (Pfeifer et al. 2018, Shah et al. 2020). Shear or torsion can cause direct, mechanical DNA damage during experimental chromatin extraction (Noll et al. 1975). But whether chromatin is ever exposed to such stark distortions while inside the nuclei of intact cells is unclear. Hypothetically, nuclear deformation could also generate mutagenic reactive oxygen species by activating mechanosensitive phospholipases or lipoxygenases (Enyedi et al. 2016, Katikaneni et al. 2020). This idea remains to be tested. Irrespective of mechanism, mechanically induced chromatin damage may contribute to laminopathic and envelopathic diseases and to the genetic instability of cancer cells.
2.3. The Nuclear Lamina
As the most resilient component of the nucleus, and target of human disease mutations, the nuclear lamina has garnered a lot of attention from cell biologists, bioengineers, and the medical community alike.
2.3.1. Structural features.
Sandwiched between the NM and chromatin, the nuclear lamina is the toughest of the three major force-bearing nuclear components. It consists of intermediate filament type V lamins (A, C, B1, and B2 in mammals). Lamin monomers have an N-terminal head domain, a coiled-coil central rod domain, and a globular C-terminal tail domain with an immunoglobulin-like fold. Recent cryoelectron tomography studies in mouse embryonic fibroblast and Xenopus laevis oocytes suggest that lamin tetramers associate with 3.5–7-nm fibers (Sapra et al. 2020, Turgay et al. 2017). These fibers are woven into a 30–100-nm-thick meshwork (Aebi et al. 1986, Turgay et al. 2017) that functions as the heavy-duty shock absorber of the nucleus (Dahl et al. 2004, Stephens et al. 2017). CROWDED NULCEI (CRWN) coiled-coil proteins seem to serve a similar function in plants (Goswami et al. 2020).
B-type lamins are conserved across all animals, although invertebrates possess only a single Lamin B gene. B-type lamins are ubiquitously expressed throughout development (Lehner et al. 1987, Schatten et al. 1985). The different lamins homotypically assemble into separate meshwork structures that interact with each other (Delbarre et al. 2006). A high-resolution study showed that the Lamin B1 meshwork forms the outermost layer of the nuclear lamina right underneath the INM. This distinct outer localization depends on Lamin B1 farnesylation (Nmezi et al. 2019). Lamin A/C, and perhaps also Lamin B2, stiffen the nuclear lamina (Lammerding et al. 2004, 2006; Vortmeyer-Krause et al. 2020). Lamin B1–deficient cells show normal nuclear stiffness but have an unstable NM that is prone to blebbing (Lammerding et al. 2006). The Lamin A/C-to-B1 ratio of the nuclear lamina is increased in regions of high negative membrane curvature, such as at the poles of ellipsoid nuclei. These regions happen to be particularly prone to nuclear blebbing and NM rupture (Funkhouser et al. 2013, Xia et al. 2018).
LMNA, which encodes for Lamin A/C, developed during vertebrate evolution from an ancestral Lamin B gene. Lamin C, which is derived from LMNA by alternative splicing, only occurs in mammals (Peter & Stick 2012). Lamin A/C are highly expressed in differentiated cells but less so in pluripotent stem cells and during early embryogenesis (Constantinescu et al. 2006, Eckersley-Maslin et al. 2013, Röber et al. 1989, Stewart & Burke 1987).
Unlike B-type lamins, Lamin A is normally not farnesylated. Non-farnesylated lamins partially reside in the nucleoplasm, where they can interact with chromatin and regulate its mobility and transcriptional activities (Bronshtein et al. 2015, Dechat et al. 2010). Mutations that result in the erroneous farnesylation of Lamin A are associated with laminopathies such as Hutchinson-Gilford progeria (Cao et al. 2007, Eriksson et al. 2003). The phosphorylation of assembled lamin filaments by cyclin-dependent kinase 1 and other kinases triggers network destabilization and disassembly at the onset of mitosis, NETosis, or apoptosis (Amulic et al. 2017, Chang et al. 2011, Gerace & Blobel 1980, Li et al. 2020, Mall et al. 2012, Ottaviano & Gerace 1985).
2.3.2. The nuclear lamina under load.
Lamin filaments absorb force through cascaded structural changes which makes them extremely resilient. Those changes comprise the elastic stretching and the partial unfolding of their coiled-coil rod domains followed by an irreversible structural transition into β-sheets, filament sliding, and eventual rupture (Qin et al. 2009, Sapra et al. 2020). Being tougher than Kevlar and about as tough as silk, lamin filaments reversibly deform at low loads (<500 pN), then stiffen with applied force and ultimately break at >2 nN (Sapra et al. 2020). Compounded by their meshwork arrangement within the lamina, they function as efficient shock absorbers for the nucleus (Dahl et al. 2004, Sapra et al. 2020).
Lamin A/C expression level, phosphorylation, and turnover correlate with ECM stiffness and cytoskeletal tension indicative of mechanochemical feedback between the lamina and actomyosin (Buxboim et al. 2014, Cho et al. 2017, Swift et al. 2013). Actomyosin-induced nuclear flattening on stiff substrates, such as bone ECM, promotes nuclear stiffening through increased Lamin A/C expression, biasing mesenchymal stem cell (MSC) differentiation toward bone (Swift et al. 2013). Adapting laminar stiffness to ECM stiffness protects against excessive nuclear deformation that may cause NM and DNA damage and associated diseases (e.g., laminopathies; inflammatory disease; cancer; and heart, muscle, and bone diseases) (Hatch & Hetzer 2014, Jaalouk & Lammerding 2009).
2.3.3. Lamina damage.
An unperturbed lamina can withstand mechanical loads throughout the entire physiological range without showing obvious structural damage, such as filament or meshwork rupture.
3. COMPONENT CONNECTIONS
The NM, chromatin, and lamina are intricately connected. Akin to the PM with its underlying actin cortex, they form an adaptive, topologically complex composite material that senses and responds to mechanical cues as a unit (Lamparter & Galic 2020). Three major types of linkages define the integrated nuclear response to mechanical perturbations: (a) lamina-associated domains (LADs), which connect chromatin to the lamina and INM; (b) the LINC complex, which couples the nucleoskeleton to the cytoskeleton; and (c) the anchoring of embedded NM proteins to underlying structures, which may modulate NM tension and flow.
The 10 kb–10 Mb-long LADs (Guelen et al. 2008) often feature high AT content, low gene density and activity, and histone methylation marks characteristic of heterochromatin (e.g., H3K9me2 and H3K9me3). Through histones, histone marks, or other chromatin-binding proteins, such as barrier-to-autointegration factor (BAF), LADs interact with lamins and lamina-associated INM proteins such as Emerin and LBR (Figure 1, right panel). Thus, chromatin is coupled both to the lamina and the INM (van Steensel & Belmont 2017). LADs help to spatially organize the genome into territories (Cremer & Cremer 2001) with potentially important, albeit not fully understood, consequences for gene regulation. In addition, membrane and lamina anchoring of chromatin stabilizes the NE against small deformations, as discussed in Section 2.2.2. (Schreiner et al. 2015, Stephens et al. 2019). In cells with little or no lamin expression, such as white blood cells or yeast, chromatin-INM contacts seem to modulate NM tension. Reducing these contacts in yeast by the deletion of LEM-family proteins was shown to augment nuclear size and shape fluctuations, perhaps by facilitating lipid flow from the ER into the NM (Kume et al. 2019, Schreiner et al. 2015).
The LINC complex is the main conduit for mechanical inputs from the ECM into the nucleus. It connects the NE to perinuclear actomyosin (i.e., the perinuclear actin cap). The disruption of the LINC complex perturbs perinuclear actin and causes nuclear shape changes and disease phenotypes that, in part, resemble LMNA disruption (Janin et al. 2017, Khatau et al. 2009).
LADs and LINC complexes couple different solids (i.e., chromatin to protein or protein to protein), which enables mechanical force transmission between these components. By contrast, the mechanical consequences of solid-liquid coupling between integral membrane proteins and their surrounding lipid bilayers are more complex. Integral membrane proteins modulate the mobility of nearby lipids by forming shells of ~50–100 lipids with reduced diffusivity around them (Niemelä et al. 2010). Anchored to underlying cytoskeletal structures, membrane-embedded proteins act as diffusion barriers (Nakada et al. 2003) and limit tension propagation within the membrane. For the PM, a small fraction of immobilized membrane proteins (~10%) is estimated to be enough to change its rheology from fluid to gel-like dynamics (Shi et al. 2018). Altering NM protein abundance or anchoring should have similar effects. For example, when the INM becomes tense upon osmotic shock, the fraction of immobilized proteins on the NM should determine how fast INM tension propagates to the distal ER to activate mechanosensitive channels or recruit additional membrane. Mechanosensitive protein redistribution is a hallmark of nuclear mechanotransduction (see Section 4.3.) and includes the redistribution and turnover of integral NM proteins such as Emerin and possibly others (Buchwalter et al. 2019, Le et al. 2016). Emerin redistribution was proposed to regulate nuclear mechanotransduction through causing actin polymerization inside or outside the nucleus (Ho et al. 2013, Holaska et al. 2004, Le et al. 2016). Conceivably, Emerin redistribution may also alter membrane mechanics, depending on the fraction of the area it occupies on the NM and its anchoring to structures underneath. Whether a deficiency of Emerin or other LEM-family proteins (or the loss of their lamina and chromatin interactions) alters the rheological properties of the NM is unclear. It would be worthwhile to test whether such perturbations cause changes in ER-NM membrane flow, as previously described for Lem2 in yeast (Kume et al. 2019).
4. MECHANISMS OF NUCLEAR MECHANOTRANSDUCTION
Intermediate nuclear mechanotransduction translates mechanical cues (e.g., substrate stiffness, adhesion strength, and others) initially sensed at the cell surface into changes in gene expression to regulate the mechanical properties of the nucleus. These mechanisms allow the adaptation of cells to their mechanical environment, for example by adjusting nuclear stiffness to substrate stiffness. In MSCs, this mechanical adaptation entails matrix-directed cell fate decisions, specifically MSC differentiation into bone or fat cells (Akter et al. 2009, Cho et al. 2017, Driscoll et al. 2015, Engler et al. 2006, Swift et al. 2013). During immediate nuclear mechanotransduction, the nucleus functions as an alternative sensory surface of the cell body that directly probes the exterior physical environment without requiring input by the cell surface or cytoplasmic actomyosin. Immediate nuclear mechanotransduction was shown to regulate cortical blebbing and cell motility, paracrine inflammatory signaling during the wound response, and cell cycle progression (Aureille et al. 2019, Enyedi et al. 2016, Lomakin et al. 2020, Venturini et al. 2020).
4.1. The Nuclear Force Sensors
On the structural level, nuclear mechanotransduction is thought to involve the force-induced masking or unmasking of sites for protein phosphorylation and protein-protein, protein-membrane, or protein-chromatin interactions that impinge on changes in biochemical pathway activity (Enyedi et al. 2016, Guilluy et al. 2014, Ihalainen et al. 2015, Tajik et al. 2016) (Figure 2A). The critical molecular intermediates of nuclear force–sensing still need to be better defined. Understanding the precise structural mechanisms of nuclear mechanosensing is complicated by the fact that some of the likely nuclear force sensors, such as Lamin A/C (Ihalainen et al. 2015), hold important structural (and other) functions in the nucleus. Delineating their structural from their mechanotransduction roles is nontrivial given the extensive feedback between the two. The same issue holds back the mechanistic interpretation of laminopathic and envelopathic disease phenotypes. The recent discovery of peripheral membrane enzymes as NM-force sensors with no obvious structural roles (Enyedi et al. 2016) may allow us to more specifically dissect nuclear mechanotransduction mechanisms and functions in the future.
4.2. Experimental Challenges and Strategies
A major experimental challenge in nuclear mechanotransduction research is to distinguish nuclear from cytoplasmic mechanotransduction mechanisms. Both may be simultaneously initiated by the same physical triggers and collaborate during intermediate nuclear mechanotransduction. A radical way to test the role of nuclear mechanotransduction in a cellular process is to remove the cytoplasm (e.g., by cell permeabilization or nuclear isolation) and check whether the mechanism still works (Enyedi et al. 2016, Guilluy et al. 2014). Conversely, one may remove the nucleus and show that the mechanism does not work anymore (Lomakin et al. 2020). Most frequently, less invasive genetic or pharmacologic techniques are used to probe the effects of altered nuclear mechanics on the pathway of interest. These include Lamin A/C depletion or overexpression to soften or stiffen the nucleus, disruption of the LINC complex to sever its cytoskeletal connection, overexpression of LBR to expand its NM, and others. Demonstrating that the process of interest tightly correlates with nuclear surface as opposed to cell surface deformation presents another line of evidence. This can be accomplished by combining high-resolution imaging of nuclear deformation with quantitative physical probing techniques such as atomic force microscopy (Lomakin et al. 2020, Venturini et al. 2020). While each of these approaches has drawbacks, together they can make a powerful case for nuclear mechanotransduction.
4.3. Mechanosensitive Protein Redistribution
The subnanometer structural changes of nuclear components during force-sensing (Figure 2A, Section 4.1.) cause the redistribution of molecular regulators between the cytoplasm, nucleoplasm and NM (Figure 2B), which can be microscopically observed in live cells and tissues.
4.3.1. Protein redistribution to the nuclear membrane.
In animals that live in fresh water or in land-living creatures whose internal epithelia are partly covered with low osmolarity solution (e.g., saliva), epithelial injury leads to local hypotonic shock that causes cell and nuclear swelling near the wound. In zebrafish, a freshwater species, hypotonic shock acts as physical danger signal that initiates early inflammatory and healing responses (Enyedi et al. 2013, Gault et al. 2014). Intravital imaging in live zebrafish larvae demonstrated that wound-induced nuclear swelling is accompanied by the rapid translocation of cPLA2 from the nucleoplasm to the INM. This required both Ca2+ ions and NM stretch (Enyedi et al. 2013). cPLA2 and another enzyme in the same pathway, 5-lipoxygenase, were proposed to sense NM tension via their C2(-like) domains (Enyedi & Niethammer 2016, 2017; Enyedi et al. 2016). Specifically, osmotic swelling and agarose pad compression of the intact nuclei of permeabilized cells triggered the rapid binding of cPLA2 to the INM in the absence of functional cytoplasm. Likewise, osmotic swelling of giant unilamellar vesicles enhanced the membrane adsorption of cPLA2’s C2 domain in the presence of Ca2+ (Enyedi et al. 2016).
cPLA2 hydrolyses AA from NM phospholipids. AA is then enzymatically converted into hormonelike lipids, the eicosanoids (Burke & Dennis 2009). Some eicosanoids are strong immune cell chemoattractants, which explains how NM mechanotransduction causes rapid leukocyte recruitment to zebrafish wounds (Enyedi et al. 2013, 2016; Katikaneni et al. 2020). Rapid biomechanical detection of wounds through NM mechanotransduction is essential for larval survival after bacterial infection (Huang & Niethammer 2018). Like osmotic shock, necrotic cell death causes cell- and nuclear swelling. Thus, the biomechanical induction of eicosanoids at the nucleus may also contribute to the devastating sterile inflammatory responses to necrosis observed in humans, for example after a stroke—an idea that will be intriguing to test.
Besides inflammation, eicosanoids control cell survival, cell differentiation, and proliferation during tissue repair, regeneration, and cancer. Prostaglandin E2, for example, modulates load-dependent bone formation, perhaps in part through regulating MSC differentiation (Robertson et al. 2006, X. Zhang et al. 2002). Bone homeostasis is an epitome of animal mechanobiology and is heavily affected by both NE and prostaglandin perturbation (Blackwell et al. 2010, Gargiuli et al. 2018).
There are numerous literature accounts of PLA2 lipid products (AA, LPC, lysophosphatidic acid, eicosanoids, and others) acting as intra- or extracellular biomechanical messengers to regulate, for instance, ion channels or transcription (Sadoshima & Izumo 1993, Vriens et al. 2004). Evidence for the role of this pathway as an important NM-mechanotransduction mechanism appears to consolidate. Recently, two independent groups (Liu et al. 2015, Lomakin et al. 2020, Ruprecht et al. 2015, Shen & Niethammer 2020, Venturini et al. 2020) reported that cell confinement mediates PM blebbing and cell motility in embryonic, cancer, and immune cells through activating cPLA2. Indicative of an immediate nuclear mechanosensing mechanism, only cell confinement strong enough to stretch the NM, triggered the blebbing response, whereas cell surface deformation alone had little effect. Contrasting AA’s classic signaling role as eicosanoid precursor, these studies raised the possibility that AA directly activates cortical actomyosin contraction through RhoA. Adjusting contractile responses to three-dimensional confinement could help cells to efficiently traverse tight tissue mazes or pores in barrier tissues without getting stuck (Lomakin et al. 2020, Venturini et al. 2020). Whether nuclear confinement or compression also activates AA-dependent paracrine signaling cascades remains to be tested.
4.3.2. Protein redistribution between the cytoplasm and the nucleus.
The nuclear accumulation of some mechanosensitive transcriptional regulators, such as of yes-associated protein 1 (YAP) and retinoic acid receptor gamma, is enhanced by cell spreading on stiff substrates (Dupont et al. 2011, Swift et al. 2013). During cell spreading, actomyosin contractility compresses the nucleus and triggers cytoplasmic and nuclear phosphorylation events that promote YAP retention in the nucleus (Codelia et al. 2014, Dupont et al. 2011, Ege et al. 2018, Pocaterra et al. 2020). This mechanism is commonly thought to involve concerted mechanotransduction events at the cell surface, in the cytoplasm, and the nucleus. Interestingly, nuclear compression alone seems to be enough to promote nuclear YAP localization. Specifically, nucleoplasmic YAP accumulation was observed after nuclear compression with an agarose pad or an atomic force microscope cantilever in cells treated with actomyosin inhibitors (Aureille et al. 2019, Elosegui-Artola et al. 2017). The underlying mechanisms remain incompletely understood. At least in part, nuclear deformation may control YAP retention by regulating the availability of factors involved in YAP nuclear export through, for example, stimulating their mechanosensitive phosphorylation. In line with this idea, the overexpression of a phosphodeficient Emerin mutant protein was shown to impair the nuclear accumulation of YAP (Guilluy et al. 2014). Emerin regulates the nuclear shuttling of at least two other transcriptional regulators, the megakaryoblastic leukaemia 1 (MLK1) protein and β-catenin (Ho et al. 2013, Markiewicz et al. 2006). In isolated nuclei, Emerin phosphorylation can be mechanically triggered by pulling on the LINC complex (Guilluy et al. 2014). Its distribution and abundance in the NM is sensitive to mechanical strain (Le et al. 2016) and the state of the nuclear lamina (Ho et al. 2013), respectively.
Nuclear deformation has also been reported to unidirectionally increase nuclear pore permeability to enhance YAP nuclear accumulation (Elosegui-Artola et al. 2017). The underlying structural mechanisms remain elusive, but they may involve the selective opening of nuclear pores to the cytoplasmic side or rearrangements within the fibrous protein structure that lines the nucleoplasmic side of the NPC, the so-called nuclear basket (Elosegui-Artola et al. 2017, García-González et al. 2018). If nuclear stretch per se was responsible for nuclear YAP accumulation, not only compression but also swelling of nuclei should promote it; however, this was not observed (Elosegui-Artola et al. 2017). Somehow, YAP can distinguish squeezed from swollen nuclei. How exactly this is accomplished requires further investigation.
4.3.3. Protein redistribution within the nuclear membrane.
Different lines of evidence suggest a central role for the NM protein Emerin as a regulator of nuclear mechanics and mechanotransduction. The nuclei of Emerin-deficient or -mutant cells show aberrant shapes and lesions, respond abnormally to some mechanical perturbations, and exhibit altered gene transcription (Fidziańska et al. 1998, Lammerding et al. 2005, Rowat et al. 2006). Human null mutations in Emerin, similar to some mutations of Lamin A/C and Nesprin-1, are associated with Emery-Dreifuss muscular dystrophy (EDMD) (Janin et al. 2017). As mice show milder phenotypes than humans, some Emerin functions might be species specific (Bengtsson & Wilson 2004).
Emerin binds to lamins, Nesprins, nuclear actin, chromatin, and gene regulators (e.g., BAF) and can promote actin polymerization in vitro (Bengtsson & Wilson 2004). In mouse embryonic fibroblasts, Lamin A/C deficiency increases Emerin mobility and reduces its INM-localization (Ho et al. 2013). The expression of some EDMD-associated Lamin A mutants leads to the loss of Emerin from the NE (Ostlund et al. 2001). Lamina and chromatin anchoring of Emerin (via its LEM domain) may promote its retention on the INM, and its turnover (Buchwalter et al. 2019). In epidermal progenitor cells, biaxial cyclic strain redistributes Emerin from the INM to the ONM (Le et al. 2016). The underlying mechanosensing mechanism is incompletely understood. Nuclear deformation may bury Emerin-binding sites on Lamin A/C (Ihalainen et al. 2015). In addition, strain-induced chromatin remodeling (Nava et al. 2020) or tyrosine phosphorylation of Emerin (Guilluy et al. 2014) may regulate its membrane mobility by, for example, modulating its interaction with the DNA-binding protein BAF (Tifft et al. 2009).
NM redistribution of Emerin after mechanical stress (Le et al. 2016) or caused by Lamin A/C deficiency (Ho et al. 2013) was reported to promote (Le et al. 2016) or impair (Ho et al. 2013) cytoplasmic actin polymerization, respectively. Cytoplasmic actin polymerization depletes nuclear globular actin (G-actin), which serves as a transcriptional coactivator (Le et al. 2016, Olson & Nordheim 2010) and nuclear export factor (Miralles et al. 2003, Vartiainen et al. 2007). Through regulating nuclear G-actin, strain-induced Emerin redistribution can therefore modulate the nuclear shuttling of transcription factors and gene expression.
4.4. Mechanosensitive Calcium Release
Ca2+ instructs a myriad of cellular signaling events either alone or together with other chemical or physical cosignals such as NM stretch (see Section 4.3.1.). Cell deformation can trigger cytoplasmic and/or nucleoplasmic elevation of the Ca2+ concentration through mechanosensitive cation channels located on the PM or the ER. As the ER is contiguous with the ONM, membrane stretch may be propagated from the NM to activate mechanosensitive channels in the ER, such as Piezo1 or others. Making the NM floppier by Lamin A/C depletion or LBR overexpression (which causes NM surface expansion) blocked confinement-induced Ca2+ transients in HeLa cells (Lomakin et al. 2020). Others showed that augmenting nuclear stiffness by Lamin A overexpression increases mechanosensitive Ca2+ release through intracellular Piezo1 in human fibrosarcoma cells (Nava et al. 2020). Whereas mechanosensitive ion channels are often connected to cell surface mechanotransduction, these studies caution that their potential intracellular functions must be carefully considered. Both studies corroborate the idea that cell deformation controls mechanosensitive cation channels in the ER via nuclear deformation. But this mechanism may be cell type specific or more complex: Work in embryonic zebrafish cells suggests that cell deformation also causes the close proximity of the ER and the PM. This may allow Ca2+ influx via stromal interaction molecule (STIM)-Orai-dependent mechanisms (Venturini et al. 2020). Whether or how nuclear mechanics regulates intracellular Ca2+ release remains an open topic for future research.
4.5. Direct Mechanical Control of Gene Transcription
Nuclear deformation may indirectly regulate transcription through the mechanosensitive redistribution mechanisms outlined in Section 4.3.2.. Alternatively, chromosome stretching could increase the accessibility of genes for the transcriptional machinery. Stretching of a fluorescently labeled reporter transgene with magnetic beads attached to integrins on the cell surface was reported to trigger rapid transgene transcription. Besides integrins, the actin cytoskeleton, the LINC complex, and lamin-chromatin contacts were required for mechanotransduction from the cell surface to chromatin (Tajik et al. 2016).
Nuclear deformation may also affect the expression of genes by changing their positions within the nucleus. Different chromosomal territories are associated with different levels of gene activity (Cremer & Cremer 2001, Shachar & Misteli 2017), and nuclear deformation could alter the relative positions of these territories. Along those lines, chromosome intermingling was found to correlate with the emergence of transcriptional programs (Maharana et al. 2016).
5. OUTLOOK
Most people probably think of the cell nucleus as a, more or less, passive vessel for genetic information. But as argued here, it is also a sensory organelle that allows cells to probe their physical environment more completely than they could via their surface alone. This notion marks a conceptual shift that will, I hope, inspire cell biologists and bioengineers to enter the field. To understand the broader physiological implications of nuclear mechanotransduction, what is dearly needed right now are better tools to selectively measure and perturb the mechanical forces that act on the nucleus in live cells and tissues. Likely, those will include non-invasive optical probes modeled after actual nuclear force sensors, such as lamins, cPLA2, and those others that still remain to be discovered. The pioneering potential of nuclear mechanotransduction research outweighs these challenges ahead.
ACKNOWLEDGMENTS
I apologize if important studies have not been mentioned here for space constraints or lack of attention. I am currently supported by the National Institutes of Health grants R01GM127356, R01GM099970, R35GM140883, and R21AI139986 and a Basic Research Innovation Award (BRIA) from the Memorial Sloan Kettering Cancer Center.
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Aebi U, Cohn J, Buhle L, Gerace L. 1986. The nuclear lamina is a meshwork of intermediate-type filaments. Nature 323(6088):560–64 [DOI] [PubMed] [Google Scholar]
- Agrawal A, Lele TP. 2019. Mechanics of nuclear membranes. J. Cell Sci 132(14):jcs229245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akter R, Rivas D, Geneau G, Drissi H, Duque G. 2009. Effect of lamin A/C knockdown on osteoblast differentiation and function. J. Bone Miner. Res 24(2):283–93 [DOI] [PubMed] [Google Scholar]
- Amulic B, Knackstedt SL, Abu Abed U, Deigendesch N, Harbort CJ, et al. 2017. Cell-cycle proteins control production of neutrophil extracellular traps. Dev. Cell 43(4):449–62.e5 [DOI] [PubMed] [Google Scholar]
- Aureille J, Buffière-Ribot V, Harvey BE, Boyault C, Pernet L, et al. 2019. Nuclear envelope deformation controls cell cycle progression in response to mechanical force. EMBO Rep. 20(9):e48084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson L, Wilson KL. 2004. Multiple and surprising new functions for emerin, a nuclear membrane protein. Curr. Opin. Cell Biol 16(1):73–79 [DOI] [PubMed] [Google Scholar]
- Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, et al. 2002. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. PNAS 99(20):13049–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigay J, Antonny B. 2012. Curvature, lipid packing, and electrostatics of membrane organelles: defining cellular territories in determining specificity. Dev. Cell 23(5):886–95 [DOI] [PubMed] [Google Scholar]
- Blackwell KA, Raisz LG, Pilbeam CC. 2010. Prostaglandins in bone: bad cop, good cop? Trends Endocrinol. Metab 21(5):294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni A, Politi AZ, Strnad P, Xiang W, Hossain MJ, Ellenberg J. 2015. Live imaging and modeling of inner nuclear membrane targeting reveals its molecular requirements in mammalian cells. J. Cell Biol 209(5):705–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronshtein I, Kepten E, Kanter I, Berezin S, Lindner M, et al. 2015. Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat. Commun 6:8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotherus J, Renkonen O. 1977. Phospholipids of subcellular organelles isolated from cultured BHK cells. Biochim. Biophys. Acta Lipids Lipid Metab 486(2):243–53 [DOI] [PubMed] [Google Scholar]
- Buchwalter A, Schulte R, Tsai H, Capitanio J, Hetzer M. 2019. Selective clearance of the inner nuclear membrane protein emerin by vesicular transport during ER stress. eLife 8:e49796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JE, Dennis EA. 2009. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res 50(Suppl.):S237–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxboim A, Swift J, Irianto J, Spinler KR, Dingal PCDP, et al. 2014. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol 24(16):1909–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. 2007. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. PNAS 104(12):4949–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca M 2002. The contribution of nuclear compartmentalization to gene regulation. Cell 108(4):513–21 [DOI] [PubMed] [Google Scholar]
- Cenni V, D’Apice MR, Garagnani P, Columbaro M, Novelli G, et al. 2018. Mandibuloacral dysplasia: a premature ageing disease with aspects of physiological ageing. Ageing Res. Rev 42:1–13 [DOI] [PubMed] [Google Scholar]
- Chang K-H, Multani PS, Sun K-H, Vincent F, de Pablo Y, et al. 2011. Nuclear envelope dispersion triggered by deregulated Cdk5 precedes neuronal death. Mol. Biol. Cell 22(9):1452–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen NY, Kim P, Weston TA, Edillo L, Tu Y, et al. 2018. Fibroblasts lacking nuclear lamins do not have nuclear blebs or protrusions but nevertheless have frequent nuclear membrane ruptures. PNAS 115(40):10100–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizmadzhev YA, Kumenko DA, Kuzmin PI, Chernomordik LV, Zimmerberg J, Cohen FS. 1999. Lipid flow through fusion pores connecting membranes of different tensions. Biophys. J 76(6):2951–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Irianto J, Discher DE. 2017. Mechanosensing by the nucleus: from pathways to scaling relationships. J. Cell Biol 216(2):305–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Vashisth M, Abbas A, Majkut S, Vogel K, et al. 2019. Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev. Cell 49(6):920–35.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codelia VA, Sun G, Irvine KD. 2014. Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr. Biol 24(17):2012–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu D, Gray HL, Sammak PJ, Schatten GP, Csoka AB. 2006. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells 24(1):177–85 [DOI] [PubMed] [Google Scholar]
- Cremer T, Cremer C. 2001. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet 2(4):292–301 [DOI] [PubMed] [Google Scholar]
- Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, et al. 2006. Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol 172(1):41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl KN, Engler AJ, Pajerowski JD, Discher DE. 2005. Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures. Biophys. J 89(4):2855–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl KN, Kahn SM, Wilson KL, Discher DE. 2004. The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber. J. Cell Sci 117(Part 20):4779–86 [DOI] [PubMed] [Google Scholar]
- Dai J, Sheetz MP. 1995. Axon membrane flows from the growth cone to the cell body. Cell 83(5):693–701 [DOI] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, et al. 2003. Lamin A truncation in Hutchinson-Gilford progeria. Science 300(5628):2055. [DOI] [PubMed] [Google Scholar]
- Dechat T, Gesson K, Foisner R. 2010. Lamina-independent lamins in the nuclear interior serve important functions. Cold Spring Harb. Symp. Quant. Biol 75:533–43 [DOI] [PubMed] [Google Scholar]
- Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, et al. 2007. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. PNAS 104(12):4955–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbarre E, Tramier M, Coppey-Moisan M, Gaillard C, Courvalin J-C, Buendia B. 2006. The truncated prelamin A in Hutchinson-Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum. Mol. Genet 15(7):1113–22 [DOI] [PubMed] [Google Scholar]
- Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, et al. 2016. Nuclear envelope rupture and repair during cancer cell migration. Science 352(6283):353–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande S, Wunnava S, Hueting D, Dekker C. 2019. Membrane tension-mediated growth of liposomes. Small 15(38):e1902898. [DOI] [PubMed] [Google Scholar]
- Diz-Muñoz A, Fletcher DA, Weiner OD. 2013. Use the force: membrane tension as an organizer of cell shape and motility. Trends Cell Biol. 23(2):47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll TP, Cosgrove BD, Heo S-J, Shurden ZE, Mauck RL. 2015. Cytoskeletal to nuclear strain transfer regulates YAP signaling in mesenchymal stem cells. Biophys. J 108(12):2783–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdz MM, Vaux DJ. 2017. Shared mechanisms in physiological and pathological nucleoplasmic reticulum formation. Nucleus 8(1):34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, et al. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474(7350):179–83 [DOI] [PubMed] [Google Scholar]
- Eckersley-Maslin MA, Bergmann JH, Lazar Z, Spector DL. 2013. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 4(1):53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ege N, Dowbaj AM, Jiang M, Howell M, Hooper S, et al. 2018. Quantitative analysis reveals that actin and Src-family kinases regulate nuclear YAP1 and its export. Cell Syst. 6(6):692–708.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, et al. 2017. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171(6):1397–410.e14 [DOI] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. 2006. Matrix elasticity directs stem cell lineage specification. Cell 126(4):677–89 [DOI] [PubMed] [Google Scholar]
- Enyedi B, Jelcic M, Niethammer P. 2016. The cell nucleus serves as a mechanotransducer of tissue damage-induced inflammation. Cell 165(5):1160–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi B, Kala S, Nikolich-Zugich T, Niethammer P. 2013. Tissue damage detection by osmotic surveillance. Nat. Cell Biol 15(9):1123–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi B, Niethammer P. 2016. A case for the nuclear membrane as a mechanotransducer. Cell Mol. Bioeng 9(2):247–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi B, Niethammer P. 2017. Nuclear membrane stretch and its role in mechanotransduction. Nucleus 8(2):156–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, et al. 2003. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423(6937):293–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidziańska A, Toniolo D, Hausmanowa-Petrusewicz I. 1998. Ultrastructural abnormality of sarcolemmal nuclei in Emery-Dreifuss muscular dystrophy (EDMD). J. Neurol. Sci 159(1):88–93 [DOI] [PubMed] [Google Scholar]
- Franke WW, Deumling B, Baerbelermen, Jarasch ED, Kleinig H. 1970. Nuclear membranes from mammalian liver. I. Isolation procedure and general characterization. J. Cell Biol 46(2):379–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker M, Hollinshead M, White N, Vaux D. 1997. Interphase nuclei of many mammalian cell types contain deep, dynamic, tubular membrane-bound invaginations of the nuclear envelope. J. Cell Biol 136(3):531–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funkhouser CM, Sknepnek R, Shimi T, Goldman AE, Goldman RD, Olvera de la Cruz M. 2013. Mechanical model of blebbing in nuclear lamin meshworks. PNAS 110(9):3248–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa T, Rochman M, Taher L, Dimitriadis EK, Nagashima K, et al. 2015. Chromatin decompaction by the nucleosomal binding protein HMGN5 impairs nuclear sturdiness. Nat. Commun 6:6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-González A, Jacchetti E, Marotta R, Tunesi M, Rodríguez Matas JF, Raimondi MT. 2018. The effect of cell morphology on the permeability of the nuclear envelope to diffusive factors. Front. Physiol 9:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargiuli C, Schena E, Mattioli E, Columbaro M, D’Apice MR, et al. 2018. Lamins and bone disorders: current understanding and perspectives. Oncotarget 9(32):22817–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault WJ, Enyedi B, Niethammer P. 2014. Osmotic surveillance mediates rapid wound closure through nucleotide release. J. Cell Biol 207(6):767–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier NC, Masters TA, Sheetz MP. 2012. Mechanical feedback between membrane tension and dynamics. Trends Cell Biol. 22(10):527–35 [DOI] [PubMed] [Google Scholar]
- Gerace L, Blobel G. 1980. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 19(1):277–87 [DOI] [PubMed] [Google Scholar]
- Goswami R, Asnacios A, Hamant O, Chabouté M-E. 2020. Is the plant nucleus a mechanical rheostat? Curr. Opin. Plant Biol 57:155–63 [DOI] [PubMed] [Google Scholar]
- Graham DM, Andersen T, Sharek L, Uzer G, Rothenberg K, et al. 2018. Enucleated cells reveal differential roles of the nucleus in cell migration, polarity, and mechanotransduction. J. Cell Biol 217(3):895–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, et al. 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453(7197):948–51 [DOI] [PubMed] [Google Scholar]
- Guilluy C, Osborne LD, Van Landeghem L, Sharek L, Superfine R, et al. 2014. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol 16(4):376–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Kim Y, Shimi T, Goldman RD, Zheng Y. 2014. Concentration-dependent lamin assembly and its roles in the localization of other nuclear proteins. Mol. Biol. Cell 25(8):1287–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider A, Wei Y-C, Lim K, Barbosa AD, Liu C-H, et al. 2018. PCYT1A regulates phosphatidylcholine homeostasis from the inner nuclear membrane in response to membrane stored curvature elastic stress. Dev. Cell 45(4):481–95.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett FR, Marsh J, Nickel BG, Wood JM. 1993. Mechanical properties of vesicles. II. A model for osmotic swelling and lysis. Biophys. J 64(2):435–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch E, Hetzer M. 2014. Breaching the nuclear envelope in development and disease. J. Cell Biol 205(2):133–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzakis NS, Bhatia VK, Larsen J, Madsen KL, Bolinger P-Y, et al. 2009. How curved membranes recruit amphipathic helices and protein anchoring motifs. Nat. Chem. Biol 5(11):835–41 [DOI] [PubMed] [Google Scholar]
- Hennekes H, Nigg EA. 1994. The role of isoprenylation in membrane attachment of nuclear lamins. A single point mutation prevents proteolytic cleavage of the lamin A precursor and confers membrane binding properties. J. Cell Sci 107(Part 4):1019–29 [DOI] [PubMed] [Google Scholar]
- Hetzer MW. 2010. The nuclear envelope. Cold Spring Harb. Perspect. Biol 2(3):a000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J. 2013. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497(7450):507–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, Pedersen SF. 2009. Physiology of cell volume regulation in vertebrates. Physiol. Rev 89(1):193–277 [DOI] [PubMed] [Google Scholar]
- Holaska JM, Kowalski AK, Wilson KL. 2004. Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLOS Biol. 2(9):E231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Niethammer P. 2018. Tissue damage signaling is a prerequisite for protective neutrophil recruitment to microbial infection in zebrafish. Immunity 48(5):1006–13.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihalainen TO, Aires L, Herzog FA, Schwartlander R, Moeller J, Vogel V. 2015. Differential basal-to-apical accessibility of lamin A/C epitopes in the nuclear lamina regulated by changes in cytoskeletal tension. Nat. Mater 14(12):1252–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irianto J, Pfeifer CR, Bennett RR, Xia Y, Ivanovska IL, et al. 2016. Nuclear constriction segregates mobile nuclear proteins away from chromatin. Mol. Biol. Cell 27(25):4011–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irianto J, Xia Y, Pfeifer CR, Athirasala A, Ji J, et al. 2017. DNA damage follows repair factor depletion and portends genome variation in cancer cells after pore migration. Curr. Biol 27(2):210–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaalouk DE, Lammerding J. 2009. Mechanotransduction gone awry. Nat. Rev. Mol. Cell Biol 10:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson EC, Perry JK, Long DS, Olins AL, Olins DE, et al. 2018. Migration through a small pore disrupts inactive chromatin organization in neutrophil-like cells. BMC Biol. 16:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin A, Bauer D, Ratti F, Millat G, Méjat A. 2017. Nuclear envelopathies: a complex LINC between nuclear envelope and pathology. Orphanet J. Rare Dis 12:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katikaneni A, Jelcic M, Gerlach GF, Ma Y, Overholtzer M, Niethammer P. 2020. Lipid peroxidation regulates long-range wound detection through 5-lipoxygenase in zebrafish. Nat. Cell Biol 22(9):1049–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandwala AS, Kasper CB. 1971. The fatty acid composition of individual phospholipids from rat liver nuclear membrane and nuclei. J. Biol. Chem 246(20):6242–46 [PubMed] [Google Scholar]
- Khatau SB, Hale CM, Stewart-Hutchinson PJ, Patel MS, Stewart CL, et al. 2009. A perinuclear actin cap regulates nuclear shape. PNAS 106(45):19017–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K, Cantwell H, Burrell A, Nurse P. 2019. Nuclear membrane protein Lem2 regulates nuclear size through membrane flow. Nat. Commun 10:1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumi A, Fujiwara TK, Chadda R, Xie M, Tsunoyama TA, et al. 2012. Dynamic organizing principles of the plasma membrane that regulate signal transduction: commemorating the fortieth anniversary of Singer and Nicolson’s fluid-mosaic model. Annu. Rev. Cell Dev. Biol 28:215–50 [DOI] [PubMed] [Google Scholar]
- Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, et al. 2006. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem 281(35):25768–80 [DOI] [PubMed] [Google Scholar]
- Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. 2005. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol 170(5):781–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, et al. 2004. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest 113(3):370–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamparter L, Galic M. 2020. Cellular membranes, a versatile adaptive composite material. Front. Cell Dev. Biol 8:684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le HQ, Ghatak S, Yeung C-YC, Tellkamp F, Günschmann C, et al. 2016. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol 18(8):864–75 [DOI] [PubMed] [Google Scholar]
- Le Roux A-L, Quiroga X, Walani N, Arroyo M, Roca-Cusachs P. 2019. The plasma membrane as a mechanochemical transducer. Philos. Trans. R. Soc. B 374(1779):20180221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner CF, Stick R, Eppenberger HM, Nigg EA. 1987. Differential expression of nuclear lamin proteins during chicken development. J. Cell Biol 105(1):577–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li M, Weigel B, Mall M, Werth VP, Liu M-L. 2020. Nuclear envelope rupture and NET formation is driven by PKCα-mediated lamin B disassembly. EMBO Rep. 21(8):e48779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-J, Le Berre M, Lautenschlaeger F, Maiuri P, Callan-Jones A, et al. 2015. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160(4):659–72 [DOI] [PubMed] [Google Scholar]
- Lomakin AJ, Cattin CJ, Cuvelier D, Alraies Z, Molina M, et al. 2020. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science 370(6514):eaba2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. 2015. Chromothripsis and kataegis induced by telomere crisis. Cell 163(7):1641–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharana S, Iyer KV, Jain N, Nagarajan M, Wang Y, Shivashankar GV. 2016. Chromosome intermingling—the physical basis of chromosome organization in differentiated cells. Nucleic Acids Res. 44(11):5148–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M, Walter T, Gorjánácz M, Davidson IF, Nga Ly-Hartig TB, et al. 2012. Mitotic lamin disassembly is triggered by lipid-mediated signaling. J. Cell Biol 198(6):981–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewicz E, Tilgner K, Barker N, van de Wetering M, Clevers H, et al. 2006. The inner nuclear membrane protein emerin regulates β-catenin activity by restricting its accumulation in the nucleus. EMBO J. 25(14):3275–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer M, Lammerding J. 2019. The driving force: nuclear mechanotransduction in cellular function, fate, and disease. Annu. Rev. Biomed. Eng 21:443–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder A, Roopa T, Basu A, Mahadevan L, Shivashankar GV. 2008. Dynamics of chromatin decondensation reveals the structural integrity of a mechanically prestressed nucleus. Biophys. J 95(6):3028–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méjat A, Misteli T. 2010. LINC complexes in health and disease. Nucleus 1(1):40–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou A-I, Treisman R. 2003. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113(3):329–42 [DOI] [PubMed] [Google Scholar]
- Miroshnikova YA, Cohen I, Ezhkova E, Wickström SA. 2019. Epigenetic gene regulation, chromatin structure, and force-induced chromatin remodelling in epidermal development and homeostasis. Curr. Opin. Genet. Dev 55:46–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CE, Homann U. 2001. Cell surface area regulation and membrane tension. J. Membr. Biol 179(2):79–102 [DOI] [PubMed] [Google Scholar]
- Nakada C, Ritchie K, Oba Y, Nakamura M, Hotta Y, et al. 2003. Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat. Cell Biol 5(7):626–32 [DOI] [PubMed] [Google Scholar]
- Nava MM, Miroshnikova YA, Biggs LC, Whitefield DB, Metge F, et al. 2020. Heterochromatin-driven nuclear softening protects the genome against mechanical stress-induced damage. Cell 181(4):800–17.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, et al. 2004. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum. Mol. Genet 13(20):2493–503 [DOI] [PubMed] [Google Scholar]
- Needham D, Nunn RS. 1990. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J 58(4):997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert E, Meyer D, Rocca F, Günay G, Kwaczala-Tessmann A, et al. 2018. Chromatin swelling drives neutrophil extracellular trap release. Nat. Commun 9:3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemelä PS, Miettinen MS, Monticelli L, Hammaren H, Bjelkmar P, et al. 2010. Membrane proteins diffuse as dynamic complexes with lipids. J. Am. Chem. Soc 132(22):7574–75 [DOI] [PubMed] [Google Scholar]
- Nmezi B, Xu J, Fu R, Armiger TJ, Rodriguez-Bey G, et al. 2019. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. PNAS 116(10):4307–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll M, Thomas JO, Kornberg RD. 1975. Preparation of native chromatin and damage caused by shearing. Science 187(4182):1203–6 [DOI] [PubMed] [Google Scholar]
- Olson EN, Nordheim A. 2010. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol 11(5):353–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostlund C, Bonne G, Schwartz K, Worman HJ. 2001. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J. Cell Sci 114(Part 24):4435–45 [DOI] [PubMed] [Google Scholar]
- Ottaviano Y, Gerace L. 1985. Phosphorylation of the nuclear lamins during interphase and mitosis. J. Biol. Chem 260(1):624–32 [PubMed] [Google Scholar]
- Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. 2010. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol 191(3):677–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter A, Stick R. 2012. Evolution of the lamin protein family: what introns can tell. Nucleus 3(1):44–59 [DOI] [PubMed] [Google Scholar]
- Pfeifer CR, Xia Y, Zhu K, Liu D, Irianto J, et al. 2018. Constricted migration increases DNA damage and independently represses cell cycle. Mol. Biol. Cell 29(16):1948–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger RC, Anderson NG, Snyder F. 1968. Lipid class and fatty acid composition of rat liver plasma membranes isolated by zonal centrifugation. Biochemistry 7(8):2826–33 [DOI] [PubMed] [Google Scholar]
- Pinot M, Vanni S, Ambroggio E, Guet D, Goud B, Manneville J-B. 2018. Feedback between membrane tension, lipid shape and curvature in the formation of packing defects. BioRxiv 389627. 10.1101/389627 [DOI] [Google Scholar]
- Piovesan A, Pelleri MC, Antonaros F, Strippoli P, Caracausi M, Vitale L. 2019. On the length, weight and GC content of the human genome. BMC Res. Notes 12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocaterra A, Romani P, Dupont S. 2020. YAP/TAZ functions and their regulation at a glance. J. Cell Sci 133(2):jcs230425 [DOI] [PubMed] [Google Scholar]
- Qin Z, Kreplak L, Buehler MJ. 2009. Hierarchical structure controls nanomechanical properties of vimentin intermediate filaments. PLOS ONE 4(10):e7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, et al. 2016. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352(6283):359–62 [DOI] [PubMed] [Google Scholar]
- Ranade SS, Syeda R, Patapoutian A. 2015. Mechanically activated ion channels. Neuron. 87(6):1162–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegman M, Sagie L, Galed C, Levin T, Steinberg N, et al. 2020. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol 22:1042–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röber RA, Weber K, Osborn M. 1989. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 105(2):365–78 [DOI] [PubMed] [Google Scholar]
- Robertson G, Xie C, Chen D, Awad H, Schwarz EM, et al. 2006. Alteration of femoral bone morphology and density in COX-2−/− mice. Bone 39(4):767–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanauska A, Köhler A. 2018. The inner nuclear membrane is a metabolically active territory that generates nuclear lipid droplets. Cell 174(3):700–15.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowat AC, Lammerding J, Ipsen JH. 2006. Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophys. J 91(12):4649–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht V, Wieser S, Callan-Jones A, Smutny M, Morita H, et al. 2015. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell 160(4):673–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. 1993. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J. 12(4):1681–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapra KT, Qin Z, Dubrovsky-Gaupp A, Aebi U, Müller DJ, et al. 2020. Nonlinear mechanics of lamin filaments and the meshwork topology build an emergent nuclear lamina. Nat. Commun 11:6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatten G, Maul GG, Schatten H, Chaly N, Simerly C, et al. 1985. Nuclear lamins and peripheral nuclear antigens during fertilization and embryogenesis in mice and sea urchins. PNAS 82(14):4727–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner SM, Koo PK, Zhao Y, Mochrie SGJ, King MC. 2015. The tethering of chromatin to the nuclear envelope supports nuclear mechanics. Nat. Commun 6:7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciaky N, Presley J, Smith C, Zaal KJ, Cole N, et al. 1997. Golgi tubule traffic and the effects of Brefeldin A visualized in living cells. J. Cell Biol 139(5):1137–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar S, Misteli T. 2017. Causes and consequences of nuclear gene positioning. J. Cell Sci 130(9):1501–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Hobson CM, Cheng S, Colville MJ, Paszek MJ, et al. 2020. Nuclear deformation causes DNA damage by increasing replication stress. Curr. Biol 31(4):753–65.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Niethammer P. 2020. A cellular sense of space and pressure. Science 370(6514):295–96 [DOI] [PubMed] [Google Scholar]
- Shi Z, Graber ZT, Baumgart T, Stone HA, Cohen AE. 2018. Cell membranes resist flow. Cell 175(7):1769–79.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto Y, Tamura S, Masumoto H, Maeshima K. 2017. Nucleosome-nucleosome interactions via histone tails and linker DNA regulate nuclear rigidity. Mol. Biol. Cell 28(11):1580–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, et al. 2011. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 25(24):2579–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staykova M, Holmes DP, Read C, Stone HA. 2011. Mechanics of surface area regulation in cells examined with confined lipid membranes. PNAS 108(22):9084–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens AD, Banigan EJ, Adam SA, Goldman RD, Marko JF. 2017. Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol. Biol. Cell 28(14):1984–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens AD, Banigan EJ, Marko JF. 2019. Chromatin’s physical properties shape the nucleus and its functions. Curr. Opin. Cell Biol 58:76–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens AD, Liu PZ, Banigan EJ, Almassalha LM, Backman V, et al. 2018. Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell 29(2):220–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C, Burke B. 1987. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell 51(3):383–92 [DOI] [PubMed] [Google Scholar]
- Strick R, Strissel PL, Gavrilov K, Levi-Setti R. 2001. Cation-chromatin binding as shown by ion microscopy is essential for the structural integrity of chromosomes. J. Cell Biol 155(6):899–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Guo SS, Fässler R. 2016. Integrin-mediated mechanotransduction. J. Cell Biol 215(4):445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PCDP, et al. 2013. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341(6149):1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajik A, Zhang Y, Wei F, Sun J, Jia Q, et al. 2016. Transcription upregulation via force-induced direct stretching of chromatin. Nat. Mater 15(12):1287–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapley EC, Starr DA. 2013. Connecting the nucleus to the cytoskeleton by SUN-KASH bridges across the nuclear envelope. Curr. Opin. Cell Biol 25(1):57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, et al. 2020. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. PNAS 117(13):7326–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tifft KE, Bradbury KA, Wilson KL. 2009. Tyrosine phosphorylation of nuclear-membrane protein emerin by Src, Abl and other kinases. J. Cell Sci 122(Part 20):3780–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbati M, Lele TP, Agrawal A. 2016. Ultradonut topology of the nuclear envelope. PNAS 113(40):11094–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay Y, Eibauer M, Goldman AE, Shimi T, Khayat M, et al. 2017. The molecular architecture of lamins in somatic cells. Nature 543(7644):261–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Björling E, Agaton C, Szigyarto CA-K, Amini B, et al. 2005. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell Proteom 4(12):1920–32 [DOI] [PubMed] [Google Scholar]
- Ungricht R, Klann M, Horvath P, Kutay U. 2015. Diffusion and retention are major determinants of protein targeting to the inner nuclear membrane. J. Cell Biol 209(5):687–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol 9(2):112–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Belmont AS. 2017. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169(5):780–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanni S, Hirose H, Barelli H, Antonny B, Gautier R. 2014. A sub-nanometre view of how membrane curvature and composition modulate lipid packing and protein recruitment. Nat. Commun 5:4916. [DOI] [PubMed] [Google Scholar]
- Vanni S, Vamparys L, Gautier R, Drin G, Etchebest C, et al. 2013. Amphipathic lipid packing sensor motifs: probing bilayer defects with hydrophobic residues. Biophys. J 104(3):575–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JD, Hatch EM, Anderson DJ, Hetzer MW. 2012. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 3(1):88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen MK, Guettler S, Larijani B, Treisman R. 2007. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316(5832):1749–52 [DOI] [PubMed] [Google Scholar]
- Vaughan A, Alvarez-Reyes M, Bridger JM, Broers JL, Ramaekers FC, et al. 2001. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J. Cell Sci 114(Part 14):2577–90 [DOI] [PubMed] [Google Scholar]
- Venturini V, Pezzano F, Català Castro F, Häkkinen HM, Jiménez-Delgado S, et al. 2020. The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science 370(6514):eaba2644 [DOI] [PubMed] [Google Scholar]
- Vortmeyer-Krause M, te Lindert M, te Riet J, te Boekhorst V, Marke R, et al. 2020. Lamin B2 follows lamin A/C- mediated nuclear mechanics and cancer cell invasion efficacy. BioRxiv 028969. 10.1101/2020.04.07.028969 [DOI] [Google Scholar]
- Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. 2004. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. PNAS 101(1):396–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Dreger M, Madrazo E, Williams CJ, Samaniego R, et al. 2018. WDR5 modulates cell motility and morphology and controls nuclear changes induced by a 3D environment. PNAS 115(34):8581–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li M, Stadler S, Correll S, Li P, et al. 2009. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol 184(2):205–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen K, Litjens SHM, Kuikman I, Tshimbalanga N, Janssen H, et al. 2005. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol 171(5):799–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Ivanovska IL, Zhu K, Smith L, Irianto J, et al. 2018. Nuclear rupture at sites of high curvature compromises retention of DNA repair factors. J. Cell Biol 217(11):3796–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Pfeifer CR, Zhu K, Irianto J, Liu D, et al. 2019. Rescue of DNA damage after constricted migration reveals a mechano-regulated threshold for cell cycle. J. Cell Biol 218(8):2545–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Ragnauth C, Greener MJ, Shanahan CM, Roberts RG. 2002. The nesprins are giant actin-binding proteins, orthologous to Drosophila melanogaster muscle protein MSP-300. Genomics 80(5):473–81 [PubMed] [Google Scholar]
- Zhang X, Schwarz EM, Young DA, Puzas JE, Rosier RN, O’Keefe RJ. 2002. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J. Clin. Invest 109(11):1405–15 [DOI] [PMC free article] [PubMed] [Google Scholar]